1. EUROPEAN MEDICINES AGENCY (EMA)
Semaglutida (Wegovy®) Novo Nordisk
Indicación: Complemento de una dieta baja en calorías y aumento de la actividad física para controlar el peso, incluida la pérdida de peso y el mantenimiento del peso, en adultos con un índice de masa corporal (IMC) inicial de ≥30 kg/m2 (obesidad), o ≥ 27 kg/m2 a <30 kg/m2 (sobrepeso) en presencia de al menos una comorbilidad relacionada con el peso, tal como disglucemia (prediabetes o diabetes mellitus tipo 2), hipertensión, dislipidemia, apnea obstructiva del sueño o enfermedad cardiovascular.
Tipo: Autorizado el 6-1-2022. Medicamento biológico, constituido por un análogo de acción prolongada del GLP-1 (péptido 1 similar al glucagón) humano. La semaglutida tiene una homología de secuencia del 94 % con el GLP-1 humano, el análogo Aib8, Arg34-GLP-1(7-37) en el que el grupo ε-amino de la lisina en la posición 26 está unido a una cadena lateral lineal formada por dos restos enlazados de ácido 8-amino-3,6-dioxaoctanoico (ADO), seguidos de ácido γ-glutámico y de ácido 1,18-octadecanodioico.
Mecanismo: Es un agonista del receptor de GLP-1, al que se une selectivamente y lo activa. El GLP-1 es un regulador fisiológico del apetito y la ingesta de calorías, que está presente en varias áreas del cerebro involucradas en la regulación homeostática de la ingesta de alimentos, como el hipotálamo y el tronco encefálico. La semaglutida puede afectar el sistema de recompensa hedonista a través de efectos directos e indirectos en áreas del cerebro que incluyen el tabique, el tálamo y la amígdala. La semaglutida reduce la ingesta de energía, aumenta la sensación de saciedad, la saciedad y el control de la alimentación, reduce la sensación de hambre y la frecuencia e intensidad de los antojos, así como la preferencia por alimentos ricos en grasas. Además, la semaglutida reduce la glucosa en sangre de forma dependiente de la glucosa al estimular la secreción de insulina y reducir la secreción de glucagón cuando la glucosa en sangre es alta; durante la hipoglucemia, la semaglutida disminuye la secreción de insulina y no altera la secreción de glucagón.
Eficacia clínica: Ensayo doble ciego, aleatorizado y controlado con placebo de 68 semanas, incluyendo a 1961 pacientes con obesidad (IMC≥30 kg/m2) o con sobrepeso (IMC≥27 kg/m2 a <30 kg/m2) y al menos una comorbilidad relacionada con el peso. Todos los pacientes siguieron una dieta baja en calorías y aumentaron la actividad física durante todo el ensayo. Al final del tratamiento (semana 68), la pérdida de peso fue superior y clínicamente significativa en comparación con el placebo (-14,9 vs. -2,4%). Además, una mayor proporción de pacientes lograron una pérdida de peso ≥5% (84 vs. 31%), ≥10% (66 vs. 12%) y ≥15% (48 vs. 5%). Entre los pacientes con prediabetes al inicio, la proporción de pacientes normoglucémicos al final del tratamiento fue del 84 vs. 48%.
Eventos adversos: Los más comunes son náusea (44%), diarrea (30%), vómitos (25%), estreñimiento (24%), dolor abdominal (>10%), cefalea (>10%) y fatiga (>10%). Un 4,3% de los pacientes suspendieron el tratamiento debidos a los eventos adversos gastrointestinales.
Voxelotor (Oxbryta®) Global Blood Therapeutics
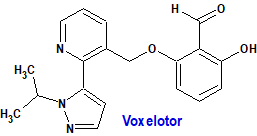
Indicación: Tratamiento de la anemia hemolítica debida a la enfermedad de células falciformes en adultos y pacientes pediátricos a partir de los 12 años de edad como monoterapia o en combinación con hidroxicarbamida.
Tipo: Autorizado el 14-2-2022, como medicamento huérfano; autorizado previamente en Estados Unidos (25-11-2019).
Mecanismo: Es un inhibidor de la polimerización de la hemoglobina S (HbS) que se une a la HbS con una estequiometría de 1:1 y muestra un reparto preferencial en los glóbulos rojos. Al aumentar la afinidad de la Hb por el oxígeno, voxelotor demuestra una inhibición dependiente de la dosis de la polimerización de la HbS. Voxelotor inhibe la formación de células falciformes en los glóbulos rojos y mejora la deformabilidad de estos.
Eficacia clínica: Ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo, incluyendo a 247 pacientes. La variable principal de eficacia fue la tasa de respuesta de Hb definida como un aumento de Hb >1 g/dl (0,62 mmol/l) desde el inicio hasta la semana 24. La tasa de respuesta para voxelotor fue del 51,1% vs 6,5 % con placebo.
Eventos adversos: Los más comunes incluyen cefalea (32%), diarrea (23%) y dolor abdominal (23%). Fueron graves cefalea (1,1%) e hipersensibilidad al fármaco (1,1%). La interrupción permanente debido a una reacción adversa se produjo en el 2,3 % de los pacientes y se produjeron modificaciones de dosis debido a una reacción adversa en el 13,6 % de los pacientes.
Finerenona (Kerendia®) Bayer
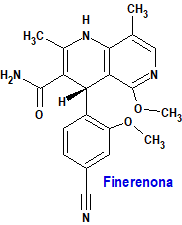
Indicación: Tratamiento de la enfermedad renal crónica (estadio 3 y 4 con albuminuria) asociada con diabetes tipo 2 en adultos.
Tipo: Autorizado el 16-2-2022; autorizado previamente en Estados Unidos (9-7-2021).
Mecanismo: Antagonista selectivo del receptor de mineralocorticoides (MR). Su unión al MR da lugar a un complejo receptor-ligando específico que bloquea el reclutamiento de coactivadores transcripcionales implicados en la expresión de mediadores proinflamatorios y profibróticos.
Eficacia clínica: Ensayo multicéntrico, aleatorizado, doble ciego y controlado con placebo incluyendo a 5674 pacientes, con un seguimiento medio de 2,6 años. La variable principal de eficacia fue una combinación del tiempo hasta la primera aparición de insuficiencia renal (definida como diálisis crónica o trasplante de riñón, o una disminución sostenida de la TFGe a <15 ml/min/1,73 m2 durante al menos 4 semanas), una disminución sostenida de la TFGe del 40% o más en comparación con el valor inicial durante al menos 4 semanas, o muerte renal: 17,8% con finerenona vs. 21,1% con placebo. La variable secundaria fue una combinación del tiempo hasta la primera aparición de muerte cardiovascular, infarto de miocardio no fatal, accidente cerebrovascular no fatal u hospitalización por insuficiencia cardíaca: reducción del 31% con respecto al placebo. Eventos adversos: Los más comunes son hiperpotasemia (18%), hipotensión, prurito y reducción de los niveles de hemoglobina (1-10%).
Lonapegsomatropina (Lonapegsomatropina Ascendis®) Ascendis
Indicación: Tratamiento del retraso del crecimiento en niños y adolescentes de 3 a 18 años debido a una secreción endógena insuficiente de la hormona del crecimiento. Tipo: Autorizado el 11-1-2022, como medicamento huérfano; autorizado previamente en Estados Unidos (25-8-21). Medicamento biológico, constituido por somatropina (hormona de crecimiento humana recombinante; hGH) conjugada con un enlazador ligado a 4 cadenas metoxipolietilenglicol (mPEG) de 10 kDa (PEG10) cada una.
Mecanismo: A pH y temperatura fisiológicos, la lonapegsomatropina liberaa (mediante autoescisión del enlazador) somatropina con el mismo modo de acción y distribución que la administración diaria de somatropina, pero requiriendo una única inyección semanal.
Eficacia clínica: Ensayo de fase 3, multicéntrico, aleatorizado, abierto, con control activo (somatropina) y de grupos paralelos, de 52 semanas de duración, en 161 pacientes pediátricos prepúberes sin tratamiento previo. La variable principal de eficacia fue la velocidad de crecimiento anualizada (cm/año): 11,2 (lonapegsomatropina semanal) vs 10,3 (somatropina diaria).
Eventos adversos: Los más comunes (≥10%) son infección del tracto respiratorio superior (23%), pirexia (16%), nasofaringitis (15%), cefalea (13%) y tos (12%).
Somatrogon (Ngenla®) Pfizer
Indicación: Tratamiento de niños y adolescentes a partir de 3 años con alteraciones del crecimiento debido a una secreción insuficiente de la hormona del crecimiento. Tipo: Autorizado el 14-2-2022 como medicamento huérfano. Medicamento biológico constituido por una proteína de fusión quimérica recombinante de la hormona del crecimiento humana (hGH) con una copia del péptido C-terminal (CTP) de la cadena β de la gonadotropina coriónica humana en el extremo N-terminal y dos copias de CTP en el extremo C-terminal.
Mecanismo: Se une al receptor de la hormona del crecimiento e inicia una cascada de transducción de señales que culmina en cambios en el crecimiento y el metabolismo. La unión de somatrogon conduce a la activación de la vía de señalización STAT5b y aumenta la concentración sérica de IGF-1 de manera dependiente. Como resultado, la GH y el IGF-1 estimulan los cambios metabólicos, el crecimiento lineal y mejoran la velocidad de crecimiento en pacientes pediátricos con GHD.
Eficacia clínica: Ensayo multicéntrico, aleatorizado y abierto controlado con somatropina, sobre 224 pacientes durante un año. La variable principal de eficacia la velocidad de altura después de 12 meses de tratamiento: 10,1 cm/año (somatrogon) vs. 9,78 (somatropina). Eventos adversos: Los más comunes son reacción en el lugar de inyección (25%), cefalea (11%) y pirexia (10%).
Nirmatrelvir / Ritonavir (Paxlovid®) Pfizer
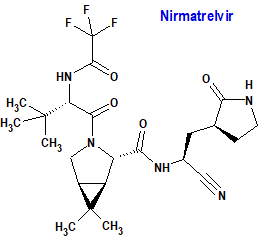
Indicación: Tratamiento de la enfermedad por coronavirus 2019 (COVID-19) en adultos que no requieren aporte de oxígeno suplementario y que tienen un riesgo alto de progresar a COVID-19 grave.
Tipo: Autorizado el 28-1-2022. La autorización fue condicional, debido a que el medicamento responde a una necesidad médica no satisfecha y el beneficio de la disponibilidad inmediata supera el riesgo de datos menos completos de lo que normalmente se requiere.
Mecanismo: Nirmatrelvir es un inhibidor peptidomimético de la proteasa principal (Mpro) del SARS-CoV-2 (proteasa 3C-like, 3CLpro o nsp5). La inhibición de la Mpro del SARS-CoV-2 impide que ésta pueda procesar precursores poliproteicos, lo que bloquea la replicación viral. Nirmatrelvir tiene actividad frente a aislados de SARS-CoV-2 pertenecientes a las variantes Alfa (B.1.1.7), Gamma (P.1), Delta (B.1.617.2), Lambda (C.37), Mu (B.1.621) y Ómicron (B.1.1.529), mientras que la variante Beta (B.1.351) es menos susceptible. Ritonavir inhibe el metabolismo de nirmatrelvir mediado por el citocromo CYP3A, lo que aumenta las concentraciones plasmáticas de nirmatrelvir.
Eficacia clínica: Ensayo clínico en fase 2/3, aleatorizado, doble ciego y controlado con placebo en 2.246 participantes adultos sintomáticos no hospitalizados con un diagnóstico de infección por SARS-CoV-2, con al menos algún factor de riesgo de evolucionar a enfermedad grave (diabetes, sobrepeso (IMC>25), enfermedad pulmonar crónica (incluido el asma), enfermedad renal crónica, fumador en activo, enfermedad inmunosupresora o tratamiento inmunosupresor, enfermedad cardiovascular, hipertensión arterial, anemia de células falciformes, trastornos del desarrollo neurológico, cáncer activo, dependencia médica de soporte tecnológico, o con 60 años de edad o más independientemente de las comorbilidades). La variable principal de eficacia fue la tasa de hospitalización relacionada con COVID-19 o muerte por cualquier causa hasta el día 28: 0,8% con nirmatrelvir/ritonavir vs. 6,3% con placebo; la mortalidad por cualquier causa hasta el día 28 fue del 0% vs. 1,1%.
Eventos adversos: Los más comunes son disgeusia (5,6%), diarrea (3,1%), cefalea (1,4%) y vómitos (1,1%).
Tixagevimab/Cilgavimab (Evusheld®) AstraZeneca
Indicación: Prevención de la COVID-19 en adultos y adolescentes a partir de 12 años que pesen al menos 40 kg, antes de una posible exposición al virus SARS-CoV -2.
Tipo: Autorizado el 25-3-2022. Medicamento biológico, compuesto por dos anticuerpos monoclonales humanos recombinantes de tipo IgG1κ. Tiene sustituciones de aminoácidos en las regiones Fc, para prolongar la semivida de los anticuerpos y reducir la función efectora de los anticuerpos y el riesgo potencial de aumento de la enfermedad dependiente de anticuerpos
Mecanismo: Tixagevimab y cilgavimab pueden unirse simultáneamente a regiones no superpuestas del dominio de unión al receptor de la proteína espiga (RBD) del SARS-CoV-2, bloqueando su interacción con el receptor ACE2 humano, lo que impide la entrada del virus en las células.
Eficacia clínica: Ensayo clínico de fase 3 (todavía en curso), aleatorizado (2:1), doble ciego, controlado con placebo para la profilaxis previa a la exposición de COVID-19 en 5.172 adultos (≥18 años). El medicamento redujo en un 77% el riesgo de enfermedad sintomática por SARS-CoV-2 (COVID-19) en comparación con el placebo (0,2 vs. 1% de incidencia), durante una mediana de 83 días de seguimiento tras la administración. La eficacia fue consistente en todos los subgrupos predefinidos, incluidos la edad, el género, el origen étnico y las comorbilidades o características iniciales asociadas con un mayor riesgo de COVID-19 grave.
Eventos adversos: Los más comunes son reacciones en el lugar de la inyección (1,3 %) e hipersensibilidad (1,0 %).
Tecovirimat (Tecovirimat SIGA®) SIGA
Indicación: Tratamiento de la viruela, viruela del mono y viruela bovina en adultos y niños con un peso corporal de al menos 13 kg. Tratamiento de las complicaciones debidas a la replicación del virus vaccinia tras la vacunación contra la viruela en adultos y niños con un peso corporal de al menos 13 kg.
Tipo: Autorizado el 6-1-2022, en «circunstancias excepcionales». Autorizado previamente en Estados Unidos (13-7-2018) como medicamento huérfano.
Mecanismo: Inhibe la actividad de la proteína VP37, codificada por un gen altamente conservado en todos los miembros del género Orthopoxvirus. El tecovirimat bloquea la interacción de VP37 con las proteínas Rab9 GTPasa y TIP47 celulares, lo que impide la formación de viriones con competencia para la diseminación del virus de célula a célula y de largo alcance.
Eficacia clínica: Por motivos éticos ligados a la indicación autorizada, la eficacia del medicamento no se pudo probar previamente en humanos, por lo que los datos considerados proceden de comparar las exposiciones a tecovirimat en sujetos humanos sanos con las observadas en modelos animales de infección por ortopoxvirus, utilizando primates no humanos (macacos del género Cynomolgus) y conejos New Zealand blancos, respectivamente infectados con el virus de la viruela del mono y el virus de la viruela del conejo. Las tasas de supervivencia a los 4 días en los diversos estudios fueron del 80-100% (tecovirimat) vs. 0% (placebo) en los macacos, y del 88-90% vs. 0% en los conejos.
Eventos adversos: Los más comunes son cefalea (12%) y náusea (4,5%).
Sotorasib (Lumykras®) Amgen
Indicación: Tratamiento de adultos con cáncer de pulmón de células no pequeñas (NSCLC) avanzado con mutación KRAS G12C y que han progresado después de al menos una línea previa de terapia sistémica.
Tipo: Autorizado el 6-1-2022; autorizado previamente en Estados Unidos (28-5-2021), como medicamento huérfano.
Mecanismo: Inhibidor selectivo del gen impulsor oncogénico KRAS G12C (homólogo del oncogén viral del sarcoma de rata de Kirsten), uniéndose de forma covalente e irreversible a la cisteína única de KRAS G12C. La inactivación de KRAS G12C por sotorasib bloquea la señalización y la supervivencia de las células tumorales, inhibe el crecimiento celular y promueve la apoptosis selectivamente en tumores que albergan el oncogén KRAS G12C.
Eficacia clínica: Ensayo multicéntrico, de un solo brazo y abierto incluyendo a 126 pacientes. La variable principal de eficacia fue la tasa de respuesta objetiva (ORR: 37,1%) definida como la proporción de pacientes que lograron una respuesta completa (2,4%) o parcial (34,7%), evaluada por un comité revisor ciego de expertos, siguiendo criterios RECIST v1.1 (Response Evaluation Criteria in Solid Tumours). La mediana de duración de la respuesta fue de 11,1 meses, siendo de ≥6 meses en el 63%.
Eventos adversos: Los más comunes son diarrea (34%), náuseas (25%) y fatiga (21%). Las reacciones adversas graves (grado ≥ 3) más comunes fueron aumento de ALT (5%), aumento de AST (4%) y diarrea (4%). Las reacciones adversas más comunes que llevaron a la interrupción permanente del tratamiento fueron aumento de ALT (1%) y aumento de AST (1%) y DILI (1%), y las que llevaron a la modificación de la dosis fueron aumento de ALT (6%), diarrea (6%), aumento de AST (6%), náuseas (3%), aumento de la fosfatasa alcalina en sangre (3%) y vómitos (2%).
Tepotinib (Tepmetko®) Merck
Indicación: Tratamiento en monoterapia de pacientes adultos con cáncer de pulmón de células no pequeñas (NSCLC) avanzado que albergan alteraciones que conducen a la omisión del exón 14 del gen del Factor de Transición Epitelial-Mesenquimatoso (METex14), que requieren terapia sistémica después de un tratamiento previo con inmunoterapia y/o quimioterapia basada en platino.
Tipo: Autorizado el 16-2-2022; autorizado previamente en Estados Unidos (3-2-2021), como medicamento huérfano.
Mecanismo: Inhibidor reversible de tipo I del Factor de Transición Epitelial-Mesenquimatoso (MET), que compite con el trifosfato de adenosina (ATP). Bloquea la fosforilación de MET y la señalización anterógrada dependiente de MET como las vías de la fosfatidilinositol 3-cinasa/proteína-cinasa B (PI3K/Akt) y de la proteína-cinasa activada por mitógenos/cinasa regulada por señales extracelulares (MAPK/ERK) de manera dependiente de la dosis.
Eficacia clínica: Estudio multicéntrico, abierto y de un solo brazo, en 275 pacientes. La variable principal de eficacia fue la respuesta objetiva confirmada (completa o parcial) conforme a los criterios de evaluación de la respuesta en tumores sólidos (RECIST v1.1) evaluada por un comité de revisión independiente (CRI): 49% global y 44% en pacientes previamente tratados. La mediana de la duración de la respuesta fue de 13,8 meses (11,1 en pacientes previamente tratados).
Eventos adversos: Los más comunes (≥20%) son edema (77%), principalmente edema periférico (66%), náuseas (30%), hipoalbuminemia (28%), diarrea (28%) y aumento de la creatinina (27%). Las reacciones adversas graves más frecuentes (≥1 %) son edema periférico (3,1%), edema generalizado (2,1%) e enfermedad intersticial pulmonar (ILD; 1,4 %). El 24% suspendieron definitivamente el tratamiento; un 49% temporalmente y un 34% requirieron una reducción de la dosis, debido a reacciones adversas.
Enfortumab Vedotina (Padcev®) Astellas
Indicación: Tratamiento de pacientes adultos con cáncer urotelial localmente avanzado o metastásico que hayan recibido previamente quimioterapia con platino y un inhibidor del receptor de muerte programada 1 (PD-1) o del ligando 1 de muerte programada (PD-L1).
Tipo: Autorizado el 13-4-2022; autorizado previamente en Estados Unidos (18-12-2019). Medicamento biológico, conjugado de un anticuerpo monoclonal tipo IgG1 completamente humano (enfortumab) con un agente citotóxico, la vedotina (monometil auristatina E, MMAE), a través de una molécula enlazadora (maleimidocaproil valina-citrulina) escindible por proteasas. La conjugación tiene lugar con los residuos de cisteína que comprenden los enlaces disulfuro intercatenarios del anticuerpo, dando lugar a un producto con una proporción de vedotina: anticuerpo de 3,8:1.
Mecanismo: El conjugado enfortumab-vedotina se une a la diana nectina-4 en la superficie celular y formar un complejo con ésta, que es rápidamente internalizado escindiéndose el conector mediante proteasas lisosómicas para permitir el suministro intracelular de vedotina, un potente agente citotóxico que actúa uniéndose a los microtúbulos para inhibir la división celular e inducir apoptosis.
Eficacia clínica: Ensayo clínico de fase 3, multicéntrico, aleatorizado y abierto, comparando enfortumab vedotina vs. quimioterapia estándar en 608 pacientes,. La variable primaria de eficacia fue la supervivencia global (mediana de duración de 12,9 vs. 9,0 meses) y las variables secundarias incluyeron la supervivencia libre de progresión (mediana de duración de 5,6 vs. 3,7 meses) y la tasa de respuesta objetiva (40,7% [4,9% completa] vs 17,9% [2,7% ], evaluadas por utilizando criterios RECIST v1.1.
Eventos adversos: Son graves en el 47% de los pacientes; las más frecuentes son infección del tracto urinario, lesión renal aguda (7%) y neumonía (5%). Mortales en el 3%, incluyendo disfunción multiorgánica (1,0%), disfunción hepática, shock séptico, hiperglucemia, neumonitis y absceso pélvico (0,3% cada uno). Conducen a la interrupción definitiva en el 17%: neuropatía periférica (5%) y erupción cutánea (4%). Llevan a la interrupción temporal en el 61%: neuropatía periférica (23%), erupción cutánea (11%) y fatiga (9%); es precisa una reducción de la dosis en el 34%: neuropatía periférica (10%), erupción cutánea (8%), disminución del apetito (3%) y fatiga (3%).
Ciltacabtagene Autoleucel (Carvykti®) Janssen Cilag
Indicación: Tratamiento de pacientes adultos con mieloma múltiple en recaída y refractario que han recibido al menos tres tratamientos previos, incluidos un agente inmunomodulador, un inhibidor del proteosoma y un anticuerpo anti-CD38 y han presentado progresión de la enfermedad al último tratamiento.
Tipo: Autorizado el 25-5-2022, como medicamento huérfano. Medicamento de terapia avanzada basado en células autólogas modificadas genéticamente, que contiene linfocitos T transducidos ex vivo mediante un vector lentiviral no replicativo que codifica un receptor antigénico quimérico (CAR) contra el antígeno de maduración de los linfocitos B (B-cell maturation antigen; BCMA), que comprende dos anticuerpos de dominio único unidos a un dominio coestimulador 4-1BB y a un dominio de señalización CD3-zeta.
Mecanismo: Inmunoterapia de linfocitos T autólogos modificados genéticamente dirigidos contra BCMA, que requiere la reprogramación de los linfocitos T del propio paciente con un transgén que codifica un receptor antigénico quimérico (CAR) que identifica y elimina las células que expresan el BCMA. El BCMA se expresa principalmente en la superficie de las células de línea B malignas del mieloma múltiple, así como en los linfocitos B y en las células plasmáticas en fase tardía. La proteína CAR del medicamento presenta dos anticuerpos de dominio único dirigidos al BCMA que se han diseñado para conferir una alta afinidad contra el BCMA humano, un dominio coestimulador 4-1BB y un dominio citoplásmico de señalización de CD3-zeta (CD3ζ). Al unirse a las células que expresan BCMA, el CAR promueve la activación y expansión de los linfocitos T y la eliminación de las células con expresión de BCMA.
Eficacia clínica: Ensayo multicéntrico, abierto y de un solo brazo, incluyendo a 113 pacientes. La variable principal de eficacia fue la tasa global de respuesta según la evaluación de un Comité de Revisión Independiente: 98% (completa en el 80%). La mediana de la duración de la respuesta fue de 21,8 meses y la mediana de tiempo transcurrido hasta obtener respuesta fue de 0,95 meses; la tasa de negatividad de enfermedad mínima residual fue del 56% en los tres meses siguientes en los pacientes que experimentaron una respuesta completa.
Eventos adversos: Los más comunes son neutropenia (91%), síndrome de liberación de citocinas (SLC, 88%), pirexia (88%), trombocitopenia (73%), anemia (72%), leucopenia (54%), linfopenia (45%), dolor musculoesquelético (43%), hipotensión (41%), fatiga (40%), aumento de transaminasas (37%), infección del tracto respiratorio superior (32%), diarrea (28%), hipocalcemia (27%), hipofosfatemia (26%), náuseas (26%), cefalea (25%), tos (25%), taquicardia (23%), escalofríos (23%), encefalopatía (22%), disminución del apetito (22%), edema (22%) e hipocalemia (20%). Se produjeron reacciones adversas graves en el 46 % de los pacientes: SLC (15%), neutropenia (6%), síndrome de neurotoxicidad asociado a células inmunoefectoras (ICANS, 4%), sepsis (3%), trombocitopenia (3%), neutropenia febril (3%) y neumonía (3%).
Mosunetuzumab (Lunsumio®) Roche
Indicación: Tratamiento de pacientes adultos con linfoma folicular (FL) en recaída o refractario que han recibido al menos dos terapias sistémicas previas.
Tipo: Autorizado condicionalmente el 3-6-2022 como medicamento huérfano. Medicamento biológico, constituido por un anticuerpo monoclonal biespecífico.
Mecanismo: Anticuerpo monoclonal biespecífico que se une simultáneamente a CD20 en los linfocitos B y a CD3 en los linfocitos T. Es un agonista condicionado, ya que la muerte celular de los linfocitos B se observa solamente bajo la unión simultánea de CD20 en los linfocitos B y CD3 en los T. La unión de ambos grupos de mosunetuzumab resulta en la formación de una sinapsis inmunológica entre un linfocito B diana y un linfocito T citotóxico que conduce a una activación de linfocitos T. La liberación subsecuente de perforina y granzimas de la activación de células T a través de la sinapsis inmunológica induce la lisis de linfocitos B.
Eficacia clínica: Ensayo abierto, multicéntrico, de multi-cohortes de fase 1/2, de monoterapia con mosunetuzumab o la combinación con atezolizumab para pacientes con linfoma folicular y leucemia linfocítica crónica. La variable principal de eficacia fue la tasa de respuesta objetiva (80%, completa: 60%), con una mediana de duración de la respuesta de 22,8 meses.
Eventos adversos: Los más comunes (≥20%) son síndrome de liberación de citocinas (SLC), neutropenia, pirexia, hipofosfatemia y cefalea. Las reacciones adversas graves más frecuentes (≥2%) son síndrome de liberación de citocinas (21%), pirexia (5%) y neumonía (3%). El 4,1% de los pacientes interrumpieron el tratamiento debido a algún efecto adverso.
Avacopan (Tavneos®) Vifor Frexenius
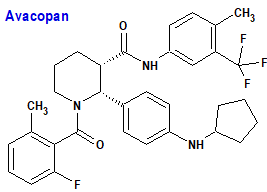
Indicación: Tratamiento de pacientes adultos con granulomatosis activa grave con poliangeítis (GPA) o poliangeítis microscópica (MPA), en combinación con un régimen de rituximab o ciclofosfamida.
Tipo: Autorizado el 11-1-2022 como medicamento huérfano; autorizado previamente en Estados Unidos (7-10-2021).
Mecanismo: Antagonista selectivo del receptor 5a del complemento humano (C5aR1, CD88) e inhibe competitivamente la interacción entre C5aR1 y la anafilatoxina C5a. El bloqueo específico y selectivo de C5aR1 por avacopan reduce los efectos proinflamatorios de C5a, que incluyen activación, migración y adherencia de neutrófilos a sitios de inflamación de vasos sanguíneos pequeños, retracción de células endoteliales vasculares y permeabilidad. En particular, avacopan bloquea la regulación al alza inducida por C5a de CD11b (integrina alfa M) en neutrófilos tomados de seres humanos que recibieron dosis de avacopan. CD11b facilita la adherencia de los neutrófilos a las superficies endoteliales vasculares, uno de los pasos en el proceso de la vasculitis.
Eficacia clínica: Ensayo multicéntrico, aleatorizado, doble ciego, doble simulación y controlado con placebo, incluyendo a 330 pacientes de 13 años o más con granulomatosis con poliangeítis (54,8 %) o poliangeítis microscópica (45,2 %) durante 52 semanas. Las variables primarias de eficacia fueron la proporción de pacientes en remisión de la enfermedad (puntuación de actividad de vasculitis de Birmingham – BVAS – de 0 y no tomar glucocorticoides para el tratamiento de la vasculitis en las 4 semanas anteriores a la semana 26); y la proporción de pacientes en remisión sostenida (remisión en la semana 26 sin recaída en la semana 52, y BVAS de 0 y sin tomar glucocorticoides para el tratamiento de vasculitis dentro de las 4 semanas anteriores a la semana 52). No hubo diferencias significativas en la remisión de la enfermedad a las 26 semanas (72,3 vs. 70,1%) pero sí las hubo a las 52 semanas (65,7 vs 54,9%).
Eventos adversos: Los más comunes son náuseas (23,5 %), cefalea (20,5 %), disminución del recuento de glóbulos blancos (18,7 %), infección del tracto respiratorio superior (14,5 %), diarrea (15,1 %), vómitos (15,1 %) y nasofaringitis (15,1%). Las reacciones adversas graves más frecuentes son alteraciones de la función hepática (5,4 %) y neumonía (4,8 %).
Inebilizumab (Uplizna®) Viela Bio
Indicación: Tratamiento de pacientes adultos con trastornos del espectro de la neuromielitis óptica (NMOSD) que son anti-acuaporina-4 inmunoglobulina G (AQP4-IgG) seropositivos.
Tipo: Autorizado el 25-4-2022; autorizado previamente en Estados Unidos (11-6-2020) como medicamento huérfano. Medicamento biológico, constituido por un anticuerpo monoclonal recombinante humanizado de tipo IgG1κ dirigido contra CD19. La línea celular de ovario de hámster chino (CHO) utilizada para la expresión de inebilizumab se diseñó para ser deficiente en fucosiltransferasa (sin capacidad para transferir fucosa) con el fin de potenciar la citotoxicidad celular dependiente de anticuerpos (ADCC) y la fagocitosis celular dependiente de anticuerpos (ADCP)
Mecanismo: Se une específicamente a CD19, un antígeno de la superficie celular presente en los linfocitos pre-B y B maduros, incluidos los plasmablastos y algunas células plasmáticas. Tras su unión en la superficie celular a los linfocitos B, inebilizumab potencia la citolisis celular dependiente de anticuerpos (ADCC) y la fagocitosis celular dependiente de anticuerpos (ADCP). Los linfocitos B parecen jugar un papel central en la patogénesis del neuromielitis óptica, donde el inebilizumab podría inducir la depleción de células B y facilitar la supresión de la secreción de anticuerpos, así como de la presentación de antígenos, la interacción de linfocitos B y T, y la producción de mediadores inflamatorios.
Eficacia clínica: Ensayo aleatorizado (3:1), doble ciego y controlado con placebo de 28 semanas de duración, que incluyó a 230 pacientes. La variable principal de eficacia fue el porcentaje de pacientes que experimentaron un ataque entre los días 1 y 197: 11,2% con inebilizumab vs. 42,3% con placebo.
Eventos adversos: Los más comunes son infección del tracto urinario (26,2%), nasofaringitis (20,9%), infección del tracto respiratorio superior (15,6%), artralgia (17,3%) y dolor de espalda (13,8%).
Anifrolumab (Saphnelo®) AstraZeneca
Indicación: Tratamiento de pacientes adultos con lupus eritematoso sistémico (LES) de moderado a grave que reciben terapia estándar
Tipo: Autorizado el 14-2-2022; autorizado previamente en Estados Unidos (30-7-2021). Medicamento biológico, constituido por un anticuerpo monoclonal tipo IgG1.
Mecanismo: Se une a la subunidad 1 del receptor de interferón tipo I (IFNAR1) con alta especificidad y afinidad, inhibiendo la señalización de IFN tipo I, bloqueando así su actividad biológica. Anifrolumab también induce la internalización de IFNAR1, lo que reduce los niveles de éste en la superficie celular disponibles para el ensamblaje del receptor. El bloqueo de la señalización de IFN de tipo I mediada por receptores inhibe la expresión génica sensible a IFN, así como los procesos inflamatorios e inmunológicos posteriores. La inhibición del IFN tipo I bloquea la diferenciación de células plasmáticas y normaliza los subconjuntos de células T periféricas. Los IFN tipo I juegan un papel en la patogenia del lupus eritematoso sistémico (LES); de hecho, el 60-80% de los pacientes adultos con LES activo expresan niveles elevados de genes inducibles por IFN tipo I.
Eficacia clínica: Tres estudios de fase 3, de 52 semanas de duración, multicéntricos, aleatorizados, doble ciego, controlados con placebo. La variable principal de eficacia del tratamiento se estableció en función de la evaluación de la respuesta clínica utilizando los criterios de valoración compuestos, la evaluación compuesta de lupus basada en el Grupo de evaluación de lupus de las Islas Británicas (BICLA) y el Índice de respuesta al LES (SRI-4). La tasa de respondedores, en diferencia de puntos porcentuales sobre el placebo osciló entre 16,3 y 28,8 (BICLA) y 6,0 y 24,0 (SRI-4).
Eventos adversos: Los más comunes son infección del tracto respiratorio superior (34 % frente al 23 % con placebo), bronquitis (11/5,2), reacciones relacionadas con la infusión (9,4/7,1), herpes zóster (6,1/1,3), tos (5,0/3,2), infección del tracto respiratorio (3,3/1,5) e hipersensibilidad (2,8/0,6).
Eptinezumab (Vyepti®) Lundbeck
Indicación: Profilaxis de la migraña en adultos que tienen al menos 4 días de migraña al mes.
Tipo: Autorizado el 24-1-2022. Medicamento biológico constituido por un anticuerpo monoclonal humanizado tipo IgG1.
Mecanismo: Se une a las formas α y β del ligando del péptido relacionado con el gen de la calcitonina humana (CGRP). Previene la activación de los receptores CGRP y, por lo tanto, la cascada posterior de eventos fisiológicos relacionados con el inicio de los ataques de migraña. Inhibe la vasodilatación y la inflamación neurogénica mediada por α y β-CGRP. Es muy selectivo (>100.000 veces) frente a otros neuropéptidos relacionados, como amilina, calcitonina, adrenomedulina e intermedina.
Eficacia clínica: Dos estudios de grupos paralelos, aleatorizados y controlados con placebo, uno en pacientes con migraña episódica (n=888) y otro en pacientes con migraña crónica (n=1072); todos ellos tenían antecedentes de migraña (con o sin aura) de al menos 12 meses. La variable principal de eficacia fue el cambio desde el inicio en la media mensual de días con migraña (DMM) durante las semanas 1 a 12, en relación al placebo. En los pacientes con migraña episódica fue de -0,7 (dosis de 100 mg) y -1,1(300 mg), mientras que en los pacientes con migraña crónica fue de -2,0 (100 mg) y -2,6 (300 mg).
Eventos adversos: Los más comunes son nasofaringitis (8%), reacciones de hipersensibilidad (4%): angioedema, urticaria, sofocos, erupciones cutáneas, prurito, etc.
Glucarpidasa (Voraxaze®) SERB SAS
Indicación: Para reducir la concentración plasmática tóxica de metotrexato (más de 1 micromol por litro) en pacientes adultos y pediátricos con aclaramiento de metotrexato retardado (concentraciones de metotrexato en plasma superiores a 2 desviaciones estándar de la curva de excreción media de metotrexato específica para la dosis de metotrexato administrada) debido una deficiente función renal.
Tipo: Autorizado el 11-1-2022 como medicamento huérfano y en circunstancias excepcionales. Autorizado previamente en Estados Unidos (17-1-2012). Medicamento biológico constituido por una proteína homodímera de 390 aminoácidos con un peso molecular de 83 kDa. Cada unidad de potencia corresponde a la escisión enzimática de 1 μmol/L de metotrexato por minuto a 37ºC.
Mecanismo: Carboxipeptidasa que hidroliza selectivamente el residuo carboxilo del glutamato terminal del ácido fólico y de los fármacos antifolatos clásicos como el metotrexato. Transforma a este último en sus metabolitos ácido 4-desoxi-4-amino-N10-metilpteroico (DAMPA) y glutamato, ambos inactivos. El tratamiento proporciona una vía alternativa no renal para la eliminación del metotrexato en pacientes con disfunción renal durante el tratamiento con dosis elevadas de metotrexato.
Eficacia clínica: Cuatro estudios abiertos, de uso compasivo y de un solo grupo, en pacientes con eliminación limitada de metotrexato (MTX) debido a disfunción renal. La variable principal de eficacia fue la “reducción clínicamente importante” (RCI) en la concentración plasmática de MTX (≤1 μmol/L tras la primera dosis de glucarpidasa). El 61,5 % de los pacientes de la población central con MTX logró una RCI que se mantuvo durante un máximo de 8 días. Se produjo una reducción mediana de más del 98% en la concentración de MTX dentro de los 15 minutos posteriores a la administración de glucarpidasa. El rebote (definido como un aumento de la concentración de MTX de al menos 1 μmol/L y al menos dos veces el nadir posterior a la glucarpidasa) se produjo en el 19,4 % de los pacientes de la población central de MTX.
Eventos adversos: Los más comunes (>1%) son parestesia, sofocos y náuseas y/o vómitos.
2. FOOD & DRUG ADMINISTRATION (FDA)
Tirzepatida (Mounjaro®) Eli Lilly
Indicación: Para mejorar el control glucémico en adultos con diabetes mellitus tipo 2, junto con dieta y ejercicio físico.
Tipo: Autorizado el 13-5-2022; no autorizado aún en la Unión Europea. Péptido de 39 aminoácidos, modificado de la secuencia del GIP natural (glucose-dependent insulinotropic peptide; polipéptido insulinotrópico dependiente de glucosa), incluyendo dos restos de ácido aminoisobutírico en las posiciones 2 y 13, una amida C-terminal y, además, la lisina de la posición 20 de la cadena peptídica está unida al ácido 1,20-eicosanodioico a través de una cadena enlazadora, con el objetivo de facilitar su unión a la albúmina plasmática y, con ello, incrementar su semivida de elminación.
Mecanismo: Agonista del receptor de GIP y del receptor de GLP-1 (glucagon-like peptide 1; péptido similar al glucagón tipo 1). Incrementa la secreción de insulina y reduce los niveles de glucagón, ambos de forma dependiente de la glucemia.
Eficacia clínica: Ensayo aleatorizado, doble ciego y controlado con placebo de 40 semanas de duración, que incluyó a 478 pacientes. La variable principal de eficacia fue la tasa de reducción de los niveles de hemoglobina glicosilada (HbA1c) en la semana 40: 1,7-1,8 vs 0,1 con placebo; además, el porcentaje de pacientes que presentaron valores de HbA1c <7% fue de 78-85 vs. 23%.
Eventos adversos: Los más comunes son gastrointestinales, principalmente náusea (12-18% según la dosis vs. 4% con placebo), diarrea (12-17 vs 9%), pérdida de apetito (5-11 vs. 1%), vómitos (5-9 vs. 2%), estreñimiento (6-7 vs. 1%), dispepsia (5-8 vs. 3%). El 3,0-6,6% de los pacientes tratados con tirzepatida interrumpieron el tratamiento debido a reacciones adversas gastrointestinales, frente al 0,4% que recibieron placebo.
Vonoprazan/Amoxicilina/Claritromicina (Voquezna Triple Pak®) Phathom
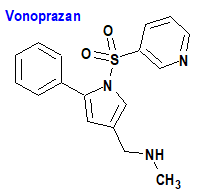
Indicación: Tratamiento de la infección por Helicobacter pylori (H. pylori) en adultos.
Tipo: Autorizado el 3-5-2022; no autorizado aún en la Unión Europea. Combinación de un agente antisecretor gástrico (vonoprazan) con un antibacteriano (amoxicilina; Voquezna Dual Pak®) o dos (amoxicilina/claritromicina: Voquezna Triple Pak®).
Mecanismo: El vonoprazan es un inhibidor del sistema enzimático (bomba) hidronio/potasio (H3O+/K+-ATPasa) de manera competitiva con el potasio, que se traduce en la supresión de la secreción basal y estimulada de ácido gástrico en la superficie secretora de la célula parietal gástrica. Vonoprazan se concentra selectivamente en las células parietales tanto en estado de reposo como estimulado, uniéndose a la bomba de hidronio/potasio activa de forma no covalente y reversible. La amoxicilina y la claritromicina son antibacterianos de tipo betalactámico y macrólido, respectivamente.
Eficacia clínica: Ensayo aleatorizado, doble ciego y controlado con lansoprazol/amoxicilina/claritromicina (LAC) de 2 semanas de duración, que incluyó a 749 pacientes. La variable principal de eficacia fue la tasa de erradicación de H. pylori confirmada mediante con una prueba negativa de aliento con urea(13C) ≥27 días después de la terapia: 80,8% (Triple Pak), 77,2% (Dual Pak) y 68,5% (LAC).
Eventos adversos: Los más comunes (2-5%) son diarrea, desgeusia, candidiasis vulvovaginal, dolor abdominal, cefalea e hipertensión. Un 2,3% de los pacientes suspendieron el tratamiento debido a alguna reacción adversa.
Mavacamten (Camzyos®) Myokardia
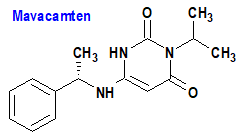
Indicación: Tratamiento de adultos con miocardiopatía hipertrófica obstructiva (MHC) clase II-III sintomática de la New York Heart Association (NYHA), para mejorar la capacidad funcional y los síntomas.
Tipo: Autorizado el 28-4-2022; no autorizado aún en la Unión Europea. Mecanismo: Inhibidor alostérico y reversible selectivo de la miosina cardíaca. Modula la cantidad de cabezas de miosina que pueden entrar en estados «en actina» (generadores de energía), lo que reduce la probabilidad de formación de puentes cruzados que producen fuerza (sistólica) y residual (diastólica). El exceso de formación de puentes cruzados de actina y miosina y la desregulación del estado súper relajado son características de la MHC. Mavacamten cambia la población general de miosina hacia un estado de ahorro de energía, reclutable y súper relajado. En pacientes con MHC, la inhibición de la miosina con mavacamten reduce la obstrucción dinámica del tracto de salida del ventrículo izquierdo y mejora las presiones de llenado cardíaco.
Eficacia clínica: Ensayo de fase 3, doble ciego, aleatorizado, controlado con placebo, multicéntrico, internacional, de grupos paralelos en 251 adultos con MCH obstructiva clase II y III de la NYHA sintomática,con fracción de eyección del ventrículo izquierdo (FEVI≥55%). La variable principal de eficacia, evaluada a las 30 semanas, fue la proporción de pacientes que lograron una mejora de la tensión venosa mixta de oxígeno (pVO2) en ≥1,5 ml/kg/min más una mejora en la clase NYHA de al menos 1 (33% con mavacamten vs. 18% con placebo) o una mejora de pVO2 por ≥3.0 ml/kg/min más sin empeoramiento en la clase NYHA (23% vs. 11%).
Eventos adversos: Los más comunes son mareos (27% vs. 18% con placebo) y síncope (6% vs. 2%).
Tapinarof (Vtama®) Dermavant
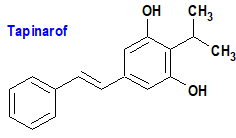
Indicación: Tratamiento tópico de la psoriasis en placas en adultos.
Tipo: Autorizado el 23-5-2022; no autorizado aún en la Unión Europea.
Mecanismo: El tapinarof se une al receptor de hidrocarburos de arilo (AhR), cuya activación conduce en pacientes con psoriasis a la regulación a la baja de las citocinas proinflamatorias, incluida la interleucina 17 (IL-17), y a la regulación de la expresión de la filagrina (proteína estructural fundamental para el desarrollo y mantenimiento de la barrera cutánea), promoviendo la normalización de esta última. El receptor de hidrocarburos de arilo (AhR) es un factor de transcripción citoplasmático dependiente de ligandos que pertenece a la familia de los factores de transcripción con dominio bHLH-PAS (basic helix loop helix-PER ARNT-SIM), que desempeñan un papel importante en el desarrollo celular y la adaptación a la hipoxia, así como en la regulación de la inmunidad y la diferenciación celular.
Eficacia clínica: Dos ensayos clínicos aleatorizados, doble ciego y controlados con placebo de 12 semanas de duración, incluyendo a 1025 pacientes. La variable principal de eficacia fue la proporción de sujetos que lograron el éxito del tratamiento, definido como una puntuación PGA (Physician’s Global Assesment) de «Claro» (0) o «Casi claro» (1), junto con una mejora de al menos 2 grados desde el inicio: 36-40% (tapinarof) vs. 6% (placebo).
Eventos adversos: Los más comunes son foliculitis (20%), nasofaringitis (11%), dermatitis de contacto (7%) y cefalea (7%).
Oteseconazol (Vivjoa®) Mycovia
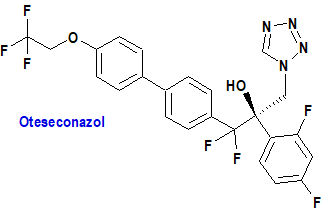
Indicación: Para reducir la incidencia de candidiasis vulvovaginal recurrente (RVVC) en mujeres con antecedentes de RVVC que NO tienen potencial reproductivo.
Tipo: Autorizado el 26-4-2022; no autorizado aún en la Unión Europea.
Mecanismo: Es un derivado azólico inhibidor de la 14α desmetilasa (CYP51), enzima que cataliza un paso inicial en la vía biosintética del ergosterol, un esterol necesario para la formación e integridad de la membrana celular fúngica. La inhibición de CYP51 da como resultado la acumulación de esteroles 14-metilados, algunos de los cuales son tóxicos para los hongos patógenos sensibles.
Eficacia clínica: Dos ensayos clínicos aleatorios y controlados con placebo, que constaron de una fase de inducción abierta y otra de mantenimiento de 11 semanas. La variable principal de eficacia fue la proporción de pacientes con ≥1 episodio agudo verificado por cultivo (positivo para especies de Candida asociado con una puntuación de signos y síntomas clínicos de ≥3) durante la fase de mantenimiento hasta la semana 48: 3,9-6,7% con oteseconazol vs. 49,7-50,8% con placebo. Un tercer ensayo aleatorizado y doble ciego evaluó la eficacia y la seguridad frente a fluconazol y placebo en adultos y mujeres pediátricas posmenárquicas; la proporción de pacientes con ≥1 episodio agudo verificado por cultivo hasta la semana 50 o no resuelto durante la fase de inducción fue del 10,3% con oteseconazol vs. 42,9% con fluconazol/placebo; asimismo, la proporcion de pacientes con ≥1 episodio agudo verificado por cultivo o que tomaron medicación hasta la semana 50 o episodio no resuelto durante la fase de inducción fue del 43,5% (oteseconazol) vs. 59,0%(fluconazol/placebo).
Eventos adversos: Los más comunes son cefalea (7,4 %) y náuseas (3,6 %).
Relatlimab/Nivolumab (Opdualag®) Bristol Myers Squibb
Indicación: Tratamiento de pacientes adultos y pediátricos de 12 años de edad o mayores con melanoma no resecable o metastásico
Tipo: Autorizado el 18-3-2022 como medicamento huérfano; no autorizado aún en la Unión Europea. Combinación de dos anticuerpos monoclonales humanos de tipo IgG4κ.
Mecanismo: Relatlimab es un anticuerpo bloqueador del gen 3 de activación de linfocitos (LAG-3) al que se une bloqueando la interacción con sus ligandos, incluido el MHC II, y reduce la inhibición de la respuesta inmunitaria mediada por la vía LAG-3; el antagonismo de esta vía promueve la proliferación de linfocitos T y la secreción de citocinas. Nivolumab se une al receptor PD-1, bloqueando la interacción con sus ligandos PD-L1 y PD-L2 y reduce la inhibición de la respuesta inmunitaria mediada por la vía PD-1 (inhibe la proliferación de linfocitos T y la producción de citocinas), incluida la respuesta inmunitaria antitumoral; la regulación al alza de los ligandos de PD-1 se produce en algunos tumores, y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmunitaria activa antitumoral de los linfocitos T. La combinación de nivolumab (anti-PD-1) y relatlimab (anti-LAG-3) produce una mayor activación de las células T en comparación con la actividad de cualquiera de los anticuerpos por separado, inhibiendo el crecimiento tumoral y promoviendo la regresión tumoral.
Eficacia clínica: Ensayo aleatorizado y doble ciego en 714 pacientes con melanoma en estadio III o IV metastásico o irresecable no tratado previamente, comparando la combinación de relatlimab con nivolumab vs. nivolumab solo. La variable principal de eficacia fue la supervivencia libre de progresión determinada por una revisión central independiente ciega (BICR) utilizando los criterios de evaluación de respuesta en tumores sólidos (RECIST v 1.1): 10,1 vs 4,6 meses (mediana).
Eventos adversos: Graves en el 36 % de los pacientes; los más frecuentes son insuficiencia suprarrenal (1,4 %), anemia (1,4 %), colitis (1,4 %), neumonía (1,4 %), infarto agudo de miocardio (1,1 %), dolor de espalda (1,1%), diarrea (1,1%), miocarditis (1,1%) y neumonitis (1,1%). El tratamiento se suspendió permanentemente debido a reacciones adversas en el 18 % de los pacientes, principalmente por miocarditis (1,7 %) y neumonitis (1,4 %). Se produjeron interrupciones de la dosis debido a una reacción adversa en el 43%.
Pacritinib (Vonjo®) CTI BioPharma
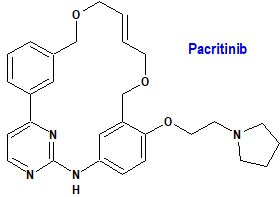
Indicación: Tratamiento de adultos con mielofibrosis primaria o secundaria (pospolicitemia vera o postrombocitemia esencial) de riesgo intermedio o alto con un recuento de plaquetas por debajo de 50 × 109/L.
Tipo: Autorizado el 28-2-2022 como medicamento huérfano; no autorizado aún en la Unión Europea.
Mecanismo: Es un inhibidor de la cinasa Jano 2 (JAK2) de tipo salvaje y del mutante JAK2V617F, así como de la tirosina cinasa 3 similar a FMS (FLT3), que contribuyen a la señalización de una serie de citocinas y factores de crecimiento que son importantes para la hematopoyesis y la función inmunológica. La mutación JAK2V617F aparece fundamentalmente en las neoplasias mieloproliferativas BCR-ABL negativas, como la policitemia vera (presente en el 90-100% de los casos), la trombocitemia esencial (50-60%) y la mielofibrosis primaria (60-70%). El FLT3 es un receptor de tirosina cinasa tipo III expresado en las células hematopoyéticas inmaduras que actúa como mediador clave de la hematopoyesis temprana.
Eficacia clínica: Ensayo multicéntrico, aleatorizado y controlado con el mejor tratamiento disponible (MTD). El porcentaje de pacientes que lograron una reducción de ≥35 % en el volumen del bazo desde el inicio hasta la semana 24 fue del 29,0% con pacritinib vs. 3,1% con MTD.
Eventos adversos: Los más comunes (>20%) son diarrea, trombocitopenia, náusea, anemia y edema periférico. Se suspendió suspendió el tratamiento en el 15%, interrumpiéndose en el 27%. Un 12% tuvieron que reducir la dosis.
Ganaxolona (Ztalmy®) Marinus
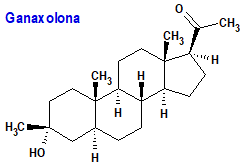
Indicación: Tratamiento de convulsiones asociadas con el trastorno por deficiencia CDKL5 (Cyclin-Dependent Kinase-Like 5) en pacientes de 2 años de edad y mayores.
Tipo: Autorizado el 18-3-2022 como medicamento huérfano; no autorizado aún en la Unión Europea.
Mecanismo: Es un análogo de la alopregnanolona, un neuroesteroide derivado de la progesterona. Su efecto anticonvulsivante se deben a la modulación alostérica positiva del receptor del ácido gamma-aminobutírico tipo A (GABAA) en el sistema nervioso central.
Eficacia clínica: Estudio doble ciego, aleatorizado y controlado con placebo en pacientes de 2 a 19 años de edad (N=101) con confirmación molecular de una mutación patogénica en el gen CDKL5, convulsiones no controladas con al menos 2 regímenes previos y un mínimo de 16 convulsiones motoras al mes. La variable principal de eficacia fue la reducción en la frecuencia mensual de convulsiones motoras graves: 31 vs. 7% (placebo).
Eventos adversos: Los más comunes (≥10%) son infección del tracto respiratorio, infección viral, diarrea, dispepsia, tos, artralgia, artritis y edema periférico. Graves (13%): sepsis estreptocócica e infección de herida estafilocócica, artralgia e infección del tracto respiratorio; ninguna condujo a la interrupción del tratamiento.
Lutecio (177Lu) Vipivotida Tetraxetano (Pluvicto®) Advanced Accelerator Applications
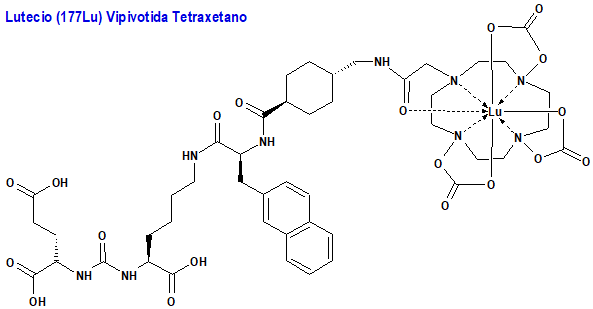
Indicación: Tratamiento de pacientes adultos con cáncer de próstata metastásico resistente a la castración (CPRCm) positivo para el antígeno de membrana específico de la próstata (PSMA) que han sido tratados con inhibición de la vía del receptor de andrógenos (AR) y quimioterapia basada en taxanos.
Tipo: Autorizado el 23-3-2022; no autorizado aún en la Unión Europea. Radiofármaco que contiene un ligando específico del PSMA unido a un agente quelante del lutecio-177, el cual se desintegra a hafnio-177 (177Hf) estable, con una vida media de 6,647 días al emitir radiación beta (β) con una energía máxima de 0,498 MeV (79%) y radiación gamma (γ) de 0,208 MeV (11%) y 0,113 MeV (6,4%). La fracción vipivotida es un ligando selectivo para la PSMA y está unida al tetraxetano (DOTA), que actúa como agente quelante del radionúclido.
Mecanismo: La fracción activa del fármaco es el radionúclido lutecio-177 (177Lu), que está unido a una fracción que se une a PSMA, una proteína transmembrana que se expresa en el cáncer de próstata. La emisión beta (β) por el lutecio-177 irradia localmente a las células tumorales que expresan PSMA, junto con el resto de células circundantes, induciendo daños en el ADN que pueden conducir a la muerte celular.
Eficacia clínica: Ensayo aleatorizado, multicéntrico y abierto que evaluó el fármaco más el mejor tratamiento estándar (BsoC) (N=551) vs. BSoC solo (N=280) en varones con CPRCm PSMA positivo progresivo. La variable principal de eficacia fue la supervivencia global (mediana): 15,3 vs. 11,3 meses; asimismo, la tasa de respuesta global fue del 30 vs 2%.
Eventos adversos: Los más comunes (≥20%) son fatiga, sequedad de boca, náuseas, anemia, disminución del apetito y estreñimiento. Los más frecuentes que dan lugar a la interrupción son anemia (5%) y trombocitopenia (3,6%).