1. INTRODUCCIÓN
De acuerdo con la definición recogida por la Organización Mundial de la Salud (OMS), “cáncer” es un término que “designa un amplio grupo de enfermedades que pueden afectar a cualquier parte del organismo, caracterizadas por la multiplicación rápida de células anormales que se extienden más allá de sus límites habituales y pueden invadir partes adyacentes del cuerpo o propagarse a otros órganos, en un proceso que se denomina «metástasis»”. Esta definición queda matizada por el Diccionario de términos médicos, de la Real Academia Nacional de Medicina, en la que se refiere a la palabra «cáncer» como sinónimo de «tumor maligno» o «neoplasia maligna», de modo que un tumor maligno es un cáncer, no un cáncer maligno.
El cáncer es una de las patologías que presenta mayor morbi/mortalidad dentro de las enfermedades no infecciosas, siendo la responsable de casi 10 millones de muertes solamente en el año 2020. Atendiendo a su incidencia, el cáncer de mama fue el más diagnosticado (2.26 millones de casos), seguido del pulmonar (2.21 millones de casos) y el colorrectal (1.96 millones de casos), de acuerdo con los datos actualizados en el portal de la OMS para 2020 (https://www.who.int/es/news-room/fact-sheets/detail/cancer). Esta elevada incidencia se traduce en un elevado coste socioeconómico, especialmente en países desarrollados con sistemas de salud afianzados y de amplias prestaciones (1).
Una neoplasia es un fenómeno originado por un crecimiento celular anómalo y descontrolado, originando un tumor en el que las células que lo constituyen no son homogéneas y presentan distintos patrones morfológicos, bioquímicos e inmunológicos. Este hecho, supone una complicación para el abordaje terapéutico de las neoplasias malignas o cáncer, que normalmente requiere de aproximaciones múltiples incluyendo cirugía y/o radioterapias coadyuvantes del tratamiento farmacológico. En cualquier caso, la quimioterapia juega un papel esencial en la terapia antineoplásica, especialmente las aproximaciones polifarmacológicas.
En este contexto, la búsqueda de fármacos para el tratamiento de los desórdenes neoplásicos sigue siendo una de las principales líneas de investigación tanto de grupos académicos como de la industria farmacéutica en todo el mundo. Prueba de ello, es el gran número de candidatos a fármaco antineoplásico que cada año entran en ensayos clínicos o se aprueban (2).
Un hito en el campo de desarrollo de fármacos anticancerígenos es el descubrimiento del imatinib (Glivec®) por los laboratorios Ciba-Geigy (actual Novartis) en 1992 y aprobado por la FDA en el 2001 (3). Inicialmente descrito como un inhibidor específico de la quinasa BCR–ABL, la c-KIT y el factor de crecimiento derivado de plaquetas (PDGF), se aprobó para el tratamiento de la leucemia mieloide crónica dependiente de ABL (3). El imatinib se considera un punto de inflexión en la quimioterapia anticancerosa y en el descubrimiento de fármacos por varios motivos. En primer lugar, se trata de uno de los primeros antineoplásicos dirigido a una diana específica y distinta del ADN, gracias a los descubrimientos impulsados por Mariano Barbacid en el campo de los oncogenes y las proteínas quinasas (4). Por otro lado, el descubrimiento del imatinib supuso un avance en el paradigma del descubrimiento de fármacos moderno, ya que se trata de uno de los primeros ejemplos de diseño basado en estructura para el cáncer, es decir, a partir del conocimiento estructural de la diana farmacológica y su interacción con el fármaco. El desarrollo de esta molécula fue posible gracias a las técnicas de cristalografía de rayos X y modelado molecular como al docking. Así pues, el descubrimiento del imatinib puso de manifiesto cómo el conocimiento de la diana específica sobre la que actúan las moléculas pequeñas es de vital importancia en el desarrollo y optimización de un fármaco, y éste conocimiento se puede obtener mediante técnicas experimentales y/o computacionales.
Con todo, la complejidad de la terapia oncológica ha impulsado la búsqueda de nuevas dianas terapéuticas que confieran mayor seguridad y eficacia al agente quimioterápico. En este sentido, en los últimos años se ha prestado especial atención al grupo de las pseudoquinasas (5). Estas proteínas pertenecen a un grupo que representa aproximadamente el 10% del quinona humano, pero en cuyos miembros falta algún motivo o aminoácido considerado esencial para la catálisis enzimática (6). De este modo, las pseudoquinasas pertenecen a una superfamilia de proteínas capaces de ejercer función de señalización celular mediante mecanismos no catalíticos, en muchos casos bastante desconocidos.
Una de las pseudoquinasas más interesantes y prometedora como diana terapéutica contra el cáncer es la quinasa ligada a integrinas (ILK). Descubierta por Dedhar y colaboradores en 1996 como una proteína de 59 kDa capaz de interaccionar con la subunidad b1 de las integrinas, la ILK se encuentra muy conservada evolutivamente y está constituida por 452 aminoácidos divididos en tres dominios bien diferenciados (7). El primero, comenzando por el extremo N-terminal, es el dominio de repeticiones de ankirina, le sigue un dominio intermedio de homología a pleckstrina (residuos 180 a 212), y finalmente el dominio globular tipo quinasa, constituido por los aminoácidos 213 a 452 (Figura 3A). En esta región es donde se encuentra el “centro pseudoactivo” de la enzima, el cual presenta diferencias significativas respecto a otras quinasas. El bucle de activación de las quinasas comprende tres motivos canónicos que participan en la catálisis y reconocimiento del sustrato: HRD, DFG y APE (8). Sin embargo, la ILK únicamente mantiene el motivo APE constituido por los residuos Ala357, Pro358 y Glu359 (9). A pesar de ello, la ILK conserva la capacidad de unión de una molécula de ATP sin hidrolizarlo (Figura 3B, PDB ID: 3KMW) y con un modo de unión distinto al de otras quinasas convencionales (10).
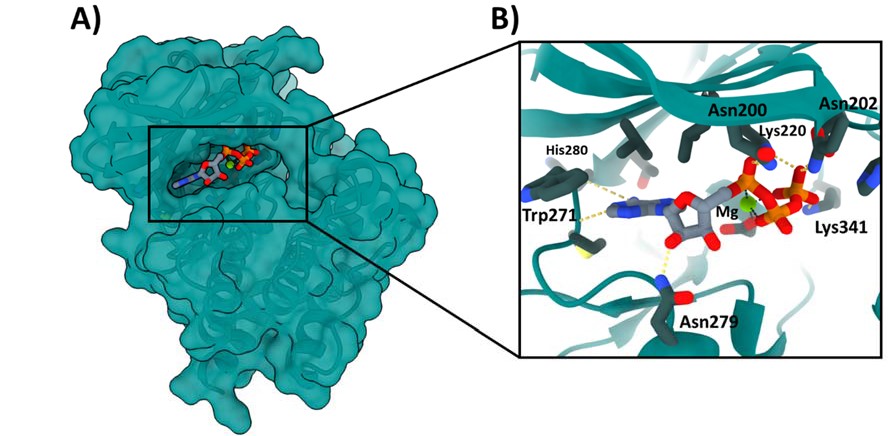
Figura 1. Representación tridimensional del dominio quinasa de ILK (PDB:3KMW). B) Ampliación del centro activo mostrando la molécula de ATP y los residuos con los que interacciona, así como el catión Mg2+.
La sobreexpresión y sobreactivación de ILK se ha relacionado con distintos tipos de cáncer incluyendo el colorrectal, pulmonar y de mama entre otros. Además, a la ILK se le atribuye una función de andamiaje o scaffolding del citoesqueleto celular, gracias a su interacción in vivo con dos proteínas adaptadoras, PINCH y parvina (11). Dado que se ha demostrado que el silenciamiento del oncogén de ILK, conduce a un mejor pronóstico y a una disminución en la invasividad y crecimiento de distintos tipos de tumores, es una diana terapéutica validada para el tratamiento del cáncer (5).
La búsqueda de inhibidores de ILK comenzó hace poco más de una década y solamente unos pocos candidatos se han descrito. Entre las “moléculas pequeñas” descritas como inhibidores de ILK destacan un alcaloide isoquinoleínico, la chelidonina, capaz de inhibir la formación del complejo ternario PINCH-ILK-parvina (12), y dos derivados de pirazol el QLT267 y CPD22 (Figura 1).
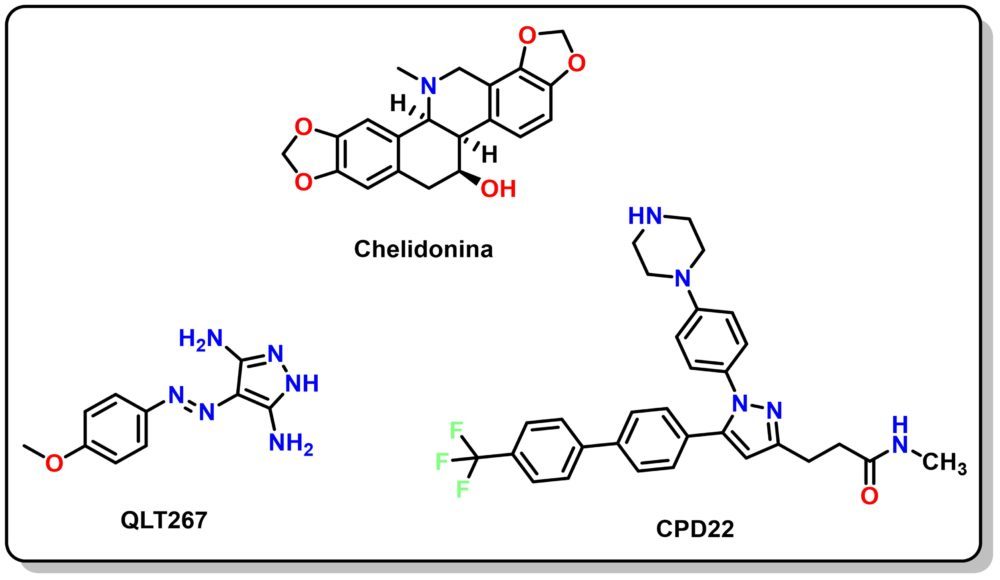
Figura 2. Principales inhibidores de ILK del tipo “moléculas pequeñas”.
El diaminopizarol QLT267 fue identificado durante una campaña de cribado de alto rendimiento para la búsqueda de nuevos antitumorales y posteriormente caracterizado como inhibidor de ILK. Presenta un efecto sinérgico y mayor actividad antitumoral en su administración conjunta con cisplatino para el tratamiento de cánceres dependientes de Her2. Por otro lado, el CPD22 fue desarrollado en 2011 a partir del esqueleto de 4- [4-(4-trifluorometilbifenil)pirazolo]anilina en un esfuerzo por hallar nuevos inhibidores de piruvato deshidrogenasa quinasas tipo 2. Éste compuesto es capaz de inhibir el crecimiento tumoral de líneas de cáncer de próstata y mama con valor de IC50 de 1-2.5 µM, vía inducción de autofagia y apoptosis y reducir los niveles fosforilación de Akt en su serina 473 y GSK-3b, dos proteínas sustrato de ILK. Esta molécula ha sido seleccionada como inhibidor de referencia de ILK y para numerosos estudios como sonda química para modular ILK (13-15). Sin embargo, sorprendentemente, no existen ningún dato estructural acerca del modo de unión y su mecanismo de acción a nivel atómico. Durante el desarrollo del CPD22 los autores hacen referencia al empleo de técnicas de docking de los hits o fragmentos iniciales, sin embargo, no se proporciona esta información ni la referente al candidato optimizado. Del mismo modo, el compuesto se describe como un inhibidor indirecto, ya que no se demuestra su acción o unión directa a la proteína recombinante.
En base a todas estas incógnitas, la búsqueda de una explicación del mecanismo de acción a nivel molecular de cómo actúa el CPD22 parece de gran utilidad. La importancia reside no sólo de cara al desarrollo de nuevos y mejores candidatos a fármacos mediante enfoques de diseño basado en estructura, sino también para poder explicar si el mecanismo de acción de esta molécula depende de su interacción directa con ILK y los efectos que puede desencadenar.
Así pues, se planteó como objetivo general del trabajo de investigación responder a la hipótesis de si el CPD22 es una molécula capaz de unirse a ILK. Por otro lado, este estudio permitirá también determinar el modo de unión de la molécula a la proteína, con el fin de poder establecer y describir aquellas interacciones clave para el desarrollo y optimización de nevos fármacos potentes y selectivos, así como explicar los resultados experimentales. Del mismo modo, esta información permitiría dilucidar el mecanismo de acción a nivel molecular del inhibidor. Para ello, se planteó el empleo de técnicas computacionales y biofísicas, claves en los procesos de desarrollo y optimización de nuevos fármacos, así como de la bioquímica estructural moderna (16).
2. MATERIAL Y MÉTODOS
2.1. Análisis quimioinformático
Las dos quimiotecas de fármacos se prepararon recopilando los códigos SMILES y nombres de 58 inhibidores comercializados de quinasas y fármacos aprobados a través del portal ChEMBl (https://www.ebi.ac.uk/chembl/). Las estructuras se procesaron a través del paquete de Python RDKit (versión 2020.09.1, http://www.rdkit.org), que se utilizó para generar las MACC fingerprints y hacer el análisis de similitud empleando el índice de Tanimoto. El análisis estadístico se llevó a cabo con la librería SciPy (17) y los gráficos con Matplolib y Seaborn (18,19).
2.2. Preparación del ligando y la proteína
Tanto para las simulaciones de docking como de DM se empleó el dominio quinasa de ILK unido a una molécula de ATP (PDB: 3KMW). Una vez descargado el archivo, se eliminó el dominio CH2 de α-parvina y a continuación se añadieron los hidrógenos, se asignaron los enlaces, cargas y estados de protonación a pH 7.2, empleando el Protein Preparation Wizard incluido en el software Maestro 2020 (Maestro, Schrödinger, LLC, New York, NY, 2021).
El CPD22 se construyó con la suite Marvin Sketch (https://chemaxon.com/products/marvin), que además se utilizó para predecir el estado de protonación del nitrógeno de la piperazina (pKa = 8.9) a 298 K empleando las correcciones de librería y el resto de los parámetros por defecto. A continuación, se exportó el archivo en formato sdf a Maestro y a través de LigPrep (Maestro, Schrödinger, LLC, New York, NY, 2021), se generó una geometría tridimensional de mínima energía, se asignó el estado de protonación con carga formal +1 a pH 7.2 y cargas parciales atómicas acordes al campo de fuerzas OPLS3 (20).
2.3 Estudios de docking
Para llevar a cabo los estudios de docking primero se generaron las grids con información del receptor a partir de la estructura tridimensional de ILK procedente de 3KMW previamente preparada y seleccionando como ligando el ATP mediante Maestro 2020. De este modo, se generó una caja con una extensión de 25 Å alrededor de la molécula de ATP donde se llevarían a cabo los cálculos.
A continuación, se realizaron los experimentos de docking utilizando Glide (21) en su modo XP ajustando el número máximo de poses a devolver por el programa a 10 (22). Los resultados se visualizaron en PyMol (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.) y UCSF ChimeraX (23). Se consideraron todas las poses devueltas por el programa (3) correspondiente a las de mejor energía libre de unión.
2.4 Simulaciones de dinámica molecular
Para llevar a cabo las simulaciones de DM la proteína se describió empleando el campo de fuerzas ff14SB (24), mientras que para el CPD22 se utilizó GAFF2 (25). Las cargas de este últimos se asignaron empleando el módulo Antechamber y ajustándolas a un nivel teórico AM1-BCC, generando así los archivos de coordenadas y topología compatibles con la suite AMBER http://ambermd.org/CiteAmber.php). Ambos complejos se solvataron en un octaedro de moléculas de agua TIP3P de 10 Å de tamaño y se adicionaron 2 cationes sodio para neutralizar el sistema empleando el módulo Leap de AmberTools. Las simulaciones se llevaron a cabo el programa OpenMM (26), en una estación de trabajo Windows 10 equipada con una tarjeta gráfica Nvidia® Gigabyte GeForce RTX 2060 GPU. Todas las simulaciones se llevaron a cabo a una temperatura de 298 K y 1 atm de presión integrando las ecuaciones de velocidad con un tiempo entre pasos o timestep de 4 fs gracias a la técnica de hydrogen mass repartitioning(27). En primer lugar, se llevó a cabo una minimización de la energía del sistema mediante el algoritmo L-BFGS (método de Broyden-Fletcher-Goldfarb-Shanno) durante 6000 pasos con el fin de eliminar contactos estrechos entre átomos y geometrías de partida torsionadas. A esta fase le siguió una de DM de equilibrado a volumen constante durante 5 ns. A continuación, se pasó a la producción de las simulaciones de DM clásica durante 500 ns para cada sistema, tardando 8 días en completar cada una de ellas. Todos los sistemas se simularon bajo las condiciones de límite periódico, y aplicando el método de Ewald para tratar las interacciones electrostáticas de corto alcance con un límite de 8 Å (28). Así mismo, se empleó el algoritmo SHAKE en todos los enlaces en los que participan átomos de hidrógeno con el fin de simplificar los cálculos durante la dinámica (32). Las trayectorias de la dinámica molecular se analizaron empleando el software MDAnalysis (29) y MDtraj (30). Para el cálculo de energía libre se utilizó el programa MM-ISMSA sobre los últimos 250 ns de las trayectorias generadas correspondientes al sistema equilibrado (31).
Las simulaciones de templado simulado también se llevaron a cabo con el software OpenMM, calentando el sistema a 310 K y enfriándolo gradualmente hasta 270 K durante 1 ns. Tras el enfriamiento, el complejo resultante se minimizó durante 2000 pasos de L-BFGS extrayéndose la estructura resultante de éste y usando las coordenadas atómicas para el inicio de un nuevo ciclo, así sucesivamente hasta 4 veces.
2.5 Experimentos de SPR
Los experimentos de SPR se realizaron en el Instituto de Química Médica del CSIC, bajo la supervisión de la Dra. Laura Lagartera empleando ILK recombinante adquirida en la casa Randox Life Sciences y el CPD22 adquirido en Calbiochem. La proteína (59.92 kDa) se inmovilizó en un chip de oro CM5 (Biacore, GE) siguiendo el protocolo estándar de inmovilización no covalente aportado por el fabricante. A continuación, se hicieron fluir distintas concentraciones del analito en un buffer (50 mM Tris pH 7.5, 50 mM NaCl, 2 mM CaCl2, 20 mM EDTA y 2% de DMSO) a una velocidad de 90 μL/min en el dispositivo Biacore X-100. Los resultados expresados en unidades de resonancia se trataron con el software X-100 (Biacore, GE Healthcare Life Science).
3. DISCUSIÓN Y RESULTADOS
3.1 Análisis quimioinformático
Una de las primeras incógnitas que surgen a la hora de abordar el problema acerca del modo de unión del CPD22 a la ILK es la zona donde puede tener lugar la interacción con la proteína. La gran mayoría de inhibidores de quinasa comercializados y aprobados están dirigidos al bolsillo de ATP de sus respectivas quinasas (32). En base a esto, parece probable que el CPD22 se dirija también al bolsillo de ATP de ILK. Si bien también existe la posibilidad de que la molécula se dirija a algún sitio alostérico, dado que el número de inhibidores desarrollados es considerablemente pequeño, entre otros factores por la dificultad de su desarrollo, únicamente se consideró la posibilidad de que el CPD22 se dirigiese al bolsillo de ATP de ILK. Para comprobar esta primera hipótesis se planteó hacer un análisis quimioinformático de similitud estructural entre distintos inhibidores de quinasas y el CPD22. Para ello, se preparó una pequeña base de datos con 56 fármacos inhibidores comercializados de quinasas, todos ellos dirigidos al bolsillo de ATP (ver material suplementario) (32). Además, como control negativo para el experimento de similitud se preparó otra quimioteca con 56 fármacos comercializados dirigidos a otras dianas (ver material suplementario).
La estructura bidimensional de las moléculas puede ser transcrita a una cadena de bits capaz de almacenar información acerca de la presencia o ausencia de ciertos patrones o motivos. Estas cadenas de bits se conocen como huellas dactilares o más comúnmente en el campo de la quimioinformática como fingerprints y se clasifican según el tipo de descripción que hacen de la molécula: estructural, topológica, circular, farmacofórica, etc (33).
Para el presente trabajo se seleccionaron las fingerprints de MACCS que se han empleado con éxito para capturar la información bidimensional de moléculas orgánicas en campañas de cribado virtual (34). Una vez generadas las huellas, se puede calcular la similitud estructural entre moléculas a través de distintos índices, siendo el de Tanimoto uno de los más empleados en el campo de descubrimiento de fármacos (Figura 3) (34).
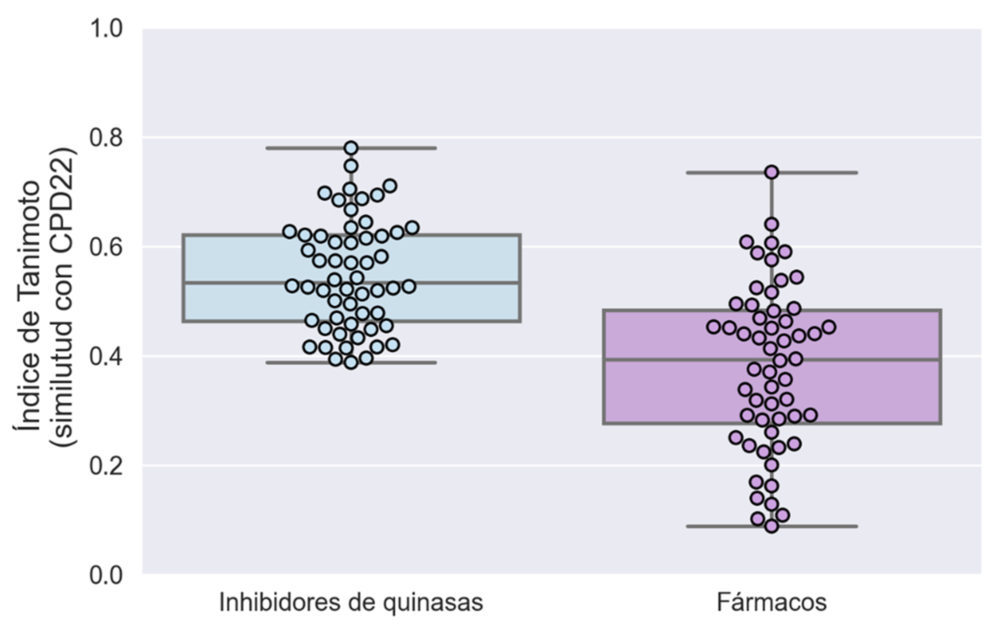
Figura 3. Diagrama de cajas y bigotes mostrando la distribución del índice de Tanimoto para los inhibidores de quinasa y el grupo de fármacos comercializados utilizando como referencia el CPD22.
Tal y como se muestra en el gráfico de la Figura 3, el grado de similitud medio entre el CPD22 y el grupo de inhibidores de quinasa es moderado, con una media de índice de Tanimoto de 0.54. Ésta es mayor que para el grupo de fármacos no inhibidores de quinasas (0.37), diferencia estadísticamente significativa cuando se realiza la prueba de t de Student (p < 0.005). Además, la distribución de valores está más concentrada en el caso de inhibidores de quinasa, apoyando la hipótesis de una mayor similitud con el CPD22. En base a este experimento se concluyó que el CPD22 tiene un grado de similitud significativo comparándolo con otros inhibidores de quinasa competitivos por lo que el resto de los estudios se llevaron a cabo en el bolsillo de ATP de ILK.
3.2. Estudios de docking
Para establecer un modo de unión entre la ILK y el CPD22 se llevó a cabo un estudio de docking o acoplamiento molecular, que permite predecir el modo de unión entre un ligando (fármaco) y su diana (macromolécula). Además, los cálculos de docking permiten hacer una estimación sencilla de la energía libre (ΔG) de unión del complejo ligando-receptor gracias a la función de scoring o tanteo que cuantifica energéticamente las interacciones establecidas.
Como herramienta de docking se empleó el programa Glide en su modo XP, el cual cuenta con una función de tanteo empírica calibrada con datasets que incluyen inhibidores de quinasas, haciendo de él una herramienta muy útil para este tipo de sistemas (21). Glide devolvió un total de tres poses, compartiendo un modo de unión bastante homogéneo y similar (Figura 4) y un rango de ΔG de entre -6.262 y -5.09 kcal/mol.
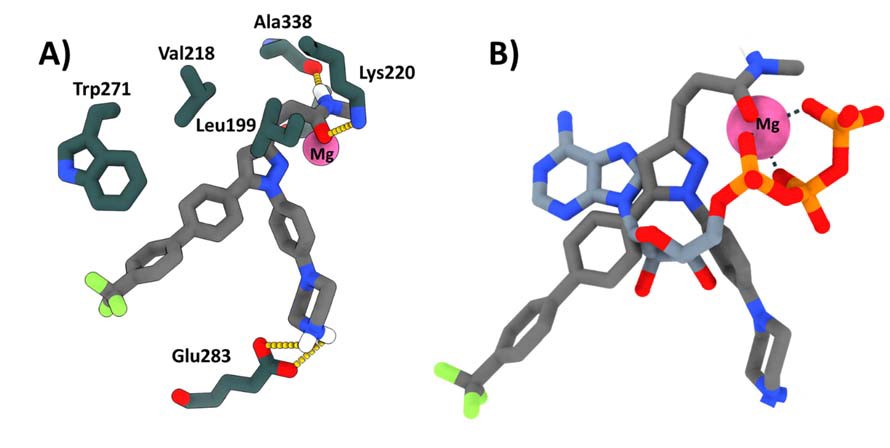
Figura 4. Modo de unión predicho mediante cálculos de docking para el CPD22 en el bolsillo de ATP de ILK. B) Comparación entre el modo de unión del CPD22 y la molécula de ATP del sitio activo (PDB: 3KMWꞏ). (Para facilitar la lectura de la imagen se muestran únicamente los hidrógenos polares del CPD22)
El CPD22 ocupa el bolsillo de ATP, sin embargo, las distintas ramificaciones que nacen del anillo de pirazol le permiten establecer interacciones diferentes. Por un lado, la cadena de 2’-carbamoiletilo se pliega hacia el bolsillo donde se aloja el catión Mg2+, con el que podría llegar a coordinarse a través del par de electrones del oxígeno del carbonilo, el cual se encuentra también unido mediante un enlace de hidrógeno a la Lys220 (Figura 4A). Además, la amida se enlaza con el carbonilo del esqueleto de la Ala338 gracias a su grupo NH. Por otro lado, el N2 del pirazol se orienta hacia el catión Mg2+, pudiendo llegar a participar en la coordinación octahédrica a través del par de electrones libre. El modo de unión se encuentra estabilizado por una interacción fuerte y altamente direccional de carácter iónico y reforzada por enlaces de hidrógeno entre el catión amonio NH2+ del anillo de piperazina, y el anión carboxilato del residuo de Glu283. Esta unión explicaría la pérdida de actividad cuando esta ramificación se sustituye con cadenas alquílicas o un anillo de morfolina (RW.ERROR – Unable to find reference: doc: 611aa1f68f08a5d2e7bd0226).
La tercera ramificación del CPD22, que se corresponde con el sustituyente 4’-trifluorometilbifenilo de la posición C5 del pirazol, establece numerosas interacciones de van der Waals, así como enlaces CH-π entre los anillos aromáticos y los residuos de Leu199, Leu207 y Trp271. Este último aminoácido también establece un apilamiento-π parcialmente desplazado con el 4’-trifluorometilbifenilo, favorecido por el fuerte efecto desactivante que ejerce el sustituyente trifluorometilo sobre el anillo de benceno. Estas observaciones concuerdan con los datos de actividad descritos por los autores, en los que la pérdida de esta ramificación o la introducción de sustituyentes aromáticos más grandes como el fenantreno, conducen a pérdida de la actividad debida a la falta de espacio (RW.ERROR – Unable to find reference:doc:611aa1f68f08a5d2e7bd0226).
Si bien las ramificaciones en N1 y C5 no se posicionan en las mismas zonas que la molécula de ATP (Figura 4B), sí que lo hacen en regiones que se han explorado con otros inhibidores de quinasas de tipo 1 y 1.5 (35). Este hecho apoya la propuesta de modo de unión del CPD22 al guardar similitud con otras poses ya descritas y caracterizadas para otras quinasas.
3.3. Simulaciones de dinámica molecular
Los estudios de docking ofrecen una primera aproximación bastante fiable acerca del modo de unión entre un fármaco y su diana, sin embargo, el uso de otras técnicas computacionales como la dinámica molecular (DM), permite apoyar la hipótesis y obtener más información. Así pues, se decidió llevar a cabo simulaciones de DM clásica empleando tres sistemas diferentes, por un lado, la ILK en su forma apo, la ILK unida a una molécula de ATP y la ILK en complejo con el CPD22 (Tabla 1). Estas simulaciones permiten, además, el estudio del mecanismo de acción del fármaco sobre la diana, ya que dependiendo de la esquela de tiempos de la simulación se puede obtener información de cambios conformacionales locales o globales sobre la proteína.
Tabla 1. Simulaciones de dinámica molecular realizadas con una duración de 500 ns.

Tras minimizar la energía de cada uno de los tres sistemas con el fin de encontrar una geometría de partida más relajada (eliminando contactos cercanos entre átomos consecuencia del empaquetamiento), los tres sistemas se calentaron gradualmente hasta una temperatura de 289 K para posteriormente llevar a cabo las simulaciones de DM sin restricciones durante 500 ns.
Uno de los parámetros más empleados para evaluar la estabilidad de los complejos en las simulaciones de DM es el cálculo de la desviación cuadrática media (RMSD, del inglés Root Mean Square Deviation) a lo largo del tiempo (Figura 5A). Este parámetro mide las fluctuaciones de la posición de aquellos átomos distintos de hidrógeno comparándola con la conformación inicial a lo largo de la simulación. Su monitorización permite comprobar si el sistema ha alcanzado el equilibrio (manteniéndose estable) o si, por el contrario, continúa evolucionando hacia otros estados.
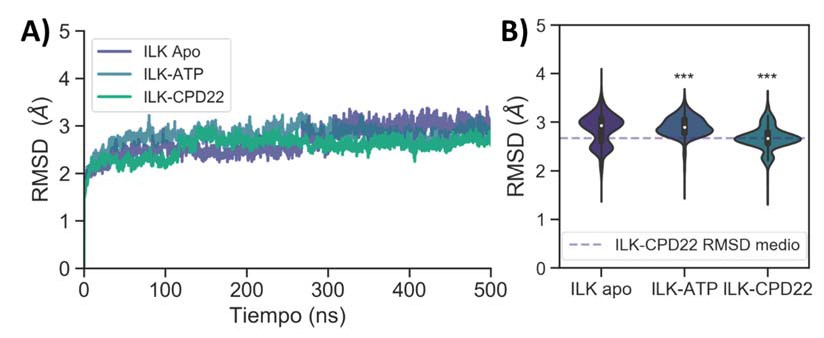
Figura 5. A) Diagrama de RMSD de los sistemas simulados mediante DM durante 500 ns. B) Diagrama de violines mostrando la distribución de los valores de RMSD. *** indica p < 0.0001 en la prueba t de Student comparando con los valores de ILK apo.
Como se observa en la figura 5A, los tres sistemas parecen alcanzar una situación más estable tras aproximadamente 200 ns, tras los cuales siguen evolucionando, pero sin grandes cambios ni diferencias notables, validando así el protocolo de dinámica. Durante este este tiempo, si se analizan más detenidamente estos datos, se puede extraer información interesante. En primer lugar, si se estudia detenidamente la evolución del RMSD a lo largo del tiempo se puede observar cómo, aunque se llega a una situación relativa de equilibrio, la ILK en su forma apo continúa evolucionando hacia valores más altos hasta el final de la dinámica, y superiores a los alcanzados por los otros dos complejos. Esto queda bien reflejado tanto en la distribución de valores (Figura 5B) como en su media, siendo de 2.67 ± 0.31, 2.79 ± 0.19 y 2.56 ± 0.21 Å para ILK en su forma apo, unida a ATP y a CPD22 respectivamente.
Si bien parece que la presencia del ATP en el bolsillo de ILK conduce a una leve disminución de la estabilidad del sistema, la unión del CPD22 produce el efecto opuesto, aumentando la estabilidad y disminuyendo su flexibilidad general en términos de RMSD. Esto es indicativo de que el CPD22 parece tener la capacidad de estabilizar la ILK en una de sus conformaciones probablemente inactiva desde el punto de vista funcional.
Mientras que el RMSD cuantifica cuánto diverge una estructura (en este caso la proteína) a lo largo del tiempo, el cálculo del RMSF (fluctuación cuadrática media, del inglés Root Mean Square Fluctuation) puede revelar qué áreas del sistema presentan mayor movilidad (Figura 6).
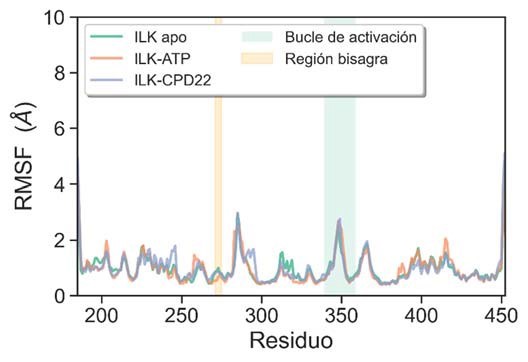
Figura 6. Gráfico de RMSF para los carbonos a de la proteína
En general el dominio tipo quinasa de la ILK presenta una movilidad moderada, con regiones especialmente flexibles como es el bucle de activación, lo que concuerda con otros estudios computacionales previos (36). De acuerdo con el análisis de RMSF, la proteína no sufre cambios importantes durante el tiempo de simulación, lo que sugiere que tanto en presencia como ausencia de ligando se encuentra en una conformación estable caracterizada por el motivo “DFG” en posición interior y la hélice αC en conformación exterior, propia de los inhibidores tipo 1.5 (36). Un segundo motivo importante para el reconocimiento molecular del sustrato es la región bisagra, ésta se encarga de establecer enlaces de hidrógeno con la base púrica del ATP con aminoácidos como el His280 y Trp271 (Figura 1B). Tal y como se observa en la Figura 6, esta región presenta mayor movilidad en el caso de la ILK apo y la ILK unida al CPD22. Esto es consecuencia del reordenamiento que sufre este bucle ante la ausencia del ligando original (ATP) o por la incorporación del CPD22. Gracias a la plasticidad estructural de la región, el residuo de Trp271 se aleja del anillo aromático del CPD22 y para compensar el apilamiento π se establece, durante gran parte de la simulación (Figura 7), un enlace de hidrógeno entre el átomo de nitrógeno no protonado del anillo de piperazina y el grupo NH2 de la Asn279, no observada durante los estudios de docking. Junto al enlace iónico entre el catión piperazinio y el residuo de Glu238 (Figura 4A) se puede observar otra interacción especialmente estable que es la de coordinación del carbonilo del grupo 2’-carbamoiletilo con el Mg2+, que se mantiene a lo largo de toda la dinámica con una distancia media de 2.1 ± 0.2Å (Figura 7).
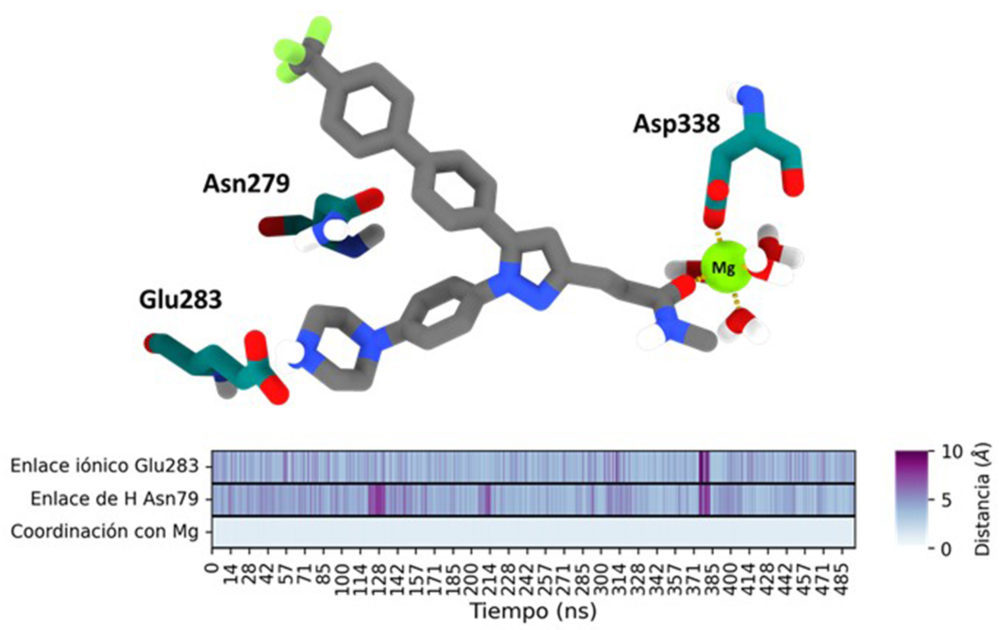
Figura 7. Arriba) Imagen mostrando la interacción entre el CPD22, las moléculas de agua y el Asp338 con el catión Mg2+ de ILK durante la simulación de DM. Abajo) Mapa de calor mostrando la distancia de las principales interacciones polares entre el CPD22 e ILK: enlace iónico (distancia entre el O del Glu238 y el N+ del anillo de piperazina), enlace de hidrógeno (distancia entre el NH2 de la Asn279 y el átomo de N de la piperazina, y la coordinación con el Mg2+ (distancia entre el átomo de Mg2+ y el átomo de O del grupo carbonilo del CPD22).
El átomo de oxígeno del grupo carbonilo pasa a formar parte de la esfera de coordinación del metal junto con otras 4 moléculas de agua, completando la coordinación octaédrica del metal gracias al átomo de oxígeno del grupo carboxilato del Asp338 (Figura 7A). La importancia de este grupo carbonilo, ilustrada por los estudios de dinámica molecular, queda bien reflejada en la actividad descrita para análogos sintetizados por Lee y colaboradores, en los que la pérdida del grupo carbonilo o la sustitución de este motivo por otros con cadenas más cortas conduce a la pérdida de actividad (RW.ERROR – Unable to find reference:doc:611aa1f68f08a5d2e7bd0226). De esta forma, el CPD22 junto con las moléculas de agua del centro pseudoactivo de la ILK pasarían a suplir el papel que desempeñan los átomos de oxígeno de los fosfatos del ATP en la coordinación observada experimentalmente (Figura 4B).
Este modo de unión se mantuvo estable durante la mayor parte de la simulación de DM (RMSD: 1.86 ± 0.31 Å) y fue bastante similar al observado en la simulación del ATP (1.03 ± 0.16). Con el fin de corroborar si efectivamente este modo de unión es el más estable, se llevó a cabo una simulación de templado simulado o simulated annealing. Estas simulaciones comienzan a temperaturas altas que permitan sobrepasar barreras energéticas del sistema para poco a poco enfriarse y alcanzar un mínimo energético local acercándose al mínimo global (37).
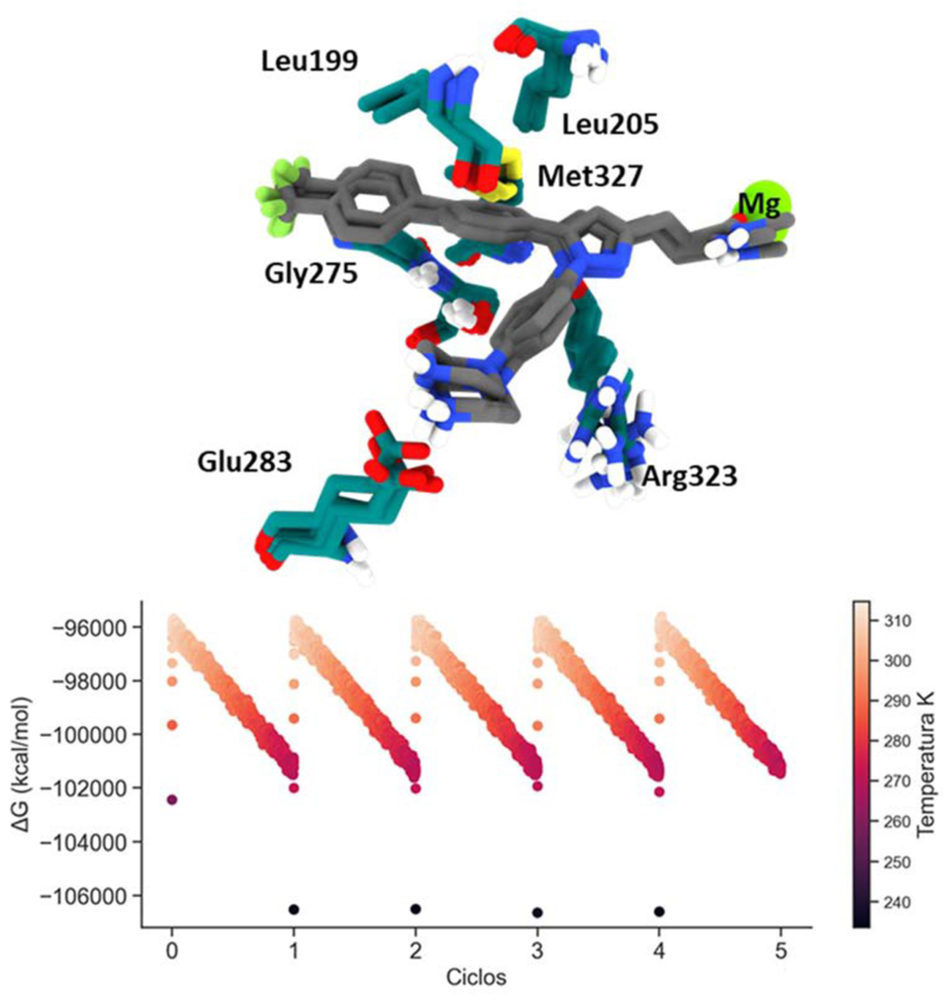
Figura 8. Arriba) Superposición de las estructuras minimizadas de los ciclos de templado simulado mostrando el modo de unión alcanzado por el CPD22 en el bolsillo de ATP de ILK. Abajo) Diagrama energético de los ciclos de templado simulado.
De esta forma, se realizaron 4 ciclos de calentamiento-enfriamiento del sistema, los cuales convergieron en el mismo modo de unión del CPD22 en el interior de la ILK. Cabe destacar que el mínimo energético alcanzado supone la pérdida del enlace de hidrógeno mediado por la Asn279 que se observa durante gran parte de la dinámica. Sin embargo, éste se ve compensado por otras interacciones como el apilamiento π-catión con el residuo de Arg323 y el anillo de benceno. Se le unen además interacciones hidrofóbicas mediadas por los residuos de Leu199, Leu207, Gly275 y Met327. Este experimento pone de manifiesto cómo de estable es termodinámicamente la unión del CPD22 en el bolsillo de ATP gracias a las interacciones previamente descritas.
La energía libre de unión y las contribuciones para los residuos más importantes dentro del bolsillo de ATP fueron estimadas teniendo en cuenta no solamente el componente de van der Waals y electrostático, sino también los componentes de desolvatación tanto del ligando como del receptor gracias al software MM-ISMSA (Tabla 2).
Tabla 2. Energía de interacción promedio (kcal/mol) y sus contribuciones para el CPD22 y la molécula de ATP en el centro activo de la ILK.

Tomando como referencia la molécula de ATP (-72.26 kcal/mol), el cálculo de ΔG demuestra que a lo largo de toda la simulación de dinámica molecular el CPD22 muestra una buena afinidad por la ILK dentro del bolsillo de ATP (-49.97 kcal/mol). Mientras que, para el ligando endógeno (ATP), la ΔG está dominada por el término electrostático, definido por un potencial de Coulomb (QQ), para el CPD22 lo hacen los componentes hidrofóbicos y de van der Waals (VDW). Esto es esperable, dado que el ATP porta un gran número de grupos polares y cargados a pH fisiológico, lo que además se traduce en una elevada energía de desolvatación en comparación al CPD22. Esto se explica porque la optimización de cabezas de serie suele estar acompañada por el anclaje de fragmentos, normalmente con componentes hidrófobicos, con el fin de establecer nuevas interacciones no covalentes que conducen a un aumento de la entalpía (38). El análisis detallado de la contribución por residuos también revela diferencias relacionadas con la propia naturaleza del ligando (Figura 9). Destaca la interacción direccional de enlace iónico reforzado por enlace de hidrógeno entre el catión piperazinio y el residuo de Glu283, que en el caso del CPD22 es una interacción clave (> 6 kcal/mol) y por tanto de vital importancia en el proceso de optimización del cabeza de serie (Figura 8). Cabe mencionar también la contribución de otros residuos polares y cargados como Asn193, Asn279 y Arg303 (Figura 9). Esta última más importante en el caso del ATP ya que compensa la carga negativa de los residuos de fosfato.
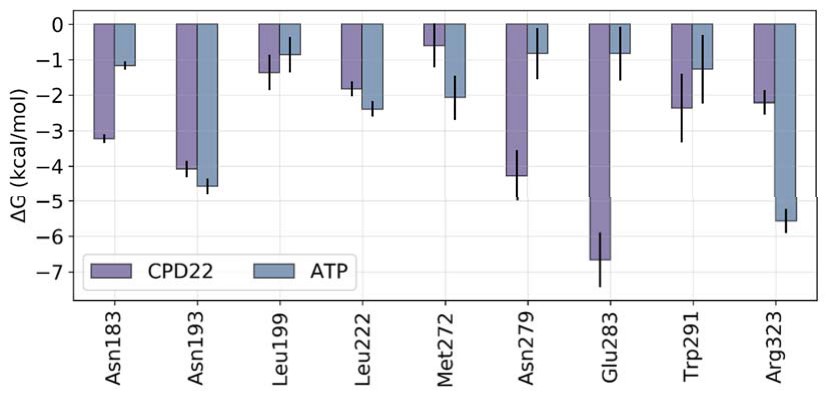
Figura 9. Contribución energética a la energía libre de unión por residuo para los complejos de ILK-ATP e ILK-CPD22.
La estimación energética que MM-ISMSA hace sobre el catión Mg2+ es de -4.69 ± 0.83 y -30.01 ± 3.88 kcal/mol para las moléculas de CPD22 y el ATP respectivamente. Esta se explicaría en gran parte porque el ATP se une fuertemente al catión gracias a los tres puntos de coordinación en los que intervienen al menos tres átomos de oxígeno del ATP cediendo densidad electrónica (Figura 4B). Este resultado sugiere que la quelación del metal es clave para la optimización y desarrollo de nuevos inhibidores a través de la incorporación de grupos funcionales capaces de coordinarse con el metal. Del mismo modo, dado que la ILK sólo posee un catión Mg2+, a diferencia de otras quinasas y pseudoquinasas que presentan más e incluso otros metales (39), también supondría una estrategia para ganar selectividad.
En general, los cálculos de energía libre apoyan también la hipótesis de que el CPD22 tiene una buena afinidad por el bolsillo de ATP de ILK, el cual constituiría su sitio de unión apoyando los resultados obtenidos en el análisis quimioinformático y de DM.
3.4. Ensayos de resonancia de plasmón superficial
La supuesta actividad quinasa de ILK es objetivo de controversia hasta el día de hoy (40-42). A pesar de que hay autores que reportan sus constantes enzimáticas, los detractores defienden que esta es una proteína obtenida de lisados no purificados correctamente con componentes desconocidos (10). Cuando se produce la proteína recombinante en Escherichia coli o se inmunoprecipita no muestra ninguna actividad enzimática (10). A pesar de todo, parece claro que de algún modo la ILK in vivo tiene capacidad de fosforilar distintos sustratos entre los que destacan Akt y GSK-3β (9,40,43).
Dado que los estudios computacionales apuntan claramente a que el CPD22 es capaz de unirse a ILK y, más en concreto a su centro pseudoactivo, se hace necesario el empleo de alguna técnica que pueda confirmar esta hipótesis y que no se base en las controvertidas capacidades catalíticas de la enzima.
Para este fin, se decidió utilizar la técnica de resonancia de plasmón de superficie (SPR, del inglés Surface Plasmon Resonance), la cual permite realizar medidas de afinidad y cinética entre una macromolécula (ILK) y un analito (CPD22) sin necesidad de marcajes enzimáticos, fluorescentes o radioactivos (44). Durante el experimento se inmovilizó en un chip de oro la ILK recombinante mediante procedimientos estándar y seguidamente se hizo fluir el CPD22 a distintas concentraciones con el fin de medir si existe unión a la proteína y su intensidad (Figura 10).
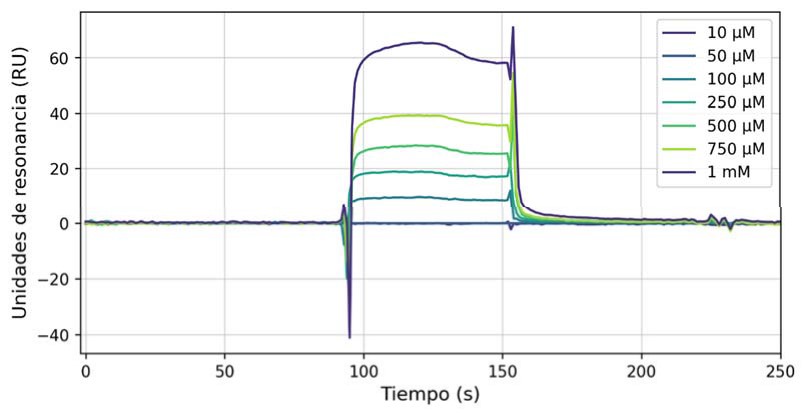
Figura 10. Sensograma de SPR mostrando la unión de CPD22 a ILK recombinante.
De este ensayo se pudo concluir que, efectivamente, el CPD22 tiene capacidad de unión directa a ILK, tal y como muestra el sensograma del experimento (Figura 10). Además, la disociación del complejo enzima-ligando es bastante rápida, lo que concuerda con el modo de unión típico de moléculas pequeñas. Si bien no se pudo determinar con exactitud la constante de afinidad, ya que a concentraciones altas el CPD22 precipita, ésta se encontraría en un rango de 450-1150 μM de acuerdo con una estequiometria 1:1 y cinética de Langmuir.
4. CONCLUSIONES
El campo de la quimioterapia antitumoral es un área muy activa de investigación farmacéutica la cual no busca únicamente mejorar tratamientos frente a dianas ya existentes, sino también la búsqueda de potenciales dianas terapéuticas novedosas que permitan un control más preciso de la terapia antitumoral.
En este sentido, si bien las proteínas quinasas han sido durante mucho tiempo la piedra angular de la búsqueda de nuevos fármacos antineoplásicos, sus homólogos no funcionales, las pseudoquinasas suponen un nuevo concepto atractivo y prometedor.
Entre las pseudoquinasas más relevantes para la terapia antineoplásica destaca la ILK, una proteína validada como diana terapéutica en distintos tipos de cáncer cuyo funcionamiento a nivel atómico y molecular es bastante desconocido. De cara a poder comprender mejor el funcionamiento de esta proteína, se hace necesario conocer cómo actúa el único inhibidor descrito hasta el momento, el CPD22. Este conocimiento permitiría establecer el mecanismo de acción de la molécula, un conocimiento vital de cara a su desarrollo como fármaco, así como racionalizar los resultados experimentales y servir de base para el diseño de nuevos fármacos.
El análisis quimioinformático de similitud entre el CPD22 y distintos inhibidores de quinasas con respecto a una familia genérica de fármacos demuestra que esta molécula presenta patrones estructurales claramente relacionados con el grupo de inhibidores de quinasas. Esto hace pensar que probablemente actúe mediante la unión al bolsillo de ATP de la ILK.
Las técnicas de modelado molecular de docking y simulaciones de dinámica molecular han permitido proponer un modelo de unión entre el CPD22 y la ILK. Éste estaría caracterizado por una serie de interacciones clave entre las que destacan el enlace iónico con el Glu283, la coordinación con el catión Mg2+ y distintos contactos hidrofóbicos (Figura 11).

Figura 11. Modelo de interacción propuesto para el CPD22 con ILK
El estudio dinámico del sistema permite no sólo estudiar las interacciones a lo largo del tiempo sino evaluar su impacto en el modo de unión, que ha sido confirmado mediante técnicas de templado simulado.
Las observaciones extraídas de estos estudios, así como del cálculo de energía libre por componentes mediante MM-ISMSA correlaciona perfectamente con los datos experimentales observados por el grupo que desarrolló el CPD22. Esto supone, por un lado, la validación de nuestro modelo y, además, la primera prueba de que probablemente el CPD22 inhibe ILK de forma directa por unión a su sitio activo, estabilizando una conformación de la enzima. Por otro lado, de cara a la optimización y búsqueda de nuevos fármacos hemos puesto de manifiesto la importancia que podría jugar la interacción con el catión Mg2+ presente en ILK y no en otras pro teínas homólogas.
Además, los estudios de SPR han demostrado que, efectivamente el CPD22 se une de forma directa a ILK, por tanto, confirman nuestra hipótesis inicial y ponen de manifiesto el potencial de las técnicas computacionales empleadas en combinación.
De este modo, este trabajo supone el primer estudio exhaustivo y de comprensión de la unión del CPD22 a la pseudoquinasa ILK demostrando, que efectivamente, la molécula actúa por unión directa a la enzima, y más concretamente a su sitio activo. De esta forma, se abre la puerta a la optimización y búsqueda de nuevos y mejores fármacos, así como aquellos estudios encaminados a comprender más en profundidad el papel de la ILK en el cuerpo humano utilizando el CPD22 como herramienta bioquímica.
Listado de abreviaturas
Organización Mundial de la Salud (OMS), quinasa ligada a integrinas (ILK), Dinámica Molecular (DM), Protein Data Bank (PDB), raíz cuadrada de la desviación cuadrática media (RMSD), Resonancia de Plasmón Superficial (SPR).
Conflicto de intereses
Los autores de este manuscrito declaran no tener ningún tipo de conflicto de intereses.
Agradecimientos
El autor agradece al Dr. Diego Rodríguez-Puyol y al Grupo de Fisiología y Fisiopatología Renal de la UAH, por ceder una muestra del CPD22 para los ensayos biofísicos. El autor agradece al Servicio de Interacciones Biofísicas del IQM por la realización de los experimentos de SPR. El autor agradece al Dr. Ramón Alajarín por la revisión del manuscrito y sus comentarios, así como al Dr. Juan J. Vaquero por su apoyo.
5. REFERENCIAS
- Luengo-Fernandez R, Leal J, Gray A, Sullivan R. Economic burden of cancer across the European Union: a population-based cost analysis. Lancet Oncol 2013 Nov;14(12):1165-1174.
- Kurzrock R, Kantarjian HM, Kesselheim AS, Sigal EV. New drug approvals in oncology. Nat Rev Clin Oncol 2020 Mar;17(3):140-146.
- Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov 2002 Jul;1(7):493-502.
- Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986;319(6056):743-748.
- Zhang H, Photiou A, Grothey A, Stebbing J, Giamas G. The role of pseudokinases in cancer. Cell Signal 2012 Jun;24(6):1173-1184.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science 2002 Dec 6;298(5600):1912-1934.
- Hannigan GE, Leung-Hagesteijn C, Fitz-Gibbon L, Coppolino MG, Radeva G, Filmus J, et al. Regulation of cell adhesion and anchorage-dependent growth by a new beta 1- integrin-linked proteinkinase. Nature 1996 Jan 4;379(6560):91-96.
- Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov 2018 May;17(5):353-377.
- Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer 2005 Jan;5(1):51-63.
- Fukuda K, Gupta S, Chen K, Wu C, Qin J. The pseudoactive site of ILK is essential for its binding to alpha-Parvin and localization to focal adhesions. Mol Cell 2009 Dec 11;36(5):819-830.
- McDonald PC, Fielding AB, Dedhar S. Integrin-linked kinase–essential roles in physiology and cancer biology. J Cell Sci 2008 Oct 1;121(Pt 19):3121-3132.
- Zhang Y, Chen K, Tu Y, Velyvis A, Yang Y, Qin J, et al. Assembly of the PINCH- ILK-CH-ILKBP complex precedes and is essential for localization of each component to cell-matrix adhesion sites. J Cell Sci 2002 Dec 15;115(Pt 24):4777-4786.
- Kim O, Hwangbo C, Kim J, Li DH, Min BS, Lee JH. Chelidonine suppresses migration and invasion of MDA-MB-231 cells by inhibiting formation of the integrin- linked kinase/PINCH/α-parvin complex. Mol Med Rep 2015 Aug;12(2):2161-2168.
- Kalra J, Warburton C, Fang K, Edwards L, Daynard T, Waterhouse D, et al. QLT0267, a small molecule inhibitor targeting integrin-linked kinase (ILK), and docetaxel can combine to produce synergistic interactions linked to enhanced cytotoxicity, reductions in P-AKT levels, altered F-actin architecture and improved treatment outcomes in an orthotopic breast cancer model. Breast Cancer Res. 009;11(3):R25.
- Lee S, Hsu E, Chou C, Chuang H, Bai L, Kulp SK, et al. Identification and characterization of a novel integrin-linked kinase inhibitor. J Med Chem 2011;54(18):6364-6374.
- Chen J, Bell J, Lau BT, Whittaker T, Stapleton D, Ji HP. A functional CRISPR/Cas9 screen identifies kinases that modulate FGFR inhibitor response in gastric cancer. Oncogenesis 2019 May 10;8(5):33-z.
- Ning Z, Zhu X, Jiang Y, Gao A, Zou S, Gu C, et al. Integrin-Linked Kinase Is Involved In the Proliferation and Invasion of Esophageal Squamous Cell Carcinoma. J Cancer 2020 Jan 1;11(2):324-333.
- Li B, Wang X, Wang R, Rutz B, Ciotkowska A, Gratzke C, et al. Inhibition of neurogenic and thromboxane A(2) -induced human prostate smooth muscle contraction by the integrin a2β1 inhibitor BTT-3033 and the integrin-linked kinase inhibitor Cpd22. Prostate 2020 Aug;80(11):831-849.
- Ou-Yang S, Lu J, Kong X, Liang Z, Luo C, Jiang H. Computational drug discovery. Acta Pharmacol Sin 2012;33(9):1131-1140.
- Attwood MM, Fabbro D, Sokolov AV, Knapp S, Schiöth HB. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat Rev Drug Discov 2021 Aug 5.
- Muegge I, Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discov 2016;11(2):137-148.
- Kuwahara H, Gao X. Analysis of the effects of related fingerprints on molecular similarity using an eigenvalue entropy approach. J Cheminform 2021 Mar 23;13(1):27-2.
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004 Mar 25;47(7):1739-1749.
- Cowan-Jacob SW, Jahnke W, Knapp S. Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med Chem 2014 Apr;6(5):541-561.
- Schwarz D, Merget B, Deane C, Fulle S. Modeling conformational flexibility of kinases in inactive states. Proteins 2019 Nov;87(11):943-951.
- Kirkpatrick S, Gelatt CD,Jr, Vecchi MP. Optimization by simulated annealing. Science 1983 May 13;220(4598):671-680.
- Klett J, Núñez-Salgado A, Dos Santos HG, Cortés-Cabrera Á, Perona A, Gil- Redondo R, et al. MM-ISMSA: An Ultrafast and Accurate Scoring Function for Protein- Protein Docking. J Chem Theory Comput 2012 Sep 11;8(9):3395-3408.
- Du X, Li Y, Xia Y, Ai S, Liang J, Sang P, et al. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. International journal of molecular sciences 2016;17(2):144.
- McClendon CL, Kornev AP, Gilson MK, Taylor SS. Dynamic architecture of a protein kinase. Proc Natl Acad Sci U S A 2014 Oct 28;111(43):4623.
- Qin J, Wu C. ILK: a pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr Opin Cell Biol 2012 Oct;24(5):607-613.
- Wickström SA, Lange A, Montanez E, Fässler R. The ILK/PINCH/parvin complex: the kinase is dead, long live the pseudokinase! EMBO J 2010 Jan 20;29(2):281-291
- Hannigan GE, McDonald PC, Walsh MP, Dedhar S. Integrin-linked kinase: not so ‘pseudo’ after all. Oncogene 2011 Oct 27;30(43):4375-4385.
- Vaynberg J, Fukuda K, Lu F, Bialkowska K, Chen Y, Plow EF, et al. Non-catalytic signaling by pseudokinase ILK for regulating cell adhesion. Nat Commun 2018 Oct 26;9(1):4465-7.
- Prabowo BA, Purwidyantri A, Liu KC. Surface Plasmon Resonance Optical Sensor: A Review on Light Source Technology. Biosensors (Basel) 2018 Aug 26;8(3):80. doi:10.3390/bios8030080.
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 2020 Mar;17(3):261-272.
- J. D. Hunter. Matplotlib: A 2D Graphics Environment. Computing in Science & Engineering 2007;9(3):90-95.
- Seaborn: statistical data visualization.
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput 2016 Jan 12;12(1):281-296.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006 Oct 19;49(21):6177-6196.
- Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 2021 Jan;30(1):70-82.
- Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J Chem Theory Comput 2020 Jan 14;16(1):528-552.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem 2004 Jul 15;25(9):1157-1174.
- Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 2017 Jul 26;13(7):e1005659.
- Hopkins CW, Le Grand S, Walker RC, Roitberg AE. Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J Chem Theory Comput 2015 Apr.14;11(4):1864-1874.
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. The Journal of chemical physics 1995 Nov 15,;103(19):8577-8593.
- Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem 2011 Jul. 0;32(10):2319-2327.
- McGibbon RT, Beauchamp KA, Harrigan MP, Klein C, Swails JM, Hernández CX, et al. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys J 2015 Oct 20;109(8):1528-1532.
- Klett J, Núñez-Salgado A, Dos Santos HG, Cortés-Cabrera Á, Perona A, Gil- Redondo R, et al. MM-ISMSA: An Ultrafast and Accurate Scoring Function for Protein- Protein Docking. J Chem Theory Comput 2012 Sep 11;8(9):3395-3408.
Material Complementario
2 Inhibidores de quinasas
1 Fármacos comercializados