1. INTRODUCCIÓN A LA EPIGENÉTICA
La pregunta sobre la herencia biológica se ha respondido a través del lenguaje de la secuencia de ADN de forma habitual. Esta visión coloca al ADN como el único material hereditario que determina los rasgos que diferencian a un organismo de otro y que se transmite de generación en generación. El ADN utiliza un lenguaje basado en la existencia de cuatro letras, que se combinan a su vez en palabras de tres letras (codones) para dar lugar a las proteínas, las moléculas que dotan de estructura y funciones específicas a los organismos.
En los últimos años, se ha evidenciado que esta visión era incompleta. Por ejemplo, aunque todas y cada una de las células de un organismo poseen la misma información almacenada en su ADN, es evidente que una célula de la piel es muy diferente a un macrófago o a un cono de la retina. ¿Entonces, cuál es la diferencia entre los distintos tipos celulares si todos comparten la misma cadencia de nucleótidos en su ADN?
La respuesta nos la brinda la Epigenética, una disciplina que se dedica a estudiar los cambios heredables que no dependen de la secuencia de bases del ADN (1, 2). El ADN, que en cada una de las células humanas forma una especie de fibra de cerca de dos metros de longitud, se encuentra empaquetado exquisitamente para ser confinado en el interior de un núcleo de diámetro un millón de veces más pequeño. La manera en que el ADN está empaquetado determina, en realidad, la forma en que este ADN funcionará. El envoltorio, que empaqueta el ADN dentro del núcleo, recibe el nombre de cromatina, y los mecanismos y modificaciones que sufre el ADN y su envoltura son las modificaciones epigenéticas, que en última instancia deciden qué funciones están activadas y cuáles inactivadas en cada tipo de célula. Cuestiones como la reprogramación en el trasplante de núcleos celulares y clonación de organismos, numerosas alteraciones en el cáncer y en otras patologías, no se pueden entender sin considerar los factores epigenéticos.
Por lo tanto, podemos definir de una forma más o menos sencilla la Epigenética como el estudio de la regulación heredable de la actividad de los genes que no está determinada por la secuencia genética. Nuestros genes son la combinación de cuatro bases o piezas denominadas A, C, G y T, que cuando sufren alteraciones las llamamos mutaciones. Hoy sabemos que los genes se controlan de muchas más formas: por ejemplo, añadiendo un grupo químico llamado metilo (CH3) a la cadena de ADN o añadiendo otro grupo químico llamado acetilo a las proteínas denominadas histonas, las “llaves” de nuestro genoma. Recordemos que son los genes: los genes, son fragmentos de ADN, se expresan originando ARN que después producirá una proteína: casi todo lo que podemos ver o tocar en nuestro cuerpo son proteínas, la insulina secretada por nuestro páncreas, la mioglobina de nuestros músculos, etc. Tiene que haber un control riguroso de los genes: no queremos que una célula del oído interno exprese una proteína de una célula del epidídimo testicular. La envoltura que permite a los genes “abrirse” (expresarse) o “cerrarse” (silenciar) es epigenética. Podemos cerrar la expresión de un gen de forma ligera, simplemente ajustando la ventana: sería lo que haría un cambio en las histonas, o bien podemos reprimir su expresión de forma más firme, cerrando la ventana con un candado: sería la metilación del ADN. Una Epigenética equilibrada, dentro de la variación fisiológica y poblacional de nuestra especia, es esencial para el ser humano: evita la expresión de secuencias de ADN endoparasitarias insertadas a lo largo de millones de años de evolución, permite la expresión correcta de un cromosoma X en mujeres, mantiene nuestro genoma estable evitando que se rompan los cromosomas, ayuda en la expresión específica de cada tejido y tipo celular, y realiza muchas otras tareas ingratas y poco reconocidas (1, 2).
Así, nuestro genoma “desnudo” no es el único responsable de nuestro comportamiento y susceptibilidad a enfermar. La secuencia de pares de bases de nucleótidos del ADN, el tema típico de estudio de la genética clásica, no puede explicar completamente la funcionalidad de nuestras células, su interrupción en enfermedades complejas o, incluso, la definición de nuestra especie. Necesitamos algo más. Parte de la explicación la proporciona el campo de la Epigenética. Waddington definió la Epigenética en 1939 como “las interacciones causales entre los genes y sus productos que dan lugar al fenotipo”. Desde nuestro conocimiento actual, podemos definir la Epigenética como “la herencia de la actividad del ADN que no depende de la secuencia de ADN desnudo”. Esta “herencia” es más fácil de entender durante la mitosis, el proceso de transmisión, cuando una célula se divide para producir células hijas durante el ciclo celular, o incluso de una manera más provocativa durante la meiosis, en las células germinales y por tanto, nuestra información epigenética será transmitida a nuestros descendientes. De esta forma, ciertos cambios químicos adquiridos en la línea germinal podrían pasar a la siguiente generación. Por lo tanto la Epigenética hace referencia a las modificaciones químicas dinámicas que ocurren en nuestro ADN, y en las proteínas reguladoras (como las histonas) asociadas al mismo. Las principales marcas epigenéticas se ilustran en la Figura 1.
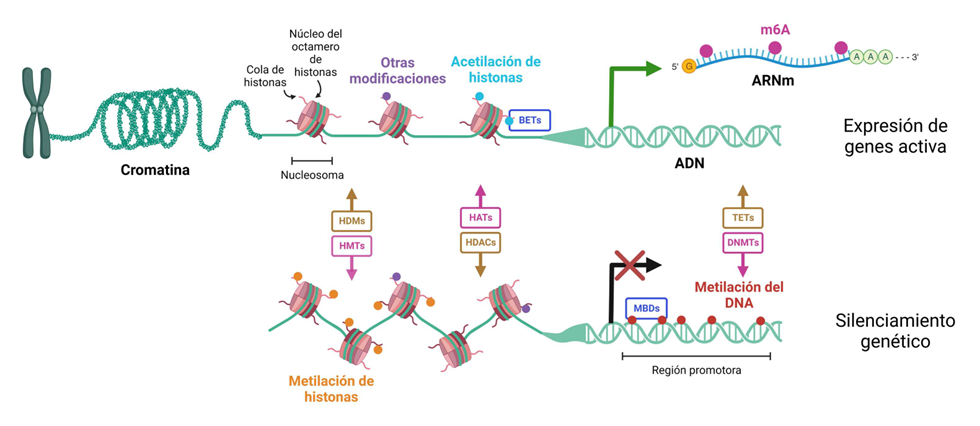
Figura 1. Ilustración de las modificaciones químicas del ARN, ARN y las histonas que determinan la actividad del genoma
La metilación del ADN tiene un papel crítico en el control de la actividad de los genes y la arquitectura nuclear (3). En los seres humanos, la metilación del ADN ocurre en la citosina en los dinucleótidos CpG. Estos sitios CpG no están distribuidos al azar en el genoma humano; las regiones ricas en CpG, conocidas como islas CpG, se asocian a menudo con la región reguladora del extremo 5’ de muchos genes y generalmente no están metiladas en células normales para los genes de expresión ubicua. Este estado no metilado se corresponde con la capacidad de la isla CpG de transcribir sus genes asociados cuando existe presencia de los activadores de la transcripción necesarios. Sin embargo, hay un subconjunto de islas CpG que están fuertemente metiladas en los tejidos normales, y estas se asocian a menudo con genes específicos tisulares, la línea germinal específica, genes con impronta genética (expresión monoalélica determinada parentalmente) y los genes que se someten a la inactivación de un cromosoma X en las mujeres. Además, las secuencias repetitivas genómicas también están muy metiladas para provocar su silenciamiento y los “saltos” entre cromosomas. Así, el mantenimiento de este estado de metilación puede tener un papel en la protección de la integridad del ADN mediante la prevención de la inestabilidad cromosómica.
La metilación del ADN no es una marca epigenética aislada. A menudo se asocia con modificaciones químicas en las colas N-terminales de las proteínas llamadas histonas (e incluso en aminoácidos localizados más internamente) (4). Años atrás consideradas solo simples proteínas empaquetadoras del ADN, las histonas ahora ocupan un lugar central como depósitos de información epigenética a través de un complejo conjunto de modificaciones post-traduccionales como la acetilación y metilación de la lisina, arginina y fosforilación de la serina, entre muchas otras más infrecuentes (por ejemplo la citrulación). Se ha propuesto que los diferentes patrones de modificaciones presentados en las colas de las histonas forman un “código de histonas” que determina la actividad del gen, usando el paralelismo del código genético.
Si alteramos la Epigenética, se producen muchas enfermedades. En realidad toda patología humana tiene un componente epigenético. Por ejemplo, una pérdida de metilación puede provocar una exposición excesiva de determinados antígenos y originar una enfermedad autoinmune, o una mutación de gen epigenético MeCP2 (que se une a la citosina metilada como un imán) puede provocar una enfermedad neurológica como el Síndrome de Rett (5), una de las principales causas de retraso mental en mujeres y que afecta a muchas familias que luchan contra esta enfermedad. No obstante, donde más se ha avanzado en el reconocimiento de la importancia de la Epigenética en patología humana es en la Oncología (6-8). La hipometilación del ADN global contribuye al origen de las células cancerosas mediante la generación de inestabilidad cromosómica, la reactivación de elementos transponible, la pérdida de la indentidad celular y la pérdida de la impronta genética. Lo más importante, y lo que se conoce como la “paradoja de la metilación del ADN”, es que hay áreas locales de ADN que aumentan la metilación CpG: las islas CpG del promotor de muchos genes supresores de tumores, como p16INK4a, p15INK4b, p14ARF, MLH1, VHL o BRCA1, lo que lleva a la inactivación de estos factores anticáncer (6). Desde el punto de vista de las histonas, los tumores humanos también presentan un código distorsionado, y para las leucemias y sarcomas, sabemos que las translocaciones patognomónicas implican genes de histona acetiltransferasas, desmetilasas, metiltransferasas y de remodelación de la cromatina. Estos mismos genes modificadores de histonas y responsables de la organización en tres dimensiones de nuestro núcleo también sufren mutaciónes en un amplio espectro de tipos tumorales.
Una de las diferencias esenciales entre la Genética y Epigenética del cáncer humano es que si la secuencia genética es fija y casi inamovible, la metilación del ADN y la modificación de las histonas son cambios reversibles en las circunstancias adecuadas. Por lo tanto, las alteraciones epigenéticas son uno de los puntos más débiles en la armadura de la célula del cáncer porque los genes supresores de tumores hipermetilados pueden despertar de su largo sueño con los regímenes de medicamentos adecuados y ejercer sus funciones normales inhibidoras de crecimiento. Dos familias de fármacos epigenéticos, agentes desmetilantes del ADN e inhibidores de las histonas desacetilasas, surgieron de inicio como los compuestos más prometedores en esta área, y otros compuestos que tenían como diana otras proteínas epigenéticas les siguieron. La excelente noticia es que nueve fármacos epigenéticos han recibido aprobación a dia de hoy para el tratamiento de subtipos de tumores humanos. Las principales dianas de los fármacos epigenéticos se ilustran en la Figura 2.
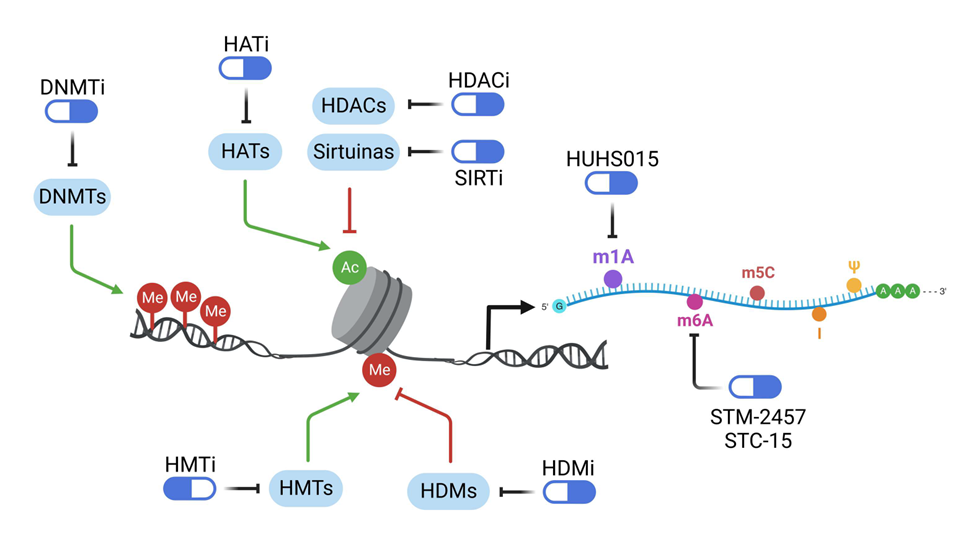
Figura 2. Ilustración de las principales dianas de los fármacos epigenéticos y epitranscriptómicos
2. INTRODUCCIÓN A LOS FÁRMACOS EPIGENÉTICOS
En tumorogénesis, es decir, en la formación de un cáncer, la inactivación epigenética de genes a través de la metilación del ADN es una fuerza motora tan importante como lo es la inactivación por mutación. Existen diversos tipos tumorales con muy pocas mutaciones y masivas lesiones epigenéticas. También sabemos que “la puerta de entrada” de muchos carcinógenos es una alteración epigenética inicial a la que luego se suman los defectos genéticos. Además, su naturaleza plástica se adapta a los cambios adaptativos que requiere la célula tumoral para sobrevivir. Así la mayor parte de investigadores aceptan que la metilación del ADN, las modificaciones de las histonas y, consecuentemente, la alteración de la cromatina, juegan un papel central en el cáncer.
La hipermetilación de las islas CpG es pues también una candidata a su tratamiento terapéutico para revertir este estado anómalo, y por ello el uso de drogas que provocan una desmetilación progresiva de las mismas han sido el punto de partida de nuevas terapias. Hasta la fecha, la atención se ha centrado principalmente en terapias basadas en la reversión de la metilación como un modo de poner en marcha los genes que han sido inactivados por metilación. Esta reversión se controlaría mediante el uso de inhibidores de las DNA metiltransferasas (DNMTs). Sin embargo, es necesario considerar si la inhibición de la metilación del ADN resulta tóxica para las células, ya que afectaría a todos los genes metilados (incluso los fisiológicos). Claramente la metilación del ADN es importante durante el desarrollo, y de hecho la pérdida de alguna de las tres DNMTs conocidas es letal en ratones, posiblemente porque la actividad de las DNMTs es vital durante el desarrollo embrionario. Sin embargo, parece que podrían ser menos necesarias en adultos, ya que están básicamente destinadas al mantenimiento del estado inactivo de la parte del genoma que debe permanecer inactiva. En el contexto del adulto, las drogas que desmetilen tendrían un efecto mayor en células en proliferación, como son las tumorales, que en células diferenciadas. Algunos de estos fármacos que desmetilan han sido empleados durante muchos años, uno de ellos es la decitabina, una molécula muy parecida estructuralmente a la citosina pero que se diferencia de esta porque cuando es incorporada al ADN mediante la replicación no puede ser convertida en 5-metilcitosina. El resultado del uso de la decitabina es una desmetilación global que resulta en una reactivación de numerosos genes. El problema de la hipometilación global que resulta del uso de estas drogas es que podría ocasionar defectos en la estabilidad cromosómica. Así, el punto de partida para el diseño de nuevas terapias basadas en la inhibición de la metilación es el patrón incrementado de metilación en las islas CpG relacionado con cáncer que no se detecta en células homeostáticas (9).
Otro gran grupo de fármacos empleados como agentes en estos nuevos acercamientos terapéuticos contra el cáncer son los bloqueantes de la modificación de la histonas (10), particularmente los inhibidores de las actividades histona deacetilasa (11). Estos compuestos afectarían directamente al grado de acetilación de las histonas. Al menos hay cinco grupos de proteínas con actividad histona acetiltransferasa, y más de tres con actividad histona deacetilasa. La inhibición de la actividad histona deacetilasa tiene como consecuencia la hiperacetilación de las histonas, ya que los inhibidores no actúan sobre las histonas acetiltransferasa (enzima que se encarga de acetilar las histonas). Debemos recordar que la acetilación de las histonas está asociada con actividad génica (mayor acetilación, mayor transcripción, y al contrario). En consecuencia, la inhibición de histona deacetilasas provoca una reactivación de la actividad genética. Como ocurría con los agentes desmetilantes, el problema es que el estado deacetilado, además de estar asociado a genes supresores de tumores, también se encuentra en muchos otros genes, y por ello el tratamiento con drogas inhibidoras de actividad histona deacetilasa tiene un componente inespecífico. La mayoría de estas sustancias afecta a la proliferación de tumores y, en algunos casos, influye en la muerte celular programada (apoptosis). Algunas de estas drogas han sido utilizadas desde hace décadas, como el butirato y el fenilbutirato. En el y cabe decir que los problemas que acompañan el uso de los fármacos como los inhibidores de metilación y de histona deacetilasa son el efecto global y la posible toxicidad.
El futuro de la terapia epigenética avanza también hacia la reactivación específica de los genes afectados, actuando sobre los genes diana que han sido silenciados por hipermetilación o hipoacetilación con fármacos que sean capaces de actuar específicamente o usando alternativas de terapia genética. Respecto a estos puntos, a día de diversas compañías farmacéuticas ya están investigando la posibilidad de “encender” o reactivar específicamente genes silenciados por hipermetilación o hipoacetilación. Una parte del futuro de la terapia epigenética para aquellos tumores más resistentes a la misma dependerá del éxito de estas estrategias, que permitirán reactivar específicamente los genes que han sido silenciados de forma aberrante sin alterar, esto es de suma importancia, el patrón de expresión de los otros genes. Por otra parte, cada vez conocemos mejor las mutaciones presentes en los tumores humanos, lo cual resulta enormemente positivo de cara a su tratamiento, y está demostrado que algunas de estas lesiones afectan a genes que regulan la epigenética. La buena noticia es que muchos de estos cánceres, portadores de lesiones en genes epigenéticos específicos, podrían ser más sensibles a los medicamentos descritos o a otros nuevos en desarrollo, como los inhibidores de histona metiltransferasas, desmetilasas y bloqueantes de los dominios “bromo” que “leen” la acetilación de las histonas, claves en el empaquetamiento del ADN en el núcleo (12). Las esperanzas de éxito de estos fármacos son elevadas tanto en neoplasias hematológicas de distinta índole como en tumores de las partes blandas, como los sarcomas, y en tumores infantiles como el neuroblastoma. El siguiente apartado describe como estas investigaciones han generado fármacos epigenéticos aprobados para su uso clínico.
3. USO LÍNICO DE LOS FÁRMACOS EPIGENÉTICOS
Hemos descrito como los mecanismos epigenéticos desempeñan un papel crucial en la función del genoma, y su desregulación en el cáncer los convierte en un objetivo prometedor para el desarrollo de fármacos. La plasticidad de las modificaciones epigenéticas ha impulsado la exploración de fármacos epigenéticos, que se dirigen a las proteínas enzimaticas involucradas en la regulación epigenética asi como a sus factores asociados. Estos medicamentos tienen como objetivo restaurar los patrones transcripcionales equilibrados de la célula sana modificando los estados de la cromatina. Los fármacos epigenéticos actuales se dirigen a enzimas conocidas como escritoras (introducen las marcas epigenéticas, sea cuales sean), lectoras (reconocen las marcas) y borradoras (eliminan las marcas) tanto en el ADN como en las histonas.
La primera clase de fármacos, por motivos históricos, fueron los inhibidores de las metiltransferasas del ADN (iDNMT). Los iDNMTi de primera generación, como azacitidina (Vidaza) y decitabina (Dacogen), son análogos de la pirimidina que se incorporan al ADN durante la replicación, lo que lleva a la generación irreversible de aductos covalentes DNMT-ADN. Este proceso resulta en la pérdida pasiva de metilación de citosina en las células hijas a medida que se van produciendo divisiones celulares, desencadenando tanto respuestas de daño al ADN y apoptosis, especialmente a concentraciones elevadas, como reactivación de genes supresores tumorales, a las concentraciones más bajas y mantenidas en el tiempo usadas actualmente en la clínica. Tanto azacitidina como decitabina han sido aprobadas para tratar diversas malignidades hematológicas, incluyendo síndrome mielodisplásico, leucemia mieloide aguda y leucemia mielomonocítica juvenil, donde suelen ser el tratamiento de elección (8).
Sin embargo, estos inhibidores de DNMTs de primera generación tienen limitaciones, como la comentada falta de selectividad de objetivo, además de cierta farmacocinética desfavorable y efectos en otras proteínas. Para superar estos desafíos, los investigadores han desarrollado iDNMTs de segunda generación como guadecitabina, un profármaco de la decitabina con mayor estabilidad molecular y captación por las células cancerosas. Además, un novísimo inhibidor de DNMT1 no nucleósido, reversible y selectivo para esta enzima, llamado GSK3685032, ha mostrado resultados prometedores en modelos preclínicos de leucemia mieloide aguda (LMA), demostrando mejor tolerabilidad y mayor actividad hipometilante del ADN que los fármacos de primera generación (13).
En adición a sus efectos anticancerígenos directos, los fármacos epigenéticos también tienen el potencial de incrementar la sensibilidad a otras terapias antineoplásicas, abriendo nuevas vías para su uso en tumores sólidos donde podrían resensibilizar a estas neoplasias. Además, las terapias combinadas de los fármacos epigenéticos com agentes inmunomoduladores podrían superar las limitaciones en la inmunoterapia al reactivar genes silenciados en las células cancerosas, causando que los tumores sean más visibles para el sistema inmunológico (incremento de exposición de antígenos cáncer- específicos). La potente actividad inmunomoduladora de los iDNMTs ha impulsado ensayos clínicos numerosos que prueban como su adición a inhibidores de puntos de control inmunológicos, como anti-PD-L1 (durvalumab, atezolizumab) o anti-PD1 (nivolumab, pembrolizumab) mejora la respuestas de diversos tumores sólidos a estos regímenes de inmunoterapia (14).
Estos enfoques innovadores ofrecen esperanza para mejorar las estrategias de tratamiento del cáncer que van más allá de la immunoterapia, y podrían también incrementar la tasa de respuesta a agentes alquilantes del ADN y a la terapia antihormonal.
La segunda gran familia de fármacos epigenéticos tiene como diana las histona desacetilasas (HDACs). Los inhibidores de histona desacetilasa (HDACi) desempeñan un papel crucial en la conformación del microambiente epigenómico celular, influenciando la expresión génica al modular la la accesibilidad de la cromatina actuando a nivel de la acetilación de histonas. Este estado de acetilación, controlado por las histona acetil transferasas (HATs) y las histona desacetilasas (HDACs), afecta la actividad de la cromatina y determina como factores de transcripción, coactivadores y correpresores pueden unirse a la misma, regulando la expresión genética. Las HDACs, clasificadas usualmente en cuatro clases, incluyen proteínas dependientes de zinc y enzimas de la clase III (sirtuinas) que utilizan NAD+ como cofactor para su actividad catalítica. Los inhibidores de HDAC interfieren con la actividad de las HDACs, evitando la desacetilación de lisina y promoviendo un estado hiperacetilado, lo que resulta en una estructura de cromatina más relajada propicia para la activación transcripcional, es decir, para devolver la expresión de genes supresores tumorales silenciados en cáncer. No obstante, es importante indicar que las HDACs tienen múltiples sustratos proteicos no histonas, ampliando el impacto de los inhibidores de HDAC para incluir otras proteínas, incluyendo factores de transcripción que también podrían ser beneficiosos para su efecto antitumoral.
Los inhibidores de HDAC de primera generación aprobados para su uso clínico fueron el vorinostat y el romidepsin, utilizados de inicio en el tratamiento del linfoma cutáneo de células T (CTCL). Vorinostat recibió la aprobación en 2006 para el tratamiento de CTCL, respaldado por ensayos que demostraron una tasa de respuesta objetiva del 31%. Romidepsin siguió en 2009 con una respuesta similar (34%) y también recibió aprobación para el tratamiento del CTCL (8). La combinación de inhibidores de HDAC con otros fármacos anticancerígenos ha mostrado efectos sinérgicos, como se ha comentado previamente para los iDNMTs, lo que ha llevado a estudios que evalúan su potencial como moduladores iniciales para la inmunoterapia. Bloqueantes de HDACs como vorinostat, en combinación con inhibidores de puntos de control inmunológicos, generan una mayor infiltración de células T en los tumores (un efecto muy deseable) y han incrementado las tasas de supervivencia en diversos modelos preclínicos.
La irrupción de los inhibidores de HDAC de segunda generación como belinostat y chidamide, ha mostrado también resultados muy esperanzadores. Belinostat recibió la aprobación acelerada en 2014 para el linfoma de células T periféricas (LCTP) recurrente/refractario, donde existia muy escaso arsenal terapéutico, mientras que chidamide obtuvo la aprobación para LCTP y cáncer de mama avanzado con HER2 negativo y receptores hormonales positivos (8). Nuevos inhibidores de HDACs como entinostat, con selectividad para miembros específicos de la subfamilias de HDACs, han mostrado también resultados clínicos prometedores. Y la combinación de los inhibidores de HDAC, azacitidina a baja dosis y terapia endocrina ha demostrado ser beneficiosa en el cáncer de mama resistente a terapia antihormonal. Si además le sumamos el efecto inmunomoduladores de los inhibidores de HDACs, los ensayos clínicos en curso para varios tipos de cáncer avanzado seguramente serán positivos en determinados subgrupos de pacientes.
Un enfoque completamente revolucionario para inhibir la actividad de las proteínas epigenéticas, entre ellas las HDACs (pero aplicable a proteínas muy diversas), implica los “proteolysis-targeting chimeras” (PROTACs) (15). Este acercamiento aprovechan el sistema ubiquitina-proteasoma para degradar proteínas diana en lugar de inhibirlas. En este sentido, los PROTACs que se dirigen a HDAC6 han demostrado una actividad antiproliferativa especial en células de mieloma múltiple, una enfermedad oncológica aún necesitada de mejores fármacos. Esta aproximación va más allá del cáncer, y aunque no sea el objetivo de este artículo, tiene el potencial de modular las enzimas epigenéticas en trastornos neurodegenerativos, enfermedades metabólicas y autoinmunidad, ofreciendo nuevas alternativas terapéuticas que antes parecían inverosímiles. Mucha investigación se está dedicando a la vía de los PROTACs en cáncer y los resultados de estos ensayos se darán a conocer en los próximos años.
Sin embargo sería magnífico poder definir subgrupos de pacientes donde esperaríamos una mayor sensibilidad para un fármaco epigenético determinado. Inaugurando una farmacogenética para iDNMTs o para los bloqueantes de las modificaciones de las histonas. Ese futuro ya está en parte aquí. Podemos pasar de reprogramadores epigenéticos amplios a una terapia más específica basada en la presencia de mutaciones activadoras en enzimas modificadoras del epigenoma, como la administración de tazemetostat para pacientes con linfoma que albergan mutaciones en la histona metiltransferasa EZH2 (16, 17). EZH2 es la subunidad catalítica de PRC2 y media la inactivación transcripcional mediante la trimetilación de la histona H3 en la lisina 27 (H3K27me3). EZH2 está sobreexpresado en un amplio abanico de tumores humanos; lo que lleva a un aumento en H3K27me3 y la represión concomitante de genes supresores de tumores donde esta marca “decora” el promotor. El hallazgo de que los niveles elevados de EZH2 se asociaban a un mal pronóstico y agresividad tumoral en diversos tipos tumorales despertó el interés inicial de EZH2 como blanco antitumoral. No obstante, fue la identificación de mutaciones activadoras en alrededor del 20-25% de los casos de linfoma difuso de células B y de linfoma folicular lo que definitivamente impulsó el interés farmacéutico en los inhibidores de EZH2 (iEZH2) (16, 17). Un primer ejemplo de medicina de precisión epigenética. Tazemetostat, el primer iEZH2 desarrollado, recibió su aprobación en 2020 para tratar el linfoma folicular al demostrarse la mejora de los pacientes con mutaciones en EZH2 en comparación con el grupo de casos con la secuencia salvaje de EZH2. Tazemestostat también recibió aprobación acelerada en 2020 para el tratamiento del sarcoma epitelioide, un subgrupo agresivo de sarcoma que muestra dependencia oncogénica de EZH2, debido a la pérdida de INI1/SMARCB1, un componente del complejo remodelador de la cromatina SWI/SNF que actúa en contraposición a EZH2 (18). Se están probando también combinaciones en la actualidad como un ensayo de fase III de tazemetostat más doxorrubicina en el entorno de primera línea en el sarcoma epitelioide. También contamos en nuestra farmacopea epigenética con inhibidores dobles para EZH2 y otra histona metiltransferasa (EZH1), como valemetostat, que reducen más H3K27me3, causando más muerte celular y superior eficacia antitumoral en modelos murinos de neoplasias hematológicas, en comparación con la inhibición selectiva de EZH2. Los resultados positivos de un ensayo de fase II de valemetostat en leucemia/linfoma de células T del adulto han respaldado la reciente aceptación de valemetostat como el primer inhibidor doble de EZH1/2 aprobado para el tratamiento de los casos refractarios. Además, la combinación de valemetostat con ipilimumab, se está estudiando en un ensayo de fase I para el tratamiento de pacientes con tumores metastásicos. Un subgrupo adicional pacientes con mayor vulnerabilidad a iEZH2 sería aquel que muestra pérdida de BAP1, resultando en un aumento de la expresión de EZH2 y elevados niveles de H3K27me3. Ocurriría en mesotelioma donde las investigaciones clínicas de tazemetostat en esta entidad maligna han observado el beneficios clínico de estabilización de la enfermedad en el 64% de los casos con defectos en BAP1 (19). Como en otros casos, la combinación de tazemetostat con el agente de inmunoterapia pembrolizumab podría ser beneficiosa en diversos tipos tumorales y los ensayos clínicos nos proporcionarán una respuesta sobre esta cuestión.
Además de EZH2, otra histona metiltransferasas (HMTs) también han sido diana de nuevos fármacos epigenéticos. Un ejemplo sería la histona lisina metilytransferasa DOT1L, la única H3K79 metiltransferasa descrita en la literatura. La atención se ha focalizado especialmente en leucemias agudas que involucran reordenaciones de otra lisina metiltransferasa (KMT2A/MLL), ya que las proteínas quiméricas involucrando KMT2A8MLL reclutan DOT1L a loci genómico aberrantes, promoviendo la expresión ectópica de oncogenes de leucemia como HOXA9 y MEIS1. Los estudios preclínicos demuestran que el inhibidor de DOT1L pinometostat sensibiliza las células de AML pediátricas al tratamiento con el inhibidor de multiquinasa sorafenib, lo que podría llevar a nuevas estrategias terapéuticas para pacientes pediátricos afectados por este tipo de leucemia.
Una tercera diana de fármacos que actúan a nivel de las modificaciones de las histonas serían los inhibidores de histona desmetilasa (iHDMi). Los más desarrollados poseen como diana la lisina-específica histona desmetilasa KDM1A/LSD1 que se encuentra alterada en varias malignidades hematopoyéticas y tumores sólidos de diversos tipos. KDM1A/LSD1 actúa principalmente como un corepresor transcripcional, desmetilando H3K4me1/2, un marcador de modificación de histonas asociado a la activación de la expresión genética. La inhibición de KDM1A/LSD1 ha demostrado inducir la reprogramación celular en las células iniciadoras del tumor ya que regula el equilibrio entre renovación y diferenciación de las células madre. Por tanto es un fármaco de especial interés en tumores quimioresistentes debido a su capacidad de atacar las células madre cancerosas. Los ensayos clínicos que evalúan inhibidores de KDM1A/LSD1, incluyendo iadademstat, pulrodemstat y seclidemstat han mostrado resultados prometedores (8). Pulrodemstat mostró una actividad antitumoral alentadora, especialmente en pacientes con neoplasias neuroendocrinas y cáncer de pulmón de células pequeñas; seclidemstat demostró actividad en pacientes con sarcomas avanzados, especialmente en sarcoma de Ewing; y Iadademstat, solo y en combinación con el desmetilante del ADN azacitidina, mostró actividad clínica en pacientes con LMA recurrente/refractaria, un grupo de especial riesgo clínico.
Existe “fuego cruzado” entre fármacos epigenéticos y la función de desmetilación proteica de KDM1A/LSD1 se extiende más allá de las histonas, incluyendo la ADN metiltransferasa DNMT1, estabilizando a la misma para mantener la metilación global del ADN. En este sentido iDNMTs e inhibidores de KDM1A/LSD1 están siendo evaluados en combinación en varios ensayos clínicos de fase I/II, particularmente en malignidades hematológicas. De igual forma que el resto de fármacos epigenéticos, los estudios preclínicos han demostrado que la inhibición de KDM1A/ LSD1 mejora la eficacia terapéutica de la inmunoterapia, lo que ha desencadenado el inicio de varios ensayos clínicos usando esta estrategia en neoplasias con respuestas limitadas al tratamiento inmune, como sería el cáncer de pulmón de células pequeñas. Una “vuelta de tuerca” final es el desarrollo a partir de química computacional de innovadores inhibidores duales de HDAC-KDM1A/LSD1 como el agente JBI-802, que demuestran actividad antitumoral sinérgica en modelos murinos lo que ha motivado el inicio de diversos ensayo clínicos de fase I/II en tumores sólidos avanzados.
Finalmente, el último grupo de fármacos epigenéticos supone un cambio de paradigma ya que no inhiben proteínas con acción enzimática sino proteínas que se unen a modificaciones de las histonas: sería el caso de los compuestos farmacológicos dirigidos a las proteínas de bromodominio y dominio extra-terminal (BET), conocidos como inhibidores de BET (iBET) que han demostrado una gran capacidad para remodelar la cromatina (12). La familia BET, que incluye entre otros BRD2, BRD3, BRD4 y BRDT, posee un bromodominio que reconoce las lisinas acetiladas, lo que lleva a cambios en la estructura de la cromatina y a la posterior activación transcripcional al reclutar factores de transcripción. Las proteínas BET desempeñan roles fundamentales en el control de la expresión de oncogenes al regular no solo los promotores clásicos de los genes sino también “enhancers”, regiones reguladoras a distancia de oncogenes críticos como el omnipresente MYC. Inicialmente identificados en el carcinoma de línea media NUT, donde el gen NUT se fusiona con BRD4 o BRD3, los inhibidores de BET como molibresib se usan para este cáncer y otros tumores pediátricos (20). Esta línea de investigación empezó con el primer inhibidor de BET denominado JQ1 que inducía la represión de oncogenes. Un compuesto que fue mejorado químicamente en la forma de TEN-010 en los ensayos clínicos actuales. Se están evaluando también combinaciones con inmunoterapias, como usar el iBET ZEN-3694 con nivolumab o pembrolizumab. La utilidad de los iBET podría darse también superando la resistencia a los inhibidores de quinasas de receptor de tirosina, una versatilidad que también ha propiciado el uso de iBETs con inhibidores de MEK o inhibidores de PARP de forma sinérgica. Como se ha descrito con otros fármacos epigenéticos anteriores, también han surgido desestabilizadores como PROTACs y “pegamentos moleculares” contra las proteínas de BET, como ARV-771, QCA570, dBET6, FHD-609 y CFT8634 que muestran eficacia antitumoral, especialmente en modelos preclínicos de leucemia linfoblástica aguda de células T, en el sarcoma sinovial y en tumores sólidos SMARCB1-negativos. Una nueva vía farmacológica antitumoral que ha despertado un gran interés académico e industrial.
Aunque no estrictamente clasificados como fármacos epigenéticos, cabe mencionar también los compuestos que interfieren con el metabolismo asociados a mutaciones en genes epigenéticos. Sería el caso de las mutaciones de ganancia de función en las isocitrato deshidrogenasas IDH1 y IDH2 (enzimas que eventualmente regulan los patrones de metilación del ADN y las histonas). Estas alteraciones son frecuentes en gliomas de bajo grado y LMA ocasionando una sobreproducción de 2-hidroxiglutarato, un potente inhibidor competitivo de diversas dioxigenasas que atenúan la actividad de enzimas como las desmetilasas de histonas y desmetilasas de ADN, afectando la metilación global de estas dianas y promoviendo la progresión del cáncer. Para contrarrestar esto, se han desarrollado varios inhibidores de las IDHs como enasidenib, vorasidenib e ivosidenib, recientemente aprobados para pacientes con leucemia recurrente/refractaria con mutaciones en IDH2 y IDH1, respectivamente; e ivosidenib también usado en colangiocarcinomas avanzados con mutación en IDH1. Se encuentran en ensayos clínicos para malignidades hematológicas y tumores sólidos, los inhibidores duales de IDH1/IDH2 mutante, como vorasidenib (21) y HMPL-306. Recientemente se están incluso ensayando vacunas de péptidos específicos para mutaciones en IDH, como IDH1R132H-DC y PEPIDH1M, en tumores cerebrales. Otro ejemplo más de la estrategias terapéuticas disruptivas basadas en el conocimiento del Epigenoma.
4. INTRODUCCIÓN A LAS MODIFICACIONES DEL ARN: LA EPITRANSCRIPTÓMICA
Más allá de las modificaciones epigenéticas (entendidas como los cambios químicos en el ADN y las proteínas que regulan su actividad, por ejemplo las histonas), existe un campo de creciente interés el campo del control de la expresión genética: el estudio de las modificaciones químicas del ARN, conocido como el Epitranscriptoma (22, 23). Conocidas en parte desde hace años, el reciente descubrimiento de su reversibilidad y el reconocimiento de su implicación en cáncer han avivado la investigación de las mismas. Así paralelamente al paradigma epigenético, los científicos han comenzado a explorar también estas modificaciones, con el objetivo de dirigirse a ellas farmacológicamente para intervenir en enfermedades humanas caracterizadas por epitranscriptomas anómalos. En las últimas cinco décadas, los investigadores han identificado más de 140 modificaciones post-transcripcionales en transcritos de ARN diversos. La mayoría de estos cambios afectan ARN no-codificantes altamente abundantes, como los ARNs ribosómicos (ARNr) y los ARNs de transferencia (ARNt) que van más allá de una función puramente estructural.
Sin embargo, el gran salto en este campo en los últimos diez años ha sido demostrar que estas modificaciones epitranscriptómicas también ocurren en el ARN mensajero (ARNm) causando importantes cambios en su actividad. En general, las bases del ARN (A, U, C, G) y el azúcar ribosa pueden albergar múltiples modificaciones químicas, desde procesos de isomerización de bases o edición hasta el uso de bases nitrogenadas altamente inusuales. Entre las modificaciones epitranscriptómicas se encuentran la N6-metiladenosina (m6A), la N1-metiladenosina (m1A) la 5-metilcitosina (m5C) (también presente en el ADN como hemos descrito anteriormente), la 5- hidroximetilcitosina (hm5C), inosina (I) y la pseudo-uridina (Ψ). Los últimos tiempos han visto una caracterización más meticulosa de estas modificaciones, desentrañando su importancia en la biogénesis y maduración del ARN, su plasticidad y dinamismo y su función en condiciones fisiológicas que requieren cambios en la estabilidad del transcrito o en su eficiencia tradicional. Además, los estudios recientes han empezado a demostrar sus implicaciones en diversas enfermedades humanas, destacando especialmente el cáncer como sucede con los cambios epigenéticos. El campo de la epitranscriptómica moderna es aún muy joven, pero ya está dando los primeros frutos prometedores que han mostrado que mucho ARN antes denominados “estructurales” (como los mencionados ARNr y ARNt) realizan muchas más funciones, que el ARN mensajero no es simplemente un paso intermedio entre el ADN y la proteína; y que han generado ya un fármaco epitranscriptómico en un ensayo clínico contra el cáncer.
Centrándonos en el potencial actuar de generar fármacos, nos focalizaremos principalmente en la N6-metiladenosina (m6A), la modificación interna más prevalente detectada en los ARN mensajeros (22, 23). La marca epitranscriptómica de la m6A se ilustra en la Figura 1. Su abundancia, que constituye del 0.2% al 0.6% de todas las adenosinas, asociada con el desarrollo de métodos de detección fiables y robustos de la misma, ha impulsado la investigación intensa de esta modificación del ARN. La modificación m6A implica la adición de un grupo metilo en la posición nitrógeno-6 de la adenosina por parte del heterodímero METTL3-METTL14, siendo METTL3 la subunidad catalítica y METTL14 actuando como andamio de unión de ARN para el reconocimiento del sustrato. Existen otras RNA metiltransferasas para m6A como METTL16 con especial afinidad por ARNs no codificantes. La deposición de m6A en el ARNm exhibe cierto patrón dependiente de la secuencia del transcrito, con mayor apetencia por regiones codificantes y las regiones 3’-no traducidas, especialmente justo antes del codón de terminación. Además existe una comunicación cruzada con las marcas epigenéticas y, por ejemplo, la trimetilación de la histona H3 en la lisina 36 regula los sitios de incorporación de m6A en secuencias de ADN específicas al reclutar el complejo METTL14, habiendo sido probado por estudios de inmunoprecipitación de cromatina seguidos de secuenciación masiva. Los microARNs, pequeños ARN no codificantes que regulan también la expresión genética y que algunos investigadores incluyen en el campo de la Epigenética, también están modificados por m6A. Y lo mismos sucede con ARNs no codificantes más largos como los ARN antisentido o el transcrito XIST que regula la inactivación de un cromosoma X en mujeres.
Y también existe una multitud de cofactores y proteínas de unión al ARN que contiene m6A. Estos lectores de m6A, incluyen proteínas de la familia YTH, proteínas IGF2BP y ribonucleoproteínas nucleares heterogéneas (hnRNP). Estas proteínas desempeñan roles fundamentales en varios aspectos de la regulación génica post-transcripcional y el ciclo de vida del ARNm, incluida la estabilidad del ARNm y la eficacia de la traducción proteica en los ribosomas. Por ejemplo, proteínas de unión a m6A como YTHDF1 y YTHDF2 regulan la estabilidad del ARNm durante la diferenciación de células madre, afectando procesos de diferenciación celular, especialmente en hematopoyesis, espermatogénesis, neurogénesis y la lipogénesis. Además, las proteínas anteriores, sumadas a YTHDF3 y IGF2BP1/2/3 también supervisan los procesos de traducción y degradación del ARN. La reversión de la marca m6A en el ARN sucede por la acción de desmetilasas como la proteína asociada a la obesidad y masa grasa (FTO) y la homóloga de AlkB 5 (ALKBH5). Tanto la adición de m6A como su eliminación ha generado un interés farmacológico creciente en el desarrollo de inhibidores contra las enzimas implicadas en estos procesos.
5. MODIFICACIONES DEL ARN: IMPLICACIONES EN LA PATOGÉNESIS DE ENFERMEDADES
Si la compleja constelación de modificaciones del ARN desempeña un papel central en la regulación del ciclo de actividad del ARN, modificando la diferenciación celular y el desarrollo, es lógico suponer que el epitranscriptoma de las enfermedades humanas estará profundamente alterado. En el campo de la patología humana, nuevamente el cáncer ha supuesto la punta de lanza de estas investigaciones revelando la asociación entre patrones de modificación del ARN anómalos y diversas enfermedades donde en Oncología se ha relacionado con procesos de invasión y metástasis, células madres del cáncer, evasión inmunológica y neoangiogénesis entre otros (24).
Focalizándonos solamente en la modificación m6A del ARN se han observado alteraciones del patrón global de la misma en tumores de todo tipo y también alteraciones específicas afectando transcritos que promueven propiedades oncogénicas o supresoras de tumores. En este sentido, la m6A RNA metiltransferasa más prominente, METTL3, ha sido implicada como un “motor” oncogénico en varios tipos de neoplasias, como en la leucemia dodne el complejo METTL3-METTL14 juega un papel crucial en mantener la capacidad de renovación de las células madre/progenitoras hematopoyéticas. La sobreexpresión de METTL3 incrementa la traducción de transcritos oncogénicos como los de los genes MYC, BCL2 y PTEN, fomentando la proliferación de las células leucémicas. Un papel similar, pero más modesto, jugaría también METTL14, con su papel oncogénico dependiente de MYC. No solo en hematopatías malignas, sino que el papel de METTL3 se extiende tumores solidos estimulando a la transición epitelio- mesenquima en la metástasis del cáncer, donde la desregulación epitranscriptómica eleva la expresión de distintos efectores mesenquimales.
En el otro lado de la ecuación y fijándonos en las RNA desmetilasas para m6A en el ARN, las alteraciones de FTO y ALKBH5 también se observan frecuentemente en tumores (24). En función del contexto celular y genómico, actuan como oncogenes o supresores de tumores. Papeles promotores de crecimiento ocurren por ejemplo en leucemia y melanoma donde la inhibición de FTO contribuye a reprimir la progresión de dichas entidades en modelos preclínicos, e incluso restableciendo cierta sensibilidad a la inmunoterapia. También ALKBH5 exhibe una acción protumorogénica en el glioblastoma, donde su sobreexpresión incrementa la auto-renovación y desdiferenciación a través de la estabilización del ARNm del factor de transcripción FOXM1. O en el cáncer de mama, donde la desmetilación de ARNm de Nanog por ALKBH5 incrementa el número de células madre del cáncer.
También las proteínas que interpretan la señal mediada por la marca m6A en el ARN contribuyen al proceso de transformación celular (24). De esta manera, la la familia de proteínas YTH, y las proteínas IGF2BP y hnRNPs se han asociado a distintos fenotipos de las células tumorales. Por ejemplo, YTHDF2 se encuentra sobreexpresada en el cáncer de colon donde promueve la estabilidad del ARNm del gen relacionado con la metástasis HIF-1α. Este hecho coloca a YTHDF2 como una potencial diana de tratamientos anti- metástasis. YTHDF2 también se asocia, cuando se sobreexpresa, a leucemias de evolución clínica adversa al regular la vida media del transcrito del gen TNFRSFy por tanto de la muerte celular programada. Otra proteína de unión a m6A, YTHDF1, suprime la presentación de antígenos en las células dendríticas, y por ello en el ámbito de la inmunoterapia, el bloqueo farmacológico de YTHDF1 mejora la eficacia de la terapia con anti- PD-L1 en modelos preclínicos de cáncer humano.
6. FÁRMACOS EPITRANSCRIPTÓMICOS
La importancia de las modificaciones químicas del ARN para determinar la actividad celular, las alteraciones de los patrones del epitranscriptoma en distintas enfermedades y la existencia de enzimas con dominios estructurales de catálisis conocidos, han propiciado una expansión enorme de la investigación en fármacos epitranscriptómicos en los últimos cinco años (25). Dichos compuestos se pueden dirigir contra diversas de las proteinas y modificaciones el RNA descritas, pero el mayor esfuerzo se ha concentrado en la inhibición de las proteínas con actividad enzimática que actúa a nivel de la m6A del ARN. El papel crucial de la m6A en muchos procesos biológicos, el ser la modificación del ARN más prevalente, junto a la existencia de perfiles de m6A anómalos vinculados al cáncer, ha llevado a la comunidad científica a focalizarse preferentemente en esta marca. La marca m6A en el ARN como diana de los fármacos epitranscriptómicos se ilustra en la Figura 2.
Al existir una sobreactivación en general de las m6A RNA metiltransferasas METTL3- METTL4, las primeras dianas de investigación farmacológica parecen claras. Estudios previos que emplearon tecnología CRISPR-Cas9 demostraron que la manipulación genética para disminuir la enzima METTL3 reduce la proliferación celular, especialmente en leucemia. Como resultado, varios laboratorios academicos y farmaceúticos, así como empresas biotecnológicas han abierto varias líneas de estudio en este campo. Las pionera en el sector privado fueron STORM Therapeutics en el Reino Unido y Gotham Therapeutics y Accent Therapeutics en Estados Unidos. Todas ellas han generado diversos inhibidores de METTL3 de uso en etapas preclínicas, listos para próximos ensayos clínicos de fase I. Destacar un paso más por parte de STORM Therapeutics, cuyo inhibidor de METTL3 denominado STM-2457 (26) ya ha entrado en 2022 en el primer ensayo clínicos de fase I. Un compuesto con eficacia probada en modelos preclínicos de leucemia mieloide aguda tanto en distintos modelos murinos como en xenotrasplantes derivados de pacientes con cáncer.
La siguiente generación de fármacos actuando a nivel de m6A procedente de los laboratorios públicos está a la vuelta de la esquina. Nuevos inhibidores de METTL3- METTL14 derivados de cribados de decenas de bibliotecas que comprende miles de análogos y derivados de la adenosina, sumados a la aplicación de la cristalografía de rayos X de las proteínas implicadas, están identificando estos nuevos compuestos con potencia en el rango micromolar. Estas moleculas pueden luego modificarse en el laboratorio y, por ejemplo, la porción de ribosa de la adenosina está empezando a ser sustituida por sistemas de anillos alternativos para crear sustancias más activas en la inhibición. Desconocemos aún muchos de los mecanismos como estos nuevos compuestos actuarán y su nivel de especificidad para la enfermedad, pero las expectativas son máximas.
Podemos atacar también la otra cara de la moneda, igual que hicimos bloqueando primero las histona metiltransferasas y luego las histona desmetilasas. De esta manera, las desmetilasas de ARN, centrandonos en aquellas que actúan a nivel de m6A, también ha sido blanco de investigación intensa (27). Muchos de estos fármacos son derivados de productos naturales como sucedió en la quimioterapia clásica del cáncer. Así una optimización química del producto natural reína permite obtener una pequeña molécula inhibidora de pequeñas de la m6A desmetilasa FTO. Este compuesto se une de forma competitiva al centro catalítico de FTO, bloquandolo e incrementando globalmente los niveles de m6A en el ARNm. Su efecto puede ser más general, quizás bloqueando la desmetilación de otras bases como m1A y m3C al inhibir también las desmetilasas ALKBH2 y ALKBH3 específicas de estas marcas. El ácido meclofenámico, previamente identificado como anti-inflamatorio, es también un buen candidato para inhibir específicamente FTO. La forma éster etílico de este ácido aumenta los niveles de m6A en el ARNm y reduce la proliferación en cultivos celulares de diversos tipos tumorales. En modelos murinos su eficacia ha sido revelada en glioblastoma, uno de los tumores con peor prognóstico. El cribado de muchas moléculas fluorescentes con estructuras similares al ácido meclofenámico reveló que la fluoresceína y dos derivados con mejor permeabilidad celular inhibían selectivamente la desmetilación de m6A mediada por FTO. Nuevos inhibidores de FTO, y en general para las subfamilias de desmetilasas ALKB, se han obtenido recientemente explorando el sitio de unión de nucleótidos al Fe(II)/2-oxoglutarato. Este oncometabolito se produce en niveles elevados en las células de glioma y leucemia con mutaciones en las isocitrato deshidrogenasas 1/2 (IDH1/2) por lo que podría originar el primer ejemplo de medicina de precisión genómica para un fármaco epitranscriptómico. Estos compuestos también reaccionan contra proteína- desmetilasas como PHD2 y JMJD2A, logrando así la inactivación de familias epigenómicas y epitranscriptómicas diversas. Dos novísimos inhibidores de FTO son FB23 y FB23-2 que se han diseñado a partir de la estructura del sitio catalítico de la proteína. Aunque ambos compuestos son inhibidores claros de FTO in vitro, el compuesto FB23-2 muestra una mayor capacidad antitumoral en modelos preclínicos con líneas celulares transformadas e induce la diferenciación celular, un fenómeno comúnmente observado para la mayoría de fármacos epigenéticos y epitranscriptómicos. Esta reprogramación celular por el fármaco es especialmente evidente en leucemias. A nivel molecular, el bloqueo de la desmetilación de m6A con el tratamiento por FB23-2 ocurre en los ARN mensajeros de multiples oncogenes. Finalmente, la última hornada de agentes inhibidores de la desmetilación de m6A ha venido de la mano de las tecnologías de cribados de alto rendimiento. De esta forma se han caracterizado varios compuestos selectivos para la inhibición de FTO que incrementan los niveles de m6A en diversas dianas de la proteína FTO en los ARN mensajeros. El inicio de los primeros ensayos clínicos con inhibidores de FTO se está planteando para el próximo año.
6. CONCLUSIONES Y EXPECTATIVAS
“El Libro de la Vida” tiene múltiples capítulos y diversas formas de escritura. De la secuenciación del ADN humano, de la descodificación de nuestro genoma, se nos dijo que era el libro entero. Las investigaciones en las modificaciones químicas del ADN, ARN y las proteínas asociadas nos sugieren que ese libro que nos entregaron estaba falto de gramática, sintaxis y ortografía. Era un inmenso telegrama sin signos de puntuación. Darle sentido a las palabras contenidas en ese texto contínuo es la tarea de la Epigenética y la Epitranscriptómica: colocar una acetilación aquí y una metilación allá, entre otras marcas químicas. El mismo Dr. Francis Collins, el líder del proyecto público de secuenciación del genoma humano comentó recientemente que la Epigenética era algo con lo que no habían contado ni Mendel (padre de la genética clásica) ni Watson ni Crick (padres de la doble cadena de ADN). Elucidar la metilación del ADN, todas las modificaciones de las histonas y los cambios químicos de nuestro ARN de forma completa en todos los tipos celulares nos permitirá una lectura más realista de nuestro “Libro de la Vida”. También nos ayudará a comprender como este escenario idílico del Epigenoma y el Epitranscriptoma se distorsiona en la enfermedad. Y por primera vez tenemos la oportunidad farmacológica de revertir los patrones de modificación química asociados a ciertas patologías: fármacos epigenéticos que, fruto de una investigación de muchos años, hoy son una realidad en los hospitales; y fármacos epitranscriptómicos que serán los siguientes en ser aprobados para su uso clínico.
7. REFERENCIAS
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010 Oct;28(10):1057-68.
- Espada J, Esteller M. DNA methylation and the functional organization of the nuclear compartment. Semin Cell Dev Biol. 2010 Apr;21(2):238-46.
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012 Oct;13(10):679-92.
- Guil S, Esteller M. DNA methylomes, histone codes and miRNAs: tying it all together. Int J Biochem Cell Biol. 2009 Jan;41(1):87-95.
- Ausió J, Martínez de Paz A, Esteller M. MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol Med. 2014 Sep;20(9):487-98.
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008 Mar 13;358(11):1148-59.
- Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011 Mar;17(3):330-9.
- Davalos V, Esteller M. Cancer epigenetics in clinical practice. CA Cancer J Clin. 2023 Jul-Aug;73(4):376-424.
- Esteller M. DNA methylation and cancer therapy: new developments and expectations. Curr Opin Oncol. 2005 Jan;17(1):55-60.
- Simó-Riudalbas L, Esteller M. Targeting the histone orthography of cancer: drugs for writers, erasers and readers. Br J Pharmacol. 2015 Jun;172(11):2716-32.
- Ropero S, Esteller M. The role of histone deacetylases (HDACs) in human cancer. Mol Oncol. 2007 Jun;1(1):19-25.
- Pérez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics. 2017 May 4;12(5):323-339.
- Pappalardi MB, Keenan K, Cockerill M, Kellner WA, Stowell A, Sherk C, Wong K, Pathuri S, Briand J, Steidel M, Chapman P, Groy A, Wiseman AK, McHugh CF, Campobasso N, Graves AP, Fairweather E, Werner T, Raoof A, Butlin RJ, Rueda L, Horton JR, Fosbenner DT, Zhang C, Handler JL, Muliaditan M, Mebrahtu M, Jaworski JP, McNulty DE, Burt C, Eberl HC, Taylor AN, Ho T, Merrihew S, Foley SW, Rutkowska A, Li M, Romeril SP, Goldberg K, Zhang X, Kershaw CS, Bantscheff M, Jurewicz AJ, Minthorn E, Grandi P, Patel M, Benowitz AB, Mohammad HP, Gilmartin AG, Prinjha RK, Ogilvie D, Carpenter C, Heerding D, Baylin SB, Jones PA, Cheng X, King BW, Luengo JI, Jordan AM, Waddell I, Kruger RG, McCabe MT. Discovery of a first-in-class reversible DNMT1-selective inhibitor with improved tolerability and efficacy in acute myeloid leukemia. Nat Cancer. 2021 Oct;2(10):1002-1017.
- Villanueva L, Álvarez-Errico D, Esteller M. The Contribution of Epigenetics to Cancer Immunotherapy. Trends Immunol. 2020 Aug;41(8):676-691.
- Chen S, Cui J, Chen H, Yu B, Long S. Recent progress in degradation of membrane proteins by PROTACs and alternative targeted protein degradation techniques. Eur J Med Chem. 2023 Oct 30;262:115911.
- Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014 Apr;13(4):842-54.
- Mondello P, Ansell SM. Tazemetostat: a treatment option for relapsed/refractory follicular lymphoma. Expert Opin Pharmacother. 2022 Feb;23(3):295-301.
- Chi SN, Yi JS, Williams PM, Roy-Chowdhuri S, Patton DR, Coffey BD, Reid JM, Piao J, Saguilig L, Alonzo TA, Berg SL, Ramirez NC, Jaju A, Mhlanga JC, Fox E, Hawkins DS, Mooney MM, Takebe N, Tricoli JV, Janeway KA, Seibel NL, Parsons DW. Tazemetostat for Tumors Harboring SMARCB1/SMARCA4 or EZH2 Alterations: Results from NCI-COG Pediatric MATCH APEC1621C. J Natl Cancer Inst. 2023 May 25:djad085.
- Zauderer MG, Szlosarek PW, Le Moulec S, Popat S, Taylor P, Planchard D, Scherpereel A, Koczywas M, Forster M, Cameron RB, Peikert T, Argon EK, Michaud NR, Szanto A, Yang J, Chen Y, Kansra V, Agarwal S, Fennell DA. EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1- inactivated malignant pleural mesothelioma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2022 Jun;23(6):758-767.
- Pearson AD, DuBois SG, Buenger V, Kieran M, Stegmaier K, Bandopadhayay P, Bennett K, Bourdeaut F, Brown PA, Chesler L, Clymer J, Fox E, French CA, Germovsek E, Giles FJ, Bender JG, Hattersley MM, Ludwinski D, Luptakova K, Maris J, McDonough J, Nikolova Z, Smith M, Tsiatis AC, Vibhakar R, Weiner S,Yi JS, Zheng F, Vassal G. Bromodomain and extra-terminal inhibitors-A consensus prioritisation after the Paediatric Strategy Forum for medicinal product development of epigenetic modifiers in children-ACCELERATE. Eur J Cancer. 2021 Mar;146:115-124.
- Mellinghoff IK, van den Bent MJ, Blumenthal DT, Touat M, Peters KB, Clarke J, Mendez J, Yust-Katz S, Welsh L, Mason WP, Ducray F, Umemura Y, Nabors B, Holdhoff M, Hottinger AF, Arakawa Y, Sepulveda JM, Wick W, Soffietti R, Perry JR, Giglio P, de la Fuente M, Maher EA, Schoenfeld S, Zhao D, Pandya SS, Steelman L, Hassan I, Wen PY, Cloughesy TF; INDIGO Trial Investigators. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. N Engl J Med. 2023 Aug 17;389(7):589-601.
- Esteller M, Pandolfi PP. The Epitranscriptome of Noncoding RNAs in Cancer. Cancer Discov. 2017 Apr;7(4):359-368.
- Davalos V, Blanco S, Esteller M. SnapShot: Messenger RNA Modifications. Cell. 2018 Jul 12;174(2):498-498.e1.
- Esteve-Puig R, Bueno-Costa A, Esteller M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 2020 Apr 1;474:127-137.
- Berdasco M, Esteller M. Towards a druggable epitranscriptome: Compounds that target RNA modifications in cancer. Br J Pharmacol. 2022 Jun;179(12):2868- 2889.
- Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG, Webster NA, Andrews B, Fosbeary R, Guest P, Irigoyen N, Eleftheriou M, Gozdecka M, Dias JML, Bannister AJ, Vick B, Jeremias I, Vassiliou GS, Rausch O, Tzelepis K, Kouzarides T. Small- molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021 May;593(7860):597-601.
- You Y, Fu Y, Huang M, Shen D, Zhao B, Liu H, Zheng Y, Huang L. Recent Advances of m6A Demethylases Inhibitors and Their Biological Functions in Human Diseases. Int J Mol Sci. 2022 May 22;23(10):5815.
