Fármacos de nueva aprobación – Newly approved drugs:
(A) TRACTO ALIMENTARIO Y METABOLISMO: Colangitis biliar primaria: Seladelpar (Livdelzi®; FDA). (D) DERMATOLOGÍA: Alopecia areata: Deuruxolitinib (Leqselvi®; FDA). Prurigo nodular: Nemolizumab (Nemluvio®; FDA). Eccema crónico de manos: Delgocitinib (Anzupgo®; EMA). (H) PREPARACIONES HORMONALES SISTÉMICAS: Hipoglucemia: Dasiglucagón (Zegalogue®; EMA). (L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES: Cáncer de pulmón no microcítico: Sugemalimab (Cejemly®; EMA). Cáncer de pulmón no microcítico: Lazertinib (Lazcluze®; FDA). Sarcoma sinovial: Afamitresgene Autoleucel (Tecelra®; FDA). Astrocitoma/Oligodendroglioma: Vorasidenib (Voranigo®; FDA). Linfoma folicular: Odronestamab (Ordspono®; EMA). Linfoma de células T: Denileukin Deftitox (Lymphir®; FDA). Enfermedad crónica de injerto contra huésped: Axatilimab (Niktimvo®; FDA). (N) SISTEMA NERVIOSO: Enfermedad de Alzheimer: Donanemab (Kisunla®; FDA); Benzgalantamina (Zunveyl®; FDA). Enfermedad de Niemann-Pick: Arimoclomol (Miplyffa®; FDA). Enfermedad de Niemann-Pick: Levacetilleucina (Aqneursa®; FDA). Esquizfrenia: Cobenfy (Xanomelina/Trospio®; FDA). (S) ÓRGANOS SENSORIALES: Queratitis por Acanthamoeba: Polihexanida (Akantior®; EMA). (V) VARIOS: Diagnóstico por imagen (PET): Galio [68Ga] + Germanio [68Ge] (GalliaFarm®; EMA). Diagnóstico por imagen (PET): Flortaucipir [18F] (Tauvid®; EMA). Diagnóstico por iomagen (PET): Flurdipiraz [18F] (Flyrcado®; FDA).
(A) TRACTO ALIMENTARIO Y METABOLISMO
Seladelpar (Livdelzi®) Cymabay (FDA, USA)
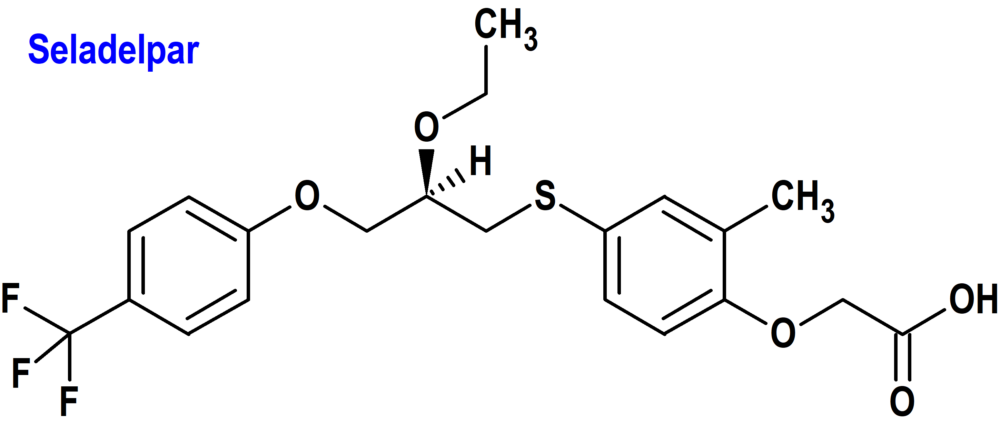
Indicación: Tratamiento de la colangitis biliar primaria (CBP) en combinación con ácido ursodesoxicólico (AUDC) en adultos que han tenido una respuesta inadecuada al AUDC, o como monoterapia en pacientes que no pueden tolerar el AUDC.
Tipo: Medicamento sintético estándar constituido por el ácido 2-[4-[(2R)-2-etoxi-3-[4-(trifluorometil)fenoxi]propil]sulfanil-2-metilfenoxi]acético. Autorizado en Estados Unidos (FDA) el 14 de agosto de 2024 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Agonista del receptor activado por el proliferador de peroxisomas delta (PPARδ). La actividad farmacológica que es potencialmente relevante para los efectos terapéuticos incluye la inhibición de la síntesis de ácidos biliares a través de la activación de PPARδ, que es un receptor nuclear expresado en la mayoría de los tejidos, incluido el hígado. La activación de PPARδ por seladelpar reduce la síntesis de ácidos biliares a través de la regulación negativa dependiente del factor de crecimiento de fibroblastos 21 (FGF21) de CYP7A1, la enzima clave para la síntesis de ácidos biliares a partir del colesterol.
Eficacia clínica: Ensayo aleatorizado, doble ciego y controlado con placebo de XX semanas de duración, que incluyó a XXX pacientes. La variable principal de eficacia fue la tasa (%) de respuesta bioquímica en el mes 12, definida como lograr una fosfatasa alcalina menor a 1,67 veces el límite superior normal (LSN), una disminución de la fosfatasa alcalina mayor o igual al 15 % desde el inicio y una bilirrubina total (TB) menor o igual al LSN (1,1 mg/dL): 62 vs. 20%. La normalización de la fosfatasa alcalina (menor o igual al LSN: 116 U/L) en el mes 12 fue un criterio de valoración secundario clave: 25 vs 0%.
Eventos adversos: Los más comunes y más frecuentes que con placebo son dolor de cabeza (8% vs 3% con placebo), dolor abdominal (7/2%), náuseas (6/5%), distensión abdominal (6/3%) y mareos (5/2%).
(D) DERMATOLOGÍA
Deuruxolitinib (Leqselvi®) Sun (FDA, USA)
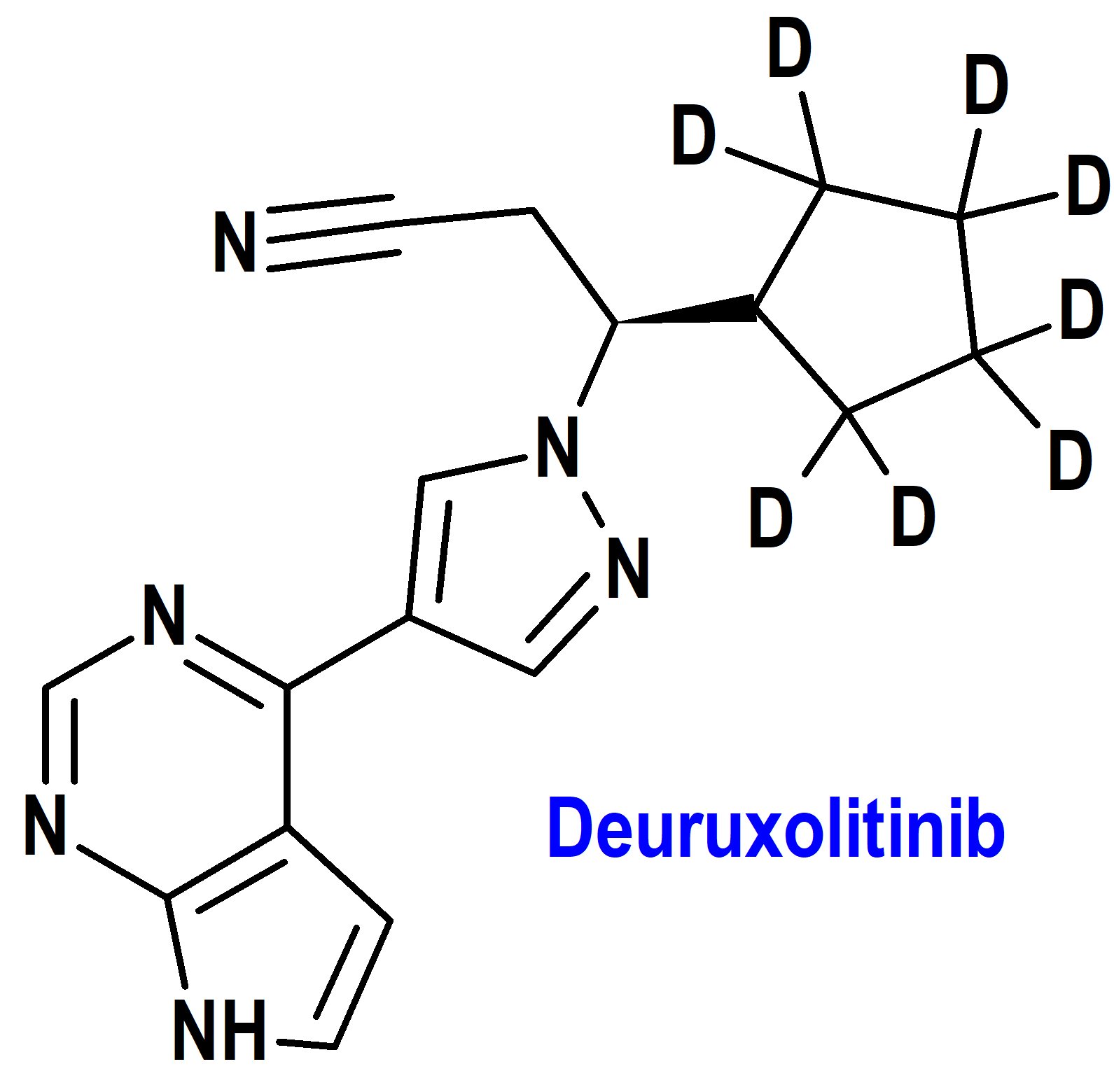
Indicación: Tratamiento de pacientes adultos con alopecia areata severa.
Tipo: Medicamento sintético estándar constituido por (3R)-3-(2,2,3,3,4,4,5,5-octadeuteriociclopentil)-3-[4-(7H-pirrolo[2,3-d]pirimidin-4-il)pirazol-1-il]propanonitrilo, una análogo del ruxolitinib en el que los átomos de hidrógeno (H) del anillo ciclopentánico han sido sustituidos por deuterio (D). Autorizado en Estados Unidos (FDA) el 25 de julio de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la cinasa Janus (JAK). Las JAK median la señalización de una serie de citocinas y factores de crecimiento que son importantes para la hematopoyesis y la función inmunitaria. La señalización de las JAK implica el reclutamiento de STAT (transductores de señales y activadores de la transcripción) a los receptores de citocinas, la activación y posterior localización de los STAT en el núcleo, lo que conduce a la modulación de la expresión génica. Las vías de señalización mediadas por la familia JAK están implicadas en la patogenia de la alopecia areata aunque por el momento no se conoce la relevancia de la inhibición de las enzimas JAK para la eficacia terapéutica.
Eficacia clínica: Dos ensayos clínicos de fase 3, multicéntricos, aleatorizados, doble ciego y controlados con placebo, en un total de 1209 sujetos adultos con alopecia areata que presentaban al menos un 50 % de pérdida de cabello en el cuero cabelludo. El criterio de valoración principal de ambos ensayos evaluó la proporción de sujetos que alcanzaron al menos un 80 % de cobertura capilar en el cuero cabelludo (puntuación SALT ≤20) en la semana 24 (29 vs 1% en el primer estudio y 32 vs. 1% en el segundo).
Eventos adversos: Los más comunes (≥5%) y con mayor frecuencia que con placebo son cefalea (12,4% vs. 9,4% con placebo), acné (10,0/4,3%), nasofaringitis (8,1/6,7%) y elevación de la creatina fosfocinasa sanguínea (5,3/2,9%).
Nemolizumab (Nemluvio®) Galderma (FDA, USA)
Indicación: Tratamiento de adultos con prurigo nodular.
Tipo: Medicamento biológico constituido por una inmunoglobulina G2 (IgG2) modificada humanizada monoclonal de origen recombinante con un peso molecular de 144 kDa. Autorizado en Estados Unidos (FDA) el 12 de agosto de 2024 mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Es un anticuerpo monoclonal humanizado que inhibe la señalización de IL-31 al unirse selectivamente al receptor de IL-31. La IL-31 es una citocina natural que está involucrada en el prurito, la inflamación, la desregulación epidérmica y la fibrosis. El nemolizumab inhibe las respuestas inducidas por IL-31, incluida la liberación de citocinas y quimiocinas proinflamatorias.
Eficacia clínica: Dos ensayos aleatorizados, doble ciego y controlados con placebo que incluyeron un total de 560 sujetos adultos con prurigo nodular (PN). La eficacia se evaluó con la proporción de sujetos con una mejoría de ≥4 desde el inicio en la escala numérica de calificación del prurito máximo (PP-NRS) y con una Evaluación Global del Investigador (EGI) de 0 o 1 (limpio o casi limpio): 22 vs. 2% y 25 vs. 4%, respectivamente; la proporción de sujetos con una EGI de 0 o 1 (limpio o casi limpio): 26/7% y 38/11%; la proporción de sujetos con una mejoría de ≥4 puntos desde el inicio (56/16% y 49/16%), y la proporción de sujetos con PP-NRS <2 (32/4% y 31/7%). Eventos adversos: Los más comunes (≥1%,) y con mayor frecuencia que con placebo son dolor de cabeza (6 vs. 3%), dermatitis atópica (4 vs. 0,5%), eccema (4 vs. 2%) y eccema numular (3 vs. 0%).
Delgocitinib (Anzupgo®) LEO (EMA, UE)
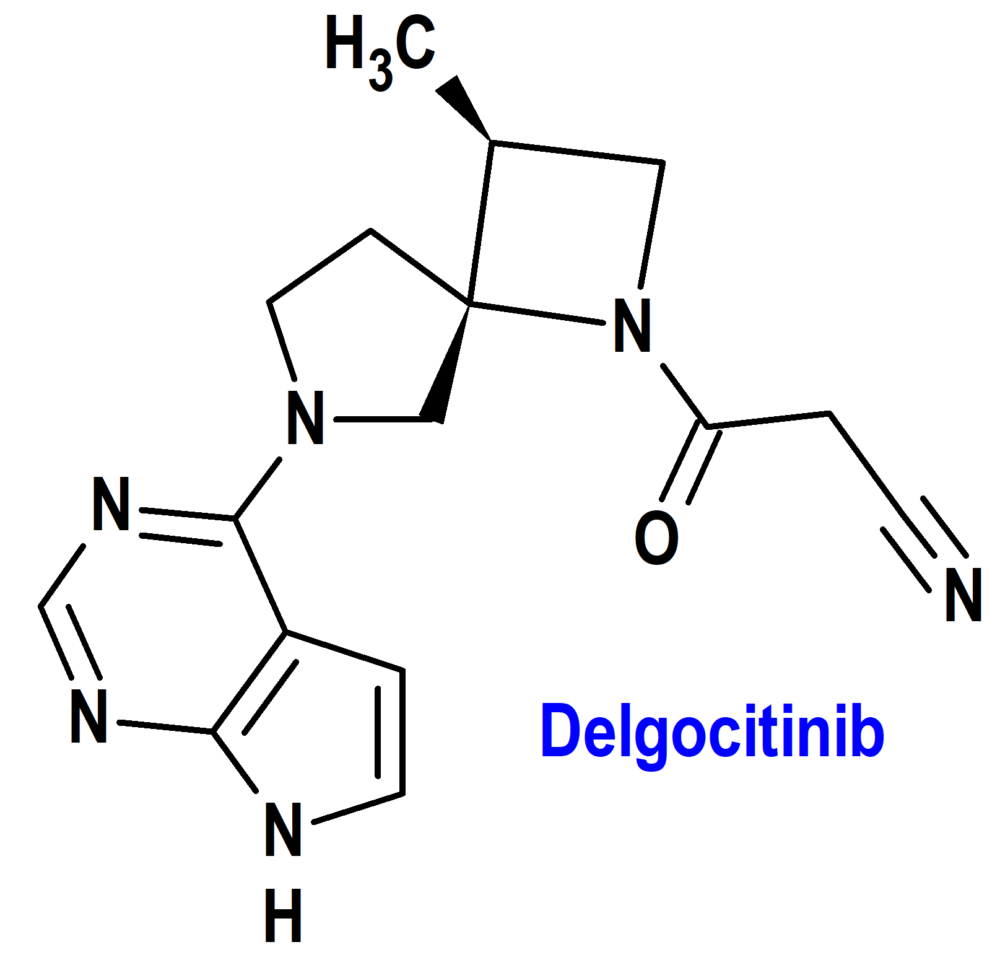
Indicación: Tratamiento del eccema crónico de manos (ECM) de moderado a grave en adultos para los que los corticoesteroides tópicos son inadecuados o inapropiados .
Tipo: Medicamento sintético estándar constituido por el 3-[(3S,4R)-3-metil-6-(7H-pirrolo[2,3-d]pirimidin-4-il)-1,6-diazaspiro[3.4]octan-1-il]-3-oxopropanonitrilo. Autorizado en la Unión Europea (EMA) el 19 de septiembre de 2024; no autorizado previamente en Estados Unidos.
Mecanismo: Inhibidor de las cinasas Janus (JAK) que se dirige a la actividad de los cuatro miembros de la familia JAK de enzimas (JAK1, JAK2, JAK3 y tirosinacinasa 2 o TYK2) de una manera dependiente de la concentración. En células humanas, la inhibición de la vía JAK-STAT por delgocitinib atenúa la señalización de varias citocinas proinflamatorias, como las interleucinas IL-2, IL-4, IL-6, IL-13, IL-21, IL-23, el factor estimulante de las colonias de granulocitos y macrófagos (FEC-GM) y el interferón (IFN)-α), lo que disminuye la respuesta inmunitaria e inflamatoria en células relevantes para la patología del eccema crónico de manos.
Eficacia clínica: Dos estudios pivotales aleatorizados, doble ciego, controlados con placebo (vehículo) y de diseño similar que incluyeron a 960 pacientes mayores de 18 años de edad con eccema crónico de manos de moderado a grave, con una puntuación de 3 o 4 según la Investigator’s Global Assessment for chronic hand eczema, IGA-CHE, y una puntuación de picor según la Hand Eczema Symptom Diary, HESD ≥4 puntos, y que ha persistido durante más de 3 meses o que ha reaparecido dos o más veces en los últimos 12 meses. La variable clínica primaria de eficacia fue la proporción de pacientes que lograron el éxito del tratamiento (puntuación IGA-CHE de 0, ausente, o 1, prácticamente ausente: eritema mínimamente perceptible; con al menos una mejoría de 2 pasos desde el inicio hasta la semana 16): 19,7 vs. 9,9% en el primer estudio y 29,1 vs. 6,9% en el segundo. Otras variables secundarias de eficacia fueron el Índice de gravedad del eccema de manos (Hand Eczema Severity Index, HECSI): la tasa de pacientes que mejorar un 90% fue del 29,5 vs. 12,3% y 31,0 vs. 8,8%, mientras que se registró una mejoría de al menos 4 puntos en el HESD en varios puntos temporales en el 47,2 vs. 24,4% y 44,5 vs. 20,9%.
Eventos adversos: Los más comunes son reacciones en la zona de aplicación (1,0 %), incluyendo dolor, parestesia, prurit y eritema.
(H) PREPARACIONES HORMONALES SISTÉMICAS
Dasiglucagón (Zegalogue®) Zealand (EMA, UE)
Indicación: Tratamiento de la hipoglucemia grave en adultos, adolescentes y niños mayores de 6 años con diabetes mellitus.
Tipo: Medicamento sintético estándar constituido por un análogo del glucagón humano que consta de 29 aminoácidos; contiene siete aminoácidos sustituidos para mejorar la estabilidad física y química en su formulación farmacéutica: (16-(2-metilalanina)(S>X),17-L-alanina(R>A),20-L-alfa-glutamil(Q>E),21-L-alfa-glutamil(D>E),24-L-lisil(Q>K),27-L-alfa-glutamil(M>E),28-L-serina(N>S))glucagón). Autorizado en la Unión Europea (EMA) el 27 de julio de 2024; autorizado previamente en Estados Unidos (FDA) el 22 de marzo de 2021.
Mecanismo: Análogo agonista del receptor glucagón, que aumenta la concentración de glucosa en sangre al activar los receptores de glucagón hepático, estimulando así la descomposición de glucógeno y la liberación de glucosa del hígado. Dasiglucagón solo es eficaz para el tratamiento de la hipoglucemia si está presente suficiente glucógeno hepático por lo que, para prevenir las recaídas de la hipoglucemia, deben administrarse carbohidratos orales a fin de restaurar el glucógeno hepático, cuando el paciente haya respondido al tratamiento. Los pacientes en estado de hambruna, con insuficiencia adrenal, abuso crónico de alcohol o hipoglucemia crónica pueden presentar niveles no adecuados de glucógeno hepático para que la administración de dasiglucagón sea eficaz y estos pacientes deben recibir tratamiento con glucosa.
Eficacia clínica: Tres ensayos multicéntricos, aleatorizados, doble ciego y controlados con placebo en pacientes con diabetes de tipo 1, dos ensayos en pacientes adultos y uno en pacientes pediátricos de 6 a 17 años. La variable principal de eficacia en los 3 ensayos fue el tiempo transcurrido hasta la recuperación de la glucosa en plasma (éxito del tratamiento), definido como un aumento de la glucosa en sangre ≥20 mg/dl desde el momento de la administración, sin intervención adicional en un plazo de 45 minutos. En los dos ensayos en adultos (170 y 44 pacientes), el tiempo de recuperación fue de 10 minutos con dasiglucagon vs. 40 y 35 minutos con placebo, y de 10 vs. 30 minutos en los pacientes pediátricos (31).
Eventos adversos: Los más comunes son náuseas (63%), vómitos (32%) y dolor de cabeza (10%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
Sugemalimab (Cejemly®) SFL (EMA, UE)
Indicación: Tratamiento de primera línea, en combinación con quimioterapia basada en platino, de adultos con cáncer de pulmón no microcítico (CPNM) metastásico sin mutaciones sensibilizantes del EGFR ni aberraciones genómicas tumorales ALK, ROS1 o RET.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal (isotipo IgG4) completamente humano contra el ligando de muerte programada 1 (PD-L1). Autorizado en la Unión Europea (EMA) el 24 de julio de 2024; no autorizado previamente en Estados Unidos (FDA).
Mecanismo: anticuerpo monoclonal inmunoglobulina G4 completamente humano. Se une específicamente al ligando de muerte celular programada 1 (PD-L1), bloqueando así su unión a la PD-1. El PD-L1, cuando se expresa en las células tumorales y en las células inmunitarias que infiltran el tumor, puede contribuir a la inhibición de la respuesta inmunitaria antitumoral. La unión del PD-L1 a la PD-1 y a los receptores CD80 (B7.1) que se encuentran en las células T y en las células presentadoras de antígenos suprime la actividad citotóxica de las células T y su proliferación, así como la producción de citocinas. El bloqueo de las interacciones PD-L1/PD-1 y PD-L1/CD80 libera la inhibición de las respuestas inmunitarias sin inducir citotoxicidad celular dependiente de anticuerpos (CCDA).
Eficacia clínica: Ensayo de fase 3 aleatorizado, doble ciego y controlado con placebo en 579 pacientes adultos ≥18 años con CPNM escamoso o no escamoso, metastásico (estadio IV), histológica o citológicamente confirmado, sin mutaciones sensibilizantes del EGFR ni fusiones ALK, ROS1 o translocaciones RET. La variable primaria de eficacia fue la supervivencia libre de progresión (9,0 vs. 4,9 meses) y la secundaria fue la supervivencia global (25,4 vs. 16,9 meses).
Eventos adversos: Los más frecuentes (≥10%) son anemia (78%), aumento de la aspartato aminotransferasa (34%), aumento de la alanina aminotransferasa (32%), erupción cutánea (26%), hiperlipemia (22%), hiperglucemia (18%), hiponatremia (17%), hipopotasemia (16%), proteinuria (14%), dolor abdominal (14%), fatiga (13%), artralgia (12%), hipoestesia (12%), hipotiroidismo (10%) e hipocalcemia (10%). La incidencia de reacciones adversas de grado ≥3 es del 33%, siendo las más frecuentes anemia (18%), hiponatremia (4,4 %), hipopotasemia (3,0%), hiperlipemia (2,3%) y aumento de la amilasa (2,1%).
Lazertinib (Lazcluze®) Janssen (FDA, USA)
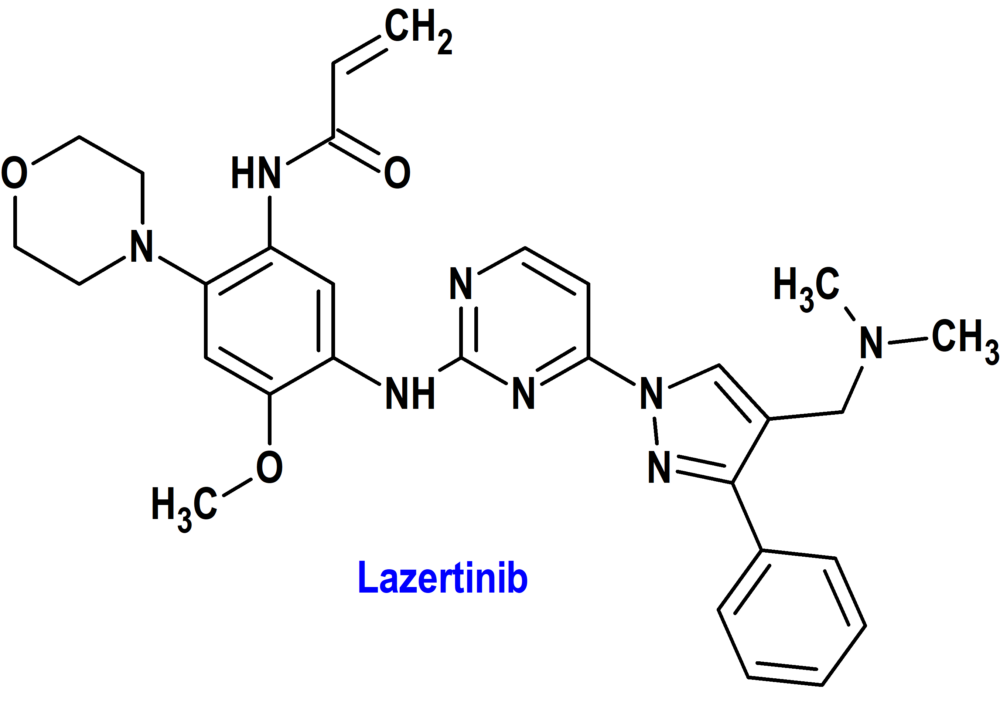
Indicación: En combinación con amivantamab, está indicado para el tratamiento de primera línea de pacientes adultos con cáncer de pulmón de células no pequeñas (CPCNP) localmente avanzado o metastásico con deleciones del exón 19 del receptor del factor de crecimiento epidérmico (EGFR) o mutaciones de sustitución del exón 21 L858R.
Tipo: Medicamento sintético estándar constituido por la N-[5-[[4-[4-[(dimetilamino)metil]-3-fenilpirazol-1-il]pirimidin-2-il]amino]-4-metoxi-2-morfolin-4-ilfenil]prop-2-enamida. Autorizado en Estados Unidos (FDA) el 19 de agosto de 2024 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la cinasa del receptor del factor de crecimiento epidérmico (EGFR) que inhibe las deleciones del exón 19 del EGFR y las mutaciones de sustitución L858R del exón 21 en concentraciones más bajas que el EGFR de tipo salvaje. El tratamiento con lazertinib en combinación con amivantamab aumentó la actividad antitumoral in vivo en comparación con cualquiera de los agentes por separado en CPNM humano con una mutación L858R del EGFR.
Eficacia clínica: Un ensayo multicéntrico, aleatorizado y controlado con placebo de 1074 pacientes con CPNM metastásico o localmente avanzado con mutación de deleción del exón 19 o de sustitución L858R del exón 21 y sin terapia sistémica previa para la enfermedad avanzada. La variable principal de eficacia fue la supervivencia libre de progresión (SLP) evaluada mediante una revisión central independiente ciega para la comparación entre lazertinib con amivantamab y osimertinib (23,7 vs. 16,6 meses).
Eventos adversos: Los más comunes (≥20%) son erupción cutánea, toxicidad en las uñas, reacción relacionada con la infusión (amivantamab), dolor musculoesquelético, edema, estomatitis, eventos tromboembólicos venosos (ETV), parestesia, fatiga, diarrea, estreñimiento, COVID-19, hemorragia, piel seca, disminución del apetito, prurito, náuseas y toxicidad ocular. Las anomalías de laboratorio de grado 3 o 4 más frecuentes (≥2%) fueron disminución de la albúmina, disminución del sodio, aumento de la ALT, disminución del potasio, disminución de la hemoglobina, aumento de la AST, aumento de la GGT y aumento del magnesio. Se produjeron reacciones adversas graves en el 49%, especialmente ETV (11%) y neumonía (4%). Se produjeron reacciones adversas fatales en el 7%. La interrupción permanente del tratamiento debido a una reacción adversa ocurrió en el 21 % y la interrupción de la dosis debido a una reacción adversa ocurrió en el 72% de los pacientes.
Afamitresgene Autoleucel (Tecelra®) Adaptaimmune (FDA, USA)
Indicación: Tratamiento de adultos con sarcoma sinovial irresecable o metastásico que han recibido quimioterapia previa, son HLA-A*02:01P, -A*02:02P, -A*02:03P, 6 o -A*02:06P positivos y cuyo tumor expresa el antígeno MAGE-A4.
Tipo: Medicamento de terapia avanzada (génica), constituido por células T autólogas modificadas genéticamente dirigidas al antígeno A4 asociado al melanoma (MAGE-A4) que consiste en células T CD4 y CD8 positivas transducidas con un vector lentiviral autoinactivante (LV) que expresa un receptor de células T (TCR) con afinidad mejorada específica para el MAGE-A4 humano; TECELRA se prepara a partir de células mononucleares de sangre periférica (PBMC) del paciente obtenidas mediante leucoféresis. Las PBMC se enriquecen con células T y luego se transducen con un lentivirus (LV) incompetente para la replicación que contiene el transgén TCR MAGE-A4. Autorizado en Estados Unidos (FDA) el 1 de agosto de 2024 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), mediante revisión prioritaria (Priority Review) y designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); no autorizado aún en la Unión Europea (EMA).
Mecanismo: El receptor de células T (TCR) reconoce un péptido MAGE-A4 restringido por HLA-A*02. MAGE-A4 es un antígeno intracelular de cáncer de testículo que tiene una expresión restringida en tejidos normales y se expresa en el sarcoma sinovial. La activación específica del antígeno de TECELRA a través del complejo TCR-péptido-HLA-A*02 da como resultado la proliferación de células T, la secreción de citocinas y la muerte de células de sarcoma sinovial que expresan MAGE-A4/HLA-A*02.
Eficacia clínica: Ensayo clínico multicéntrico, de un solo brazo y abierto. Hubo 44 pacientes con sarcoma sinovial que recibieron una única infusión de TECELRA. La principal variable clínica de eficacia fue la tasa de respuesta general evaluada por un comité de revisión independiente (43%; completa en el 4,5% y parcial en el 39%). Adicionalmente, la mediana de la duración de la respuesta fue de 6 meses (≥6 meses: 46%; ≥12 meses: 39%).
Eventos adversos: Se produjeron reacciones adversas graves en el 52% de los pacientes con sarcoma sinovial. Las reacciones adversas graves más frecuentes (≥5%) incluyeron síndrome liberación de citocinas (CRS, 9%) y derrame pleural (7%).
Vorasidenib (Voranigo®) Servier (FDA, USA)
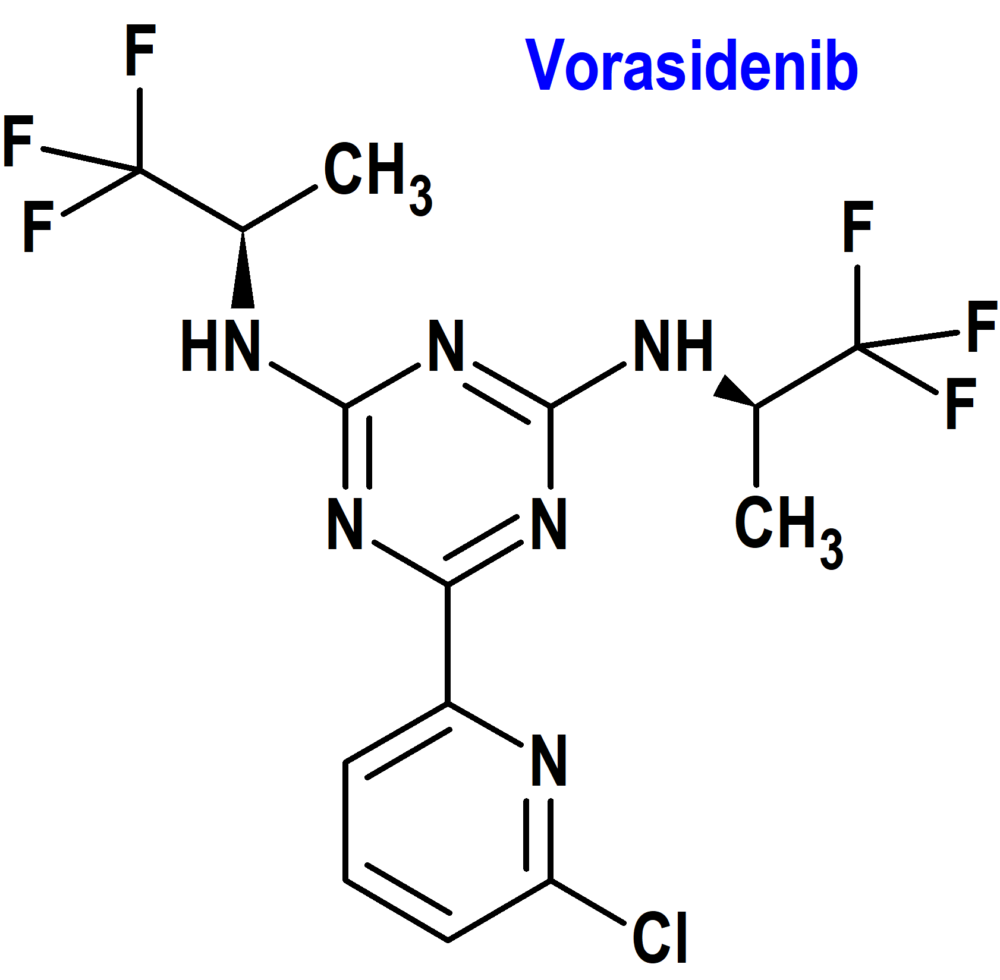
Indicación: Tratamiento de pacientes adultos y pediátricos de 12 años o más con astrocitoma de grado 2 u oligodendroglioma con una mutación susceptible de isocitrato deshidrogenasa-1 (IDH1) o isocitrato deshidrogenasa-2 (IDH2) después de una cirugía que incluya biopsia, resección subtotal o resección total macroscópica.
Tipo: Medicamento sintético estándar constituido por la 6-(6-cloropiridin-2-il)-2-N,4-N-bis[(2R)-1,1,1-trifluoropropan-2-il]-1,3,5-triazina-2,4-diamina. Autorizado en Estados Unidos (FDA) el 6 de agosto de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la enzima isocitrato deshidrogenasa-1 y 2 (IDH1 e IDH2), tanto a las variantes de tipo salvaje y mutante de IDH1, incluidas R132H, como las variantes de tipo salvaje y mutante de IDH2. En modelos tumorales in vivo y basados en células que expresan proteínas mutadas de IDH1 o IDH2, vorasidenib disminuyó la producción de 2-hidroxiglutarato (2-HG) y restauró parcialmente la diferenciación celular. Vorasidenib reduce entre un 64% y un 95% las concentraciones tumorales de 2-HG en pacientes con gliomas con mutación IDH1 o IDH2. Los tumores con alteraciones de IDH1 e IDH2 ocurren en la población pediátrica como los gliomas de grado bajo (grado 2 de la OMS), los gliomas de grado alto (3 y 4), y los oligodendrogliomas con codeleción de 1p y 19q.
Eficacia clínica: Estudio aleatorizado, multicéntrico, doble ciego, controlado con placebo en 331 pacientes con astrocitoma u oligodendroglioma de grado 2 con mutación IDH1 o IDH2, con cirugía previa que incluyó biopsia, resección subtotal o resección total macroscópica. La variable principal de eficacia fue la supervivencia libre de progresión (SLP) evaluada por un comité de revisión independiente ciego (72 vs. 46%). La variable secundaria fue el tiempo hasta la siguiente intervención (tiempo desde la aleatorización hasta el inicio de la primera terapia anticancerosa posterior o la muerte por cualquier causa): la mediana de tiempo no se alcanzó para los pacientes del grupo de vorasidenib y fue de 17,8 meses para los pacientes del grupo placebo.
Eventos adversos: Los más comunes (≥15%) fueron fatiga (37%), COVID-19 (33%), dolor musculoesquelético (26%), diarrea (25%) y convulsiones (16%). Las anomalías de laboratorio de grado 3 o 4 (≥2%) fueron aumento de ALT (10%), aumento de AST (4,8%), aumento de GGT (3%) y disminución de neutrófilos (2,4%). Se produjeron reacciones adversas graves en el 7 % de los pacientes, y entre ellas las más frecuentes (≥2%) de los pacientes fueron convulsiones (3%). La interrupción permanente del tratamiento debido a una reacción adversa se produjo en el 3,6 %, debido mayoritariamente a un aumento de ALT (3%). Se produjeron interrupciones temporales de la dosis debido a una reacción adversa en el 30% de los pacientes.
Odronestamab (Ordspono®) Regeneron (EMA, UE)
Indicación: Tratamiento en monoterapia de pacientes adultos con linfoma folicular en recaída o refractario (LF R/R) después de dos o más líneas de tratamiento sistémico; también en monoterapia de pacientes adultos con linfoma B difuso de células grandes en recaída o refractario (LBDCG R/R) después de dos o más líneas de tratamiento sistémico.
Tipo: Medicamento biológico constituido por un anticuerpo biespecífico recombinante basado en la inmunoglobulina (Ig)G4 humana que se une a CD20 y CD3. Autorizado en la Unión Europea (EMA) el 22 de agosto de 2024 condicionalmente (Conditional marketing authorisation); no autorizado previamente en Estados Unidos (FDA).
Mecanismo: Anticuerpo biespecífico que se une a CD20, un antígeno de superficie de las células B presente en células B normales y malignas, y CD3, un antígeno de las células T asociado al complejo receptor de las células T. La interacción simultánea de los dos brazos de odronextamab da lugar a la formación de una sinapsis entre la célula T y la célula que expresa CD20, lo que provoca la activación de las células T y la generación de respuesta de las células T citotóxicas policlonales, con la consiguiente lisis redirigida de las células diana, incluyendo las células B malignas.
Eficacia clínica: Un estudio abierto, multicéntrico y no aleatorizado de varias cohorte en 128 pacientes con linfoma folicular en recaída o refractario (LF R/R). La variable principal de eficacia fue la tasa de respuesta objetiva (TRO: 80%; 73% completa) y la variable secundaria fue la duración de la respuesta (DR: mediana de 23 meses; 25 en pacientes con respuesta completa) evaluado por un comité de revisión independiente. La eficacia en linfoma B difuso de células grandes en recaída o refractario (LBDCG R/R) se evaluó en dos estudios abiertos, multicéntricos y no aleatorizados de varias cohortes. El primero se hizo en 127 pacientes sin tratamiento previo con T-CAR (TRO: 52%, 31% completa; DR: 11 meses, 18 en pacientes con respuesta completa) y el segundo en 60 pacientes tras un tratamiento con T-CAR (TRO: 48%, 32% completa; DR: 15 meses).
Eventos adversos: Los más comunes son síndrome de liberación de citocinas (54%), neutropenia (41%), pirexia (39%), anemia (38%), trombocitopenia (27%), diarrea (24%) y COVID19 (22%). Los de intensidad grave más frecuentes (grado ≥3 según los CTCAE del NCI) son neutropenia (34%), anemia (19%), trombocitopenia (13%), linfopenia (12%), neumonía (10%), leucopenia (9%), COVID-19 (8%), hipopotasemia (6%) e hiperglucemia (5 %). Las reacciones adversas graves más frecuentes son síndrome de liberación de citocinas (14%), neumonía (9%), COVID-19 (9%) y pirexia (6%). La frecuencia de la interrupción de la infusión IV debido a una reacción adversa es del 16% y la de interrupción del tratamiento debido a una reacción adversa es del 14%, más frecuentemente COVID-19 (2,4%), neumonía (1,3%) y encefalopatía (0,8%).
Denileukin Deftitox (Lymphir®) Citius (FDA, USA)
Indicación: Tratamiento de pacientes adultos con linfoma cutáneo de células T (CTCL) en estadio 4 recidivante o refractario después de al menos una terapia sistémica previa.
Tipo: Medicamento biológico constituido por una proteína de fusión derivada de ADN recombinante compuesta por las secuencias de aminoácidos de los fragmentos A y B de la toxina de la difteria (Met1-Thr387)-His y la secuencia de la interleucina-2 humana (IL-2; Ala1-Thr133); tiene un peso molecular de 58 kD. Autorizado en Estados Unidos (FDA) el 7 de agosto de 2024 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Proteína de fusión diseñada para dirigir la acción citocida de la toxina diftérica (DT) a las células que expresan el receptor IL-2. Después de la captación en la célula, el fragmento DT se escinde y los fragmentos DT libres inhiben la síntesis de proteínas, lo que produce la muerte celular. Denileukin diftitox demostró la capacidad de agotar los linfocitos T reguladores inmunosupresores (Tregs) y la actividad antitumoral a través de una acción citocida directa sobre los tumores que expresan IL-2R.
Eficacia clínica: Un ensayo multicéntrico, de un solo brazo y abierto en 69 pacientes con linfoma cutáneo de células T en estadio I a IV recidivante o refractario, con una expresión de CD25 en ≥20% de las células malignas biopsiadas mediante inmunohistoquímica. La variable principal de eficacia fue la tasa de respuesta objetiva (ORR), según la puntuación de respuesta global (GRS) de ISCL/EORTC por parte del Comité de Revisión Independiente: 36% (9% completa y 27% parcial).
Eventos adversos: Los más comunes (≥20%), incluidos las anomalías de laboratorio, fueron aumento de las transaminasas (70%), disminución de la albúmina (53 %), náuseas (40%), edema (35%), disminución de la hemoglobina (34%), fatiga (30%), dolor musculoesquelético (26%), erupción cutánea (23%), escalofríos (22%), estreñimiento (22%), pirexia (21%) y síndrome de extravasación capilar (20%). Se produjeron reacciones adversas graves en el 38%, principalmente síndrome de extravasación capilar (10 %), reacción relacionada con la infusión (9 %), sepsis (7 %), infección cutánea (2,9 %), pirexia (2,9 %) y erupción cutánea (2,9 %). La interrupción permanente del tratamiento debido a una reacción adversa se produjo en el 12% de los pacientes, y las interrupciones de la dosis debido a una reacción adversa ocurrieron en el 38% de los pacientes.
Axatilimab (Niktimvo®) Incyte (FDA, USA)
Indicación: Tratamiento de la enfermedad de injerto contra huésped crónica (EICHc) tras el fracaso de al menos dos líneas previas de terapia sistémica en pacientes adultos y pediátricos que pesen al menos 40 kg.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal humanizado IgG4 (cadena ligera kappa) recombinante con un peso molecular aproximado de 150 kDa. Autorizado en Estados Unidos (FDA) el 14 de agosto de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo monoclonal que se une a los receptores del factor estimulante de colonias 1 (CSF-1R) expresados en monocitos y macrófagos. El bloqueo del CSF-1R con axatilimab reduce los niveles de estos monocitos circulantes proinflamatorios y profibróticos y macrófagos derivados de monocitos, e inhibe la actividad de los macrófagos patógenos en los tejidos.
Eficacia clínica: Ensayo aleatorizado, doble ciego y controlado con placebo de XX semanas de duración, que incluyó a XXX pacientes. La variable principal de eficacia fue
Eventos adversos: Los más frecuentes (≥15%), son aumento de AST, infección, aumento de ALT, disminución de fosfato, disminución de hemoglobina, infección viral, aumento de gamma glutamil transferasa (GGT), dolor musculoesquelético, aumento de lipasa, fatiga, aumento de amilasa, aumento de calcio, aumento de CPK, aumento de fosfatasa alcalina (ALP), náuseas, dolor de cabeza, diarrea, tos, infección bacteriana, pirexia y disnea. Se produjeron reacciones adversas graves en el 44 % de los pacientes, mientras que la interrupción permanente del tratamiento debido a una reacción adversa ocurrió en el 10% de los pacientes y la reducción de la dosis debido a este motivo en el 8 %. Las interrupciones de la dosis debido a una reacción adversa ocurrieron en el 44 % de los pacientes.
(N) SISTEMA NERVIOSO
Donanemab (Kisunla®) Lilly (FDA, USA)
Indicación: Tratamiento de la enfermedad de Alzheimer. El tratamiento debe iniciarse en pacientes con deterioro cognitivo leve o en etapa de demencia leve de la enfermedad.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal de inmunoglobulina gamma 1 (IgG1) humanizado dirigido contra el beta amiloide, con un peso molecular de 145 kDa. Autorizado en Estados Unidos (FDA) el 2 de julio de 2024 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo monoclonal dirigido contra el beta-amiloide con piroglutamato N-terminal. Las especies de beta-amiloide (Aβ) modificadas con piroglutamato (pE) truncadas en el extremo N (Aβ pE3 ) tienen un mayor potencial de agregación y una elevada propensión a formar oligómeros tóxicos, supuestamente relacionados con la aparición de la enfermedad de Alzheimer.
Eficacia clínica: Estudio doble ciego, controlado con placebo, de grupos paralelos en 1.736 pacientes con enfermedad de Alzheimer, presencia confirmada de patología amiloide y deterioro cognitivo leve o etapa de demencia leve de la enfermedad. Los pacientes tratados con donanemab demostraron una reducción estadísticamente significativa en el deterioro clínico en la Escala Integrada de Calificación de la Enfermedad de Alzheimer (iADRS) en comparación con el placebo en la Semana 76 en la población general (2,92, p<0,0001), así como en las escalas del componente iADRS, el Escala de evaluación de la enfermedad de Alzheimer-subescala cognitiva (ADAS-Cog13) (-1,33, p=0,0006), en la escala del Estudio cooperativo de la enfermedad de Alzheimer – actividades instrumentales de la vida diaria (ADCS-iADL) (1,70, p=0,0001) y en el deterioro clínico en la Escala de Calificación Clínica de Demencia – Suma de Cajas (CDR-SB) (-0,70, p<0,0001).
Eventos adversos: Los más comunes son anomalías de imagen relacionadas con amiloide (ARIA), especialmente ARIA-H (con deposición de siderina), siendo ARIA-H con microhemorragia del 25% (donanemab) vs 11% (placebo), ARIA-H con siderosis superficial del 15 vs. 3%; ARIA-E (con edema) en el 24 vs. 2%; cefalea (13 vs. 10%) y reacciones asociadas a la infusión (9 vs. 0,5%). El porcentaje de pacientes que interrumpen el tratamiento debido a una reacción adversa es del 13% (donanemab) vs. 4% con placebo, debido fundamentalmente a reacciones asociadas con el procedimiento de infusión (4 vs. 0%).
Benzgalantamina (Zunveyl®) Alpha Cognition (FDA, USA)
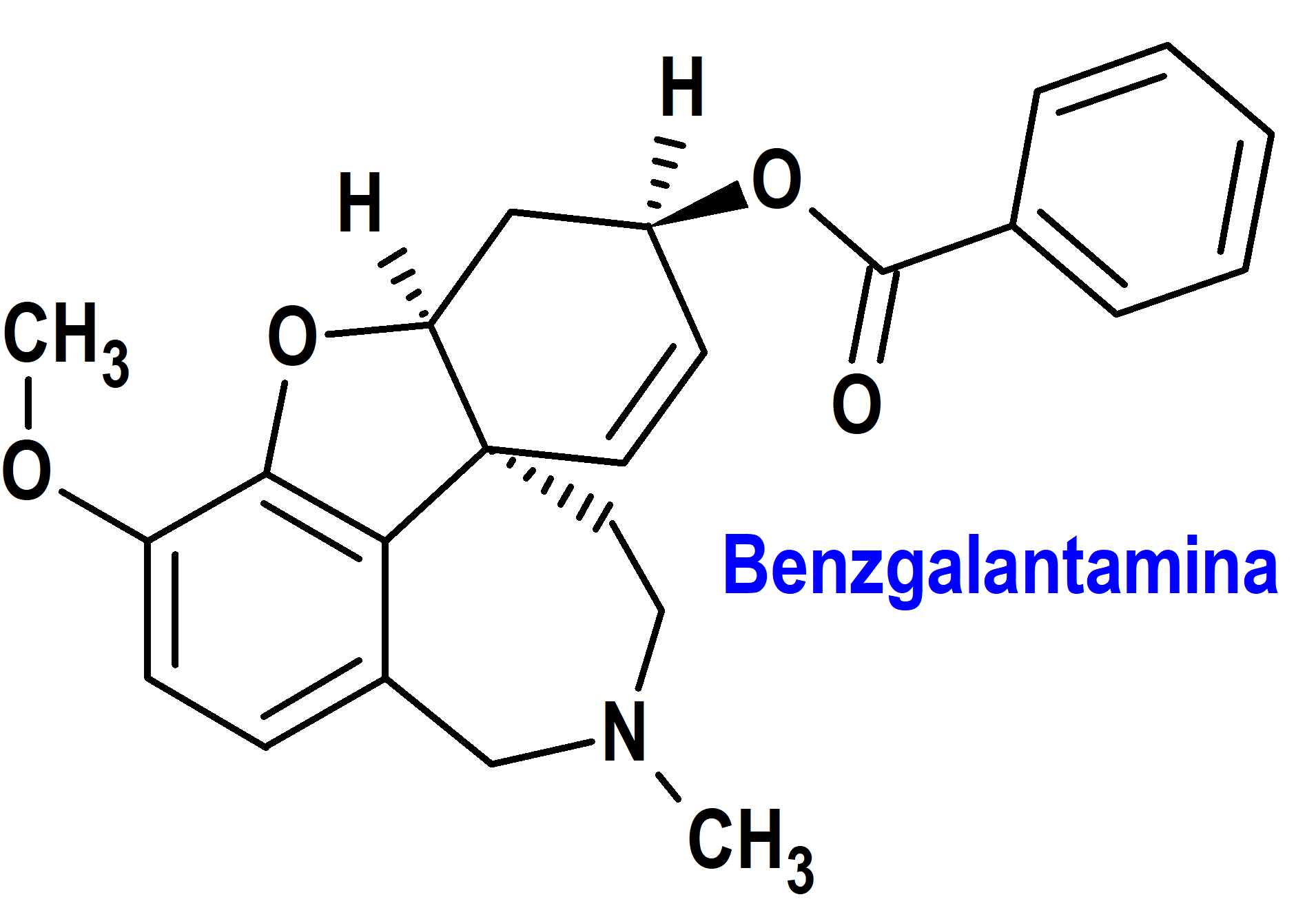
Indicación: Tratamiento de la demencia leve a moderada del tipo Alzheimer en adultos.
Tipo: Medicamento sintético estándar constituido por un profármaco de galantamina: benzoato de (4aS,6R,8aS)-11-metil-3-metoxi-4a,5,9,10,11,12-hexahidro-6H-[1]benzofuro[3a,3,2-ef][2]benzazepin-6-ilo. Autorizado en Estados Unidos (FDA) el 26 de julio de 2024 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD) y designado como terapia innovadora (Breakthrough Therapy), designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT), designado como producto calificado para enfermedades infecciosas (Qualified Infectious Disease Product); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Profármaco de la galantamina, un inhibidor competitivo y reversible de la acetilcolinesterasa. Las neuronas productoras de acetilcolina se degeneran en los cerebros de los pacientes con enfermedad de Alzheimer. El grado de esta pérdida colinérgica se ha correlacionado con el grado de deterioro cognitivo y la densidad de las placas amiloides (una característica neuropatológica de la enfermedad de Alzheimer). Si bien se desconoce el mecanismo preciso de acción de la galantamina, se postula que ejerce su efecto terapéutico al mejorar la función colinérgica. Esto se logra al aumentar la concentración de acetilcolina a través de la inhibición reversible de su hidrólisis por la colinesterasa. Si este mecanismo es correcto, el efecto de la galantamina puede disminuir a medida que avanza el proceso de la enfermedad y menos neuronas colinérgicas permanecen funcionalmente intactas. No hay evidencia de que la galantamina altere el curso del proceso de demencia subyacente.
Eficacia clínica: La eficacia de ZUNVEYL se basa en tres estudios de biodisponibilidad en adultos sanos comparando los comprimidos de liberación inmediata de galantamina y las cápsulas de liberación prolongada de galantamina con ZUNVEYL.
Eventos adversos: Los más comunes (≥5%) son náuseas (21 vs 5,5% con placebo), vómitos (10,5 vs. 2,3%), diarrea (7,4 vs. 4,9%), mareos (7,5 vs. 3,4%), dolor de cabeza (7,1 vs. 5,5%) y disminución del apetito (7,4 vs. 2,1%).
Arimoclomol (Miplyffa®) Zevra (FDA, USA)
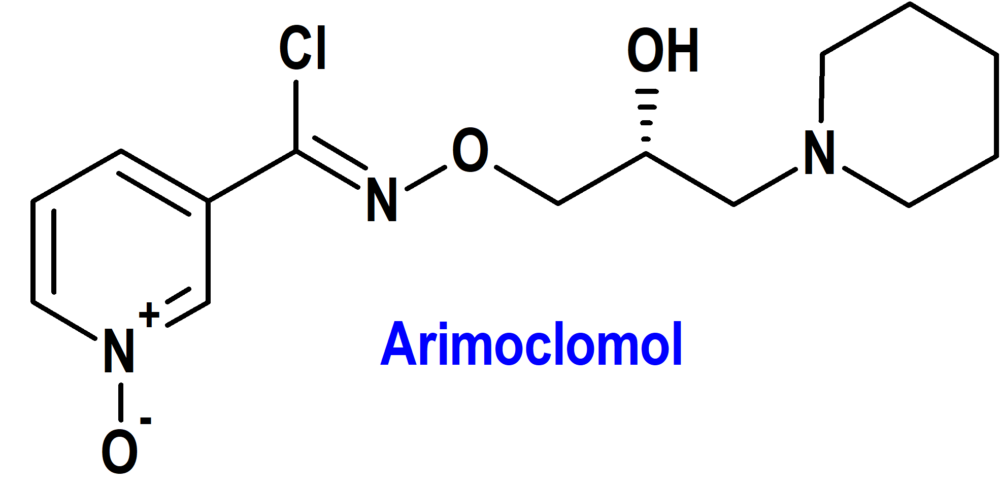
Indicación: Tratamiento en combinación con miglustat de las manifestaciones neurológicas de la enfermedad de Niemann-Pick tipo C (NPC) en pacientes adultos y pediátricos de 2 años de edad y mayores.
Tipo: Medicamento sintético estándar constituido por el cloruro de N-[(2R,Z)-2-hidroxi-3-(1-piperidil)propoxi]piridina-3-carboximidoilo, 1-óxido. Autorizado en Estados Unidos (FDA) el 20 de septiembre de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Permite tratar los síntomas neurológicos asociados con la NPC, aunque se desconocen los mecanismos por los cuales el arimoclomol ejerce sus efectos clínicos en estos pacientes. La enfermedad de Niemann-Pick tipo C (NPC) es una enfermedad genética rara que provoca síntomas neurológicos progresivos y disfunción orgánica. Está causada por cambios en el gen NPC1 o NPC2, que afectan el transporte necesario de colesterol y otros lípidos dentro de una célula. En promedio, las personas afectadas por esta enfermedad solo viven unos 13 años.
Eficacia clínica: Un ensayo clínico aleatorizado, doble ciego, controlado con placebo, de 12 meses de duración en 50 pacientes de 2 a 19 años de edad que tenían un diagnóstico confirmado molecularmente de NPC. Se realizaron evaluaciones de eficacia, incluida la puntuación de la Escala de gravedad clínica de NPC de 4 dominios (R4DNPCCSS) al inicio y reevaluada cada 3 meses hasta los 12 meses de tratamiento. La R4DNPCCSS es una medida de la progresión de la enfermedad de NPC que consta de los cuatro elementos que evalúan la deambulación, el habla, la deglución y las habilidades motoras más relevantes, donde las puntuaciones más altas representan una mayor gravedad de la enfermedad. A los 12 meses, la variación de la puntuación R4DNPCCSS fue de -0,2 puntos con arimoclomol/miglustat vs. +2,0 con placebo/miglustat.
Eventos adversos: Los más comunes son infección de las vías respiratorias superiores (31 vs 15% con placebo), diarrea (23/23%), pérdida de peso (15/0%), disminución del apetito (12/0%), temblor (12/0%), urticaria (12/0%), dolor de cabeza (12/8%), infección de las vías respiratorias inferiores 3(12/8%) y convulsiones (12/8%).
Levacetilleucina (Aqneursa®) Intrabio (FDA, USA)
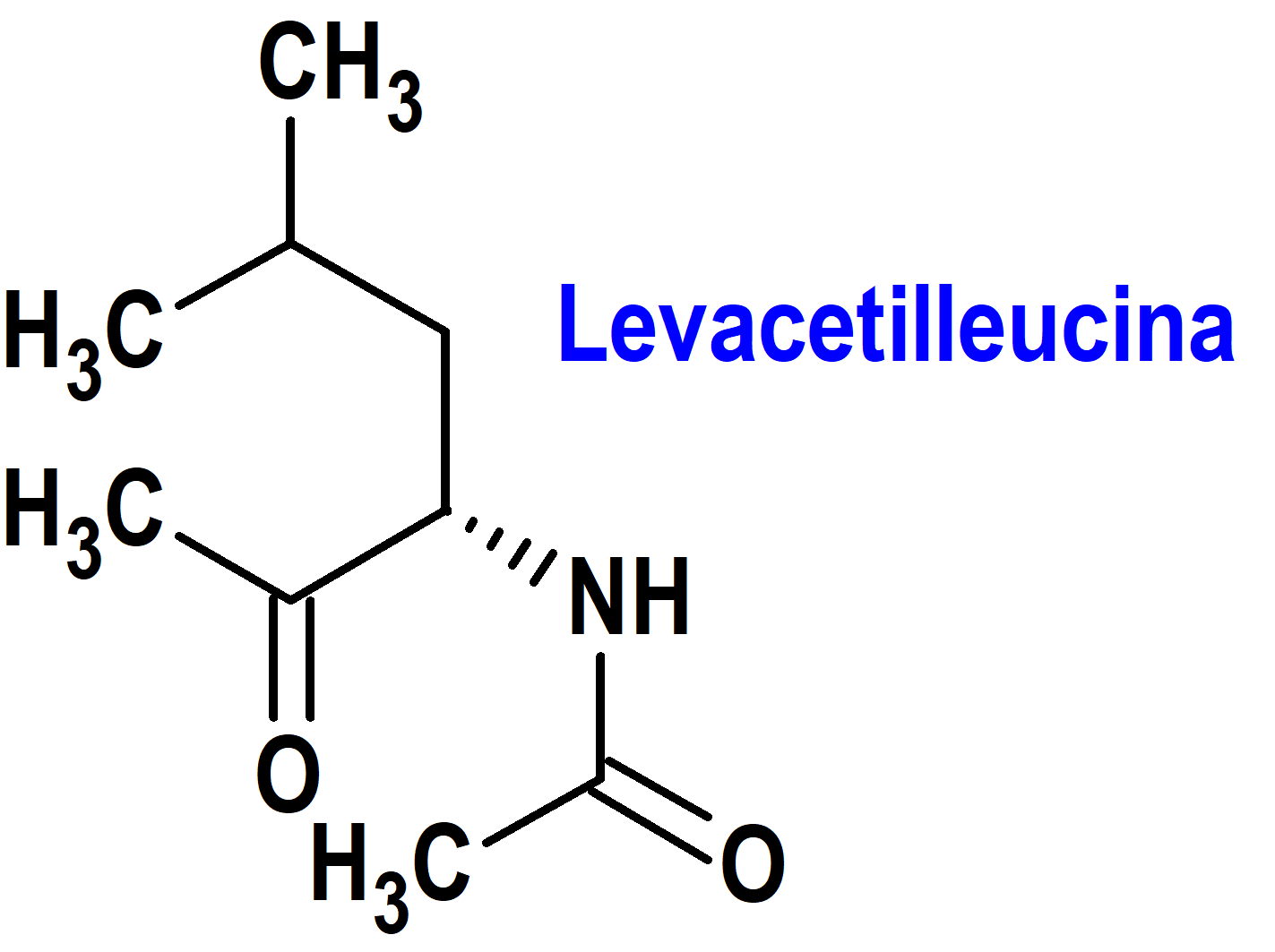
Indicación: Tratamiento de las manifestaciones neurológicas de la enfermedad de Niemann-Pick tipo C (NPC) en pacientes adultos y pediátricos con peso ≥15 kg.
Tipo: Medicamento sintético estándar constituido por el ácido L-2-acetamido-4-metilpentanoico (N-acetil-L-leucina). Autorizado en Estados Unidos (FDA) el 24 de septiembre de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Desconocido.
Eficacia clínica: Estudio aleatorizado, doble ciego, controlado con placebo, de dos periodos, cruzado, de 24 semanas de duración (12 semanas para cada periodo de tratamiento) en 60 pacientes con 4 años de edad o más y con un diagnóstico confirmado de NPC y al menos síntomas neurológicos leves relacionados con la enfermedad. La variable principal de eficacia fue una versión modificada de la Escala para la Evaluación y Calificación de la Ataxia (SARA), conocida como SARA funcional (fSARA), que consta de los dominios de alteración de la marcha, sedestación, postura y habla. La puntuación media estimada de fSARA por tratamiento fue de 5,1 con AQNEURSA frente a 5,6 con placebo, con una diferencia de tratamiento (IC95%) de -0,4 puntos.
Eventos adversos: Los más comunes son infección del tracto respiratorio superior (17 vs. 3% con placebo), dolor abdominal (7 vs. 0%), disfagia (7 vs. 0%) y vómitos (7 vs. 0%).
Xanomelina + Trospio (Cobenfy®) Bristol Myers Squibb (FDA, USA)
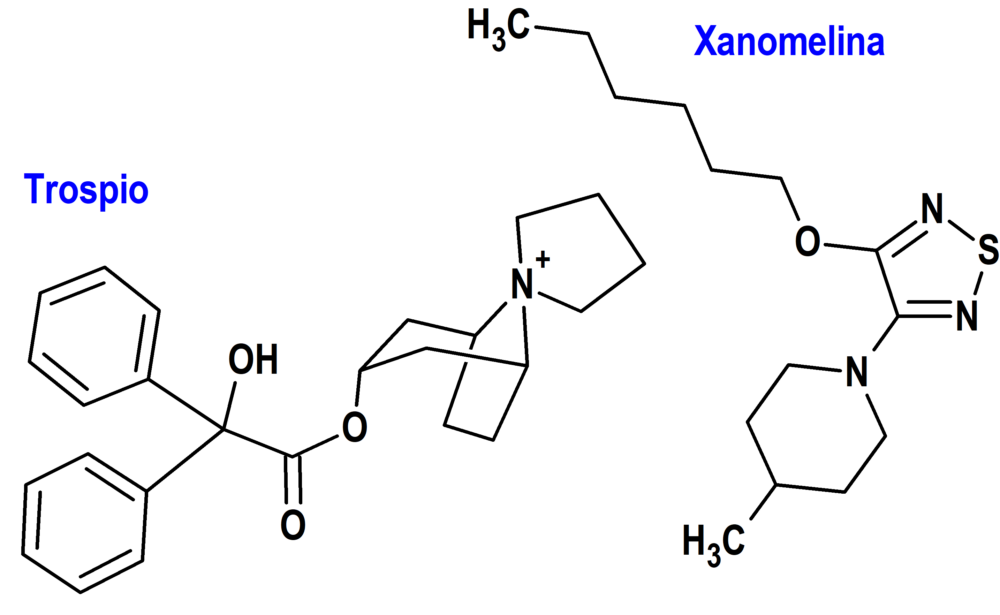
Indicación: Tratamiento de la esquizofrenia en adultos.
Tipo: Medicamento sintético estándar constituido por el ácido 3-hexoxi-4-(1-metil-3,6-dihidro-2H-piridin-5-il)-1,2,5-tiadiazol-(2R,3R)-2,3-dihidroxibutanodioico (xanomelina) y por el cloruro de (1alfa,3beta,5alfa)-3-((2-hidroxi-2,2-difenilacetil)oxi)-espiro(8-azoniabiciclo(3.2.1)octano-8,1’-pirrolidinio) (cloruro de trospio). Autorizado en Estados Unidos (FDA) el 26 de septiembre de 2024 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Agonista de los receptores colinérgicos muscarínicos M1 y M4 del sistema nervioso central. El cloruro de trospio es un antagonista muscarínico que antagoniza los receptores muscarínicos principalmente en los tejidos periféricos, a fin de evitar o paliar los efectos colinérgicos periféricos de la xanomelina.
Eficacia clínica: Dos estudios controlados con placebo con diseños idénticos de cinco semanas, aleatorizados, doble ciego, controlados con placebo y multicéntricos en un total de 470 pacientes adultos con diagnóstico de esquizofrenia según los criterios del DSM-5. La medida principal de eficacia fue el cambio desde el inicio en la puntuación total de la Escala de Síndromes Positivos y Negativos (PANSS) en la Semana 5. La PANSS es una escala de 30 ítems que mide los síntomas de la esquizofrenia; cada ítem es calificado en una escala de siete puntos, desde 1 (ausencia de síntomas) 7 (síntomas extremadamente graves), donde la puntuación total de la PANSS puede variar de 30 a 210 (mayor gravedad). La diferencia (media de mínimos cuadrados) entre el tratamiento activo y el placebo fue de -9,6 y -8,4 puntos en los dos estudios.
Eventos adversos: Los más comunes son náuseas (19 vs. 4% con placebo), dispepsia (18/5%), estreñimiento (17/7%), vómitos (15/1%), hipertensión (11/2%), dolor abdominal (8/4%), diarrea (6/2%), taquicardia (5/2%), mareos (5/2%), enfermedad por reflujo gastroesofágico (5/<1%).
(S) ÓRGANOS SENSORIALES
Polihexanida (Akantior®) SIFI (EMA, UE)

Indicación: Tratamiento de la queratitis por Acanthamoeba en adultos y niños a partir de 12 años de edad.
Tipo: Medicamento sintético estándar constituido por el hidrocloruro de poli(iminocarbonimidoiliminocarbonimidoilimino-1,6-hexanodiilo). Autorizado en la Unión Europea (EMA) el 22 de agosto de 2024 como medicamento huérfano (Orphan drug); no autorizado previamente en Estados Unidos (FDA).
Mecanismo: Polímero policatiónico compuesto de unidades de hexametilenobiguanida que actúa tanto sobre los trofozoítos activos como sobre las formas quísticas inactivas de Acanthamoeba. La polihexanida altera las membranas celulares de Acanthamoeba debido a su carga positiva, uniéndose a la bicapa fosfolipídica de la membrana de los trofozoítos, que tiene carga negativa, provocando la lisis de la membrana la muerte y muerte celular; también es capaz de penetrar el ostiolo de Acanthamoeba enquistada para ejercer el mismo efecto. Esta acción solo afecta mínimamente a los fosfolípidos neutros de la membrana celular de los mamíferos. Asimismo, la polihexanida se une al ADN lesionando los cromosomas de Acanthamoeba; en concreto, interacciona con la estructura de fosfato del ADN para bloquear el proceso de replicación. Este mecanismo se limita a las células de Acanthamoeba, ya que polihexanida no es capaz de penetrar el núcleo de las células de los mamíferos.
Eficacia clínica: Un un ensayo clínico de fase 3, aleatorizado, con enmascaramiento doble y controlado con tratamiento activo con datos de controles históricos sobre sujetos que no recibieron tratamiento (identificados mediante una revisión bibliográfica sistemática; n = 56); la tasa de resolución clínica sin cirugía en este control histórico fue del 19,6 % , mientras que el 80,4 % restante de los pacientes requirió cirugía. El efecto del tratamiento (porcentaje de pacientes curados sin cirugía) de polihexanida frente a la ausencia de tratamiento (control histórico) fue del 84,8 vs. 19,6%.
Eventos adversos: Los más comunes son dolor ocular (13%) e hiperemia ocular (12%). Las más graves son perforación corneal (1,4%), necesidad de trasplante de córnea (1,4%) y deficiencia visual (1,4%), que también forman parte de la evolución natural de la enfermedad.
(V) VARIOS
Galio [68Ga] + Germanio [68Ge] (GalliaFarm®) Eckert Ziegler (EMA, UE)
Indicación: Eluido estéril (solución de cloruro de galio [68Ga]) para el radiomarcaje in vitro de varios kits de preparación radiofarmacéutica desarrollados y aprobados para el radiomarcaje con dicho eluido, que se utilizarán para la obtención de imágenes de tomografía por emisión de positrones (PET). Este generador de radionúclidos no debe utilizarse directamente en los pacientes.
Tipo: Medicamento sintético estándar constituido por un generador de radionúclidos que contiene germanio (68Ge) como núclido original, que se desintegra en el núclido derivado galio (68Ga). El germanio (68Ge) usado para la producción del generador (68Ge/68Ga) no tiene ningún portador añadido. Autorizado en la Unión Europea (EMA) el 1 de agosto de 2024; no autorizado previamente en Estados Unidos (FDA).
Mecanismo: El generador de radionúclidos GalliaPharm 1,11-3,70 GBq es un sistema para la elución de solución de cloruro de galio (68Ga) estéril. Esta solución se eluye de una columna en la que se ha fijado el núclido original germanio (68Ge), que es la sustancia precursora del galio (68Ga). El período de semidesintegración del germanio-68 [68Ge] es de 270,95 días mediante captura de electrones, mientras que el del galio-68 [68Ga] es 67,71 minutos, emitiendo positrones, empleándose en las pruebas de diagnóstico por imagen mediante PET (tomografía por emisión de positrones).
Eficacia clínica: Para potencias iniciales en el generador de radionúclidos de 1,11 y 3,70 Gbq, la actividad obtenida mediante elución al final del periodo de validez es no menor de 0,16 y 0,55 GBq, respectivamente.
Eventos adversos: Las posibles reacciones adversas debidas al uso de un radiofármaco marcado con 68Ga dependerán del kit específico para la preparación del radiofármaco específico que se está utilizando. La exposición a la radiación ionizante está relacionada con la inducción de cáncer y la posibilidad de desarrollar anomalías congénitas.
Flortaucipir [18F] (Tauvid®) Lilly (EMA, UE)

Indicación: Para la obtención de imágenes del cerebro mediante tomografía por emisión de positrones (PET) para evaluar la distribución neocortical de los ovillos neurofibrilares de tau agregado (NFTs, por sus siglas en inglés) en pacientes adultos con deterioro cognitivo que están siendo evaluados por enfermedad de Alzheimer.
Tipo: Medicamento sintético estándar constituido por el 7-(6-[18F]fluoranilpiridin-3-il)-5H-pirido[4,3-b]indol. Autorizado en la Unión Europea (EMA) el 22 de agosto de 2024; autorizado previamente en Estados Unidos (FDA) el 28 de mayo de 2020.
Mecanismo: Flortaucipir (18F) se une a los agregados de la proteína tau. En los cerebros de pacientes con EA, el filamento helicoidal emparejado (PHF) de tau forma agregados que se combinan para formar ovillos neurofibrilares (NFTs), un componente necesario del diagnóstico neuropatológico de la enfermedad de Alzheimer. Flortaucipir (18F) se retiene diferencialmente en áreas neocorticales que contienen agregados de tau y no se une al amiloide. El flúor [18F] decae a oxígeno [18O] estable con un periodo de semidesintegración de 110 minutos mediante emisión de positrones con una energía máxima de 634 keV, seguida de una radiación de aniquilación de positrones de 511 keV. Los principales fotones útiles para el diagnóstico por imágenes son los fotones gamma de 511 keV, resultantes de la interacción del positrón emitido con un electrón.
Eficacia clínica: Un estudio pivotal de correlación neuropatológica de la EA y otro estudio adicional con lectores, que compararon el rendimiento diagnóstico de la utilización de flortaucipir (18F) para estimar la distribución de los NFTs de tau agregado con el examen post-mortem. En cada estudio, 5 lectores independientes, ciegos a la información clínica, interpretaron las imágenes de flortaucipir (18F) como positivas o negativas. Se evaluó el rendimiento de las imágenes del patrón de flortaucipir (18F) de la enfermedad de Alzheimer para distinguir la patología tau B3 (verdadero positivo) de la B0-B2 (verdadero negativo). El rendimiento diagnóstico del escáner con flortaucipir (18F) en pacientes sometidos a autopsia (B3) mostró una sensibilidad del 89-92% y una especificidad del 76-77%.
Eventos adversos: Los más comunes son dolor de cabeza (0,9%), dolor en el lugar de la inyección (0,6%) y aumento de la presión arterial (0,5%).
Flurdipiraz [18F] (Flyrcado®) GE Healthcare (FDA, USA)
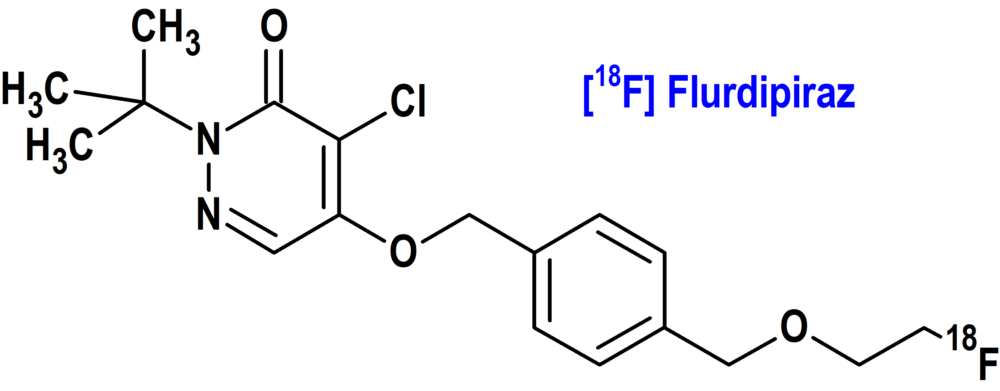
Indicación: Tomografía por emisión de positrones (PET) y la obtención de imágenes de perfusión miocárdica (MPI) en reposo o estrés (farmacológico o ejercicio) en pacientes adultos con enfermedad coronaria (CAD) conocida o sospechada para evaluar la isquemia y el infarto de miocardio.
Tipo: Medicamento sintético estándar constituido por la 2-terc-butil-4-cloro-5-[[4-(2-(18F)fluoraniletoximetil)fenil]metoxi]piridazin-3-ona. Autorizado en Estados Unidos (FDA) el 27 de septiembre de 2024 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: El flurpiridaz [18F] es un análogo del inhibidor del complejo mitocondrial 1 (MC-1), el piridabeno. El flurpiridaz [18F] es extraído por el miocardio de manera proporcional al flujo sanguíneo y se une al tejido cardíaco que tiene mitocondrias biológicamente activas. Por lo tanto, la radiactividad en el miocardio viable es mayor que en el tejido infartado. El flúor [18F] decae a oxígeno [18O] estable con un periodo de semidesintegración de 110 minutos mediante emisión de positrones con una energía máxima de 634 keV, seguida de una radiación de aniquilación de positrones de 511 keV. Los principales fotones útiles para el diagnóstico por imágenes son los fotones gamma de 511 keV, resultantes de la interacción del positrón emitido con un electrón.
Eficacia clínica: Dos estudios clínicos prospectivos, multicéntricos y abiertos en adultos con enfermedad coronaria (CAD) sospechada o conocida. El Estudio 1 evaluó la sensibilidad y la especificidad para la detección de CAD significativa en sujetos con CAD sospechada que estaban programados para una angiografía coronaria invasiva (ACI). En tres lectores de imágenes, las estimaciones de sensibilidad variaron del 74 % al 89 % y las estimaciones de especificidad variaron del 53 % al 70 % para la CAD definida como al menos un 50 % de estrechamiento de una arteria. El Estudio 2 evaluó la sensibilidad y especificidad para la detección de CAD significativa en sujetos con CAD conocida o sospechada que tuvieron una ACI sin intervención dentro de los 60 días previos a la obtención de imágenes o estaban programados para una ACI. En tres lectores de imágenes, las estimaciones de sensibilidad variaron del 63% al 77% y las estimaciones de especificidad variaron del 66% al 86% para la CAD.
Eventos adversos: Los más comunes son disnea (17%), dolor de cabeza (15%), angina de pecho (10%), dolor torácico (8%), fatiga (7%), cambios en el segmento ST (6%) y sofocos (5%).
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE)
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
Producto Calificado para Enfermedades Infecciosas. (Qualified Infectious Disease Product): un medicamento antibacteriano o antifúngico para uso humano destinado a tratar infecciones graves o potencialmente mortales, incluidas aquellas causadas por un patógeno resistente a antibacterianos o antifúngicos, incluidos patógenos infecciosos nuevos o emergentes.
