1. EUROPEAN MEDICINES AGENCY (EMA)
(L) Agentes Antineoplásicos e Inmunomoduladores
Loncastuximab Tesirina (Zynlonta®) ADC Therapeutics
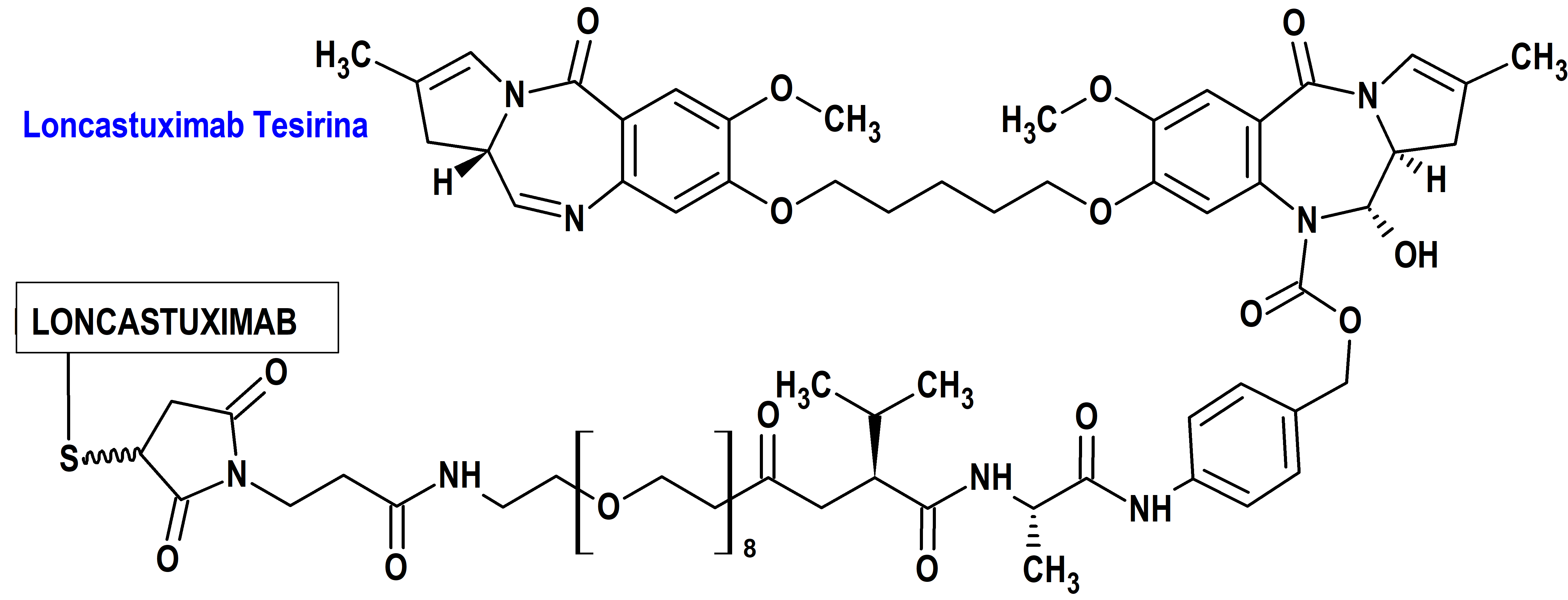
Indicación: Tratamiento en monoterapia de pacientes adultos con linfoma B difuso de células grandes (LBDCG) y linfoma B de alto grado (LBAG), en recaída o refractario después de dos o más líneas de tratamiento sistémico.
Tipo: Autorizado el 20-12-2022 condicionalmente (Conditional marketing authorisation); autorizado previamente en Estados Unidos en 23-4-2021. Medicamento biológico constituido compuesto de un anticuerpo monoclonal IgG1 kappa humanizado, conjugado con SG3199, un alquilante citotóxico (dímero de pirrolobenzodiazepina; PBD), mediante un conector valina-alanina escindible por proteasa. La combinación de SG3199 acoplado al conector se designa SG3249, conocido como tesirina.
Mecanismo: Es un conjugado anticuerpo-fármaco dirigido a CD19. El componente anticuerpo monoclonal IgG1 kappa se une a la proteína transmembrana CD19 expresada sobre la superficie de las células de linaje B. El componente molecular pequeño es un agente alquilante, SG3199, dímero de PBD. Después de unirse a CD19, loncastuximab tesirina se interioriza y se produce la liberación de SG3199 mediante escisión proteolítica. El SG3199 liberado se une al surco menor del ADN y forma entrecruzamientos intercatenarios del ADN altamente citotóxicos, induciendo así la muerte celular.
Eficacia clínica: Estudio abierto con un solo grupo en 145 pacientes adultos con linfoma B difuso de células grandes en recaída o refractario después de como mínimo 2 terapias sistémicas previas; los pacientes estuvieron en tratamiento durante 1 año, o más si estaban obteniendo un beneficio clínico o hasta el empeoramiento de la enfermedad o una toxicidad inaceptable (el 60 % recibió tres o más ciclos y el 34 %, cinco o más). La variable principal de eficacia fue la tasa global de respuesta (48,3%; 24,8% completa) según la valoración de un comité de revisión independiente (CRI) usando los criterios de Lugano 2014; la mediana de tiempo hasta la respuesta fue de 1,3 meses y la mediana de la duración de la respuesta global fue de 13,4 meses.
Eventos adversos: Los más comunes son aumento de la gammaglutamil transferasa (36%), neutropenia (35%), fatiga (30%), anemia (29%), trombocitopenia (28%), náuseas (27%), edema periférico (23%) y exantema (20%). Las reacciones adversas graves más frecuentes fueron neutropenia febril (3,3%), dolor abdominal, disnea y derrame pleural (1,9% cada una).
2. FOOD & DRUG ADMINISTRATION (FDA)
(A) Tracto Alimentario y Metabolismo
Bexagliflozina (Brenzavy®) Theraxobio
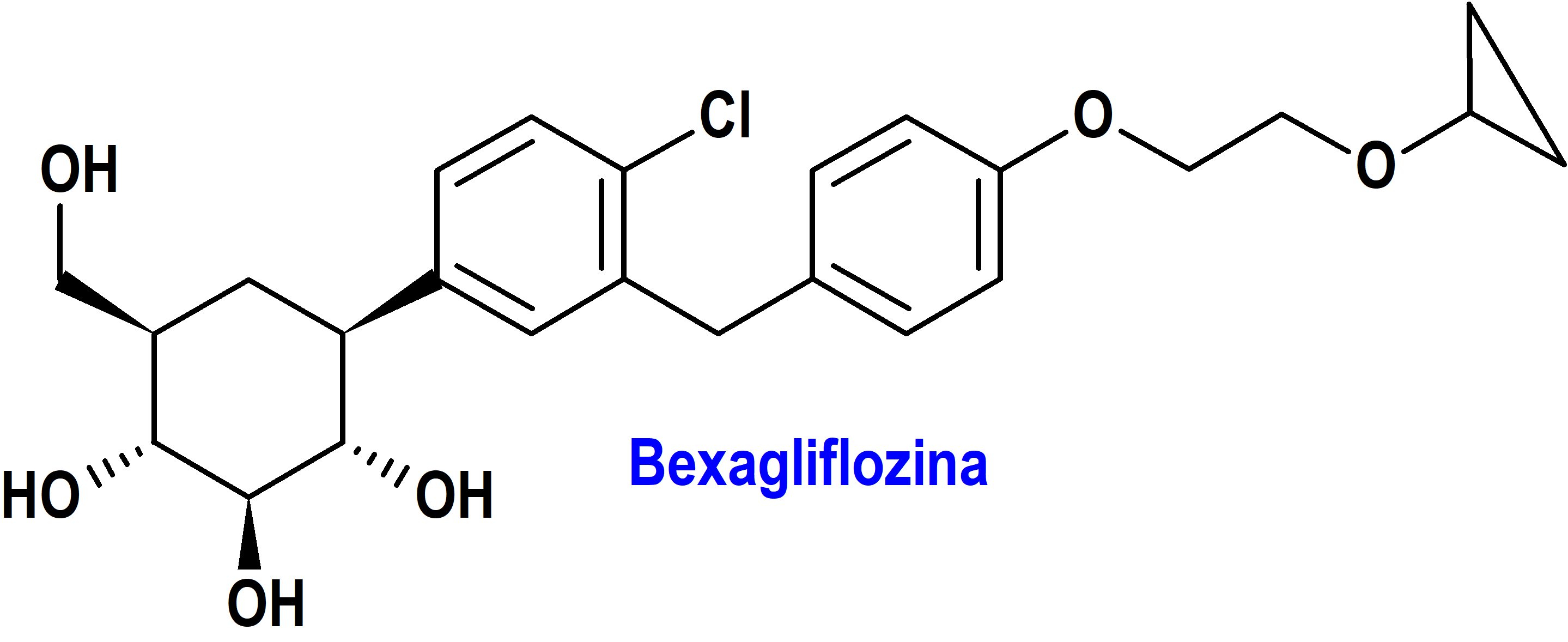
Indicación: Como complemento de la dieta y el ejercicio para mejorar el control glucémico en adultos con diabetes mellitus tipo 2
Tipo: Autorizado el 20-1-2023; no autorizado aún en la Unión Europea.
Mecanismo: Inhibidor del cotransportador de sodio-glucosa 2 (SGLT2), responsable de la reabsorción de la mayoría de la glucosa del filtrado glomerular renal en el túbulo proximal renal. Al inhibir SGLT2, la bexagliflozina reduce la reabsorción renal de la glucosa filtrada y reduce el umbral renal para la glucosa y, por lo tanto, aumenta la excreción urinaria de glucosa.
Eficacia clínica: Seis estudios clínicos aleatorizados, doblemente ciegos y multicéntricos, en los que las variables principales de eficacia fueron la reducción de la tasa de hemoglobina glicosilada (HbA1c), la proporción de pacientes que alcanzaron una valor de Hb1Ac inferior al 7% y la reducción de la glucemia en ayunas. Un estudio como monoterapia (vs. placebo): 0,5 vs. 0,1% (31 vs. 20% de los pacientes consiguió reducir la tasa por debajo del 7%), mientras que la glucemia en ayunas fue reducida en un 9,5 vs. 5,9%. Tres en combinación con metformina (vs. placebo, glimepirida y sitagliptina). En comparación con placebo, 1,0 vs. 0,5% (26 vs. 10%), la glucemia en ayunas fue reducida en un 22,6 vs. 10,5%. En comparación con glimepirida y con sistagliptina, los resultados obtenidos con bexagliflozina no fueron significativamente diferentes. En un estudio en adultos con diabetes mellitus tipo 2 con insuficiencia renal moderada en terapia aditiva con el régimen estándar y en comparación con placebo, 0,6 vs. 0,3% (33 vs. 22%), la glucemia en ayunas fue reducida en un 14,1 vs. 9,0%. En otro estudio en adultos con diabetes mellitus tipo 2 con patología cardiovascular establecida, en terapia aditiva con el régimen estándar y en comparación con placebo, 0,8 vs. 0,4% (29 vs. 17%), la glucemia en ayunas fue reducida en un 13,9 vs. 2,5%; además, la proporción de pacientes que experimentaron al menos un evento de cardiovascular grave fue del 10,1% en el grupo de placebo y del 7,9 % con bexagliflozina (4,2 vs. 3,3 eventos por 100 años-persona).
Eventos adversos: Los más comunes (>1%) son incremento de la frecuencia de micciones urinarias (2%), infección urinaria (2%) e infecciones micóticas del tracto genital femenino (2%).
(B) Sangre y Sistema Hematopoyético
Daprodustat (Jesduvroq®) GalxoSmithKline
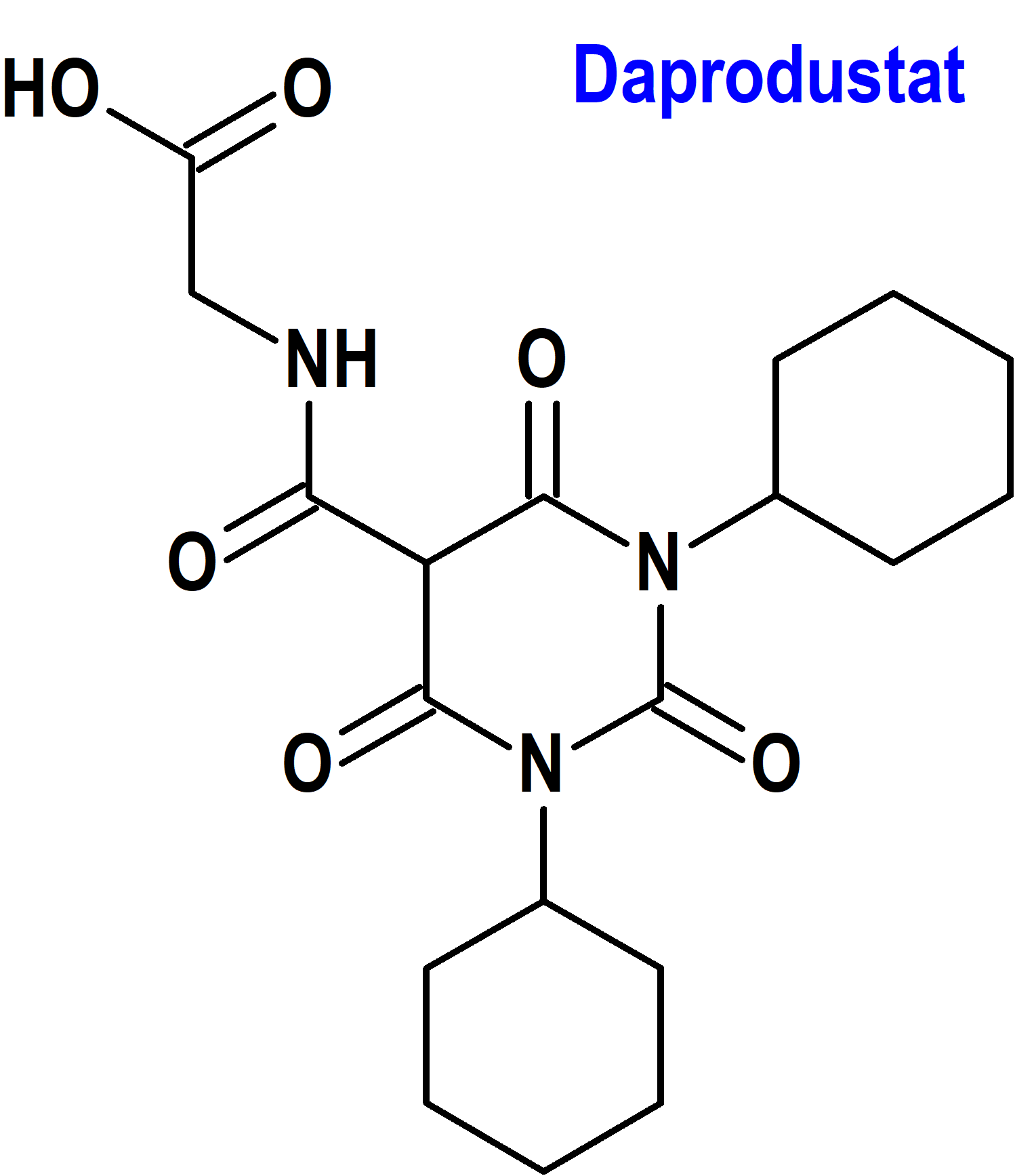
Indicación: Tratamiento de anemia debida a enfermedad renal crónica en adultos que han estado recibiendo diálisis durante al menos cuatro meses.
Tipo: Autorizado el 1-2-2023; no autorizado aún en la Unión Europea.
Mecanismo: Inhibidor reversible de las prolilhidroxilasas del factor inducible por hipoxia HIF-PH1, PH2 y PH3 (hypoxia inducible factor [HIF]; prolyl hydroxylases 1, 2, 3 [PH1, PH2, PH3]). Esta actividad da como resultado la estabilización y la acumulación nuclear de los factores de transcripción HIF-1a y HIF-2a, lo que conduce a una mayor transcripción de los genes que responden a HIF, incluida la eritropoyetina. La PH1 es una hidroxilasa dependiente de alfa-cetoglutarato, una superfamilia de proteínas que no contienen hierro hemo; por su parte, el factor inducible por hipoxia (HIF) es un complejo transcripcional que está implicado en la homeostasis del oxígeno. Daprodustat aumenta la eritropoyetina endógena de forma dependiente de la dosis dentro de las 6 a 8 horas posteriores a la administración; con dosis repetidas, los aumentos máximos en los recuentos de reticulocitos ocurren entre los días 7 y 15, con aumentos posteriores en la producción de glóbulos rojos.
Eficacia clínica: Ensayo clínico aleatorizado, patrocinador ciego, con control activo (epoetina alfa o darbepoetina), global, multicéntrico y basado en eventos, en 2964 adultos con anemia debido a la insuficiencia real crónica en diálisis. La eficacia y la seguridad se evaluaron como criterios de valoración coprimarios: el cambio medio en la hemoglobina desde el inicio hasta el período de evaluación, en las semanas 28 a 52 (+0,3 g/dL daprodustat vs. 0,1 epoetina) e incidencia de eventos cardiovasculares graves (mortalidad por todas las causas, infarto de miocardio no mortal o accidente cerebrovascular no fatal): 11,1 vs. 11,9 por 100 pacientes-año.
Eventos adversos: Los más comunes son hipertensión (24%), dolor abdominal (11%), vértigo (7%) y reacciones de hipersensibilidad (7%): erupciones exantemáticas, urticaria, dermatitis).
(C) Sistema Cardiovascular
Sparsentan (Filspari®) Travere
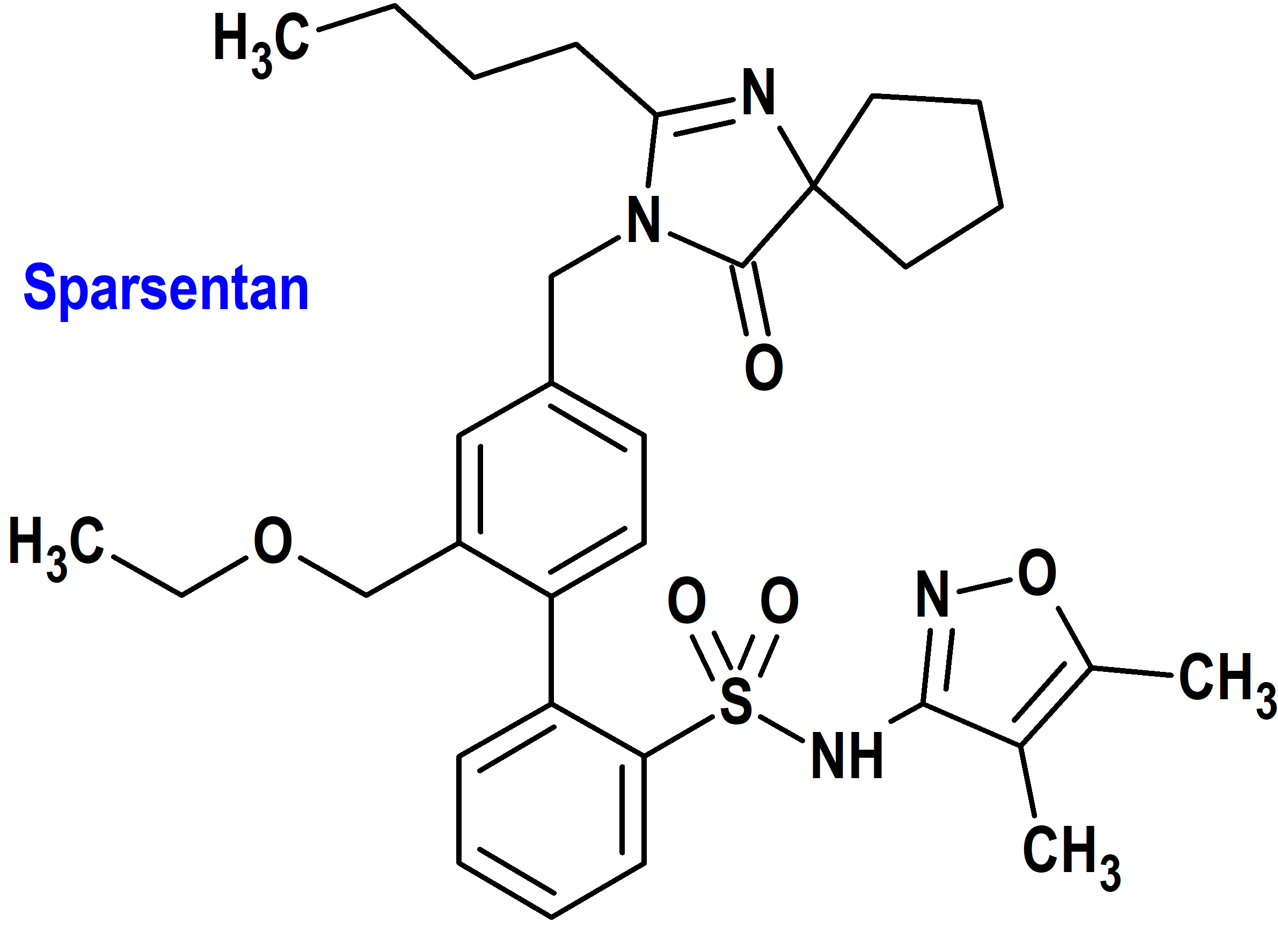
Indicación: Para reducir la proteinuria en adultos con nefropatía primaria por inmunoglobulina A (IgAN) con riesgo de progresión rápida de la enfermedad, generalmente una relación proteína/creatinina en orina (UPCR) ≥1,5 g/g.
Tipo: Autorizado el 17-2-2023 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Antagonista del receptor de endotelina tipo A (ETAR) y el receptor de angiotensina II tipo 1 (AT1R). Sparsentan tiene una alta afinidad tanto por ETAR (Ki = 12,8 nM) como por AT1R (Ki = 0,36 nM), y una selectividad de más de 500 veces por estos receptores sobre los receptores de endotelina tipo B y angiotensina II subtipo 2. Se cree que la endotelina-1 y la angiotensina II contribuyen a la patogenia de la IgAN a través de ETAR y AT1R, respectivamente.
Eficacia clínica: Estudio aleatorizado, doble ciego, con control activo (irbesartan), multicéntrico, global en 281 adultos con IgAN comprobada por biopsia, eGFR ≥30 ml/min/1,73 m2 y proteína total en orina ≥1,0 g/ día en una dosis estable máxima de tratamiento con inhibidor del sistema renina-angiotensina que fue al menos el 50% de la dosis máxima. El criterio principal de valoración fue el cambio relativo desde el inicio en la relación proteína/creatinina en orina UPCR en la semana 36, que de -4% para sparsentan vs. -15 para irbesartan.
Eventos adversos: Los más comunes (>5%) son edema periférico (14%), hipotensión (incluida la hipotensión ortostática) (14%), mareos (13%), hiperpotasemia (13%) y anemia (5%).
(L) Agentes Antineoplásicos e Inmunomoduladores
Elacestrant (Orserdu®) Stemline
Indicación: Tratamiento de mujeres posmenopáusicas u hombres adultos con cáncer de mama avanzado o metastásico con receptor de estrógeno (ER) positivo, receptor del factor de crecimiento epidérmico humano 2 (HER2) negativo, mutado en ESR1 con progresión de la enfermedad después de al menos una línea de terapia endocrina.
Tipo: Autorizado el 27-1-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: antagonista del receptor de estrógeno-alfa (ERα). En células de cáncer de mama ERpositivas (ER+) y HER2-negativas (HER2-), elacestrant inhibe la proliferación celular mediada por 17β-estradiol en concentraciones que inducen la degradación de la proteína ERa mediada a través de la vía proteasómica. Elacestrant demostró actividad antitumoral in vitro e in vivo, incluso en modelos de cáncer de mama ER+ HER2- resistentes a fulvestrant y a los inhibidores de la cinasa 4/6 dependiente de ciclina y en aquellos que albergan mutaciones en el gen del receptor de estrógeno 1 (ESR1).
Eficacia clínica: Ensayo aleatorizado, abierto, con control activo (fulvestrat o un inhibidor de aromatasa) y multicéntrico en 478 mujeres posmenopáusicas y hombres con cáncer de mama avanzado o metastásico ER+/HER2-, de los cuales 228 pacientes tenían mutaciones en ESR1. La variable resultado de eficacia fue la supervivencia libre de progresión, evaluada por un comité ciego de revisión de imágenes (BIRC), con una mediana 3,8 meses (elacestrant) vs. 1,9 meses (control activo). No hubo diferencias estilísticamente significativas en la supervivencia general.
Eventos adversos: Los más comunes (>10 %) son dolor musculoesquelético, náuseas, aumento del colesterol, aumento de la AST, aumento de los triglicéridos, fatiga, disminución de la hemoglobina, vómitos, aumento de la ALT, disminución del sodio, aumento de la creatinina, disminución del apetito, diarrea, dolor de cabeza, estreñimiento, dolor abdominal, sofocos y dispepsia. Se produjeron reacciones adversas graves en el 12 % de los pacientes, siendo mortales en el 1,7 % de los pacientes La discontinuación permanente del tratamiento debido a una reacción adversa ocurrió en el 6% de los pacientes.
Pirtobrutinib (Jaypirica®) Loxo
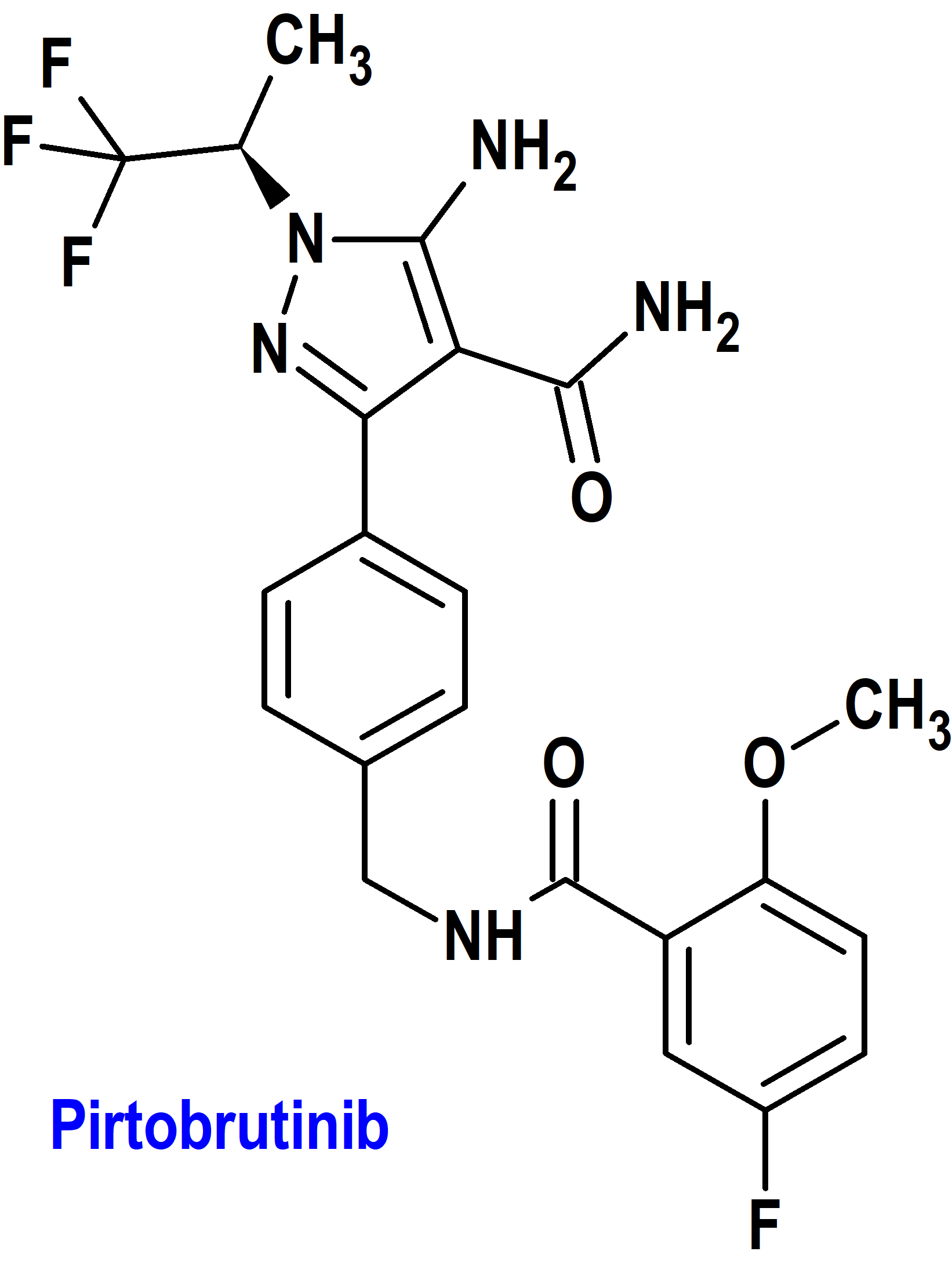
Indicación: Tratamiento de pacientes adultos con linfoma de células del manto (MCL) en recaída o refractario después de al menos dos líneas de terapia sistémica, incluido un inhibidor de BTK.
Tipo: Autorizado el 27-1-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Inhibidor no covalente de la tirosina cinasa de Bruton (BTK; Bruton’s tyrosine kinase), una proteína de señalización del receptor de antígeno de células B (BCR) y las vías del receptor de citocinas. En las células B, la señalización de BTK da como resultado la activación de las vías necesarias para la proliferación, el tráfico, la quimiotaxia y la adhesión de las células B. Pirtobrutinib se une a BTK de tipo salvaje y BTK que albergan mutaciones C481, lo que lleva a la inhibición de la actividad de la cinasa BTK. Pirtobrutinib inhibe la expresión de CD69 de células B mediada por BTK y la proliferación de células B malignas.
Eficacia clínica: Estudio abierto, internacional, de un solo brazo de pirtobrutinib como monoterapia. Las variables principales de eficacia fueron la tasa de respuesta general (ORR: 50%; completa 13%, parcial 38%) y la duración de la respuesta (DOR; 8,3 meses), según lo evaluado por un comité de revisión independiente utilizando los criterios de Lugano de 2014. La estimación de Kaplan-Meier para la tasa de DOR a los 6 meses fue del 65%.
Eventos adversos: Los más comunes (≥20%) son disminución del recuento de neutrófilos (41%), disminución de la hemoglobina (37%), disminución del recuento de plaquetas (27%), fatiga (27%), dolor musculoesquelético (26%), disminución recuento de linfocitos (24%), hematomas (20%) y diarrea (20%).
(N) Sistema Nervioso
Lecanemab (Leqembi®) Eisai
Indicación: Tratamiento de la enfermedad de alzheimer en pacientes que se encuentren en la etapa de la enfermedad caracterizadas por deterioro cognitivo leve o demencia leve.
Tipo: Autorizado el 6-1-2023 de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea. Medicamento biológico constituido por un anticuerpo monoclonal de inmunoglobulina gamma 1 humanizada recombinante (IgG1) dirigido contra formas agregadas de amiloide beta.
Mecanismo: La acumulación de placas de amiloide beta en el cerebro es una característica fisiopatológica posiblemente definitoria de la enfermedad de Alzheimer. Lecanemab está dirigido contra las formas agregadas solubles e insolubles de amiloide beta.
Eficacia clínica: Estudio doble ciego, controlado con placebo, de grupos paralelos, de búsqueda de dosis en 856 pacientes con enfermedad de Alzheimer y presencia confirmada de patología amiloide y deterioro cognitivo leve. La variable principal de eficacia fue el cambio desde el inicio en una puntuación compuesta ponderada que constaba de elementos seleccionados de CDR-SB (Clinical Dementia Rating), MMSE (Mini-Mental State Examination) y ADAS-Cog 14 (Alzheimer’s Disease Assessment Scale–Cognitive Subscale) en la semana 53, dando lugar a un 64 % de probabilidad de un 25 % o más de ralentización de la progresión, en relación con el placebo.
Eventos adversos: Los más comunes son reacciones relacionadas con la infusión IV (20% vs. 3% con placebo), cefalea (14 vs. 10%), anomalías de imagen relacionadas con amiloide (10 vs 1%), tos (9 vs. 5%) y diarrea (8 vs. 5%). El 15% de los pacientes tratados con lecanemab, en comparación con el 6% de los pacientes tratados con placebo, interrumpieron el tratamiento del estudio debido a una reacción adversa.
Trofinetida (Daybue®) Acadia
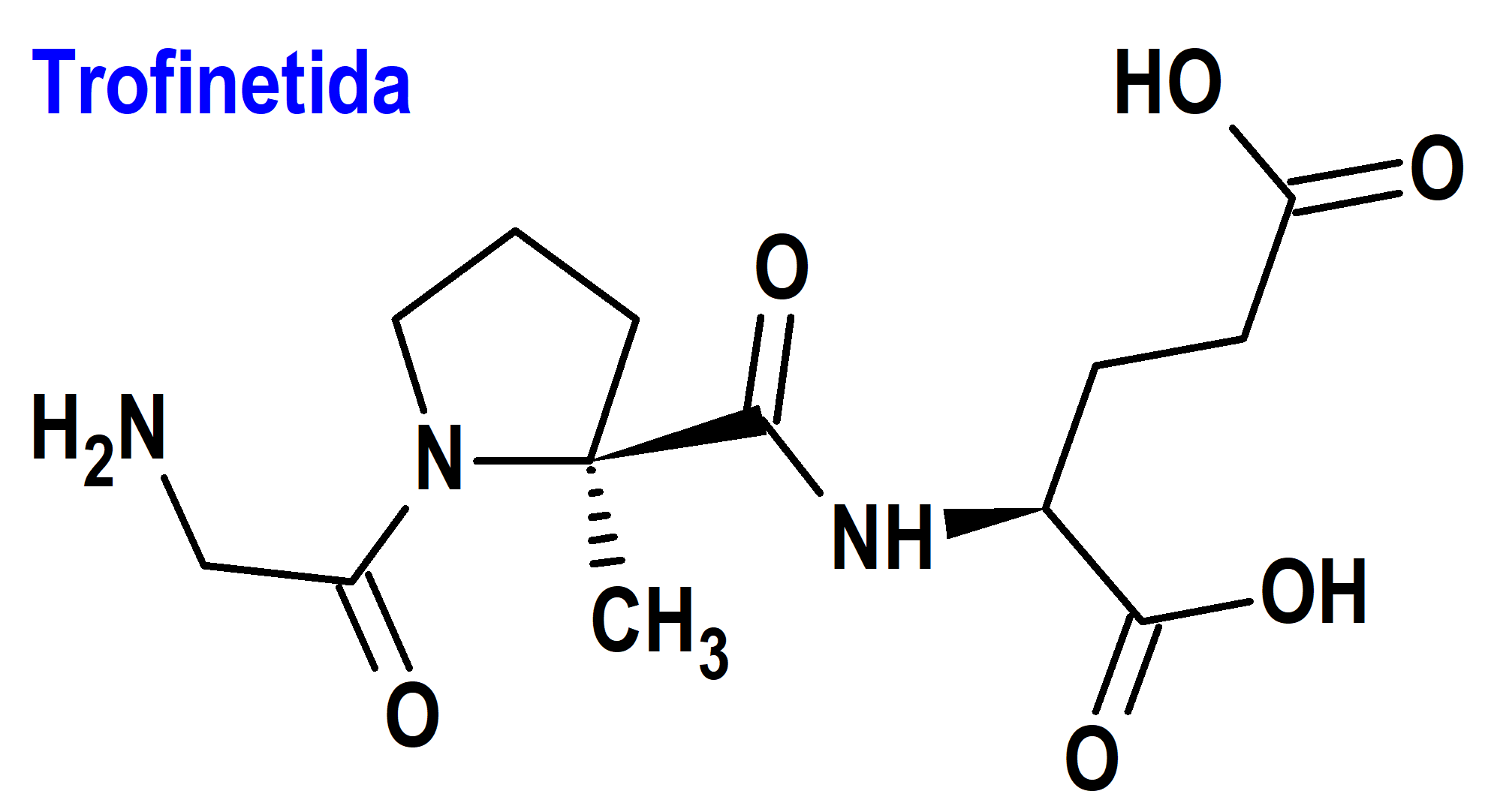
Indicación: Tratamiento del síndrome de Rett en adultos y pacientes pediátricos a partir de los 2 años de edad.
Tipo: Autorizado el 10-3-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea.
Mecanismo: Es un análogo del neuropéptido (1-3) IGF-1, constituido por un tripéptido simple con secuencia Gly-Pro-Glu obtenido por escisión enzimática del factor de crecimiento IGF-1 dentro del cerebro. Se desconoce el mecanismo por el cual la trofinetida ejerce efectos terapéuticos en pacientes con síndrome de Rett, el cual se debe a una mutación genética en el gen MECP2 en el cromosoma X, casi siempre adquirida (menos del 1% de los casos tienen carácter hereditario).
Eficacia clínica: Estudio aleatorizado, doble ciego, controlado con placebo, de 12 semanas de 187 pacientes con síndrome de Rett de cinco a 20 años de edad. Las variables coprimarias de eficacia fueron el cambio desde el inicio en la puntuación total del Cuestionario de comportamiento del síndrome de Rett (Rett Syndrome Behavior Questionnaire, RSBQ) (-4,9 vs. -1,7) y la puntuación de Mejora de la impresión clínica global (CGI-I) en la semana 12 (3,5 vs. 3,8). El RSBQ es una escala de calificación de 45 elementos que evalúa una variedad de signos y síntomas del síndrome de Rett, cuyas puntuaciones más bajas representan una menor gravedad en los signos y síntomas; la CGI-I es una escala de 7 puntos, cuya disminución indica una mejora.
Eventos adversos: Los más comunes son diarrea (82%), vómitos (29%), fiebre (9%), convulsiones (9%), ansiedad (8%), disminución del apetito (8%), fatiga (8%) y nasofaringitis (5%).
Deutetrabenazina (Austedo XR®) Teva
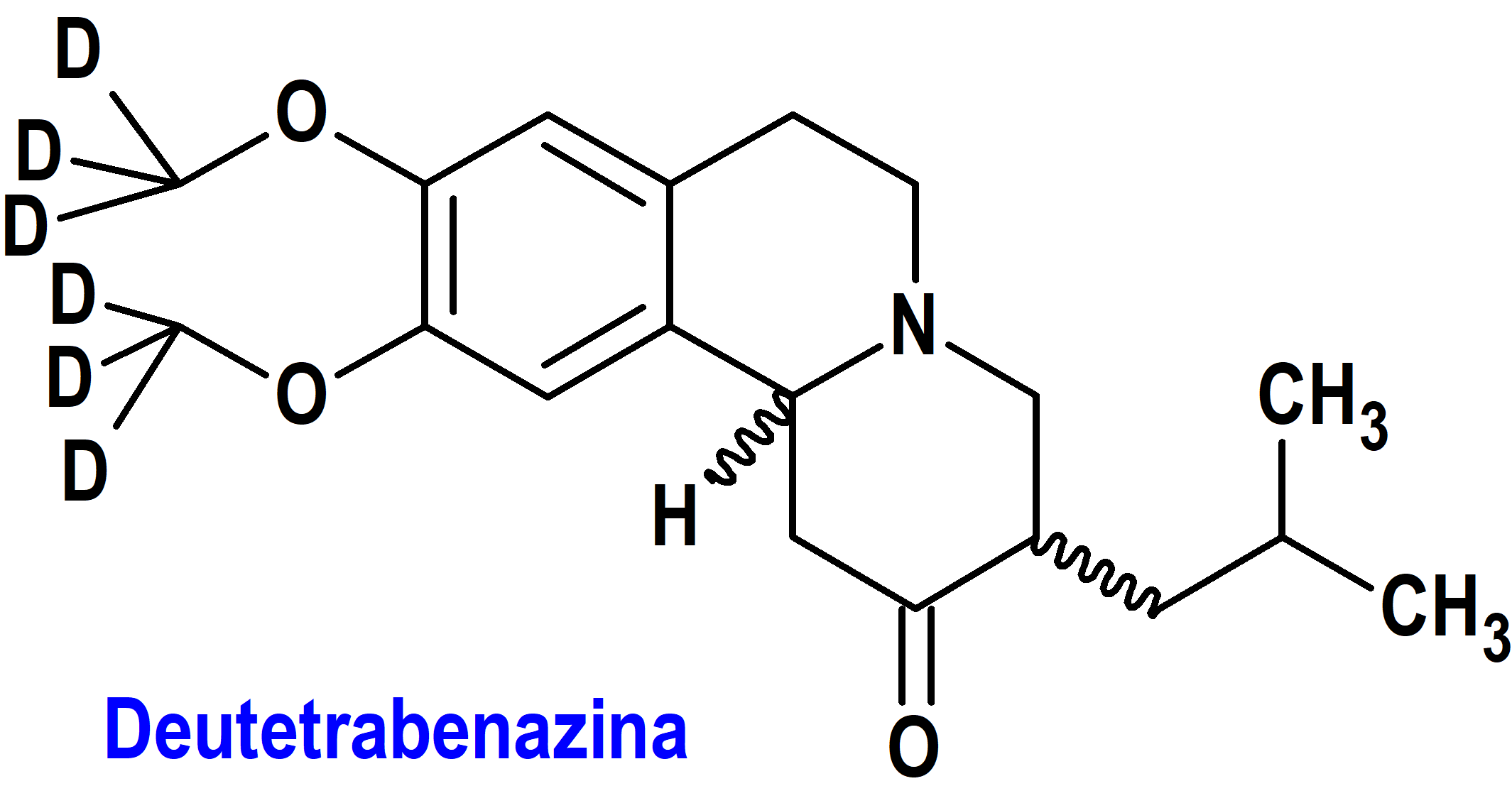
Indicación: Tratamiento en adultos de la corea asociada con la enfermedad de Huntington y de la discinesia tardía.
Tipo: Autorizado el 17-2-2023 como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea.
Mecanismo: Eliminación reversible de monoaminas (dopamina, serotonina, norepinefrina e histamina) de las terminales nerviosas. Los principales metabolitos circulantes (alfa y beta-dihidrotetrabenazina) de la deutetrabenazina son inhibidores reversibles de VMAT2 (transportador de la monoamina vesicular de tipo 2), lo que provoca una disminución de la captación de monoaminas en las vesículas sinápticas y el agotamiento de las reservas de monoamina.
Eficacia clínica: Ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo, en 90 pacientes ambulatorios con corea manifiesta asociada con la enfermedad de Huntington. La duración del tratamiento fue de 12 semanas, incluido un período de titulación de dosis (desde 6 hasta un máximo de 48 mg(día) de 8 semanas y un período de mantenimiento de 4 semanas, seguido de un lavado de 1 semana. El criterio principal de valoración de la eficacia fue la puntuación máxima total de corea, un elemento de la Escala Unificada de Calificación de la Enfermedad de Huntington (Unified Huntington’s Disease Rating Scale, UHDRS), en la que la corea se califica de 0 a 4 para 7 partes diferentes del cuerpo con una puntuación total que va de 0 a 28. Las puntuaciones máximas totales de corea para los pacientes que recibieron deutetrabenazina mejoraron en 4,4 unidades vs. 1,9 unidades en el grupo de placebo; en la visita de seguimiento de la semana 13 (1 semana después de la interrupción del medicamento del estudio), las puntuaciones máximas totales de corea de los pacientes que habían recibido deutetrabenazina volvieron a las iniciales de partida. La eficacia en el tratamiento de la discinesia tardía se estableció en dos ensayos multicéntricos, aleatorizados, doble ciego, controlados con placebo y de 12 semanas de duración realizados en 335 pacientes adultos ambulatorios con discinesia tardía provocada por el uso de antagonistas de los receptores de dopamina, siendo la principal variable de eficacia la variación de la puntuación en la escala de movimiento involuntario anormal (Abnormal Involuntary Movement Scale, AIMS; cuya puntuación total, suma de los ítems 1 a 7, va de 0 a 28, y una disminución en la puntuación indica una mejora). En el Estudio 1 se utilizaron dosis fijas controlado con placebo (12, 24 o 36 mg/día, o placebo), de 12 semanas. En el Estudio 2, un ensayo de dosis flexible (de 12 a un máximo de 49 mg/día), controlado con placebo, de 12 semanas. En ambos estudios la diferencia (estadísticamente significativa) entre los grupos con deutetrabenazina y placebo fueron de 1,4-1,8 puntos.
Eventos adversos: Los más comunes son (>5%) somnolencia (11%), diarrea (9%), boca seca (9%), fatiga (9%), infección del tracto urinario (7%) e insomnio (7%).
Omaveloxolona (Skyclarys®) Reata
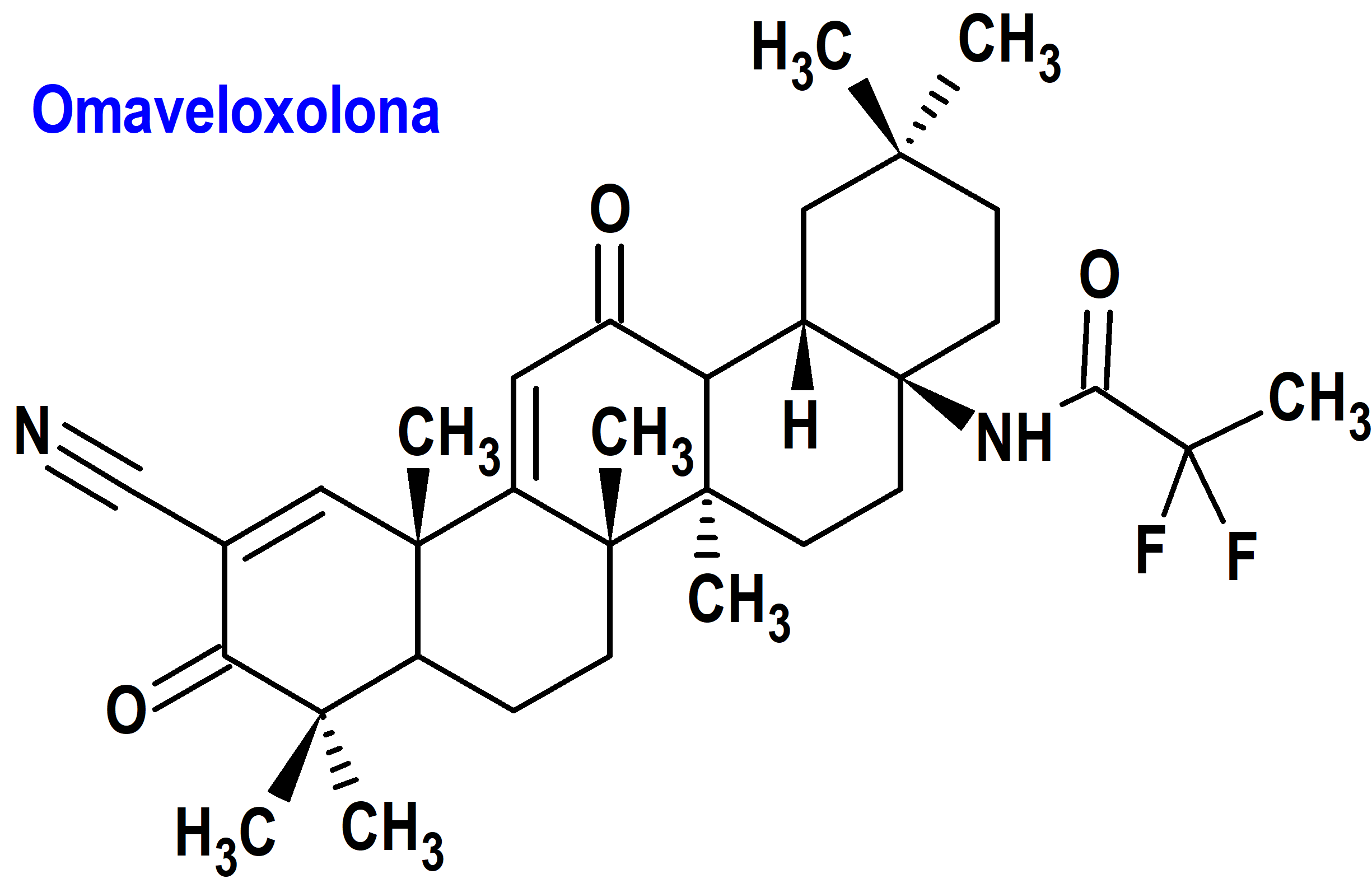
Indicación: Tratamiento de la ataxia de Friedreich en adultos y adolescentes mayores de 16 años.
Tipo: Autorizado el 28-2-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea.
Mecanismo: Activa la vía del factor nuclear eritroide 2 (Nrf2), un factor de transcripción que actúa como regulador maestro del estrés oxidativo, manteniendo el equilibrio entre las reacciones de reducción y oxidación en las células. La activación de una proteína Nrf2 podría prevenir la desintegración de las células nerviosas característica de la ataxia de Friedreich. Los inductores de Nrf2 contrarrestan la neurodegeneración en las neuronas motoras silenciadas por la proteína frataxina. Precisamente, las mutaciones del gen responsable de la frataxina que limitan o impiden su producción, son la causa de ataxia de Friedreich. Se trata de una deficiencia mitocondrial que impide que la célula genere suficiente energía para mantener su propio funcionamiento; de hecho, el estrés oxidativo es un sello distintivo de la ataxia de Friedreich, producido por un desequilibrio entre la producción de especies de oxígeno reactivas nocivas y la capacidad del sistema antioxidante para controlarlas.
Eficacia clínica: Estudio de 48 semanas, aleatorizado, doble ciego, controlado con placebo en 103 pacientes de 16 a 40 años de edad. La variable principal de eficacia fue el cambio desde el inicio en la puntuación de la Escala de Calificación de Ataxia de Friedreich modificada (modified Friedreich’s Ataxia Rating Scale, mFARS; con una puntuación máxima de 99, indicando los valores más altos una mayor discapacidad física) en comparación con el placebo en la semana 48 en la población de pacientes sin pie cavo (n=82), mostrando una diferencia media de mínimos cuadrados entre grupos de tratamiento de -2,41 (-1,56 vs. +0,85). Considerando a toda la población del estudio (N=103), independientemente del estado del pie cavo, la diferencia fue de -1,94.
Eventos adversos: Los más comunes son elevación de las enzimas hepáticas elevadas (AST/ALT) (37%), dolor de cabeza (37%), náuseas (33 %), dolor abdominal (29%), fatiga (24%), diarrea (20%), dolor musculoesquelético (20%) , dolor orofaríngeo (18%), síntomas gripales (16%), vómitos (16%), espasmos musculares (14%), dolor de espalda (13%), disminución del apetito (12%) y sarpullido (10%).
Zavegepant (Zavzpret®) Pfizer
Indicación: Tratamiento agudo de la migraña con o sin aura en adultos.
Tipo: Autorizado el 9-3-2023; no autorizado aún en la Unión Europea.
Mecanismo: Es un antagonista del receptor del péptido relacionado con el gen de la calcitonina (CGRP), aunque se desconoce la relación entre la actividad farmacodinámica y el mecanismo por el que zavegepant ejerce sus efectos clínicos.
Eficacia clínica: Dos ensayos aleatorizados, doble ciego y controlados con placebo sobre un total de 2.061 pacientes, que fueron aleatorizados para recibir una dosis única. La covariable principal de eficacia consistió en la combinación de ausencia de dolor (24 vs. 15% en el estudio 1; 23 vs. 16% en el estudio 2) y de los síntomas más molestos (fotofobia, náusea y fonofobia) (40 vs. 31%; 41 vs. 34%) a las 2 horas después de una dosis única, en comparación con el placebo.
Eventos adversos: Los más comunes son disgeusia/ageusia (18%), náusea (4%), molestias nasales (3%) y vómito (2%).
3. PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA) como la Food & Drug Administration (FDA) de Estados Unidos disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
