1. INTRODUCCIÓN
El diseño (de novo) o modelado (optimización) de fármacos engloba a un conjunto de técnicas matemáticas combinatorias, físicas, químicas y farmacológicas que, mediante complejos procedimientos computacionales, permiten proponer teóricamente nuevas moléculas de potencial interés farmacológico u optimizar – mediante determinadas modificaciones estructurales – aquellas otras previamente conocidas, con el fin de potenciar su efectividad, limitar su toxicidad o mejorar otros aspectos biomédicos relevantes. Tradicionalmente, existe un doble enfoque para el diseño y modelado molecular, según se centre en el análisis de los ligandos biológicos potencialmente útiles o en la estructura molecular de fármacos previamente conocidos de los que se dispone algún análisis de relación estructura-actividad cualitativa (Structure-Activity Relationship, SAR) o cuantitativa (Quantitative Structure-Activity Relationship, QSAR); es decir, la elaboración de modelos matemáticos que relaciona cualitativa o cuantitativamente una actividad biológica o una propiedad físicoquímica, o una medida numérica cuantitativa de ella, con la estructura química.
Las técnicas y métodos computacionales utilizados en el diseño molecular farmacológico forman un amplio conjunto de técnicas que incluyen, entre otras, química cuántica, termodinámica, mecánica estadística, química computacional, fisiología y bioquímica, abarcando desde sistemas químicos pequeños a grandes moléculas biológicas. Actualmente, la inteligencia artificial (IA) se ha convertido en un elemento central el descubrimiento de nuevos fármacos, que no solo abarca el diseño de novo y la optimización molecular de nuevos presuntos fármacos, sino que también explora aspectos relativos a las interacciones fármaco-diana y a las posibles vías de síntesis química, entre otros. Por todo ello, la IA se ha convertido en una herramienta irreemplazable en la predicción de la toxicidad, la bioactividad, las propiedades farmacocinéticas y fisicoquímicas, etc. A título de ejemplo, una de las plataformas de IA más populares utilizadas en el diseño de fármacos es el SwissDrugDesign, un conjunto de aplicaciones de software desarrollado por el Instituto Suizo de Bioinformática, de acceso libre y gratuito (1). Por su parte, AlphaFold, desarrollado por DeepMind y EMBL-EBI, es considerado como el primer método computacional efectivo para predecir con extremada precisión la estructura 3D de proteínas, únicamente a partir la secuencia líneal de aminoácidos (2). En definitiva, la IA es capaz de recopilar y analizar en poco tiempo enormes cantidades de datos, permitiendo seleccionar dianas y ligandos complementarios, así diseñar pruebas adecuadas (3).
2. VARIABLES ESTRUCTURALES EN QUÍMICA FARMACOLÓGICA COMPUTACIONAL: ISÓMEROS, ISÓSTEROS E ISOTOPÓLOGOS
Sin duda, entre los procedimientos empleados en el modelado y optimización molecular de fármacos los más ampliamente utilizados son aquellos en los que se sustituyen o modifican átomos o partes de las moléculas farmacológicas de referencia, con el fin de adecuar sus propiedades a los objetivos previamente definidos. En este sentido, los isómeros y los isósteros juegan un papel capital.
La isomería es una propiedad de algunas moléculas – particularmente en la química del carbono – que pese a tener el mismo número de átomos – igual fórmula molecular global, con idénticas proporciones relativas de los átomos – presentan estructuras químicas espacialmente (3D) diferentes, lo que se traduce en propiedades físicas y químicas distintas en mayor o menor grado.
Tradicionalmente, en química orgánica y bioquímica se distinguen los isómeros estructurales (con una diferente distribución de los enlaces entre sus átomos), que pueden ser de cadena o esqueleto, de posición, de función o tautómeros (transposición de un átomo entre dos estructuras, habitualmente H, siendo común la existencia de un fácil equilibrio entre ambas formas tautómeras); por otro lado están los isómeros espaciales o estereoisómeros, cuyos átomos presentan la misma distribución (misma forma de la cadena; mismos grupos funcionales y sustituyentes situados en la misma posición), pero su disposición – orientación – en el espacio es distinta.
Dentro de la estereoisomería, se distingue la isomería configuracional, cuyos isómeros son aislables, ya que no es posible – o se requiere mucha energía – para que dos isómeros configuracionales puedan convertirse uno en otro. En esta forma de isomería se distingue la geométrica (cis-trans), compuestos que tienen sus átomos conectados en el mismo orden pero con una diferente orientación tridimensional con relación a un eje o plano de la molécula (cis o Z, con los dos sustituyentes más voluminosos del mismo lado y trans o E, con los dos sustituyentes más voluminosos en posiciones opuestas). La otra forma de estereoisomería configuracional es la isomería óptica o enantiomería, (L/D, S/R), existente cuando hay al menos un átomo de carbono (C) asimétrico o quiral (con cuatro sustituyentes diferentes), dando lugar los estereoisómeros ópticos (enantiómeros, enantiomorfos o formas quirales). El otro tipo de estereoisomería es la isomería conformacional, cuya diferencia más práctica es que la conversión de una forma en otra sí es factible – en muchos casos – en un medio químico o biológico convencional hasta el punto en que los confórmeros raramente pueden ser separados o aislados, debido a la facilidad de interconversión aún a temperaturas relativamente bajas. Hay confórmeros rotámeros (que presentan rotación libre en torno al eje del enlace formado por dos átomos de carbono) y confórmeros estructurales, generalmente referidos a anillos idénticos con diferentes formas espaciales (como las típicas conformaciones en forma de silla o de bote del ciclohexano).
La química computacional utiliza modelos informatizados para facilitar el estudio y resolver problemas químicos a través de la aplicación de técnicas y simulaciones computacionales de sistemas moleculares, siendo ampliamente utilizada desde hace varias décadas en el diseño de nuevos medicamentos y materiales. Así, por ejemplo, la importancia de los cálculos teóricos de la densidad electrónica en las moléculas farmacológicas viene determinada porque es responsable en gran medida de muchas de sus propiedades físico-químicas más relevantes biológicamente, particularmente en lo que respecta a la formación y estabilidad de los enlaces con otras estructuras (receptores biológicos, membranas, etc.), arquitectura molecular (distancias interatómicas), polaridad de los enlaces, constantes de disociación, índice de reactividad, etc. En este sentido, las características conformacionales de cada molécula biológica interactúan con la propia identidad atómica y la arquitectura molecular de cara a la distribución molecular de densidad electrónica.
El análisis conformacional es una técnica de investigación con una larga tradición en el diseño molecular. A título de ejemplo, hace más de 40 años se estableció mediante procedimientos de química cuántica computacional que en los fármacos antiinflamatorios con estructura de ácido aril alfa-metilpropiónico (ibuprofeno, naproxeno, ketoprofeno, etc.) la mayor actividad antiinflamatoria se corresponde a las posiciones conformacionales rotacionales más probables (menos energéticas) en las que el plano del grupo carboxílico (-COOH) se encuentra separado del plano del sistema aromático en un ángulo de mayor de 60º y menor de 300º, con diferencias totales entre las conformaciones más y menos energéticas que superan con los 5 o 10 kcal/mol e incluso algunas llegan a ser de más de 45 kcal/mol, valores que pueden ser considerados como auténticas “barreras rotacionales”. Además, la introducción de sustituyentes en el anillo aromático en posición orto (proximal) con respecto al grupo alfa-metilcarboxílico, incluso con volúmenes atómicos pequeños (como el del flúor), reduce drásticamente la actividad biológica. Asimismo, la incorporación de un segundo grupo metilo (-CH3) en la posición alfa respecto del grupo carboxílico altera radicalmente el comportamiento conformacional (rotacional), hasta el punto que la conformación más estable (menos energética) del derivado dimetilado se corresponde con la menos estable (más energética) del dimetilado (4,5).
Es amplísima la bibliografía relativa a las implicaciones farmacológicas – entre otros ámbitos – de la estereoisomería ya que, teóricamente, cada enantiómero podría tener un comportamiento específico en términos de farmacodinámica, farmacocinética y toxicidad. Por tanto, la separación de enatiómeros podría mejorar la bioactividad de un fármaco, así como reducir la incidencia e intensidad de los efectos adversos. En comparación con finales del siglo pasado, cuando alrededor del 55% de los fármacos utilizados clínicamente eran quirales y la mitad de ellos se utilizaban como racematos (mezclas de estereoisómeros), la tendencia actual en el desarrollo de nuevos fármacos es principalmente hacia sustancias que contienen una única forma enantiomérica (6).
Un ejemplo trágico la importancia de la enantiomería en farmacología es el de la talidomida, un sedante de amplio uso en Europa (nunca de comercializó en Estados Unidos) desde su comercialización en 1956, que en pruebas animales mostró aparentemente un margen muy amplio de seguridad. Sin embargo, pocos años después su uso por mujeres embarazadas durante el primer trimestre del embarazo fue asociado con el nacimiento de bebés con graves malformaciones congénitas, particularmente focomelia (ausencia de elementos óseos y musculares en los miembros superiores o inferiores). El involuntario – pero trágico – error consistió en fiarse de las pruebas de embriotoxicidad y teratogenicidad en ratas, que no mostraron resultados perjudiciales, y además emplear una mezcla racémica de los dos enatiómeros (R y S); hoy sabemos que la forma R es la responsable del efecto sedante mientras que la S fue la que provocó los efectos teratogénicos. Aunque, como veremos posteriormente, tampoco el empleo del enantiómero R puro hubiera evitado la tragedia.
En la mayoría de los medicamentos enantioméricos autorizados hoy día no hay una ventaja terapéutica real sobre las mezclas racémicas, ya que el isómero no farmacológico inactivo carece de relevancia biológica o cinética y, por tanto, el empleo de enatiómeros farmacológicos puros no resuelve ningún problema, más allá de reducir las unidades de dosificación a la mitad. Sin embargo, ello puede tener un importante coste, dada la dificultad inherente a la síntesis y separación de enatiómeros puros y toda la investigación clínica posterior y, en este sentido, un estudio publicado en 2019 comprobó que entre 2011 y 2017, el gasto de la Parte D de Medicare (Estados Unidos) en 12 medicamentos de enantiómeros únicos totalizó $ 19,3 mil millones de dólares que, si hubieran sido sustituidos por las correspondientes mezclas racémicas (con idéntica efectividad farmacológica y riesgo toxicológico) Medicare podría haber ahorrado 16,6 mil millones de dólares (112,43 $ por receta, o un 86% del total) y los beneficiarios de la Parte D de Medicare, que recibieron 104,3 millones de recetas para estos 12 medicamentos de enantiómeros individuales, gastaron 1,6 mil millones de su bolsillo, que si se hubieran reemplazado por los productos racémicos, podrían haber ahorrado 1,1 mil millones (11,02 $ por receta, un 69% del gasto) (7).
Los isósteros son átomos, iones, fracciones moleculares y sistemas anulares (generalmente aromáticos) que tienen muy similares propiedades electrónicas y que, en el ámbito de la biomedicina, se denominan bioisósteros cuando se relacionan con iguales o similares actividades biológicas. En este caso, el propósito de intercambiar un bioisóstero por otro es adecuar determinadas propiedades biológicas o físicas deseadas en un compuesto sin necesidad de realizar cambios notables en la estructura química, con el fin de modular la toxicidad, adecuar la biodisponibilidad o modificar la actividad del compuesto original, así como alterar el metabolismo de éste.
Finalmente, los isotopólogos son moléculas que difieren sólo en su composición isotópica; es decir, un isotopólogo de una especie química que contiene un isótopo diferente de al menos uno de los átomos de la molécula de referencia. El ejemplo más obvio es la sustitución de átomos de hidrógeno convencional o protio (1H) por deuterio (D o 2H), que es un isótopo estable (no radiactivo); pero también se han desarrollado radioisotopólogos que sustituyen a sus isótopos naturales en moléculas biológicas y que emiten determinada radiación y partículas al desintegrarse, que resultan útiles en pruebas de diagnóstico por imagen, como en la PET (tomografía por emisión de positrones), para marcar determinados fármacos o trazadores biológicos: 11C, 13N, 15O y 18F.
3. ISOTOPOLOGÍA HIDRÓGENO-DEUTERIO: ASPECTOS GENERALES
El hidrógeno tiene tres isótopos que existen de forma natural: protio o hidrógeno convencional (1H, que tiene un protón y un electrón, pero carece de neutrones), con una abundancia natural del 99,9844%), deuterio (2H o D, que lleva un neutrón añadido en su núcleo, con una abundancia del 0,0156%) y tritio (3H o T, que lleva dos neutrones añadidos, presente en la naturaleza solo en mínimas trazas). Este último, el tritio, es el único isótopo radiactivo del hidrógeno, desintegrándose mediante la emisión de un electrón – desintegración beta – para transformarse en 3He1+; sin embargo, tiene una vida media relativamente larga, con un periodo de semidesintegración de 4500 ± 8 días (12,3 años). Tanto el hidrógeno convencional (H, protio) como el deuterio (D) son estables – no son radiactivos – por lo que su presencia en una molécula no comporta riesgo biológico alguno.
La sustitución de H por D es uno de los ejemplos más conservadores de isotopología, dado que los dos isótopos son muy similares en términos de propiedades fisicoquímicas, aunque el deuterio tiene un volumen algo menor y los enlaces con el carbono (C–D) son ligeramente más cortos que los enlaces C–H. Sin embargo, dado que la masa atómica del D es doble que la del H, el enlace C–D está asociado con una menor frecuencia de estiramiento vibracional en comparación con el enlace C–H, así como una energía de estado fundamental menor y, a su vez, una mayor energía de activación para la escisión; en resumen: el enlace carbono-deuterio (C-D) es más estable el del carbono-hidrógeno (C-H), con una diferencia de 1,2–1,5 kcal/mol, y su escisión se produce de forma más lenta. Esta diferencia de reactividad se cuantifica mediante el denominado efecto isotópico cinético, es decir, la variación en la velocidad de una reacción química cuando en uno de los reactivos, un átomo es sustituido por uno de sus isótopos.
Todo lo anterior justifica que la sustitución isotopológica de D por H podría modificar significativamente la velocidad de reacción, cuando la sustitución se produzca en un enlace C-H implicado en algún aspecto relevante de la interacción con receptores biológicos o con la degradación química pura o mediada por enzimas metabolizadoras. Esta sustitución isotopológica es relevante cuando el isótopo sustituyente tiene un impacto significativo sobre la masa, como ocurre con la sustitución de un átomo de hidrógeno por deuterio, ya que representa un 100% de incremento en masa, mientras que en la sustitución de 12C con 13C, la masa aumenta sólo un 8%; así, la velocidad de una reacción que implique un enlace C-H es 6 a 10 veces mayor que la correspondiente a un enlace C-D, mientras que una reacción de 12C es apenas 1,04 veces más rápida que la equivalente con 13C.
En el caso del deuterio el efecto isotópico cinético (DKIE, deuterium kinetic isotope effect), se expresa como una relación de las constantes de velocidad (kH/kD) y cuanto mayor sea el valor de DKIE, más lenta será la velocidad de ruptura del enlace C–D en comparación con la de C–H. Los DKIE pueden alcanzar valores significativos en algunos casos y podrían ralentizar la tasa de escisión de enlaces, con un profundo impacto en los procesos químicos – puros o catalizados por enzimas – y, por lo tanto, pueden emplearse para optimizar los perfiles de los fármacos en diferentes maneras. Sin embargo, en la mayoría de los casos, es muy difícil o casi imposible sintetizar un compuestos 100% isotópicamente puros, lo cual plantea importantes cuestiones sintéticas, analíticas e incluso regulatorias (8,9).
En resumen, en determinados casos la isotopología hidrógeno-deuterio puede ser relevante en términos físico-químicos y, por ende, afectar al comportamiento farmacológico de los medicamentos deuterados.
4. OBJETIVOS FARMACOLÓGICOS DE LA ISOTOPOLOGÍA HIDRÓGENO-DEUTERIO
Cambiar unos algunos átomos de hidrógeno por deuterio podría ser una forma de ralentizar la transformación de un fármaco en el organismo; es decir, prolongar su presencia y, en consecuencia, aumentar la duración de su efecto farmacológico (aunque también el toxicológico, no hay que olvidarlo). La idea no es nueva, ya que hace más de sesenta años que se registraron patentes de fármacos deuterados, si bien no se les pudo atribuir ninguna ventaja terapéutica, más allá de sentar registros de patente potencialmente útiles en el futuro y, ciertamente, esto es lo que ocurrió al verse como una forma de soslayar la patentabilidad de algunos fármacos. Sin embargo, cuando la investigación clínica orilló en costes a la química y a la preclínica, la motivación económica pasó – aparentemente – a un segundo plano y el potencial valor farmacológico de la deuteración volvió a cobrar un protagonismo creciente hasta llegar a la actualidad.
Dado que la deuteración (sustitución de uno o más átomos de H por D) puede afectar a la farmacocinética de un buen número de fármacos que se metabolizan por vías que implican la rotura del enlace C-H (C-D, en su caso), podría mejorar algunas limitaciones de fármacos no deuterados (originales), reduciendo el metabolismo en sitios sensibles de la molécula del fármaco, lo que se conoce como “puntos blandos” o “puntos calientes”, optimizando las propiedades farmacocinéticas del compuesto, que podría desembocar en una menor o menos frecuente dosificación y, lo que es aún más importante, limitar o disminuir la formación de metabolitos no selectivos del fármaco y, con ello, mejorar su selectividad o su perfil toxicológico; todo ello también podría redundar en facilitar ensayos clínicos más rápidos, selectivos y económicos con la versión deuterada (10).
Un aspecto farmacocinético en el que las versiones deuteradas de fármacos terapéuticamente importantes puede ser particularmente relevante es de la metabolización a través de isoenzimas del citocromo P450 (CYP450), dado que la activación metabólica de fármacos a electrófilos químicamente reactivos por estas enzimas puede ser un evento iniciador molecular clave que sustenta la lesión hepática inducida por fármacos (hepatopatía iatrogénica). Además, estos intermediarios lábiles pueden ser secuestrados dentro del sitio activo del CYPP450 y generar una forma única de inhibición irreversible que tiene profundas implicaciones clínicas a nivel de interacciones fármaco-fármaco, autoinhibición de la eliminación hepática o farmacocinética dependiente del tiempo y/o no lineal, entre otras. Desafortunadamente, el metabolismo oxidativo puede estar asociado con la formación de intermediarios y/o metabolitos inestables, reactivos, no selectivos y tóxicos; aparición de variabilidad entre pacientes debido a polimorfismos genéticos; y saturación, inducción o inhibición de determinadas isoenzimas del CYP450, lo que lleva a interacciones fármaco-fármaco potencialmente dañinas. Por ello, la deuteración de una molécula farmacológica podría reducir o evitar la bioactivación en su “punto caliente”, desviando el metabolismo de estas vías aberrantes y reducir la formación de metabolitos reactivos (11).
No debe minusvalorarse la importancia de la potencial utilidad de la deuteración de fármacos para soslayar los posibles efectos deletéreos del metabolismo mediado por enzimas del sistema citocromo P450, ya que se estima que más de la mitad de los fármacos comercializados son metabolizados por enzimas de la familia del citocromo P450 (12). Generalmente, la O-desalquilación es la reacción más sensible a la sustitución de hidrógeno por deuterio, seguida de la N-desalquilación de amida y la oxidación de grupos alquilo, mientras que la N-desalquilación de aminas es la menos sensible, y la hidroxilación de anillos aromáticos prácticamente no se ve influenciada.
Así pues, la sustitución de H por D, es una estrategia particularmente perseguida por los desarrolladores de fármacos para atenuar el metabolismo mediado por CYP y soslayar sus desventajas inherentes. El efecto principal del cambio de H a D es la mejora del rendimiento farmacocinético, principalmente a nivel de la semivida de eliminación (t½), área bajo la curva concentración/tiempo (AUC) y concentración plasmática máxima (Cmax).
Sin embargo, debe tenerse en cuenta que la deuteración en un sitio de la molécula podría desviar el compuesto a otras rutas de biotransformación poco conocidas o imprevisibles y aumentar el metabolismo en otros sitios, lo que se conoce como derivación metabólica. Así, por ejemplo, una disminución en la tasa de formación de metabolitos después de la deuteración de puntos blandos oxidables de una molécula podría aumentar la tasa de oxidación en uno o más puntos blandos alternativos no deuterados, todo lo cual exige la realización de detallados estudios cinéticos con los fármacos deuterados.
El deuterio no solo puede afectar los procesos metabólicos mediados enzimáticamente, sino que también puede influir en los eventos químicos puros que implican un paso de escisión C–H (C–D), como la enantiomerización, un fenómeno importante en el contexto de los cambios quirales dado que la incorporación de D en el átomo de carbono quiral podría modificar la tasa de abstracción de átomos de hidrógeno y estabilizar los estereoisómeros interconvertidos. Si el estereocentro está adyacente a un grupo carbonilo enolizable, el enlace quiral carbono-hidrógeno puede romperse para formar un intermedio enólico y facilitar una rápida interconversión que impide el aislamiento de los estereoisómeros correspondientes. La abstracción o transferencia de hidrógeno implica un proceso por el cual se extrae un radical libre de hidrógeno de un sustrato, tal como ocurre – entre otras localizaciones biológicas – en el citocromo P450; el agente abstractor suele ser una especie radical en sí misma y el proceso puede tener lugar a través de la transferencia de electrones acoplados a protones.
Este procedimiento se ha venido utilizando como un medio para estabilizar, aislar, caracterizar y también administrar el estereoisómero preferido, mejorando así la utilidad terapéutica de los fármacos configuracionalmente inestables. Este es el caso de la talidomida, en el que la rápida interconversión no permite el uso seguro del enantiómero (R) no teratogénico. Por ello, la deuteración de la talidomida reduciría la enolización y estabilizaría la configuración del estereocentro en la configuración R, no teratógena.
Atendiendo a todo ello, la capacidad de aumentar la resistencia de una molécula a la escisión de alguno de sus enlaces sin alterar significativamente su impedimento estérico o sus propiedades electrónicas, es la mayor ventaja de la incorporación de deuterio, lo que claramente hace que este enfoque se destaque de otros bloqueadores metabólicos, como los haluros, especialmente el flúor (F). Además, en algunos casos, el deuterio podría representar un bioisóstero más seguro en comparación con el flúor, que aumenta la inestabilidad y puede producir metabolitos que son peligrosos tanto para la salud humana como para el medio ambiente (13,14).
Entre los agrupamientos químicos más universalmente presentes en las moléculas de interés biológico el grupo metilo (-CH3) es quizá el más común y, por tanto, su relevancia electrónica y estérica se hace evidente para la actividad farmacológica, toxicológica y cinética de los medicamentos. De hecho, alrededor del 80% de los fármacos más empleado contienen un grupo metilo, por lo que infravalorar la importancia de la metilación selectiva del sitio en la química médica. Por ello, en el campo de la química farmacéutica, la incorporación precisa de un grupo trideuterometilo (-CD3) está ganando cada vez más atención, lo ha venido en llamarse el “metilo mágico” deuterado. Por otro lado, la metilación es una de las transformaciones químicas más frecuentes e importantes en biología. Baste recordar que un solo metilo distingue la timina (ADN) del uracilo (ARN), mientras que cinco aminoácidos – de los 20 esenciales – solo difieren en el número y la posición de grupos metilo (15).
Aunque hasta ahora se consideraba que la sustitución de H a D influye poco en la potencia de interacción bioquímica o en el perfil de selectividad de una molécula, dado que las propiedades electrónicas, la forma, el tamaño y la flexibilidad estérica del análogo deuterado son muy similares a las de su contraparte no deuterada. Sin embargo, algunos informes recientes han cuestionado esto, argumentando que el D en realidad podría ser capaz de modular las interacciones fármaco-diana. Por ejemplo, un estudio reciente que muestra que los humanos son capaces de distinguir el sabor del agua pesada (D2O), más dulce que el del del agua normal (H2O), lo que implica una diferente reactividad en determinadas papilas gustativas (17).
Asimismo, el deuterio podría influir en la unión a los receptores de células B, modulando así el reconocimiento de células B. En este sentido, se ha comparado recientemente en experimentación anaimal (ratones) la respuesta inmune a las vacunas contra la heroína (contra el abuso de opioides, destinadas a bloquear los efectos de la heroína y el fentanilo en pacientes con trastorno por abuso de estas sustancias) que albergan un hapteno no deuterado con la de su análogo deuterado (d6), observándose que los animales vacunados con la versión deuterada títulos de anticuerpos más altos que los observados en los grupos tratados con la forma no deuterada (18).
Todo esto sugiere que la sustitución de algunos átomos de H por D no solo tiene un potencial modulador farmacocinético sino también farmacodinámico, lo que puede ser una herramienta para modular y optimizar las propiedades de algunos fármacos al inducir efectos estereoelectrónicos.
5. DEUTERIO EN FÁRMACOS ANTICANCEROSOS
Tras el desarrollo hace más de un cuarto de siglo del imatinib, el primer inhibidor de la tirosina cinasa (TKI) BCR-ABL para el tratamiento de la leucemia mielocítica, se crearon grandes expectativas terapéuticas la quimioterapia del cáncer; sin embargo, pronto se comprobó que muchas células tumorales son capaces de desarrollar mecanismos de resistencia también en esta línea farmacológica, observándose la aparición de mutaciones puntuales mutación puntual dentro del dominio de la cinasa ABL1 de BCR-ABL1, que reducía drásticamente la efectividad antineoplásica de esta línea de quimioterapia.
Como es obvio, rápidamente se desarrollaron modificaciones moleculares para hacer frente a esta ésta y a otras mutaciones que posteriormente han ido apareciendo a las diferentes líneas específicas de TKI. Una de las líneas de investigación en la búsqueda de mejores agentes anticancerosos ha sido la deuteración de algunos de TKI, con el objetivo de modificar las propiedades fisicoquímicas de la molécula y retrasar el desarrollo de resistencia tumoral (19).
El primer fármaco antitumoral deuterado oficialmente autorizado es el donafenib, Desarrollado por Suzhou Zelgen, el donafenib fue aprobado por la Administración Nacional de Productos Médicos de China en 2021 para el tratamiento de primera línea de pacientes con carcinoma hepatocelular irresecable que no habían recibido previamente tratamiento sistémico y se está evaluando, solo o en combinación, para el tratamiento de otros cánceres (20).
El donafenib es el resultado de la sustitución de deuterio en el sorafenib, un inhibidor de múltiples cinasas que está autorizado en la Unión Europea desde 2006 para el tratamiento del carcinoma hepatocelular, el cáncer de células renales y el cáncer de tiroides. En particular, el donafenib proporciona mejores propiedades farmacocinéticas, mayor eficacia y efectos adversos menos frecuentes en comparación directa con sorafenib en estudios clínicos. El donafenib (d3 sorafenib) incorpora tres átomos de D en la amida secundaria del anillo de piridina del sorafenib (figura 1). Curiosamente, no se puede encontrar en la literatura una justificación clara para la deuteración del sorafenib, probablemente debido al hecho de que el principal metabolito circulante del sorafenib es el igualmente potente N-óxido de la piridina (21).
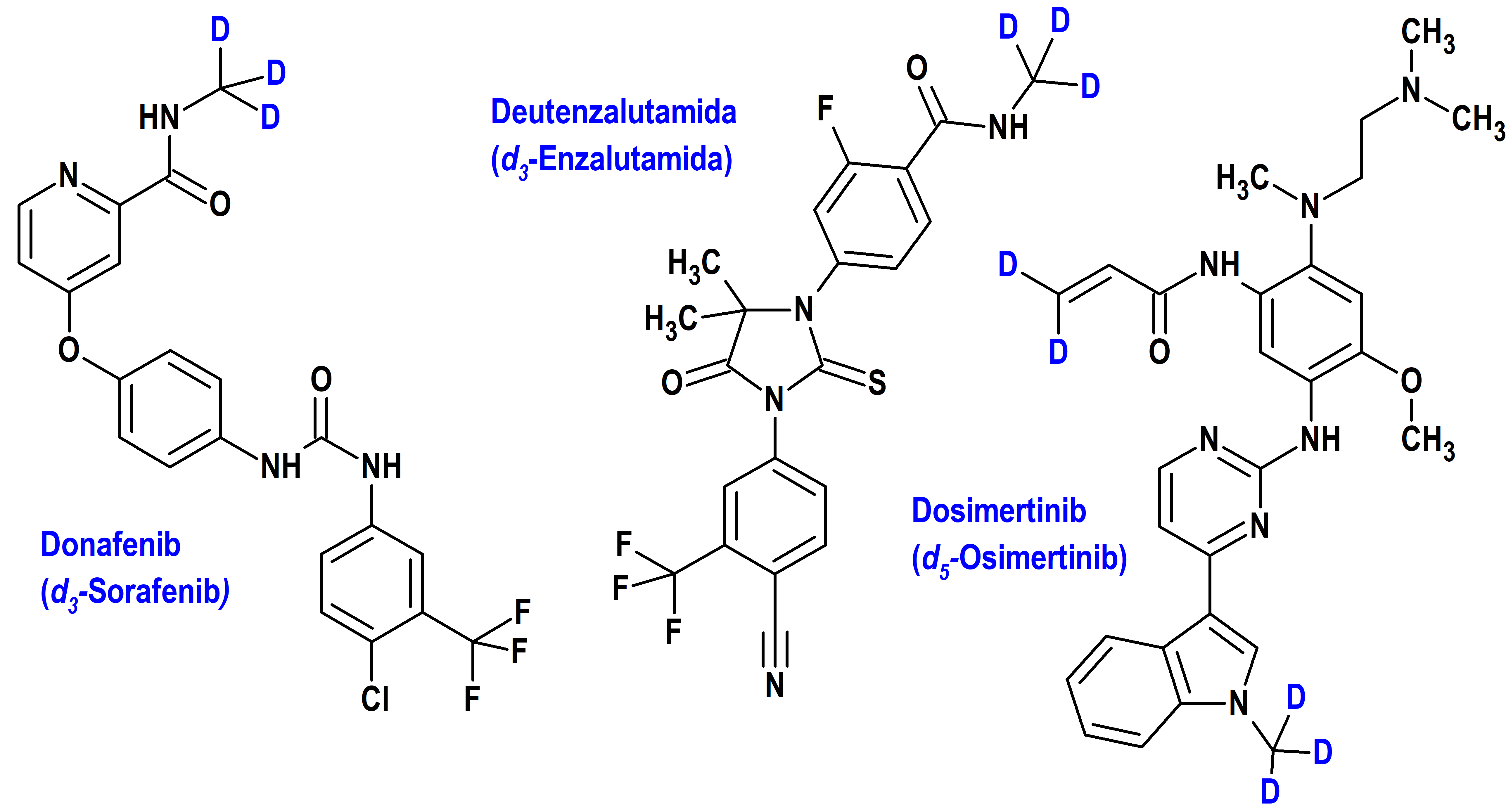
Figura 1.
Otro buen número de fármacos anticancerosos deuterados está siendo objeto de ensayos preclínicos y clínicos, entre los que cabe destacar la deutenzalutamida (HC‐1119), un derivado deuterado de la enzalutamida, capaz de unirse a los receptores de andrógenos (AR) expresados en las células del cáncer de próstata e inhibe su actividad, lo que altera la translocación nuclear y la unión al ADN, lo que da lugar a la apoptosis de las células del cáncer de próstata, reduciendo el crecimiento de las células del cáncer de próstata. El reemplazo del hidrógeno por deuterio en la fracción N-CH3 de la amida, disminuye el metabolismo de la enzalutamida y permite un mejor perfil farmacocinético, mejorando así su eficacia antitumoral en comparación con la enzalutamida no deuterada. Como la forma deuterada no puede atravesar la barrera hematoencefálica, la forma deuterada también reduce los efectos secundarios neurológicos no deseados de la enzalutamida, mejorando así su perfil de seguridad (35).
El dosimertinib es un derivado pentadeuterado (d5) del osimertinib (autorizado en la Unión Europea en 2016); como su antecesor, el dosimertinib está siendo estudiado (fase 1) en cáncer de pulmón de células no pequeñas con mutación EGFR positiva. Otros isotopólogos deuterados de TKI desarrollados y en fase de investigación preclínica son Hc-1144 (tivozanib), para el carcinoma de células renales metastásico y el BRP800 (dasatinib) para la leucemia mieloide crónica y cáncer de pulmón de células no pequeñas. Asimismo, también están en fase preclínica los derivados deuterados Mbri-001 (plinabulina) en cáncer de pulmón, CTP-221 (S-lenalidomida) en síndromes mielodisplásicos, mieloma múltiple, linfoma de células del manto, linfoma folicular y linfoma de la zona marginal y el tamoxifeno deuterado, en cáncer de mama con receptor de estrógeno positivo (ER+) (16).
A pesar de sus temibles efectos teratogénicos, desde hace tres décadas la talidomida está siendo objeto de un creciente interés por su potencial terapéutico en multitud de patologías graves. Es inhibidor del factor de necrosis tumoral alfa (TNF), produce una degradación del ARN mensajero, lo que conlleva que este fármaco tenga efectos antiinflamatorios e inmunomoduladores; además, es inhibidor de la angiogénesis. Disminuye la fagocitosis monocitaria y estimula las células T, influyendo sobre la quimiotaxis leucocitaria. En las décadas de los 80 y 90 del siglo pasado se empezó a utilizar la talidomida en algunos tipos de cánceres por sus propiedades antiangiogénicas, particularmente en mieloma múltiple, aunque son numerosos los ensayos clínicos en los que ha demostrado eficacia terapéutica en enfermedad de Crohn, síndrome de Behçet, psoriasis, artritis reumatoide, enfermedad de injerto contra huésped y en determinadas patologías asociadas al SIDA, carcinoma hepatocelular, epilepsia refractaria e incluso lepra; sin embargo, en la actualidad la única indicación autorizada en la Unión Europea es el mieloma múltiple. Dado que los estereosómeros (R y S) de la talidomida pueden experimentar una rápida interconversión, ello no permite el uso seguro del enantiómero R puro, no teratogénico. Por este motivo, se ha especulado que la talidomida deuterada podría experimentar un menor grado de enolización y ello estabilizaría la configuración del estereocentro en la configuración R, no teratógena.
6. DEUTERIO EN FARMACOLOGÍA DEL SISTEMA NERVIOSO
La deutetrabenazina (Austedo®) ha sido el primero medicamento deuterado autorizado oficialmente. En concreto, fue aprobado por la FDA de Estados Unidos el 3 de abril de 2017, aunque posteriormente fue reformulada en forma de liberación oral prolongada (Austedo XR®) el 17 de febrero de 2023. La indicación oficialmente autorizada fue el tratamiento en adultos de la corea asociada con la enfermedad de Huntington y de la discinesia tardía (33). Se trata del isotopólogo deuterado de la tetrabenazina (Xenazine®), autorizada por la FDA el 15 de agosto de 2008 para esa misma indicación.
La enfermedad de Huntington es un trastorno neurodegenerativo progresivo y hereditario caracterizado por disfunción motora, deterioro cognitivo y trastornos neuropsiquiátricos; su síntoma más típico y limitante es la corea, un movimiento involuntario y repentino que puede afectar a cualquier músculo y afectar aleatoriamente a cualquier parte del cuerpo. Los síntomas psicomotores, como la corea, están relacionados con la neurotransmisión dopaminérgica hiperactiva.
La deutetrabenazina – como su antecesor, la tetrabenazina – provoca una eliminación reversible de monoaminas (dopamina, serotonina, norepinefrina e histamina) de las terminales nerviosas. Los principales metabolitos circulantes (alfa y beta-dihidrotetrabenazina) de la deutetrabenazina son inhibidores reversibles de VMAT2, el transportador de la monoamina vesicular de tipo 2, responsable de la captación de dopamina en vesículas presinápticas en neuronas monoaminérgicas y la liberación exocitótica, lo que provoca una disminución de la captación de monoaminas en las vesículas sinápticas y el agotamiento de las reservas de monoaminas. Se trata de un fármaco solo útil para aliviar los síntomas motores de la enfermedad de Huntington y no parece afectar a la evolución o progresión de la enfermedad.
La presencia de deuterio en la deutetrabenazina aumenta la semivida del metabolito activo y prolonga su actividad farmacológica al ralentizar el metabolismo a través de la isoenzima CYP2D6 del citocromo P450, lo que permite una dosificación menos frecuente y una dosis diaria más baja con una mejor tolerabilidad, a la que parece colaborar la menor fluctuación de los niveles plasmáticos de la deutetrabenazina debido al metabolismo atenuado. La deutetrabenazina es una mezcla racémica que contiene RR-Deutetrabenazina y SS-Deutetrabenazina (figura 2).
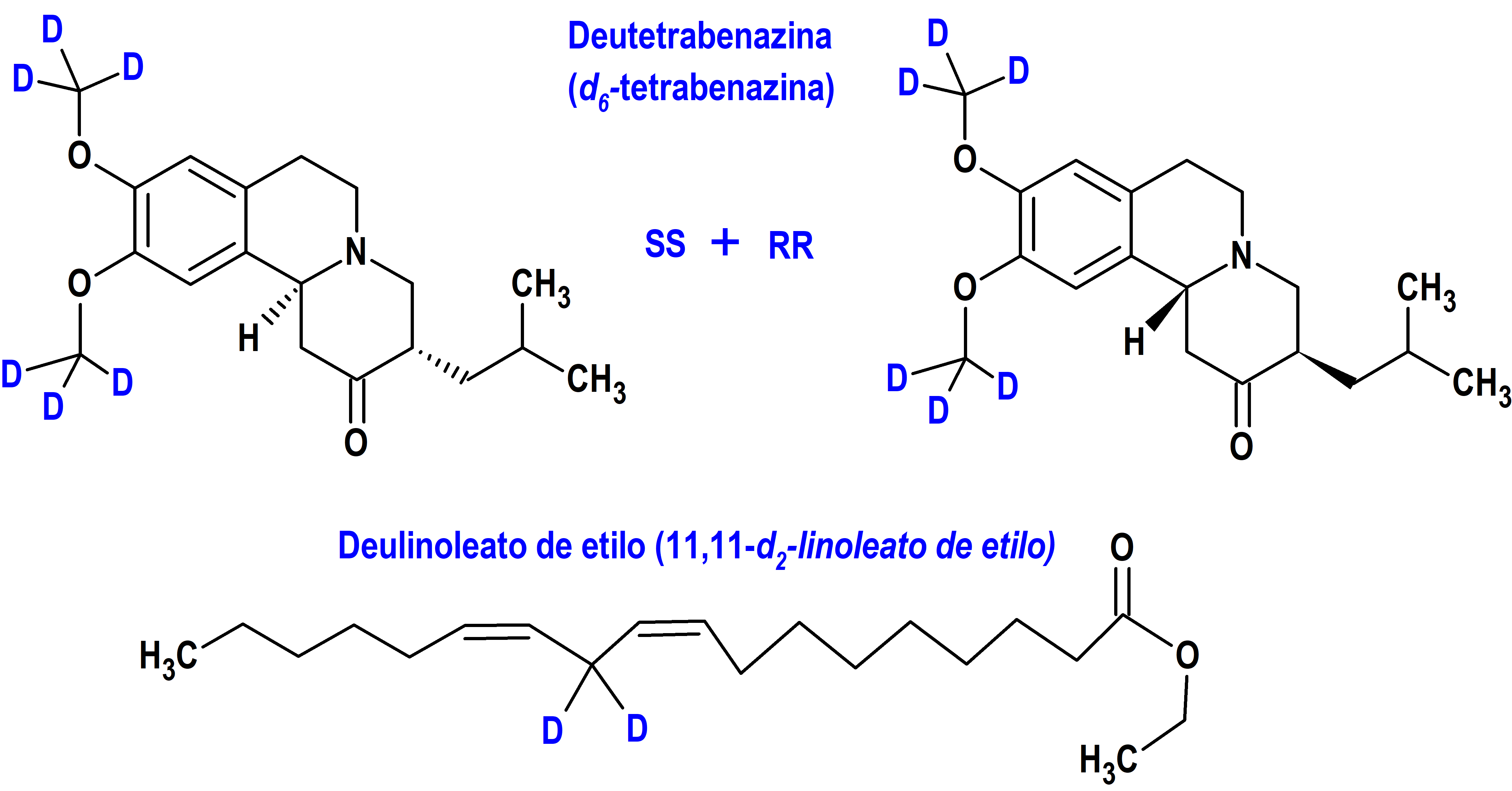
Figura 2.
La autorización oficial de la deutetrabenazina se fundamenta en los resultados obtenidos en un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo, en 90 pacientes ambulatorios con corea manifiesta asociada con la enfermedad de Huntington. La duración del tratamiento fue de 12 semanas, incluido un período de titulación de dosis de 8 semanas y un período de mantenimiento de 4 semanas, seguido de un lavado de 1 semana. El criterio principal de valoración de la eficacia fue la puntuación máxima total de corea, un elemento de la Escala Unificada de Calificación de la Enfermedad de Huntington (Unified Huntington’s Disease Rating Scale, UHDRS), en la que la corea se califica de 0 a 4 para 7 partes diferentes del cuerpo con una puntuación total que va de 0 a 28. Las puntuaciones máximas totales de corea para los pacientes que recibieron deutetrabenazina mejoraron en 4,4 unidades vs. 1,9 unidades en el grupo de placebo; en la visita de seguimiento de la semana 13 (1 semana después de la interrupción del medicamento del estudio), las puntuaciones máximas totales de corea de los pacientes que habían recibido deutetrabenazina volvieron a las iniciales de partida. Por su parte, la eficacia en el tratamiento de la discinesia tardía se estableció en dos ensayos multicéntricos, aleatorizados, doble ciego, controlados con placebo y de 12 semanas de duración realizados en 335 pacientes adultos ambulatorios con discinesia tardía provocada por el uso de antagonistas de los receptores de dopamina, siendo la principal variable de eficacia la variación de la puntuación en la escala de movimiento involuntario anormal (Abnormal Involuntary Movement Scale, AIMS; cuya puntuación total, suma de los ítems 1 a 7, va de 0 a 28, y una disminución en la puntuación indica una mejora). En el Estudio 1 se utilizaron dosis fijas controlado con placebo (12, 24 o 36 mg/día, o placebo), de 12 semanas. En el Estudio 2, un ensayo de dosis flexible, controlado con placebo, de 12 semanas. En ambos estudios la diferencia (estadísticamente significativa) entre los grupos con deutetrabenazina y placebo fueron de 1,4-1,8 puntos (33).
El deuterio se ha incorporado también a agentes nutricionales para ralentizar procesos como la dimerización o la peroxidación lipídica, y proporcionar así nuevas actividades farmacológicas. En este sentido, el ácido linoleico, uno de los ácidos grasos poliinsaturados (PUFA) dietéticos más abundantes, es altamente susceptible a la oxidación impulsada por especies reactivas de oxígeno (ROS) debido a su posición bis-alílica lábil, donde la abstracción de hidrógeno produce un radical que puede desencadenar una reacción en cadena de oxidación de PUFA autopropagante e iniciar la peroxidación lipídica. La deuteración en esta posición blanda condujo a la generación del deulinoleato de etilo (RT001), un producto más resistente a la oxidación, que se está ensayando en diversas patologías neurodegenerativas donde la peroxidación lipídica puede tener un papel patológico crucial. Sobre la base de datos satisfactorios de farmacocinética y seguridad en humanos, RT001 está siendo estudiado ya en ensayos clínicos para el tratamiento de la ataxia de Friedrich (27), así como en la distrofia neuroaxonal infantil, la esclerosis lateral amiotrófica (ELA) y la parálisis supranuclear progresiva (16).
En definitiva, el reemplazo selectivo de hidrógeno por deuterio en lípidos esenciales está creando una nueva clase de fármacos que previenen el daño oxidativo celular y vascular que causa diversas patologías, no solo la neurodegeneración, sino también la aterosclerosis y la degeneración macular (28).
Merece la pena citar algunos otros medicamentos deuterados que están siendo testados en ensayos clínicos de fase 2 o incluso 3, como el AVP-786 (una combinación de d6-dextrometorfano y quinidina) en la enfermedad de Alzheimer y en otras patologías neuropsiquiátricas como la esquizofrenia, el síndrome de desinhibición y la depresión (36); el CYB003, un análogo deuterado de la psilocibina, es un psicodélico serotoninérgico que se encuentra en investigación clínica (fase 3) para el tratamiento del trastorno depresivo mayor, el alcoholismo y otros trastornos psiquiátricos (37); la deudomperidona (CIN-102 ; d4-domperidona) en gastroparesia diabética, actúa como un antagonista selectivo de los receptores de dopamina D2 y D3 y selectividad periférica, con una eficacia, tolerabilidad y farmacocinética mejoradas en comparación con la domperidona (38). Finalmente, la deutarserina (d-D-serina, CTP-692) estudiada en esquizofrenia, aunque los ensayos clínicos han sido abandonados por no mostrar una mejora estadísticamente significativa con respecto al placebo.
7. DEUTERIO EN PATOLOGÍAS AUTOINMUNES
En este apartado destacan dos fármacos deuterados que ya han recibido la autorización de comercialización en Estados Unidos, deurixolitib y deucravacitinib, este último autorizado recientemente también en la Unión Europea.
El deuruxolitinib (Leqselvi®) fue autorizado en Estados Unidos (FDA) el 25 de julio de 2024 para el tratamiento de pacientes adultos con alopecia areata severa. Se trata de un isotopólogo del ruxolitinib en el que los átomos de hidrógeno (H) del anillo ciclopentánico han sido sustituidos por deuterio (D) (figura 3). Se trata de un inhibidor de la cinasa Janus (JAK). Las JAK median la señalización de una serie de citocinas y factores de crecimiento que son importantes para la hematopoyesis y la función inmunitaria. La señalización de las JAK implica el reclutamiento de STAT (transductores de señales y activadores de la transcripción) a los receptores de citocinas, la activación y posterior localización de los STAT en el núcleo, lo que conduce a la modulación de la expresión génica. Las vías de señalización mediadas por la familia JAK están implicadas en la patogenia de la alopecia areata aunque por el momento no se conoce la relevancia de la inhibición de las enzimas JAK para la eficacia terapéutica (31).

Figura 3.
Su autorización por la FDA está soportado por dos ensayos clínicos de fase 3, multicéntricos, aleatorizados, doble ciego y controlados con placebo, en un total de 1209 sujetos adultos con alopecia areata que presentaban al menos un 50 % de pérdida de cabello en el cuero cabelludo. El criterio de valoración principal de ambos ensayos evaluó la proporción de sujetos que alcanzaron al menos un 80 % de cobertura capilar en el cuero cabelludo (puntuación SALT ≤20) en la semana 24 (29 vs 1% en el primer estudio y 32 vs. 1% en el segundo).
Por su parte el deucravacitinib (Sotyktu®) ha sido autorizado para el tratamiento de adultos con psoriasis en placas de moderada a grave que son candidatos para terapia sistémica o fototerapia (34), tanto por la FDA de Estados Unidos (9 de septiembre de 2022) como por la EMA de la Unión Europea (24 de marzo de 2023).
Se trata del primer fármaco aprobado que incorpora deuterio sin tener una contraparte no deuterada en el mercado (figura 4). En este caso, la incorporación de deuterio evita la formación de un metabolito no selectivo y preserva la exquisita especificidad del fármaco original para TYK2 frente a las otras enzimas que pertenecen a la familia de las cinasas Janus (JAK) (24).
Deucravacitinib se une al dominio regulador de TYK2, estabilizando una interacción inhibidora entre los dominios regulador y catalítico de la enzima, dando como resultado la inhibición alostérica de la activación mediada por el receptor de TYK2 y de la activación de los transductores de señal y activadores de la transcripción (STAT). Las cinasas JAK, incluida la TYK2, funcionan como pares de homodímeros o heterodímeros en las vías JAK-STAT, donde TYK2 se empareja con JAK1 para mediar en múltiples vías de citocinas y con JAK2 para transmitir señales. El deucravacitinib reduce la expresión génica asociada a la psoriasis en la piel psoriásica de manera dependiente de la dosis, incluidas reducciones en los genes regulados por la vía de la IL-23 y la vía del IFN tipo I.
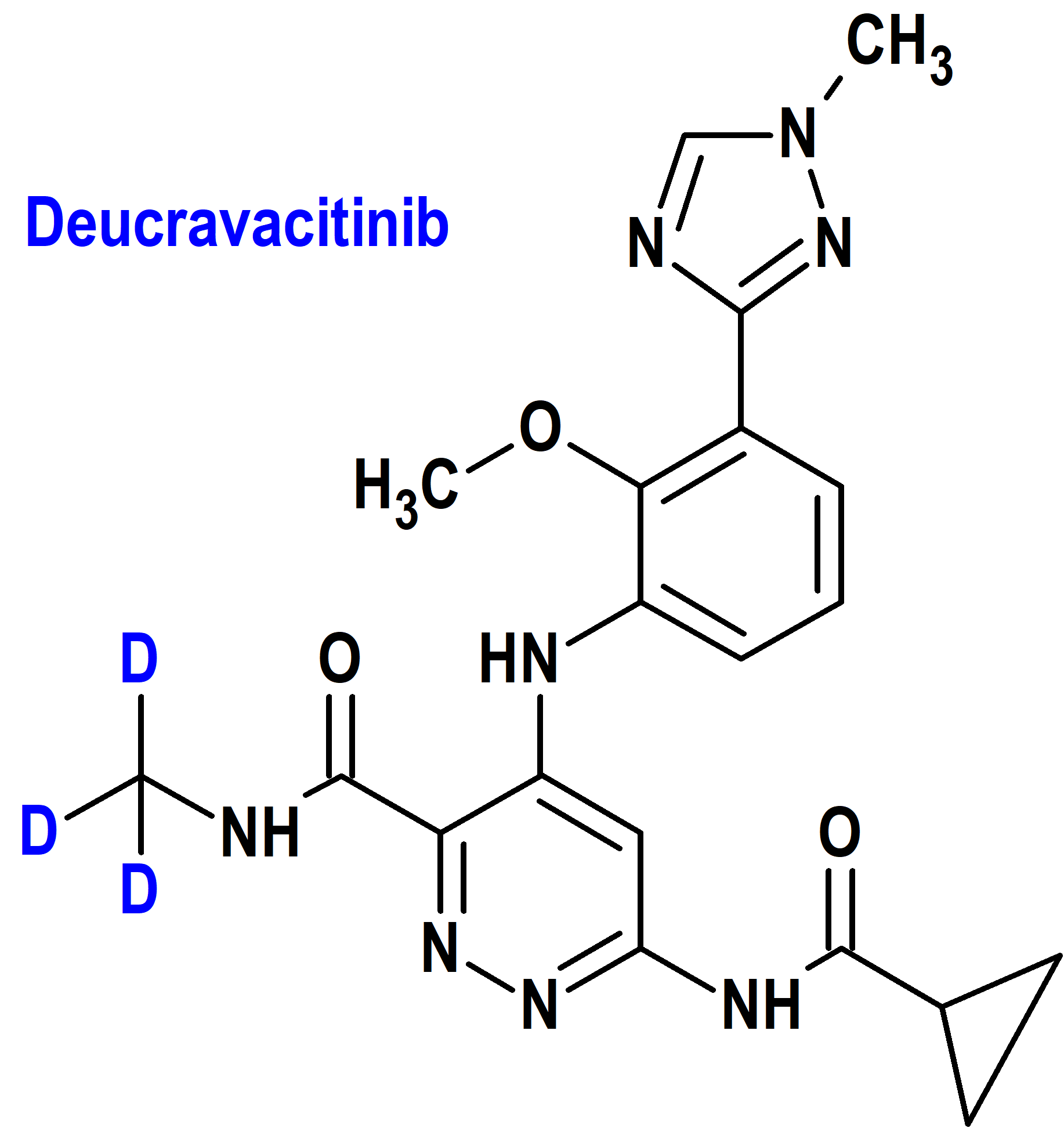
Figura 4.
Su autorización se fundamenta en los resultados de dos estudios de fase 3 de 52 semanas de duración, multinacionales, multicéntricos, aleatorizados, doble ciego, controlados con placebo y con un comparador activo (apremilast), incluyendo a un total de 1.684 pacientes con psoriasis en placas de moderada a grave, candidatos para fototerapia o terapia sistémica, con una afectación del área de superficie corporal ≥10%, una puntuación del índice de gravedad y área de psoriasis (PASI) ≥12 y una evaluación global del médico (sPGA) estática ≥3 (moderada o grave). Las variables principales de eficacia fueron el porcentaje de pacientes que alcanzaron el Índice de gravedad y área de psoriasis de 75 (PASI 75; reducción de al menos un 75% en la puntuación PASI basal) y el porcentaje de pacientes que alcanzaron una puntuación estática de 0 o 1 en la Evaluación global del médico (sPGA 0/1; claro o casi claro) en la semana 16 frente a placebo: (PASI 75) 53-58% (deucravacitinib) vs. 35-40% (apremilast) vs. 9-13% (placebo); (sPGA 0/1) 50-54% (deucravacitinib) vs. 32-34% (apremilast) vs. 7-9% (placebo). Los criterios de valoración secundarios clave incluyeron el porcentaje de pacientes que lograron PASI 75, PASI 90 y sPGA 0/1 en comparación con apremilast en semana 24: (PASI 75) 58-69% (deucravacitinib) vs. 38% (apremilast); (PASI 90) 32-42% (deucravacitinib) vs. 20-22% (apremilast); (sPGA 0/1) 49-59% (deucravacitinib) vs. 30-31% (apremilast) (34).
Como se ha comentado en epígrafes anteriores, se ha demostrado que la deuteración estabiliza los enantiómeros y epímeros de algunos fármacos. Los metabolitos del éster etílico del ácido araquidónico son mediadores importantes en muchos procesos fisiológicos y patofisiológicos; de hecho el éster etílico del ácido araquidónico es un componente principal de las bicapas lipídicas y el sustrato clave para las cascadas de eicosanoides. Este producto se hidroliza inicialmente a la forma ácida por la enzima fosfolipasa A2, antes de la oxidación por las enzimas del citocromo P450, y los productos metabólicos inducen respuestas inflamatorias en casi todos los tejidos, incluidos los pulmones. Por lo tanto, una estrategia para interferir con el metabolismo es la deuteración en el punto de oxidación y de hecho se ha demostrado que la deuteración en las posiciones bisalílicas disminuye sustancialmente la tasa general de oxidación cuando la abstracción de hidrógeno es un evento iniciador (figura 5). De hecho, muchos de los beneficios y toxicidades tanto de los glucocorticoides como de los fármacos antiinflamatorios no esteroides se deben al bloqueo de la producción de metabolitos beneficiosos y perjudiciales del éster etílico del ácido araquidónico. La alteración del metabolismo a través de la deuteración en puntos específicos de la estructura química podría tener una amplia variedad de efectos (26).
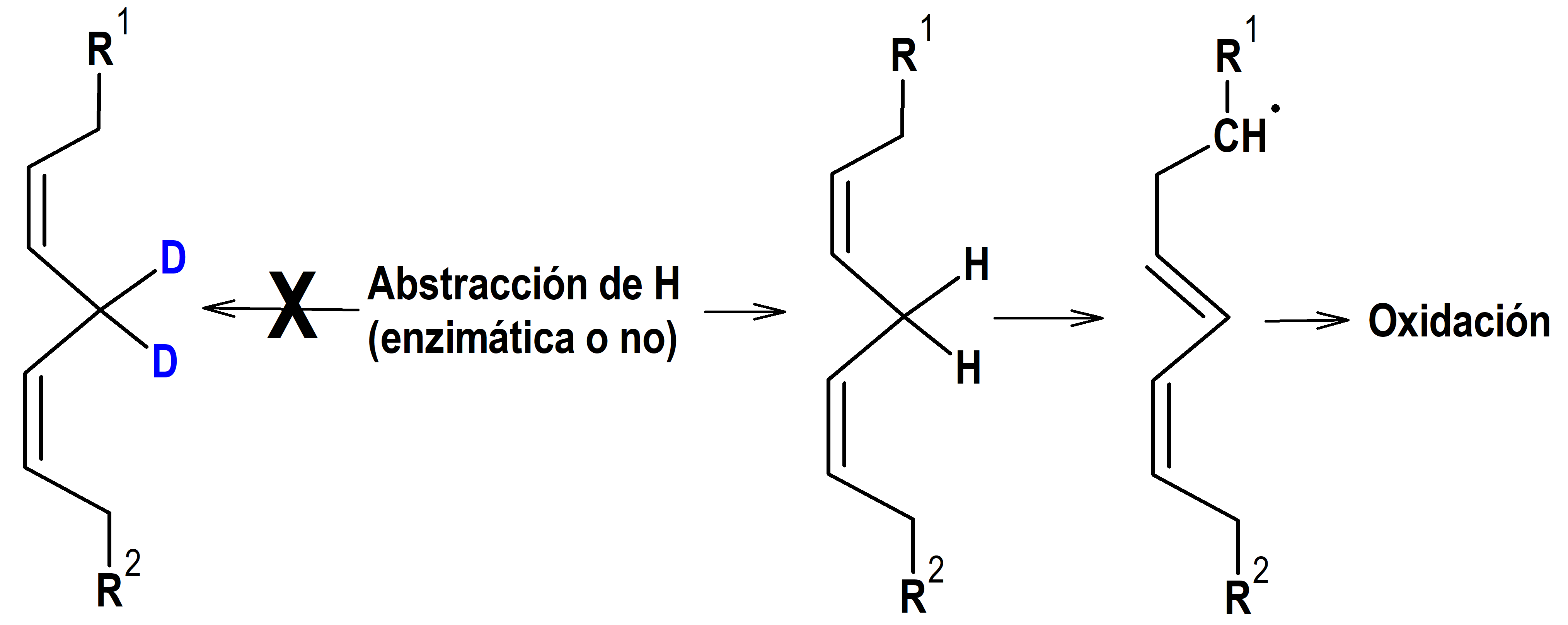
Figura 5.
En la actualidad, otros nuevos compuestos deuterados se encuentran bajo investigación clínica dentro de este grupo farmacológico, entre ellos el deucrictibant, un antagonista del receptor de bradicinina B2, que ha demostrado tener un significativo potencial para tratar y prevenir de forma eficaz y segura los ataques de angioedema debidos a la deficiencia adquirida del inhibidor de C1 (25). Asimismo, la forma deuterada de la pirfenidona, la deupirfenidona (d3-pirfenidona) también está siendo objeto de investigación clínica en fibrosis pulmonar idiopática (16).
8. DEUTERIO EN FÁRMACOS PARA PATOLOGÍAS METABÓLICAS
Alyftrek® es un medicamento constituido por la combinación a dosis fija de venzacaftor, tezacaftor y deutivacaftor, que ha sido autorizado en Estados Unidos (FDA) el 20 de diciembre de 2024 para el tratamiento de la fibrosis quística (mucoviscidosis) en pacientes de 6 años de edad y mayores que tienen al menos una mutación F508del u otra mutación sensible en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR). El deutivacaftor es la forma deuterada del ivacaftor (d9-ivacaftor), el primer medicamento (Kalydeco®) de la serie que fue autorizado (enero de 2012 por la FDA y julio de 2012 por la EMA) para el tratamiento etiológico de la fibrosis quística (figura 6).
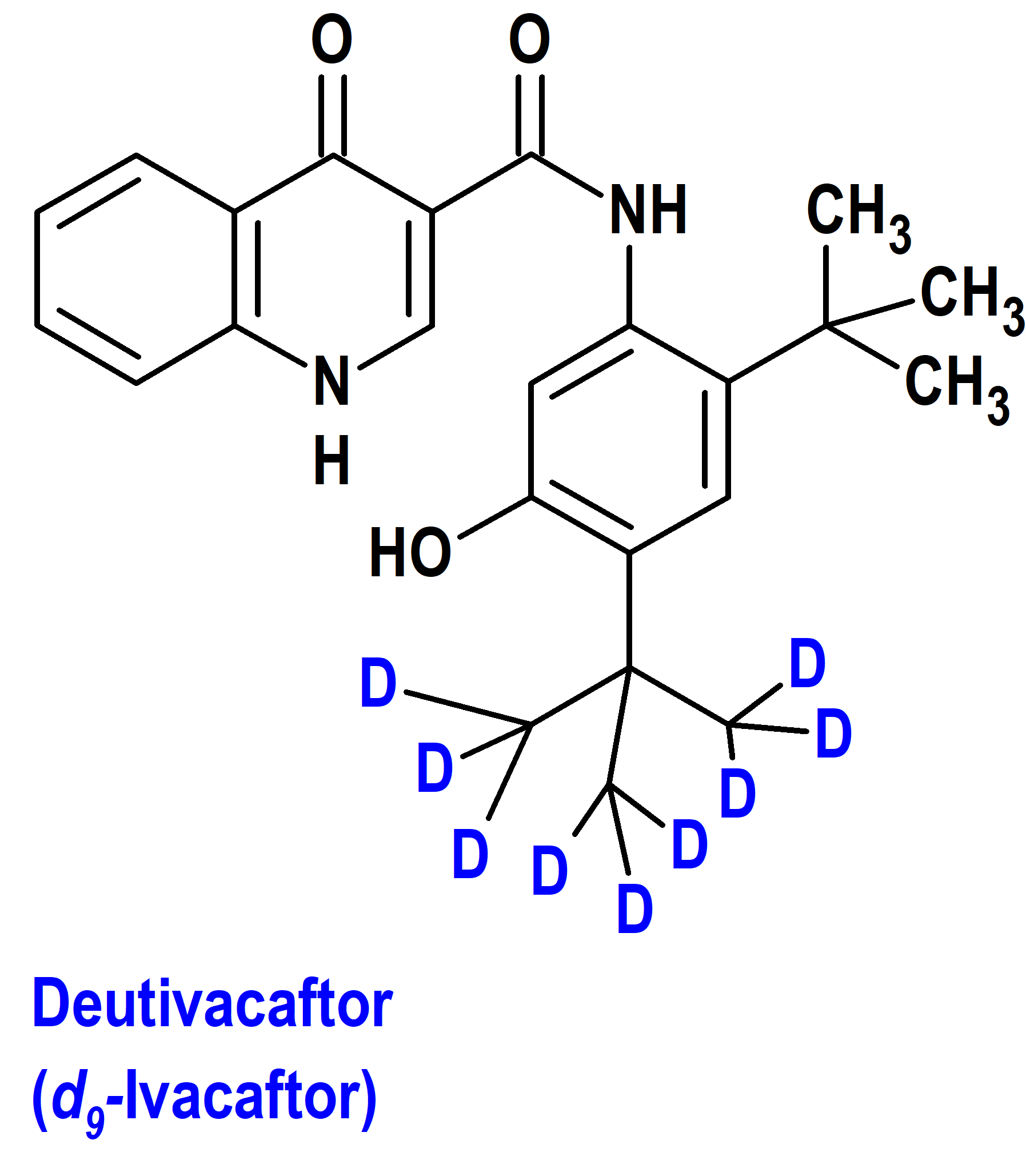
Figura 6.
El vanzacaftor y el tezacaftor se unen a diferentes sitios de la proteína CFTR y tienen un efecto aditivo para facilitar el procesamiento celular y el tráfico de formas mutantes seleccionadas de CFTR (incluido F508del-CFTR) para aumentar la cantidad de proteína CFTR entregada a la superficie celular en comparación con cada molécula sola. Por su arte, el deutivacaftor potencia la probabilidad de apertura del canal (o activación) de la proteína CFTR en la superficie celular. El efecto combinado de vanzacaftor, tezacaftor y deutivacaftor es un aumento de la cantidad y la función de CFTR en la superficie celular, lo que resulta en un aumento de la actividad de CFTR medida tanto por el transporte de cloruro mediado por CFTR como por el cloruro en el sudor en pacientes con fibrosis quística.
Su autorización oficial está apoyada en dos ensayos aleatorizados, doble ciego y controlados con fármaco activo de 52 semanas de duración que compararon Alyftrek® y una combinación a dosis fija que contenía elexacaftor, tezacaftor e ivacaftor (ELX/TEZ/IVA). En los dos ensayos participaron un total de 971 pacientes de 12 años o más con fibrosis quística que tenían al menos una mutación F508del u otras mutaciones que respondían a ELX/TEZ/IVA en el gen CFTR. En ambos ensayos, el criterio de valoración principal evaluó la no inferioridad en el cambio absoluto medio en ppFEV1 (porcentaje previsto del volumen espiratorio forzado en 1 segundo) desde el inicio hasta la semana 24 y un criterio de valoración secundario clave evaluó el cambio absoluto medio desde el inicio en el cloruro en el sudor hasta la semana 24 en los grupos de tratamiento con Alyftrek® y ELX/TEZ/IVA. En el ensayo 1, el tratamiento con ALYFTREK dio como resultado una diferencia media de mínimos cuadrados de 0,2 puntos porcentuales en el cambio absoluto en el ppFEV1 desde el inicio hasta la semana 24 en comparación con ELX/TEZ/IVA, junto con una diferencia de -8,4 puntos en cloruro en el sudor. En el ensayo 2, la diferencia media de mínimos cuadrados fue de 0,2 puntos porcentuales en el cambio absoluto en el ppFEV1 y una diferencia de -2,8 puntos en cloruro en el sudor (32).
PXL065 es una forma deuterada de pioglitazona (d1-(R)-pioglitazona), un nuevo agente oral que ha demostrado mantener la eficacia de la pioglitazona en modelos preclínicos de esteatohepatitis no alcohólica, con un potencial reducido de efectos secundarios impulsados por PPARγ. Los resultados de un estudio de fase 2 mostraron que PXL065 mejoró varias características clave de la enfermedad con un perfil de seguridad favorable (39). Otros medicamentos aprobados y candidatos clínicos con isotópologos de deuterio son ALK001 (d3-vitamina A), con interesantes perspectivas en atrofia geográfica y enfermedad de Stargardt (40), y CTP-499 (un deuteroderivado de pentoxifilina), que ha mostrado resultados clínicos prometedores para la nefropatía diabética (41).
9. DEUTERIO EN FÁRMACOS ANTIINFECCIOSOS
El derivado deuterado oral de remdesivir VV116 fue aprobado para el tratamiento de emergencia de pacientes con enfermedad por coronavirus 2019 (COVID-19) en Uzbekistán. En este caso, la incorporación de deuterio combinada con el diseño de profármacos permitió el desarrollo de un agente antiviral biodisponible por vía oral con el mismo mecanismo de acción que el remdesivir (22).
Remdesivir, un agente antiviral de amplio espectro desarrollado originalmente por Gilead Sciences como un agente potencial para la infección por el virus del Ébola, fue reutilizado para COVID-19 y aprobado por la FDA en 2020 para uso de emergencia. Es un profármaco, un inhibidor de la ARN polimerasa dependiente de ARN de los coronavirus que requiere administración intravenosa debido a las reacciones de oxidación y apertura de anillo que involucran su fracción de pirrolotriazina que imita a la adenina. GS-441524 es el metabolito predominante del remdesivir e incluso más activo que el propio remdesivir frente al SARS-Cov-2, el virus causante de la COVID-19 (42). En un esfuerzo por sortear estas limitaciones farmacocinéticas, se deuteró el punto blando de GS-441524 para reducir la degradación del anillo y se esterificaron los grupos hidroxilo del fragmento de ribosa para proporcionar un profármaco y mejorar la biodisponibilidad oral, lo que dio lugar al VV116 (figura 7). Colateralmente, se ha descubierto recientemente que VV116 es también muy eficaz contra el virus respiratorio sincitial (23).
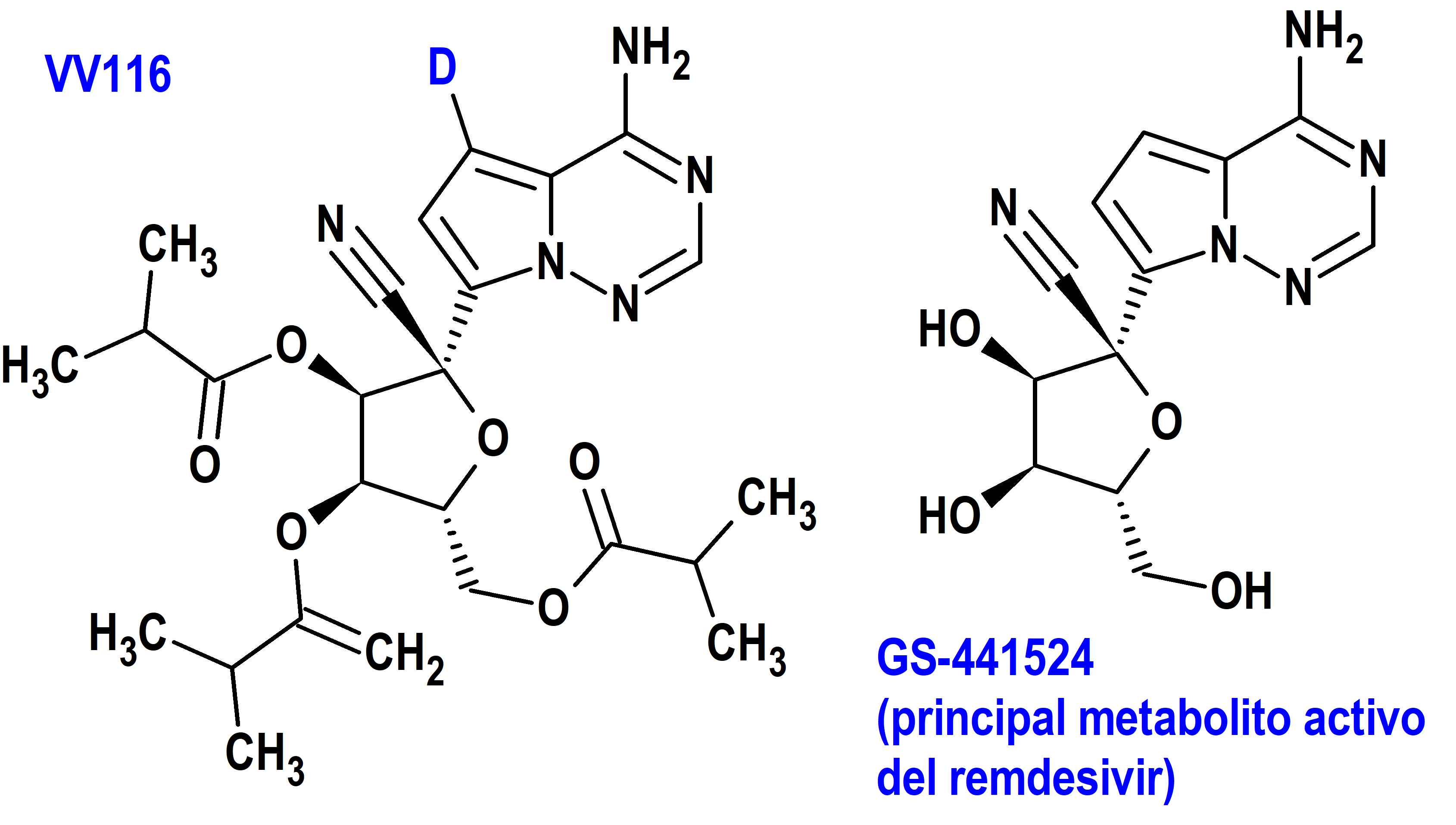
Figura 7.
10. ALGUNOS PROBLEMAS CON EL DEUTERIO Y LA DEUTERACIÓN
Más allá de su potencialidad en el diseño de nuevos fármacos, unos niveles precisos de deuterio son fundamentales para mantener el funcionamiento biológico normal, ya que su desequilibrio por exceso o por defecto puede afectar a los sistemas vivos en múltiples niveles. Así, por ejemplo, el agua ligera o agua empobrecida en deuterio (deuterium-depleted water, DDW), es capaz de alterar el metabolismo celular e incluso inhibir el crecimiento de células cancerosas. Este efecto antiproliferativo de la DDW se ha confirmado en modelos celulares, al provocar un desequilibrio de las especies reactivas de oxígeno (ROS) en las mitocondrias, de lo que se deduce que la DDW tiene potencial como adyuvante en la terapia antitumoral (43). También parece tener propiedades ansiolíticas y mejorar la memoria a largo plazo en experimentos en ratas, reduciendo la oxidación de radicales libres y regulando el metabolismo de lípidos, armonizando los índices relacionados con la diabetes y el síndrome metabólico, y aliviando los efectos tóxicos causados por el cadmio, el manganeso y otras sustancias nocivas, lo que implica su tremendo potencial anticancerígeno, neuroprotector, antienvejecimiento, antioxidante, alivio de la obesidad, tratamiento de la diabetes y el síndrome metabólico, antiinflamatorio y desintoxicante, atrayendo así una gran atención de los investigadores (29).
El deuterio transportado en algunas de las moléculas de agua de varios compartimentos celulares estabiliza las vacunas de ARNm con efectos transformadores del ADN no deseados. Los deuterones no solo preservan las plantillas de ARN para que persistan en los tejidos y/o la circulación utilizando varios mecanismos, sino que también desencadenan cambios conformacionales en los subdominios catalíticos de la enzima polimerasa que pueden afectar negativamente la incidencia y los resultados del cáncer, entre otras enfermedades, después de inyecciones de ARNm estables (30).
11. CONCLUSIONES
Los isotopólogos son especies químicas que contienen un isótopo diferente de al menos uno de los átomos de la molécula de referencia. El ejemplo más obvio es la sustitución de uno o más átomos de hidrógeno convencional o protio (¹H) por deuterio (D o ²H), que es un isótopo estable (no radiactivo). Ambos isótopos son muy similares en términos de propiedades fisicoquímicas, aunque el enlace carbono-deuterio (C-D) es más estable el del carbono-hidrógeno (C-H) y su escisión se produce de forma más lenta, por lo que la sustitución isotopológica de D por H podría modificar significativamente la velocidad de reacción, cuando la sustitución se produzca en un enlace C-H implicado en algún aspecto relevante de la interacción con receptores biológicos o con enzimas metabolizadoras.
El efecto isotópico cinético del deuterio – la deuteración (sustitución de uno o más átomos de H por D) – puede afectar a la farmacocinética de un buen número de fármacos que se metabolizan por vías que implican la rotura del enlace C-H (C-D, en su caso), lo que podría mejorar algunas limitaciones de fármacos no deuterados (originales), optimizando las propiedades farmacocinéticas para desembocar en una menor o menos frecuente dosificación y, lo que es aún más importante, limitar o disminuir la formación de metabolitos no selectivos del fármaco y, con ello, mejorar su selectividad del fármaco o mejorar su perfil toxicológico. Un aspecto farmacocinético en el que las versiones deuteradas de fármacos terapéuticamente importantes puede ser particularmente relevante es de la metabolización a través de isoenzimas del citocromo P450 (CYP450), ya que esta importante vía de metabolismo oxidativo puede estar asociado con la formación de intermediarios y/o metabolitos inestables, reactivos, no selectivos y tóxicos, dando lugar a la aparición de variabilidad entre pacientes debido a polimorfismos genéticos; y saturación, inducción o inhibición de determinadas isoenzimas del CYP450, lo que lleva a interacciones fármaco-fármaco potencialmente dañinas. Por ello, la deuteración de una molécula farmacológica podría reducir o evitar la bioactivación en su “punto caliente”, desviando el metabolismo de estas vías aberrantes y reducir la formación de metabolitos reactivos. Sin embargo, debe tenerse en cuenta que la deuteración en un sitio de la molécula podría desviar el compuesto a otras rutas de biotransformación poco conocidas o previsibles y aumentar el metabolismo en otros sitios, lo que se conoce como derivación metabólica.
El deuterio también puede influir en los eventos químicos puros que implican un paso de escisión C–H (C–D), como la enantiomerización, un fenómeno importante en el contexto de los cambios quirales. La deuterización se ha venido utilizando como un medio para estabilizar, aislar, caracterizar y también administrar estereoisómero específicos, mejorando así la utilidad terapéutica de los fármacos que originalmente eran configuracionalmente inestables.
Aunque hasta ahora se consideraba que la sustitución de H a D influye poco en la potencia de interacción bioquímica o en el perfil de selectividad de una molécula, dado que las propiedades electrónicas, la forma, el tamaño y la flexibilidad estérica del análogo deuterado son muy similares a las de su contraparte no deuterada. Sin embargo, algunos informes recientes han cuestionado esto, argumentando que el D en realidad podría ser capaz de modular las interacciones fármaco-diana. Todo esto sugiere que la sustitución de algunos átomos de H por D no solo tiene un potencial modulador farmacocinético sino también farmacodinámico, lo que puede ser una herramienta para modular y optimizar las propiedades de algunos fármacos al inducir efectos estereoelectrónicos.
Hasta el momento se han autorizado 6 medicamentos deuterados aunque el número de fármacos en fases avanzadas de investigación clínica es amplio y está aumentando rápidamente, especialmente en las áreas de cáncer, neurología y psiquiatría, así como en patologías autoinmunes, metabólicas e infecciosas. Todo ello ofrece una perspectiva esperanzadora en el desarrollo de nuevos medicamentos más efectivos, seguros y predecibles.
Sin embargo, en ocasiones, el impacto de la deuteración podría no ser el esperado. Las razones detrás del fracaso farmacológico de algunos análogos deuterados son diversas e incluyen la posibilidad de cambios metabólicos multidireccionales promovidos por el deuterio (como ocurre con la d7 -doxofilina), eficacia reducida inducida por metabolitos in vivo (por ejemplo, d1 -N-desmetildiazepam, d6 -δ-tocotrienol); enmascaramiento del efecto isotópico cinético del deuterio (DKIE) por enzimas conjugantes competitivas o mecanismos de eliminación no metabólicos (como ocurre con algunos análogos deuterados de propofol); y variabilidad interespecies que pone en peligro la predictibilidad de las pruebas preclínicas (d3 -imatinib) (13).
Por lo indicado anteriormente, parece constatarse que los isotopólogos deuterados de fármacos ya conocidos están empezando a estar “pasados de moda”. Durante varias décadas algunas compañías farmacéuticas se aprestaron a deuterar moléculas de eficacia probada y con una alta probabilidad de ser seguras y eficaces, como una vía rápida y relativamente segura de desarrollo farmacológico, fruto de los cual obtuvieron varios centenares de patentes de moléculas deuteradas, con la única finalidad de obtener un rápido y cómodo beneficio. Sin embargo, la mayoría de las grandes compañías farmacéutico tomaron el camino de la deuteración de novo, convirtiéndola en una parte integral de los procedimientos de diseño molecular de nuevos fármacos para optimizar no solo las condiciones farmacocinéticas sino también otros aspectos como la liberación y absorción, ya desde las etapas tempranas del desarrollo farmacológico. Otros aspectos muy interesantes son la estabilización de estereosiómeros puros para incrementar la actividad y el conocimiento mecanístico de fármacos y, al mismo tiempo, impedir la interconversión a estereoisómeros potencialmente tóxicos o divergentes farmacológicamente.
En resumen, la deuteración es un interesante enfoque que está en constante evolución, con múltiples y – en ocasiones – imprevistas aplicaciones.
12. REFERENCIAS
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017; 7: 42717. https://doi.org/10.1038/srep42717).
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Aug; 596(7873): 583-589. doi:10.1038/s41586-021-03819-2)
- Doytchinova I. Drug Design—Past, Present, Future. Molecules. 2022; 27(5):1496. https://doi.org/10.3390/molecules27051496
- Cuéllar Rodríguez S, Smeyers Guillemin YG (dir.), Gálvez Ruano E (dir.). Estudio mecano-cuántico de la actividad antagonista sobre la prostaglandina ciclooxigenasa: Aplicaciones al diseño de nuevos fármacos: Tesis doctoral. Facultad de Farmacia; Universidad Complutense de Madrid (1983) https://ucm.on.worldcat.org/oclc/1123924630
- Smeyers YG, Cuéllar Rodríguez S, Gálvez Ruano E, Arias Pérez MS. Conformational Analysis of Some α-Phenylpropionic Acids with Anti-inflammatory Activity. J Pharm Sci. 1985; 74 (1): 47-49. https://doi.org/10.1002/jps.2600740113
- Valentová J, Lintnerová L, Miklášová N, Oboňová B, Habala L. Analogues of Anticancer Natural Products: Chiral Aspects. Int J Mol Sci. 2023 Mar 16; 24(6): 5679. doi:10.3390/ijms24065679
- Egilman AC, Zhang AD, Wallach JD, Ross JS. Medicare Part D Spending on Single-Enantiomer Drugs Versus Their Racemic Precursors. Ann Intern Med. 2019 Oct 1; 171(7): 521-523. doi:10.7326/M19-1085
- Czeskis B, Elmore CS, Haight A, Hesk D, Maxwell BD, Miller SA, Raglione T, Schildknegt K, Traverse JF, Wang P. Deuterated active pharmaceutical ingredients: A science-based proposal for synthesis, analysis, and control. Part 1: Framing the problem. J Labelled Comp Radiopharm. 2019 Sep; 62(11): 690-694. https://doi.org/10.1002/jlcr.3743
- Wood WW. Deuterated Drugs: Isotope Distribution and Impurity Profiles. J Med Chem. 2024 Oct 10; 67(19): 16991-16999. https://doi.org/10.1021/acs.jmedchem.4c01694
- Belete TM. Recent Updates on the Development of Deuterium-Containing Drugs for the Treatment of Cancer. Drug Des Devel Ther. 2022 Oct 4; 16: 3465-3472. https://doi.org/10.2147/DDDT.S379496
- Tang LWT, Chan ECY. Metabolic activation of drugs by cytochrome P450 enzymes: Biochemical insights into mechanism-based inactivation by fibroblast growth factor receptor inhibitors and chemical approaches to attenuate reactive metabolite formation. Biochem Pharmacol. 2022 Dec; 206: 115336. doi:10.1016/j.bcp.2022.115336
- Furge LL, Guengerich FP. Cytochrome P450 enzymes in drug metabolism and chemical toxicology: an introduction. Biochem. Mol Biol Educ. 2006; 34: 66–74. doi:10.1002/bmb.2006.49403402066
- Di Martino RMC, Maxwell BD, Pirali T. Deuterium in drug discovery: progress, opportunities and challenges. Nat Rev Drug Discov. 2023 Jul; 22(7): 562-584. doi:10.1038/s41573-023-00703-8
- Du Y, Chen Y. The Application of Deuteration Strategy in Drug Design. ChemMedChem. 2024 Dec 23: e202400836. doi:10.1002/cmdc.202400836
- Steverlynck J, Sitdikov R, Rueping M. The Deuterated “Magic Methyl” Group: A Guide to Site-Selective Trideuteromethyl Incorporation and Labeling by Using CD3 Reagents. Chemistry. 2021 Aug 16; 27(46): 11751-11772. doi:10.1002/chem.202101179
- Chandra Mouli HM, Vinod A, Kumari S, Tiwari AK, Kathiravan MK, Ravichandiran V, Peraman R. Deuterated driven new chemical entities: An optimistic way to improve therapeutic efficacy. Bioorg Chem. 2023 Jun; 135: 106490. Doi:10.1016/j.bioorg.2023.106490
- Ben Abu N, et al. Sweet taste of heavy water. Commun. Biol. 2021;4:440. doi:10.1038/s42003-021-01964-y
- Belz TF, et al. Enhancement of a heroin vaccine through hapten deuteration. J. Am. Chem. Soc. 2020; 142: 13294–13298. doi:10.1021/jacs.0c05219
- Mourya A, Prajapati N. Precision Deuteration in Search of Anticancer Agents: Approaches to Cancer Drug Discovery. Cancer Biother Radiopharm. 2024 Feb; 39(1): 1-18. doi:10.1089/cbr.2023.0031
- Keam SJ, Duggan S. Donafenib: first approval. Drugs. 2021; 81: 1915–1920. doi:10.1007/s40265-021-01603-0
- Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 7, 6 (2021). https://doi.org/10.1038/s41572-020-00240-3. A Correction to this paper has been published: https://doi.org/10.1038/s41572-024-00500-6
- Qian Hj, Wang Y, Zhang Mq, et al. Safety, tolerability, and pharmacokinetics of VV116, an oral nucleoside analog against SARS-CoV-2, in Chinese healthy subjects. Acta Pharmacol Sin 43, 3130–3138 (2022). https://doi.org/10.1038/s41401-022-00895-6
- Xie Y, Yin W, Zhang Y, et al. Design and development of an oral remdesivir derivative VV116 against SARS-CoV-2. Cell Res 31, 1212–1214 (2021). https://doi.org/10.1038/s41422-021-00570-1
- Wrobleski ST, Moslin R, Lin S, Zhang Y, Spergel S, Kempson J, et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J Med Chem. 2019 Oct 24; 62(20): 8973-8995. doi:10.1021/acs.jmedchem.9b00444
Lesage A, Marceau F, Gibson C, Loenders B, Katzer W, Ambrosi HD, Saupe J, Faussner A, Pardali E, Knolle J. In vitro pharmacological profile of PHA-022121, a small molecule bradykinin - B2 receptor antagonist in clinical development. Int Immunopharmacol. 2022 Apr; 105: 108523. doi:10.1016/j.intimp.2022.108523
- Jansen-van Vuuren RD, Jedlovčnik L, Košmrlj J, Massey TE, Derdau V. Deuterated Drugs and Biomarkers in the COVID-19 Pandemic. ACS Omega. 2022 Nov 13; 7(46): 41840-41858. doi:10.1021/acsomega.2c04160
- Zesiewicz T, Heerinckx F, De Jager R, Omidvar O, Kilpatrick M, Shaw J, Shchepinov MS. Randomized, clinical trial of RT001: Early signals of efficacy in Friedreich’s ataxia. Mov Disord. 2018 Jul; 33(6): 1000-1005. doi:10.1002/mds.27353
- Demidov VV. Site-specifically deuterated essential lipids as new drugs against neuronal, retinal and vascular degeneration. Drug Discov Today. 2020 Aug; 25(8): 1469-1476. doi:10.1016/j.drudis.2020.03.014
- Qu J, Xu Y, Zhao S, Xiong L, Jing J, Lui S, Huang J, Shi H. The biological impact of deuterium and therapeutic potential of deuterium-depleted water. Front Pharmacol. 2024 Jul 22; 15: 1431204. doi:10.3389/fphar.2024.1431204
- Boros LG, Kyriakopoulos AM, Brogna C, Piscopo M, McCullough PA, Seneff S. Long-lasting, biochemically modified mRNA, and its frameshifted recombinant spike proteins in human tissues and circulation after COVID-19 vaccination. Pharmacol Res Perspect. 2024 Jun; 12(3): e1218. doi:10.1002/prp2.1218
- Cuéllar Rodríguez S. Novel drugs recently authorized by EMA and FDA (Q3, 2024). ANALES RANF [Internet]. Real Academia Nacional de Farmacia; An Real Acad Farm · Año 2024 · volumen 90 · número 03 pp: 369-384. https://doi.org/10.53519/analesranf.2024.90.03.06
- Cuéllar Rodríguez S. Novel drugs recently authorized by EMA and FDA (Q4, 2024). ANALES RANF [Internet]. Real Academia Nacional de Farmacia; An. Real Acad. Farm. · Año 2024 · volumen 90 · número 04 [in press]
- Cuéllar Rodríguez S. Fármacos novedosos autorizados recientemente por EMA y FDA (1º trimestre de 2023). ANALES RANF [Internet]. Real Academia Nacional de Farmacia; An Real Acad Farm Año 2023. Volumen 89 Número 1 pp. 127-134. DOI: https://doi.org/10.53519/analesranf.2023.89.01.07
- Cuéllar Rodríguez S. Fármacos novedosos autorizados recientemente por EMA y FDA (3º trimestre de 2022). ANALES RANF [Internet]. Real Academia Nacional de Farmacia; An Real Acad Farm Año 2022. Volumen 88 Número 3. pp. 323-335. DOI: https://doi.org/10.53519/analesranf.2022.88.03.06
- National Cancer Institute. deuterated enzalutamide. https://www.cancer.gov/publications/dictionaries/cancer-drug/def/deutenzalutamide (consultado el 6 de enero de 2025)
- Cao Y, Yu F, Lyu Y, Lu X. Promising candidates from drug clinical trials: Implications for clinical treatment of Alzheimer’s disease in China. Front Neurol. 2022 Nov 15; 13: 1034243. doi:10.3389/fneur.2022.1034243
- Palfreyman M, Krakowsky J, Morgan M, Canal C, Pathare P, Avery K, et al. . “ACNP 61st Annual Meeting: Poster Abstracts P271-P540: P361. In Vitro and In Vivo Profile of CYB003: A Novel, Deuterated Psilocybin Analog for the Potential Treatment of Major Depressive Disorder” (PDF). Neuropsychopharmacology. 2022; 47 (Suppl 1): 220–370 (271). doi:10.1038/s41386-022-01485-0
- Wo JM, McCallum RW, Gonzalez Z . “Antiemetic therapy for gastroparesis”. In McCallum RW, Parkman HP (eds.). Gastroparesis. Elsevier. (2021) pp. 341–359. doi:10.1016/B978-0-12-818586-5.00025-9
- Harrison SA, Thang C, Bolze S, Dewitt S, Hallakou-Bozec S, Dubourg J, et al. Evaluation of PXL065 – deuterium-stabilized (R)-pioglitazone in patients with NASH: A phase II randomized placebo-controlled trial (DESTINY-1). J Hepatol. 2023 May; 78(5): 914-925. doi:10.1016/j.jhep.2023.02.004
- Xu XL, Zhang W, Rao GW. Clinical Application and Synthesis Methods of Deuterated Drugs. Curr Med Chem. 2023; 30(36): 4096-4129. doi:10.2174/0929867330666221122123201
- Pérez Gómez MV, Sánchez Niño MD, Sanz AB, Zheng B, Martín Cleary C, Ruiz Ortega M, et al. Targeting inflammation in diabetic kidney disease: early clinical trials. Expert Opin Investig Drugs. 2016 Sep; 25(9): 1045-58. doi:10.1080/13543784.2016.1196184
- Goto S, Kamiyoshi T, Iwasaki R. Predictive factors associated with short-term mortality in cats with feline infectious peritonitis treated with remdesivir or GS-441524 or both. J Vet Intern Med. 2025 Jan-Feb; 39(1): e17249. doi:10.1111/jvim.17249
- Zhang X, Gaetani M, Chernobrovkin A, Zubarev RA. Anticancer Effect of Deuterium Depleted Water – Redox Disbalance Leads to Oxidative Stress*. Molecular & Cellular Proteomics. 2019; 18(12): 2373-2387. https://doi.org/10.1074/mcp.RA119.00145
