1. INTRODUCCIÓN
Las enfermedades cardiovasculares (ECV) son la principal causa de mortalidad en todo el mundo (1). Tanto la hipertensión como la obesidad son los factores de riesgo más importantes para desarrollar un futuro evento cardiovascular. Además, particularmente la hipertensión, es un factor de riesgo importante para el desarrollo de los aneurismas.
La hipertensión es una patología crónica caracterizada por niveles elevados de presión arterial continuados en el tiempo, lo que aumenta significativamente el riesgo de desarrollar ECV. Se calcula que 1.130 millones de personas en todo el mundo padecen hipertensión, sin embargo, menos del 20% de ellos tiene la presión arterial controlada. Además, la hipertensión se asocia con altas tasas de morbilidad y mortalidad y es la principal causa de muerte prematura en todo el mundo (1). La etiología de esta enfermedad no está del todo clara, sin embargo, se sabe que la hipertensión es una enfermedad multifactorial que resulta de la interacción entre factores genéticos, fisiológicos y ambientales (2).
La hipertensión puede ser primaria o secundaria. En la hipertensión primaria o esencial no hay una causa específica que explique el aumento de la presión arterial. La hipertensión esencial representa alrededor del 90-95% del total (3, 4). El término esencial proviene de la creencia inicial de que el aumento de la presión arterial era esencial para mantener una perfusión tisular suficiente (5). Una de las principales características de la hipertensión esencial es el aumento de la resistencia periférica (6), que se explica con más detalle a continuación. En la hipertensión secundaria existe una causa específica de elevación de la presión arterial (malformación de los vasos sanguíneos, tumores secretores de esteroides o catecolaminas en la corteza suprarrenal, coartación de la aorta, enfermedad renal o problemas endocrinos como el aldosteronismo primario) (5).
Los valores óptimos de presión arterial para un adulto son <120 mm Hg para la presión arterial sistólica (PAS) y <80 mm Hg para la presión arterial diastólica (PAD) (120/80). La hipertensión se refiere a una PAS igual o superior a 140 mm Hg y a una PAD igual o superior a 90 mm Hg, aunque estos límites pueden variar en función de la edad o de la presencia de otros problemas concomitantes como la enfermedad renal o la diabetes. Los valores entre 120/80 mm Hg y 129/84 mm Hg se consideran «normales», mientras que los valores entre 130/85 mm Hg y 139/89 mm Hg se conocen como «normal-alto». Sin embargo, en un estudio publicado recientemente en JAMA con datos de más de 8 millones de personas en el mundo, PAS por encima de 110 mm Hg, aumenta el riesgo se sufrir eventos cardiovasculares, y la tendencia observada en todo el mundo es que cada vez hay un mayor número de personas con PAS por encima de 110 mm Hg, incrementándose a su vez tanto el número de eventos cardiovasculares, así como las muertes asociadas a ellos, siendo la cardiopatía isquémica y el accidente cerebrovascular los eventos mayoritarios (7).
La obesidad es también un importante problema de salud en todo el mundo, con importantes consecuencias sobre la morbilidad y la mortalidad (8). De hecho, está bien aceptado que el riesgo de ECV aumenta con el índice de masa corporal (IMC, en kg/m2) (9). Un IMC superior o igual a 25 indica sobrepeso, mientras que un IMC superior o igual a 30 puntos define la obesidad. En el caso de los niños, hay que tener en cuenta la edad a la hora de definir el sobrepeso y la obesidad.
La acumulación excesiva de grasa en el cuerpo se conoce como aumento de la adiposidad. Según la Organización Mundial de la Salud, en 2016, el 13% de los adultos eran obesos, y el 39% de los adultos tenían sobrepeso con una alta probabilidad de desarrollar obesidad o al menos de sufrir los daños causados por un exceso de grasa (9). La obesidad infantil también sigue siendo motivo de gran preocupación, ya que 38 millones de niños menores de 5 años tenían sobrepeso u obesidad en 2019 y su prevalencia ha aumentado drásticamente del 4% en 1975 a más del 18% (9). Los factores relacionados con el estilo de vida son los que más contribuyen, pero cada vez se conocen mejor las vías genómicas y metabólicas implicadas en la obesidad (8) y en las complicaciones derivadas de ella (10).
Tanto la hipertensión como la obesidad y la ECV tienen cierto grado de trasfondo genético. Comparten algunos polimorfismos dentro de los genes que potencian la expresión de proteínas que influyen en la ECV. Hay al menos 5 genes (FTO, ADCY3, BDNF, MC4R, TBX15-WARS2) con diferentes variantes genéticas que pertenecen a la obesidad y la hipertensión al mismo tiempo (10). Entre otras funciones, estos genes están implicados en la regulación de la masa grasa y los niveles de insulina, la adipogénesis y la homeostasis energética. Además, otras enfermedades que comparten algunos polimorfismos con la obesidad y la hipertensión son la diabetes tipo 2 y la dislipidemia.
El aneurisma aórtico (AA) es una patología vascular potencialmente letal que se caracteriza por una dilatación permanente de la aorta de más de 3 cm (11). Sin tratamiento, la pared del vaso sigue debilitándose y puede llegar a ser incapaz de compensar la presión ejercida por la sangre intraluminal, produciéndose la rotura de la aorta. Debido a la falta de síntomas clínicos, la muerte por rotura de la aorta es frecuente y se produce en aproximadamente el 80% de los pacientes que padecen AA (11, 12). Los aneurismas pueden ser AA abdominales (AAA) o AA torácicos (TAA) (13). La aorta, independientemente de la localización, depende de las capas fibromusculares (unidades laminares) para distribuir la tensión y proporcionar elasticidad. La capa media de la aorta torácica está compuesta por unas 60 unidades laminares, mientras que la aorta abdominal consta de 30 unidades laminares (14). Esta diferencia en el número de unidades laminares es, probablemente, una de las causas que hacen que la aorta abdominal sea más propensa a la degeneración aneurismática (13).
La etiología del AAA sigue siendo objeto de investigación, pero algunas causas incluyen trastornos inflamatorios, infecciones y traumatismos. Los factores de riesgo asociados a los AAA incluyen la hipertensión, la dislipidemia, los antecedentes familiares de AAA, la enfermedad arterial periférica, la edad avanzada, el sexo masculino y el tabaquismo (11, 12). La fisiopatología de esta enfermedad está relacionada con una lesión arterial inicial que provoca una cascada inflamatoria y una alteración de las proteínas de la matriz extracelular (MEC) que conduce al debilitamiento de la pared arterial (12). Sin embargo, los AAT suelen ser el resultado de mutaciones genéticas en enfermedades raras y procesos degenerativos como el síndrome de Marfan (15). Cuando se identifican de forma precoz, los aneurismas se controlan en función del tamaño, la tasa de crecimiento y la presencia de propiedades biomecánicas aberrantes del saco aneurismático, factores con alta probabilidad de aumentar el riesgo de rotura (12).
A continuación, revisaremos diferentes conceptos de la función, la estructura y las propiedades mecánicas arteriales, y cómo la hipertensión, la obesidad y los AAA influyen en estos parámetros. En las últimas décadas, se ha hecho evidente la relación entre la hipertensión, la obesidad, los AAA y la inflamación. Revisaremos el papel del sistema inmune y de algunas citoquinas específicas como el interferónγ (IFNγ) en las alteraciones vasculares asociadas a estas patologías. En este sentido, nos centraremos en un gen estimulado por el IFN (ISG15) y en la modificación postraduccional que produce esta proteína (ISGilación). Por último, discutiremos brevemente los aspectos farmacológicos para el tratamiento de la hipertensión, la obesidad y los aneurismas.
2. ESTRUCTURA Y FUNCIÓN VASCULAR
2.1 Remodelado vascular
El remodelado vascular se refiere al proceso que se produce cuando la pared vascular cambia su estructura para mantener una resistencia vascular adecuada. El remodelado suele ser un proceso adaptativo que se produce en respuesta a cambios a largo plazo en las condiciones hemodinámicas, pero también puede contribuir a la fisiopatología de las enfermedades vasculares y los trastornos circulatorios (16). Así, se observa un remodelado vascular fisiológico durante el embarazo o el envejecimiento y en muchas situaciones patológicas, como la hipertensión, la obesidad o los aneurismas. El remodelado vascular es un proceso activo que implica cambios en el crecimiento, la muerte o la migración de las células y en la síntesis, la reorganización o la degradación de la MEC (17).
En términos generales, el remodelado vascular puede clasificarse como remodelado hipertrófico, que se asocia a un crecimiento vascular, remodelado hipotrófico, con disminución del área de la pared del vaso, o remodelado eutrófico, en el que no se observan cambios en la cantidad de material de la pared vascular. Además, el remodelado puede ser hacia adentro (inward) cuando se asocia con diámetros menores, mientras que el remodelado hacia afuera (outward) o compensado se asocian con un tamaño mayor o similar del vaso, respectivamente (18,19) (Figura 1).
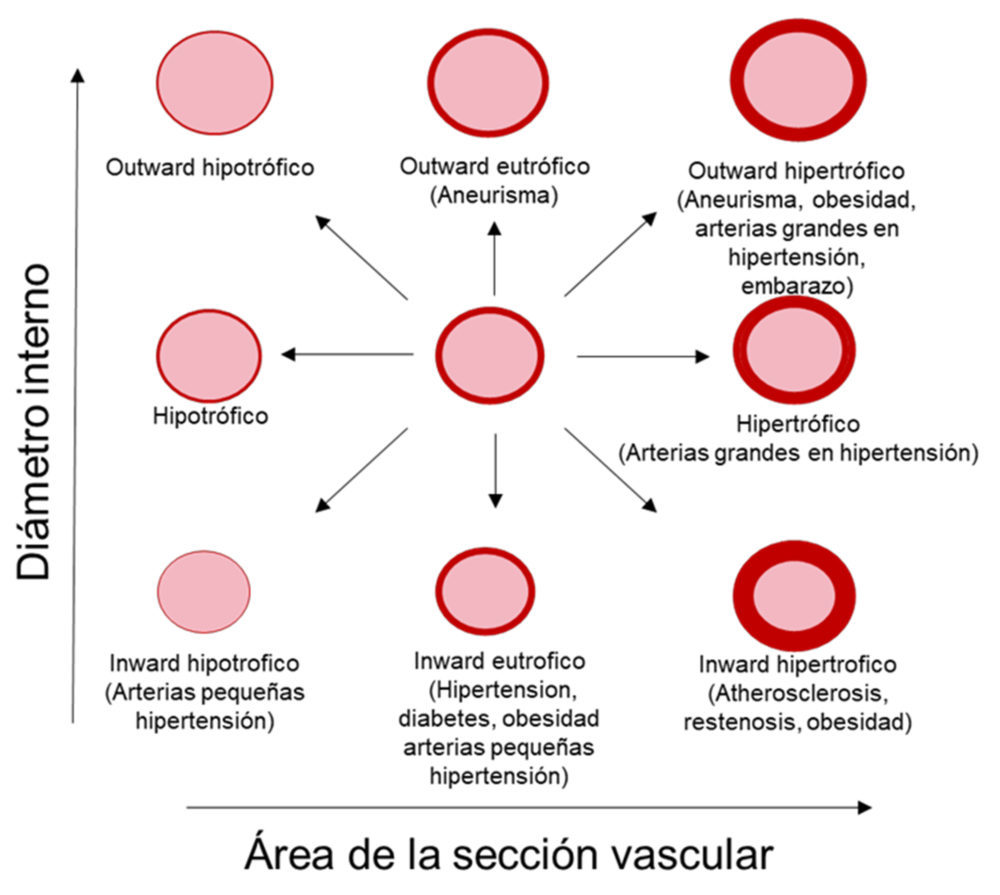
Figura 1. Tipos de remodelado vascular. Modificada de (18).
Más concretamente:
- Remodelado hipertrófico: generalmente se asocia con un aumento del grosor de la media debido a cambios en el crecimiento o en el número de células. Al mismo tiempo, aumentan la relación media/lumen y el área de la sección transversal vascular (CSA) (18). Este tipo de remodelado es característico de las arterias elásticas con el envejecimiento y la hipertensión (20), la obesidad (21), la aterosclerosis y la reestenosis (17) y predomina en la hipertensión secundaria (22).
- Remodelado hipotrófico: se caracteriza por una disminución de la cantidad de material de la pared vascular (menor CSA) (18). El remodelado hipotrófico puede producirse debido a apoptosis o a una reorganización del material de la pared vascular (23). Se ha observado en las arteriolas aferentes renales de ratas espontáneamente hipertensas (SHR) (24) y en las arterias mesentéricas de resistencia de ratas hipertensas inducidas por ouabaína (25).
- Remodelado eutrófico: se asocia a una disminución de los diámetros externo e interno y a un aumento del grosor de la media y de la relación media/lumen sin cambios en el CSA (18). Los procesos subyacentes a este tipo de remodelado son menos conocidos, pero se ha sugerido una combinación de crecimiento de la pared vascular hacia el interior y apoptosis en la zona periférica.
Asimismo, el remodelado eutrófico puede ser la consecuencia de una vasoconstricción prolongada de las células vasculares una MEC expandida (20, 26). Las arterias de resistencia de pacientes con hipertensión esencial, los modelos genéticos de hipertensión en ratas como la SHR o los modelos de infusión de angiotensina II (Ang II) en ratones presentan un remodelado inward predominantemente eutrófico, aunque estos últimos también pueden asociarse a un remodelado hipertrófico (27). Además, algunas arterias de modelos animales diabéticos u obesos también pueden mostrar este tipo de remodelado (21).
Asimismo, el remodelado outward se produce en los aneurismas, en las arterias de conductancia durante la hipertensión y en vasos de algunos modelos de obesidad, mientras que el remodelado inward puede observarse en la obesidad, la aterosclerosis, la reestenosis o las arterias de resistencia en la hipertensión (17, 21, 28) (Figura 1).
2.2. Rigidez vascular
Los componentes de las arterias que participan en la mayor parte de sus propiedades mecánicas son el colágeno y la elastina, que se encuentran depositados en la capa media (29). A la presión sanguínea fisiológica, depositados menos del 10% de las fibras de colágeno están comprometidas, mientras que, a presiones más altas, el vaso se vuelve progresivamente menos distensible a medida que las fibras de colágeno se reclutan para soportar la tensión pasiva de la pared y restringir la distensión aórtica. Con aumentos adicionales en la tensión de la pared, hay poco cambio en el radio a medida que se reclutan más fibras de colágeno, lo que explica la naturaleza no lineal de la elasticidad vascular (29).
El estrés vascular es la fuerza aplicada a una arteria dividida por la superficie sobre la que se aplica dicha fuerza. El estrés de rozamiento procedente del flujo sanguíneo, el estrés longitudinal procedente del tejido circundante y el estrés circunferencial procedente de la presión sanguínea son los responsables del estrés ejercido sobre la pared del vaso (Figura 2). A su vez, las fuerzas que generan estrés también pueden inducir deformaciones en la pared del vaso, convirtiéndose en tensión vascular. La tensión relaciona las dimensiones deformadas de un objeto con respecto a sus dimensiones no deformadas y es, por tanto, una medida de la deformación independientemente de la geometría original (29). De forma que la relación estrés-tensión es una medida de la rigidez vascular.
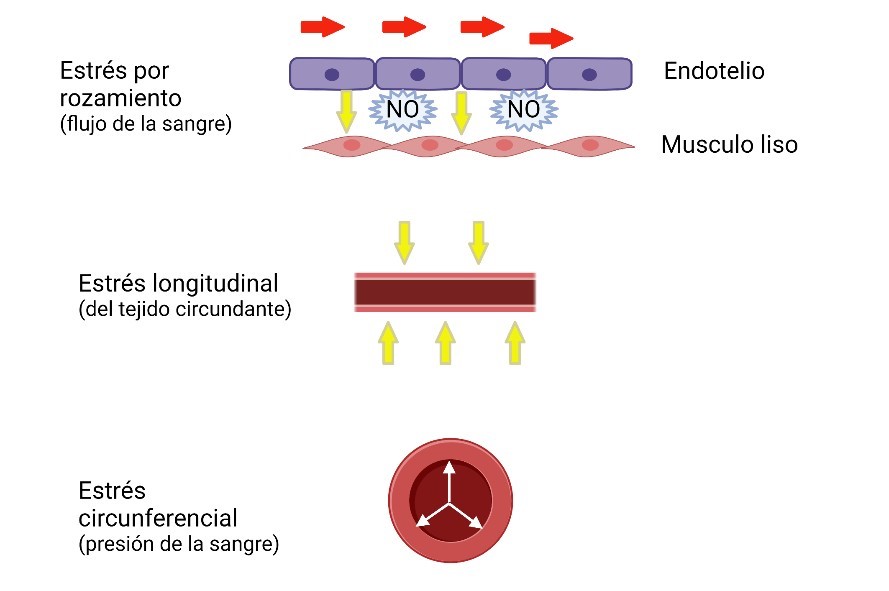
Figura 2. Principales responsables de las tensiones ejercidas sobre la pared del vaso sanguíneo
La rigidez arterial modifica las propiedades de la pared de los vasos. La rigidez arterial tiene un valor predictivo independiente para la morbilidad y la mortalidad cardiovascular y es responsable, al menos en parte, del remodelado vascular en la hipertensión (27, 30). El aumento de la rigidez de la vasculatura de resistencia contribuye, al menos en parte, al aumento de la resistencia periférica observado en la hipertensión (27). Los cambios en la rigidez arterial están determinados por alteraciones cuantitativas y cualitativas en la MEC vascular debido a factores hemodinámicos, genéticos o humorales (27, 31).
2.3. Disfunción endotelial
Un endotelio sano en reposo libera continuamente potentes vasodilatadores en respuesta al flujo sanguíneo (32). La función endotelial se considera un «barómetro» de la salud cardiovascular y es útil en la evaluación de la enfermedad y en el manejo terapéutico del paciente. Los factores de riesgo cardiovascular, pero también varias enfermedades inmunometabólicas y neuroendocrinas, cambian el fenotipo endotelial de saludable hacia la disfunción endotelial causante de enfermedad (33). La disfunción endotelial se asocia con un desequilibrio en la biodisponibilidad de las sustancias activas, lo que da lugar a una vasodilatación dependiente de endotelio atenuada y a una activación inflamatoria endotelial (34), ambos procesos relacionados con el remodelado y la rigidez vascular (32, 35).
Los mecanismos moleculares que subyacen a la alteración de la homeostasis endotelial siguen sin estar claros (36). La disfunción de las células endoteliales se considera un evento temprano antes del desarrollo de enfermedades vasculares graves como la aterosclerosis, la trombosis o las fugas vasculares en los capilares (37, 38). Cuando la disfunción endotelial aparece, suele considerarse una condición sistémica (39), que afecta tanto a la vasculatura periférica como a las arterias coronarias. Pero, aunque está bien aceptado que la disfunción endotelial es un factor predictivo del desarrollo de la aterosclerosis y que se observa tempranamente en la obesidad, su papel en la elevación de la presión arterial es menos conocido. En este sentido, la contribución exacta de la liberación atenuada de sustancias vasodilatadoras al desarrollo de la hipertensión no se ha establecido del todo, y no podemos olvidar que los factores metabólicos y nerviosos locales, así como los mecanismos renales y centrales, pueden controlar directamente la presión arterial.
3. REGULACIÓN DEL TONO VASCULAR
Diversas moléculas regulan el tono vascular actuando tanto como señales endocrinas o paracrinas. Los principales factores vasodilatadores son el óxido nítrico (NO), la prostaglandina I2 (PGI2) y la prostaglandina E2 (PGE2), el factor hiperpolarizante derivado del endotelio (EDHF) y algunas especies reactivas del oxígeno (ROS). Los principales factores vasoconstrictores son el tromboxano A2 (TXA2) y otros prostanoides, la endotelina-1 (ET-1), algunos componentes del sistema renina-angiotensina-aldosterona (SRAA) y algunas ROS. La mayoría de estos factores pueden ser generados por las tres capas de la pared vascular. En concreto, el endotelio vascular actúa como fuente de varios mediadores químicos potentes (Figura 3), que en su mayoría inhiben la contracción del músculo liso adyacente y evitan la adhesión excesiva de leucocitos, el crecimiento de las células musculares lisas vasculares (CMLV) y la agregación de plaquetas. Muchos otros tipos de células pueden producir estos mediadores y afectar al tono vascular.
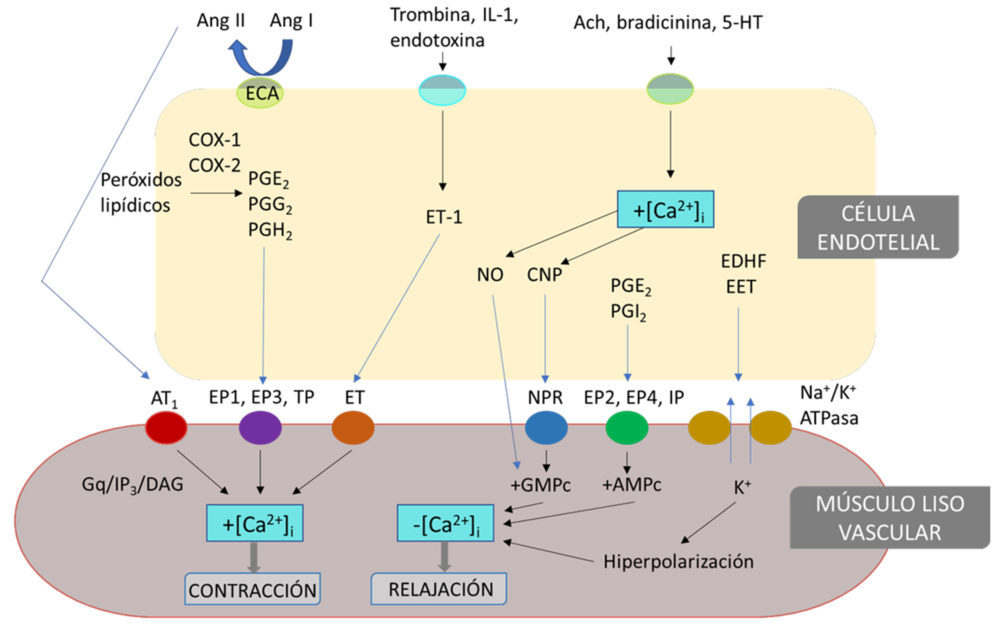
Figura 3. Mediadores vasoconstrictores y vasodilatadores derivados del endotelio. En situaciones fisiológicas o tras la activación de las células endoteliales (CE) con diferentes estímulos como la angiotensina II (Ang II), la trombina, la interleucina-1 (IL-1), la endotoxina, la acetilcolina (Ach), la bradicinina o la 5-hidroxitriptamina (5-HT), la CE puede liberar varios factores químicos, como prostaglandinas (PG), NO, endotelina-1 (ET-1) o factores hiperpolarizantes derivados del endotelio (EDHF), que se unen a sus receptores en las células musculares lisas vasculares (CMLV), o activan los canales de K+ desencadenando respuestas de relajación o contracción. ECA, enzima convertidora de angiotensina; AT1, receptor de angiotensina 1; CNP, péptido natriurético C; DAG, diacilglicerol; EET, ácido epoxieicosatetraenoico; ET, receptores de endotelina; EP, receptor de prostanoides P; Gq, proteínas G; IP, receptor de prostanoides I; IP3, inositol 1,4,5-trifosfato; NPR, receptor de péptidos natriuréticos; TP, receptor de prostanoides T. Modificado de (5).
3.1. Prostanoides
Los prostanoides son moduladores críticos del tono vascular y la agregación plaquetaria en condiciones fisiológicas y patológicas. La biosíntesis de los prostanoides depende de la acción de las ciclooxigenasas 1 y 2 (COX-1 y COX-2, respectivamente) sobre el ácido araquidónico liberado de los fosfoglicéridos de la membrana por las fosfolipasas. Ambas isoformas de la COX transforman el ácido araquidónico en dos endoperóxidos muy inestables, PGG2 y PGH2, que luego se transforman en prostaglandinas específicas (PGE2, PGI2, PGD2, PGF2α) y TXA2 por la acción de isomerasas específicas. La prostaglandina I2 (PGI2 o prostaciclina) fue el primer componente descubierto de este grupo, por Bunting, Gryglewski, Moncada y Vane (40).
A diferencia de la COX-1, la proteína COX-2 no está normalmente presente, aunque la COX-2 constitutiva se encuentra en algunos órganos como el riñón. Es un gen de respuesta inmediata-temprana, y su expresión puede regularse por mecanismos transcripcionales y post-transcripcionales. El promotor de la COX-2 contiene una caja TATA, con varios sitios de unión para factores de transcripción como el factor nuclear kappa de las células B activadas (NFκB), la proteína de unión que responde al AMP cíclico (CREB), el factor nuclear de las células T activadas (NFAT) o la proteína activadora-1 (AP-1) que son los responsables de su expresión en condiciones patológicas. La COX-2 es inducida por el lipopolisacárido y por citoquinas inflamatorias como la interleucina (IL)-1 y el factor de necrosis tumoral-α (TNFα ), entre otras. Además, en los últimos años se ha demostrado que otros factores como Ang II, ET-1 o ROS pueden inducir su expresión a nivel cardiovascular (5, 41).
Como se ha mencionado, la producción de los diferentes prostanoides depende de la actividad de sintasas específicas. La PGE2 es el prostanoide más abundante en el cuerpo humano. Existen tres PGE sintasas, la PGE2 sintasa citosólica (cPGES) y dos sintasas diferentes unidas a la membrana (mPGES-1 y mPGES-2). La cPGES y la mPGES-2 son constitutivas, mientras que la mPGES-1 es inducible por estímulos inflamatorios de forma similar a la COX-2 (41).
Los prostanoides se unen a receptores acoplados a proteínas G (GPCRs) específicos. La PGE2 puede unirse a cuatro subtipos de receptores (EP1-EP4), en los que el EP1 y el EP3 inducen la vasoconstricción, mientras que el EP2 y el EP4 median la vasodilatación. Los receptores EP también están implicados en la agregación plaquetaria, la migración de monocitos y macrófagos, la proliferación y migración de CML, la producción de citoquinas vasculares o la activación de metaloproteinasas de matriz (MMP) (42). La PGI2 es el principal PG liberado por el endotelio vascular, y actúa sobre los receptores de prostanoides de tipo I (PI), induciendo la vasodilatación e inhibiendo la activación plaquetaria (41). El receptor de tromboxano (TP) está implicado en la agregación plaquetaria, la contracción del músculo liso, la expresión de moléculas de adhesión y la infiltración de monocitos/macrófagos. Los intermedios endoperóxidos inestables PGG2 y PGH2 son factores de contracción derivados del endotelio y, concretamente, PGH2 también puede activar los receptores TP (Figura 4) (41, 43).
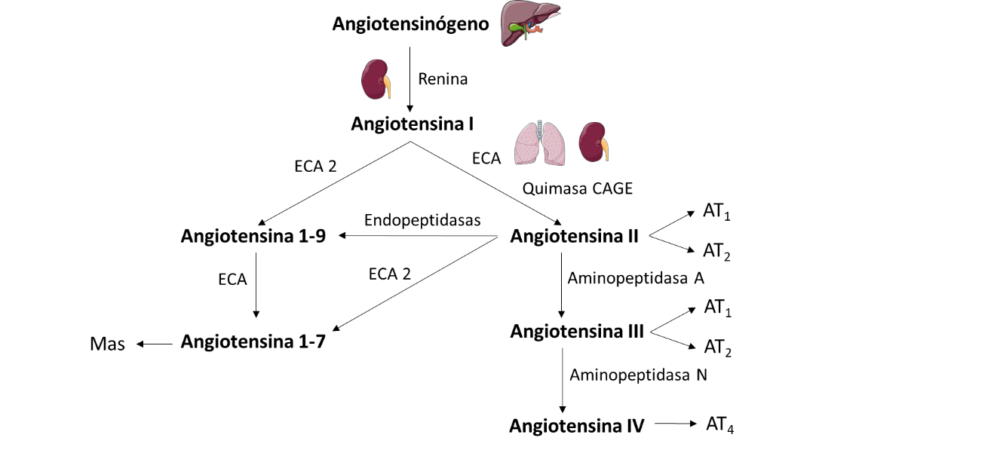
Figura 4. Esquema del sistema renina-angiotensina (SRA). Modificado de (60).
3.2. Óxido nítrico (NO)
En 1980, Furchgott y Zawadzki descubrieron un factor relajante derivado del endotelio, que posteriormente se identificó como NO (44, 45). Existen diferentes NO sintasas (NOS), dos constitutivas y una inducible. La eNOS constitutiva está presente sobre todo en las CE de los vasos, y puede ser regulada por el estrés por rozamiento, el ejercicio crónico o el embarazo, la nNOS constitutiva se expresa en el cerebro y el sistema nervioso periférico, y la NOS inducible (iNOS) se activa por estímulos inmunológicos o inflamatorios como el lipopolisacárido o la IL-1b. El NO se libera continuamente desde el endotelio vascular y se difunde a las CMLV subyacentes, donde activa la guanilato ciclasa y la producción de GMPc que produce vasodilatación al disminuir la concentración de calcio intracelular e interviene en el control fisiológico de la presión arterial (Figura 3). Además, el NO inhibe la proliferación de las CMLV, la adhesión y agregación de las plaquetas y la adhesión y migración de los monocitos, por lo que el NO protege en gran medida los vasos sanguíneos del desarrollo de ateromas y trombosis (5, 46).
3.3. Endotelina-1 (ET-1)
La ET-1 es un factor vasoconstrictor secretado por las CE. La ET-1 se une a los GPCR ETA y ETB (Figura 4). Los receptores ETA y ETB en las CMLV producen contracción y proliferación celular, mientras que los ETB en las CE desencadena la relajación al estimular la liberación de PGI2 y NO endoteliales (47). Las CMLV, los cardiomiocitos y los fibroblastos expresan ETA que activa la fosfolipasa C (PLC), lo que conduce a la generación de inositol 1,4,5-trifosfato (IP3) y diacilglicerol (DAG). La secreción de ET-1 se ve potenciada por factores físicos como el estrés por rozamiento, o por estímulos como la trombina, la IL-1, la endotoxina, la epinefrina, la Ang II, los factores de crecimiento, las citoquinas y las ROS. En cambio, la liberación de ET-1 endógena se ve reducida por mediadores como el NO, el GMPc, el péptido natriurético auricular y la PGI2. En condiciones fisiológicas, los efectos de la ET-1 se regulan cuidadosamente mediante la inhibición o la estimulación de la liberación de ET-1 desde el endotelio. Así, la desregulación del sistema de ET-1 es importante en la patogénesis de varias enfermedades como la hipertensión y la resistencia a la insulina (47).
3.4. Factores hiperpolarizantes derivados del endotelio (EDHF)
No se conoce la naturaleza exacta del EDHF y se han sugerido varios factores. Entre ellos se encuentran los ácidos epoxieicosatrienoicos (EET, derivados de las enzimas del citocromo P450), varios productos de la lipoxigenasa (LOX), el peróxido de hidrógeno (H2O2), el monóxido de carbono (CO), el sulfuro de hidrógeno (H2S) y el péptido natriurético C (CNP). Estas sustancias provocan la vasodilatación mediante la hiperpolarización de las CMLV a través de la activación de los canales de K+ (Figura 3). Además, el EDHF también produce una hiperpolarización de las CE, lo que aumenta la [Ca2+] intracelular. La mejora de la vía del EDHF contribuye al efecto beneficioso de algunas intervenciones terapéuticas, como los inhibidores de la enzima convertidora de la angiotensina (ECA), los bloqueadores del AT1R y los inhibidores de la fosfodiesterasa 3 (48).
Además de secretar mediadores vasoactivos, las CE expresan muchas enzimas, mecanismos de transporte y receptores que se unen a las hormonas circulantes y a otras sustancias, contribuyendo a las diferentes funciones del endotelio, pero eso no se tratará en esta revisión.
3.5. Sistema renina-angiotensina (SRA)
El SRA actúa de forma sinérgica con el sistema nervioso simpático y tiene un papel fundamental en el desarrollo de varias patologías cardiovasculares, principalmente a través de la regulación sobre la excreción de Na<sup+ y el tono vascular (49). La hiperactivación de este sistema se ha descrito en la aterosclerosis (50), la diabetes (51), la obesidad (52) y la hipertensión (53-55). El SRA fue considerado inicialmente como un sistema estrictamente circulante, sin embargo, ahora se acepta que hay miembros locales del SRA en el cerebro, el riñón, la corteza suprarrenal, el tejido adiposo o la propia pared vascular (49, 55-57).
El péptido principal de este sistema es la Ang II, que se sintetiza por la acción consecutiva de dos enzimas: la renina y la ECA. Además, el sistema convencional se está haciendo más complejo debido a la aparición de otras enzimas capaces de participar en la síntesis de Ang II, como la enzima generadora de Ang II sensible a la quimostatina (quimasa CAGE) (58) (Figura4). La renina se produce en el riñón, concretamente en el aparato yuxtaglomerular, en respuesta a diferentes estímulos fisiológicos, como la disminución de la presión de perfusión renal o la disminución de la [Na+] en el líquido del túbulo distal. Además, los agonistas de los receptores b-adrenérgicos y la PGI2 estimulan la secreción de renina directamente, mientras que la Ang II actúa como inhibidor de la vía por autorregulación. El sustrato de la renina es el angiotensinógeno circulante producido por el hígado, para formar angiotensina I (Ang I), un péptido de 10 aminoácidos sin actividad biológica. La ECA, sintetizada principalmente en el endotelio pulmonar y en el riñón, actúa sobre esta Ang I, para producir Ang II, un potente vasoconstrictor (Figura 4). El paso limitante de esta vía es la producción de renina en las células granulares del aparato yuxtaglomerular (59). Además, existe una segunda ECA (ECA 2) capaz de realizar la hidrólisis de Ang I a Ang 1-9 y la hidrólisis de Ang II a Ang 1-7. La formación de Ang 1-9 a partir de Ang I por la ECA 2 es considerablemente más lenta que la hidrólisis de Ang II para dar Ang 1-7 por la misma enzima. La Ang 1-7 también puede formarse a partir de la Ang 1-9 por la acción de la ECA (60, 61) (Figura 5). La Ang II puede transformarse en Ang III y Ang IV por la acción de dos enzimas (aminopeptidasas A y N, respectivamente), y la Ang II también puede transformarse en Ang 1-9 por la acción de endopeptidasas (Figura 4).
La Ang II es esencial para la integridad funcional y estructural de la pared vascular, y desempeña un papel importante en los procesos fisiológicos que regulan la presión arterial y en los procesos patológicos implicados en las enfermedades vasculares (49). En concreto, entre los numerosos efectos de la Ang II a nivel vascular se encuentran la modulación del tono vasomotor, la regulación del crecimiento celular y la apoptosis, la regulación de la migración celular y la deposición de la MEC, la estimulación de la generación de ROS por parte de las oxidasas de nicotinamida-adenina dinucleótido fosfato (NADPH), la estimulación de la producción de factores de crecimiento específicos como el factor de crecimiento derivado de las plaquetas (PDGF) o el factor de crecimiento transformante b (TGF-b), y la síntesis de factores vasoconstrictores como la ET-1, moléculas de adhesión como la molécula de adhesión intracelular, (ICAM-1 ), la E- selectina, las integrinas o citoquinas como el TNF α , el IFN γ o las ILs (62).
Las principales acciones de la Ang II están mediadas por los GPCR AT1 o AT2 (Figura 4). Los efectos de los receptores AT1 incluyen la vasoconstricción generalizada, el aumento de la liberación de noradrenalina que potencia los efectos simpáticos, la estimulación de la reabsorción tubular proximal de Na+, la secreción de aldosterona en la corteza suprarrenal y el crecimiento celular en el corazón y las arterias a través de vías de señalización intracelular, como JAK-STAT o ERK1/2. También es una fuente reconocida de estrés oxidativo en el sistema cardiovascular (63). Los receptores AT2 se expresan sobre todo durante la vida fetal y en ciertas áreas del cerebro adulto. Participan en el crecimiento, el desarrollo y la muerte celular programada. Los efectos cardiovasculares de los receptores AT2 incluyen la inhibición del crecimiento celular, la disminución de la presión arterial y la vasodilatación, pero estos efectos son relativamente débiles, especialmente en comparación con los efectos del receptor AT1 (62). A pesar de ello, existen evidencias actuales que demuestran claramente los efectos protectores de un agonista selectivo del receptor AT2 no peptídico a nivel vascular (64).
La Ang III y la IV se han considerado de menor importancia, pero la Ang III también se une a los receptores AT1 y AT2, desencadenando respuestas fisiológicas similares a las de la Ang II (Figura 4), mientras que la Ang IV estimula la liberación del inhibidor del activador del plasminógeno I por el endotelio. Los receptores de Ang IV (AT4) tienen una distribución peculiar, que incluye el hipotálamo (62). Por su parte, la Ang 1-7 se une al receptor Mas (Figura 4) y tiene acciones opuestas a la Ang II: induce la vasodilatación, a través del aumento de la producción de NO, EDHF y prostanoides vasodilatadores, y tiene efectos antiproliferativos (62).
3.6. Especies reactivas de oxígeno (ROS)
Las ROS son producidas por casi todos los tipos de células y, en concreto, por todas las células de la pared vascular, incluidas las CMLV, las CE y las células adventicias, junto con las células circulantes, como las plaquetas, los leucocitos y los glóbulos rojos. Actúan como segundos mensajeros intracelulares que interactúan con múltiples vías de señalización y afectan a funciones celulares básicas como la proliferación, la migración, la muerte celular, la modulación y la degradación de la MEC, la inactivación del NO y la expresión de genes proinflamatorios (65). El término ROS engloba especies reactivas derivadas del oxígeno, entre los que destacan por sus implicaciones biológicas el anión superóxido (O2.-), el peróxido de hidrógeno (H2O2) y el peroxinitrito (ONOO–). Otros son el radical hidroxilo (OH) y el ácido hipocloroso (HOCl).
El O2.- se forma por la reducción univalente del oxígeno molecular. Las principales fuentes de O2.- son la NADPH oxidasa (NOX), la xantina oxidasa, la lisil oxidasa, la COX, las isoformas del CYP450, las monoxigenasas y la eNOS desacoplada. El O2.- también puede ser generado de forma no enzimática por la cadena de transporte de electrones mitocondrial, el retículo endoplásmico (RE) y los peroxisomas (66).
El O2.- tiene un papel fundamental en la biología redox porque, además de tener numerosas funciones, es el precursor de las demás ROS. Por ejemplo, el ONOO– se forma tras la reacción química del O2.- con el NO, reduciendo también la biodisponibilidad del NO (67). La mayor parte del O2.- generado se convierte rápidamente en H2O2 por las tres superóxido dismutasas (Mn-, extracelular-, Cu-Zn-SOD) que, a diferencia del O2.-, penetra fácilmente en las membranas celulares, y funciona como un segundo mensajero que activa múltiples vías de señalización (66).
En cuanto a las funciones del O2.-, es capaz de activar diferentes vías de señalización como Akt o MAPK, que conducen a la activación de factores de transcripción proinflamatorios como AP-1 y NFκB (68, 69), y está implicado en la síntesis de genes proinflamatorios como la proteína quimioatrayente de monocitos 1 (MCP-1) o las moléculas de adhesión ICAM-1 y VCAM-1, entre muchas otras (68).
El desequilibrio entre la generación y la eliminación de ROS determina el estrés oxidativo. Actualmente se acepta que el estrés oxidativo está implicado en muchos procesos celulares y tisulares en relación con la ECV. De hecho, todos los factores de riesgo cardiovascular establecidos, como la hipercolesterolemia, la hipertensión, la diabetes mellitus, la obesidad y el tabaquismo, potencian la generación de ROS. En el contexto de la ECV, no sólo las células vasculares producen y liberan ROS. Las células inflamatorias infiltradas a nivel (peri)vascular se reconocen ahora como una fuente potencial de ROS en diferentes ECV, incluyendo la aterosclerosis, la hipertensión, la obesidad y el AAA (66, 67).
3.6.1. NADPH oxidasa
La familia NOX es la principal fuente de ROS en la pared vascular, tanto en condiciones fisiológicas como patológicas (67, 70, 71). La principal actividad catalítica de la NOX es la producción de ROS (NADPH + 2 O2 → O2.-– + NADP+ + H+), mientras que el resto de enzimas generadoras de ROS tienen otra función principal y sólo producen ROS como subproducto o cuando funcionan mal. La NOX es un complejo enzimático formado por diferentes subunidades citosólicas y transmembrana. Las subunidades transmembrana son p22phox y las 7 isoformas diferentes de la NOX: NOX1-5 y DUOX1-2, también llamadas NOX6-7. NOX5 y DUOX carecen de subunidades adicionales (72). Las subunidades transmembrana de la NOX son responsables del transporte de electrones a través de las membranas biológicas, reduciendo el O2 a O.-2 , y utilizando el NADPH como donante de electrones. Las subunidades citosólicas (organizador NOX 1 (NOXO1), activador NOX 1 (NOXA1), Rac1/2, p67phox, p47phox y p40phox) participan en el ensamblaje del complejo a la membrana y en la activación de la enzima (73).
La NOX2 fue la primera en ser caracterizada en los fagocitos. NOX1, NOX2, NOX4 y NOX5 se expresan en el sistema cardiovascular, incluidas las CE y las CMLV. Además, NOX1, NOX2 y NOX4 se encuentran en los fibroblastos adventiciales (67). Todas las isoformas producen O2.- en las células vasculares con la excepción de NOX4, que produce preferentemente H2O2. La NOX5 está presente en formas inferiores y en mamíferos superiores, pero no en roedores (72).
A nivel vascular, la expresión basal de NOX1/2 suele ser baja, pero varios estímulos como el estrés mecánico, la Ang II, la ET-1 o la aldosterona aumentan su expresión (73). La NOX4 está implicada en la producción constitutiva de ROS pero ciertos estímulos como la hipoxia, el TGF-b1, la Ang II o el estrés mecánico también pueden aumentar su expresión (75, 76). En particular, se ha puesto de manifiesto que, en algunas condiciones, NOX4 tiene un papel protector a nivel vascular, impidiendo la activación o proliferación celular (77). A diferencia de otras NOX, la NOX5 está regulada por la concentración intracelular de Ca2+, ya que esta subunidad consta de 4 dominios similares a la calmodulina con capacidad para unirse al Ca2+ (67). NOX5 también está estrechamente regulada a través de numerosas modificaciones postraduccionales y se activa por agentes vasoactivos, factores de crecimiento y citoquinas proinflamatorias (78). Por ejemplo, la NOX5 vascular se activa por la trombina, el PDGF y la ionomicina, a través de la proteína quinasa C (PKC) y AMPc (79, 80). Además, en las CE humanas, la Ang II y la ET-1 inducen la señalización redox y la activación de MAPK de forma dependiente de NOX5 (81).
Las diferentes funciones de las NOX son muy complejas y dependen de las distintas condiciones fisiopatológicas. En general, los datos procedentes de pacientes con enfermedad arterial coronaria o hipertensión, y de modelos animales de hipertensión, diabetes o aterosclerosis, sugieren que NOX1 y NOX2 promueven la disfunción endotelial y la inflamación, mientras que NOX4 podría tener un papel vasoprotector en determinadas situaciones al aumentar la biodisponibilidad del NO y suprimir las vías de muerte celular (82). Además, especialmente NOX1 y NOX4 están implicadas en el remodelado vascular en diferentes condiciones patológicas como la hipertensión, la reestenosis, la aterosclerosis, la dilatación aórtica o la hipertensión pulmonar (83). NOX5 está implicada en la regulación de diferentes funciones vasculares, incluyendo la contracción y relajación vascular y el remodelado vascular, aunque debido a que no se expresa en roedores, se dispone de menos información sobre esta isoforma de NOX (78).
3.6.2. Sistemas antioxidantes
Una buena función vascular depende del equilibrio entre los mecanismos oxidativos y antioxidantes. Varios sistemas antioxidantes regulan los niveles de ROS. Entre las enzimas se encuentran la catalasa, las SOD, la glutatión peroxidasa, las glutatión S- transferasas, la tiorredoxina reductasa y la epóxido hidrolasa 2 (84). Las SODs son las más importantes y como se ha mencionado, su función principal es transformar el O2.- en H2O2. Existen 3 isoformas de SOD (Cu/Zn-SOD, Mn-SOD y extracelular-SOD), que difieren en el cofactor que utilizan y en su localización celular. La catalasa, la glutatión peroxidasa o la tiorredoxina reductasa catalizan la conversión de H2O2 en H2O y O2(66).
El aumento de los niveles de ROS activa el factor nuclear derivado de los eritroides 2 (Nrf2), un regulador maestro de la respuesta antioxidante, que se activa para contrarrestar el estrés oxidativo. Nrf2 controla la expresión de unos 250 genes, incluidos los que codifican enzimas antioxidantes como los sistemas de glutatión y tiorredoxina, las SOD, la catalasa y la hemoxigenasa-1, entre muchos otros. Además de la degradación enzimática de las ROS, varios compuestos de bajo peso molecular pueden reaccionar directamente con las ROS. Estas moléculas pueden ser sintetizadas endógenamente u obtenidas de la dieta e incluyen las vitaminas C y E, el ácido úrico, el glutatión, los flavonoides y los tioles (66).
3.7. Factores vasoactivos liberados por el tejido adiposo perivascular (PVAT)
Los estudios experimentales realizados en las dos últimas décadas han identificado los factores vasoactivos liberados por el PVAT como importantes reguladores del tono vascular, el crecimiento arterial y el remodelado (85). En situaciones fisiológicas, el PVAT muestra un efecto anticontráctil neto. Así la respuesta contráctil a varios agonistas como la fenilefrina, la Ang II, la serotonina y la noradrenalina se encuentra disminuida en arterias montadas con PVAT o tras la incubación de las arterias con medios condicionados de PVAT (85).
Entre los factores relajantes liberados por el PVAT, el factor relajante derivado de los adipocitos (ADRF) ha recibido especial atención. El ADRF fue el primer factor que se sugirió que se liberaba y transfería desde el PVAT a la pared vascular subyacente para ejercer un efecto anticontráctil paracrino (86). Aunque el ADRF aún no ha sido identificado, se sabe que puede producir relajación arterial al abrir los canales de K+ activados por voltaje de las CMLV. La liberación y la acción del ADRF dependen de la [Ca2+] externa y es independiente de las CE (86). Además, existen otros factores relajantes derivados del PVAT que causan relajación a través de mecanismos endoteliales (85). Se desconoce la naturaleza precisa de las vías de señalización intracelular responsables de la producción de factores relajantes derivados del PVAT. Sin embargo, se sabe que numerosos factores vasoactivos derivados del tejido adiposo actúan a través de la vía de la fosfoinositida 3-quinasa (PI3K) que participa en la síntesis de NO (87). Curiosamente, la señalización PI3K también se ha implicado en la resistencia a la insulina y la señalización inflamatoria en el tejido adiposo en situaciones de obesidad (85).
La leptina y la adiponectina son las adipoquinas derivadas del PVAT más conocidas y también se han propuesto como factores relajantes derivados del PVAT en diferentes lechos vasculares, como las arterias coronarias y las arterias mesentéricas. Otras adipoquinas, como la omentina, la visfatina, la irisina y la apelina, se han sugerido como posibles factores relajantes derivados del PVAT, ya que también son capaces de relajar los vasos sanguíneos mediante vías principalmente dependientes del endotelio (88). Es importante destacar que una creciente lista de estudios sugiere que el PVAT también puede ser una fuente de sustancias contráctiles, como la noradrenalina, la Ang II, las adipoquinas o las ROS, especialmente en un estado patológico (85). Esto se discutirá con más detalle en otras secciones de esta revisión.
4. SISTEMA INMUNE
El sistema inmune es conocido clásicamente por ser capaz de discriminar entre lo propio y lo ajeno para luego luchar contra los microorganismos patógenos. Las células del sistema inmunitario se originan, y algunas también maduran, en la médula ósea, y luego, migran a diferentes tejidos periféricos. Las células madre pluripotentes hematopoyéticas de la médula ósea pueden transformarse en dos células más especializadas: el progenitor mieloide común y el progenitor linfoide común (Figura 5) (89).
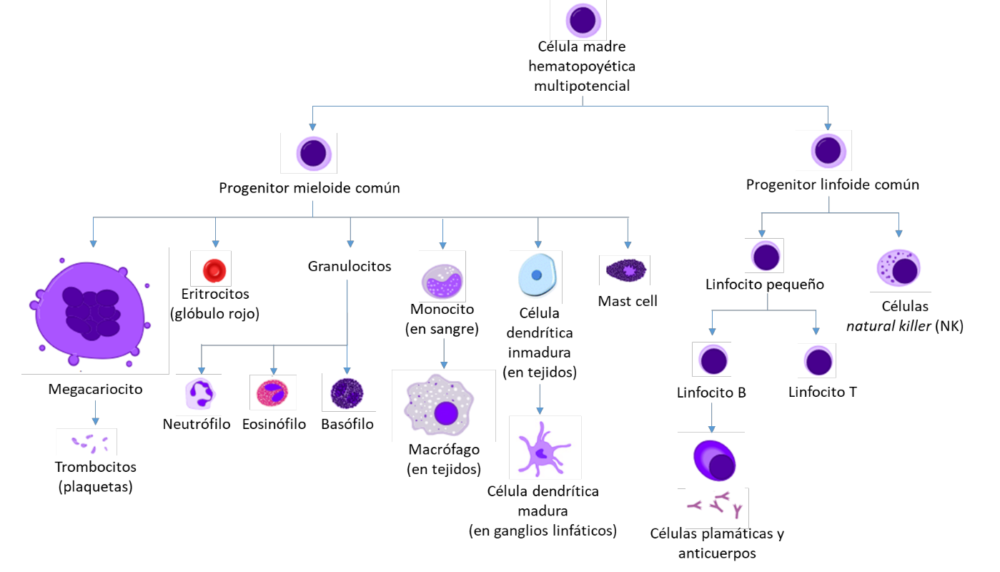
Figura 5. Células mieloides y linfoides. Modificado de A. Rad y Mikael Häggström, M.D. Diagrama de hematopoyesis (humana).
El progenitor mieloide es el precursor de los eritrocitos, las plaquetas, los granulocitos (incluidos los neutrófilos, los eosinófilos y los basófilos), los macrófagos, las células dendríticas y los mastocitos (Figura5) (89). Los granulocitos tienen una vida relativamente corta y sus niveles aumentan cuando salen de la sangre para migrar a los focos de infección o donde tiene lugar un proceso de inflamación. Los neutrófilos son granulocitos fagocíticos y representan el componente celular más numeroso de la respuesta inmunitaria innata, la respuesta más rápida pero sencilla del sistema inmunitario. Los eosinófilos y los basófilos son también granulocitos con un papel importante en la defensa contra las infecciones parasitarias y la inflamación alérgica. Las células dendríticas se encargan de presentar el antígeno para que sea reconocido por los linfocitos. Los mastocitos residen principalmente cerca de los pequeños vasos sanguíneos y pueden liberar sustancias que afectan a la permeabilidad vascular, aunque son más conocidos por su papel en las respuestas alérgicas. Pueden reclutar eosinófilos y basófilos, que también son exocíticos.
Los macrófagos son fagocitos que están ampliamente distribuidos en los tejidos del cuerpo y desempeñan un papel fundamental en la inmunidad innata. También pueden reclutar otras células fagocíticas de la sangre. Los macrófagos son la forma madura de los monocitos, que circulan por la sangre y se diferencian continuamente en macrófagos tras su migración a diferentes tejidos. En términos generales, una vez que alcanzan el tejido diana y en respuesta al microambiente, los macrófagos pueden polarizarse hacia un fenotipo M1 (proinflamatorio) y/o M2 (antiinflamatorio), aunque actualmente se acepta la existencia de un sofisticado marco de diferenciación funcional y fenotípica (90). Los macrófagos M1 activan y guían a los linfocitos T Th1 y los macrófagos M2 están asociados a la inducción de respuestas de linfocitos T Th2 (90). Además, los macrófagos M1 son capaces de producir ROS, que pueden limitar la biodisponibilidad de NO. Clásicamente, el IFNγ induce la diferenciación de los macrófagos M1, mientras que la IL-4 induce los macrófagos M2 antiinflamatorios (90). Aparte de estos estímulos clásicos, se están describiendo nuevos estímulos para la polarización de los macrófagos, incluidos algunos estrechamente relacionados con la ECV (véase más adelante).
El progenitor linfoide produce linfocitos, las células responsables de la inmunidad adaptativa (Figura 5) (89). Hay dos tipos principales de linfocitos: Los linfocitos B, que tras su activación se diferencian en la médula ósea en células plasmáticas que secretan anticuerpos; y los linfocitos T, que se diferencian en el timo en células T efectoras con una variedad de funciones. Una vez que han completado su maduración, ambos tipos de linfocitos pasan al torrente sanguíneo, para finalmente migrar a los órganos linfoides periféricos. Existen dos tipos principales de linfocitos T, diferenciados por sus proteínas de membrana, los linfocitos T cooperadores son CD4+ mientras que los linfocitos T citotóxicos son CD8+. Las células natural killer (NK) son un tercer tipo de células linfoides pero que forman parte del sistema inmunitario innato. Son capaces de reconocer y eliminar algunas células anormales, como las células tumorales y las infectadas por virus.
Nuestra comprensión del sistema inmunitario ha evolucionado en las últimas décadas hasta reconocer que es un órgano regulado dinámicamente que desempeña un papel fundamental en varios procesos fisiopatológicos, como el desarrollo de los órganos y la homeostasis y reparación tisular (91). Los tejidos dañados liberan señales de alarma que activan las células presentadoras de antígenos. Las señales de peligro son reconocidas por el sistema inmunitario innato a través de diferentes receptores (receptores de reconocimiento de patrones (PRR)), como los receptores tipo Toll (TLR), que activan la producción de citoquinas y atraen a los leucocitos al lugar de la lesión (33). Durante la respuesta celular al daño, las células dañadas y el sistema inmunitario activan al inflamasoma, que es responsable de la activación de los procesos inflamatorios. La aparición de enfermedades crónicas se explica por la activación anormal de la respuesta celular al daño más allá de la resolución de la lesión, alterando el metabolismo de todo el cuerpo, lo que lleva a la disfunción de múltiples órganos (92-95) y a la fibrosis (91). En este sentido, cada vez hay más evidencias sobre un flujo continuo de intercambio molecular entre el sistema cardiovascular y el inmunitario. Sin embargo, no se comprende del todo cómo se produce esta comunicación (36).
El papel de las células inmunitarias innatas (monocitos, macrófagos y neutrófilos) junto con los linfocitos en el inicio y desarrollo de la aterosclerosis ha sido bien explorado debido a su contribución a la inestabilidad de la placa (91). Además, cada vez hay más pruebas que apoyan el papel de los macrófagos y linfocitos tisulares en la hipertensión (90, 96) y en las enfermedades metabólicas asociadas a la obesidad, como la resistencia a la insulina (97). Además, los neutrófilos han surgido recientemente como importantes moduladores de la ECV (98, 99).
En términos generales, el aumento de la inflamación suele comenzar tras la activación endotelial por parte del sistema inmunitario y la expresión de diversas moléculas de adhesión para atraer a diferentes células inmunitarias (100). Estas células inmunitarias, así como las células residentes en el tejido, amplifican la señal inflamatoria mediante el aumento de la expresión de citoquinas (100, 101), que engloba una amplia categoría de pequeñas proteínas solubles, como IFN, IL, quimioquinas, linfoquinas y TNF. Curiosamente, estas citoquinas son producidas principalmente por los linfocitos B y T, pero también por las CE activadas, las CMLV, los fibroblastos, las neuronas o los adipocitos (102). Las principales citoquinas proinflamatorias son el IFNγ, el TNFα , la IL-1, la IL-6, la IL-8 o la IL-12, mientras que los compuestos antiinflamatorios son el TGF-β, la IL-4, la IL-10, la IL-11 o la IL-13, entre otros (Figura 6). El desequilibrio entre los mediadores pro y antiinflamatorios se considera un mecanismo fisiopatológico común a diferentes ECV, y parece estar estrechamente relacionado con la disfunción endotelial (101, 103, 104) (Figura 6). Los principales factores de transcripción implicados en la generación de citoquinas a nivel vascular son NFκB y NFAT. Además de los estímulos inflamatorios clásicos, son activados por la Ang II o la ET-1 y participan en la disfunción endotelial y el remodelado vascular observados en la ECV (105, 106). El papel de las citoquinas específicas en el daño vascular en la hipertensión y la obesidad se analizará en las secciones 6.2. y 7.2., respectivamente.
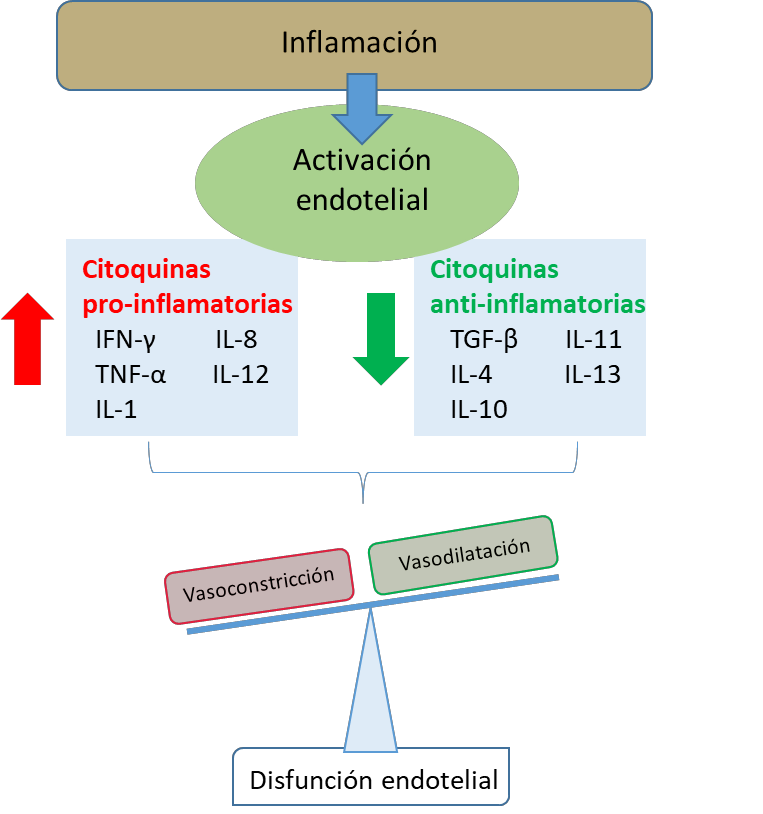
Figura 6. Efecto de la descompensación entre citoquinas pro y antiinflamatorias tras la activación endotelial en una situación inflamatoria.
5. GEN ESTIMULADO POR INTERFERON-15 (ISG15)
El gen 15 estimulado por el IFN (ISG15) codifica una proteína similar a la ubiquitina que se produce como un precursor de 17 kDa con dos dominios similares a la ubiquitina (Figura 9). Hasta ahora, ISG15 sólo se ha encontrado en vertebrados (107). Se expresa principalmente en monocitos, linfocitos y neutrófilos, pero también en células dendríticas, células NK, líneas celulares derivadas del epitelio, fibroblastos y en varios tumores celulares (108-111). También se ha observado la expresión de ISG15 en células endoteliales microvasculares humanas (HMECs) tras la infección con Rickettsia conorii, el agente causante de la fiebre botonosa mediterránea (112) y en cardiomiocitos, donde la expresión de ISG15 estaba aumentada en pacientes con cardiomiopatía viral (113). Sin embargo, hasta la fecha se sabe muy poco sobre el papel del sistema ISG15/USP18 a nivel vascular.
La proteína ISG15 puede encontrarse en dos estados diferentes: como proteína libre (intracelular o extracelular) o conjugada a proteínas sustrato, tras una modificación postraduccional denominada ISGilación (Figura 7). Tanto ISG15 como las enzimas implicadas en la ISGilación se inducen en respuesta a diferentes IFNs, principalmente de tipo I (a y b) pero también de tipo II (IFNγ), y de tipo III (IFNl), así como en respuesta a lipopolisacáridos o TNFα (107, 111, 114-118).
La ISGilación es una modificación postraduccional reversible de las proteínas, llevada a cabo en proteínas sintetizadas de novo. La ISGilación requiere una cascada de reacciones enzimáticas para unir ISG15 a un residuo de lisina de la proteína sustrato (107, 111) (Figura 9). La ISGilación se asemeja a la ubiquitinación y requiere una enzima activadora E1 (UbE1L), una enzima conjugadora E2 (UbCH8) y ligasas E3 (HERC5 en humanos, HERC6 en ratones) (119). Además, el sistema ISG15 opera de forma similar o incluso solapado con el sistema de ubiquitina (120, 121). Aunque la unión a ISG15 no dirige a sus sustratos para su degradación, parece haber una interacción con el proteasoma, ya que se observa un aumento de los conjugados de ISG15 tras la inhibición del proteasoma (122). La ISGilación es una modificación reversible, la proteasa USP18, que es una proteasa específica de ISG15 se encarga de la de ISGilación (123). Además, USP18 actúa como un regulador negativo de las respuestas inducidas por el IFN de tipo I (124) (Figura 9). La proteína ISG15 intracelular humana actúa como estabilizadora de esta acción de USP18, ya que la falta de ISG15 intracelular conduce a niveles inestables de USP18 (125, 126). Esto da lugar a una señalización persistente de las vías desencadenadas por el receptor de IFN tipo I, especialmente JAK-STAT (127). A nivel clínico, este aumento de la señalización de IFN tipo I debido a la falta de ISG15 intracelular parece ser el responsable de las calcificaciones cerebrales (126) y de las lesiones cutáneas ulcerosas (128) observadas en algunos pacientes con deficiencia heredada de ISG15. La proteína USP18 se expresa en el hígado, el bazo, el timo, la médula ósea, el tejido adiposo y los pulmones.
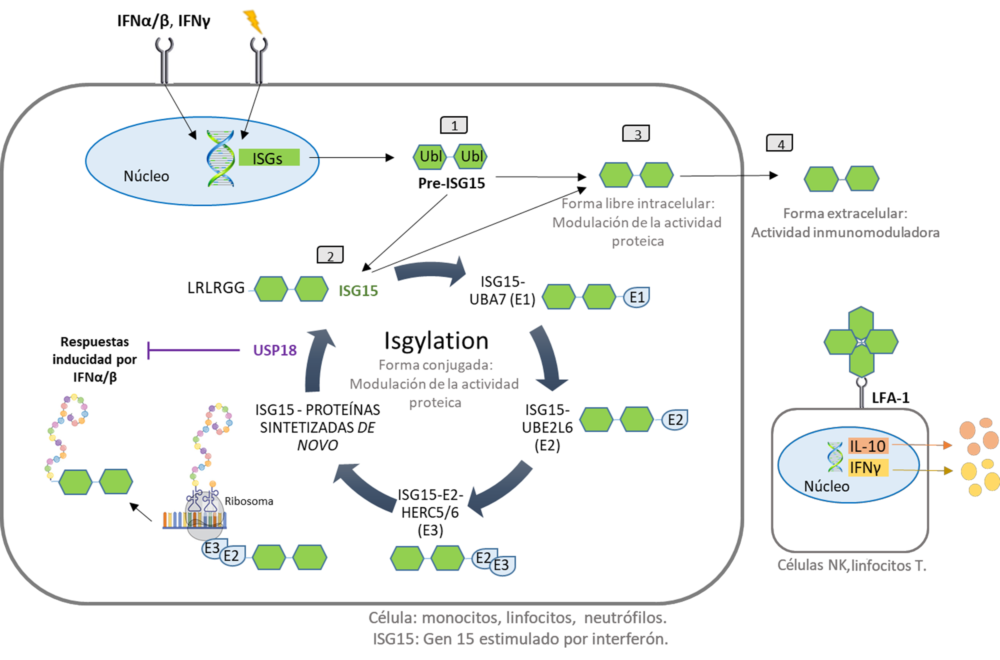
Figura 7. Proceso de ISGilación e ISG15 intracelular/extracelular. Diferentes estímulos, como los IFN, desencadenan la expresión de ISG15, que se produce como un precursor de 17 kDa con dos dominios similares a la ubiquitina (1). La proteína ISG15 intracelular puede estar libre (3) o conjugarse con proteínas sintetizadas de novo en el proceso denominado ISGilación (2). La ISGilación puede ser reversible por la acción de la proteasa USP18, que también regula la señalización mediada por IFN (2). La forma libre también puede ser secretada por la célula y actúa como citoquina, uniéndose al receptor de integrina LFA-1 en la superficie de las células NK o los linfocitos T, provocando la liberación de IFNγ e IL-10 (4). Esta forma extracelular de ISG15 puede formar dímeros o multímeros con el objetivo de modular los niveles de citoquinas. Modificado de (111)
Además, los macrófagos peritoneales y los macrófagos derivados de monocitos expresan altos niveles de USP18 (123).
Como se ha señalado anteriormente, la forma ISG15 libre es secretada por las células y se ha detectado en sangre y orina (129, 130). Esto ocurre a pesar de que ISG15 carece de un péptido señal para la secreción. Los mecanismos implicados en la secreción de ISG15 se desconocen, aunque se han identificado aminoácidos específicos en la secuencia de ISG15(L72A, S83A y L85F) implicados en la secreción (131). Además, se ha observado que en los macrófagos infectados por Mycobacterium tuberculosis, ISG15 puede liberarse en microvesículas (132) y, en el contexto de la infección por el VIH, la proteína ISG15 se encontró en los exosomas liberados por las CE cerebrales microvasculares activadas por TLR3 (133). Más recientemente, se ha publicado que NS1B, una proteína que se une a ISG15 y a las proteínas ISGiladas (134, 135), está implicada en el bloqueo de la secreción de ISG15 porque la unión de NS1B ocluye L72, uno de los residuos que son críticos para la secreción de ISG15 (131). Curiosamente, la secreción de ISG15 aumenta en los macrófagos M2 y en los macrófagos asociados a tumores (136). Además, los linfocitos y las células epiteliales también son capaces de secretar ISG15 (131), lo que sugiere que el mecanismo de secreción de ISG15 funciona en una amplia gama de tipos de células y condiciones.
La forma libre extracelular de ISG15 actúa como modulador de neutrófilos, monocitos, células NK y células dendríticas (Figura 9), facilitando la liberación de citoquinas como IL-8 e IFNγ, o la antiinflamatoria IL-10, generando así una retroalimentación positiva en la expresión de ISG15 (137).
5.1. Receptor de ISG15 libre
Recientemente se ha descubierto un receptor para la forma ISG15 libre. Se trata del antígeno-1 asociado a la función leucocitaria (LFA-1), el clásico receptor de ICAM-1 (138), que participa en el reclutamiento y la adhesión de las células inmunitarias y se expresa principalmente en los linfocitos T y B, los macrófagos, los neutrófilos y las células NK (139). Específicamente, en las células NK y los linfocitos T la vía ISG15/LFA-1 promueve la secreción de IFNγ e IL-10 (Figura 9) a través de la actividad quinasa de la familia Src (138). Se observaron resultados similares en macrófagos asociados a tumores (140), en los que ISG15 indujo un fenotipo M2 a través de la interacción con LFA-1, la acción de las quinasas de la familia Src y la posterior secreción de CCL18. Además, la vía ISG15/LFA-1 desempeña un papel en la susceptibilidad de las córneas de ratón a la infección por Pseudomonas aeruginosa (141). La identificación de LFA-1 como un receptor de ISG15 ha revelado la base de la señalización de ISG15, pero aún es necesario seguir investigando para identificar los tipos de células y las señales inmunológicas que están implicadas en las respuestas mediadas por ISG15 en el espacio extracelular. En particular, hay una escasez de información sobre el papel de ISG15 en las células no inmunes.
5.2. Funciones de ISG15
Las funciones biológicas de ISG15 son muy complejas y diversas. En general, ISG15 se ha estudiado ampliamente en las infecciones virales, donde tiene un papel protector (134, 142-145). Además, las proteínas ISGiladas parecen estar implicadas en la formación de tumores (107) y, más recientemente, se ha descrito la implicación de ISG15 en la obesidad (146). ISG15 puede unirse a cientos de proteínas, pero solo se conoce su consecuencia biológica para un pequeño número de estas interacciones (111, 137). Por ejemplo, la primera diana molecular de la ISGilación que se identificó es el inhibidor de serina proteasa 2a, que tiene un papel importante en la regulación de las proteasas intracelulares en las células presentadoras de antígenos (147), proporcionando así información sobre la relación de la ISG15 con la inflamación. Además, la ISGilación puede inhibir o activar la función de muchas proteínas implicadas en la infección viral pero también en la progresión del cáncer, en la respuesta a la hipoxia o en la secreción de exosomas, entre otros procesos (111, 148). A nivel celular, las proteínas ISGiladas afectan al metabolismo de aminoácidos, proteínas y carbohidratos, al ciclo celular, a la proliferación y diferenciación celular, a la estructura y motilidad celular, a la contracción muscular, al tráfico intracelular de proteínas, a la traducción de proteínas, a la ubiquitinación o a la autofagia (148, 149). La identificación de las proteínas ISGiladas requiere estudios específicos de espectrometría de masas. Así, algunas proteínas conjugadas a ISG15 incluyen proteínas inducidas por IFNs de tipo I, como PKR y RIG-I, y algunas proteínas reguladoras implicadas en la señalización de IFN, como Jasus Kinase 1/2 (JAK1/2) y el transductor de señales y activador de la transcripción 1 (STAT1) (115, 150). Curiosamente, en el sistema cardiovascular, la vía JAK-STAT es activada por la Ang II, mediando varios de sus efectos deletéreos (151). Como se ha mencionado, existe poca información sobre el papel de la vía ISG15 a nivel cardiovascular. La ISGilación fue descrita como un mecanismo crítico en la respuesta del sistema inmune innato de los cardiomiocitos frente a infecciones virales, disminuyendo la cardiomiopatía inflamatoria, el daño cardíaco y la mortalidad (113). Además, la expresión específica en cardiomiocitos de la quinasa IκB 2 constitutivamente activa fue suficiente para activar la vía ISG15 y causó la ISGilación generalizada de proteínas de manera dependiente de NFκB, pero no se describió la implicación fisiopatológica de este proceso (152).
El papel de la proteína ISG15 libre es menos conocido. Como se mencionó anteriormente, en los seres humanos, la proteína ISG15 libre intracelular parece prevenir la autoinflamación dependiente de IFNα/β a través de potenciar el papel de USP18 como regulador negativo de esta vía (126). En cuanto a la ISG15 extracelular, varios estudios han definido esta forma secretada como una molécula inductora de IFNγ, específicamente en linfocitos CD4+ y CD8+ de sangre periférica (PBMC) (108, 129, 153, 154), y esta producción de IFNγ es independiente de la ISGilación (109). Es importante destacar que la falta de ISG15 secretada en los pacientes con deficiencia de ISG15 explica sus bajos niveles de secreción de IFNγ ex vivo y, por tanto, su susceptibilidad a las enfermedades micobacterianas (109). Más recientemente, se ha observado el mismo efecto en las células NK (109, 138) y en las PBMC humanas, donde ISG15 libera citoquinas proinflamatorias relevantes como CXCL1, CXCL5, CXCL8, CCL20, IL-1, IL-6, TNF e IFNγ (155). Además, en pacientes infectados por el VIH, la proteína ISG15 liberada en los exosomas de las CE microvasculares cerebrales activadas parece estar implicada en el transporte de moléculas antivirales (133).
Se ha descubierto que varias de-ISGilasas virales invierten la ISGilación intracelular, potenciando así la secreción extracelular de ISG15. Entre los virus conocidos que utilizan este mecanismo se encuentran los coronavirus (como el SARS-CoV2), los nairovirus y los virus de la fiebre aftosa humana, lo que sugiere que la secreción extracelular de ISG15 y la posterior señalización de ISG15 asociada a la liberación de IL-8 e IFNγ podrían ser los responsables, al menos en parte, del síndrome de liberación de citoquinas o «tormenta de citoquinas» observado tras la infección por SARS-Cov2 que provoca la COVID-19 (131). La explicación más sencilla para el hecho de que la conjugación intracelular de ISG15 inhiba la secreción, es que la ISGilación limita el pool de ISG15 libre disponible para la secreción (131). En particular, el papel de la vía de la ISG15 en la ECV ha sido explorado recientemente por nuestro grupo de investigación, como describiremos más adelante.
6. HIPERTENSIÓN Y ALTERACIONES VASCULARES FUNCIONALES Y ESTRUCTURALES
La hipertensión induce alteraciones en la función y la estructura de la vasculatura que, a su vez, pueden contribuir al aumento de la presión arterial. Estas alteraciones incluyen principalmente el remodelado vascular, la disfunción endotelial y el aumento de las respuestas vasoconstrictoras (20), que tienen valor pronóstico para la ECV (156, 157).
El fenotipo aórtico hipertensivo clásico se caracteriza por la degeneración y calcificación de la pared vascular y el aumento del diámetro de la aorta (20), pero los cambios vasculares con la hipertensión son más complejos y dependen del lecho vascular. En la hipertensión primaria, el remodelado de las grandes arterias se caracteriza por un aumento del grosor de la íntima-media (IMT) (alrededor de +15-40%). Además, en las arterias elásticas proximales es común un agrandamiento del lumen, y no se suelen observar cambios en el diámetro del lumen de las arterias musculares distales (31). Durante la hipertensión, en las arterias grandes, se produce una aceleración del remodelado hipertrófico hacia el exterior y también se observa un aumento de la rigidez con el envejecimiento (20, 158). En la hipertensión avanzada, las láminas elásticas de las grandes arterias se duplican y fragmentan y aumenta la deposición de colágeno y fibronectina, lo que contribuye a aumentar la rigidez, y también se ha observado la hipertrofia de CMLV en la aorta (31). En las arterias de resistencia, se encuentra hiperplasia, hipoplasia o la ausencia de cambios de las CMLV incrustadas en una pared vascular más gruesa, dependiendo de las diferentes arterias estudiadas en distintos modelos de roedores hipertensos (20, 159, 160).
Como se ha mencionado anteriormente, el remodelado eutrófico suele encontrarse en la hipertensión primaria, tanto en humanos como en SHR y en arterias mesentéricas de ratones infundidos con Ang II, probablemente debido al crecimiento hacia el interior con apoptosis periférica o a la vasoconstricción incrustada en una MEC expandida (20, 26, 83, 159, 161). De hecho, existe una deposición de colágeno y fibronectina con aumento de la relación colágeno: elastina en pequeños vasos de humanos y roedores hipertensos (20) que puede ser inducida por ET-1 (162), Ang II y aldosterona (163, 164). El remodelado hipertrófico se ha descrito en la hipertensión secundaria como en la hipertensión renovascular, el aldosteronismo primario o en el feocromocitoma, pero también en la hipertensión asociada a la diabetes mellitus. En la hipertensión por mineralocorticoides en roedores y en las ratas Dahl sensibles a la sal el remodelado de las arterias pequeñas también es hipertrófico (20). El remodelado de las arterias pequeñas puede ser la primera manifestación del daño en el órgano diana, al menos en la hipertensión humana, ya que, en una serie de pacientes, el 100% de los sujetos hipertensos en fase I mostraban remodelado de las arterias pequeñas, mientras que solo el 60% presentaban disfunción endotelial (165).
La disfunción del endotelio se asocia a menudo con la elevación de la presión arterial, pero no está del todo claro si es una causa o una consecuencia. Hay pruebas abrumadoras de que, en la hipertensión, la disfunción endotelial se manifiesta como una reducción de la vasodilatación dependiente del endotelio, sin modificaciones generales de las respuestas vasculares al NO exógeno (32). La disfunción endotelial también se caracteriza por un fenotipo inflamatorio de las CE con un aumento de la proliferación, la muerte celular programada, la alteración de la morfología, la producción de proteína C reactiva (PCR) y otros mediadores inflamatorios y trombogénicos, como MCP-1 y el inhibidor del activador del plasminógeno 1, la regulación al alza de las moléculas de adhesión y el aumento de la trombogenicidad y la adhesividad para las células circulantes (166). Esta disfunción endotelial es paralela a una mayor respuesta frente a diversos agonistas vasoconstrictores, como los agonistas alfa adrenérgicos o los TP, entre otros, aunque esto depende claramente del lecho vascular y del modelo animal estudiado (167, 168).
Actualmente se acepta que la activación del SRA, el estrés oxidativo y los procesos proinflamatorios contribuyen a las alteraciones funcionales y estructurales asociadas a la hipertensión (169-171).
Esto se discutirá en detalle en las siguientes secciones.
6.1. Papel de las ROS en la función y el remodelado vascular en la hipertensión
Varios factores implicados en la hipertensión, como la Ang II, el aumento de sodio, las catecolaminas y la alteración de las fuerzas mecánicas, aumentan la producción celular de ROS (171). Se ha observado un aumento de los niveles de ROS en pacientes hipertensos (172-176), y en el plasma y los vasos sanguíneos de modelos animales de hipertensión como las ratas SHR, las ratas con acetato de desoxicorticosterona (DOCA) y los ratones C57Bl6 infundidos con Ang II (167, 177-180).
Como se ha mencionado anteriormente, la Ang II es capaz de inducir la producción de ROS principalmente a través de las NOX y las especies de oxígeno generadas activan vías de señalización como quinasas (MAPK, Akt, c-Src o ERK1/2), factores de transcripción (NFκB, AP-1, STAT3 o Nrf2), MMP, canales iónicos (canales de Ca2+ o canales de Na+) o varios genes que codifican citoquinas, quimioquinas o factores de crecimiento. Estas vías de señalización desempeñan un papel importante en el crecimiento celular, la angiogénesis, la migración y la proliferación, promoviendo la disfunción vascular (180). Aparte de las NOX, las mitocondrias producen un exceso de ROS en pacientes hipertensos y en modelos animales, y existen mecanismos de retroalimentación por los que las ROS de las NOX pueden estimular la formación de radicales en las mitocondrias y viceversa (181). Nuestro grupo también describió una relación recíproca entre las ROS y la COX-2 que tuvo consecuencias en la disfunción vascular en la hipertensión (167). Además, la NO sintasa desacoplada y la xantina oxidasa también han sido implicadas en la formación de ROS en la hipertensión (171).
La literatura que demuestra que las ROS están implicadas en las alteraciones funcionales de la vasculatura observadas en la hipertensión es abrumadora. Así, los estudios que utilizan enfoques farmacológicos con antioxidantes o inhibidores de las diferentes fuentes de ROS, y los estudios que utilizan modelos de ratones transgénicos para las subunidades NOX y otras enzimas implicadas en la generación de ROS, parecen demostrar que, en general, estas estrategias normalizan las respuestas vasoconstrictoras aumentadas, la vasodilatación dependiente del endotelio deteriorada o ambas (180, 182, 183). Por ejemplo, nuestro grupo demostró en estudios ex vivo e in vivo con modelos animales de hipertensión que quelantes de ROS, como la apocinina, el tempol o el mito-tempo, reducían las respuestas vasoconstrictoras aumentadas inducidas por la fenilefrina o la serotonina y/o mejoraban la función endotelial en SHR o ratones infundidos con Ang II (164, 167, 184, 185). Asimismo, varios estudios sugieren que las ROS producidas por NOX1 y NOX5 disminuyen la relajación endotelial, y NOX5 también aumenta la vasoconstricción (180). Curiosamente, las ROS de NOX4 podrían aumentar la relajación endotelial, atribuible a la producción de H2O2 (180). Mecánicamente, la teoría más aceptada supone que el exceso de O2.- reacciona con el NO facilitando la formación de ONOO– que reduce la biodisponibilidad del NO y produce estrés nitrosativo a nivel vascular (65, 186).
Como se ha comentado, las ROS activan las vías celulares responsables de la proliferación y migración de las CMLV, la generación de proteínas de la matriz y la activación de las MMP (41). Este impacto en el remodelado y en la alteración de la mecánica vascular se ha demostrado en muchos estudios utilizando de nuevo estrategias farmacológicas o genéticas (63, 83, 180, 187). Por ejemplo, en respuesta a la Ang II, los ratones NOX-1-/- mostraron una marcada reducción de la hipertrofia aórtica debida a una disminución de la acumulación de MEC y no a cambios en el número de CMLV (188). En general, las ROS derivadas de NOX1 facilitan el remodelado vascular al provocar la desdiferenciación de las CMLV e inducir su proliferación y migración. Sin embargo, NOX4 parece preservar el fenotipo del músculo liso, aunque también se ha descrito un papel en la proliferación y la migración. Así, dependiendo del modelo de enfermedad o de la localización celular concreta, se pueden encontrar efectos beneficiosos o deletéreos de NOX4 (83).
6.2. Papel de la inflamación en la función y el remodelado vascular en la hipertensión
En las últimas décadas se ha demostrado de forma convincente el papel de diferentes componentes de los sistemas inmunitarios innato y adaptativo como factores que contribuyen al desarrollo de la hipertensión. En concreto, se ha demostrado el papel de los linfocitos T en la hipertensión, así como en la disfunción endotelial que la acompaña (96, 189-191). Utilizando modelos murinos de hipertensión, Guzik et al. fueron los primeros en demostrar la dependencia de la hipertensión por Ang II de la presencia de linfocitos T (190). Además, la hipertensión inducida por la Ang II se redujo en torno a un 50% en ratones CD8-/- (192), y en un pequeño estudio de 45 pacientes hipertensos, Ji et al. demostraron un aumento significativo de los linfocitos T Th1 y Th17 circulantes, en contraste con un descenso drástico de los linfocitos T Th2 (193). El mecanismo por el que los linfocitos T promueven el daño vascular en la hipertensión se está investigando activamente. Se sabe que los linfocitos T CD4+ se activan por diversos estímulos hipertensivos, como la alta concentración de sal, las ROS o la Ang II (191). En general, una vez que estas células se han activado, se diferencian en fenotipos T-cooperadores 1, 2, o 17 (TH1, TH2 o TH17). Las células polarizadas hacia el fenotipo TH1 producen citoquinas proinflamatorias IFNγ, IL-2, TNFα y TNFβ(191). Por su parte, la IL-17, citoquina clave de la respuesta Th17, también participa en el desarrollo de la hipertensión. Así, la infusión de IL-17 aumenta la presión arterial y produce un remodelado inward de las arterias mesentéricas, y el tratamiento con un anticuerpo neutralizante de IL-17A disminuyó el remodelado vascular en un modelo de infusión de Ang II (194). Además, aunque menos estudiado, varias pruebas sugieren un papel para la activación de las células B y la producción de IgG en el remodelado vascular y la disfunción endotelial que contribuyen a la rigidez vascular y la exacerbación de la hipertensión (96).
Los monocitos/macrófagos también participan en el desarrollo de la hipertensión, puesto que se ha encontrado un aumento de los marcadores M1 en los tejidos vasculares de ratones infundidos con Ang II durante dos (195) y cuatro semanas (196, 197). Además, estos monocitos/macrófagos tienen un papel causal en el desarrollo del remodelado vascular y la disfunción endotelial, puesto que diferentes tipos de ratones deficientes en macrófagos (98, 198-200) están protegidos frente al desarrollo del remodelado vascular, la disfunción endotelial y la hipertensión. Además, recientemente hemos publicado que los medios condicionados de macrófagos de ratones infundidos con Ang II inducen la disfunción endotelial a través de la liberación de IL-1b y prostaglandinas derivadas de la COX-2 (201). Los macrófagos tipo M2 también se encontraron dentro de la pared vascular después de la infusión de Ang II (202) y parecen promover la rigidez vascular y el remodelado de la MEC, incluyendo la deposición de colágeno, la fibrosis adventicial y la pérdida de elastina (202), confirmando que los macrófagos tipo M1 o M2 tienen un papel en la disfunción vascular en la hipertensión.
Una gran familia de receptores cuya activación conduce a la producción de citoquinas inflamatorias a través de NFκB, es la familia de los PRR. Dentro de esta familia destaca el TLR4, ya que su expresión está aumentada en varios modelos de hipertensión y su inhibición con un anticuerpo neutralizante previene el remodelado vascular asociado a la hipertensión, la rigidez, la hipercontractilidad y la disfunción endotelial, a través de la inhibición del estrés oxidativo (185, 203).
En respuesta a diversos estímulos, como las ROS, las citoquinas inflamatorias, las fuerzas mecánicas y las catecolaminas, las CE expresan mayores niveles de quimioquinas, selectinas y moléculas de adhesión, como ICAM-1 y VCAM-1, producidas no sólo por las CE, sino también por las CMLV. Los monocitos poseen ligandos, como el antígeno 4 muy tardío (VLA4), el LFA-1 y el antígeno 1 de los macrófagos (MAC1), que se unen a los receptores de la superficie de las CE y promueven inicialmente el rodamiento, luego la adhesión y finalmente la transmigración. Los monocitos transmigrados pueden transformarse en macrófagos inflamatorios, células dendríticas derivadas de monocitos, o pueden existir en un estado mínimamente diferenciado pero activado y posteriormente resurgir como monocitos circulantes activados (171). La hipertensión inducida por la Ang II se asoció con un aumento de la expresión vascular de ICAM-1, y esto se atenuó al inhibir la NOX (204). Recientemente, Lang et al. observaron que un anticuerpo neutralizante de ICAM-1 redujo notablemente la hipertensión, mejoró la función vascular, redujo la hipertrofia vascular y atenuó la inflamación vascular en ratones infundidos con Ang II (205), confirmando el papel de estas moléculas de adhesión en el daño vascular derivado de la hipertensión. La expresión endotelial de VCAM-1 es estimulada por las ROS y las fuerzas mecánicas alteradas, y es inhibida por el NO (171). Por lo tanto, sería concebible que la expresión de VCAM-1 por parte de las CE también esté aumentada en la hipertensión. De hecho, se han notificado correlaciones entre la presión arterial y los niveles circulantes de VCAM-1 en humanos (206, 207). Sin embargo, el papel específico de VCAM-1 en el daño vascular en la hipertensión no se ha dilucidado experimentalmente, probablemente porque la supresión embrionaria de VCAM-1 es letal (171).
6.2.1. Papel de IFNγ TNFα e ISG15 en la función y el remodelado vascular en la hipertensión
Diversos estudios han demostrado que la infusión de Ang II en ratones se asocia con una elevada expresión de IFNγ en las lesiones vasculares (191), el corazón (208, 209), el endotelio vascular (210), el músculo liso (210) y el riñón (211), con importantes funciones en el daño causado por la hipertensión. Además, el IFNγ induce el estrés oxidativo y la disfunción endotelial (212) y los ratones knockout para IFNγ están protegidos contra el daño cardíaco y la disfunción endotelial inducidos por la Ang II (208-210). El aumento del TNFα se observa en muchas condiciones fisiopatológicas, incluida la hipertensión (213) y tiene un papel en la función vascular, ya que un anticuerpo neutralizante del TNFα disminuyó la formación de ROS y mejoró la vasodilatación mediada por el NO (213). Estos hallazgos ponen de manifiesto el papel del IFNγ y del TNFα en el daño cardiovascular asociado a la hipertensión. La relación entre las ROS y la inflamación a nivel vascular está aceptada, no sólo porque estas citoquinas proinflamatorias inducen estrés oxidativo en los tejidos vasculares, sino también porque las ROS liberadas por las células inflamatorias pueden afectar a la homeostasis vascular. Uno de nuestros trabajos recientes muestra esta relación. En primer lugar, en este trabajo (214) identificamos mediante análisis bioinformático al TNFα y al IFNγ como reguladores maestros implicados en el desarrollo de la hipertensión. Como ya hemos mencionado previamente, ambas citoquinas inducen la expresión de ISG15 en diversos tipos celulares (111). Encontramos una correlación positiva entre los niveles de PAS y los niveles de expresión de ISG15 y entre el grado de remodelado vascular carotideo y la expresión de ISG15 en PBMCS provenientes de pacientes asintomáticos. Describimos también el aumento, dependiente de IFNγ, en la expresión de ISG15 a nivel vascular, en respuesta a la infusión con Ang II.
Observamos como ratones deficientes en ISG15 están protegidos frente a la hipertensión, la rigidez vascular, la alteración en la matriz de elastina y la disfunción endotelial inducida por Ang II, y que esta protección, depende, en gran medida, de la disminución en la inflamación y en la producción de ROS. Finalmente, evaluamos también, como ISG15 induce per se, disfunción endotelial a través de la producción vascular de ROS y la producción de prostaglandinas liberadas de COX-2 (Figura 8).
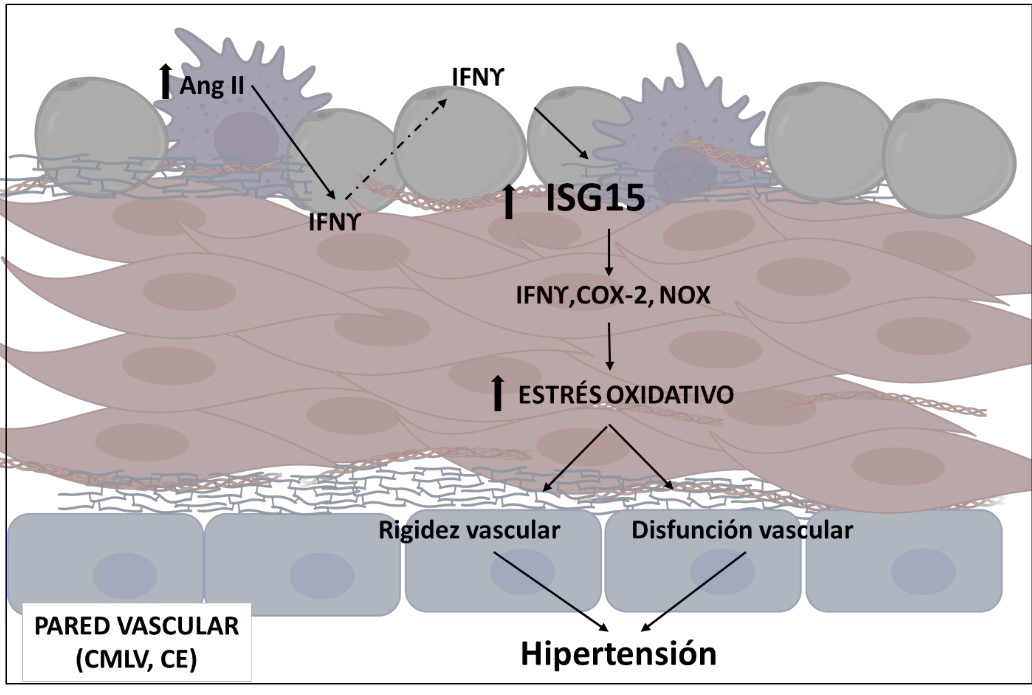
Figura 8. Modelo propuesto para el papel de ISG15 en el daño vascular en hipertensión. Ang II estimula la expresión de ISG15 a través de la producción de IFNγ en células endoteliales, musculares lisas vasculares y aorta. ISG15 libre produce IFNγ y mediadores inflamatorios como especies reactivas de oxígeno derivadas de NADPH oxidasa y derivados de la COX-2 que son responsables de la disfunción endotelial inducida por ISG15. ISG15 también participa en la rigidez vascular inducida por Ang II. Estas alteraciones vasculares podrían contribuir al desarrollo de la hipertensión. Modificado de (214).
7. OBESIDAD Y ALTERACIONES VASCULARES FUNCIONALES Y ESTRUCTURALES
El riesgo de ECV aumenta con el IMC (9). La dislipidemia, la intolerancia a la glucosa, la insensibilidad a la insulina, la hipertensión y los entornos protrombóticos y proinflamatorios desempeñan un papel importante en la fisiopatología de la obesidad (215). Una elevada proporción de pacientes obesos desarrolla hipertensión, disfunción endotelial y remodelado vascular (216) (Figura 9).
La disfunción endotelial es una de las primeras alteraciones vasculares observadas en la obesidad. Numerosos estudios han descrito la disfunción endotelial en diferentes modelos de obesidad, incluyendo la obesidad genética, la obesidad inducida por la dieta o la inducción de alteraciones neuroendocrinas (216). En pacientes obesos, se ha observado una disfunción endotelial junto con hiperglucemia, inflamación y estrés oxidativo (217). De hecho, la mayor parte de los estudios reportan el estrés oxidativo y la inflamación como mecanismos subyacentes. Este tema se tratará más adelante. La disfunción endotelial se ha demostrado incluso en adolescentes y niños con obesidad. Además, esta alteración de la función endotelial no sólo afecta a las arterias de conducción, como la aorta, sino también a las arterias pequeñas, como las mesentéricas, las coronarias, las renales o las del pene. En la disfunción endotelial causada por la obesidad existe un desequilibrio entre los factores vasodilatadores (como NO, EDHF, PGI2) y vasoconstrictores (como Ang II, ET-1, TXA2) y también una reducción de los niveles o la actividad de la eNOS (Figura 9), que parece mejorar en respuesta al ejercicio o a los suplementos dietéticos (216, 218). Además, un estudio reciente demostró que la disfunción endotelial y el engrosamiento de la pared observados en la aorta de ratones alimentados con un alto contenido de fructosa estaban asociados a una reducción de la diversidad de la microbiota intestinal y a una disminución de la abundancia de bacterias beneficiosas (219). Todos estos datos apoyan la complejidad de los mecanismos implicados en las alteraciones funcionales vasculares que se producen en la obesidad.
La obesidad se asocia con el remodelado vascular, caracterizado principalmente por el engrosamiento de la media y la rigidez arterial, no solo en las arterias de conductancia como la aorta, sino también en las pequeñas como las mesentéricas, renales y coronarias (21, 216, 220, 221). En este sentido, el IMT, un marcador del remodelado vascular, es un buen predictor de eventos cardiovasculares en adultos obesos (222, 223). El remodelado vascular también se observa en las pequeñas arterias subcutáneas de pacientes hipertensos con sobrepeso u obesidad, y se acompaña de un aumento de la fibrosis o de una reducción de la elasticidad (224). La alteración de la estructura vascular implica diferentes mecanismos que incluyen el remodelado de la MEC o la hiperplasia de las CMLV (21, 216). De hecho, la proliferación de las CMLV es una característica común reportada en los vasos en el contexto de la obesidad (221). Esta proliferación excesiva puede contribuir al engrosamiento de la media y parece estar facilitada por el fenotipo sintético de las CMLV (caracterizado por una alta tasa de proliferación y síntesis de MEC y factores vasoactivos) (216). En este sentido, la fibrosis vascular debida a acumulación de colágeno tipo I es también una característica común asociada a la obesidad (216). Los cambios en los niveles de expresión de elastina no se han asociado con la obesidad, pero si se han descrito cambios en su estructura 3D, con una reducción en el número de fenestras en la lámina elástica interna en arterias mesentéricas de ratones obesos (221). Esta alteración afecta a las propiedades mecánicas vasculares, produciendo así la rigidez del vaso (Figura 9) (225, 226).
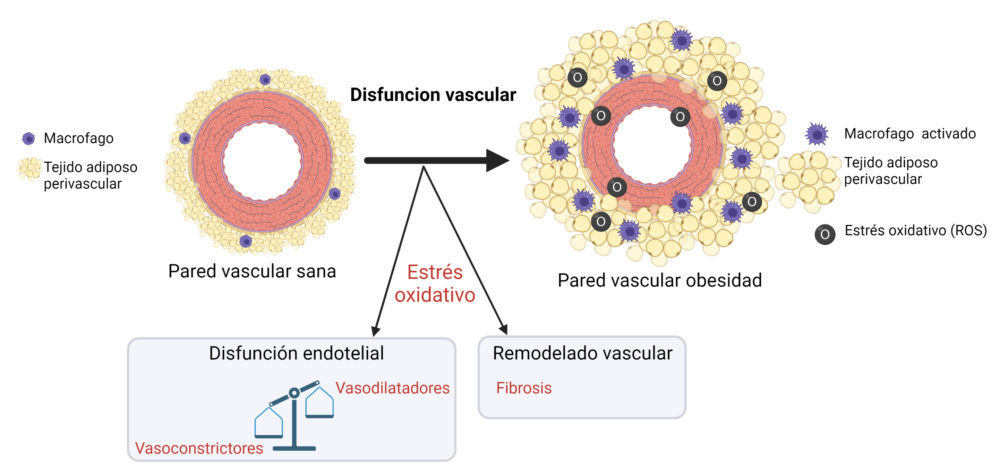
Figura 9. Papel del estrés oxidativo en diferentes mecanismo implicados en as alteraciones vasculares asociadas a la obesidad. En condiciones de obesidad, se observa un aumento del tejido adiposo perivascular (PVAT), del estrés oxidativo (ROS) y de los macrófagos del vaso. El estrés oxidativo contribuye a la disfunción endotelial y al remodelado vascular observado en la obesidad. Modificado de (216).
La obesidad induce profundos cambios en el PVAT. En animales y humanos obesos, tanto el volumen del PVAT como el tamaño de los adipocitos aumentan en todos los lechos vasculares. Además, la fracción estromal de este tejido cambia hacia un perfil más inflamatorio (85). Este fenotipo obeso cambia el perfil secretor del PVAT afectando a la función vascular (Figura 9). Aunque podría haber algunas diferencias entre los distintos lechos vasculares, probablemente, el efecto más común de la disfunción del PVAT en la obesidad es una pérdida de los efectos anticontráctiles inducidos por el PVAT, y un mayor fenotipo proinflamatorio (85). Además, se ha descrito un deterioro de las respuestas vasodilatadoras dependientes de endotelio en las aortas de animales obesos en presencia de PVAT (227). Curiosamente, se ha sugerido que, durante los pasos iniciales de la obesidad inducida por la dieta, el aumento de los niveles de leptina conduce a una sobreproducción de NO en el PVAT, lo que podría preservar la función vascular. Sin embargo, a largo plazo, el aumento de los niveles de leptina se correlaciona con una pérdida en la expresión de NO y eNOS derivada del PVAT, probablemente debido al desarrollo de resistencia a la leptina (228). Los mecanismos subyacentes responsables de la falta de efectos beneficiosos del PVAT en la obesidad son el aumento en la producción de citoquinas proinflamatorias, como se comenta a continuación.
7.1. Papel de las ROS en el daño vascular en la obesidad
Las ROS desempeñan un papel clave en el daño vascular causado por la obesidad, ya que afectan tanto a la disfunción endotelial como al remodelado y la rigidez vascular (Figura 9). El aumento de las ROS podría ser un acontecimiento temprano antes del desarrollo de las alteraciones funcionales vasculares. Así, en un modelo animal de dieta alta en grasa durante seis semanas, las ratas obesas presentaron fibrosis aórtica e inflamación vascular incluso en ausencia de alteraciones funcionales vasculares, y estas alteraciones estructurales se acompañaron de un aumento de los niveles de O2.- (220).
Un gran número de pruebas experimentales y humanas han mostrado que la obesidad se asocia con la disfunción endotelial en diferentes lechos vasculares y esto se debe principalmente a la biodisponibilidad comprometida del NO debido al incremento en los niveles de estrés oxidativo por el aumento de todas sus fuentes principales (216, 218, 229, 230). El estrés oxidativo observado en la obesidad también podría participar en el remodelado vascular y la fibrosis, aunque existen menos estudios sobre esta implicación (21, 216). Aunque las ROS generadas localmente pueden afectar claramente a la función y estructura vascular, en las dos últimas décadas se ha hecho evidente que, en la obesidad, el PVAT que rodea todo tipo de arterias es una fuente importante de ROS que deteriora las vías de señalización del NO. De hecho, los estudios que utilizan arterias montadas con PVAT, medios condicionados derivados de la incubación del PVAT y fenotipado molecular del PVAT de obesos, han demostrado claramente que el estrés oxidativo derivado del PVAT participa en la disfunción vascular en la obesidad (85, 231). Por ejemplo, los animales tratados con dieta alta en grasa mostraron el desacoplamiento de la eNOS y la reducción de la producción de NO y el aumento de la generación de O2.- en el PVAT, induciendo los efectos pro-contráctiles (228, 232).
La incubación ex vivo con SOD y catalasa restaura la función anticontráctil del PVAT de individuos obesos (233, 234). Otro estudio descubrió que el PVAT de ratones obesos mostraba una mayor formación de H2O2 y O2 que repercute en el endotelio vascular, ya que la alteración de la vasodilatación dependiente del endotelio inducida por el PVAT en las aortas obesas se restablecía tras la incubación con el antioxidante tirón o tras la eliminación de H2O2 mediante polietilenglicol-catalasa (227). Además de este efecto, el TNFα derivado del PVAT parece tener un papel clave. Así, un estudio reciente descubrió que el PVAT de ratas obesas inducía una disfunción endotelial que estaba mediada por una mayor producción de TNFα que a su vez activaba la NOX2 (235), lo que sugiere una interacción entre la inflamación y el estrés oxidativo en el daño vascular dependiente del PVAT.
7.2. Papel de la inflamación en el daño vascular en la obesidad
Se sabe que la obesidad y otros estados de desnutrición alteran la función inmunitaria, modificando las poblaciones de leucocitos y las respuestas inmunitarias mediadas por células. En 2003, dos estudios informaron simultáneamente de que la obesidad induce la infiltración de macrófagos en el tejido adiposo tanto en ratones como en humanos (236, 237). De hecho, el tejido adiposo de los individuos delgados sólo contiene entre un 5% y un 10% de macrófagos, pero estas células representan hasta el 60% del total de las células del tejido adiposo en los pacientes con sobrepeso inducido por la dieta (238). La obesidad no sólo aumenta la infiltración de macrófagos en el tejido adiposo, sino que provoca un cambio de los subtipos de macrófagos de M2 a M1, lo que conduce a un aumento de los niveles de citoquinas proinflamatorias (como TNFα e IL-6) y de ROS, que inducen la resistencia a la insulina (239). De hecho, Hotamisligil y sus colaboradores describieron que el tejido adiposo de los ratones obesos segrega TNFα, que tiene un papel directo en la resistencia a la insulina inducida por la obesidad (240). Este fue el primer vínculo funcional entre la obesidad y la inflamación, que evolucionó hacia el concepto de inflamación metabólica, que ha sido ampliamente aceptado como una importante conexión mecánica entre la obesidad y sus complicaciones (241). Después del TNFα, se demostró que el tejido adiposo produce una serie de citoquinas y quimioquinas como IL-6 y MCP-1, entre muchas otras, que regulan el metabolismo sistémico de la glucosa y los lípidos (242). Desde entonces, se han identificado casi todos los tipos principales de células inmunitarias en el tejido adiposo y se ha descubierto que participan en la regulación metabólica (243-245).
La producción local de adipoquinas es paralela al aumento de los niveles circulantes de proteínas proinflamatorias en adultos, adolescentes y niños (246-248). En concreto, en los adultos con sobrepeso y obesidad, se han notificado mayores niveles de TNFα, IL-6 o PCR (249, 250). Varios mecanismos parecen contribuir al desequilibrio del sistema inmunitario/inflamatorio en la obesidad (Figura 10). Por ejemplo, la obesidad se asocia a un deterioro de la producción de adipoquinas, con un aumento de los niveles de leptina proinflamatoria y una reducción de la adiponectina antiinflamatoria (Figura 11). Los ácidos grasos no esterificados, que pueden ser liberados en respuesta a la lipólisis, también pueden inducir la inflamación a través de varios mecanismos, como la modulación de la producción de adipoquinas, o la activación de los TLR o de los receptores activados por proliferadores de peroxisomas (PPAR) (Figura 10). Concretamente, la activación de los TLR induce la síntesis de marcadores inflamatorios en los macrófagos y agrava la resistencia a la insulina (251).
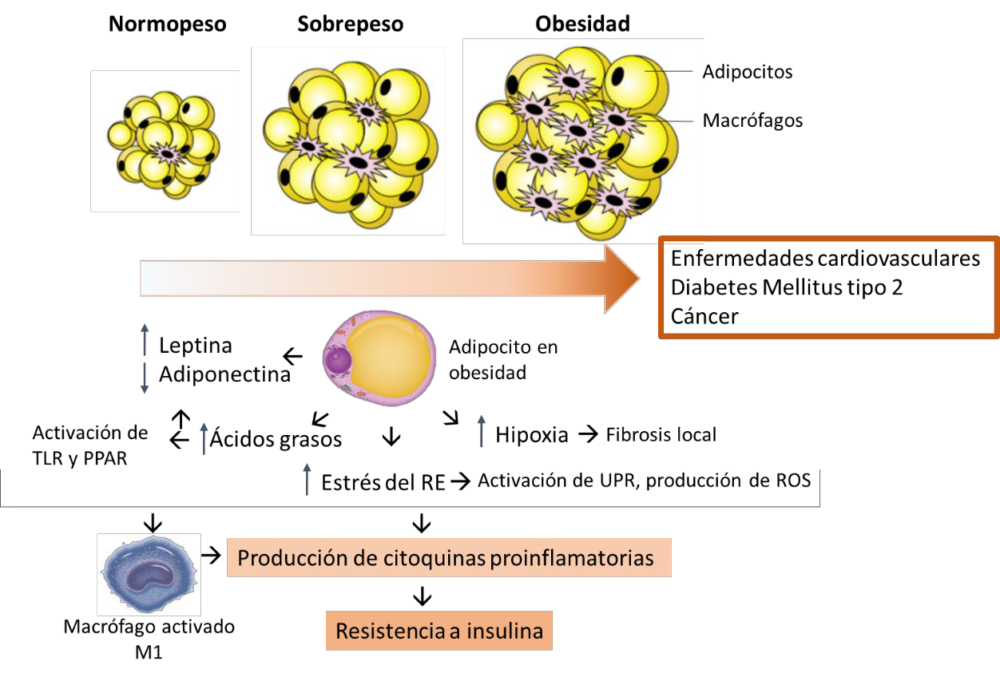
Figura 10. Mecanismos propuestos implicados en las alteraciones inmunológicas de la obesidad. Con la obesidad aumenta la cantidad y el tamaño de los adipocitos, pero también el número de células inmunitarias como los macrófagos. Los adipocitos hipertrofiados muestran una secreción alterada de adipoquinas y ácidos grasos, acompañada de un aumento del estrés del RE y de la hipoxia. En este escenario, los macrófagos aumentan su actividad liberando citoquinas proinflamatorias que provocan resistencia a la insulina. RE: retículo endoplásmico; TLR: receptores tipo Toll; PPAR: receptores activados por proliferadores de peroxisomas; UPR: respuesta a proteínas desplegadas; ROS: especies reactivas de oxígeno.
El exceso de nutrientes y la expansión de los adipocitos desencadenan el estrés del RE. Esto activa un mecanismo de seguridad conocido como respuesta a proteínas desplegadas (UPR), que se asocia a un aumento de la producción de ROS y a la expresión de diferentes citoquinas como IL-8, IL-6, MCP-1 y TNFα (Figura 10) (252). Además, durante la expansión del tejido adiposo en la obesidad llega un punto en el que el desarrollo de la vasculatura local y el suministro de oxígeno son insuficientes para las demandas del tejido, lo que promueve un entorno hipóxico. Se planteó la hipótesis de que los adipocitos hipóxicos producirían señales inflamatorias con el fin de estimular la angiogénesis (102, 253). Pero se ha observado que la respuesta a la hipoxia no consigue aumentar la vascularización del tejido adiposo, sino que desencadena una situación de fibrosis local, contribuyendo a la disfunción del tejido adiposo (254) (Figura 10).
Este estado inflamatorio crónico también se refleja localmente por la presencia continua de linfocitos T y macrófagos proinflamatorios en el PVAT y, posteriormente, por la elevación de citoquinas proinflamatorias, afectando a la fisiología vascular (85). Por ejemplo, Apovian et al. encontraron que la infiltración de macrófagos en el tejido adiposo subcutáneo se asocia con la disfunción endotelial sistémica y la resistencia a la insulina en pacientes obesos (255). Al igual que con el estrés oxidativo, una amplia literatura ha demostrado un papel deletéreo de la inflamación en la disfunción endotelial y el remodelado vascular en la obesidad (216, 218, 221, 229, 230).
7.2.1 Papel del TNFα e ISG15 en la obesidad
El TNFα es la citoquina más importante liberada por el PVAT obeso. Existe una correlación positiva entre los niveles de TNFα circulante y la disfunción endotelial en pacientes obesos (256). Es importante destacar que la hipoxia indujo una disfunción del PVAT que afectó a la función vascular, y este efecto se normalizó mediante la incubación in vitro con un anticuerpo anti-TNFα (257). En concordancia, la incubación ex vivo con el anticuerpo anti-TNFα infliximab mejora la producción de NO en pequeñas arterias aisladas de pacientes obesos y este efecto es más pronunciado en los vasos que contienen PVAT que en las arterias sin PVAT (258). Además, como se ha mencionado, TNFα puede activar las ROS en el PVAT e inducir la disfunción endotelial (235).
Datos no publicados llevados a cabo en nuestro laboratorio muestran que la expresión de ISG15 esta aumentada en arterias de ratones obesos inducidos por dieta alta en grasa en paralelo a un aumento en la expresión de TNFα. Además, hemos encontrado una correlación positiva entre el índice de masa corporal, los niveles de glucosa plasmática y la expresión de ISG15 en los PBMCs de pacientes asintomáticos, lo que podría indicar un papel de ISG15 en la obesidad y la diabetes (214). De acuerdo con esto, dos estudios recientes correlacionan a ISG15 con la obesidad (146) o la diabetes mellitus (259). Lo que es más importante, los ratones deficientes en ISG15 tienen un gasto energético elevado y son resistentes a la obesidad inducida por la dieta (146) y estos efectos se atribuyeron a la capacidad de ISG15 de producir ISGilación en casi todas las enzimas glucolíticas en los adipocitos. Estos datos apuntan a un posible papel de ISG15 en el desarrollo de las alteraciones asociadas a la obesidad.
8. ANEURISMAS AÓRTICOS ABDOMINALES (AAA)
El AAA es un agrandamiento y debilitamiento permanente de la pared aórtica. El desarrollo del AAA requiere un primer paso de aumento de la inflamación y la alteración de las proteínas de la MEC que conducen al debilitamiento de la pared arterial. Diferentes estudios demuestran que las paredes de las arterias aneurismáticas tienen menos elastina y colágeno de tipo I que las arterias normales. El desarrollo del AAA comienza con la neovascularización de la túnica media a través de los vasa vasorum. Esto puede iniciarse por la hipertensión, el tabaquismo o factores genéticos, causando lesiones en la pared aórtica. Tras la neovascularización, las células inflamatorias, como los macrófagos, migran a la pared del vaso liberando citoquinas proinflamatorias y MMP que estimulan a las CMLV a producir más MMP y otras proteinasas. Esto conduce a la descomposición del colágeno y la elastina, y promueve la migración de más células inflamatorias, estableciendo así una cascada de catabolismo de la pared aórtica asociada con la disfunción contráctil de las CMLV, el comportamiento mecánico anormal y la disfunción endotelial (12, 260, 261). Los mecanismos moleculares que subyacen a la formación de AAA no se conocen con claridad, pero las pruebas experimentales demuestran un papel importante del incremento de estrés oxidativo por NOX (262, 263), y la implicación del aumento de mediadores inflamatorios (264, 265), como se discutirá más adelante.
Algunos estudios han observado la implicación de un SRA desregulado en la formación y progresión del AAA, a través del receptor AT1 (266, 267). De hecho, el modelo animal más utilizado para el AAA es la infusión de Ang II en ratones hiperlipidémicos, probablemente por su simplicidad técnica y cierto grado de similitud con el AAA humano, incluyendo una marcada regulación al alza de la inflamación y el remodelado de la matriz extracelular (268, 269). Sin embargo, en algunos modelos genéticos de ratón o en ciertas condiciones experimentales, la Ang II conduce a la dilatación y disección aórtica sin coexistir con la ApoE o la deleción del LDLR (270, 271), lo que proporciona un gran potencial para la investigación de los mecanismos celulares y moleculares implicados en el AAA.
8.1. Papel de las ROS en el daño vascular en el AAA
Tanto el aumento de la producción de ROS procedente de diferentes fuentes como la desregulación de los sistemas antioxidantes se han propuesto como responsables del aumento del estrés oxidativo en el AAA, en humanos y modelos animales experimentales (272, 273). Los estímulos para la generación de ROS en la patología del AAA no están del todo definidos y probablemente actúan en combinación para aumentar el estrés oxidativo vascular. Así, los factores de riesgo clásicos del AAA como el tabaquismo, la edad avanzada, los factores prohipertensivos y proinflamatorios como la Ang II o las fuerzas mecánicas, podrían desencadenar reacciones enzimáticas para producir ROS.
Además, las citoquinas proinflamatorias, que son abundantes en el contexto del AAA, son un estímulo bien conocido para la generación de ROS a nivel vascular.
En general, se acepta que la desregulación de las ROS conduce a cambios generalizados en la pared arterial que juegan un papel principal en el proceso de desarrollo del AAA. Estos incluyen el aumento de la expresión de productos génicos proinflamatorios, la apoptosis del músculo liso y el aumento de la expresión y la activación de las MMP (272, 273). Lo que es más importante, diferentes enfoques dirigidos a las ROS han tenido eficacia para prevenir la formación de AAA en modelos animales experimentales. Por ejemplo, la administración de vitaminas antioxidantes (274), la supresión de las isoformas de NOX (275), la reversión del desacoplamiento de la eNOS (276), la supresión de la mieloperoxidasa de los neutrófilos o la sobreexpresión de la catalasa (277), entre muchos otros, han sido eficaces para prevenir la formación de AAA (272). Sin embargo, la información sobre la eficacia de estos tratamientos en los AAA establecidos es limitada y, lo que es más importante, hasta la fecha no se han notificado efectos beneficiosos de los tratamientos antioxidantes en ensayos clínicos bien diseñados.
8.2. Papel de la inflamación en el daño vascular en el AAA
La inflamación de la pared aórtica es un proceso de participación multicelular que incluye la infiltración de células mononucleares, la secreción de inmunoglobulinas (Ig) y la producción de citoquinas, lo que sugiere que están implicadas tanto las respuestas inmunitarias innatas como las adaptativas (265). A pesar de ser un aspecto clave, el papel de las células inflamatorias infiltradas en el AAA no se conoce completamente. Células T, macrófagos, células dendríticas, neutrófilos, células B y mastocitos se infiltran en la pared del AAA (265). Las interacciones entre estas células proporcionan el microambiente inflamatorio de las paredes aórticas. Por ejemplo, las citoquinas secretadas por las células T son esenciales para la activación de los macrófagos, mientras que las células dendríticas y los macrófagos pueden presentar antígenos a las células T para estimular las respuestas de las células T primarias (278). Asimismo, no hay consenso sobre cuáles son las citoquinas inflamatorias clave implicadas en el AAA, aunque se ha observado una mayor presencia de las citoquinas proinflamatorias IFNγ, IL-6, TNFα, IL-17 o de la antiinflamatoria IL-10 (279). Estudios recientes destacan la contribución de la inflamación del PVAT a la patología del AAA. Sagan et al. descubrieron que las células T, más que los macrófagos, son el principal subconjunto de leucocitos en el AAA y que se acumulan principalmente en el PVAT. Curiosamente, observaron que solo la infiltración de células T en el PVAT estaba fuertemente relacionada con el tamaño del AAA (280). En otro estudio, Meekel y colaboradores describieron que el PVAT de pacientes con AAA muestra un aumento de las MMP y de la expresión de genes proinflamatorios (PTPRC, CXCL8, LCK y CCL5), así como una disminución de la expresión del gen PPARG, un gen antiinflamatorio (281). Hay muchos estudios que muestran los efectos beneficiosos de la supresión génica de diferentes subtipos de células inflamatorias o citoquinas en la prevención de la formación de AAA (265, 282), aunque lamentablemente, en general, la regresión del AAA es un reto y no se consigue con éxito. Esto no se comentará en detalle en esta revisión.
8.2.1 Papel del IFNγ e ISG15 en el daño vascular en el AAA
Sharma et al. mostraron una elevada expresión de IFNγ en los aneurismas del modelo de infusión de elastasa (283). Del mismo modo, Zhou et al. demostraron el papel clave del IFNg producido por las células T CD8+ en el mismo modelo, y los resultados de varios artículos, incluidos dos metaanálisis (284-287), mostraron una mayor expresión de IFNγ en los aneurismas humanos. Sin embargo, se han descrito niveles circulantes de IFNγ disminuidos (279) o inalterados (288) en pacientes. Además, se ha sugerido un papel protector del IFNγ en la formación y ruptura de aneurismas (289). Sin embargo, también se ha descrito una falta de efecto tras el bloqueo del IFNγ en el modelo de aneurisma de ratón ApoE-/- infundido con Ang II (290). Se desconocen las razones de esta variabilidad, pero hay que tener en cuenta las diferencias en los modelos animales, en los pacientes o en la detección de citoquinas locales o circulantes. Por otro lado, existen estudios recientes que parecen sugerir un posible papel de los IFN de tipo I en la patología del aneurisma, tanto en modelos animales como en humanos (291).
Datos recientes de nuestro laboratorio (214) muestran el papel de ISG15 en el desarrollo de los AAA. Utilizando un modelo de ganancia de función examinamos el efecto de Ang II en el ratón transgénico USP18C61A, que selectivamente carece de la actividad proteasa, y que por tanto presenta un exceso de ISGilación. En estos ratones, Ang II indujo la disección letal aórtica en el 40% de los ratones y un incremento en la presión arterial mayor que la de sus correspondientes ratones controles. Los ratones USP18C61A infundidos con Ang II presentaron además un aumento en el diámetro de la aorta tanto torácica como abdominal que fue mayor que el de los ratones silvestres. Además, encontramos también un aumento en la expresión de ISG15 tanto en muestras de aortas de ratones ApoE-/- infundidos con Ang II como en muestras de aorta abdominal procedentes de pacientes. Este papel de la ISGilación es dependiente del estrés oxidativo, puesto que el tratamiento con el antioxidante TEMPOL mejoró la supervivencia y el remodelado vascular producido por la Ang II en los ratones USP18C61A, lo que nuevamente muestra una relación entre la inflamación y las ROS en el daño vascular e identifica a ISG15 como un mediador clave en el desarrollo de los aneurismas en una situación de hipertensión (Figura 11).
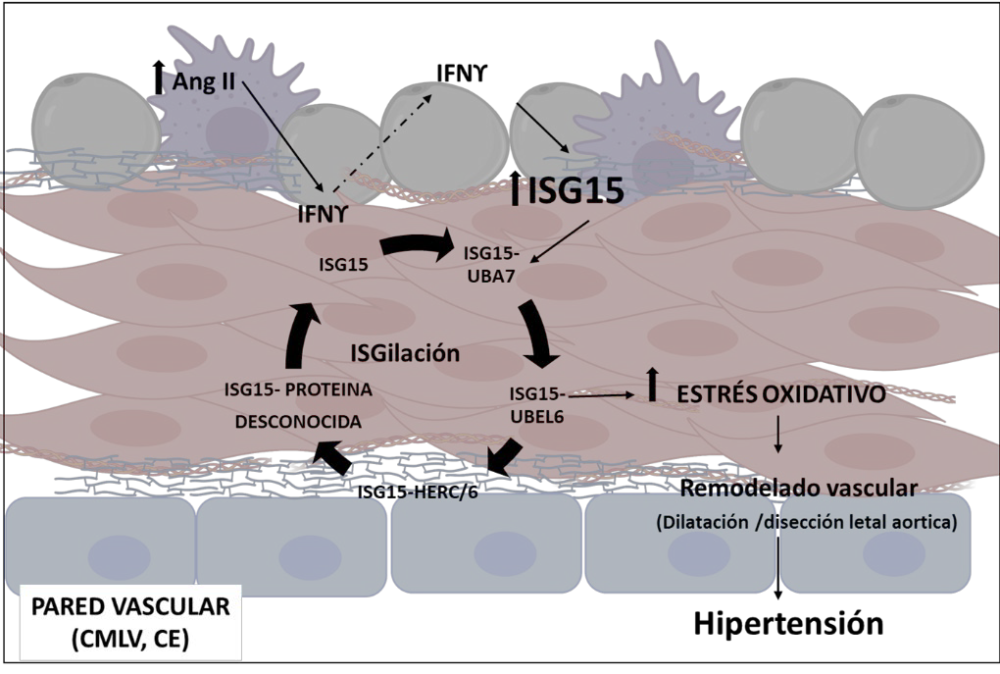
Figura 11. Modelo propuesto para el papel de ISG15 en el daño vascular en el desarrollo de aneurismas en hipertensión. Ang II estimula la expresión de ISG15 a través de la producción de IFNγ en células endoteliales, musculares lisas vasculares y aorta. Ang II induce también la ISGilación de proteínas desconocidas, lo que incrementa también el estrés oxidativo y participa en la dilatación y disección letal aórtica. Estas alteraciones vasculares podrían contribuir al desarrollo de la hipertensión. Modificado de (214).
9. CONCLUSIONES
Como ya se ha comentado, tanto la hipertensión, la obesidad y los AAA comparten alteraciones vasculares funcionales, estructurales y mecánicas y son importantes factores de riesgo de ECV. La obesidad es un factor de riesgo para el desarrollo de la hipertensión, al menos en parte debido a la liberación de sustancias vasoactivas del tejido adiposo que repercuten en la vasculatura subyacente (216), y ambos son factores de riesgo del desarrollo de los AAA. Actualmente se acepta que la inflamación de bajo grado y el aumento del estrés oxidativo tanto en el tejido adiposo (perivascular) como en la vasculatura son características de la hipertensión, la obesidad y los AAA. De hecho, abundantes pruebas en modelos animales preclínicos demuestran que el bloqueo de la inflamación o del estrés oxidativo tiene, en general, efectos beneficiosos sobre la homeostasis vascular en las tres patologías. Otra similitud entre ellas es que células inmunitarias como los macrófagos y los linfocitos T se infiltran en el tejido adiposo (perivascular) y en los vasos y producen mediadores inflamatorios como el TNFα y el IFNγ, que son efectores clave del mayor daño vascular observado en estas patologías. Ambas citoquinas inducen la expresión de ISG15 en diferentes células (111). A nivel vascular, la expresión de ISG15 se indujo por la Ang II de forma dependiente del IFNγ, se incrementó en vasos de modelos animales de hipertensión y correlacionó positivamente con la presión arterial sistólica en PBMCs humanas (214). La expresión de ISG15 está aumentada también en arterias de animales con obesidad inducida por dieta alta en grasa (datos no publicados) y existe una correlación entre ISG15, el índice de masa corporal y la glucemia en PBMCs (214), la obesidad (146), o la diabetes mellitus (259). Por último, la expresión de ISG15 está también aumentada en muestras de AAA procedentes de modelos animales de la enfermedad y en pacientes. Todos estos datos sitúan a ISG15 en el escenario de las tres patologías.
Uno de los rasgos más característicos del daño vascular en el contexto de la hipertensión y la obesidad es la disfunción endotelial, en la que influyen el IFNγ, el TNFα y las ROS liberadas localmente o desde el tejido adiposo perivascular subyacente (212, 235). En este contexto, la deleción de ISG15 mejoró la disfunción endotelial en la hipertensión al disminuir la inflamación y el estrés oxidativo (214). El remodelado y la rigidez vascular también son característicos de la hipertensión, la obesidad y el AAA. ISG15 y la ISGilación parecen claves en la formación de AAA y en el aumento de rigidez asociada a la hipertensión, nuevamente a través de mecanismos relacionados con la inflamación y la producción de ROS. No sabemos si los ratones deficientes en ISG15 estarán protegidos frente a las alteraciones vasculares inducidas por la obesidad, lo que justifica una mayor investigación. Sin embargo, todos estos datos apuntan a un posible papel del sistema IFNγ/TNFα/ISG15 como mediador común del daño vascular en la ECV.
10. PERSPECTIVAS
La eficacia de los principales fármacos antihipertensivos (diuréticos, inhibidores de la ECA, antagonistas AT1, bloqueadores de los canales de calcio y betabloqueantes) para disminuir la presión arterial en los pacientes hipertensos está establecida. Y lo que es más importante, muchos de estos enfoques farmacológicos disminuyen el riesgo cardiovascular a largo plazo (292). Sin embargo, a pesar del amplio conocimiento sobre los mecanismos responsables del desarrollo de la hipertensión y su relación con los aneurismas, la obesidad o incluso el infarto de miocardio y el ictus, sigue habiendo un número masivo de pacientes hipertensos que no tienen la presión arterial controlada, muchos son resistentes a los efectos reductores de la presión arterial de los fármacos mencionados, y parece haber un riesgo cardiovascular residual en estos pacientes. Además, no existe una relación directa y unívoca entre la elevación de la presión arterial y el daño de los órganos diana. Hasta la fecha, no existen tratamientos farmacológicos para ralentizar o hacer retroceder el desarrollo de los aneurismas, aunque en la actualidad se está actuando en estos pacientes sobre el SRAA o se utilizan inhibidores de la hidroximetilglutaril-CoA reductasa (estatinas), o de betabloqueantes. Los inhibidores del SRAA y las estatinas no solo disminuyen la presión arterial y los niveles lipídicos, sino que también podrían conferir cierto grado de protección vascular probablemente a través de la atenuación de la inflamación (293, 294). Los betabloqueantes, además de reducir la presión arterial y la frecuencia cardíaca, parecen tener un efecto positivo al aumentar la resistencia de la aorta al estiramiento (295). Del mismo modo, actualmente no existen enfoques farmacológicos para tratar la obesidad, y la protección cardiovascular se obtiene en pacientes seleccionados con los fármacos cardiovasculares clásicos. En conjunto, estos estudios sugieren que podría haber mecanismos adicionales implicados y que no están siendo eficazmente regulados por los tratamientos farmacológicos actuales.
Es interesante que, aunque numerosos estudios preclínicos demuestran la participación de las ROS en la hipertensión (296), la obesidad (297) y el AAA (272), el uso de sustancias para reducir el estrés oxidativo como tratamiento de estas enfermedades no ha demostrado su eficacia hasta el momento. Diferentes estudios epidemiológicos concluyeron que los individuos con mayor ingesta de antioxidantes tienen menor incidencia de riesgo cardiovascular (298, 299), y también se observaron efectos positivos al combinar los antioxidantes con otro tratamiento antihipertensivo (300, 301). Sin embargo, no se observa un efecto antihipertensivo claro en la mayoría de los estudios que analizan el efecto de los antioxidantes a largo plazo (302, 303). Las razones de estas discrepancias incluyen falta de consistencia, carencias en los criterios de selección, falta de información sobre las enfermedades concomitantes, defectos de la metodología utilizada para medir el estrés oxidativo y la omisión de aspectos científicos necesarios como las propiedades farmacocinéticas o la biodisponibilidad de los antioxidantes utilizados (304-306). De hecho, actualmente no existen inhibidores de NOX/ROS o antioxidantes aprobados clínicamente para su uso en el tratamiento de la hipertensión u otras enfermedades vasculares. Además, es necesario completar el conocimiento sobre los mecanismos moleculares que subyacen al estrés oxidativo y los beneficios de inhibir la formación de ROS frente a aumentar la desintoxicación de las mismas. En cualquier caso, se ha sugerido que, aunque es necesario llevar a cabo más estudios bien realizados para obtener resultados concluyentes sobre la eficacia de los antioxidantes en la ECV, la utilidad clínica de los agentes que reducen el estrés oxidativo para controlar la hipertensión y el daño de órganos diana es prometedora (180).
Como se expone en esta revisión, las crecientes pruebas en modelos animales y en pacientes apuntan a un posible papel de la inflamación como mecanismo adicional de inicio y mantenimiento de la ECV. Citoquinas específicas como TNFα o IL-1 se asocian a un alto riesgo cardiovascular, lo que apoya la teoría de que los tratamientos farmacológicos antiinflamatorios pueden ser una estrategia prometedora para reducir el riesgo cardiovascular en el ámbito de la terapia médica tradicional (307). De hecho, algunas de las terapias utilizadas actualmente en la clínica, por ejemplo, las estatinas y los antagonistas AT1, tienen efectos antiinflamatorios que podrían contribuir a sus efectos protectores en el sistema cardiovascular. Sin embargo, los resultados de los ensayos clínicos con antiinflamatorios son variables. Los primeros estudios en los que se utilizó un anticuerpo monoclonal frente a TNFα (infliximab, ensayo ATTACH) o un receptor soluble del TNF (etanercept, ensayos RECOVER y RENAISSANCE) mostraron falta de efecto o incluso efectos perjudiciales en pacientes con insuficiencia cardíaca (308). Sin embargo, los datos recientes del ensayo CANTOS han proporcionado resultados alentadores. En este estudio, los pacientes con antecedentes de infarto de miocardio y niveles elevados de PCR se trataron con un anticuerpo anti-interleucina-1b (canakinumab). Los autores mostraron una clara reducción de las tasas de eventos cardiovasculares que no se atribuyó a los cambios en la presión arterial o la hipertensión incidente (309, 310).
Tratar la inflamación podría ser un enfoque difícil debido a la abrumadora cantidad de vías y mediadores proinflamatorios descritos. Por un lado, es probable que la inhibición de un mediador específico no sea eficaz. Por otro lado, los fármacos antiinflamatorios generales podrían producir inmunosupresión y susceptibilidad a las infecciones. En este sentido, no podemos olvidar el papel esencial de la inflamación en la lucha contra los patógenos. Prueba de esta complejidad es el hecho de que los fármacos antiinflamatorios no esteroideos, que inhiben la actividad de la isoforma COX-2 implicada en la síntesis del mediador proinflamatorio PGE2, se asocian a un mayor riesgo cardiovascular (311). Entonces, identificar claramente los mecanismos moleculares específicos implicados en la ECV en relación con la inflamación podría contribuir a una optimización en el desarrollo de fármacos útiles para este propósito. En este sentido, nuestros recientes datos al respecto del papel de ISG15 como nuevo mediador del daño de vascular lo sitúan como una interesante nueva diana terapéutica.
Abreviaturas
AA aneurisma aórtico
AAA aneurisma de aorta abdominal
ADRF factor relajante derivado de los adipocitos
Ang angiontensina
AP-1 proteína activadora 1
AT1R receptor de angiotensina II tipo 1
CMLVs células musculares lisas vasculares
COX ciclooxigenasa
cPGES PGE2 sintasa citosólica
CSA área de la sección transversal
CXCL ligando de quimioquina (motivo C-X-C)
ECA enzima convertidora de angiotensina
ECV enfermedades cardiovasculares
EDHF factor hiperpolarizante derivado del endotelio
eNOS sintasa endotelial de óxido nítrico
ET-1 endotelina-1
GPCRs receptores acoplados a proteínas G
ICAM-1 molécula de adhesión intracelular 1
IFNs interferones
ILs interleuquinas
IMC índice de masa corporal
IMT espesor de la íntima-media
ISG15 gen 15 estimulado por interferón
JAK1/2 janus kinase 1/2 antígeno asociado a la función leucocitaria-1
LFA-1 antígeno asociado a la función leucocitaria
MCP-1 proteína quimioatrayente de monocitos 1
MEC matriz extracelular
MMP metaloproteinasas de la matriz
mPGES PGE2 sintasa ligada a la membrana
NADP nicotinamida adenina dinucleótido fosfato
NFAT factor nuclear de las células T activadas
NFκB factor nuclear kappa-cadena ligera reforzador activadas de células B
nNOS óxido nítrico sintasa neuronal
NOX NADPH oxidasa
Nrf2 factor nuclear (derivado de los eritroides 2) similar a 2
O2.- anión superóxido
ONOO- peroxinitrito
PAD presión arterial diastólica
PAS presión arterial sistólica
PBMCs células mononucleares de sangre periférica
PCR Proteína C-reactiva
PDGF factor de crecimiento derivado de las plaquetas
PG prostaglandina
PI3K fosfoinositida 3-quinasa
PPAR receptor activado por el proliferador de peroxisomas
PRR receptor de reconocimiento de patrones
PVAT tejido adiposo perivascular
RE retículo endoplásmico
ROS especies reactivas de oxígeno
SHR ratas espontáneamente hipertensas
SODs superóxido-dismutasas
SRA sistema renina-angiotensina
STAT transductor de señales y activador de la transcripción
Tempol 4-hidroxi-2,2,6,6-tetrametilpiperidin-1-oxilo
TGF-β factor de crecimiento transformador β
TLR receptores tipo toll
TNFs factores de necrosis tumoral
TXA2 tromboxano A2
VCAM-1 molécula de adhesión celular vascular 1
Conflicto de interés
Los autores declaran que no tienen ningún conflicto de interés.
Financiación
Este trabajo ha sido financiado por la Comunidad de Madrid (CM)-Universidad Autónoma de Madrid (SI1-PJI-2019-00321) y MGA ha sido subvencionada por una beca FPI-UAM.
11. REFERENCIAS
- Organización Mundial de la Salud. World Health Organization 2019: https://www.who.int/news-room/fact-sheets/detail/hypertension.
- Kumar V. Robbins and Cotran’s pathologic basis of disease. Philadelphia, Pa. London: Saunders. xiv; 2010.
- Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation. 2000; 101(3): 329-35.
- Kakar P, Lip GY. Towards understanding the aetiology and pathophysiology of human hypertension: where are we now? J Hum Hypertens. 2006; 20:833-36.
- Rang HP, Dale MM. Farmacología. 7ª edición. 2012. Elsevier. ISBN: 978-84-8086-908-9.
- Bund SJ, Lee RM. Arterial structural changes in hypertension: a consideration of methodology, terminology and functional consequence. J Vasc Res. 2003;40(6):547–57.
- Forouzanfar MH, Liu P, Roth GA, et al. Global Burden of Hypertension and Systolic Blood Pressure of at Least 110 to 115 mm Hg, 1990-2015. JAMA. 2017;317(2):165-182. Erratum in: JAMA. 2017;317(6):648.
- Regan JA, Shah SH. Obesity Genomics and Metabolomics: a Nexus of Cardiometabolic Risk. Curr Cardiol Rep. 2020;22(12):174.
- Organización Mundial de la Salud. World Health Organization2020:https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
- Sanghera DK, Bejar C, Sharma S, Gupta R, Blackett PR. Obesity genetics and cardiometabolic health: Potential for risk prediction. Diabetes Obes Metab. 2019;21(5):1088-100.
- Chaikof EL, Dalman RL, Eskandari MK, et al. The Society for Vascular Surgery practice guidelines on the care of patients with an abdominal aortic aneurysm. J Vasc Surg. 2018;67(1):2–77.e2.
- Anagnostakos J, Lal BK. Abdominal aortic aneurysms. Prog Cardiovasc Dis. 2021:S0033-0620(21)00037-2.
- Kuivaniemi H, Ryer EJ, Elmore JR, Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13(9):975–87.
- Wolinsky H, Glagov S. Comparison of abdominal and thoracic aortic medial structure in mammals. Deviation of man from the usual pattern. Circ Res. 1969;25(6):677‐86.
- Davis FM, Rateri DL, Daugherty A. Mechanisms of aortic aneurysm formation: translating preclinical studies into clinical therapies. Heart. 2014;100(19):1498–505.
- Gibbons GH, Dzau VJ. The emerging concept of vascular remodelling. N Engl J Med. 1994;330(20):1431–38.
- Renna NF, de Las Heras N, Miatello RM. Pathophysiology of vascular remodelling in hypertension. Int J Hypertens. 2013;2013:808353.
- Mulvany MJ. Vascular remodelling of resistance vessels: can we define this?. Cardiovasc Res. 1999;41(1):9–13.
- van Varik BJ, Rennenberg RJ, Reutelingsperger CP, Kroon AA, de Leeuw PW, Schurgers LJ. Mechanisms of arterial remodelling: lessons from genetic diseases. Front Genet. 2012;3:290.
- Schiffrin EL. Vascular remodelling in hypertension: mechanisms and treatment. Hypertension. 2012;59(2):367–74.
- Briones AM, Aras-López R, Alonso MJ, Salaices M. Small artery remodelling in obesity and insulin resistance. Curr Vasc Pharmacol. 2014;12(3):427-37.
- Mulvany MJ. Small artery remodelling in hypertension: causes, consequences and therapeutic implications. Med Biol Eng Comput. 2008;46:461–67.
- Intengan HD, Schiffrin EL. Vascular remodelling in hypertension: roles of apoptosis,inflammation, and fibrosis. Hypertension. 2001;38(3 Pt 2):581–87.
- Nørrelund H, Christensen KL, Samani NJ, Kimber P, Mulvany MJ, Korsgaard N. Early narrowed afferent arteriole is a contributor to the development of hypertension. Hypertension. 1994;24(3):301–08.
- Briones AM, Xavier FE, Arribas SM, et al. Alterations in structure and mechanics of resistance arteries from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol. 2006;291(1):H193–H201.
- Bakker EN, van der Meulen ET, van den Berg BM, Everts V, Spaan JA, VanBavel E. Inward remodelling follows chronic vasoconstriction in isolated resistance arteries. J Vasc Res. 2002;39(1):12–20.
- Briones AM, Arribas SM, Salaices M. Role of extracellular matrix in vascular remodelling of hypertension. Curr Opin Nephrol Hypertens. 2010;19(2):187-94.
- Dorrance AM, Matin N, Pires PW. The effects of obesity on the cerebral vasculature. Curr Vasc Pharmacol. 2014 May;12(3):462-72.
- Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89(3):957–89.
- Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37(5):1236-41.
- Laurent S, Boutouyrie P. The structural factor of hypertension: large and small artery alterations. Circ Res. 2015;116(6):1007-21.
- Brandes RP. Endothelial dysfunction and hypertension. Hypertension. 2014;64(5):924-8.
- Dal Lin C, Tona F, Osto E. Coronary Microvascular Function and Beyond: The Crosstalk between Hormones, Cytokines, and Neurotransmitters. Int J Endocrinol. 2015;2015:312848.
- Watson T, Goon PK, Lip GY. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid Redox Signal. 2008;10(6):1079-88.
- Duprez DA, Hearst MO, Lutsey PL, et al. Associations among lung function, arterial elasticity, and circulating endothelial and inflammation markers: the multiethnic study of atherosclerosis. Hypertension. 2013;61(2):542-8.
- Dal Lin C, Tona F, Osto E. The crosstalk between the cardiovascular and the immune system. Vasc Biol. 2019;1(1):H83-H88.
- Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007;27(2):266–74.
- Husain K, Hernandez W, Ansari RA, Ferder L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J Biol Chem. 2015;6(3):209–17.
- Anderson TJ, Gerhard MD, Meredith IT, et al. Systemic nature of endothelial dysfunction in atherosclerosis. Am J Cardiol. 1995;75(6):71B-74B.
- Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature. 1976;263(5579):663‐65.
- Hernanz R, Briones AM, Salaices M, Alonso MJ. New roles for old pathways? A circuitous relationship between reactive oxygen species and cyclo-oxygenase in hypertension. Clin Sci (Lond). 2014;126(2):111–121.
- Félétou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011;164(3):894-912.
- Nakahata N. Thromboxane A2: physiology/pathophysiology, cellular signal transduction and pharmacology. Pharmacol Ther. 2008;118(1):18-35.
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265‐69.
- Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524‐26.
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357(Pt 3):593-615.
- Marasciulo FL, Montagnani M, Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem. 2006;13(14):1655-65.
- Félétou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond). 2009;117(4):139–55.
- Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52(4):639–72.
- Hammoud RA, Vaccari CS, Nagamia SH, Khan BV. Regulation of the renin- angiotensin system in coronary atherosclerosis: a review of the literature. Vasc Health Risk Manag. 2007;3(6):937–45.
- McGuire DK, Winterfield JR, Rytlewski JA, Ferrannini E. Blocking the renin- angiotensin-aldosterone system to prevent diabetes mellitus. Diab Vasc Dis Res.2008;5(1):59–66.
- Cabandugama PK, Gardner MJ, Sowers JR. The Renin Angiotensin Aldosterone System in Obesity and Hypertension: Roles in the Cardiorenal Metabolic Syndrome. Med Clin North Am. 2017;101(1):129-37.
- Boos CJ, Lip GY. Is hypertension an inflammatory process? Curr Pharm Des.2006;12(13):1623-35.
- Ruiz-Ortega M, Esteban V, Rupérez M, et al. Renal and vascular hypertension- induced inflammation: role of angiotensin II. Curr Opin Nephrol Hypertens.2006;15(2):159–166.
- Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59(3):251–87.
- Bader M, Peters J, Baltatu O, Müller DN, Luft FC, Ganten D. Tissue renin-angiotensin systems: new insights from experimental animal models in hypertension research. J Mol Med (Berl). 2001;79(2-3):76–102.
- Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, Teboul M, Massiéra F, Sharma AM. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003 Jun;35(6):807-25.
- Lorenz JN. Chymase: the other ACE?. Am J Physiol Renal Physiol. 2010;298(1):F35– F36.
- Gradman AH, Kad R. Renin inhibition in hypertension. J Am Coll Cardiol.2008;51(5):519–28.
- Rüster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17(11):2985–91.
- Ocaranza MP, Jalil JE. Protective Role of the ACE2/Ang-(1-9) Axis in Cardiovascular Remodelling. Int J Hypertens. 2012;2012:594361.
- Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R, Eguchi S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol Rev. 8;98(3):1627-738.
- Touyz RM, Tabet F, Schiffrin EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol. 2003;30(11):860–66.
- Sumners C, Peluso AA, Haugaard AH, Bertelsen JB, Steckelings UM. Anti-fibrotic mechanisms of angiotensin AT2 -receptor stimulation. Acta Physiol (Oxf). 2019;227(1):e13280.
- Paravicini TM, Touyz RM. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31Suppl 2:S170–S180.
- Martín-Ventura JL, Rodrigues-Diez R, Martinez-Lopez D, Salaices M, Blanco-Colio LM, Briones AM. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int J Mol Sci. 2017;18(11):2315.
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10(6):453–71.
- Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators- induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84(5):581–90.
- Tsai KH, Wang WJ, Lin CW, et al. NADPH oxidase-derived superoxide anion- induced apoptosis is mediated via the JNK-dependent activation of NF-κB in cardiomyocytes exposed to high glucose. J Cell Physiol. 2012;227(4):1347–57.
- Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110(10):1364–90.
- Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal. 2014;20(1):164–82.
- Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. 2020;17(3):170-94.
- Touyz RM, Briones AM, Sedeek M, Burger D, Montezano AC. NOX isoforms and reactive oxygen species in vascular health. Mol Interv. 2011;11(1):27–35.
- Santillo M, Colantuoni A, Mondola P, Guida B, Damiano S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front Physiol. 2015;7;6:194.
- Cucoranu I, Clempus R, Dikalova A, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97(9):900–07.
- Lee DY, Wauquier F, Eid AA, et al. Nox4 NADPH oxidase mediates peroxynitrite- dependent uncoupling of endothelial nitric-oxide synthase and fibronectin expression in response to angiotensin II: role of mitochondrial reactive oxygen species. J Biol Chem. 2013;288(40):28668–86.
- Touyz RM, Montezano AC. Vascular Nox4: a multifarious NADPH oxidase. Circ Res. 2012;110(9):1159–61.
- Touyz RM, Anagnostopoulou A, Camargo LL, Rios FJ, Montezano AC. Vascular Biology of Superoxide-Generating NADPH Oxidase 5-Implications in Hypertension and Cardiovascular Disease. Antioxid Redox Signal. 2019;30(7):1027-40.
- Serrander L, Jaquet V, Bedard K, Plastre O, Hartley O, Arnaudeau S, Demaurex N, Schlegel W, Krause KH. NOX5 is expressed at the plasma membrane and generates superoxide in response to protein kinase C activation. Biochimie. 2007;89(9):1159-67.
- Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassègue B, Griendling KK. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. 2008;45(3):329-35.
- Montezano AC, Burger D, Paravicini TM, et al. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res. 2010;106(8):1363-73.
- Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease?. Trends Endocrinol Metab. 2014;25(9):452–63.
- García-Redondo AB, Aguado A, Briones AM, Salaices M. NADPH oxidases and vascular remodelling in cardiovascular diseases. Pharmacol Res. 2016;114:110-20.
- Snezhkina AV, Kudryavtseva AV, Kardymon OL, et al. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid Med Cell Longev. 2019;2019:6175804.
- Fernández-Alfonso MS, Somoza B, Tsvetkov D, Kuczmanski A, Dashwood M, Gil-Ortega M. Role of Perivascular Adipose Tissue in Health and Disease. Compr Physiol. 2017;8(1):23-59.
- Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM.Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16(9):1057-63.
- Zhang Y, Wang SJ, Han ZH, Li YQ, Xue JH, Gao DF, Wu XS, Wang CX. PI3K/AKT signaling pathway plays a role in enhancement of eNOS activity by recombinant human angiotensin converting enzyme 2 in human umbilical vein endothelial cells. Int J Clin Exp Pathol. 2014;7(11):8112-7.
- Gollasch M. Adipose-Vascular Coupling and Potential Therapeutics. Annu Rev. Pharmacol Toxicol. 2017;57:417-436.
- Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science. 2001.
- Harwani SC. Macrophages under pressure: the role of macrophage polarization in hypertension. Transl Res. 2018;191:45-63.
- Sattler S and Kennedy-Lydon T. The Immunology of Cardiovascular Homeostasis and Pathology. Springer. 2017.
- Ehlers S, Kaufmann SH; Participants of the 99(th) Dahlem Conference. Infection, inflammation, and chronic diseases: consequences of a modern lifestyle. Trends Immunol. 2010;31(5):184-90.
- Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13(4):260-70.
- Naviaux RK. Metabolic features of the cell danger response. Mitochondrion. 2014;16:7-17.
- Tang WH, Kitai T, Hazen SL. Gut Microbiota in Cardiovascular Health and Disease. Circ Res. 2017;120(7):1183-96.
- Drummond GR, Vinh A, Guzik TJ, Sobey CG. Immune mechanisms of hypertension. Nat Rev Immunol. 2019;19(8):517-32.
- Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738-49.
- Kossmann S, Hu H, Steven S, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289(40):27540-50.
- Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. 2020;17(6):327-40.
- Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83(2):456S–60S.
- Kofler S, Nickel T, Weis M. Role of cytokines in cardiovascular diseases: a focus on endothelial responses to inflammation. Clin Sci (Lond). 2005;108(3):205–13.
- Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92(3):347-55.
- Vicenová B, Vopálenský V, Burýsek L, Pospísek M. Emerging role of interleukin-1 in cardiovascular diseases. Physiol Res. 2009;58(4):481–98.
- Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation. Curr Pharm Des.2014;20(22):3579–94.
- Esteban V, Méndez-Barbero N, Jiménez-Borreguero LJ, et al. Regulator of calcineurin 1 mediates pathological vascular wall remodelling. J Exp Med. 2011;208(10):2125-39.
- Su C, Xue J, Ye C, Chen A. Role of the central renin-angiotensin system in hypertension (Review). Int J Mol Med. 2021;47(6):95.
- Zhang D and Zhang D-E. Interferon-stimulated gene 15 and the protein ISGylation System. J Interf Cytok Res. 2011; 31(1):119-130.
- Knight E Jr, Cordova B. IFN-induced 15-kDa protein is released from human lymphocytes and monocytes. J Immunol. 1991; 146:2280-84.
- Bogunovic D, Byun M, Durfee LA, Abhyankar A, Sanal O, Mansouri D et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. 2012; 337:1684–88.
- Tecalco Cruz AC, Mejía-Barreto K. Cell type-dependent regulation of free ISG15 levels and ISGylation. J Cell Commun Signal. 2017;11(2):127–35.
- Albert M, Bécares M, Falqui M, Fernández-Lozano C, Guerra S. ISG15, a Small Molecule with Huge Implications: Regulation of Mitochondrial Homeostasis. Viruses. 2018;10(11):629.
- Colonne PM, Sahni A, Sahni SK. Rickettsia conorii infection stimulates the expression of ISG15 and ISG15 protease UBP43 in human microvascular endothelial cells. Biochem Biophys Res Commun. 2011;416(1-2):153-8.
- Rahnefeld A, Klingel K, Schuermann A, et al. Ubiquitin-like protein ISG15 (interferon-stimulated gene of 15 kDa) in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation. 2014;130(18):1589-600.
- Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JE. Synergistic interaction between interferon-a and interferon-g through induced synthesis of one subunit of the transcription factor ISGF3. The EMBO Journal. 1990;9(4):1105-11.
- Jeon YJ, Yoo HM, Chung CH. ISG15 and immune diseases. Biochim biophys acta. 2010; 1802:485-96.
- Chairatvit K, Wongnoppavich A, Choonate S. Up-regulation of interferon- stimulated gene15 and its conjugates by tumor necrosis factor-a via type I interferon- dependent and -independent pathways. Mol Cell Biochem. 2012;368(1-2):195-201.
- MacParland SA, Ma XZ, Chen L, et al. Lipopolysaccharide and Tumor Necrosis Factor Alpha Inhibit Interferon Signaling in Hepatocytes by Increasing Ubiquitin-Like Protease 18 (USP18) Expression. J Virol. 2016;90(12):5549–60.
- Lertsooksawat W, Wongnoppavich A, Chairatvit K. Up-regulation of interferon- stimulated gene 15 and its conjugation machinery, UbE1L and UbcH8 expression by tumor necrosis factor-a through p38 MAPK and JNK signaling pathways in human lung carcinoma. Mol Cell Biochem. 2019;462(1-2):51–59.
- Durfee LA, Huibregtse JM. The ISG15 conjugation system. Methods Mol. Biol.2012;832:141–49.
- Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. Herc5, an interferon- induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J Biol Chem. 2006;281(7):4334‐38.
- Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J Biol Chem. 2006;281(7):3989‐94.
- Liu M, Li XL, Hassel BA. Proteasomes modulate conjugation to the ubiquitin-like protein, ISG15. J Biol Chem. 2003;278(3):1594‐1602.
- Honke N, Shaabani N, Zhang DE, Hardt C, Lang KS. Multiple functions of USP18.Cell Death Dis. 2016;7(11):e2444.
- Basters A, Knobeloch KP, Fritz G. USP18 – a multifunctional component in the interferon response. Biosci Rep. 2018;38(6):BSR20180250.
- Francois-Newton V, Livingstone M, Payelle-Brogard B, Uzé G, Pellegrini S. USP18 establishes the transcriptional and anti-proliferative interferon a /β differential. Biochem J. 2012;446(3):509-16.
- Zhang X, Bogunovic D, Payelle-Brogard B, et al. Human intracellular ISG15 prevents interferon-a /β over-amplification and auto-inflammation. Nature. 2015;517(7532):89-93.
- Stark GR, Darnell JE Jr. The JAK-STAT pathway at twenty. Imunity.2012;36(4):503-14.
- Martín-Fernández M, Bravo García-Morato M, Gruber C, et al. Systemic Type I IFN Inflammation in Human ISG15 Deficiency Leads to Necrotizing Skin Lesions. Cell Rep. 2020;31(6):107633.
- D’Cunha J, Knight E Jr, Haas AL, Truitt RL, Borden EC. Immunoregulatory properties of ISG15, an interferon-induced cytokine. Proc Natl Acad Sci USA.1996;93(1):211–15.
- Hoan NX, Van Tong H, Giang DP, et al. Interferon-stimulated gene 15 in hepatitis B-related liver diseases. Oncotarget. 2016;7(42):67777–87.
- Swaim CD, Canadeo LA, Monte KJ, Khanna S, Lenschow DJ, Huibregtse JM. Modulation of Extracellular ISG15 Signaling by Pathogens and Viral Effector Proteins. Cell Rep. 2020;31(11):107772.
- Hare NJ, Chan B, Chan E, Kaufman KL, Britton WJ, Saunders BM. Microparticles released from Mycobacterium tuberculosis-infected human macrophages contain increased levels of the type I interferon inducible proteins including ISG15. Proteomics. 2015;15(17):3020-9.
- Sun L1, Wang X2, Zhou Y3, Zhou RH1, Ho WZ4, Li JL5. Exosomes contribute to the transmission of anti-HIV activity from TLR3-activated brain microvascular endothelial cells to macrophages. Antiviral Res. 2016;134:167-71.
- Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20(3):362-71.
- Zhao C, Sridharan H, Chen R, Baker DP, Wang S, Krug RM. Influenza B virus non- structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat Commun. 2016;7:12754.
- Sainz B Jr, Martín B, Tatari M, Heeschen C, Guerra S. ISG15 is a critical microenvironmental factor for pancreatic cancer stem cells. Cancer Res. 2014;74(24):7309–20.
- Dos Santos PF, Mansur DS. Beyond ISGlylation: Functions of Free Intracellular and Extracellular ISG15. J Interferon Cytokine Res. 2017;37(6):246-53.
- Swaim CD, Scott AF, Canadeo LA, Huibregtse JM. Extracellular ISG15 signals cytokine secretion through the LFA-1 integrin receptor. Mol Cell. 2017;68(3):581-90.
- Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339-62.
- Chen RH, Xiao ZW, Yan XQ, et al. Tumor Cell-Secreted ISG15 Promotes Tumor Cell Migration and Immune Suppression by Inducing the Macrophage M2-Like Phenotype. Front Immunol. 2020;11:594775.
- Gao N, Me R, Dai C, Yu FX. ISG15 Acts as a Mediator of Innate Immune Response to Pseudomonas aeruginosa Infection in C57BL/6J Mouse Corneas. Invest Ophthalmol Vis Sci. 2020;61(5):26.
- Kunzi MS, Pitha PM. Role of interferon-stimulated gene ISG15 in the interferon- omega-mediated inhibition of human immunodeficiency virus replication. J Interferon Cytokine Res. 1996; 16(11):919–27.
- Lenschow DJ, Lai C, Frias-Staheli N, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci USA. 2007;104(4):1371–76.
- Guerra S, Caceres A, Knobeloch KP, Horak I, Esteban M. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 2008; 4(7):e1000096.
- Hsiang TY, Zhao C, Krug RM. Interferon-induced ISG15 conjugation inhibits influenza A virus gene expression and replication in human cells. J Virol. 2009; 83(12):5971–77.
- Yan S, Kumari M, Xiao H, et al. IRF3 reduces adipose thermogenesis via ISG15- mediated reprogramming of glycolysis. J Clin Invest. 2021;131(7):e144888.
- Hamerman JA, Hayashi F, Schroeder LA, et al. Serpin 2a is induced in activated macrophages and conjugates to a ubiquitin homolog. J Immunol. 2002;168(5):2415‐23.
- Villarroya-Beltri C, Guerra S, Sánchez-Madrid F. ISGylation – a key to lock the cell gates for preventing the spread of threats. J Cell Sci. 2017;130(18):2961–69.
- Giannakopoulos NV, Luo JK, Papov V, et al. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem Biophys Res Commun. 2005;336(2):496-506.
- Hsiao NW, Chen JW, Yang TC, Orloff GM, Wu YY, Lai CH, Lan YC, Lin CW. ISG15 over-expression inhibits replication of the Japanese encephalitis virus in human medulloblastoma cells. Antiviral Res. 2010; 85(3):504-11.
- Satou R, González-Villalobos RA. JAK-STAT and the renin-angiotensin system: The role of the JAK-STAT pathway in blood preassure and intrarenal renin-angiotensin system regultaion. JAKSTAT. 2012; 1(4):250-6.
- Maier HJ, Schips TG, Wietelmann A, et al. Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2012;109:11794-9.
- Recht M, Borden EC, Knight E Jr. A human 15-kDa IFN-induced protein induces the secretion of IFN-gamma. J Immunol. 1991;147(8):2617–23.
- D’Cunha J, Ramanujam S, Wagner RJ, Witt PL, Knight E Jr, Borden EC. In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokine. J Immunol. 1996a;157(9):4100–8.
- Østvik AE, Svendsen TD, Granlund AVB, et al. Intestinal Epithelial Cells Express Immunomodulatory ISG15 During Active Ulcerative Colitis and Crohn’s Disease. J Crohns Colitis. 2020;14(7):920-34.
- Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104(2):191-6.
- Rizzoni D, Porteri E, Boari GE, et al. Prognostic significance of small-artery structure in hypertension. Circulation. 2003;108(18):2230-5.
- Mitchell GF, Lacourcière Y, Ouellet JP, et al. Determinants of elevated pulse pressure in middle-aged and older subjects with uncomplicated systolic hypertension: the role of proximal aortic diameter and the aortic pressure-flow relationship. Circulation. 2003;108(13):1592-8.
- Briones AM, Rodríguez-Criado N, Hernanz R, et al. Atorvastatin prevents angiotensin II-induced vascular remodelling and oxidative stress. Hypertension. 2009;54(1):142-9.
- Roque FR, Briones AM, García-Redondo AB, et al. Aerobic exercise reduces oxidative stress and improves vascular changes of small mesenteric and coronary arteries in hypertension. Br J Pharmacol. 2013;168(3):686-703.
- Marchesi C, Rehman A, Rautureau Y, et al. Protective role of vascular smooth muscle cell PPARγ in angiotensin II-induced vascular disease. Cardiovasc Res. 2013;97(3):562-70.
- Pu Q, Neves MF, Virdis A, Touyz RM, Schiffrin EL. Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodelling. Hypertension. 2003;42(1):49-55.
- Neves MF, Virdis A, Schiffrin EL. Resistance artery mechanics and composition in angiotensin II-infused rats: effects of aldosterone antagonism. J Hypertens. 2003;21(1):189-98.
- Avendaño MS, García-Redondo AB, Zalba G, et al. mPGES-1 (Microsomal Prostaglandin E Synthase-1) Mediates Vascular Dysfunction in Hypertension Through Oxidative Stress. Hypertension. 2018;72(2):492-502.
- Park JB, Schiffrin EL. Small artery remodelling is the most prevalent (earliest?) form of target organ damage in mild essential hypertension. J Hypertens. 2001;19(5):921-30.
- Endemann DH, Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. 2004;15(8):1983-92.
- Martínez-Revelles S, Avendaño MS, García-Redondo AB, et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal. 2013;18(1):51-65.
- Wang H, Gao XY, Rao F, et al. Mechanism of contractile dysfunction induced by serotonin in coronary artery in spontaneously hypertensive rats. Naunyn Schmiedebergs Arch Pharmacol. 2020;393(11):2165-76.
- Belmonte SL, Blaxall BC. G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ Res. 2011;109:309-19.
- Savoia C, Burger D, Nishigaki N, Montezano A, Touyz RM. Angiotensin II and the vascular phenotype in hypertension. Expert Rev Mol Med. 2011;13:e11.
- Xiao L, Harrison DG. Inflammation in Hypertension. Can J Cardiol. 2020;36(5):635-47.
- Redón J, Oliva MR, Tormos C, et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41(5):1096–1101.
- Minuz P, Patrignani P, Gaino S, et al. Determinants of platelet activation in human essential hypertension. Hypertension. 2004;43(1):64–70.
- Ahmad A, Singhal U, Hossain MM, Islam N, Rizvi I. The role of the endogenous antioxidant enzymes and malondialdehyde in essential hypertension. J Clin Diagn Res. 2013;7(6):987–90.
- Carrizzo A, Puca A, Damato A, et al. Resveratrol improves vascular function in patients with hypertension and dyslipidemia by modulating NO metabolism. Hypertension. 2013;62(2):359–66.
- Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: clinical evidence and therapeutic implications. Trends Cardiovasc Med. 2014;24(4):165–9.
- Álvarez Y, Pérez-Girón JV, Hernanz R, et al. Losartan reduces the increased participation of cyclooxygenase-2-derived products in vascular responses of hypertensive rats. J Pharmacol Exp Ther. 2007;321(1):381–8.
- Viel EC, Benkirane K, Javeshghani D, Touyz RM, Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295(1):H281–H288.
- Agarwal D, Haque M, Sriramula S, Mariappan N, Pariaut R, Francis J. Role of proinflammatory cytokines and redox homeostasis in exercise-induced delayed progression of hypertension in spontaneously hypertensive rats. Hypertension. 2009;54(6):1393–400.
- Griendling KK, Camargo LL, Rios FJ, Alves-Lopes R, Montezano AC, Touyz RM. Oxidative Stress and Hypertension. Circ Res. 2021;128(7):993-1020.
- Dikalov SI, Dikalova AE. Crosstalk Between Mitochondrial Hyperacetylation and Oxidative Stress in Vascular Dysfunction and Hypertension. Antioxid Redox Signal. 2019;31(10):710-21.
- Daiber A, Chlopicki S. Revisiting pharmacology of oxidative stress and endothelial dysfunction in cardiovascular disease: Evidence for redox-based therapies. Free Radic Biol Med. 2020;157:15-37.
- Touyz RM, Rios FJ, Alves-Lopes R, Neves KB, Camargo LL, Montezano AC. Oxidative Stress: A Unifying Paradigm in Hypertension. Can J Cardiol. 2020;36(5):659-70.
- Álvarez Y, Briones AM, Hernanz R, Pérez-Girón JV, Alonso MJ, Salaices M. Role of NADPH oxidase and iNOS in vasoconstrictor responses of vessels from hypertensive and normotensive rats. Br J Pharmacol. 2008;153(5):926–35.
- Hernanz R, Martínez-Revelles S, Palacios R, et al. Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol. 2015;172(12):3159-76.
- Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda). 2006;21:269–80.
- Li MW, Mian MO, Barhoumi T, et al. Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2013;33(10):2306–15.
- Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580(2):497-504.
- Seaberg EC, Munoz A, Lu M, Detels R, Margolick JB, Riddler SA, Williams CM, Phair JP. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to2003. Aids. 2005; 19:953-60.
- Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449-60.
- Harrison DG, Vinh A, Lob Heinrich, Madhur MS. Role of the adaptive immune system in hypertension. Curr Opin Pharmacol. 2010;10(2):203-207.
- Trott DW, Thabet SR, Kirabo A, et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension. 2014;64(5):1108-15.
- Ji Q, Cheng G, Ma N, et al. Circulating Th1, Th2, and Th17 Levels in Hypertensive Patients. Dis Markers. 2017;2017:7146290.
- Orejudo M, García-Redondo AB, Rodrigues-Diez RR, et al. Interleukin-17A induces vascular remodelling of small arteries and blood pressure elevation. Clin Sci (Lond). 2020;134(5):513-27.
- Qian L, Li X, Fang R, et al. Class A scavenger receptor deficiency augments angiotensin II-induced vascular remodelling. Biochem Pharmacol. 2014;90(3):254-64.
- Qi D, Wei M, Jiao S, et al. Hypoxia inducible factor 1a in vascular smooth muscle cells promotes angiotensin II-induced vascular remodelling via activation of CCL7- mediated macrophage recruitment. Cell Death Dis. 2019;10(8):544.
- Ye J, Que B, Huang Y, et al. Interleukin-12p35 knockout promotes macrophage differentiation, aggravates vascular dysfunction, and elevates blood pressure in angiotensin II-infused mice. Cardiovasc Res. 2019;115(6):1102-113.
- De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodelling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25(10):2106-13.
- Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, Schiffrin EL. Resistance artery remodelling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol. 2007;292(4):H1789-95.
- Wenzel P, Knorr M, Kossmann S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation.2011;124(12):1370-81.
- Olivencia MA, Martínez-Casales M, Peraza DA, et al. KV 1.3 channels are novel determinants of macrophage-dependent endothelial dysfunction in angiotensin II-induced hypertension in mice. Br J Pharmacol. 2021;178(8):1836-54.
- Moore JP, Vinh A, Tuck KL, et al. M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol. 2015;309(5):H906-17.
- De Batista PR, Palacios R, Martín A, et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS One. 2014;9(8):e104020.
- Liu J, Yang F, Yang XP, Jankowski M, Pagano PJ. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler Thromb Vasc Biol. 2003;23(5):776-82.
- Lang PP, Bai J, Zhang YL, Yang XL, Xia YL, Lin QY, Li HH. Blockade of intercellular adhesion molecule-1 prevents angiotensin II-induced hypertension and vascular dysfunction. Lab Invest. 2020;100(3):378-86.
- Zhang HG, Guo W, Gu HF, et al. Correlation of VCAM-1 expression in serum, cord blood, and placental tissue with gestational hypertension associated with fetal growth restriction in women from Xingtai Hebei, China. Genet Mol Res. 2016;15(3).
- Ciobanu DM, Mircea PA, Bala C, Rusu A, Vesa Ş, Roman G. Intercellular adhesion molecule-1 (ICAM-1) associates with 24-hour ambulatory blood pressure variability in type 2 diabetes and controls. Cytokine. 2019;116:134-38.
- Han YL, Li YL, Jia LX, Cheng JZ, Qi YF, Zhang HJ, Du J. Reciprocal interaction between macrophages and T cells stimulates IFN-γ and MCP-1 production in Ang II- induced cardiac inflammation and fibrosis. Plos One. 2012;7(5):e35506.
- Markó L, Kvakan H, Park JK, et al. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60(6): 1430-1436.
- Kossmann S, Schwenk M, Hausding M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-γ-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. 2013;33(6):1313-1319.
- Kamat NV, Thabet SR, Xiao L, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension. 2015;65(3):569-76.
- Mikolajczyk TP, Nosalski R, Szczepaniak P, et al. Role of chemokine RANTES in the regulation of perivascular inflammation, T-cell accumulation, and vascular dysfunction in hypertension. FASEB J. 2016;30(5):1987-99.
- Zhang H, Park Y, Wu J, et al. Role of TNF-alpha in vascular dysfunction. Clin Sci (Lond). 2009;116(3):219-30.
- González-Amor M, García-Redondo AB, Jorge I, Zalba G, et al. Interferon stimulated gene 15 pathway is a novel mediator of endothelial dysfunction and aneurysms development in angiotensin II infused mice through increased oxidative stress. Cardiovasc Res. 2021:cvab321.
- Stapleton PA, James ME, Goodwill AG, Frisbee JC. Obesity and vascular dysfunction. Pathophysiology. 2008;15(2):79-89.
- Martínez-Martínez E, Souza-Neto FV, Jiménez-González S, Cachofeiro V. Oxidative Stress and Vascular Damage in the Context of Obesity: The Hidden Guest. Antioxidants (Basel). 2021;10(3):406.
- Dimassi S, Chahed K, Boumiza S, Canault M, Tabka Z, Laurant P, Riva C. Role of eNOS- and NOX-containing microparticles in endothelial dysfunction in patients with obesity. Obesity (Silver Spring). 2016;24(6):1305-12.
- Prieto D, Contreras C, Sánchez A. Endothelial dysfunction, obesity and insulin resistance. Curr Vasc Pharmacol. 2014;12(3):412-26.
- Wang G, Zhang Y, Zhang R, Pan J, Qi D, Wang J, Yang X. The protective effects of walnut green husk polysaccharide on liver injury, vascular endothelial dysfunction and disorder of gut microbiota in high fructose-induced mice. Int J Biol Macromol. 2020;162:92-106.
- Martínez-Martínez E, Miana M, Jurado-López R, et al. The potential role of leptin in the vascular remodelling associated with obesity. Int J Obes (Lond). 2014;38(12):1565-72.
- Gil-Ortega M, Martín-Ramos M, Arribas SM, et al. Arterial stiffness is associated with adipokine dysregulation in non-hypertensive obese mice. Vascul Pharmacol. 2016;77:38-47.
- Heiss G, Sharrett AR, Barnes R, Chambless LE, Szklo M, Alzola C. Carotid atherosclerosis measured by B-mode ultrasound in populations: associations with cardiovascular risk factors in the ARIC study. Am J Epidemiol. 1991;134(3):250-6.
- Ciccone M, Vettor R, Pannacciulli N, et al. Plasma leptin is independently associated with the intima-media thickness of the common carotid artery. Int J Obes Relat Metab Disord. 2001;25(6):805-10.
- Elfimova EM, Litvin AY, Chazova IE. The effectiveness of combination antihypertensive therapy in patients with arterial hypertension and additional risk factors: obesity and obstructive sleep apnea syndrome. Ter Arkh. 2018;90(12):28-33.
- Briones AM, González JM, Somoza B, et al. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J. Physiol. 2003;552(Pt 1):185–95.
- González JM, Briones AM, Somoza B, et al. Postnatal alterations in elastic fiber organization precede resistance artery narrowing in SHR. Am J Physiol Heart Circ Physiol. 2006;291(2):H804-12.
- Ketonen J, Shi J, Martonen E, Mervaala E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ J. 2010;74(7):1479-87.
- Gil-Ortega M, Condezo-Hoyos L, García-Prieto CF, et al. Imbalance between pro and anti-oxidant mechanisms in perivascular adipose tissue aggravates long-term high- fat diet-derived endothelial dysfunction. PLoS One. 2014;9(4):e95312.
- Virdis A, Masi S, Colucci R, et al. Microvascular Endothelial Dysfunction in Patients with Obesity. Curr Hypertens Rep. 2019;21(4):32.
- Muñoz M, López-Oliva ME, Rodríguez C, et al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020;28:101330.
- Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol. 2017;174(20):3496-513.
- Xia N, Horke S, Habermeier A, et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler Thromb Vasc Biol. 2016;36(1):78-85.
- Aghamohammadzadeh R, Greenstein AS, Yadav R, et al. Effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J Am Coll Cardiol. 2013;62(2):128-35.
- Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM. Effects of Obesity on Perivascular Adipose Tissue Vasorelaxant Function: Nitric Oxide, Inflammation and Elevated Systemic Blood Pressure. J Vasc Res. 2015;52(5):299-305.
- DeVallance E, Branyan KW, Lemaster K, et al. Aortic dysfunction in metabolic syndrome mediated by perivascular adipose tissue TNFa- and NOX2-dependent pathway. Exp Physiol. 2018;103(4):590-603.
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796-808.
- Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821-30.
- Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939-49.
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175-84.
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87-91.
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860-7.
- Cao H. Adipocytokines in obesity and metabolic disease. J Endocrinol.2014;220(2):T47-59.
- Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930-9.
- Liu J, Divoux A, Sun J, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15(8):940-5.
- Wu D, Molofsky AB, Liang HE, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332(6026):243-7.
- Weiss R, Dziura J, Burgert TS, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350(23):2362-74.
- Wärnberg J, Nova E, Romeo J, Moreno LA, Sjöström M, Marcos A. Lifestyle- related determinants of inflammation in adolescence. Br J Nutr. 2007;98 Suppl 1:S116-20.
- Gøbel RJ, Jensen SM, Frøkiaer H, Mølgaard C, Michaelsen KF. Obesity, inflammation and metabolic syndrome in Danish adolescents. Acta Paediatr. 2012;101(2):192-200.
- Festa A, D’Agostino R Jr, Williams K, Karter AJ, Mayer-Davis EJ, Tracy RP, Haffner SM. The relation of body fat mass and distribution to markers of chronic inflammation. Int J Obes Relat Metab Disord. 2001;25(10):1407-15.
- Bulló M, García-Lorda P, Megias I, Salas-Salvadó J. Systemic inflammation, adipose tissue tumor necrosis factor, and leptin expression. Obes Res. 2003;11(4):525-31.
- Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol. 2009;20(5):379-85.
- Gregor MF, Hotamisligil GS. Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48(9):1905-14.
- Trayhurn P, Pérez de Heredia F, Wang B et al. Hypoxia – role in adipocyte function and dysfunction. In Novel Insight into Adipose Cell Functions. Clement K, Spiegelman BM and Christen Y (eds). Berlin, Heidelberg: Springer-Verlag. 2010:45-60.
- Halberg N, Khan T, Trujillo ME, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29(16):4467-83.
- Apovian CM, Bigornia S, Mott M, et al. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler Thromb Vasc Biol. 2008;28(9):1654-9.
- Winkler G, Lakatos P, Salamon F, et al. Elevated serum TNF-alpha level as a link between endothelial dysfunction and insulin resistance in normotensive obese patients. Diabet Med. 1999;16(3):207-11.
- Greenstein AS, Khavandi K, Withers SB, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119(12):1661-70.
- Virdis A, Duranti E, Rossi C, et al. Tumour necrosis factor-alpha participates on the endothelin-1/nitric oxide imbalance in small arteries from obese patients: role of perivascular adipose tissue. Eur Heart J. 2015;36(13):784-94.
- Sun Y, Xiaoyan H, Yun L, et al. Identification of Key Candidate Genes and Pathways for Relationship between Ovarian Cancer and Diabetes Mellitus Using Bioinformatical Analysis. Asian Pac J Cancer Prev. 2019;20(1):145-55.
- Vorp DA. Biomechanics of abdominal aortic aneurysm. J Biomech. 2007;40(9):1887–902.
- Wu D, Ren P, Zheng Y, et al. NLRP3 (Nucleotide Oligomerization Domain-Like Receptor Family, Pyrin Domain Containing 3)-Caspase-1 Inflammasome Degrades Contractile Proteins: Implications for Aortic Biomechanical Dysfunction and Aneurysm and Dissection Formation. Arterioscler Thromb Vasc Biol. 2017;37(4):694–706.
- Fan LM, Douglas G, Bendall JK, et al. Endothelial cell-specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation. 2014;129(25):2661–72.
- Siasos G, Mourouzis K, Oikonomou E, et al. The Role of Endothelial Dysfunction in Aortic Aneurysms. Curr Pharm Des. 2015;21(28):4016–34.
- Wang M, Lee E, Song W, et al. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117(10):1302–9.
- Yuan Z, Lu Y, Wei J, Wu J, Yang J, Cai Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front Immunol. 2021;11:609161.
- Malekzadeh S, Fraga-Silva RA, Trachet B, Montecucco F, Mach F, Stergiopulos N. Role of the renin-angiotensin system on abdominal aortic aneurysms. Eur J Clin Invest. 2013;43(12):1328-38.
- Silverberg D, Younis A, Savion N, et al. Long-term renin-angiotensin blocking therapy in hypertensive patients with normal aorta may attenuate the formation of abdominal aortic aneurysms. J Am Soc Hypertens. 2014;8(8):571-7.
- Trollope A, Moxon JV, Moran CS, Golledge J. Animal models of abdominal aortic aneurysm and their role in furthering management of human disease. Cardiovasc Pathol. 2011;20(2):114-23.
- Golledge J, Krishna SM, Wang Y. Mouse models for abdominal aortic aneurysm. Br J Pharmacol. 2020.
- Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29(10):1458-64.
- Villahoz S, Yunes-Leites PS, Méndez-Barbero N, et al. Conditional deletion of Rcan1 predisposes to hypertension-mediated intramural hematoma and subsequent aneurysm and aortic rupture. Nat Commun. 2018;9(1):4795.
- Emeto TI, Moxon JV, Au M, Golledge J. Oxidative stress and abdominal aortic aneurysm: potential treatment targets. Clin Sci (Lond). 2016;130(5):301-15.
- Quintana RA, Taylor WR. Cellular Mechanisms of Aortic Aneurysm Formation. Circ Res. 2019;124(4):607-18.
- Gavrila D, Li WG, McCormick ML, et al. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25(8):1671-7.
- Thomas M, Gavrila D, McCormick ML, et al. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E- deficient mice. Circulation. 2006;114(5):404-13.
- Gao L, Siu KL, Chalupsky K, et al. Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension. 2012;59(1):158-66.
- Maiellaro-Rafferty K, Weiss D, Joseph G, Wan W, Gleason RL, Taylor WR. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am J Physiol Heart Circ Physiol. 2011;301(2):H355-62.
- Guerriero JL. Macrophages: Their Untold Story in T Cell Activation and Function. Int Rev Cell Mol Biol. 2019;342:73-93.
- Liao M, Liu CL, Lv BJ, et al. Plasma cytokine levels and risks of abdominal aortic aneurysms: A population-based prospective cohort study. Ann Med. 2015;47(3):245–52.
- Sagan A, Mikolajczyk TP, Mrowiecki W, et al. T Cells Are Dominant Population in Human Abdominal Aortic Aneurysms and Their Infiltration in the Perivascular Tissue Correlates With Disease Severity. Front Immunol. 2019;10:1979.
- Meekel JP, Dias-Neto M, Bogunovic N, et al. Inflammatory Gene Expression of Human Perivascular Adipose Tissue in Abdominal Aortic Aneurysms. Eur J Vasc Endovasc Surg. 2021:S1078-5884(21)00186-6.
- Takahashi M. NLRP3 Inflammasome as a Common Denominator of Atherosclerosis and Abdominal Aortic Aneurysm. Circ J. 2021.
- Sharma AK, Lu G, Jester A, et al. Experimental abdominal aortic aneurysm formation is mediated by IL-17 and attenuated by mesenchymal stem cell treatment. Circulation. 2012;126(Suppl 1):S38-45.
- Lindeman JH, Abdul-Hussien H, Schaapherder AF, Van Bockel JH, Von der Thüsen JH, Roelen DL, Kleemann R. Enhanced expression and activation of pro-inflammatory transcription factors distinguish aneurysmal from atherosclerotic aorta: IL-6- and IL-8- dominated inflammatory responses prevail in the human aneurysm. Clin Sci (Lond). 2008;114(11):687-97.
- Golledge AL, Walker P, Norman PE, Golledge J. A systematic review of studies examining inflammation associated cytokines in human abdominal aortic aneurysm samples. Dis Markers. 2009;26(4):181-8.
- Zhou HF, Yan H, Cannon JL, Springer LE, Green JM, Pham CT. CD43-mediated IFN-γ production by CD8+ T cells promotes abdominal aortic aneurysm in mice. J Immunol. 2013;190(10):5078-85.
- Stather PW, Sidloff DA, Dattani N, Gokani VJ, Choke E, Sayers RD, Bown MJ. Meta-analysis and meta-regression analysis of biomarkers for abdominal aortic aneurysm. Br J Surg. 2014;101(11):1358-72.
- Wang SK, Green LA, Gutwein AR, et al. Description of human AAA by cytokine and immune cell aberrations compared to risk-factor matched controls. Surgery. 2018;164(2):354-8.
- King VL, Lin AY, Kristo F, Anderson TJ, et al. Interferon-gamma and the interferon- inducible chemokine CXCL10 protect against aneurysm formation and rupture. Circulation. 2009;119(3):426-35.
- Wang Y, Ait-Oufella H, Herbin O, et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest. 2010;120(2):422-32.
- Yan H, Zhou HF, Akk A, Hu Y, Springer LE, Ennis TL, Pham CTN. Neutrophil Proteases Promote Experimental Abdominal Aortic Aneurysm via Extracellular Trap Release and Plasmacytoid Dendritic Cell Activation. Arterioscler Thromb Vasc Biol. 2016;36(8):1660-69.
- Williams B, Mancia G, Spiering W, et al. 2018 Practice Guidelines for the management of arterial hypertension of the European Society of Hypertension and the European Society of Cardiology: ESH/ESC Task Force for the Management of Arterial Hypertension. J Hypertens. 2018;36(12):2284-309.
- Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet. 2006;368(9536):659‐65.
- Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res. 2017;120(1):229‐43.
- Slaiby JM, Ricci MA, Gadowski GR, Hendley ED, Pilcher DB. Expansion of aortic aneurysms is reduced by propranolol in a hypertensive rat model. J Vasc Surg.1994;20:178–83.
- Sinha N, Dabla PK. Oxidative stress and antioxidants in hypertension-a current review. Curr Hypertens Rev. 2015;11(2):132–42.
- DeVallance E, Li Y, Jurczak MJ, Cifuentes-Pagano E, Pagano PJ. The Role of NADPH Oxidases in the Etiology of Obesity and Metabolic Syndrome: Contribution of Individual Isoforms and Cell Biology. Antioxid Redox Signal. 2019;31(10):687-709.
- Moran JP, Cohen L, Greene JM, et al. Plasma ascorbic acid concentrations relate inversely to blood pressure in human subjects. Am J Clin Nutr. 1993;57(2):213–17.
- Nawrot TS, Staessen JA, Roels HA, et al. Blood pressure and blood selenium: a cross-sectional and longitudinal population study. Eur Heart J. 2007;28(5):628–33.
- Barrios V, Calderón A, Navarro-Cid J, Lahera V, Ruilope LM. N-acetylcysteine potentiates the antihypertensive effect of ACE inhibitors in hypertensive patients. Blood Press. 2002;11(4):235–39.
- On YK, Kim CH, Sohn DW, et al. Improvement of endothelial function by amlodipine and vitamin C in essential hypertension. Korean J Intern Med. 2002;17(2):131–7.
- Hajjar IM, George V, Sasse EA, Kochar MS. A randomized, double-blind, controlled trial of vitamin C in the management of hypertension and lipids. Am J Ther. 2002;9(4):289–93.
- Juraschek SP, Guallar E, Appel LJ, Miller ER 3rd. Effects of vitamin C supplementation on blood pressure: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2012;95(5):1079–88.
- Ginter E. Vplyv vol’ných radikálov a antioxidantov na cievnu stenu [Effect of free radicals and antioxidant on the vascular wall]. Vnitr Lek. 2000;46(6):354–9.
- Antoniades C, Tousoulis D, Tentolouris C, Toutouzas P, Stefanadis C. Oxidative stress, antioxidant vitamins, and atherosclerosis. From basic research to clinical practice. Herz. 2003;28(7):628–38.
- Rodrigo R, Guichard C, Charles R. Clinical pharmacology and therapeutic use of antioxidant vitamins. Fundam Clin Pharmacol. 2007;21(2):111–27.
- Golia E, Limongelli G, Natale F, et al. Inflammation and cardiovascular disease: from pathogenesis to therapeutic target. Curr Atheroscler Rep. 2014;16(9):435.
- Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Anti-TNF Therapy Against Congestive Heart Failure Investigators. Randomized, double-blind, placebo- controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):3133-40.
- Ridker PM, Everett BM, Thuren T, et al. CANTOS Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377(12):1119-31.
- Rothman AM, MacFadyen J, Thuren T, et al. Effects of Interleukin-1b Inhibition on Blood Pressure, Incident Hypertension, and Residual Inflammatory Risk: A Secondary Analysis of CANTOS. Hypertension. 2020;75(2):477-82.
- Khan S, Andrews KL, Chin-Dusting JPF. Cyclo-Oxygenase (COX) Inhibitors and Cardiovascular Risk: Are Non-Steroidal Anti-Inflammatory Drugs Really Anti- Inflammatory? Int J Mol Sci. 2019;20(17):4262.