1. EUROPEAN MEDICINES AGENCY (EMA)
(A) Tracto Alimentario y Metabolismo
Fosdenopterina (Nulibry®) Comharsa
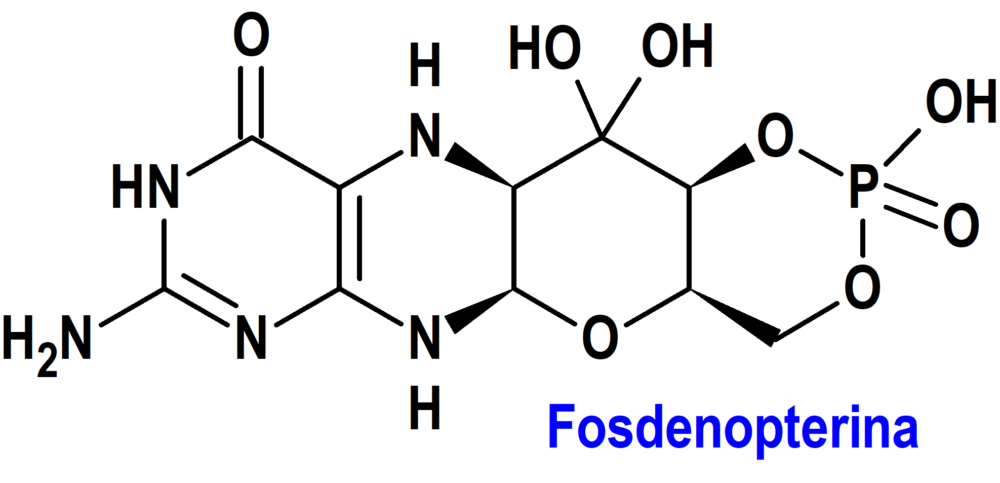
Indicación: Tratamiento de pacientes con deficiencia de cofactor de molibdeno (DCMo) tipo A.
Tipo: Autorizado el 15-9-2022, como medicamento huérfano (Orphan drug) y en condiciones excepcionales (Exceptional circumstances); autorizado previamente en Estados Unidos el 26-2-2021.
Mecanismo: Los pacientes con DCMo tipo A tienen mutaciones en el gen de la proteína 1 de biosíntesis del cofactor de molibdeno (MOCS1), que dan lugar a una deficiencia en la síntesis del sustrato intermedio, la cPMP, dependiente de MOCS1A/B. El tratamiento de reposición del sustrato con fosdenopterina proporciona una fuente exógena de cPMP, que es transformada a molibdopterina. La molibdopterina es transformada en cofactor molibdeno, el cual es necesario para la activación de las enzimas dependientes del molibdeno, como la sulfito oxidasa (SOX), una enzima que reduce la concentración de sulfitos neurotóxicos.
Eficacia clínica: Análisis combinado de 15 pacientes con DCMo tipo A confirmada mediante diagnóstico genético que recibieron tratamiento de reposición de sustrato con fosdenopterina y/o rcPMP, la cual tiene el mismo grupo activo que fosdenopterina y se considera su equivalente desde el punto de vista terapéutico. La probabilidad de supervivencia según Kaplan-Meier a 1 año fue del 93 % y a 3 años del 86%; la media y la mediana del tiempo de supervivencia fueron de 73 y 64 meses, respectivamente.
Eventos adversos: Los más comunes (>20%) son complicaciones asociadas al catéter empleado en la administración y no a la fosdenopterina. Ningún paciente tuvo que interrumpir el tratamiento debido a eventos adversos.
Maralixibat (Livtencity®) Mirum
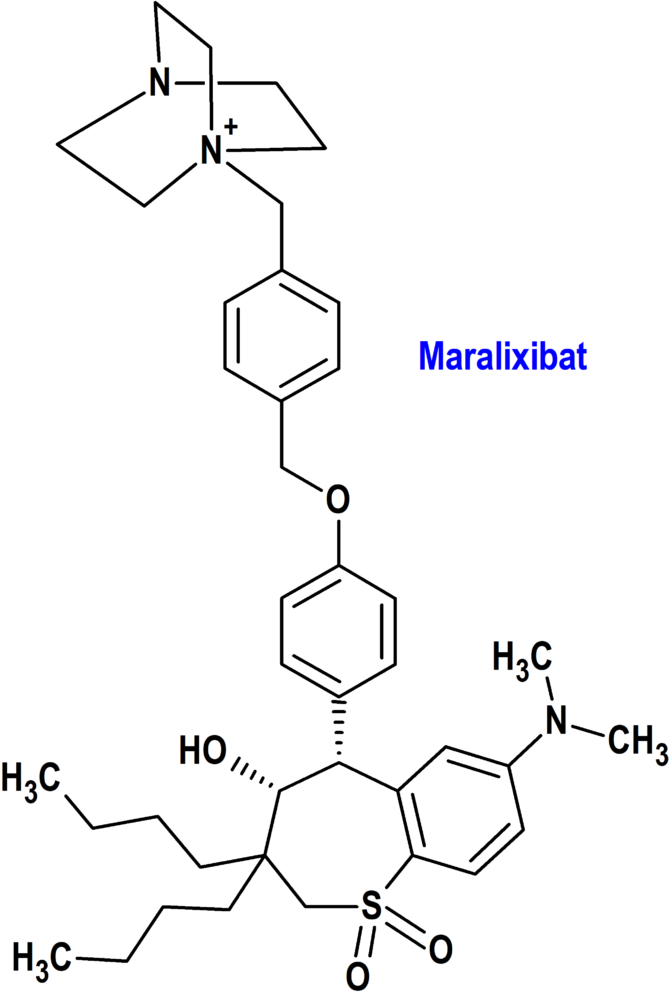
Indicación: Tratamiento del prurito colestático en pacientes con síndrome de Alagille (SALG) de 2 meses de edad y mayores.
Tipo: Autorizado el 9-12-2022 como medicamento huérfano (Orphan drug) y en condiciones excepcionales (Exceptional circumstances); autorizado previamente en Estados Unidos el 29-9-2021.
Mecanismo: Inhibidor selectivo, potente y reversible del transportador ileal de ácidos biliares (IBAT), que se absorbe en cantidades mínimas, actuando localmente en el íleon distal para reducir la recaptación de ácidos biliares y aumentar su eliminación a través del colon, lo que disminuye la concentración de ácidos biliares en sangre.
Eficacia clínica: Ensayo con 29 pacientes con SALG acompañado colestasis y prurito aleatorizados para recibir maralixibat o placebo durante el tiempo de espera aleatorizado y doble ciego de 4 semanas en las semanas. Posteriormente, todos recibieron maralixibat en régimen abierto durante un máximo de 48 semanas. Se observó una reducción media estadísticamente significativa en los ácidos biliares en comparación con los valores iniciales de 88 (120) y 96 (166,6) μmol/l en la semanas 18 y 48 tras la administración de maralixibat. Al final del periodo controlado con placebo, se demostró una diferencia en la media por mínimos cuadrados estadísticamente significativa entre maralixibat y placebo con respecto al cambio en los ABS desde la semana 18 hasta la semana 22 (−114 [48,0] µmol/l). Los pacientes que recibieron maralixibat presentaron un cambio clínicamente significativo y reducciones estadísticamente significativas en la escala ItchRO(Obs) [escala validada de 0 a 4 cumplimentada por los cuidadores, donde 0=nada y 4=muy grave), en la que los cambios ≥ 1,0 clínicamente significativos] de −1,7 y −1,6 puntos con respecto al inicio en las semanas 18 y 48, respectivamente.
Eventos adversos: Los más comunes son diarrea (36%) y dolor abdominal (29%), ninguno grave.
(H) Preparaciones Hormonales Sistémicas
Abaloparatida (Eladynos®) Radius Health
Indicación: Tratamiento de la osteoporosis en mujeres posmenopáusicas que presentan un aumento del riesgo de fractura..
Tipo: Autorizado el 12-12-2022; autorizado previamente en Estados Unidos el 28-4-2017.
Mecanismo: Péptido de 34 aminoácidos que comparte una homología del 41% con la hormona paratiroidea (PTH[1-34]) y una homología del 76% con el péptido relacionado con la hormona paratiroidea (PTHrP[1-34]). Es un activador de la vía de señalización del receptor PTH1 que estimula la formación de hueso nuevo en las superficies óseas trabeculares y corticales mediante la estimulación de la actividad osteoblástica, causando aumentos transitorios y limitados de la resorción ósea y aumenta la densidad ósea.
Eficacia clínica: Ensayo clínico multicéntrico, aleatorizado, con doble enmascaramiento, controlado con placebo y con un producto activo no enmascarado (teriparatida) durante 18 meses de tratamiento con 1 mes de seguimiento en 2070 mujeres posmenopáusicas de entre 50 y 86 años de edad (media de 69). La variable principal de eficacia fue la incidencia de fracturas vertebrales nuevas en las pacientes tratadas con abaloparatida, en comparación con las que recibieron el placebo: a los 18 meses, la abaloparatida (0,5%) y la teriparatida (0,7%) redujeron la tasa fracturas vertebrales nuevas en comparación con el placebo (4,2%); a los 19 meses, la incidencia de fracturas no vertebrales fue similar entre los grupos de la abaloparatida (2,7%) y de la teriparatida (2,0%) y no fue estadísticamente diferente en comparación con el placebo (3,6%).
Eventos adversos: Los más comunes son hipercalciuria (15,6 %), mareo (11,1 %), dolor de espalda (8,6 %), náuseas (8,5 %), cefalea (8,5 %), artralgia (8,4 %), hipertensión (6,8 %), reacción en el lugar de inyección (6,2 %) y palpitaciones (5,6 %).
(J) Antiinfecciosos Sistémicos
Nirsevimab (Beyfortus®) AstraZeneca
Indicación: Prevención de la enfermedad de las vías respiratorias inferiores producida por el Virus Respiratorio Sincitial (VRS) en neonatos y lactantes durante su primera temporada del VRS.
Tipo: Autorizado el 31-10-2022. Medicamento biológico, constituido por un anticuerpo monoclonal IgG1 kappa humano recombinante de larga duración neutralizante de la conformación de prefusión de la proteína VRS F que ha sido modificada con una sustitución triple de aminoácidos (YTE) en la región Fc para extender la semivida en suero.
Mecanismo: Nirsevimab se une a un epítopo altamente conservado en el sitio antigénico Ø de la proteína de prefusión del VRS. Inhibe el paso esencial de fusión de membrana en el proceso de entrada viral, neutralizando el virus y bloqueando la fusión célula-célula.
Eficacia clínica: Dos ensayos multicéntricos aleatorizados, doble ciego, controlados con placebo para la prevención de la infección de las vías respiratorias inferiores por VRS en 2.933 lactantes a término y prematuros (≥29 semanas) expuestos a su primera temporada del VRS. La variable primaria de eficacia fue la incidencia de infección de las vías respiratorias inferiores atendidas médicamente (incluida la hospitalización) causada por el VRS confirmado por RT-PCR, caracterizada predominantemente como bronquiolitis o neumonía, hasta 150 días después de la dosificación: 2,6 vs. 9,5% (efectividad del 70,1%) en muy prematuros y moderadamente prematuros (≥29 a <35 semanas) y 1,2 vs. 5,0% (74,5%) en nacidos a término y prematuros tardíos (≥35 semanas). La variable secundaria fue la incidencia de hospitalización hasta 150 días después de administrar el medicamento: 0,8 vs. 4,1% (efectividad del 78,4%) en muy prematuros y moderadamente prematuros (≥29 a <35 semanas) y 0,6 vs. 1,6% (62,1%) en nacidos a término y prematuros tardíos (≥35 semanas).
Eventos adversos: Los más comunes son erupción (0,7%), que se produjo en los 14 días posteriores a la dosis. La mayoría de los casos fueron de intensidad leve a moderada. Además, se notificaron pirexia y reacciones en el lugar de la inyección en una tasa de 0,6% y 0,4% dentro de los 7 días posteriores a la dosis, respectivamente. Las reacciones en el lugar de la inyección no fueron graves.
Vacuna Covid-19 Recombinante (VidPrevtyn Beta®) Sanofi Pasteur
Indicación: Como refuerzo de la inmunización activa para prevenir la COVID-19 en adultos que hayan recibido previamente una vacuna contra la COVID-19 de ARNm o vector adenoviral.
Tipo: Autorizado el 10-11-2022. Medicamento biológico, constituido por una vacuna con adyuvante compuesta por la proteína de punta (S) recombinante trimérica soluble SARS-CoV-2 (cepa B.1.351) estabilizada en su conformación de prefusión y delecionada de sus dominios transmembrana e intracelular. Utiliza como adyuvante AS03, compuesto por escualeno, DL-a-tocoferol y polisorbato 80.
Mecanismo: La combinación de antígeno y adyuvante mejora la magnitud de la respuesta inmunitaria, lo que puede contribuir a la protección contra la COVID-19.
Eficacia clínica: Estudio clínico aleatorizado, simple ciego, multicéntrico, que evaluó la respuesta inmunitaria inducida por una dosis de refuerzo de VidPrevtyn Beta o la vacuna de ARNm de COVID19 (nucleósido modificado/tozinamerán) en personas vacunadas previamente con 2 dosis de Vacuna de ARNm de COVID-19 (tozinamerán). La población de análisis por protocolo incluyó a 143 participantes de 18 años de edad y mayores que recibieron 2 dosis de vacuna de ARNm de COVID-19 (tozinameran) de 3 a 7 meses antes de recibir VidPrevtyn Beta (N=67) o vacuna de ARNm de COVID-19 (tozinameran ) (N=76). Se compararon los títulos (media geométrica) de los anticuerpos neutralizantes 28 días después: 1.327,5 vs 524,0 (ratio de 2,53 favorable a VidPrevtyn Beta).
Eventos adversos: Los más comunes son dolor en el lugar de la inyección (76,2%), dolor de cabeza (41,4%), mialgia (37,8%), malestar general (33,0%), artralgia (28,7%) y escalofríos (19,9%). La mediana de duración de las reacciones adversas locales y sistémicas es de 1 a 3 días. La mayoría ocurrieron dentro de los 3 días posteriores a la vacunación y fueron de gravedad leve a moderada.
Vacuna Dengue Tetravalente (Qdenga®) Takeda
Indicación: Prevención del dengue en personas a partir de los 4 años de edad.
Tipo: Autorizado el 5/-12/2022; autorizada previamente en Estados Unidos el 1-5-2019. Medicamento biológico, constituido por Virus del dengue vivos y atenuados de los serotipos 1, 2, 3 y 4, producidos en células Vero mediante tecnología de ADN recombinante.
Mecanismo: Replicar de forma local y provocar respuestas inmunitarias humorales y celulares contra los cuatro serotipos del virus del dengue.
Eficacia clínica: Ensayo clínico de fase 3, doble ciego, aleatorizado y controlado con placebo realizado en 5 países de América Latina (Brasil, Colombia, República Dominicana, Nicaragua y Panamá) y en 3 países de Asia (Sri Lanka, Tailandia y Filipinas). Un total de 20.099 niños de entre 4 y 16 años fueron aleatorizados (en una proporción de 2:1) para recibir la vacuna o placebo, indistintamente de la infección previa por dengue. La variable primaria de eficacia fue la tasa de sujetos con fiebre manifestada entre 30 días y 12 meses después de la segunda dosis de vacuna: 0,5% (vacuna) vs. 2,4% (placebo), con una eficacia del 80,2%.
Eventos adversos: Los más comunes son dolor en el lugar de inyección (50%), cefalea (35%), mialgia (31%), eritema en el lugar de inyección (27%), malestar (24%), astenia (20%) y fiebre (11%). Generalmente ocurrieron en los 2 días posteriores a la inyección, fueron de intensidad leve a moderada, tuvieron una duración breve (de 1 a 3 días) y fueron menos frecuentes tras la segunda inyección que después de la primera.
Maribavir (Livtencity®) Takeda
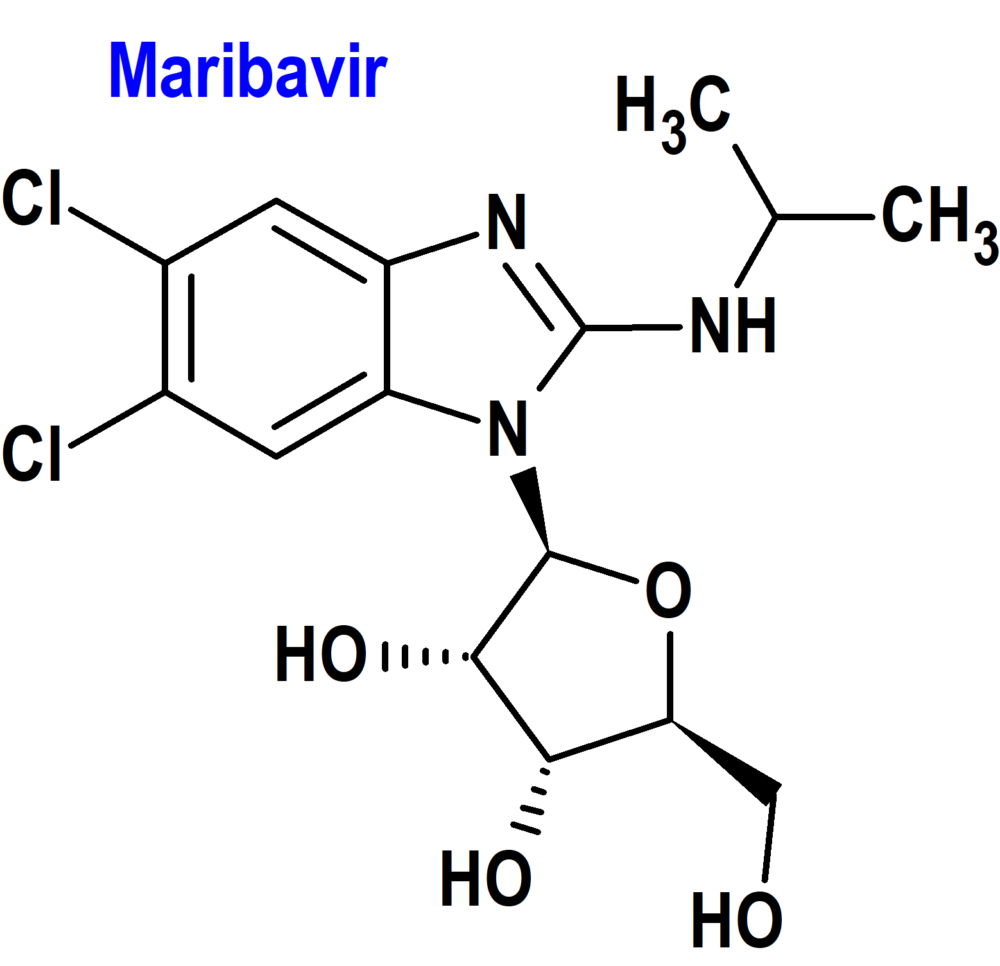
Indicación: Tratamiento de la infección y/o enfermedad por citomegalovirus (CMV) que es refractaria (con o sin resistencia) a uno o más tratamientos previos, incluyendo ganciclovir, valganciclovir, cidofovir o foscarnet en pacientes adultos que se han sometido a un trasplante de células madre hematopoyéticas (TCMH) o a un trasplante de órganos sólidos (TOS).
Tipo: Autorizado el 9-11-2022 como medicamento huérfano (Orphan drug) y con revisión prioritaria (Priority Medicines; PRIME); autorizado previamente en Estados Unidos el 23-11-2021.
Mecanismo: Inhibidor competitivo de la proteína cinasa UL97 en la fase de replicación del ADN viral, inhibiendo la serina/treonina cinasa de la UL97 mediante la inhibición competitiva de la unión de ATP al sitio de unión de ATP de la cinasa, sin afectar al proceso de maduración del concatenador, aboliendo la fosfotransferasa que inhibe la replicación, maduración, encapsidación y salida nuclear del ADN del CMV.
Eficacia clínica: Estudio clínico de fase 3, multicéntrico, aleatorizado, abierto y con control activo (comparación con el tratamiento asignado por el investigador, IAT) en 352 receptores de TCMH y TOS con infecciones por CMV refractarias al tratamiento con ganciclovir, valganciclovir, foscarnet o cidofovir, incluidas las infecciones por CMV con o sin resistencia confirmada a 1 o varios agentes anti-CMV. La variable principal de eficacia fue la reducción de la viremia confirmada por CMV (concentración plasmática de ADN del CMV por debajo del límite inferior de cuantificación <137 UI/ml) en la semana 8 de tratamiento, independientemente de que se interrumpiera el tratamiento asignado por el estudio antes del final de las 8 semanas estipuladas de terapia: 56 vs. 24%. La variable secundaria fue la reducción de la viremia por CMV y el control de los síntomas de la infección por CMV en la semana 8, con el mantenimiento de este efecto del tratamiento hasta la semana 16: 19 vs. 10%. Eventos adversos: Los más comunes (>10%) son alteraciones del gusto (46%), náuseas (21%), diarrea (19%), vómitos (14%) y fatiga (12%). Las reacciones adversas graves notificadas con mayor frecuencia fueron la diarrea (2%) y las náuseas, la disminución de peso, la fatiga, el aumento del nivel de concentración de fármacos inmunosupresores y los vómitos (todos se produjeron en más del 1%).
(L) Agentes Antineoplásicos e Inmunomoduladores
Voclosporina (Lupkynis®) Otsuka

Indicación: Tratamiento de pacientes adultos con nefritis lúpica (NL) activa de clase III, IV o V (incluidas las clases mixtas III/V y IV/V) en combinación con micofenolato de mofetilo.
Tipo: Autorizado el 15-9-2022; autorizado previamente en Estados Unidos el 22-01-2021.
Mecanismo: Es un inmunosupresor que inhibe la calcineurina de forma dependiente de la dosis. La activación de los linfocitos implica un aumento de las concentraciones de calcio intracelular. La calcineurina es una fosfatasa dependiente de calcio/calmodulina cuya actividad es necesaria para la inducción de la producción y proliferación de linfocinas en los linfocitos T. La actividad inmunosupresora se traduce en la inhibición de la proliferación de linfocitos, la producción de citocinas de linfocitos T y la expresión de antígenos de superficie para la activación de linfocitos T.
Eficacia clínica: Dos ensayos clínicos controlados con placebo en pacientes con nefritis lúpica (NL) de clase III o IV (solas o en combinación con la clase V) o de clase V pura. El primer estudio fue de fase 3, prospectivo, aleatorizado y con doble enmascaramiento, durante un periodo de tratamiento de 52 semanas, que incluyó a 357 pacientes. La proporción total de pacientes que alcanzaron cada uno de los criterios que componían la variable primaria en la semana 52 fueron: cociente proteína/creatinina en orina (CPCo) ≤0,5 mg/mg (45,3% vs. 23,0%); con función renal normal y estable, definida como TFGe ≥60 ml/minuto/1,73 m2 o sin disminución confirmada de la TFGe desde el inicio >20% (82,1% vs. 75,8 %); en presencia de dosis bajas sostenidas de corticosteroides (no más de 10 mg durante ≥3 días consecutivos o durante ≥7 días en total durante las semanas 44 a 52 (87,2% vs. 85,4%); y que no recibieron medicación de rescate para la nefritis lúpica (91,1% vs. 86,5%). El otro estudio fue un estudio de continuación (2 años más) para evaluar la seguridad y la eficacia a largo plazo en 216 pacientes que completaron el tratamiento en el estudio anterior, complentando el estudio más del 85 % de los pacientes. La proporción de pacientes con respuesta renal en el mes 36 fue del 51% vs. 39 %.
Eventos adversos: Los más comunes son la disminución de la tasa de filtración glomerular (TFGe; 26%) y la hipertensión (19%); son también muy frecuentes (>10%) las infecciones del tracto respiratorio superior, anemia, cefalea, tos, diarrea y dolor abdominal. Las reacciones adversas graves notificadas más frecuentemente son las infecciones (10,1%), lesión renal aguda (3 %) e hipertensión (1,9 %).
Tabelecleucel (Ebvallo®) Atara Biotherapeutics
Indicación: Monoterapia para el tratamiento de pacientes adultos y pediátricos de 2 años de edad y mayores con enfermedad linfoproliferativa postrasplante positiva asociada al virus de Epstein-Barr (EBV+ PTLD) en recaída o refractaria que han recibido al menos un tratamiento previo. En el caso de los pacientes con trasplante de órganos sólidos, el tratamiento previo incluye quimioterapia, a menos que no sea adecuada
Tipo: Autorizado el 16-12-2022 como medicamento huérfano (Orphan drug), con revisión prioritaria (Priority Medicines; PRIME) y en condiciones excepcionales (Exceptional circumstances). Medicamento biológico de terapia avanzada, consistente en inmunoterapia alogénica de células T específicas del virus de EpsteinBarr (EBV) dirigida a las células positivas para el EBV y eliminándolas de forma restringida por medio del antígeno leucocitario humano o antígeno de histocompatibilidad (HLA). Tabelecleucel se produce a partir de células T obtenidas de donantes humanos.
Mecanismo: Inmunoterapia alogénica de células T específicas del EBV que se dirige a las células infectadas por el EBV y las elimina de forma restringida por medio del antígeno leucocitario humano (HLA). El mecanismo de acción es equivalente al demostrado por las células T endógenas circulantes en los donantes de los que se deriva el medicamento. El receptor de células T de cada población clonal dentro del medicamento reconoce un complejo peptídico del EBV con una molécula HLA específica en la superficie de las células diana (el alelo HLA restrictivo) y permite que el medicamento ejerza una actividad citotóxica contra las células infectadas por el EBV.
Eficacia clínica: Ensayo multicéntrico de fase 3, abierto, de un grupo y realizado en 43 pacientes adultos y pediátricos con EBV+ PTLD después de un trasplante de órganos sólidos (TOS) o alogénico de células hematopoyéticas (TPH) tras el fracaso de la terapia previa. El medicamento se administró los días 1, 8 y 15, evaluándose la respuesta al día 28. La variable principal de eficacia fue la tasa de respuesta objetiva (ORR: 56%, 31% respuesta completa en TOS; 50%, 43% respuesta completa en TPH) según la evaluación de la adjudicación de la respuesta oncológica independiente (IORA), utilizando los criterios de clasificación de Lugano con la modificación de los criterios de respuesta del linfoma al tratamiento inmunomodulador (LYRIC); también se determinó la duración de la respuesta (DOR: 15,2 meses en TOS y 23,0 meses en TPH).
Eventos adversos: Los más comunes son fiebre (31,1%), diarrea (26,2%), cansancio (23,3%), náuseas (18,4%), anemia (16,5%), disminución del apetito (15,5%), hiponatremia (15,5%), dolor abdominal (14,6%), disminución del recuento de leucocitos (14, 6%), disminución del recuento de neutrófilos (14,6%), aumento de aspartato aminotransferasa (13,6%), estreñimiento (12,6%), aumento de alanina aminotransferasa (11,7%), aumento de fosfatasa alcalina en sangre (11,7%), hipoxia (11,7%), deshidratación (10,7%), hipotensión (10,7%), congestión nasal (10,7%) y exantema (10,7%).
Sutimlimab (Enjaymo®) Genzyme
Indicación: Tratamiento de la anemia hemolítica por crioaglutininas (cold agglutinin disease, CAD) en pacientes adultos.
Tipo: Autorizado el 15-11-2022, como medicamento huérfano (Orphan drug); autorizado previamente en Estados Unidos en 4-2-2022. Medicamento biológico, constituido por un anticuerpo monoclonal de inmunoglobulina G4 (IgG4).
Mecanismo: Inhibe la vía clásica del complemento y se une específicamente al componente de la proteína del complemento 1, subcomponente s (C1s), una serina proteasa que escinde C4; sin embargo, sutimlimab no inhibe las actividades de la lectina y las vías alternativas del complemento. La inhibición de la vía clásica del complemento a nivel de C1s impide el depósito de opsoninas del complemento en la superficie de los glóbulos rojos, lo que provoca la inhibición de la hemólisis en pacientes con anemia hemolítica por crioaglutininas, previene la generación de anafilotoxinas proinflamatorias C3a y C5a y el complejo terminal del complemento posterior C5b.
Eficacia clínica: Dos estudios clínico de fase 3, el primero aleatorizado, doble ciego y controlado con placebo en 42 pacientes, y el segundo abierto y de un solo grupo en 24 pacientes durante un periodo de 26 semanas. La variable principal de eficacia consistió en la proporción de pacientes que cumplieron los criterios de la variable primaria: un aumento desde el momento basal en el nivel de Hg ≥1,5 g/dl en el momento de la evaluación del tratamiento (semanas 23-26) [16 vs. 3%] y la media de transfusiones semanales desde la semana 5 hasta la semana 26 [0,05 vs. 0,5%]. Las variables secundarias consistieron en el cambio medio de la hemoglobina [+2,56 vs. +0,09 g/dl], puntuación de fatiga según la FACIT para evaluar el cambio en la calidad de vida [+10,83 vs. +1,91] y cambio medio de la bilirrubina total [-22,13 vs. -1,83 micromol/l].
Eventos adversos: Los más comunes (≥10%) son infección del tracto urinario, cistitis, infecciones del tracto respiratorio superior, nasofaringitis, gastroenteritis, rinitis, cefalea, hipertensión, cianosis (notificada como acrocianosis), denómeno de Raynaud, dolor abdominal y náuseas.
(R) Sistema Respiratorio
Tezepelumab (Tezspire®) AstraZeneca
Indicación: Tratamiento de mantenimiento adicional en adultos y adolescentes a partir de 12 años con asma grave que no están adecuadamente controlados a pesar de la administración de corticosteroides inhalados a dosis altas en combinación con otro medicamento para el tratamiento de mantenimiento.
Tipo: Autorizado el 19-9-2022; autorizado previamente en Estados Unidos el 17-12-2021. Medicamento biológico, constituido por un anticuerpo monoclonal humano inmunoglobulina G2-lambda producido mediante tecnología de ADN recombinante.
Mecanismo: Anticuerpo monoclonal dirigido contra la linfopoyetina estromal tímica (TSLP), impidiendo su interacción con el receptor heterodimérico de TSLP. En el asma, tanto los desencadenantes alérgicos como los no alérgicos inducen la producción de TSLP. El bloqueo de TSLP con tezepelumab reduce un amplio espectro de biomarcadores y citoquinas asociados con la inflamación de las vías respiratorias (eosinófilos, IgE, FeNO, IL-5 e IL-13).
Eficacia clínica: Dos ensayos clínicos aleatorizados, doble ciego, de grupos paralelos, controlados con placebo, de 52 semanas de duración en los que participaron un total de 1609 pacientes a partir de 12 años con asma grave. La variable principal para ambos ensayos clínicos fue la tasa de exacerbaciones asmáticas graves medidas durante 52 semanas (empeoramiento del asma que precisó el uso o aumento de corticosteroides orales o sistémicos durante al menos 3 días o una dosis única inyectable de corticosteroides, y/o visitas al servicio de urgencias que requerían el uso de corticosteroides orales o sistémicos y/u hospitalización): 0,93 vs. 2,10 en el primer estudio y 0,20 vs. 0,72 en el segundo.
Eventos adversos: Los más comunes son artralgia (3,8%) y faringitis (4,1%).
(V) Varios
Gozetotida (Locametz®) Novartis

Indicación: Tras el marcaje radiactivo con galio-68, está indicado para la detección de lesiones positivas al antígeno prostático de membrana (PSMA) mediante tomografía por emisión de positrones (PET) en adultos con cáncer de próstata (CaP) en los siguientes escenarios clínicos: Estadificación inicial de pacientes con CaP de alto riesgo antes de la terapia curativa inicial; sospecha de recidiva de CaP en pacientes con niveles crecientes de antígeno prostático específico (PSA) en suero después de la terapia curativa inicial o identificación de pacientes con cáncer de próstata resistente a la castración metastásico progresivo positivo a PSMA (CPRCm) para los que está indicada la terapia dirigida con PSMA.
Tipo: Autorizado el 9-12-2022; autorizado previamente en Estados Unidos el 1-12-2020. Medicamento constituido por un equipo de reactivos para preparación radiofarmacéutica, aunque el radionucleido (galio-68) no está incluido en el equipo.
Mecanismo: El galio-68-gozetotida se une a las células que expresan PSMA, incluidas las células malignas del cáncer de próstata, que sobreexpresan PSMA. El galio-68 es un radionucleido con un rendimiento de emisión que permite obtener imágenes PET que indican la presencia de la proteína PSMA en los tejidos.
Eficacia clínica: Un estudio de 103 pacientes varones adultos con cáncer de próstata con confirmación por biopsia y características de riesgo intermedio y alto indicados para una disección de los ganglios linfáticos pélvicos extendida (DGLPe), sometidos a imágenes PET/TC con galio (68)-gozetotida.; la sensibilidad fue del 42%, la especificidad del 91%, el valor predictivo positivo (VPP) del 77% y el negativo (VPN) del 68%. Otro estudio en 635 pacientes varones adultos con cáncer de próstata con recidiva bioquímica con confirmación histopatológica tras prostatectomía, radioterapia o ambas, se sometieron a PET/TC con galio (68Ga)-gozetotida o imágenes PET/MRI; la detección de lesiones positivas para PSMA ocurrió en el 75% pacientes que recibieron galio (68Ga)-gozetotida y la tasa de detección aumentó significativamente con los niveles de PSA. La sensibilidad fue del 92% y los valores predictivos positivo y negativo fueron del 84%.
Eventos adversos: Los más comunes son fatiga (1,2%), náuseas (0,8%), estreñimiento (0,5%) y vómitos (0,5%), todos ellos leves o moderados.
2. FOOD & DRUG ADMINISTRATION (FDA)
(A) Tracto Alimentario y Metabolismo
Teplizumab (Tzield®) Provention Bio
Indicación: Para retrasar la aparición de la diabetes tipo 1 en estadio 3 en adultos y pacientes pediátricos de 8 años de edad y mayores con diabetes tipo 1 en estadio 2.
Tipo: Autorizado el 17/11/2022; no autorizado aún en la Unión Europea. Medicamento biológico constituido por un anticuerpo monoclonal humanizado (IgG1 kappa).
Mecanismo: Se une a CD3 (antígeno de superficie presente en los linfocitos T) y retrasa la aparición de la diabetes tipo 1 en estadio 3 en adultos y pacientes pediátricos de 8 años o más con diabetes tipo 1 en etapa 2. El mecanismo puede implicar la señalización agonista parcial y la desactivación de los linfocitos T autorreactivos de las células beta pancreáticas, provocando un aumento de la proporción de linfocitos T reguladores y de linfocitos T CD8+ agotados en sangre periférica.
Eficacia clínica: Estudio aleatorizado, doble ciego, basado en eventos y controlado con placebo en 76 pacientes, de 8 a 49 años de edad con diabetes tipo 1 en estadio 2. Se diagnosticó diabetes tipo 1 en etapa 3 en el 45% (teplizumab) vs. 72% (placebo). Según un modelo de riesgos proporcionales de Cox, estratificado por edad y estado de la prueba de tolerancia a la glucosa oral inicial, demostró que la mediana de tiempo desde la aleatorización hasta el diagnóstico de diabetes tipo 1 en etapa 3 fue de 50 meses (teplizumab) vs. 25 meses (placebo).
Eventos adversos: Los más comunes son linfopenia (73%), erupción (36%), leucopenia (21%), cefalea (11%), neutropenia (5%), aumento de alanina aminotransferasa (11%), náusea (5%), diarrea (5%) y nasofaringitis (5%).
Microbiota Fecal Viva (Rebyota®) Ferring
Indicación: Prevención de la recurrencia de la infección por Clostridioides difficile (CDI) en personas de 18 años de edad y mayores después del tratamiento con antibióticos para la CDI recurrente.
Tipo: Autorizado el 30-11-2022 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea. Medicamento biológico constituido una suspensión de microbiota fecal para administración rectal producida a partir de materia fecal humana procedente de donantes seleccionados, en una solución de polietilenglicol (PEG) 3350 y solución salina.
Mecanismo: Cada dosis contiene entre 1 x 10(8) y 5 x 10(10) unidades formadoras de colonias (CFU) por ml de microbios fecales, incluidos >1 x 10(5) CFU/ml de Bacteroides. No se conoce el mecanismo de acción.
Eficacia clínica: Un estudio de fase 3 multicéntrico, aleatorizado, doble ciego, controlado con placebo, y otro estudio de fase 2 también controlado con placebo. Se incluyeron en los dos estudios un total de 344 adultos con diagnóstico confirmado de CDI recurrente. El éxito del tratamiento se definió como la ausencia de diarrea CDI dentro de las 8 semanas de tratamiento. En el análisis bayesiano de los datos procedentes de ambos estudios, la tasa estimada de éxito fue del 70,6 % (tratamiento activo) vs. 57,5% (placebo).
Eventos adversos: Los más comunes (≥3%) son dolor abdominal (8,9%), diarrea (7,2%), distensión abdominal (3,9%), flatulencia (3,3%) y náuseas (3,3%).
(B) Sangre y Sistema Hematopoyético
Etranacogene Dezaparvovec (Hemgenix®) CSL Behring
Indicación: Tratamiento de adultos con hemofilia B (deficiencia congénita del factor IX) que actualmente usan terapia de profilaxis con factor IX, o tienen hemorragia actual o histórica que pone en peligro la vida, o tienen episodios repetidos de sangrado espontáneo grave.
Tipo: Autorizado el 22/11/2022 como medicamento huérfano (Orphan drug) mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea. Medicamento biológico de terapia avanzada (génica), basado en vectores de virus adenoasociados.
Mecanismo: Se trata de un virus adenoasociado de tipo 5 (AAV5) recombinante y no replicante que contiene una secuencia de ADN de codones optimizados de la variante Padua con ganancia de función del Factor IX humano (variante R338L), bajo el control de un promotor 1 específico del hígado (LP1), consistente en elementos de núcleo específicos de hígado humano. La infusión intravenosa única del medicamento da como resultado la transducción celular y aumenta la actividad del factor IX circulante en pacientes con hemofilia B. Los niveles medios de actividad del factor IX fueron del 39% a los seis meses y del 36,7% a los 24. El inicio de la expresión de la proteína del factor IX después de la dosis fue detectable en la semana 3.
Eficacia clínica: Ensayo multinacional abierto, de dosis única, de un solo brazo (N = 54). Tras un periodo inicial de seis meses con tratamiento convencional, los pacientes recibieron una dosis intravenosa única, usándose los datos hasta 18 meses después del tratamiento para la evaluación de eficacia. La variable principal de eficacia fue una prueba de no inferioridad de la tasa de sangrado anualizada (TSA) durante los meses 7 a 18 después en comparación con la TSA durante los seis meses previos, registrándose todos los episodios hemorrágicos, independientemente de la evaluación del investigador. La TSA media estimada durante los meses 7 a 18 después del tratamiento fue de 1,9 hemorragias/año, en comparación con una TSA media estimada de 4,1 durante el período inicial. La proporción de TSA (meses 7 a 18/ inicio) fue de 0,46, lo que demuestra la no inferioridad de TSA durante los meses 7 a 18 en comparación con en el período de inicio.
Eventos adversos: Los más comunes (≥5%) son elevaciones de ALT (42%), dolor de cabeza (18%), elevaciones de creatina cinasa en sangre (42%), síntomas gripales (14%), reacciones relacionadas con la infusión IV (33%), fatiga (12%), malestar general (12%) y elevaciones de AST (42%).
Betibeglogene Autotemcel (Zynteglo®) Bluebird Bio
Indicación: Tratamiento de pacientes adultos y pediátricos con b-talasemia que requieren transfusiones periódicas de glóbulos rojos.
Tipo: Autorizado el 17-8-2022 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); autorizado en la Unión Europea el 29-5-2019, pero retirado a petición del titular de la autorización de comercialización. Medicamento biológico de terapia avanzada (génica), constituido por células CD34+ autólogas, que se obtienen mediante procedimientos de aféresis y que contienen células madre hematopoyéticas, transducidas con BB305 LVV.
Mecanismo: Las células autólogas se enriquecen con células CD34+ y luego se transducen ex vivo con BB305 LVV, que codifica la globina b A-T87Q, con un vector lentiviral (LVV) autoinactivante. El promotor, un elemento regulador del LVV que controla la expresión del transgén seleccionado para BB305 LVV, es un promotor celular (no viral) que controla la expresión génica específica de las células del linaje eritroide.
Eficacia clínica: Dos estudios en fase 3, abiertos, de un solo brazo, multicéntricos, de 24 meses de duración, en 41 pacientes de 4 a 34 años con b-talasemia que requerían transfusiones de sangre con regularidad. La variable principal de eficacia en ambos estudios fue el logro de la independencia transfusional (IT), definida como un promedio ponderado de Hb≥9 g/dL sin transfusiones durante un período continuo de ≥12 meses: 89%.
Eventos adversos: Los más comunes (≥20%) son mucositis, neutropenia febril, vómitos, pirexia, alopecia, epistaxis, dolor abdominal, dolor musculoesquelético, tos, dolor de cabeza, diarrea, erupción, estreñimiento, náuseas, disminución del apetito, trastorno de la pigmentación y picor. Se produjeron reacciones adversas graves en el 37% de los pacientes, siendo las más frecuentes (>3%) pirexia, trombocitopenia, enfermedad venooclusiva del hígado, neutropenia febril, neutropenia y estomatitis.
(L) Agentes Antineoplásicos e Inmunomoduladores
Nadofaragene Firadenovec (Adstiladrin®) Ferring
Indicación: Tratamiento de pacientes adultos con cáncer de vejiga sin invasión muscular de alto riesgo que no responde al bacilo de Calmette-Guérin (BCG) (NMIBC) con carcinoma in situ (CIS), con o sin tumores papilares.
Tipo: Autorizado el 16-12-2022 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea. Medicamento de terapia avanzada (génica), basado en un vector adenoviral (adenovirus recombinante del serotipo 5) no replicante, para uso en instilación intravesical.
Mecanismo:Terapia génica basada en un vector adenoviral no replicante diseñada para administrar una copia de un gen que codifica un interferón-alfa 2b humano (IFNα2b) en el urotelio de la vejiga. La instilación intravesical del medicamento da como resultado la transducción celular y la expresión local transitoria de la proteína IFNa2b que se prevé que tenga efectos antitumorales.
Eficacia clínica: Estudio clínico multicéntrico que incluyó a 98 pacientes con NMIBC de alto riesgo y carcinoma in situ (CIS) que no responde a BCG, con o sin tumores papilares. Los pacientes recibieron el tratamiento una vez cada tres meses hasta por 12 meses, o hasta toxicidad inaceptable para la terapia o NMIBC de alto grado recurrente. El 51% de los pacientes tratados lograron una respuesta completa (desaparición de todos los signos de cáncer vistos en la cistoscopia, biopsia de tejido y orina). La mediana de duración de la respuesta fue de 9,7 meses; el 46% de los pacientes que respondieron permanecieron en respuesta completa durante al menos un año.
Eventos adversos: Los más comunes (>10%), son aumento de la glucosa, secreción en el lugar de la instilación, aumento de los triglicéridos, fatiga, espasmo de la vejiga, urgencia miccional, aumento de la creatinina, hematuria, disminución de fosfato, escalofríos, disuria y fiebre. Se produjeron reacciones adversas graves en el 11 % de los pacientes, siendo las más frecuentes (>1%) enfermedad arterial coronaria y hematuria. La interrupción permanente del tratamiento debido a una reacción adversa se produjo en el 1,9% de los pacientes, debido a secreción del sitio de instilación, espasmo vesical o neoplasia benigna de la vejiga.
Tremelimumab (Imjudo®) AstraZeneca
Indicación: Tratamiento de pacientes adultos con carcinoma hepatocelular irresecable.
Tipo: Autorizado el 21-10-2022, como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea. Medicamento biológico constituido por un anticuerpo monoclonal IgG2 humano.
Mecanismo: Tremelimumab se une al antígeno 4 asociado a linfocitos T citotóxicos (CTLA-4), un regulador negativo de la actividad de los linfocitos T citotóxicos, y bloquea la interacción con sus ligandos CD80 y CD86, liberando la inhibición de la activación de los linfocitos T mediada por CTLA-4; el bloqueo de la actividad de CTLA-4 da como resultado una disminución del crecimiento tumoral y un aumento de la proliferación de linfocitos T en los tumores.
Eficacia clínica: Estudio aleatorizado (1:1:1), abierto, multicéntrico en pacientes con cáncer hepactocelular confirmado que no habían recibido tratamiento sistémico previo para el mismo. Los pacientes recibieron tremelimumab más durvalumab, como una infusión intravenosa única en combinación con durvalumab el mismo día, seguido de durvalumab cada 4 semanas; o sorafenib por vía oral dos veces al día, hasta progresión de la enfermedad o toxicidad inaceptable. La variable principal de eficacia fue la supervivencia global: 16,4 vs. 13,8 meses; como variables secundarias se determinaron la supervivencia libre de progresión evaluada por el investigador: 3,8 vs 4,1 meses, la tasa de respuesta objetiva: 3,1 vs 0% (respuesta completa) y 17,0 vs 5,1% (respuesta parcial); y la duración de la respuesta según RECIST v1.1: 22,3 vs. 18,4 meses (mediana), 65,8 vs 63,2% durante al menos 12 meses.
Eventos adversos: Los más comunes (≥20% de los pacientes que recibieron tremelimumab en combinación con durvalumab) son erupción cutánea, diarrea, fatiga, prurito, dolor musculoesquelético y dolor abdominal. Se produjeron reacciones adversas graves en el 41%: hemorragia (6%), diarrea (4%), sepsis (2,1%), neumonía (2,1%), erupción cutánea (1,5%), vómitos (1,3%) , lesión renal aguda (1,3%) y anemia (1,3 %). Ocurrieron reacciones adversas fatales en el 8 % de los pacientes: hemorragia intracraneal (0,5 %), paro cardíaco (0,5 %), neumonitis (0,5 %), insuficiencia hepática (0,5 %) y hepatitis inmunomediada (0,5 %). La interrupción permanente del régimen de tratamiento debido a una reacción adversa ocurrió en el 14 %: hemorragia (1,8%), diarrea (1,5%), aumento de AST (1%) y hepatitis (1%).
Mirvetuximab Soravtansina (Elahere®) Immunogen

Indicación: Tratamiento de pacientes adultos con cáncer epitelial de ovario, de trompas de Falopio o peritoneal primario resistente al platino, positivo para el receptor de folato alfa (FRa), que han recibido de uno a tres regímenes de tratamiento sistémico previos.
Tipo: Autorizado el 14-11-2022 como medicamento huérfano (Orphan drug) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea. Medicamento biológico constituido por un conjugado de anticuerpo-fármaco (ADC) que consta de tres componentes unidos covalentemente por conjugación química del mirbetuximab, un anticuerpo monoclonal quimérico de subtipo IgG1, con el agente antitubulina DM4 (un derivado de maitansina) a través de un conector (sulfo-SPDB), con un promedio de 3,4 moléculas de DM4 por cada molécula de mirbetuximab.
Mecanismo: Es un conjugado de anticuerpo y fármaco, en el que el anticuerpo – mirvetuximab – está dirigido contra el receptor de folato alfa (FRa). El DM4 es un inhibidor de microtúbulos unido al anticuerpo a través de un conector escindible. Al unirse a FRa el conjugado mirvetuximab soravtansina se internaliza, tras lo que se produce la liberación intracelular de DM4 a través una escisión proteolítica; el DM4 interrumpe la red de microtúbulos dentro de la célula, lo que provoca la detención del ciclo celular e induce la muerte celular por apoptosis.
Eficacia clínica: Ensayo de un solo grupo (brazo único) de pacientes con cáncer peritoneal primario, de trompas de Falopio o de ovario epitelial FRa positivo y resistente al platino (n = 106) que habían recibido hasta tres líneas previas de terapia sistémica; todos habían recibido bevacizumab. Las variables de eficacia fueron la tasa de respuesta global (ORR: 31,7%, 4,8% completa y 26,9% parcial) y la duración de la respuesta (DOR: mediana de 6,9 meses), ambas evaluadas por el investigador de acuerdo con los Criterios de Evaluación de Respuesta en Tumores Sólidos (RECIST), versión 1.1.
Eventos adversos: Se produjeron reacciones adversas graves en el 31% de los pacientes, siendo las más comunes (≥2%) obstrucción intestinal (8%), ascitis (4%), infección (3%) y derrame pleural (3%). Se produjeron reacciones adversas mortales en el 2% de los pacientes, incluida la obstrucción del intestino delgado (1%) y la neumonitis (1%). La discontinuación permanente del tratamiento debido a reacciones adversas ocurrió en el 11% de los pacientes, siendo las más frecuentes (≥2%) obstrucción intestinal (2%) y trombocitopenia (2%). Se produjeron retrasos en la dosificación debido a una reacción adversa en el 39% de los pacientes tratados, siendo las más comunes (≥3%) discapacidad visual (15%), queratopatía (11%), neutropenia (6%), ojo seco (5%), cataratas (3%) y aumento de la gamma-glutamiltransferasa (3%).
Olutasidenib (Rezlidhia®) Forma Therapeutics
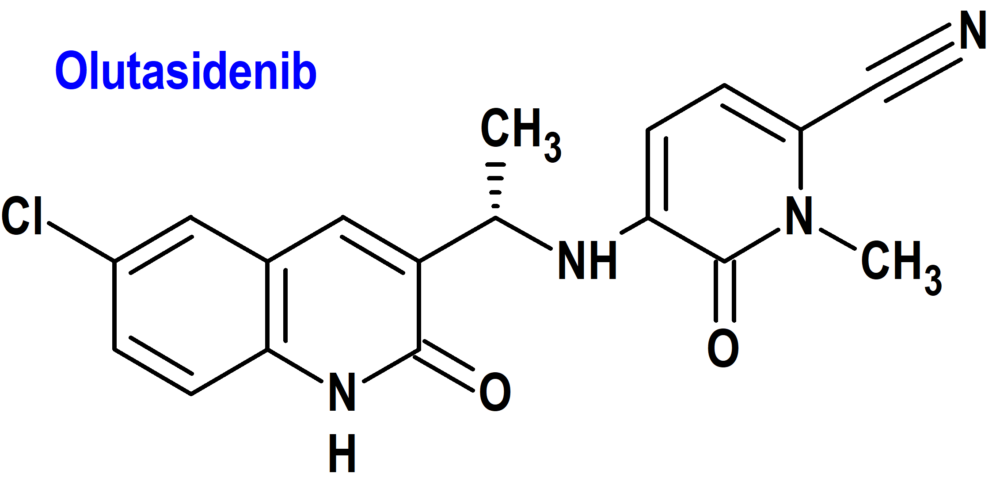
Indicación: Tratamiento de pacientes adultos con leucemia mieloide aguda (LMA) recidivante o refractaria con una mutación susceptible de isocitrato deshidrogenasa-1 (IDH1) detectada mediante una prueba aprobada por la FDA.
Tipo: Autorizado el 1-12-2022 , como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea.
Mecanismo: Es un inhibidor de la isocitrato deshidrogenasa-1 (IDH1) mutada. En pacientes con LMA, las mutaciones susceptibles de IDH1 se definen como aquellas que conducen a un aumento de los niveles de 2-hidroxiglutarato (2-HG) en las células leucémicas y cuya eficacia se predice mediante remisiones clínicamente significativas con la dosis recomendada de olutasidenib y/o la inhibición de la actividad enzimática de IDH1 mutante a concentraciones de olutasidenib sostenibles con la dosis recomendada según métodos validados. Las más comunes de tales mutaciones en pacientes con LMA son las sustituciones R132H y R132C.
Eficacia clínica: Ensayo clínico abierto, de un solo brazo, multicéntrico en 147 pacientes adultos con LMA recidivante o refractaria con una mutación IDH1. La eficacia se estableció sobre la base de la tasa de remisión completa (RC: 47%) más la remisión completa con recuperación hematológica parcial (Rch: 4%; CR+CRh: 51%), la duración de CR+CRh (mediana de 25,9 meses) y la tasa de conversión de dependencia de transfusión a independencia de transfusión: entre los 86 pacientes que dependían de transfusiones de glóbulos rojos y/o plaquetas al inicio del estudio, el 34% se volvieron independientes de las transfusiones durante cualquier período de 56 días posterior al inicio del tratamiento. De los pacientes que eran independientes de las transfusiones al inicio del estudio, el 64% permanecieron independientes de las transfusiones durante ese mismo periodo.
Eventos adversos: Los más comunes (≥20%), aumento de la aspartato aminotransferasa, aumento de la alanina aminotransferasa, disminución del potasio, disminución del sodio, aumento de la fosfatasa alcalina, náuseas, aumento de la creatinina, fatiga/malestar general, artralgia, estreñimiento, aumento de los linfocitos, aumento de bilirrubina, leucocitosis, aumento de ácido úrico, disnea, pirexia, exantema, aumento de lipasa, mucositis, diarrea y transaminitis. Se producen reacciones adversas graves en el 25% de los pacientes; las más frecuentes (≥5%) son el síndrome de diferenciación (9%) y transaminitis (6%). Se produjeron reacciones adversas mortales en el 1% de los pacientes, debido al síndrome de diferenciación. La interrupción permanente del tratamiento debido a una reacción adversa ocurre en el 8% de los pacientes, siendo las más comunes (≥1%) transaminitis, síndrome de diferenciación y trastornos de la vesícula biliar.
Adagrasib (Krazati®) Mirati Therapeutics

Indicación: Tratamiento de pacientes adultos con cáncer de pulmón de células no pequeñas (CPCNP) localmente avanzado o metastásico, con mutación en KRAS G12C, determinada mediante una prueba aprobada por la FDA, que hayan recibido al menos una terapia sistémica previa .
Tipo: Autorizado el 12-12-2022 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval); no autorizado aún en la Unión Europea.
Mecanismo: Inhibidor irreversible del KRAS G12C (homólogo del oncogén viral del sarcoma de rata de Kirsten), que actúa uniéndose covalentemente a la cisteína mutante en KRAS G12C y bloquea la proteína KRAS mutante en su estado inactivo que evita la señalización posterior sin afectar la proteína KRAS de tipo salvaje. La inactivación de KRAS G12C bloquea la señalización y la supervivencia de las células tumorales, inhibe el crecimiento celular y promueve la apoptosis selectivamente en tumores que albergan el oncogén KRAS G12C.
Eficacia clínica: Estudio de cohorte de expansión abierto, de un solo brazo y multicéntrico, a partir de 112 pacientes con NSCLC y mutación KRAS G12C localmente avanzado o metastásico que previamente recibieron tratamiento con un régimen basado en platino y un inhibidor del punto de control inmunitario. Los pacientes recibieron adagrasib hasta una toxicidad inaceptable o progresión de la enfermedad. Las evaluaciones del tumor se realizaron cada 6 semanas y las variables principales de eficacia fueron la tasa de respuesta objetiva (ORR) confirmada [43%; 0,9% completa y 42% parcial] y la duración de la respuesta (DOR) [8,5 meses de mediana y al menos 6 meses en el 58% de los pacientes], evaluadas mediante una revisión central independiente ciega de acuerdo con RECIST v1.1.
Eventos adversos: Los más comunes (≥25%) son náuseas (70%), diarrea (69%), vómitos (57%), fatiga (55%), dolor musculoesquelético (38%), hepatotoxicidad (37%), insuficiencia renal (33% ), edema (30%), disnea (26%) y disminución del apetito (29%). Las anomalías de laboratorio de Grado 3 o 4 más comunes (≥2%) son disminución de linfocitos (20%), de hemoglobina (7%), de leucocitos (2,5%) y de neutrófilos (2,3 %); aumento de alanina aminotransferasa (4,5%), de aspartato aminotransferasa (4,2%), de lipasa (2,5%), y de fosfatasa alcalina (2,0 %); hipopotasemia (3,6%) e hiponatremia (3,4%).
Futibatinib (Lytgobi®) Taiho Oncology
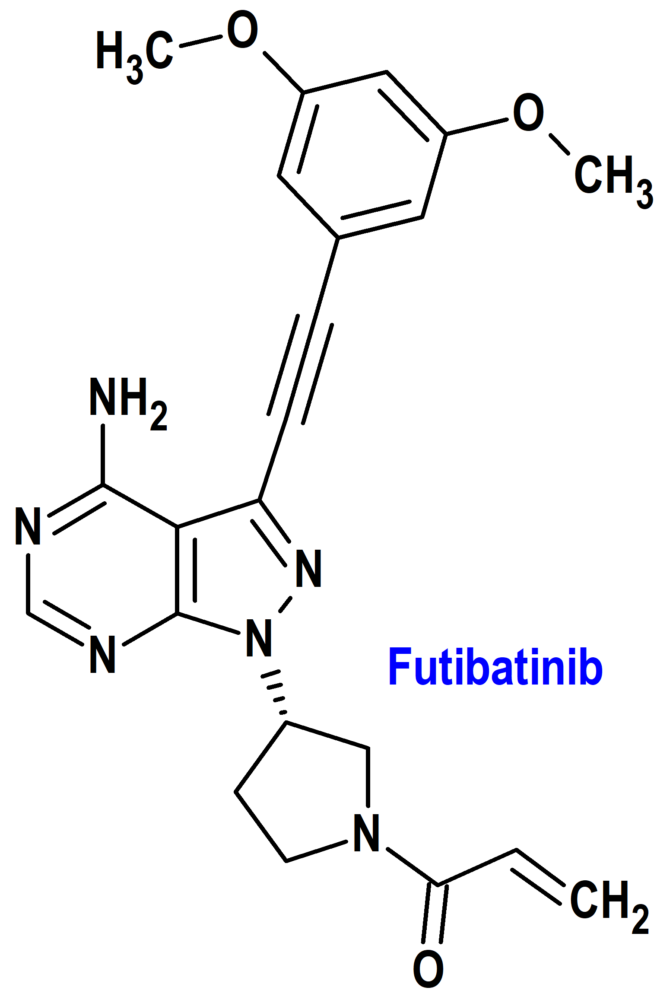
Indicación: Tratamiento de pacientes adultos con colangiocarcinoma intrahepático previamente tratado, no resecable, localmente avanzado o metastásico que alberga fusiones del gen receptor 2 del factor de crecimiento de fibroblastos (FGFR2) u otros reordenamientos.
Tipo: Autorizado el 30-9-2022 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Inhibidor de cinasa de los receptores 1, 2, 3 y 4 del factor de crecimiento de fibroblastos (FGF), cuya señalización puede facilitar la proliferación y supervivencia de células neoplásicas. La unión covalente de futibatinib al receptor 2 (FGFR2) inhibe la fosforilación de éste y la subsiguiente señalización, disminuyendo la viabilidad celular de células cancerosas con alteraciones de FGFR, incluidas fusiones/reordenamientos, amplificaciones y mutaciones de FGFR.
Eficacia clínica: Ensayo multicéntrico, abierto y de un solo brazo sobre 103 pacientes con colangiocarcinoma intrahepático previamente tratado, irresecable, localmente avanzado o metastásico que albergaba una fusión del gen FGFR2 u otro reordenamiento; el tratamiento se mantuvo hasta la progresión de la enfermedad o toxicidad inaceptable. Las variables principales de eficacia fueron la tasa de respuesta general (ORR: 43%, todas respuestas parciales) y la duración de la respuesta (DoR: mediana de 9.7 meses) según lo determinado por un comité de revisión independiente de acuerdo con RECIST v1.1.
Eventos adversos: Los más comunes (≥20%) son toxicidad en las uñas, dolor musculoesquelético, estreñimiento, diarrea, fatiga, boca seca, alopecia, estomatitis, dolor abdominal, piel seca, artralgia, disgeusia, ojo seco, náuseas, disminución del apetito, infección del tracto urinario , síndrome de eritrodisestesia palmar-plantar y vómitos. Las anomalías de laboratorio más comunes (≥20%) fueron aumento de fosfato, creatinina, glucosa, calcio, alanina aminotransferasa, fosfatasa alcalina, tiempo de tromboplastina parcial activada, aspartato aminotransferasa, creatina cinasa, bilirrubina e índice internacional normalizado de protrombina; disminución de hemoglobina, sodio, fosfato, linfocitos, plaquetas, leucocitos, albúmina, neutrófilos, glucosa y potasio. Se produjeron reacciones adversas graves en el 39% de los pacientes, siendo las más comunes pirexia (3,9%), hemorragia gastrointestinal (3,9%), ascitis (2,9%), dolor musculoesquelético (2,9%) y obstrucción de las vías biliares (2,9%). Se suspendió permanentemente el tratamiento 4,9% por reacciones adversas: esofagitis, disestesia oral, obstrucción del conducto biliar, mareos y anemia. Se produjeron interrupciones de la dosis en el 66% debido hiperfosfatemia, síndrome de eritrodisestesia palmar-plantar, aumento de alanina aminotransferasa, aumento de aspartato aminotransferasa y fatiga. Se produjeron reducciones de dosis debido a una reacción adversa en el 58% debido a hiperfosfatemia, síndrome de eritrodisestesia palmar-plantar, fatiga, aumento de alanina aminotransferasa, aumento de aspartato aminotransferasa, toxicidad ungueal y estomatitis.
Ublituximab (Briumvi®) TG Therapeutics
Indicación: Tratamiento de formas recidivantes de esclerosis múltiple, para incluir síndrome clínicamente aislado, enfermedad remitente-recidivante y enfermedad progresiva secundaria activa, en adultos.
Tipo: Autorizado el 28-12-2022 como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea. Medicamento biológico constituido por un anticuerpo IgG1 monoclonal quimérico recombinante con contenido reducido de fucosa, dirigido contra las células B que expresan CD20.
Mecanismo: Actúa uniéndose a CD20, un antígeno de la superficie celular presente en los linfocitos pre-B y B maduros. Después de la unión de la superficie celular a los linfocitos B, ublituximab provoca la lisis celular a través de mecanismos que incluyen la citolisis celular dependiente de anticuerpos y la citólisis dependiente del complemento. Con ello, se busca eliminar la población de linfocitos B en la sangre periférica y así controlar la actividad de la enfermedad.
Eficacia clínica: Dos ensayos clínicos aleatorizados, doble ciego, con doble simulación, de grupos paralelos, controlados con un comparador activo (teriflunomida), de diseño idéntico, sobre un total de 1.094 (549+545) pacientes tratados durante 96 semanas. Las variables primarias de eficacia clínica en ambos estudios fueron la tasa de recaída anualizada durante el período de tratamiento: 0,076 vs. 0,188 (-59%, favorable a ublituximab vs. teriflunomida) en el primer estudio y 0,091 vs. 0,178 (-49%) en el segundo, y la proporción de pacientes con progresión de la discapacidad confirmada a las 12 semanas: 5,2 vs. 5,9%. Las variables secundarias fueron el número medio de lesiones T1 hiperintensas en imágenes de resonancia magnética (MRI) en la semana 96: 0,016 vs 0,419 (-97%) y 0,009 vs 0,259 (-97%); y el de lesiones hiperintensas en T2 en MRI nuevas o en aumento en la semana 96: 0,213 vs 2,789 (-92%) y 0,282 vs 2,813 (-90%).
Eventos adversos: Los más comunes son reacciones a la infusión IV (48%), infecciones del tracto respiratorio superior (45%), infecciones del tracto respiratorio inferior (9%), infecciones asociadas al virus del herpes (6%), dolor en las extremidades (6%), insomnio (6%) y fatiga (5%).
(N) Sistema Nervioso
Taurursodiol/Fenilbutirato (Relyvrio®) Amylyx

Indicación: Tratamiento de la esclerosis lateral amiotrófica (ELA) en adultos.
Tipo: Autorizado el 29-9-2022 como medicamento huérfano (Orphan drug) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea. Combinación de dos principios activos, el fenilbutirato sódico y el taurursodiol (ácido tauroursodesoxicólico) sódico, un ácido biliar ambifílico producto de la conjugación de taurina con ursodiol (ácido ursodesoxicólico).
Mecanismo: Parece reducir la muerte neuronal al mitigar tanto el estrés del retículo endoplásmico como la disfunción mitocondrial. La disfunción conjunta de estos dos orgánulos dentro de las neuronas motoras se ha reconocido como un factor patógeno potencial en la ELA. Hay datos preclínicos que respaldan un efecto mitigador de ambos fármacos tanto individualmente como en combinación sobre la muerte neuronal y otras características específicas de la enfermedad en modelos de enfermedades neurodegenerativas y disfunción mitocondrial.
Eficacia clínica: Ensayo clínico multicéntrico, aleatorizado, doble ciego, controlado con placebo, de grupos paralelos y 24 semanas de duración, que incluyó a 137 pacientes adultos con un diagnóstico definitivo de ELA esporádica o familiar según la definición de los criterios revisados de El Escorial, con inicio de síntomas en los últimos 18 meses y una capacidad vital lenta (SVC) superior al 60 % de lo previsto. La variable principal de eficacia fue la comparación de la tasa de reducción en las puntuaciones totales de ALSFRS-R ( ALS Functional Rating ScaleRevised) desde el inicio hasta la semana 24: 29,03 vs 26,73 (diferencia de 2,32 estadísticamente significativa).
Eventos adversos: Los más comunes son diarrea (25% vs. 19% placebo), dolor abdominal (21 vs. 13%), náusea (18 vs. 13%), infección del tracto respiratorio superior (18 vs, 10%), fatiga (12 vs. 6%), hipersecreción salival (11 vs. 2%) y mareo (10 vs. 4%).
Elivaldogene Autotemcel (Skysona®) Bluebird Bio
Indicación: Para retrasar la progresión de la disfunción neurológica en niños de 4 a 17 años de edad con adrenoleucodistrofia cerebral activa temprana que sean asintomáticos o levemente sintomáticos (puntuación de función neurológica, NFS≤1) que tienen realce de gadolinio en imágenes de resonancia magnética (IRM) del cerebro y puntuaciones de Loes de 0,5 a 9.
Tipo: Autorizado el 16-9-2022 mediante revisión prioritaria (Priority Review) y con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); fue autorizado en la Unión Europea el 16-7-2021, pero fue retirada el 18-11-2021 a solicitud del titular de autorización. Medicamento biológico de terapia avanzada (génica) preparado a partir de las células hematopoyéticas del propio paciente, que se recolectan mediante aféresis. Las células autólogas se enriquecen con células CD34+, luego se transducen ex vivo con un vector lentiviral (Lenti-D LVV) y se cultivan con factores de crecimiento. Lenti-D LVV es autoinactivante, incompetente para la replicación, que lleva ADNc de ABCD1 que codifica ALDP normal. El gen ABCD1 está bajo el control de un promotor interno MNDU3, que es un promotor viral modificado y se ha demostrado que controla la expresión del transgén en las células hematopoyéticas y su progenie en todos los linajes.
Mecanismo: El medicamento agrega copias funcionales del ADNc de ABCD1 en las células madre hematopoyéticas (HSC) de los pacientes a través de la transducción de células CD34+ autólogas con Lenti-D LVV. Después de la infusión, las HSC CD34+ transducidas se injertan en la médula ósea y se diferencian en varios tipos de células, incluidos los monocitos (CD14+) capaces de producir ALDP funcional. La ALDP funcional puede entonces participar en la degradación local de los ácidos grasos de cadena muy larga (VLCFA), que se cree que ralentiza o posiblemente previene una mayor inflamación y desmielinización.
Eficacia clínica: Dos estudios abiertos de un solo brazo de 24 meses, totalizando 65 pacientes. Se observó una progresión más lenta de la disfunción neurológica o muerte desde el momento del inicio de los síntomas para los pacientes tratados en comparación con una historia natural similar de la enfermedad, estimándose que la supervivencia sin discapacidad funcional fue del 72 % para la subpoblación sintomática tratada en el mes 24 desde el momento de la primera dosis y del 43 % para la población de historia natural.
Eventos adversos: Se produjeron reacciones adversas graves en el 54% de los pacientes, siendo las más comunes (incidencia ≥3 %) neutropenia febril (18%), pirexia (18%), convulsiones (7%), síndrome mielodisplásico (4%), bacteriemia por Pseudomonas (3%), pancitopenia (3%), infección de dispositivos vasculares (3%), mucositis (3%) y vómitos (3%).
(S) Órganos Sensoriales
Omidenepag Isopropilo (Omlonti®) Santen

Indicación: Reducción de la presión intraocular (PIO) elevada en pacientes con glaucoma de ángulo abierto o hipertensión ocular.
Tipo: Autorizado el 22-9-2022; no autorizado aún en la Unión Europea. Se trata de un profármaco que libera el agente activo (omidenepag) mediante hidrólisis, tras su absorción a través de la córnea.
Mecanismo: Agonista selectivo del receptor de la prostaglandina E2 (EP2), que disminuye la presión intraocular (PIO), reduciendo por tanto la probabilidad de daño del nervio óptico y de la correspondiente pérdida del campo visual glaucomatoso.
Eficacia clínica: Tres ensayos clínicos aleatorizados y controlados con timolol y latanoprost en sujetos con glaucoma de ángulo abierto o hipertensión ocular con una PIO inicial promedio de 24-26 mm Hg. La duración del tratamiento con doble enmascaramiento fue de 3 meses en los 3 estudios, el último de los cuales incluyó un período de tratamiento abierto de 9 meses después del período de tratamiento doble ciego de 3 meses. En los tres estudios, se observaron reducciones de la PIO en el brazo de omidenepag entre 5 y 7 mm Hg en los tres estudios. Las reducciones correspondientes para los brazos de timolol y latanoprost fueron de 5 a 7 mm Hg y de 6 a 8 mm Hg, respectivamente.
Eventos adversos: Los más comunes son hiperemia conjuntival (9%), fotofobia (5%), visión borrosa (4%), ojo seco (3%), dolor en el lugar de la instilación (3%), dolor ocular (2%), hiperemia ocular (2%), puntiforme queratitis (2%), dolor de cabeza (2%), irritación ocular (1%) y discapacidad visual (1%).
(V) Varios
Tiosulfato Sódico (Pedmark®) Fennec
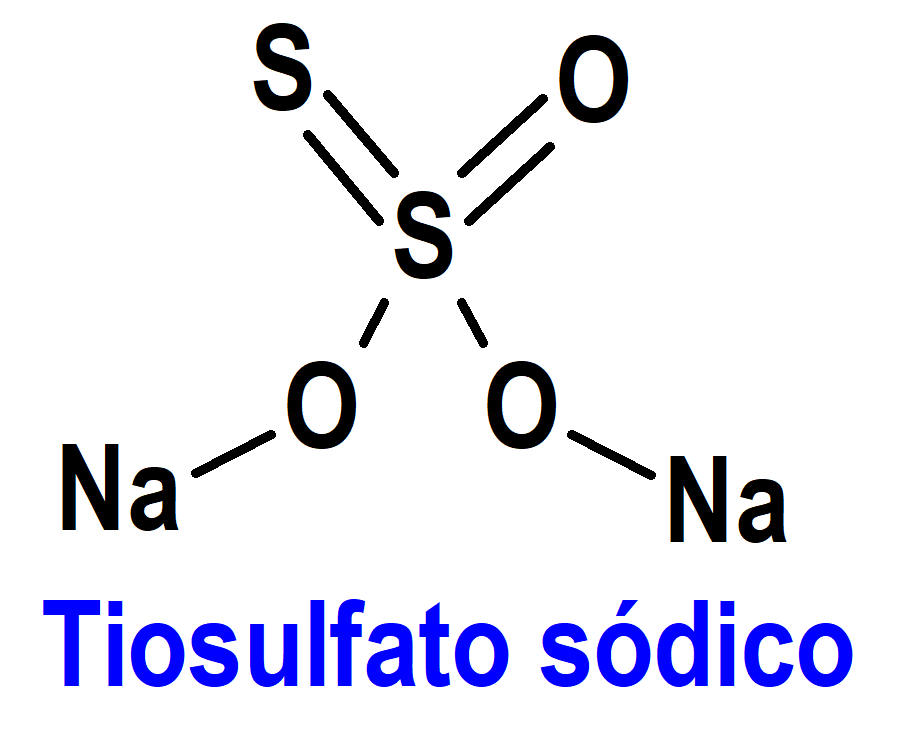
Indicación: Reducción del riesgo de ototoxicidad asociada con el cisplatino en pacientes pediátricos de 1 mes de edad y mayores con tumores sólidos localizados no metastásicos.
Tipo: Autorizado el 20-9-2022 como medicamento huérfano (Orphan drug) y con revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea para esta indicación.
Mecanismo: La ototoxicidad inducida por el cisplatino es causada por un daño irreversible a las células ciliadas en la cóclea, posiblemente como consecuencia de la combinación de la producción de especies reactivas de oxígeno (ROS) y de la reacción de alquilación directa de ADN, lo que conduce a la muerte celular. El tiosulfato de sodio interactúa directamente con el cisplatino para producir una especie de platino inactiva; además, el tiosulfato de sodio puede ingresar a las células a través del cotransportador de sulfato de sodio 2 y causar efectos intracelulares como el aumento de los niveles de glutatión antioxidante y la inhibición del estrés oxidativo intracelular. Ambas actividades pueden contribuir a la capacidad del tiosulfato de sodio para reducir el riesgo de ototoxicidad.
Eficacia clínica: Dos estudios multicéntricos, aleatorizados, controlado y abiertos, totalizando 239 pacientes entre 1 mes y 18 años de edad recibieron 6 ciclos de quimioterapia perioperatoria basada en cisplatino, con o sin tiosulfato. La variable principal de eficacia fue la tasa de pacientes que experimentaron pérdida auditiva evaluada según los criterios de la American SpeechLanguage-Hearing Association (ASHA): 39-44% (con tiosulfato) vs. 58-68% (sin tiosulfato).
Eventos adversos: Los más comunes (significativamente más que con cisplatino solo) son vómitos (85%), náusea (40%), reducción de los niveles de hemoglobina (34%), hipernatremia (26%), hipopotasemia (15%), hiperfosfatemia (15%), pirexia (15%) e hipomagnesemia (11%). Las reacciones adversas de grado 3 o 4 más comunes son reducción de los niveles de hemoglobina (19%), hiperfosfatemia (9%), pirexia (9%), hipomagnesemia (9%) y vómitos (8%).
Gadopiclenol (Elucirem®) Guerbet

Indicación: En pacientes adultos y pediátricos a partir de los 2 años de edad para usar con resonancia magnética nuclear (RMN) para detectar y visualizar lesiones con vascularización anormal en el sistema nervioso central (cerebro, columna y tejidos asociados) y en el cuerpo (cabeza y cuello, tórax, abdomen, pelvis y sistema musculoesquelético).
Tipo: Autorizado el 21-9-2022, mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea.
Mecanismo: Es un agente de contraste a base de gadolinio, que forma un complejo no iónico macrocíclico de carácter paramagnético, que desarrolla un momento magnético cuando se coloca en un campo magnético, el cual altera las tasas de relajación de los protones del agua en su vecindad en el cuerpo, lo que lleva a un aumento en la intensidad de la señal (brillo) de los tejidos.
Eficacia clínica: Dos estudios clínicos prospectivos, doble ciego, aleatorizados y cruzados. El estudio 1 se realizó en 256 adultos con lesiones conocidas o muy sospechadas del SNC con áreas focales de alteración de la barrera hematoencefálica y el estudio 2 se realizó en 304 adultos con presuntas anomalías realzadas en al menos una región del cuerpo entre la cabeza y el cuello, el tórax, el abdomen, la pelvis y el sistema musculoesquelético. En ambos estudios, los pacientes recibieron gadopiclenol y gadobutrol (como comparador activo) en orden aleatorio separados por 2 a 14 días. Se realizaron imágenes de resonancia magnética antes y después de la administración de cada agente de contraste. Tres lectores centrales que desconocían la identidad del agente de contraste evaluaron de forma independiente conjuntos de imágenes previas al contraste y pareadas (previas y posteriores al contraste para el mismo fármaco). Los lectores puntuaron hasta tres lesiones por paciente para la delimitación de los bordes, la morfología interna y la mejora del contraste, cada una en una escala del 1 al 4. También se informó el número total de lesiones. Un lector central independiente adicional realizó el seguimiento de lesiones para permitir la comparación de lesiones entre imágenes previas al contraste y emparejadas. El análisis comparó la puntuación promedio a nivel del paciente para las lesiones coincidentes para cada parámetro de visualización entre conjuntos de imágenes previas al contraste y pareadas. Las puntuaciones de visualización de lesiones con gadopiclenol y el número de lesiones identificadas por paciente fueron similares a las del gadobutrol en los dos estudios.
Eventos adversos: Los más comunes son dolor en el lugar de la inyección (0,7%), dolor de cabeza (0,7%), náuseas (0,4%), calor en el lugar de la inyección (0,4%), frío en el lugar de la inyección (0,3%), mareos (0,3%) e inflamación localizada (0,3%).
Xenón [Xe-129] Hiperpolarizado (Xenoview®) Polarean
Indicación: Obtención de imágenes por resonancia magnética para la evaluación de la ventilación pulmonar en adultos y pacientes pediátricos de 12 años de edad y mayores.
Tipo: Autorizado el 23-12-2022; no autorizado aún en la Unión Europea. El xenón Xe-129 hiperpolarizado es químicamente idéntico al xenón Xe-129 no polarizado; es un gas transparente, incoloro, inerte y estable que está presente de forma natural en el aire a un nivel de 0,087 ppm (partes por millón), formando parte de un conjunto de nueve isótopos no radiactivos, de los cuales el xenón Xe-129 supone el 26,4 %. El medicamento consiste en un cilindro con una mezcla de gases que contiene xenón con pureza isotópica Xe-129 >80% (1%), junto con nitrógeno (10%) y helio (89%). La hiperpolarización del Xe-129 es producida in situ por el sistema de hiperpolarización HPX.
Mecanismo: La inhalación de gas xenón Xe-129 hiperpolarizado se utiliza junto con un escáner de resonancia magnética con capacidad multinuclear, para obtener imágenes de la ventilación pulmonar. Los núcleos de xenón Xe-129 hiperpolarizados se detectan directamente mediante una bobina de resonancia magnética específica.
Eficacia clínica: Dos ensayos clínicos prospectivos, multicéntricos, aleatorizados, abiertos y cruzados que compararon la resonancia magnética con xenón Xe-129 con gammagrafía con xenón Xe-133 en pacientes adultos con trastornos pulmonares. La dosis media de utilizada fue de 99 ml de DE de xenón Xe-129 hiperpolarizado en el momento de la medición dentro de los 5 minutos posteriores a la administración. En cada prueba se calculó en ambos brazos de estudio la fracción de la señal total en los pulmones en cada una de las seis zonas (regiones superior, media e inferior en cada pulmón). Estos valores se usaron para estimar el porcentaje posoperatorio de ventilación pulmonar que se predijo que permanecería después de la resección planificada de un área pulmonar especificada previamente. En el primer estudio (31 pacientes), los porcentajes de pacientes que tenían diferencias estandarizadas dentro de ±10%, ±15% y ±20% fueron 81%, 94% y 94 %, respectivamente; en el segundo estudio (49 pacientes), los porcentaje respectivos del ±10%, ±15% y ±20% fueron del 65% , 80% y 96%.
Eventos adversos: Los más comunes son dolor orofaríngeo (4,8%), dolor de cabeza (2,4%) y mareos (2,4%).
3. PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA) como la Food & Drug Administration (FDA) de Estados Unidos disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA
- Revisión prioritaria (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
- Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
- Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
- Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA
- Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
- Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
- Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
- Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
- Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
- Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
