1. INTRODUCCIÓN
La enfermedad del hígado graso no alcohólico (EHGNA) es la enfermedad hepática crónica más común en el mundo, afectando aproximadamente al 25 % de la población adulta, especialmente en los países industrializados (1). Al mismo tiempo, la EHGNA es también la forma más prevalente de enfermedad hepática crónica en la infancia, afectando aproximadamente al 10 % de la población pediátrica general (2, 3). La definición clásica de la EHGNA excluye el consumo excesivo de alcohol, que causa la llamada enfermedad hepática alcohólica. Numerosas evidencias apoyan la hipótesis de que la EHGNA es la manifestación hepática del síndrome metabólico (SM), siendo la resistencia a la insulina el factor patogénico común entre ambas (4). Aunque se han descrito algunos factores genéticos como elementos de riesgo para la EHGNA (5), el aumento del peso corporal, así como la presencia de varias características del SM, como la adiposidad, la hiperglucemia, la dislipidemia o la hipertensión, pueden ser determinantes clave en la patogenia de la EHGNA.
La EHGNA engloba un amplio espectro de lesiones en el hígado, en el que se incluyen la esteatosis simple, la esteatohepatitis no alcohólica (EHNA), la fibrosis y finalmente la cirrosis. Se estima que alrededor de un 7% de estos pacientes en estadio cirrótico desarrollarán un carcinoma hepatocelular (CHC) que puede llegar a causar una insuficiencia hepática crónica y la necesidad de trasplante de hígado en aproximadamente el 50% de los casos. Mientras que en la esteatosis hepática simple no hay signos de daño hepatocelular ni de fibrosis, la EHNA se caracteriza por la presencia de esteatosis e inflamación lobulillar con degeneración balonizante de los hepatocitos, con o sin fibrosis (6, 7).
Los factores desencadenantes, así como los mecanismos asociados al desarrollo y la progresión de la EHGNA son complejos y multifactoriales. Originalmente, la “la teoría del doble impacto” se formuló para explicar la progresión de la esteatosis simple a la EHNA. Según esta visión tradicional, la acumulación intrahepática de lípidos provocada por el sedentarismo, las dietas hipercalóricas, la obesidad y la resistencia a la insulina, actúa como el «primer impacto» favoreciendo el aumento de la sensibilidad de los hepatocitos a sucesivos daños o agresiones. La acumulación de citoquinas proinflamatorias, adipoquinas, endotoxinas bacterianas, junto con la disfunción mitocondrial y a la aparición de estrés oxidativo y/o estrés del retículo endoplásmico (RE) representan el «segundo impacto» en la progresión hacia estadios más avanzados. Así, este segundo impacto provoca un efecto deletéreo en los hepatocitos, provocando mayor inflamación y fibrosis. Hoy en día, la hipótesis más aceptada es el «modelo de múltiples impactos” (8), ya que otros elementos adicionales como las hormonas/adipoquinas secretadas del tejido adiposo, diversos factores nutricionales, la microbiota intestinal y numerosos factores genéticos y epigenéticos también contribuyen en mayor o menor medida a la progresión de la enfermedad (9-13). A pesar de esta diferencia, la acumulación de grasa en el hígado, causada por la obesidad y la resistencia a la insulina, aún representa el primer impacto en ambas hipótesis. En el tejido adiposo, la resistencia a la insulina conduce a una lipólisis periférica excesiva, provocando la degradación de los triglicéridos (TG) y una acumulación masiva de ácidos grasos libres (AGL) y glicerol. En condiciones normales, los AGL circulantes son captados por los hepatocitos y de nuevo esterificados en TG. Sin embargo, una captación excesiva de AGL puede saturar la capacidad celular para almacenarlos y esterificarlos en TG, lo que conllevaría la acumulación masiva de AGL, provocando el daño celular (lipotoxicidad) y la muerte celular apoptótica (lipoapoptosis) de los hepatocitos, procesos fuertemente asociados con la progresión de la EHGNA a EHNA.
En la mayoría de los casos, la EHNA se desarrolla de forma asintomática hasta que la enfermedad progresa a etapas más avanzadas en las que el trasplante de hígado es la única opción terapéutica disponible. Por tanto, la detección temprana de esta enfermedad sería útil para identificar a aquellos individuos con EHGNA en estadios iniciales, donde la enfermedad es aún silenciosa. Solo en un reducido número de pacientes, se sospecha de enfermedad tras la detección de una elevación, que resulta difícil de explicar, de los niveles de enzimas hepáticas, tales como la fosfatasa alcalina, la alanina aminotransferasa (ALT) o la aspartato aminotransferasa (AST), o de alguna evidencia de esteatosis por imagen mediante tomografía computarizada y resonancia magnética (RM). No obstante, estas pruebas no permiten diferenciar la esteatosis simple de la EHNA con o sin fibrosis.
Por ello, hasta la fecha, la biopsia hepática sigue siendo el método más fiable para el diagnóstico de la EHGNA ya que permite clasificar el estado de la enfermedad por evaluación histológica. Sin embargo, este procedimiento altamente invasivo y dañino para el paciente no puede predecir la progresión de la enfermedad y a menudo, el diagnóstico es tardío.
Hasta ahora, la FDA (U.S. Food and Drug Administration) no ha aprobado ningún agente farmacológico para el tratamiento de la EHNA. Hoy en día, cambios en el estilo de vida y dietas para perder peso son la única estrategia para revertir la EHGNA. Desafortunadamente, se ha demostrado que un gran número de pacientes recuperan la mayor parte del peso después de un período de pérdida de peso exitoso, un efecto probablemente debido a la falta de disponibilidad de un programa multidisciplinar completo y personalizado centrado en el mantenimiento del peso corporal a largo plazo para cada paciente (14, 15). Por este motivo, existe una necesidad urgente de identificar biomarcadores no invasivos que sean fiables y específicos para el diagnóstico de la EHGNA y la EHNA en las fases iniciales, donde las intervenciones en el estilo de vida y los posibles fármacos desarrollados se puedan utilizar con cierta garantía de éxito a largo plazo.
Hasta la fecha, se considera que la ausencia de un tratamiento eficaz para la EHGNA podría deberse a la heterogeneidad en los factores desencadenantes que presentan los pacientes provocados por la coexistencia de otras patologías asociadas como las enfermedades cardiovasculares (ECV) o la diabetes mellitus tipo 2 (DM2). Teniendo esto en consideración, recientemente ha surgido el término actualizado “MAFLD” (del inglés, metabolic-dysfunction-associated fatty liver disease), que significa en español enfermedad de hígado graso asociada a disfunción metabólica, para describir mejor la patogénesis de la EHGNA, definiéndose así como la manifestación hepática de un trastorno multisistémico que es heterogéneo en su origen, desarrollo y progresión (16, 17). De acuerdo con esta nueva definición, la EHGNA asociada a disfunción metabólica englobaría múltiples subtipos y se clasificaría en función del factor dominante. Según los artículos publicados, los criterios para el diagnóstico se basan en la evidencia de esteatosis hepática además de uno de los siguientes tres elementos, sobrepeso/obesidad, presencia de DM2 o evidencia de desregulación metabólica, independientemente de la cantidad de alcohol consumida. Así, la identificación de este factor predominante en cada paciente podría ayudar en su estratificación y podría permitir la implementación de un tratamiento personalizado para asegurar la mejor respuesta terapéutica con pocos efectos secundarios adversos.
Recientes avances en la investigación básica y traslacional han proporcionado información sobre los mecanismos patogénicos que impulsan la progresión de la EHGNA, involucrando a las células hepáticas parenquimales y no parenquimales (18). Los hepatocitos afectados tras un daño lipotóxico liberan moléculas intracelulares denominadas patrones moleculares asociados al daño (DAMPs, del inglés damage-associated molecular patterns) que activan varios tipos celulares, como las células de Kupffer (KCs, del inglés Kupffer cells) -los macrófagos residentes en el hígado-, los neutrófilos y las células estelares hepáticas, también llamadas células de Ito (HSC, del inglés hepatic stellate cells), provocando un aumento de la inflamación y de la fibrosis. Cabe destacar que la ubicación de las HSCs y KCs dentro del espacio de Disse facilita su contacto directo con otros tipos celulares, incluidos los hepatocitos y las células endoteliales sinusoidales hepáticas (LSECs, del inglés liver sinusoidal endothelial cells), promoviendo así el transporte intercelular de mediadores solubles y citoquinas. De acuerdo con esto, la progresión de EHNA a estadios más avanzados es el resultado de un interactoma intrahepático complejo entre los diferentes tipos celulares a través de diversos mediadores secretados, lo que ilustra la complejidad de la señalización célula-célula en la fisiología normal y patológica del hígado. Ante este escenario, considerando a la EHGNA como una enfermedad multifactorial, no existe una única diana terapéutica para su tratamiento, lo que explica la falta de una terapia eficaz para la enfermedad.
2. PAPEL EMERGENTE DE LAS VESÍCULAS EXTRACELULARES EN LA COMUNICACIÓN INTERCELULAR
Durante años se ha pensado que la comunicación intercelular estaba mediada únicamente por la interacción directa célula-célula o la secreción de factores solubles. Sin embargo, actualmente está ampliamente reconocido que las células son capaces también de secretar, de una manera conservada evolutivamente, diversos tipos de vesículas de membrana que constituyen una tercera variedad de comunicación intercelular. A estas vesículas se las conoce generalmente como vesículas extracelulares o “VEs” (19).
El término genérico «VE» comprende una población heterogénea de vesículas de tamaño nanométrico conformadas por una membrana de tipo bicapa lipídica que son liberadas por las células. Actualmente, las VEs se pueden clasificar en las siguientes tres categorías según su tamaño y biogénesis celular: exosomas, microvesículas y cuerpos apoptóticos (19, 20). Brevemente, los exosomas son las vesículas más pequeñas (30-150 nm) y se originan como vesículas intraluminales (ILV, del inglés intraluminal vesicles) dentro de los cuerpos multivesiculares (MVB, del inglés multivesicular bodies) de la célula que por fusión con la membrana plasmática se liberan al espacio extracelular. Las microvesículas (50-1000 nm) y los cuerpos apoptóticos (100-5000 nm) son más grandes y se forman directamente por escisión de la membrana plasmática o por fragmentación de células apoptóticas, respectivamente. La Figura 1 muestra las características más comunes que se utilizan para diferenciar los diferentes subtipos de VEs.
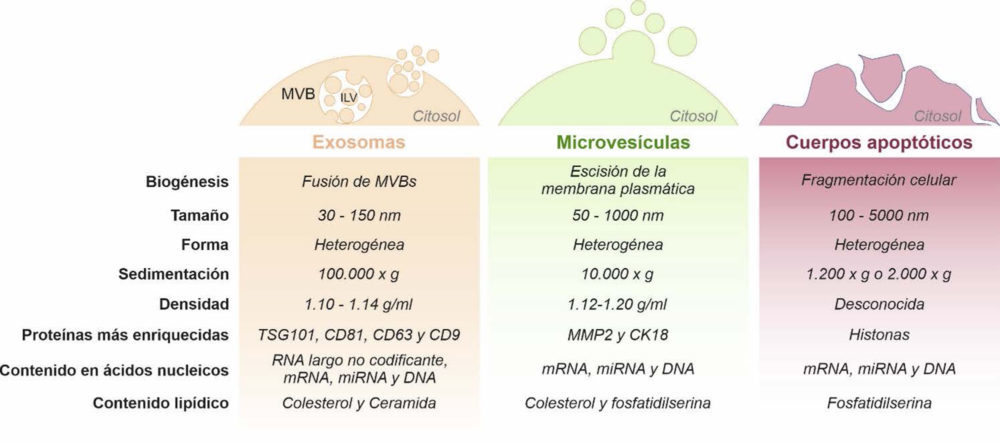
Figura 1. Clasificación y características representativas de los subtipos de vesículas extracelulares (VEs).
Por otro lado, aunque la biogénesis de los exosomas y de las microvesículas ocurre en distintos sitios dentro de la célula, estas muestran una morfología y tamaño similar, y además comparten la maquinaria intracelular en su formación lo que hace difícil su diferenciación. Este hecho junto con la falta de estandarización tanto de los procedimientos de aislamiento como de los métodos para su caracterización complica en la actualidad el diseño de una nomenclatura más precisa (20, 21). En este contexto y dado que el número de publicaciones científicas en el campo ha aumentado exponencialmente en la última década (22), la Sociedad Internacional de Vesículas Extracelulares (ISEV: www.isev.org) publicó en 2014 una guía con la información básica para los estudios con vesículas extracelulares (MISEV, del inglés Minimal Information for Studies of Extracellular Vesicles) (23) con el objetivo de unificar la nomenclatura y las metodologías utilizadas en todos los laboratorios. La “MISEV” fue actualizada en 2018 (24).
A pesar de que las VEs se describieron por primera vez en los años 80 (25, 26), es ahora cuando han cobrado interés en el campo de la investigación biomédica a causa del descubrimiento de su capacidad de transportar entre las células, moléculas bioactivas específicas como ácidos nucleicos (DNAs, RNAs y miRNAs), proteínas, lípidos, azúcares y otros metabolitos en respuesta a estímulos celulares concretos. Además, cabe destacar que, aunque se conoce que el proceso por el que se selecciona el cargo que formará parte de las EVs está altamente regulado, los mecanismos implicados siguen siendo aún en gran parte desconocidos (27). En consecuencia, cada tipo de célula podrá regular la producción de EVs tanto cuantitativa como cualitativamente dependiendo de su estado fisiológico o patológico, pudiendo secretar una misma célula poblaciones heterogéneas de VEs en función del estímulo (19, 20, 28).
Tras la secreción de las VEs al espacio extracelular, éstas pueden interaccionar con células cercanas (comunicación célula-célula) o difundir por el torrente sanguíneo u otros fluidos biológicos para actuar sobre órganos distantes (comunicación entre órganos). En definitiva, las VEs transmitirán un paquete único de información desde la célula donadora a la célula receptora, capaz de desencadenar en ésta última, respuestas funcionales y promover cambios fenotípicos que afectarán finalmente a su estado fisiológico o patológico (19, 20). No obstante, las VEs necesitan acoplarse selectivamente a la membrana plasmática de sus células diana para desencadenar los efectos fenotípicos. Para ello, todas las VEs portan moléculas de superficie que les permiten ser reconocidas por las células receptoras y, de hecho, son varios los estudios que indican que los patrones de glicosilación de su superficie y los receptores y ligandos expuestos como por ejemplo, las integrinas, pueden ser relevantes para que las VEs se unan a las células diana y, por lo tanto, para su biodistribución (29, 30).
Finalmente, una vez unidas a su célula diana, las VEs pueden iniciar diversas vías de señalización intracelular mediante la simple interacción con receptores o ligandos de superficie. Por ejemplo, las VEs que portan ligandos de superficie como FasL, perforina o el ligando inductor de apoptosis relacionado con el factor de necrosis tumoral (TRAIL, del inglés tumor necrosis factor-related apoptosis-inducing ligand) son completamente funcionales a la hora de inducir la apoptosis mediada por receptores de muerte (31-33). No obstante, la transferencia de su cargo mediante la internalización vesicular (endocitosis) o la fusión con la célula diana es generalmente necesaria para desencadenar respuestas celulares específicas. Las VEs pueden liberar dentro de la célula receptora las proteínas, los lípidos bioactivos o incluso las moléculas de señalización activas que contienen, incluidas enzimas, y activar múltiples vías de señalización. Además, las VEs también pueden transferir a las células huésped mRNA y miRNAs activos que regulan la expresión génica a nivel traduccional o producen modificaciones postraduccionales de mRNAs diana, respectivamente (20, 34).
La comunicación intercelular mediada por VEs es necesaria para el mantenimiento de la homeostasis celular y las funciones fisiológicas, mientras que las alteraciones en este proceso pueden indicar estados patológicos. El hecho de que el cargo de las VEs pueda modificarse en condiciones patológicas, plantea la pregunta de si las VEs podrían tener un papel biológico distinto en la salud o la enfermedad y, por tanto, ser una posible diana terapéutica en el tratamiento de diversas patologías. Además, debido a su cargo asociado a la enfermedad y a su presencia ubicua y estabilidad en los fluidos biológicos humanos, las VEs también pueden tener relevancia clínica como biomarcadores no invasivos para la detección temprana y el prognosis de muchas enfermedades (35).
Por otro lado, las VEs ya sea sin modificaciones o manipuladas, también han generado una atención considerable en la comunidad científica debido a su uso potencial con fines terapéuticos (36). Las VEs son biodisponibles, biocompatibles y resistentes a las RNasas y proteasas (37), características que las convierten en vehículos ideales para la administración de fármacos, proteínas, miRNA, RNAs silenciadores (siRNAs, del inglés silencing RNAs) y otras moléculas. Con respecto a las enfermedades hepáticas, el foco principal de esta revisión, los esfuerzos se han centrado en dos áreas principales, por un lado, el uso de las VEs como vehículos de administración de medicamentos directamente al hígado (38) y, por otro lado, el uso de las mismas VEs como agentes terapéuticos para estimular la regeneración hepática, modular la inflamación, reducir la fibrosis hepática o bloquear la hepatocarcinogénesis (39, 40). No obstante, aún deben resolverse múltiples desafíos antes de que se pueda llevar a cabo estudios clínicos controlados, sobre la estandarización de la metodología y la selección del tipo de VEs para la administración de fármacos (41, 42).
La identificación y el análisis de patrones moleculares específicos de células/tejidos son prometedores para fines diagnósticos, pronósticos y terapéuticos. La composición de proteínas tejido- específicas de las VEs brinda la oportunidad de identificar patrones celulares específicos para utilizarlos como marcadores de diagnóstico. Sin embargo, los métodos de cribado de proteínas comunes actualmente, como la electroforesis en geles bidimensionales y la espectrometría de masas, son técnicas que requieren mucho tiempo por lo que no son adecuados para un cribado de alto rendimiento cuando se manejan muchas muestras. Además, su sensibilidad y reproducibilidad pueden ser en algunos casos limitadas. En esta línea, Larsen y colaboradores demostraron que los ensayos de extensión de proximidad multiplex (PEA, del inglés proximity extension asays) son una herramienta poderosa de detección de proteínas en la investigación de las VEs (43). Esta tecnología permite la identificación, análisis y validación de posibles marcadores asociados a las VEs para identificar con alta especificidad y sensibilidad los perfiles de proteínas de las VEs de diferente origen. Es importante destacar que la capacidad de esta tecnología para rastrear el origen celular de las VEs podría extenderse a las VEs circulantes en plasma, facilitando estrategias de diagnóstico eficientes y no invasivas en etapas tempranas de enfermedad. Sin embargo, este estudio presenta una limitación y es el hecho de que los paneles PEA todavía no están disponibles para todas las células secretoras de VEs. Por tanto, se necesita más investigación y optimización de los PEA para su futuro uso en la detección en cohortes de pacientes mayores y en otros fluidos corporales.
Muchas evidencias respaldan el papel de las VEs en una amplia gama de enfermedades humanas, incluido el espectro de enfermedades asociadas con la obesidad y el SM (44). Además, se ha reportado que el número y el fenotipo de las VEs circulantes en la sangre cambian en la obesidad y en los estados patológicos asociados, incluida la resistencia a la insulina, la DM2 y la EHGNA (45). Varios mecanismos implicados en la progresión de la EHGNA, como la inflamación, la fibrosis y la angiogénesis, todos ellos relacionados con la lipotoxicidad asociada al SM, desencadenan la producción y liberación de VEs en el hígado (45). Por un lado, las VEs median la comunicación intercelular local entre las células hepáticas, lo que impulsa la patogénesis de la enfermedad y, por otro lado, las VEs derivadas del hígado podrían afectar tejidos y órganos distantes tras su liberación al torrente sanguíneo. Por tanto, las VEs derivadas del hígado tienen alto potencial como biomarcadores para fines de diagnóstico y pronóstico en pacientes (46). Sin embargo, la identificación de VEs de origen hepático en la circulación como indicadoras de alteraciones metabólicas en este órgano sigue siendo un desafío para los investigadores básicos y clínicos.
Como se ha mencionado previamente, la EHGNA no es una condición aislada y, en términos generales, esta enfermedad se presenta como una complicación de otros trastornos metabólicos. Por esta razón, pueden verse afectados múltiples tejidos y, en consecuencia, no se puede excluir la contribución de las VEs extrahepáticas durante la EHGNA, como por ejemplo las VEs derivadas de adipocitos o células inmunes. Además, la mayoría de las células hepáticas producen VEs, incluidos los hepatocitos, colangiocitos, HSC y LSEC (35). No obstante, como el 80% del volumen del hígado está compuesto por hepatocitos, su participación en el conjunto total de VEs hepáticas es probablemente la más relevante. Por ello, en esta revisión nos centraremos en las VEs derivadas de hepatocitos (Hep-VEs) como impulsores de la patogénesis de la EHGNA o biomarcadores no invasivos para su diagnóstico y pronóstico.
3. PAPEL DE LAS VEs HEPÁTICAS EN LA EHGNA
La lipotoxicidad es uno de los desencadenantes de la progresión de la EHGNA, ya que es un proceso mediante el cual se acumulan lípidos tóxicos en los hepatocitos, como los AGL saturados, que activan las vías moleculares relacionadas con el estrés celular que pueden provocar la muerte celular (47, 48). En esta sección analizaremos diferentes estudios con evidencias sobre como la lipotoxicidad en los hepatocitos afecta, o incluso desencadena diversas respuestas de las células hepáticas circundantes a través de la liberación de Hep-VEs. Por otro lado, destacaremos los estudios actualmente disponibles sobre la participación de las Hep-VEs en las complicaciones asociadas a la EHGNA.
3.1 Papel clave de las Hep-VEs en la progresión de EHGNA a EHNA
Como hemos mencionado anteriormente, hay varios eventos clave estrechamente interconectados que están involucrados en la progresión de EHGNA a EHNA tales como la inflamación, la fibrosis, la apoptosis de hepatocitos y la angiogénesis alterada. Todos estos signos están relacionados con la lipotoxicidad asociada al SM que desencadena la producción y liberación de VEs por el hígado (45). A continuación, revisaremos en detalle varios estudios que recopilan el papel de los Hep-VEs proinflamatorios, proangiogénicos y profibróticos como mediadores patogénicos durante la lipotoxicidad en la EHGNA (Figura 2).
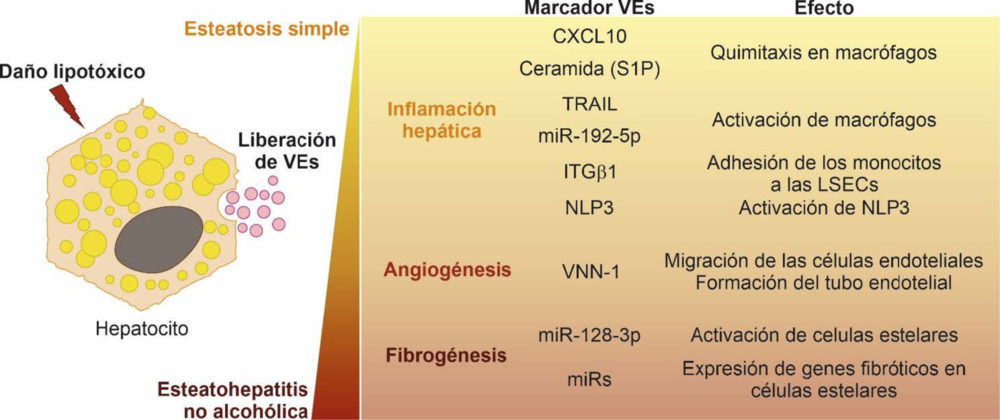
Figure 2. Eventos de señalización mediados por las vesículas extracelulares durante la lipotoxicidad de los hepatocitos.
3.1.1 Hep-VEs implicadas en la inflamación del hígado
El reclutamiento de macrófagos en el hígado contribuye a la respuesta inflamatoria durante la EHNA (49). Sin embargo, no está claro cómo la lipotoxicidad de los hepatocitos promueve la quimiotaxis, la activación y la inflamación hepática de los macrófagos, siendo todos estos procesos patogénicos esenciales en la progresión de la EHGNA.
Ibrahim y colaboradores demostraron que la señalización lipotóxica proapoptótica desencadenada por la “MLK3” (del inglés mixed lineage kinase 3) induce la liberación de Hep-VEs proinflamatorias enriquecidas en un potente ligando, el “CXCL10” (del inglés C-X-C motif chemokine ligand 10) que, a su vez, conduce a la quimiotaxis de los macrófagos al hígado pudiendo activar a las KCs durante la progresión de la EHNA (50). Además, los ratones deficientes en MLK3 alimentados con una dieta rica en grasas, fructosa y colesterol (dieta FFC, del inglés fat-, fructose- and cholesterol-enriched diet) estaban protegidos contra el desarrollo de esteatohepatitis. Este efecto beneficioso se asoció con una reducción en el número de VEs totales en plasma y VEs que contenían CXCL10 en comparación con los ratones de tipo salvaje. En otro estudio, Kakazu y colaboradores demostraron que las Hep- VEs liberadas tras la estimulación con palmitato estaban enriquecidas en ceramida C16:0, una especie lipídica bioactiva generada a partir del palmitato (51). Los Hep-VEs enriquecidos con ceramida C16:0 se liberaban desde los hepatocitos dañados en respuesta a la lipotoxicidad, un efecto mediado por una proteína sensora de estrés en el RE, la “IRE1α” (del inglés, inositol requiring enzyme 1α). Las VEs inducidas por el tratamiento con palmitato además contenían esfingosina 1- fosfato (S1P, del inglés sphingosine 1-phosphate), un metabolito de la ceramida que activa su receptor en los macrófagos, por lo que también actuaban como quimioatrayantes de estos. Igualmente, estos autores evidenciaron un aumento de ceramida C16: 0 en la sangre de ratones y humanos con EHNA. En resumen, estos datos proporcionan una asociación mecanística entre el estrés lipotóxico del RE y la patogénesis de la enfermedad a través de las VEs y sugieren que el contenido de ceramida C16:0 y S1P en las VEs podría usarse como biomarcador en pacientes con EHNA. En la misma línea, Hirsova y colaboradores publicaron que ante la sobrecarga de lípidos tóxicos, los hepatocitos pueden iniciar la cascada de señalización de la proteína homóloga C/EBP (CHOP, del inglés C/EBP homologous protein) /receptor de muerte 5 (DR5, del inglés death receptor 5)/caspasa-8/caspasa-3 que resulta en la activación de la proteína quinasa asociada a Rho 1 (ROCK1, del inglés Rho-associated protein kinase 1) y la liberación de VEs que expresan TRAIL en su superficie (31). Estas VEs al llevar TRAIL en su superficie eran capaces de activar macrófagos derivados de la médula ósea de ratón hacia un fenotipo inflamatorio (M1) a través de la señalización mediada por el factor nuclear κb (NFκb, del inglés nuclear factor-κb). Los autores también demostraron que la liberación de Hep-VEs y, por lo tanto, la activación de los macrófagos disminuía al inactivar la vía de señalización de DR5 o al usar inhibidores de ROCK1. Asimismo, encontraron que la inhibición de ROCK1 en ratones con EHNA conducía a una reducción de los niveles de VEs circulantes lo que iba asociado a menor daño hepático, inflamación y fibrosis.
Por otro lado, Guo y colaboradores realizaron un estudio que proporciona datos sobre el mecanismo por el cual las Hep-VEs lipotóxicas pueden regular la adhesión de los monocitos de sangre periférica a las LSECs y su reclutamiento y retención en el hígado durante la EHNA (52). Estos autores descubrieron que la integrina b1 (ITGb1), una molécula de adhesión celular altamente expresada en los hepatocitos, juega un papel importante en la progresión de la EHNA. El daño lipotóxico en los hepatocitos activa la ITGb1 y facilita su tráfico endocítico y su liberación en VEs promoviendo así la adhesión de los monocitos a las LSEC, evento esencial en la inflamación hepática. También demostraron que el bloqueo de ITGb1 en ratones alimentados con una dieta FFC mejora la inflamación, las lesiones y la fibrosis del hígado. Por tanto, estos autores proponen que reducir la capacidad de las LSEC para reclutar monocitos proinflamatorios dañinos mediante la inhibición de ITGb1 puede servir como una estrategia terapéutica antiinflamatoria para combatir la EHNA. Por otro lado, Cannito y colaboradores probaron que las VEs secretadas por los hepatocitos cargados de grasa que experimentan lipotoxicidad pueden activar directamente el sistema inflamasoma “NLRP3” (del inglés, nucleotide-binding oligomerization domain-like receptor protein 3) tanto en hepatocitos como en macrófagos, lo que resulta en la activación de la caspasa 1 y producción de pro- interleuquina-1β (IL-1β) y pro-interleuquina-18, lo que finalmente conduce a una liberación significativa de IL-1β (53). Dado que se ha sugerido que la liberación de VEs por las células lipotóxicas y la posterior activación del inflamasoma NLRP3 contribuyen a la progresión de la EHGNA, estos datos destacan la existencia de un novedoso vínculo entre la lipotoxicidad y las respuestas inflamatorias.
Cabe mencionar también el estudio reciente realizado por Liu y colaboradores que muestra que los hepatocitos lipotóxicos liberan exosomas enriquecidos en el miR-192-5p que activan a los macrófagos proinflamatorios y producen una inflamación hepática a través de la regulación negativa de la vía de señalización Rictor/Akt/FoxO1 (54). Además, en pacientes con EHGNA, los niveles séricos de miR-192-5p se correlacionaron positivamente con la inflamación hepática y la progresión de la enfermedad. Igualmente, los niveles séricos de miR-192-5p y el número de macrófagos M1, así como los niveles de expresión de mediadores proinflamatorios hepáticos, se correlacionaron con la progresión de la enfermedad en la EHGNA inducida por una dieta alta en grasas y colesterol (HFHCD, del inglés high-fat, high-cholesterol diet) en ratas. Por tanto, este estudio propone el miR-192-5p exosómico sérico como potencial biomarcador no invasivo y diana terapéutica para la EHNA.
3.1.2 Hep-VEs implicadas en la angiogénesis
La angiogénesis es una característica patológica de la EHNA y juega un papel central en la progresión de la enfermedad hacia estadios más avanzados. Sin embargo, los mecanismos moleculares y de señalización que inducen la angiogénesis, así como el vínculo potencial entre la lipotoxicidad y la angiogénesis, siguen sin entenderse completamente.
El estudio publicado por Povero y colaboradores nos proporciona evidencias de que los hepatocitos lipotóxicos sobrecargados de AGL saturados secretan señales proangiogénicas (55). Estos autores identificaron Hep-VEs cargadas con Vanin-1 (VNN1), una ectoenzima epitelial con propiedades reconocidas en la migración y adherencia celular, que inducían la migración de células endoteliales y la formación de tubos vasculares, dos procesos necesarios para la angiogénesis. Cabe destacar que las VEs secretadas por células HepG2 deficientes en VNN1 fracasaron en inducir la migración de células endoteliales y la formación de tubos endoteliales. Asimismo, la administración de siRNA dirigidos a VNN1 a ratones alimentados con una dieta deficiente en metionina y colina protegió contra la angiogénesis patológica inducida por la EHNA en el hígado. En conjunto, estos hallazgos describen un mecanismo que vincula la lipotoxicidad de los hepatocitos con la angiogénesis e identifica una potencial diana terapéutica para desarrollar nuevas estrategias antiangiogénicas para el tratamiento de la EHNA, así como un biomarcador circulante de daño hepático.
3.1.3 Hep-VEs implicadas en la fibrosis
Las HSCs desempeñan un papel crucial durante la fibrosis hepática en la EHGNA avanzada (56). En el desarrollo de la esteatosis hepática, las HSCs se activan y expresan varios marcadores de fibrosis como el fator “TGF-b” (del inglés, transforming growth factor b), “TIMP-1” y “TIMP-2” (del inglés, tissue inhibitor of metalloproteinases 1 and 2) y “MMP2” (del inglés, matrix-metallo-proteinase-2) (57). Sin embargo, el desencadenante de la activación de las HSCs en la EHGNA aún está siendo investigado. Los estudios que se mencionan a continuación sugieren que las VEs pueden desempeñar funciones importantes en la comunicación entre los hepatocitos y las HSCs durante la progresión de simple esteatosis a EHNA, y las identifican como posibles dianas moleculares para terapias antifibróticas.
En otro estudio Povero y colaboradores demuestran mediante la inducción genes profibrogénicos, como el colágeno-I, la α-actina del músculo liso y TIMP2, conjuntamente con respuestas de proliferación, quimiotaxis y cicatrización de heridas, que las VEs derivadas de hepatocitos cargados de lípidos se internalizan en las HSCs induciendo el cambio fenotípico de células quiescentes a activadas, paso necesario para el desarrollo de la fibrosis hepática (55). Estos autores además determinaron que estos cambios iban asociados con la presencia del miR-128-3p como cargo de las VEs, el cual está descrito que regula la expresión de varias proteínas involucradas en la fibrosis hepática y la activación de las HSCs, así como la de “PPAR-γ” (del inglés, peroxisome proliferator- activated receptor gamma) el cual se ha propuesto con anterioridad como mediador en el mantenimiento de una fenotipo inactivo de las HSCs en el hígado normal (58). Es interesante señalar que la exposición de las HSCs a VEs carentes de miR-128-3p resultó en la regulación negativa de los marcadores profibrogénicos y en la regulación positiva de PPAR-γ. Igualmente, las VEs carentes de miR-128-3p atenuaron la proliferación y migración de las HSC. En esta misma línea, Lee y colaboradores demostraron que los hepatocitos tratados con ácido palmítico (PA, del inglés palmitic acid) liberaban más exosomas con un perfil de miRNAs alterado capaces de aumentar los niveles de expresión de genes profibróticos en las HSCs. Además, se confirmó que en los exosomas de células tratadas con PA, la expresión del miRNA-122, uno de los miRNAs más abundantes en el hígado (59, 60), aumentaba conjuntamente con la expresión del miRNA-192 también asociado con la progresión de la EHNA y la fibrosis (61, 62). Además, en el trabajo también se demuestra que la transfección directa del miRNA-192 en las HSCs aumentaba la expresión de marcadores de fibrosis. Por otro lado, los autores encontraron que la expresión de los miRNA-122 y miRNA-192 se veía incrementada en los exosomas circulantes de pacientes con EHGNA avanzada en comparación con aquellos pacientes en etapas tempranas de la enfermedad (63). Por tanto, en este artículo finalmente se sugiere que el perfil de miRNAs de los exosomas circulantes podría ser utilizado como biomarcador para el diagnóstico de EHGNA o EHNA avanzada.
En conclusión, estos estudios señalan la relevancia de las Hep-VEs en la modulación de las respuestas de las células no parenquimales del hígado, incluidas las LSECs, las HSCs y las KCs, en un modelo de múltiples impactos que finalmente resulta en la progresión acelerada de la EHGNA.
3.2 Hep-VEs y complicaciones asociadas a la EHGNA
El efecto nocivo de la EHGNA no solo está limitado al daño de la función hepática en los procesos metabólicos y de detoxificación entre otros, sino que también proporciona un riesgo independiente para el desarrollo de aterosclerosis y otras ECV relacionadas, que suponen la principal causa de muerte de estos pacientes (64). Aunque las evidencias clínicas relacionan la EHGNA y la ECV, aún es necesario describir los mecanismos moleculares subyacentes. Jiang y colaboradores (65) han sido pioneros en abordar el papel potencial que desempeñan las Hep-VEs en la inflamación endotelial y la aterogénesis en el contexto de la EHGNA. Estos autores proponen el miR-1 como mediador del efecto proinflamatorio de las VEs ya que induce la regulación negativa del factor “KLF4” (del inglés, Kruppel-like factor 4), un regulador transcripcional de la homeostasis vascular, y la activación de la vía mediada por el factor de transcripción NF-κB en las células endoteliales. Además, la inhibición del miR-1 con un antagonista específico en un modelo animal de aterosclerosis con hígado graso, (ratones deficientes en ApoE alimentados con una dieta alta en grasa (HFD, del inglés high fat diet)), conseguía suprimir el crecimiento de las células del músculo liso vascular, estabilizó las placas aterogénicas y redujo la inflamación endotelial, lo que en conjunto conducía a una notable mitigación en la formación de la placa aterogénica. Este estudio proporciona una evidencia convincente que implica a las Hep-VEs en la comunicación entre el hígado y la vasculatura en la EHGNA, y también describe un mecanismo molecular subyacente en el desarrollo de la enfermedad cardiometabólica.
Tal como se ha mencionado previamente, la esteatosis hepática a través de la acumulación aberrante de TG en los hepatocitos, constituye el primer impacto en el desarrollo de la EHGNA. La comunicación entre tejidos con importante función metabólica como el hígado y el tejido adiposo regula la distribución de TGs en el organismo, lo cual es fundamental para mantener la homeostasis metabólica (66). Un estudio reciente sugiere que, en el contexto de la sobrecarga de lípidos, el hígado se comunica con el tejido adiposo a través de VEs que contienen miRNAs específicos (67). Los autores proponen un mecanismo de comunicación entre órganos donde el hígado en respuesta a la sobrecarga de lípidos envía la señal al tejido adiposo de modular adaptaciones metabólicas con el objetivo de contrarrestar el excesivo depósito de lípidos e impulsar la redistribución de TGs para mantener la homeostasis sistémica. A nivel molecular, el trabajo responsabiliza a una enzima de la vía del mevalonato, la “GGPPS” (del inglés, geranylgeranyl diphosphate synthase) importante en la regulación de la homeostasis de la glucosa y la sensibilidad a la insulina, en la secreción de Hep-VEs que contienen los miRNAs específicos. Además, en este estudio se demuestra que la expresión de Ggpps inducida por el consumo agudo y crónico de una dieta HFD permite la geranilgeranilación de la Rab-GTPAsa Rab27A que, a su vez, aumenta la secreción de VEs. Entre los miRNAs de estas VEs se encuentra let-7e-5p que aumenta el almacenamiento de lípidos en los adipocitos mediante el incremento de la lipogénesis y la inhibición de la oxidación de lípidos a través del eje alet-7e-5p-Pgc1α. Además, este fenómeno se inhibe en ratones deficientes para Ggpps en el hígado como consecuencia de una secreción disminuida de Hep-VEs. Por tanto, este estudio sugiere la existencia de un eje de señalización tejido adiposo-hígado mediado por Hep-VEs que podría ser necesario en las adaptaciones metabólicas de los adipocitos en respuesta a la sobrecarga de lípidos con el fin de mantener la homeostasis sistémica durante la EHGNA.
4. VEs CIRCULANTES COMO BIOMARCADORES EN EL DIAGNÓSTICO DE LA EHGNA
Como se ha indicado con anterioridad, la biopsia hepática sigue siendo el procedimiento empleado en el diagnóstico, la estratificación y el seguimiento de los pacientes con EHGNA. Sin embargo, esta es cara, altamente invasiva e inexacta debido a errores en la toma de la muestra y, además, conlleva cierta morbilidad y un, aunque poco común, riesgo de mortalidad. En conjunto, todas estas características convierten a la biopsia hepática en inadecuada para su uso rutinario en personas con riesgo de EHGNA (68, 69). Es importante señalar que la falta de rastreo sistemático de la EHGNA ha conducido a un infradiagnóstico masivo de pacientes que progresan hacia una EHNA avanzada o cirrosis, etapas más graves e irreversibles de la enfermedad.
Actualmente, se están utilizando varios métodos no invasivos en la práctica clínica para evaluar la EHGNA con el fin de mitigar la necesidad de la biopsia hepática. Estos incluyen técnicas de imagen como la RM y la elastografía basada en ultrasonido, y el análisis de las enzimas hepáticas séricas como marcadores subyacentes a la inflamación y función hepática. No obstante, estas técnicas carecen de sensibilidad y especificidad suficiente para la detección de las primeras etapas de la EHGNA y no siempre se correlacionan con la gravedad o extensión de la lesión y/o inflamación hepatocelular (70, 71). Los marcadores ideales de la enfermedad deberían reflejar no solo la presencia de EHGNA, sino también su gravedad, lo que es vital para el diagnóstico temprano y la adecuada estatificación de la enfermedad (45). Esta utilidad diagnóstica podría proyectarse además en un tratamiento temprano de la EHGNA dirigido a disminuir la incidencia de EHNA y cirrosis.
En este contexto, las VEs circulantes pueden representar un biomarcador sanguíneo no invasivo óptimo proporcionando una biopsia líquida para el diagnóstico de la EHGNA (35). La Tabla 1 recoge los posibles biomarcadores propuestos basados en las VEs para el diagnóstico de la EHGNA.
Hoy día se han descrito varios biomarcadores basados en proteínas de las VEs para el daño hepático de la EHGNA (72), EHNA (31, 50) o CHC (73-76), aunque hasta la fecha la mayoría de los estudios se han centrado en caracterizar los ácidos nucleicos asociados a las VEs, especialmente los miRNAs y particularmente en el CHC (77-80). Además de su utilidad en el reconocimiento de neoplasias malignas del hígado, los miRNAs asociados a las VEs también pueden servir como biomarcadores en enfermedades hepáticas no malignas como la fibrosis inducida por la EHNA (81).
Además, debido a las interacciones específicas entre las VEs y las células/tejido diana, los biomarcadores de las VEs basados en lípidos también podrían ser importantes en el diagnóstico de la EHGNA, aunque menos del 3% de los lípidos circulantes se transportan en las VEs (82). Los únicos datos disponibles en este área se muestran en el estudio antes mencionado realizado por Kakazu y colaboradores que muestra un aumento de las VEs enriquecidos en ceramida C16:0 en ratones y humanos con EHNA (51). No obstante, las VEs circulantes son heterogéneas y no reflejan exclusivamente la contribución específica del hígado. Las VEs extrahepáticas pueden, de hecho, enmascarar las VEs derivadas del hígado que, en última instancia, como hemos revisado, tienen un papel relevante en la patogénesis de la EHGNA. Por tanto, la identificación de marcadores específicos del hígado en las VEs podría facilitar la detección de cargos poco abundantes que generalmente no se detectan, proporcionando así información directa sobre la progresión de la enfermedad, la recuperación y la respuesta al tratamiento. En este contexto, el enriquecimiento de las VEs basado en marcadores específicos del hígado, seguido del análisis del cargo podría representar una buena estrategia para el descubrimiento de biomarcadores en la EHGNA.
4.1 VEs circulantes de origen hepático
Uno de los primeros estudios en abordar este desafío fue realizado por Povero y colaboradores en modelos murinos experimentales de EHGNA inducida por dieta. Los autores observaron que los niveles de VEs circulantes incrementaban con el tiempo durante la progresión de la EHNA y se correlacionaban con varias características histológicas como muerte celular, angiogénesis y fibrosis (83). De acuerdo con hallazgos previos de otros grupos (84, 85), en un análisis posterior, Povero y colaboradores revelaron que las VEs circulantes aisladas de ratones con EHGNA estaban enriquecidas en miR-122 y miR-192, dos miRNAs expresados abundantemente en los hepatocitos. De acuerdo con estos resultados, propusieron que los hepatocitos eran probablemente la principal fuente de VEs circulantes, concluyendo que los Hep-VEs circulantes aumentaban en ratones con EHGNA (83). Lee y colaboradores validaron posteriormente los miR-122 y miR-192 asociados a VEs como biomarcadores de la progresión de la EHNA en sueros de pacientes con EHGNA (63). No obstante, las Hep-VEs, como se señaló anteriormente, también estaban enriquecidas en la enzima VNN-1, que promovió la angiogénesis e indujo daño hepático durante la EHNA, por lo que representa otro biomarcador potencial (55). Además, en el estudio de Liu y colaboradores que se ha mencionado también se observó un incremento de Hep-VEs y de miR-192-5p asociado a las VEs en el suero en otro modelo de EHGNA basado en ratas alimentadas con dieta HFHCD y en pacientes con EHNA, proponiendo por tanto el miR-192-5p como otro biomarcador (54). En un estudio anterior, encontramos que las VEs circulantes derivadas de hepatocitos que contienen DNA mitocondrial (mitoDNA) también aumentaron en el plasma tanto de ratones como de pacientes con EHNA y, lo que es más importante, este trabajo implicó a las VEs en la activación de los macrófagos que estaba mediada por el receptor tipo toll 9 (TLR9, del inglés toll-like receptor 9) (86).
Brodsky y colaboradores abordaron un enfoque distinto a la hora de identificar las VEs de origen hepático en la circulación (87). Los autores aislaron las VEs circulantes enriquecidas en proteínas de origen hepático en pacientes con CHC mediante citometría de flujo e inmunomarcaje contra la carbamoil fosfato sintetasa 1 con el anticuerpo monoclonal “Hep Par 1” (del inglés, hepatocyte paraffin 1), utilizado rutinariamente como marcador tisular del CHC. En este estudio encontraron que los niveles aumentados de las VEs circulantes derivadas de hígado en pacientes con CHC se correlacionaban positivamente con el tamaño de los tumores hepáticos. También se estudiaron las VEs de origen endotelial y se encontró la misma correlación (87). En esta línea, más recientemente Li y colaboradores identificaron las VEs derivadas de los hepatocitos utilizando citometría de flujo a nanoescala, técnica que detecta marcadores de superficie selectivos de los hepatocitos en las VEs como el receptor de asialoglicoproteína 1 (ASGR1, del inglés asialoglycoprotein receptor 1) y el citocromo “CYPE2E1” (del inglés, cytochrome P450 family 2 subfamily E member 1) (88). Al emplear esta tecnología, se encontró un incremento en las Hep-VEs en ratones macho y hembra en una etapa temprana de la EHGNA (12 y 10 semanas de alimentación con dieta FFC) antes de la apreciación histológica de la inflamación, y permanecieron elevadas con el tiempo (24, 36 y 48 semanas). También se analizaron las VEs derivadas de macrófagos y neutrófilos debido al importante papel del sistema inmune en la EHNA. Las VEs derivadas de macrófagos y neutrófilos se elevaron significativamente a las 24 semanas de alimentación con dieta FFC concomitante con la inflamación histológica en el hígado. Además, las VEs derivadas de hepatocitos, macrófagos y neutrófilos se correlacionaron fuertemente con la evaluación histológica de la EHNA y los biomarcadores de EHNA no invasivos basados en RM. También se cuantificaron las VEs derivadas de plaquetas que mostraron un dimorfismo sexual, ya que estaban elevadas en ratones macho a las 12, 24 y 48 semanas de dieta FFC, mientras que en ratones hembra solo se encontraron elevaciones a las 24 semanas (88). Este trabajo constituye el primer informe descriptivo de los cambios cinéticos en las VEs derivadas de hepatocitos, macrófagos, neutrófilos y plaquetas en un modelo de ratón con EHNA.
4.2 VEs circulantes derivadas de células inmunes
Con respecto al papel del sistema inmunológico en la progresión de la EHGNA, es importante destacar el estudio pionero de diagnóstico de la EHGNA en pacientes basado en las VEs publicado por Kornek y colaboradores (89). Estos autores propusieron por primera vez la existencia de una correlación entre la abundancia en circulación de las VEs específicas derivadas de leucocitos y la gravedad de la enfermedad, determinada por los niveles de transaminasas hepáticas y el grado de biopsia (en inglés NAFLD activity score). Hasta la fecha, estos hallazgos aún representan el estudio más convincente con muestras clínicas para el diagnóstico y la estratificación de la EHGNA basada en las VEs. De manera consistente, este estudio ha sido confirmado recientemente por Welsh y colaboradores quienes también reportaron las VEs derivadas de leucocitos como un marcador de la gravedad de la fibrosis hepática en la EHGNA (90). Cabe mencionar que Kornek y colaboradores también observaron que los pacientes con hepatitis C crónica podrían diferenciarse de los pacientes con EHNA utilizando las VEs derivadas de las células inmunes (89). Esta evidencia ha sido respaldada por otro estudio en el que el análisis transcriptómico reveló que los miRNAs derivados de las VEs en el suero estaban regulados positiva o negativamente de acuerdo a las características histológicas de la enfermedad, como la inflamación y fibrosis, diferenciando así múltiples etiologías de enfermedad hepática (91). En otro estudio, se analizaron tanto las Hep-VEs circulantes como las VEs derivadas de células inmunes (92). Se encontró que las VEs derivadas de hepatocitos o de origen mieloide aumentaban en los pacientes con cirrosis en comparación con los individuos sanos. En pacientes con cirrosis, las Hep-VEs plasmáticas contenían niveles elevados de citoqueratina 18 (CK18) en comparación con las de individuos sanos. Además, la gravedad de la cirrosis se correlacionó con los niveles de VEs leucoendoteliales y Hep-VEs (92).
En conjunto, todos estos estudios han establecido una base sólida para el descubrimiento de biomarcadores basados en las VEs para el diagnóstico de la EHGNA. Sin embargo, debido a las notables limitaciones actuales, todavía queda un largo camino por recorrer antes de que las investigaciones con las VEs tengan utilidad traslacional. Además de la especificidad de la enfermedad y el tejido, la falta de una estandarización de los métodos para el aislamiento de las VEs y las directrices relacionadas con la recolección y el manejo de muestras, pueden interferir con el análisis posterior, lo que genera una alta variabilidad que complica la reproducibilidad y validación de las VEs como biomarcadores (35, 45).
5. CONCLUSIONES
En esta revisión, hemos resumido algunos de los estudios más recientes y originales que investigan el papel clave de las VEs liberadas por hepatocitos dañados (Hep-VEs) al dirigirse a células no parenquimales como las HSCs, LSECs y macrófagos. Este interactoma vincula la lipotoxicidad con la inflamación y la angiogénesis, eventos relevantes en la progresión de la EHGNA a EHNA. Además, compilamos varios estudios sobre el interés creciente de las Hep-VEs liberadas a la circulación sistémica como posibles biomarcadores para el diagnóstico y la estatificación de la EHGNA. Sin embargo, aún se necesitan más estudios que examinen los mecanismos moleculares adicionales involucrados en la biogénesis y liberación de las VEs y las alteraciones que causan en las células diana, así como la identificación de cargos con valor potencial como biomarcadores para el diagnóstico no invasivo y el seguimiento de la progresión de la enfermedad.
6. Abreviaturas
AGLs, ácidos grasos libres; ASGR1, del inglés asialoglycoprotein receptor 1; CHC, carcinoma hepatocelular; CHOP, del inglés C/EBP homologous protein; CK18, citoqueratina 18; CXCL10, del inglés C-X-C motif chemokine ligand 10; CYP2E1, del inglés cytochrome P450 family 2 subfamily E member 1; DAMPs, del inglés damage-associated molecular patterns; DR5, del inglés death receptor 5; D2M, diabetes mellitus tipo 2; ECV, enfermedad cardiovascular; EHGNA, enfermedad de hígado graso no alcohólica; EHNA, esteatohepatitis no alcohólica; FFC diet, del inglés fat-, fructose- and cholesterol-enriched diet; GGPPS, del inglés geranylgeranyl diphosphate synthase; Hep-VEs, VEs derivadas de hepatocitos; Hep Par 1, del inglés hepatocyte paraffin 1; HFD, del inglés high fat diet; HFHCD, del inglés high-fat, high-cholesterol diet; HSCs, del inglés hepatic stellate cells; IRE1α, del inglés inositol requiring enzyme 1α; ITGβ1, integrina β1; IL-1β, interleuquina-1β; ILV, del inglés intraluminal vesicle; KCs, del inglés Kupffer cells; KLF4, del inglés Kruppel-like factor 4; LSECs, del inglés liver sinusoidal endothelial cells; miR, del inglés mature form of the miRNA; MISEV, del inglés Minimal Information for Studies of Extracellular Vesicles; mitoDNA, DNA mitocondrial; MLK3, del inglés mixed lineage kinase 3; MMP2, del inglés, matrix-metallo- proteinase-2; MVBs, del inglés multivesicular bodies; NLRP3, del inglés nucleotide-binding oligomerization domain-like receptor protein 3; NFκB, del inglés nuclear factor-κB; PA, del inglés palmitic acid; PPAR-γ, del inglés peroxisome proliferator-activated receptor gamma; RE, retículo endoplásmico; RM, resonancia magnética; ROCK1, del inglés rho-associated protein kinase 1; S1P, del inglés sphingosine 1-phosphate; TGF-β, del inglés transforming growth factor β; TG, triglicéridos; TIMP, del inglés tissue inhibitor of metalloproteinases; TLR9, del inglés toll-like receptor 9; TRAIL, del inglés tumor necrosis factor-related apoptosis-inducing ligand; VEs, del inglés extracellular vesicles; VNN1, vanin-1.
Conflicto de intereses
Los autores declaran que este artículo de revisión se llevó a cabo en la ausencia de cualquier relación comercial o financiera que pudiera suponer un potencial conflicto de interés.
7. REFERENCIAS
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73-84.
- Fitzpatrick E, Dhawan A. Childhood and Adolescent Nonalcoholic Fatty Liver Disease: Is It Different from Adults? J Clin Exp Hepatol. 2019;9(6):716-22.
- Berardis S, Sokal E. Pediatric non-alcoholic fatty liver disease: an increasing public health issue. Eur J Pediatr. 2014;173(2):131-9.
- Mundi MS, Velapati S, Patel J, Kellogg TA, Abu Dayyeh BK, Hurt RT. Evolution of NAFLD and Its Management. Nutr Clin Pract. 2020;35(1):72-84.
- Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol. 2017;23(1):1-12.
- Adams LA, Ratziu V. Non-alcoholic fatty liver – perhaps not so benign. J Hepatol. 2015;62(5):1002-4.
- Pai RK. NAFLD Histology: a Critical Review and Comparison of Scoring Systems. Current Hepatology Reports. 2019;18(4):473-81.
- Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038-48.
- Tsochatzis EA, Papatheodoridis GV, Archimandritis AJ. Adipokines in nonalcoholic steatohepatitis: from pathogenesis to implications in diagnosis and therapy. Mediators Inflamm. 2009;2009:831670.
- Spencer MD, Hamp TJ, Reid RW, Fischer LM, Zeisel SH, Fodor AA. Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology. 2011;140(3):976-86.
- Ahrens M, Ammerpohl O, von Schonfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18(2):296-302.
- Kirpich IA, Marsano LS, McClain CJ. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin Biochem. 2015;48(13-14):923-30.
- Tilg H, Burcelin R, Tremaroli V. Liver tissue microbiome in NAFLD: next step in understanding the gut-liver axis? Gut. 2020.
- Jensen MD, Ryan DH, Apovian CM, Ard JD, Comuzzie AG, Donato KA, et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol. 2014;63(25 Pt B):2985-3023.
- Montesi L, El Ghoch M, Brodosi L, Calugi S, Marchesini G, Dalle Grave R. Long-term weight loss maintenance for obesity: a multidisciplinary approach. Diabetes Metab Syndr Obes. 2016;9:37-46.
- Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol. 2020.
- Eslam M, Sanyal AJ, George J, International Consensus P. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology. 2020;158(7):1999-2014 e1.
- Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61(5):1294-303.
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
- van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213-28.
- Mathieu M, Martin-Jaular L, Lavieu G, Thery C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol. 2019;21(1):9-17.
- Wang Y, Wang Q, Wei X, Shao J, Zhao J, Zhang Z, et al. Global scientific trends on exosome research during 2007-2016: a bibliometric analysis. Oncotarget. 2017;8(29):48460-70.
- Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913.
- Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
- Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. 1983;97(2):329-39.
- Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985;101(3):942-8.
- Anand S, Samuel M, Kumar S, Mathivanan S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim Biophys Acta Proteins Proteom. 2019;1867(12):140203.
- Lasser C, Jang SC, Lotvall J. Subpopulations of extracellular vesicles and their therapeutic potential. Mol Aspects Med. 2018;60:1-14.
- Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329-35.
- Menck K, Scharf C, Bleckmann A, Dyck L, Rost U, Wenzel D, et al. Tumor-derived microvesicles mediate human breast cancer invasion through differentially glycosylated EMMPRIN. J Mol Cell Biol. 2015;7(2):143-53.
- Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology. 2016;150(4):956-67.
- Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, et al. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. 2012;189(6):2833-42.
- Cai Z, Yang F, Yu L, Yu Z, Jiang L, Wang Q, et al. Activated T cell exosomes promote tumor invasion via Fas signaling pathway. J Immunol. 2012;188(12):5954-61.
- Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3.
- Szabo G, Momen-Heravi F. Extracellular vesicles in liver disease and potential as biomarkers and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2017;14(8):455-66.
- Murphy DE, de Jong OG, Brouwer M, Wood MJ, Lavieu G, Schiffelers RM, et al. Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking. Exp Mol Med. 2019;51(3):1-12.
- Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles – endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014;1846(1):75-87.
- Villa F, Quarto R, Tasso R. Extracellular Vesicles as Natural, Safe and Efficient Drug Delivery Systems. Pharmaceutics. 2019;11(11).
- Balaphas A, Meyer J, Sadoul R, Morel P, Gonelle-Gispert C, Buhler LH. Extracellular vesicles: Future diagnostic and therapeutic tools for liver disease and regeneration. Liver Int. 2019;39(10):1801-17.
- Borrelli DA, Yankson K, Shukla N, Vilanilam G, Ticer T, Wolfram J. Extracellular vesicle therapeutics for liver disease. J Control Release. 2018;273:86-98.
- Gao J, Dong X, Wang Z. Generation, purification and engineering of extracellular vesicles and their biomedical applications. Methods. 2020;177:114-25.
- Momen-Heravi F, Balaj L, Alian S, Mantel PY, Halleck AE, Trachtenberg AJ, et al. Current methods for the isolation of extracellular vesicles. Biol Chem. 2013;394(10):1253-62.
- Larssen P, Wik L, Czarnewski P, Eldh M, Lof L, Ronquist KG, et al. Tracing Cellular Origin of Human Exosomes Using Multiplex Proximity Extension Assays. Mol Cell Proteomics. 2017;16(3):502-11.
- Samuelson I, Vidal-Puig AJ. Fed-EXosome: extracellular vesicles and cell-cell communication in metabolic regulation. Essays Biochem. 2018;62(2):165-75.
- Ban LA, Shackel NA, McLennan SV. Extracellular Vesicles: A New Frontier in Biomarker Discovery for Non-Alcoholic Fatty Liver Disease. Int J Mol Sci. 2016;17(3):376.
- Masyuk AI, Masyuk TV, Larusso NF. Exosomes in the pathogenesis, diagnostics and therapeutics of liver diseases. J Hepatol. 2013;59(3):621-5.
- Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut. 2018;67(5):963-72.
- Hirsova P, Ibrahim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res. 2016;57(10):1758-70.
- Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. 2012;302(11):G1310-21.
- Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. 2016;63(3):731-44.
- Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched proinflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res. 2016;57(2):233-45.
- Guo Q, Furuta K, Lucien F, Gutierrez Sanchez LH, Hirsova P, Krishnan A, et al. Integrin beta1-enriched extracellular vesicles mediate monocyte adhesion and promote liver inflammation in murine NASH. J Hepatol. 2019;71(6):1193-205.
- Cannito S, Morello E, Bocca C, Foglia B, Benetti E, Novo E, et al. Microvesicles released from fat-laden cells promote activation of hepatocellular NLRP3 inflammasome: A pro-inflammatory link between lipotoxicity and non-alcoholic steatohepatitis. PLoS One. 2017;12(3):e0172575.
- Liu XL, Pan Q, Cao HX, Xin FZ, Zhao ZH, Yang RX, et al. Lipotoxic Hepatocyte-Derived Exosomal miR-192-5p Activates Macrophages via Rictor/Akt/FoxO1 Signaling in NAFLD. Hepatology. 2019.
- Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat du Jeu X, Mulya A, et al. Lipid-induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin-1 for uptake by endothelial cells. Sci Signal. 2013;6(296):ra88.
- McKee C, Sigala B, Soeda J, Mouralidarane A, Morgan M, Mazzoccoli G, et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci Rep. 2015;5:8812.
- Wobser H, Dorn C, Weiss TS, Amann T, Bollheimer C, Buttner R, et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009;19(8):996-1005.
- Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119(2):466-78.
- Jopling C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012;9(2):137-42.
- Castoldi M, Vujic Spasic M, Altamura S, Elmen J, Lindow M, Kiss J, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest. 2011;121(4):1386-96.
- Pirola CJ, Fernandez Gianotti T, Castano GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015;64(5):800-12.
- Becker PP, Rau M, Schmitt J, Malsch C, Hammer C, Bantel H, et al. Performance of Serum microRNAs -122, -192 and -21 as Biomarkers in Patients with Non-Alcoholic Steatohepatitis. PLoS One. 2015;10(11):e0142661.
- Lee YS, Kim SY, Ko E, Lee JH, Yi HS, Yoo YJ, et al. Exosomes derived from palmitic acidtreated hepatocytes induce fibrotic activation of hepatic stellate cells. Sci Rep. 2017;7(1):3710.
- Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363(14):1341-50.
- Jiang F, Chen Q, Wang W, Ling Y, Yan Y, Xia P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol. 2020;72(1):156-66.
- Azzu V, Vacca M, Virtue S, Allison M, Vidal-Puig A. Adipose Tissue-Liver Cross Talk in the Control oef Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology. 2020.
- Zhao Y, Zhao MF, Jiang S, Wu J, Liu J, Yuan XW, et al. Liver governs adipose remodelling via extracellular vesicles in response to lipid overload. Nat Commun. 2020;11(1):719.
- Sumida Y, Nakajima A, Itoh Y. Limitations of liver biopsy and non-invasive diagnostic tests for the diagnosis of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2014;20(2):475-85.
- Sanyal AJ, Friedman SL, McCullough AJ, Dimick-Santos L, American Association for the Study of Liver D, United States F, et al. Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases-U.S. Food and Drug Administration Joint Workshop. Hepatology. 2015;61(4):1392-405.
- Kim WR, Flamm SL, Di Bisceglie AM, Bodenheimer HC, Public Policy Committee of the American Association for the Study of Liver D. Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology. 2008;47(4):1363-70.
- Eguchi A, Feldstein AE. Extracellular vesicles in non-alcoholic and alcoholic fatty liver diseases. Liver Res. 2018;2(1):30-4.
- Moratti E, Vezzalini M, Tomasello L, Giavarina D, Sorio C. Identification of protein tyrosine phosphatase receptor gamma extracellular domain (sPTPRG) as a natural soluble protein in plasma.PLoS One. 2015;10(3):e0119110.
- Taleb RSZ, Moez P, Younan D, Eisenacher M, Tenbusch M, Sitek B, et al. Quantitative proteome analysis of plasma microparticles for the characterization of HCV-induced hepatic cirrhosis and hepatocellular carcinoma. Proteomics Clin Appl. 2017;11(11-12).
- Arbelaiz A, Azkargorta M, Krawczyk M, Santos-Laso A, Lapitz A, Perugorria MJ, et al. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology. 2017;66(4):1125-43.
- Abbate V, Marcantoni M, Giuliante F, Vecchio FM, Gatto I, Mele C, et al. HepPar1-Positive Circulating Microparticles Are Increased in Subjects with Hepatocellular Carcinoma and Predict Early Recurrence after Liver Resection. Int J Mol Sci. 2017;18(5).
- Julich-Haertel H, Urban SK, Krawczyk M, Willms A, Jankowski K, Patkowski W, et al. Cancer-associated circulating large extracellular vesicles in cholangiocarcinoma and hepatocellular carcinoma. J Hepatol. 2017;67(2):282-92.
- Wang H, Hou L, Li A, Duan Y, Gao H, Song X. Expression of serum exosomal microRNA-21 in human hepatocellular carcinoma. Biomed Res Int. 2014;2014:864894.
- Sugimachi K, Matsumura T, Hirata H, Uchi R, Ueda M, Ueo H, et al. Identification of a bona fide microRNA biomarker in serum exosomes that predicts hepatocellular carcinoma recurrence after liver transplantation. Br J Cancer. 2015;112(3):532-8.
- Sohn W, Kim J, Kang SH, Yang SR, Cho JY, Cho HC, et al. Serum exosomal microRNAs as novel biomarkers for hepatocellular carcinoma. Exp Mol Med. 2015;47:e184.
- Fornari F, Ferracin M, Trere D, Milazzo M, Marinelli S, Galassi M, et al. Circulating microRNAs, miR-939, miR-595, miR-519d and miR-494, Identify Cirrhotic Patients with HCC. PLoS One. 2015;10(10):e0141448.
- Chen L, Chen R, Kemper S, Charrier A, Brigstock DR. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am J Physiol Gastrointest Liver Physiol. 2015;309(6):G491-9.
- Watt MJ, Miotto PM, De Nardo W, Montgomery MK. The Liver as an Endocrine Organ-Linking NAFLD and Insulin Resistance. Endocr Rev. 2019;40(5):1367-93.
- Povero D, Eguchi A, Li H, Johnson CD, Papouchado BG, Wree A, et al. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS One. 2014;9(12):e113651.
- Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, et al. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology. 2012;56(5):1946-57.
- Csak T, Bala S, Lippai D, Satishchandran A, Catalano D, Kodys K, et al. microRNA-122 regulates hypoxia-inducible factor-1 and vimentin in hepatocytes and correlates with fibrosis in dietinduced steatohepatitis. Liver Int. 2015;35(2):532-41.
- Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest. 2016;126(3):859-64.
- Brodsky SV, Facciuto ME, Heydt D, Chen J, Islam HK, Kajstura M, et al. Dynamics of circulating microparticles in liver transplant patients. J Gastrointestin Liver Dis. 2008;17(3):261-8.
- Li J, Liu H, Mauer AS, Lucien F, Raiter A, Bandla H, et al. Characterization of Cellular Sources and Circulating Levels of Extracellular Vesicles in a Dietary Murine Model of Nonalcoholic Steatohepatitis. Hepatol Commun. 2019;3(9):1235-49.
- Kornek M, Lynch M, Mehta SH, Lai M, Exley M, Afdhal NH, et al. Circulating microparticles as disease-specific biomarkers of severity of inflammation in patients with hepatitis C or nonalcoholic steatohepatitis. Gastroenterology. 2012;143(2):448-58.
- Welsh JA, Scorletti E, Clough GF, Englyst NA, Byrne CD. Leukocyte extracellular vesicle concentration is inversely associated with liver fibrosis severity in NAFLD. J Leukoc Biol. 2018;104(3):631-9.
- Murakami Y, Toyoda H, Tanahashi T, Tanaka J, Kumada T, Yoshioka Y, et al. Comprehensive miRNA expression analysis in peripheral blood can diagnose liver disease. PLoS One. 2012;7(10):e48366.
- Rautou PE, Bresson J, Sainte-Marie Y, Vion AC, Paradis V, Renard JM, et al. Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis. Gastroenterology. 2012;143(1):166-76 e6.
