1. EUROPEAN MEDICINES AGENCY (EMA)
(A) Tracto Alimentario y Metabolismo
Cipaglucosidasa Alfa (Pombiliti®) Amicus
Indicación: Tratamiento de sustitución enzimática a largo plazo que se utiliza en combinación con el estabilizador enzimático miglustat para el tratamiento de adultos con enfermedad de Pompe (deficiencia de alfaglucosidasa ácida) de inicio tardío.
Tipo: Medicamento biológico, constituido por alfa-glucosidasa ácida humana con N-glicanos bisfosforilados (bis-M6P) y producido mediante técnicas de ADN recombinante. Autorizado el 20-3-2023.
Mecanismo: La enfermedad de Pompe está causada por una deficiencia de alfaglucosidasa ácida (GAA), que descompone el glucógeno lisosomal en glucosa. Cipaglucosidasa alfa suple la enzima endógena afectada o ausente; es estabilizada por miglustat, lo cual minimiza la pérdida de actividad enzimática en la sangre durante la perfusión de esta enzima hidrolítica específica del glucógeno, enriquecida con N-glicanos bis-M6P que confieren una unión de alta afinidad al receptor de manosa-6-fosfato independiente de cationes (CI-MPR). Después de la unión, es internalizada en el lisosoma, donde es objeto de rotura proteolítica y escisión del N-glicano, ambas necesarias para producir la forma activa y más madura de la enzima GAA. A continuación, cipaglucosidasa alfa ejerce su actividad enzimática hidrolizando el glucógeno y reduciendo la concentración intramuscular de este, y de este modo atenúa el daño tisular.
Eficacia clínica: Ensayo clínico en fase 3, aleatorizado, doble ciego, controlado con comparador activo (alglucosidasa alfa), internacional y multicéntrico de 52 semanas de duración, en 122 adultos (edad ≥18 años). Las variables de eficacia principales fueron la distancia caminada en 6 minutos (6MWD; variable primaria) y la capacidad vital forzada (CVF) prevista expresada en porcentaje y medida en sedestación. La 6MWD mejoró en 15,9 metros (cipoglucosidasa alfa más miglustat) vs. 1,0 m (alfaglucosidasa alfa más placebo), mientras la capacidad vital forzada (CVF) prevista experimentó un cambio medio de -1,4 vs. -3,7%.
Eventos adversos: Los más comunes son escalofríos (4,0%), mareos (2,6%), rubefacción (2,0%), somnolencia (2,0%), molestias torácicas (1,3%), tos (1,3%), inflamación en el lugar de la perfusión (1,3%) y dolor (1,3%). Las reacciones adversas graves notificadas son urticaria (2,0%), anafilaxia (1,3%), fiebre (0,7%), presíncope (0,7%), disnea (0,7%), edema faríngeo (0,7%), sibilancias (0,7%) e hipotensión (0,7%).
Pegunigalsidasa Alfa (Elfabrio®) Chiesi
Indicación: Para la terapia de reemplazo enzimático a largo plazo en pacientes adultos con diagnóstico confirmado de enfermedad de Fabry (deficiencia de alfa-galactosidasa).
Tipo: Medicamento biológico constituido una forma recombinante pegilada de la α-galactosidasa A humana. Autorizado el 4-5-2023.
Mecanismo: La pegunigalsidasa alfa complementa o reemplaza a la α-galactosidasa-A, la enzima que cataliza la hidrólisis de las fracciones terminales α-galactosilo de los oligosacáridos y polisacáridos en el lisosoma, reduciendo la cantidad de acumulación de globotriaosilceramida (Gb3) y globotriaosilesfingosina (Lyso-Gb3), responsables de la enfermedad de Fabry.
Eficacia clínica: Se evaluó en 142 pacientes (94 hombres y 48 mujeres). Los apacientes sin tratamiento previo con pegunigalsidasa alfa mostraron una reducción del sustrato globotriaosilceramida (Gb3) de los capilares peritubulares renales, medido con BLISS (Barisoni Lipid Inclusion Scoring System) del 68 % tras 6 meses de tratamiento. En 11 de 13 sujetos con biopsias disponibles se registró una reducción sustancial (≥50%) en su puntaje BLISS. Los niveles plasmáticos de Lyso-Gb3 disminuyeron en un 49 % tras 12 meses de tratamiento (n=16) y en un 83 % tras 60 meses (n=10). En un estudio de fase 3, en el que los pacientes cambiaban de agalsidasa beta a pegunigalsidasa alfa, los valores plasmáticos de Lyso-Gb3 se mantuvieron estables después de 24 meses de tratamiento (media de +3,3 nM, n=48).
Eventos adversos: Los más comunes son reacciones relacionadas con la infusión IV (6,3%) de los pacientes, hipersensibilidad y astenia (5,6%).
Velmanasa Alfa (Lamzede®) Chiesi
Indicación: Tratamiento enzimático sustitutivo para controlar las manifestaciones no neurológicas de los pacientes con formas leves a moderadas de alfa-manosidosis.
Tipo: Medicamento biológico constituido por una forma recombinante de la alfa-manosidasa humana. Autorizado el 23-3-2023 como medicamento huérfano (Orphan drug); autorizado previamente en Estados Unidos el 17-2-2023.
Mecanismo: Es una forma recombinante de la alfa-manosidasa humana. Se pretende complementar o sustituir a la alfa-manosidasa natural, una enzima que cataliza la degradación secuencial de los oligosacáridos híbridos y complejos con alto contenido de manosa en los lisosomas, reduciendo la cantidad de oligosacáridos ricos en manosa acumulados.
Eficacia clínica: Estudio multicéntrico fase 3 de grupos paralelos, doble ciego, aleatorizado y controlado con placebo, a lo largo de 52 semanas en un total de 25 pacientes, de ellos 12 sujetos pediátricos. Los efectos globales del tratamiento se evaluaron en los dominios farmacodinámico (reducción de los oligosacáridos séricos: -5,11 vs. -1,61) y funcional: prueba de subida de escaleras de tres minutos (PSE3M: +0,46 vs. -2,16), prueba de marcha de seis minutos (PM6M: +3,74 vs. -3,61) y de la capacidad vital forzada predicha (CVF: +8,2% vs. +2,3%).
Eventos adversos: Los más comunes son aumento de peso (18%), RRP (reacciones relacionadas con la perfusión IV: hipersensibilidad, náuseas, vómitos, pirexia, escalofríos, sensación de calor, malestar general, urticaria, reacción anafilactoide e hiperhidrosis) (9%), diarrea (12%), cefalea (9%), artralgia (9%), aumento del apetito (6%) y dolor en las extremidades (6%).
(B) Sangre y Sistema Hematopoyético
Vadadustat (Vafseo®) Akebia
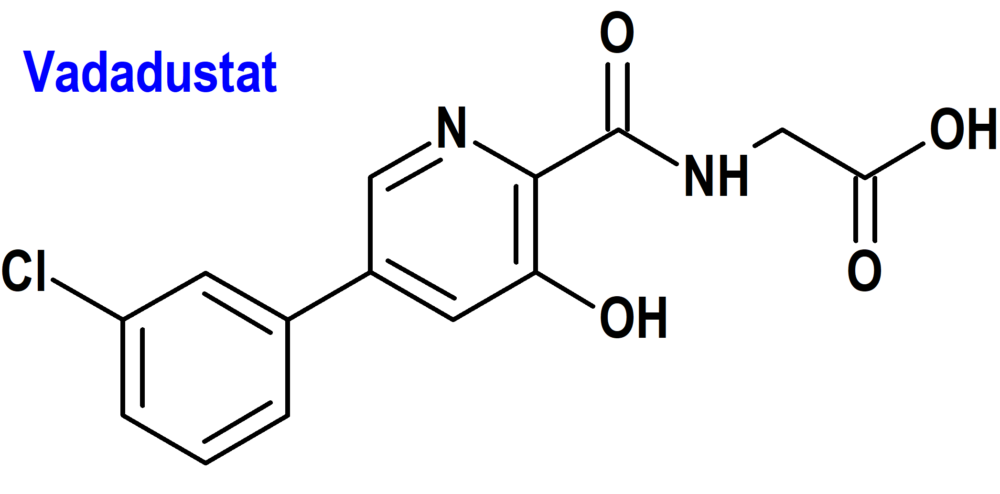
Indicación: Tratamiento de la anemia sintomática asociada a enfermedad renal crónica (ERC) en adultos en diálisis de mantenimiento crónica.
Tipo: Medicamento sintético estándar derivado de fenilpiridina. Autorizado el 24-4-2023.
Mecanismo: Inhibidor de la prolilhidroxilasa del factor inducible por hipoxia que produce un aumento de los niveles celulares del factor inducible por hipoxia (HIF), estimulando así la producción endógena de eritropoyetina (EPO), lo que aumenta la movilización del hierro y la producción de eritrocitos, y que, a su vez, da lugar a una tasa gradual de aumento de la hemoglobina (Hb).
Eficacia clínica: Dos estudios multicéntricos, aleatorizados, controlados con tratamiento activo (darbopoetina alfa), de no inferioridad y abiertos en pacientes dependientes de diálisis, durante un año. La variable primaria de eficacia en ambos estudios fue la diferencia en el cambio medio de la Hb desde el inicio hasta el periodo de evaluación primario (semanas 24 a 36): 1,26 g/dl (vadadustat) vs. 1,58 (darbopoetina alfa) y 0,19 vs. 0,36 g/dl ; la variable secundaria de eficacia fue la diferencia en el cambio medio de la Hb desde el inicio hasta el periodo de evaluación secundario (semanas 40 a 52): 1,42 vs. 1,50 g/dl y 0,23 vs. 0,41 g/dl. Se consideró como no inferioridad de vadadustat frente a darbepoetina alfa si el límite inferior del intervalo de confianza (IC) del 95% para la diferencia en el cambio medio estimado en la hemoglobina media desde el inicio en los dos grupos de tratamiento era mayor que el margen de no inferioridad preespecificado de −0,75 g/dl.
Eventos adversos: Los más comunes son acontecimientos tromboembólicos (13,7%), diarrea (12,7%) e hipertensión (11,1%). Las reacciones adversas graves más frecuentes (≥1 %) son acontecimientos tromboembólicos (10,0%), hipotensión (1,6%) e hipertensión (1,1%).
(D) Dermatología
Ruxolitinib (Opzelura®) Incyte
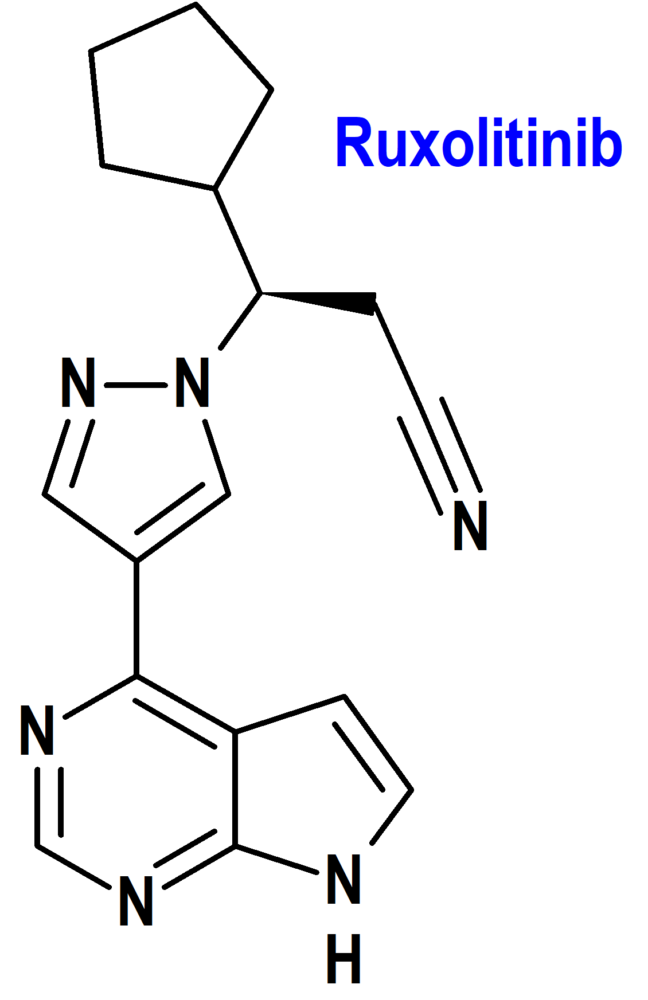
Indicación: Tratamiento del vitíligo no segmentario con localización facial en adultos y adolescentes a partir de 12 años.
Tipo: Medicamento sintético estándar derivado de pirrolopirimidina. Autorizado el 19-4-2023; autorizado previamente en Estados Unidos el 19-7-2022.
Mecanismo: Inhibidor selectivo de las quinasas asociadas a Janus (JAK) JAK1 y JAK2. La señalización intracelular de JAK implica la fosforilación y activación de transductores de señal y activadores de la transcripción (STAT), lo que activa la expresión genética dentro de la célula. Los linfocitos T citotóxicos productores de IFNγ autoinmunes parecen ser los responsables directos de la destrucción de melanocitos en el vitíligo humano. El reclutamiento de linfocitos citotóxicos en la piel lesionada está mediada a través de quimiocinas dependientes del IFNγ, como la CXCL10 (motif chemokine ligand 10, también conocido como Proteína 10 inducida por interferón gamma, IP10). La señalización descendente de IFNγ depende de JAK1/2 y el tratamiento con ruxolitinib reduce los niveles de CXCL10 en pacientes con vitíligo.
Eficacia clínica: Dos estudios aleatorizados, doble ciego, controlados con placebo (vehículo) sobre un total de 674 pacientes con vitíligo facial y corporal total que no superaban el 10% de la superficie corporal. La variable de eficacia principal fue la proporción de pacientes que lograron una repigmentación del 75% en el índice de puntuación del área de vitíligo 7 facial en la semana 24: 29,8-30,9% (ruxolitinib) vs. 7,4-11,4% (placebo). Las variables secundarias principales incluían las proporciones de pacientes que lograron una repigmentación del 90% (15,3-16,3% vs 1,3-2,2%); una mejora del 50% en el índice de puntuación del área de vitíligo corporal total (20,5-23,9 vs. 5,1-6,8%); y una puntuación en la escala de notoriedad del vitíligo de 4 o 5 (vitíligo “mucho menos perceptible” o “ya no perceptible”): 20,5-24,5 vs. 1,9-3,3%.
Eventos adversos: El más común es acné en el sitio de aplicación (5,8%).
(J) Antiinfecciosos Sistémicos
Vacuna COVID-19 recombinante (Bimervax®) Hipra
Indicación: Como refuerzo para la inmunización activa para prevenir la COVID-19 en personas de 16 años de edad y mayores que hayan recibido previamente una vacuna de ARNm contra la COVID-19.
Tipo: Medicamento biológico, constituido por una proteína heterodimérica que contiene el sitio de unión al receptor (RBD) de la proteína de espiga (spike) recombinante (S) del virus SARS-CoV-2 (cepas B.1.351 y B.1.1.7), producida por tecnología de ADN recombinante utilizando un vector de expresión de plásmido en una línea celular CHO; adyuvado con SQBA (escualeno, polisorbato 80, trioleato de sorbitán, citrato de sodio y ácido cítrico), que incrementa la respuesta inmune. Autorizado el 30-3-2023.
Mecanismo: Vacuna de proteína recombinante cuyo antígeno es el heterodímero de fusión B.1.351-B.1.1.7 del dominio de unión al receptor (RBD) de proteína de espiga (spike) recombinante (S) del virus SARS-CoV-2. Tras su administración se genera una respuesta inmune, tanto a nivel humoral como celular, frente al antígeno. Los anticuerpos neutralizantes contra el dominio RBD del SARS-CoV-2 evitan que éste se una a su diana celular ACE2, bloqueando así la fusión de membranas y la infección viral. Además, la vacuna induce una respuesta inmunitaria de linfocitos T específica de antígeno, lo que puede contribuir a la protección contra la COVID-19.
Eficacia clínica: Ensayo clínico en curso de fase 2b, doble ciego, aleatorizado, con control activo, multicéntrico, de no inferioridad para evaluar la inmunogenicidad y la seguridad de una vacunación de refuerzo con esta vacuna en comparación con la vacuna de ARNm COVID-19 (tozinameran), en 751 adultos vacunados completamente contra COVID-19 con una vacuna de ARNm al menos 6 meses antes. La inmunogenicidad de una dosis de refuerzo de BIMERVAX se basó en una evaluación de los títulos medios geométricos (GMT) de anticuerpos neutralizantes, medidos mediante un ensayo de neutralización basado en pseudoviriones (PBNA) contra las cepas Beta, Delta y la variante BA.1 de Omicron del SARS-CoV-2 (D614G). Se consideró como no inferioridad de BIMERVAX frente a la vacuna de ARNm de COVID-19 (tozinamerán) si el límite superior del intervalo de confianza (IC) bilateral del 95 % de la relación GMT fuese menor de 1,4 y la superioridad si ésta es inferior a 1,0; a los 14 días fueron de 1,71 (cepa D614G), 0,62 (Beta), 1,02 (Delta) y 0,60 (Omicron BA.1), mientras que a los 182 días fueron de 0,62 (D614G), 0,70 (Beta), 0,55 (Delta) y 0,76 (Omicron BA.1). Otro ensayo clínico, en este caso de fase 3, multicéntrico, de un solo grupo, abierto y actualmente en curso en sujetos vacunados con vacunas primarias (de ARNm: tozinamerán y elasomerán, y de vectores de adenovirus: ChAdOx1-S y Ad26.COV2-S), con o sin antecedentes de COVID-19 no grave, en el que BIMERVAX se administró al menos 91 días después de la última dosis o al menos 30 días después de la infección por COVID-19. El informe provisional incluye datos de un total de 2.646 sujetos que fueron vacunados con BIMERVAX como dosis de refuerzo en individuos sanos (>15 años) previamente vacunados con diferentes vacunas contra el COVID-19. La inmunogenicidad fue medida por ensayo de neutralización basado en pseudovirión (PBNA) contra la cepa SARS-CoV-2 (D614G) y contra Beta, Delta y Omicron BA.1 Datos sobre GMT (título medio geométrico: ID50) al inicio (antes de la administración) y 2 semanas después de la administración de la dosis de refuerzo, siendo la relación entre los datos del día 14 y los del inicio de 6,6 (D614G), 18,7 (Beta), 9,4 (Delta) y 22,3 (Omicron BA.1) sobre tozinameran; de 6,8 (D614G), 13,8 (Beta), 6,4 (Delta) y 19,7 (Omicron BA.1) sobre elasomeran; y de 8,0 (D614G), 9,3 (Beta), 9,2 (Delta) y 11,6 (Omicron BA.1) sobre ChAdOx1-S.
Eventos adversos: Los más comunes (>10%) son cefalea, mialgia, fatiga y dolor en el punto de inyección; también son frecuentes (1-10%): fiebre, linfadenopatía, diarrea, náusea y vómito.
Vacuna Virus Respiratorio Sincitial (Arexvy®) GlaxoSmithKline
Indicación: Inmunización activa para la prevención de la enfermedad del tracto respiratorio inferior causada por el virus respiratorio sincitial en personas de 60 años de edad y mayores.
Tipo: Medicamento biológico, constituido por la glicoproteína F recombinante del virus respiratorio sincitial (VRS) estabilizada en conformación previa a la fusión (RSVPreF3) como componente antigénico y adyuvado por AS01E, que está compuesto por 3-O-desacil-4′-monofosforil lípido A (MPL) de Salmonella minnesota y QS-21, una saponina purificada procedente del extracto de Quillaja saponaria, combinados en una formulación liposomal. Autorizado el 6-6-2023; autorizado en Estados Unidos el 3-5-2023, mediante revisión prioritaria (Priority Review).
Mecanismo: Induce una respuesta inmune protectora frente a la enfermedad del tracto respiratorio inferior causada por el virus respiratorio sincitial (VRS).
Eficacia clínica: Estudio clínico en curso, aleatorizado, controlado con placebo en 25,000 personas de 60 años de edad y mayores. Entre los participantes que recibieron la vacuna y los que recibieron placebo, la vacuna redujo en un 82,6 % el riesgo de desarrollar la enfermedad del tracto respiratorio inferior causada por el virus respiratorio sincitial, y un 94,1% de los cuadros graves.
Eventos adversos: Los más comunes son dolor en el lugar de la inyección (61%), fatiga (334%), mialgia (29%), dolor de cabeza (27%) y artralgia (18%).
(L) Agentes Antineoplásicos e Inmunomoduladores
Ivosidenib (Tibsovo®) Servier

Indicación: Tratamiento, en combinación con azacitidina, de pacientes adultos con leucemia mieloide aguda (LMA) de nuevo diagnóstico con mutación de la isocitrato deshidrogenasa-1 (IDH1) R132, que no son candidatos a quimioterapia de inducción estándar. En monoterapia, está indicado para el tratamiento de pacientes adultos con colangiocarcinoma localmente avanzado o metastásico con mutación IDH1 R132 que hayan recibido al menos una línea previa de tratamiento sistémico.
Tipo: Medicamento sintético estándar derivado de glicinamida. Autorizado el 4-5-2023 como medicamento huérfano (Orphan drug); autorizado previamente en Estados Unidos el 20-7-2018.
Mecanismo: Inhibidor de la enzima isocitrato deshidrogenasa-1 (IDH1) mutada, la cual convierte el alfa-cetoglutarato en 2-hidroxiglutarato (2-HG), que bloquea la diferenciación celular y favorece la carcinogénesis en neoplasias malignas tanto hematológicas como no hematológicas. En la médula ósea de los pacientes con neoplasias malignas hematológicas y en la biopsia tumoral de los pacientes con colangiocarcinoma, la reducción media (% del coeficiente de variación [%CV]) de las concentraciones de 2-HG fue del 93,1% (11,1%) y del 82,2% (32,4%), respectivamente.
Eficacia clínica: Estudio clínico multicéntrico, aleatorizado, doble ciego y controlado con placebo de 146 pacientes adultos con leucemia mieloide aguda no tratada previamente con una mutación IDH1, que no eran candidatos para recibir quimioterapia de inducción intensiva. La variable primaria de eficacia fue la supervivencia libre de eventos (SLE), medida desde la fecha de aleatorización hasta el fracaso del tratamiento (imposibilidad de alcanzar la remisión completa en la semana 24), recaída tras remisión o muerte por cualquier causa: 64 vs. 84% (fracaso: 58 vs. 80%; recaída: 4,2 vs. 2,7% y muerte: 1,4 vs. 1,4%). La mediana de la supervivencia global fue de 24,0 vs. 7,9 meses.
Eventos adversos: Los más comunes son vómitos (40%), neutropenia (31%), trombocitopenia (28%), prolongación del intervalo QT del electrocardiograma (21%) e insomnio (19%). Las reacciones adversas graves más frecuentes fueron el síndrome de diferenciación (8%) y trombocitopenia (3%). En los pacientes tratados con ivosidenib en combinación con azacitidina, la frecuencia de suspensión del tratamiento con ivosidenib debido a reacciones adversas fue del 6%. Las reacciones adversas que condujeron a la suspensión fueron prolongación del intervalo QT del electrocardiograma (1%), insomnio (1%), neutropenia (1%) y trombocitopenia (1%).
Mirikizumab (Omvoh®) Lilly
Indicación: Tratamiento de la colitis ulcerosa activa, de moderada a grave, en pacientes adultos que hayan tenido una respuesta inadecuada, presenten pérdida de respuesta o intolerancia al tratamiento convencional o a un tratamiento biológico.
Tipo: Medicamento biológico, constituido por un anticuerpo monoclonal IgG4 humanizado producido mediante tecnología de ADN recombinante. Autorizado el 26-5-2023.
Mecanismo: Anticuerpo monoclonal que se une de forma selectiva a la subunidad p19 de la citocina IL-23 humana e inhibe su interacción con el receptor de IL-23. La IL-23, una citocina reguladora que actúa sobre la diferenciación, expansión y supervivencia de subconjuntos de células T (Th17 y Tc17) y subconjuntos de células inmunitarias innatas, que representan fuentes de citocinas efectoras (incluyendo IL-17A, IL-17F e IL-22) que desencadenan la enfermedad inflamatoria. En humanos, el bloqueo selectivo de la IL-23 demostró normalizar la producción de estas citocinas.
Eficacia clínica: Dos estudios aleatorizados, doble ciego, controlados con placebo y multicéntricos. El primero fue un estudio de inducción por vía intravenosa de hasta 12 semanas en 1,162 pacientes, seguido de un estudio de mantenimiento subcutáneo, de retirada aleatorizada, de 40 semanas en 544 pacientes, totalizando al menos 52 semanas de tratamiento. La variable primaria para el estudio de inducción fue la proporción de pacientes en remisión clínica en la semana 12 (puntuación Mayo modificada, MMS, definida como: subpuntuación de frecuencia de deposiciones = 0 o 1 con una disminución de ≥ 1 punto con respecto al valor basal, subpuntuación de sangrado rectal = 0, y subpuntuación endoscópica = 0 o 1): 30,9% (mirikizumab) vs. 15,8% (placebo) en pacientes naif, y 15,2% vs. 8,5% en pacientes no naif . En el estudio de mantenimiento, los porcentajes de pacientes que obtuvieron remisión clínica tras 40 semanas fueron del 51,5% vs. 30,7% en pacientes naif, y del 46,1% vs. 15,6% en pacientes no naif.
Eventos adversos: Los más comunes son infecciones del tracto respiratorio superior (7,9%, con mayor frecuencia nasofaringitis), cefalea (3,3%), erupción cutánea (1,1%) y reacciones en el lugar de inyección (8,7%).
2. FOOD & DRUG ADMINISTRATION (FDA)
(A) Tracto Alimentario y Metabolismo
Esporas de microbiota fecal viva (Vowst®) Seres
Indicación: Para prevenir la recurrencia de la infección por Clostridioides difficile (CDI) en personas de 18 años de edad y mayores que siguen un tratamiento antibacteriano para la CDI recurrente (rCDI).
Tipo: Medicamento biológico constituido por esporas de microbiota fecal viva, fabricado a partir de materia fecal humana procedente de donantes cualificados. La suspensión de esporas se genera al tratar la materia fecal con etanol para eliminar los organismos que no son esporas. Cada cápsula contiene entre 1×10(6) y 3×10(7) unidades formadoras de colonias de esporas. Autorizado el 26-4-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea.
Mecanismo: No se conoce el mecanismo de acción, aunque genéricamente se pretende restaurar la diversidad de microorganismos en la microbiota colónica y eliminar el crecimiento del Clostridioides difficile.
Eficacia clínica: Estudio clínico aleatorizado, controlado con placebo en 182 pacientes adultos con un diagnóstico confirmado de CDI recurrente (≥3 episodios de CDI en el último año). Durante las 8 semanas posteriores al tratamiento, la recurrencia de CDI en los tratados con el medicamento fue menor que con placebo (12,4 vs. 39,8%).
Eventos adversos: Los más comunes son distensión abdominal (31,1%), fatiga (22,2%), estreñimiento (14,4%), escalofríos (11,1%) y diarrea (10,0%).
(B) Sangre y Sistema Hematopoyético
Efanesostocog alfa (Altuviiio®) Bioverativ
Indicación: Uso en adultos y niños con hemofilia A (deficiencia congénita del factor VIII) para profilaxis de rutina para reducir la frecuencia de episodios hemorrágicos; tratamiento a demanda y control de episodios hemorrágicos; manejo perioperatorio de hemorragias.
Tipo: Medicamento biológico, constituido por una proteína recombinante correspondiente al factor VIII, unida a tres componentes adicionales que evitan su unión al factor von Willebrand y aumentan su semivida de eliminación. El Factor de coagulación VIII humano (FVIII, factor antihemofílico, AHF, componente procoagulante) en el dominio B reemplazado (746-1648) [dominios A1-a1-A2-a2 del FVIII (1-740) y fragmento N-terminal del dominio B (741-745), fusionado a través de un péptido sintético de 291 aminoácidos, hecho de 24 péptidos de 12 aminoácidos cada uno, que se repiten (4 tipos) (746-1033), y de un tripéptido ASS (10341036), con la parte C-terminal de los dominios a3-A3-C1-C2 16492332 del FVIII (1037-1720)], fusionado con el fragmento Fc C-terminal K> del de la inmunoglobulina G1 humana (1721-1946), unido por puentes disulfuro con el fragmento que contiene el dominio TIL3-D3-TIL4 del factor de von Willebrand humano 742-1218 (1′-477′) fusionado a través de un péptido sintético de 148 aminoácidos, hecho de 12 péptidos de 12 aminoácidos cada uno, que se repiten (4 tipos), además un tetrapéptido GASS (478′-625′), con un fragmento del FVIII escindible por la trombina 712-743 (626′-657′) [región 2 ácido escindible por la trombina además el (1-3)-péptido del dominio B3] fusionado con el fragmento Fc C-terminal K>del de la inmunoglobulina G1 humana (658′-883′), producido en las células renales embrionarias humanas 293 (HEK293), glicoforma alfa. Autorizado el 22-2-2023; no autorizado aún en la Unión Europea.
Mecanismo: Reemplaza temporalmente el factor VIII de coagulación faltante necesario para una hemostasia eficaz. Efanesostocog ha demostrado una semivida de 3 a 4 veces más prolongada en relación con otros productos de FVIII.
Eficacia clínica: Dos estudios clínicos multicéntricos, prospectivos, abiertos (un estudio en 133 adultos y adolescentes ≥12 años y un estudio pediátrico en niños <12 años) en pacientes previamente tratados (PTP) con hemofilia A severa (<1% actividad endógena del factor VIII o una mutación genética documentada compatible con hemofilia A grave). En el primer estudio (adultos y adolescentes), la tasa anualizada de hemorragias en uso de profilaxis fue de 0,7 (64-77% de los pacientes sin ninguna hemorragia) y en uso bajo demanda de 21,4. En el estudio en pediátrico, la tasa anualizada de hemorragias en profilaxis fue del 0,5 en pacientes pretratados y de 3,6 en total (tratados y no tratados).
Eventos adversos: Los más comunes son cefalea (21%), artralgia (16%) y dolor de espalda (6%).
Omidubicel (Omisirge®) Gamida
Indicación: Uso en adultos y pacientes pediátricos de 12 años y mayores con neoplasias hematológicas que están programados para un trasplante de sangre de cordón umbilical después de un acondicionamiento mieloablativo, para reducir el tiempo de recuperación de neutrófilos y la incidencia de infección.
Tipo: Medicamento biológico de terapia avanzada (celular somática), administrado como una dosis intravenosa única, constituido por células madre alogénicas humanas de la sangre del cordón umbilical que se procesan y cultivan con nicotinamida (vitamina B3). Cada dosis es específica para el paciente y contiene células madre sanas de un donante alogénico preseleccionado. Consta de 2 fracciones celulares; una fracción cultivada, consistente en una suspensión de células progenitoras hematopoyéticas CD34+ alogénicas y una fracción no cultivada, ambas derivadas de la misma unidad de sangre del corazón específica del paciente. La fracción no cultivada está formada por una suspensión que contiene células linfoides y mieloides maduras hematopoyéticas alogénicas. Autorizado el 17/4/2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea.
Mecanismo: El trasplante de células progenitoras (madre) hematopoyéticas es un tratamiento común para los cánceres hematológicos, consistente en incorporar células progenitoras sanas en el cuerpo para ayudar a restaurar la producción y el funcionamiento normales de las células sanguíneas. Una fuente de células madre sanas es la sangre del cordón umbilical. Generalmente, antes de recibir este tipo de trasplante, el paciente debe ser sometido a un ciclo de tratamientos (radioterapia o quimioterapia) para eliminar sus propias células progenitoras y preparar el cuerpo para las nuevas células progenitoras, tratamientos que pueden debilitar el sistema inmunológico de un individuo. El cultivo ex-vivo de células progenitoras hematopoyéticas derivadas de la sangre del cordón umbilical en presencia de omidubicel conduce a la preservación de su tallo, a la localización en la médula ósea y a la retención de la capacidad del injerto celular.
Eficacia clínica: Estudio multicéntrico aleatorizado comprando el trasplante de omidubicel con el de sangre de cordón umbilical, en 125 sujetos de 12 a 65 años de edad, con cánceres hematológicos confirmados. El criterio principal de consistió en la cantidad de tiempo necesario para la recuperación de los neutrófilos del sujeto y la incidencia de infecciones después del trasplante. El 87% tratados con omidubicel lograron la recuperación de neutrófilos en una mediana de 12 días después del tratamiento, en comparación con el 83 % de los que recibieron un trasplante de sangre del cordón umbilical y que lograron la recuperación de neutrófilos con una mediana de 22 días. Se observaron infecciones bacterianas o fúngicas a lo largo de 100 días después del trasplante en el 39% omidubicel vs. 60 % del grupo de control que recibieron sangre del cordón umbilical.
Eventos adversos: Los más comunes son dolor (38% vs. 18% con sangre de cordón umbilical), mucositis (31/34%), hipertensión (25/38%), hemorragia (12/18%), disfagia (12/13%), insuficiencia respiratoria (12/30%), insuficiencia renal (12/5%), disnea (8/16%), fatiga (4/21%) y fiebre (2/11%). Las reacciones adversas de grado 3 o superior fueron: relacionadas con el procedimiento de infusión IV (17/21%), infecciones virales (8/27%) y reacciones de injerto contra huésped agudas (15/21%) y crónicas (23/20%). La incidencia de reacciones adversas fatales fue del 17% y 21%, respectivamente.
(D) Dermatología
Beremagene Geperpavec (Vyjuvek®) Krystal
Indicación: Tratamiento de heridas en pacientes de 6 meses de edad y mayores con epidermolisis ampollosa distrófica con mutaciones en el gen de la cadena alfa 1 del colágeno tipo VII (COL7A1).
Tipo: Medicamento biológico de terapia avanzada (génica), constituido por un vector vivo basado en virus del Herpes simplex tipo 1 (HSV-1) con replicación defectuosa (modificado para eliminar su capacidad de replicarse en células normales), que ha sido modificado genéticamente para expresar la proteína de colágeno humano tipo VII (COL7). Autorizado el 19-5-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: La epidermólisis ampollosa distrófica es un trastorno genético que afecta el tejido conectivo en la piel y las uñas y es el resultado de mutaciones en el gen COL7A1. Este gen codifica el colágeno tipo VII (COL7), una proteína esencial que se organiza en haces largos y delgados que forman fibrillas de anclaje que mantienen unidas la epidermis (piel) y la dermis, lo cual es esencial para mantener la integridad de la piel. Cuando COL7A1 es deficiente, las capas de la piel pueden separarse, causando la formación de ampollas y de heridas dolorosas y debilitantes. Beremagene Geperpavec permite administrar copias normales del gen COL7A1 en las heridas.
Eficacia clínica: Ensayo aleatorizado, doble ciego, intrasujeto, controlado con placebo, que incluyó a 31 sujetos. La eficacia se estableció sobre el porcentaje de pacientes que experimentaron una mejora de la cicatrización de heridas, definida como la diferencia en la proporción de cierre completo (100%) de la herida a las 10 (68% con el tratamiento vs. 23% con placebo) y 24 semanas (65 vs 26%). Eventos adversos: Los más comunes son (>5%) son picazón, escalofríos, enrojecimiento, sarpullido, tos y secreción nasal.
(G) Sistema Genitourinario y Hormonas Sexuales
Fezolinetant (Veozah®) Astellas
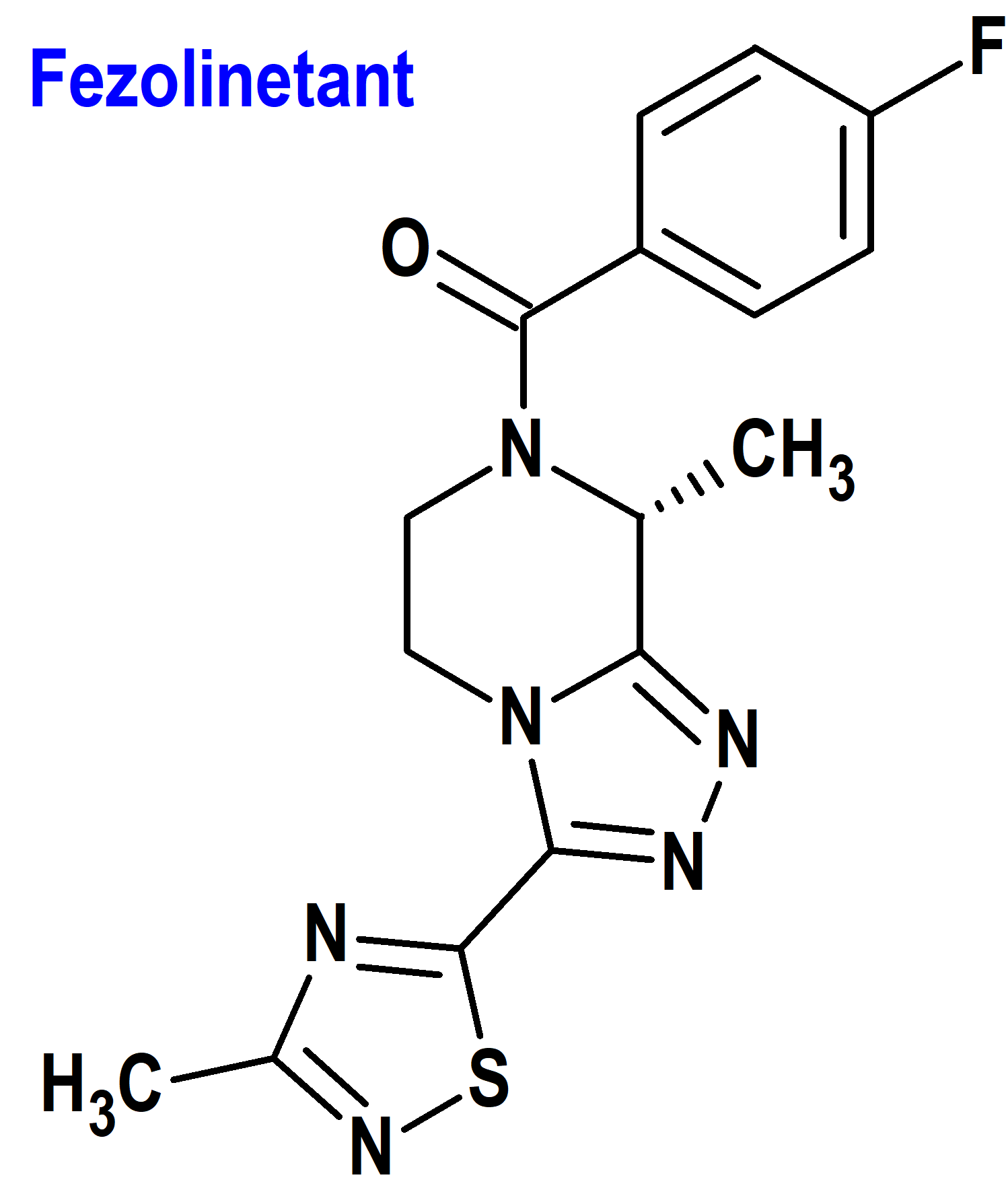
Indicación: Tratamiento de los síntomas vasomotores moderados a severos debidos a la menopausia.
Tipo: Medicamento sintético estándar derivado de triazolopirazina. Autorizado el 12-5-2023 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Antagonista del receptor de neurocinina 3 (NK3) que bloquea la unión de la neurocinina B (NKB) en la neurona kisspeptina/neurocinina B/dinorfina (KNDy) para modular la actividad neuronal en el centro termorregulador. Fezolinetant tiene una gran afinidad por el receptor NK3 (>450 veces mayor que la afinidad de unión a los receptores NK1 o NK2).
Eficacia clínica: Dos ensayos clínicos de fase 3 y 12 semanas de duración, aleatorizados, controlados con placebo y doble ciego, sobre un total de 1022 mujeres (522 y 500) que tenían un promedio mínimo de 7 síntomas vasomotores de moderados a graves por día. En cada uno de estos dos ensayos, después de las primeras 12 semanas, las mujeres que recibieron placebo se volvieron a asignar al azar a fexolinetant durante una extensión de 40 semanas para evaluar la seguridad de hasta 52 semanas de exposición total. La variable principal de eficacia para ambos ensayos consistió en el cambio medio desde el inicio en la frecuencia y la gravedad de los síntomas vasomotores de moderados a graves hasta las semanas 4 y 12. Los datos de cada ensayo demostraron una reducción estadística y clínicamente significativa (≥2 sofocos en 24 horas) desde el inicio en la frecuencia de síntomas vasomotores de moderados a graves para fezolinetant 45 mg en diferencia con el placebo: -2,1 a -2,6 en la semana 4 y -2,5 a -2,6 en la semana 12.
Eventos adversos: Los más comunes son dolor abdominal (4,3 vs. 2,1%, frente a placebo), diarrea (3,9/2,6%), insomnio (3,9/1,8%), dolor de espalda (3,0/2,1%), sofocos (2,5/1,6%), elevación de transaminasas hepáticas (2,3/0,8%).
(J) Antiinfecciosos Sistémicos
Rezafungina (Rezzayo®) Cidara

Indicación: Tratamiento de candidemia y candidiasis invasiva en pacientes de 18 años de edad o mayores que tienen opciones alternativas limitadas o nulas.
Tipo: Medicamento sintético estándar derivado del grupo de equinocandinas. Autorizado el 22-3-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Antifúngico del grupo de las equinocandinas. Inhibe el complejo enzimático 1,3-β-D-glucano sintasa, que está presente en las paredes celulares de los hongos pero no en las células de los mamíferos, provocando la inhibición de la formación de 1,3-β-D-glucano, un componente esencial de la pared celular fúngica de muchos hongos. Se ha demostrado que es activa tanto in vitro como en infecciones clínicas contra Candida albicans, C. glabrata, C. parapsilosis y C. tropicalis.
Eficacia clínica: Estudio multicéntrico, aleatorizado, doble ciego y controlado con comparador activo (caspofungina) sobre 199 pacientes. La eficacia se evaluó por la mortalidad por cualquier causa en el día 30: 23,7% (rezafungina) vs. 21,3 % (caspofungina). Los resultados de eficacia adicionales fueron la curación clínica a los 30 días (54,8 vs. 55,3%) y la erradicación micológica a los 14 días (67,7 vs. 66,0%).
Eventos adversos: Los más comunes son hipopotasemia (15%), fiebre (12%), anemia (9%) diarrea (9%), vómito (9%), náusea (9%), hipomagnesemia (8%), dolor abdominal (7%) y estreñimiento (5%).
Durlobactam/Sulbactam (Xacduro®) Entasis
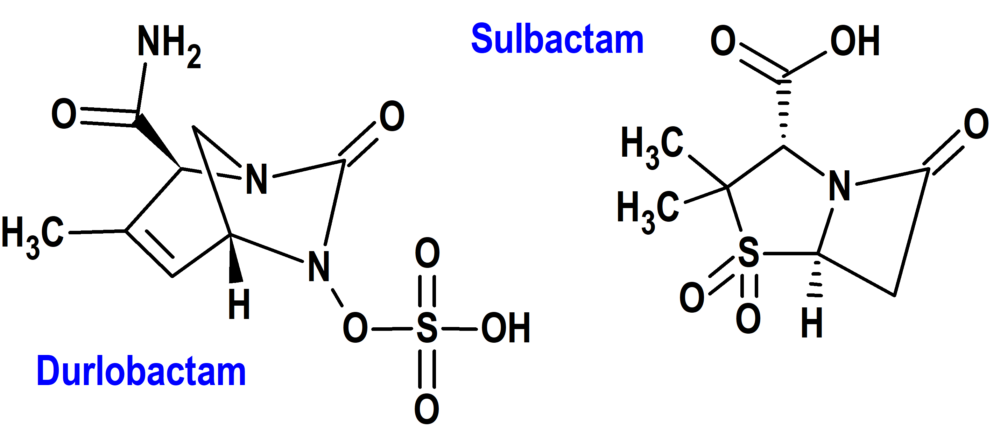
Indicación: Tratamiento de la neumonía bacteriana adquirida en el hospital y la neumonía bacteriana asociada al ventilador (HABP/VABP), causada por cepas susceptibles del complejo Acinetobacter baumannii-calcoaceticus, en pacientes de 18 años de edad y mayores.
Tipo: Medicamento sintético estándar formada por la combinación de durlobactam y sulbactam; el durlobactam es un derivado del núcleo de diazabiciclooctano y el sulbactam tiene estructura betalactámica. Autorizada el 23-5-2023 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Antibacteriano formado por la combinación de sulbactam sódico, un inhibidor de beta-lactamasa y antibacteriano betaláctámico derivado de la penicilina, y durlobactam un inhibidor beta-lactamasa. Sulbactam es un inhibidor de serina betalactamasas de Clase A que tiene actividad bactericida debido a su inhibición de las proteínas de unión a penicilina PBP1 y PBP3 del complejo Acinetobacter baumannii-calcoaceticus (ABC), que son enzimas esenciales necesarias para la síntesis de la pared celular bacteriana. Durlobactam es un diazabiciclooctano no beta-lactámico, inhibidor de beta-lactamasa, que protege al sulbactam de la degradación por ciertas serina-betalactamasas. Durlobactam solo no tiene actividad antibacteriana frente a ABC.
Eficacia clínica: Ensayo de fase 3 multicéntrico, aleatorizado, con control activo (colistina), de no inferioridad, no cegado por el investigador pero sí para el evaluador independiente, sobre 177 adultos hospitalizados con infecciones documentadas del complejo Acinetobacter baumannii-calcoaceticus. Ambos brazos de tratamiento también recibieron imipenem/cilastatina. Los pacientes recibieron hasta 14 días de terapia. El criterio principal de valoración de la eficacia fue la mortalidad por todas las causas a los 28 días: 19,0 vs. 32,3%. Las tasas de curación clínica (resolución completa o la mejoría significativa de los signos y síntomas) a los 7 (±2) días tras el final del tratamiento fueron del 61,9 vs. 40,3%.
Eventos adversos: Los más comunes (>10%) son anomalías en las pruebas hepáticas, diarrea, anemia e hipopotasemia.
(L) Agentes Antineoplásicos e Inmunomoduladores
Ratifanlimab (Zynyz®) Incyte
Indicación: Tratamiento de pacientes adultos con carcinoma de células de Merkel (CCM) localmente avanzado metastásico o recurrente.
Tipo: Medicamento biológico, constituido por un anticuerpo monoclonal IgG4 kappa humanizado dirigido a bloquear el receptor de muerte programada 1 (PD-1). Autorizado el 22-3-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: La unión de los ligandos de PD-1, PD-L1 y PD-L2, al receptor de PD-1 que se encuentra en los linfocitos T, inhibe la proliferación estos y la producción de citocinas. La regulación al alza de los ligandos de PD-1 se produce en algunos tumores, y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmunitaria activa de los linfocitos T de los tumores. Retifanlimab se une al receptor PD-1, bloquea la interacción con sus ligandos PD-L1 y PD-L2 y potencia la actividad de los linfocitos T.
Eficacia clínica: Estudio abierto, multirregional, de un solo brazo que evaluó a 65 pacientes con CCM localmente avanzado metastásico o recurrente que no habían recibido tratamiento sistémico previo para la enfermedad avanzada. Las variables primarias de eficacia clínica fueron la tasa de respuesta objetiva (ORR) y la duración de la respuesta (DOR) evaluadas por un comité de revisión central independiente de acuerdo con RECIST v1.1. El ORR fue del 52 %, con una tasa de respuesta completa del 18 %; el 76 % tuvieron una DOR ≥6 meses y el 62% ≥12 meses.
Eventos adversos: Los más comunes (≥10%) son fatiga, dolor musculoesquelético, prurito, diarrea, erupción cutánea, pirexia y náuseas. Se produjeron reacciones adversas graves en el 22% de los pacientes.
Leniolisib (Joenja®) Pharming Technologies
Indicación: Tratamiento del síndrome de la fosfoinositida 3-cinasa delta activada (PI3Kδ) (APDS) en pacientes adultos y pediátricos a partir de los 12 años de edad.
Tipo: Medicamento sintético estándar derivado del núcleo de piridopirimidina. Autorizado el 24-3-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea.
Mecanismo: Inhibe fosfoinositida 3-cinasa delta (PI3Kδ) al bloquear su sitio de unión activo. Leniolisib es selectivo para PI3K-delta frente a PI3K-alfa, PI3K-beta y PI3K-gamma. El síndrome de la fosfoinositida 3-cinasa delta activada (APDS) es una inmunodeficiencia primaria genética y poco frecuente (<1/1.000.000) caracterizada por una mayor susceptibilidad a infecciones virales y bacterianas graves y/o recurrentes, linfoproliferación crónica benigna y/o enfermedad autoinmune; las variantes de ganancia de función en el gen que codifica la subunidad catalítica p110-delta (PIK3CD) o las variantes de pérdida de función en el gen que codifica la subunidad reguladora p85-alfa causan hiperactividad de PI3K-delta. Leniolisib inhibe las vías de señalización que conducen a una mayor producción de PIP3, a la hiperactividad de la vía mTOR/AKT y a la desregulación de los linfocitos B y T. Eficacia clínica: Estudio ciego, aleatorizado, controlado con placebo de 12 semanas de duración de 31 pacientes de 12 años de edad y mayores con PI3Kδ genético asociado con APDS confirmado y mutación con una variante documentada en PIK3CD o PIK3R1. Los criterios de valoración coprimarios de la eficacia fueron la mejora de la linfoproliferación (reducción del tamaño de los ganglios linfáticos) y la normalización del inmunofenotipo (porcentaje de linfocitos B vírgenes o naïf del total de linfocitos B). Al día 85, los pacientes tratados con leniolisib experimentaron una reducción del 25% en el tamaño de los ganglios linfáticos y una mejora del 37% en los recuentos de células B naïf, ambos en comparación con el placebo.
Eventos adversos: Los más comunes (>10%) son cefalea, sinusitis y dermatitis atópica.
Epcoritamab (Epkinly®) Genmab
Indicación: Tratamiento de pacientes adultos con linfoma difuso de células B grandes (DLBCL, por sus siglas en inglés) recidivante o refractario, no especificado, incluido DLBCL que surge de un linfoma indolente y linfoma de células B de alto grado después de dos o más líneas de terapia sistémica.
Tipo: Medicamento biológico constituido por un anticuerpo IgG1 humanizado producido mediante tecnología de ADN recombinante, con un peso molecular aproximado de 149 kDa. Autorizado el 19-5-2023 de forma acelerada (Accelerated Approval), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Anticuerpo biespecífico que se acopla a las células T y se une al receptor CD3 expresado en la superficie de las células T y al CD20 expresado en la superficie de las células de linfoma y las células de linaje B sanas. Epcoritamab activa los linfocitos T, provocando la liberación de citocinas proinflamatorias e induciendo la lisis de los linfocitos B, reduciendo su recuento hasta niveles indetectables (<10 células/microlitro).
Eficacia clínica: Ensayo abierto, multicohorte, multicéntrico, de un solo brazo en 157 pacientes con linfoma de células B grandes en recaída o refractario después de dos o más líneas de terapia sistémica. La eficacia se estableció en función de la tasa de respuesta general (ORR) determinada por los criterios de Lugano 2014 evaluados por el Comité de revisión independiente (61%; 38% completa y 23% parcial) y la duración de la respuesta (mediana de 15,6 meses y estimación a 9 meses del 63%).
Eventos adversos: Los más comunes (≥20%) son síndrome de liberación de citocinas (CRS), fatiga, dolor musculoesquelético, reacciones en el lugar de la inyección, pirexia, dolor abdominal, náuseas y diarrea; las anomalías de laboratorio de Grado 3 a 4 más comunes (≥10%) fueron disminución del recuento de linfocitos, disminución del recuento de neutrófilos, disminución del recuento de glóbulos blancos, disminución de la hemoglobina y disminución de las plaquetas. Se produjeron reacciones adversas graves en el 54% de los pacientes y reacciones adversas mortales en el 3,8%. La interrupción permanente del tratamiento debido a una reacción adversa se produjo en el 3,8% de los pacientes.
Glofitamab (Columvi®) Roche
Indicación: Tratamiento de pacientes adultos con linfoma difuso de células B grandes en recaída o refractario, no especificado o linfoma de células B grandes derivado de linfoma folicular, después de dos o más líneas de terapia sistémica.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal recombinante humanizado tipo IgG1 producido mediante tecnología de ADN recombinante, con un peso molecular aproximado de 197 kDa. Autorizado el 15-6-2023, de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Anticuerpo monoclonal biespecífico anti-CD20 y anti-CD3ɛ; es decir, se une al CD20 expresado en la superficie de las células B y al receptor CD3 expresado en la superficie de las células T, provocando la activación y proliferación de células T, la secreción de citocinas y la lisis de células B que expresan CD20.
Eficacia clínica: Un ensayo clínico abierto, multicéntrico, de múltiples cohortes y de un solo brazo que incluyó a 132 pacientes con linfoma difuso de células B grandes en recaída o refractario después de dos o más líneas de terapia sistémica. La eficacia se basó en la tasa de respuesta objetiva (ORR: 56%, 43% completa) y la duración de la respuesta (DOR: mediana de 11,4 meses), según lo determinado por un Comité de revisión independiente utilizando los criterios de Lugano de 2014. La mediana del tiempo hasta la primera respuesta fue de 42 días.
Eventos adversos: Los más comunes son síndrome de liberación de citocinas (70%), dolor musculoesquelético (21%), erupción (20%) y fatiga (20%). Las anomalías de laboratorio de Grado 3 a 4 más comunes (≥20%) son disminución del recuento de linfocitos, de fosfato, del recuento de neutrófilos y de fibrinógeno, y aumento del ácido úrico. Se produjeron reacciones adversas graves en el 48% de los pacientes , que fueron mortales en el 5%. Un 7% de los pacientes experimentaron reacciones adversas que llevaron a la suspensión permanente de tratamiento.
(N) Sistema Nervioso
Tofersen (Qalsody®) Biogen
Indicación: Tratamiento de la esclerosis lateral amiotrófica (ELA) en adultos que tienen una mutación en el gen superóxido dismutasa 1 (SOD1).
Tipo: Medicamento sintético estándar constituido por un oligonucleótido antisentido de esqueleto mixto de 20 bases. De los 19 enlaces internucleotídicos, quince son diésteres de fosforotioato de 3′-O a 5′-O y cuatro son diésteres de fosfato de 3′-O a 5′-O. Diez de los veinte residuos de azúcar son 2-desoxi-D-ribosa y el resto son 2′-O-(2-metoxietil)-D-ribosa (MOE). Los residuos están dispuestos de manera que haya cinco nucleósidos MOE en los extremos 5′ y 3′ de la molécula que flanquean un espacio de diez 2′-desoxinucleósidos. Las bases de citosina y uridina están metiladas en la posición 5. Autorizado el 25-4-2023 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Es un oligonucleótido antisentido que provoca la degradación del ARNm de SOD1 mediante la unión al ARNm de SOD1, lo que da como resultado una reducción de la síntesis de la proteína SOD1. El gen SOD1 es uno de los genes asociadosa la esclerosis lateral amiotrófica tipo 1 (ELA1) y codifica para la enzima superóxido dismutasa tipo 1 (SOD1), cuya función es proteger del daño mediado de los radicales libres derivados del oxígeno; su mecanismo fisiopatológico en ELA1 se relaciona con isquemia. Las mutaciones en el gen SOD1 se dan tanto en las formas esporádicas (90-95% de los casos) de la ELA como en las familiares (5-10%).
Eficacia clínica: Estudio clínico aleatorizado, doble ciego, controlado con placebo de 28 semanas en 108 pacientes (23 a 78 años) con debilidad atribuible a la ELA y una mutación SOD1 confirmada . Se permitió el uso concomitante de riluzol y/o edaravona para los pacientes. La variable primaria de eficacia fue el cambio desde el inicio hasta la semana 28 en la puntuación total de ALSFRS-R (ALS Functional Rating Scale–Revised), analizado mediante la prueba de clasificación conjunta, para tener en cuenta la mortalidad junto con la imputación múltiple (IM) para tener en cuenta otros datos que pudieran faltar: diferencia de 0,33 puntos entre tofersen y placebo en la relación media geométrica (IC 95%; p<0,0001).
Eventos adversos: Los más comunes (≥10%) son dolor (42% con tofersen vs. 22% placebo), fatiga (17/6%), artralgia (16/6%), aumento de leucocitos en el LCR (14/0%) y mialgia (14/6%).
(S) Órganos Sensoriales
Perfluorohexiloctano (Miebo®) Bausch & Lomb

Indicación: Tratamiento de los signos y síntomas de la enfermedad del ojo seco
Tipo: Medicamento sintético estándar derivado de tetradecano, en la que los átomos de hidrógeno de los seis primeros seis átomos de carbono han sido completamente sustituidos por átomos de flúor. Autorizado el 18-5-2023; no autorizado aún en la Unión Europea.
Mecanismo: Forma una monocapa en la interfaz aire-líquido de la película lagrimal que se espera que reduzca la evaporación.
Eficacia clínica: Dos ensayos aleatorizados, multicéntricos, doble ciego, controlados con solución salina (0,6%) en un total de 1217 pacientes con antecedentes de ojo seco y signos clínicos de disfunción de las glándulas de Meibomio. La tinción con fluoresceína corneal total (tCFS) se registró en cada visita del estudio mediante un sistema de calificación estandarizado de 0 a 3 para cada una de las cinco áreas de la córnea (inferior, superior, central, nasal y temporal), totalizando una puntuación máxima de tCFS para cada ojo de 15. El tCFS de referencia promedio fue de aproximadamente 6,7 y 7,0 en los dos estudios. En los días 15 y 57, se observó una reducción estadísticamente significativa en tCFS a favor del tratamiento activo en ambos estudios (-1,7 vs. -1,1 y -1,9 vs. -1,3 a los 15 días y -2,0 vs. -1,0 y -2,3 vs. -1,1 a los 57 días).
Eventos adversos: Los más comunes son visión borrosa y enrojecimiento conjuntival (1-3%).
(V) Varios
Flotufolastat F-18 (Posluma®) Blue Earth Diagnostics

Indicación: Para la tomografía por emisión de positrones (PET) de lesiones positivas al antígeno prostático específico de membrana (PSMA) en hombres con cáncer de próstata con sospecha de metástasis que son candidatos para una terapia inicial definitiva o con sospecha de recurrencia basada en un nivel sérico elevado de antígeno prostático específico (PSA).
Tipo: Medicamento sintético estándar consistente en un heptapéptido ligado a un núcleo de tetraazaciclododecano acomplejado con un átomo de galio (Ga+3) no radiactivo, con presencia de un átomo de silicio (Si) ligado a un átomo de flúor radiactivo (F-18), un radionúclido – producido por un ciclotrón – que se desintegra por emisión de positrones (desintegración β+, 96,7 %) y captura de electrones orbitales (3,3 %) a oxígeno-18 estable con una vida media física de 109,8 minutos; los principales fotones útiles para el diagnóstico por imagen PET son el par coincidente de fotones gamma de 511 keV, resultantes de la interacción del positrón emitido con un electrón. Autorizado el 25-5-2023; no autorizado aún en la Unión Europea.
Mecanismo: Flotufolastat F 18 se une al antígeno prostático específico de membrana (PSMA) que es sobreexpresado en las células de cáncer de próstata, y se internaliza.
Eficacia clínica: Estudio multicéntrico, abierto, de un solo brazo en 356 pacientes con cáncer de próstata de riesgo intermedio desfavorable (32%) o de riesgo alto/muy alto (68%) que eran candidatos para prostatectomía radical y disección de ganglios linfáticos pélvicos. El rendimiento de POSLUMA se evaluó frente a la histopatología después de emparejar por hemipelvis (al menos una región hemipélvica positiva verdadera está definida por un paciente positivo verdadero): sensibilidad 23-27%, especificidad 93-97%, valor predictivo positivo 57-70% y negativo 80-81%.
Eventos adversos: Los más comunes son diarrea (0,7%), aumento de la presión arterial (0,5%) y dolor en el lugar de la inyección (0,4%).
3. PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA) como la Food & Drug Administration (FDA) de Estados Unidos disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA
- Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
- Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
- Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
- Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
- Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA
- Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
- Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
- Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
- Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
- Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
- Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
