1. PROTACs, una alternativa a las aproximaciones basadas en ácidos nucleicos
Tradicionalmente en el descubrimiento de fármacos se han desarrollado moléculas pequeñas que actúan en sitios específicos de determinadas proteínas, activando o inhibiendo una acción determinada. Sin embargo, la presencia de proteínas que carecen de sitios bien definidos impide el desarrollo de moléculas pequeñas a través de este enfoque tradicional. Como alternativa a esta estrategia, han surgido con fuerza nuevas aproximaciones basadas en ácidos nucleicos que han permitido modificar la expresión de proteínas o inhibirla por completo como es el caso del CRISP/Cas9 o el empleo de siARN. Sin embargo, estas nuevas aproximaciones no están exentas de limitaciones como pueden ser los problemas de liberación, biodisponibilidad y estabilidad.
Recientemente, se han desarrollado compuestos basados en moléculas pequeñas, capaces de producir la degradación selectiva de proteínas, principalmente a través del sistema ubiquitina-proteasoma (UPS). Esta estrategia se basa en el empleo de compuestos quiméricos heterobifuncionales (PROTACs, en inglés Protein Targeting Chimeras), capaces de reconocer por un lado la proteína diana que debe de ser degradada y por el otro la ligasa E3 encargada de marcar a esta proteína para su eliminación por parte del proteasoma. Esta estrategia permite silenciar las diferentes proteínas de modo similar a los oligonucleótidos antisentido (ASO) o los siARN, a través de su degradación por el proteasoma, pero evitando los inconvenientes que presentan las terapias basadas en ácidos nucleicos (1).
El funcionamiento UPS está basado en un proceso secuencial, en el que una enzima activadora de ubiquitina (E1) tras el empleo de una molécula de ATP liga la ubiquitina al sitio activo de una ligasa E2. Finalmente, una ligasa E3 se encarga de la transferencia del residuo de ubiquitina desde la ligasa E2 al residuo de lisina adecuado en la proteína a degradar. Posteriormente, la adición de cadenas de varias unidades de ubiquitina sobre una proteína promueve el reconocimiento de esta por parte del proteasoma. El proteasoma es el encargado de degradar estas proteínas poliubiquitinadas y está formado por un complejo de proteínas organizado en dos tipos de subunidades:
Una subunidad 20S central, conformada por cuatro anillos apilados en forma de barril. Cada anillo esta a su vez conformado por siete proteínas y se organizan quedando las dos subunidades α en el centro y las dos β en los extremos. Estas últimas son las que presentan la actividad catalítica en su cara interior.
Dos subunidades 19S, que se apilan sobre la parte exterior de los anillos β de la subunidad 20S. Estas subunidades más externas se encargan del proceso de reconocimiento de las cadenas de poliubiquitina y se encargan de hidrolizar el ATP.
La intervención farmacológica sobre el proteasoma ha demostrado ser una estrategia eficaz para evitar la degradación aberrante de proteínas que es el detonante de ciertas enfermedades. Un ejemplo es la degradación exacerbada mediada por el proteasoma de p53, uno de los principales reguladores de la apoptosis y del ciclo celular, que termina por desencadenar una división celular descontrolada y está relacionada con algunos tipos de cáncer. Esta degradación irregular del p53 está provocada por MDM2, una ligasa E3 que al encontrarse sobreexpresada en algunos tipos de tumores promueve una elevada degradación de p53 al marcarlo con cadenas de ubiquitina. La solución a esta eliminación masiva es el empleo de inhibidores del proteasoma.
Estos inhibidores presentan una estructura de péptidos con grupos altamente electrófilos en su extremo terminal, como pueden ser derivados del tipo ácido borónico en el bortezomib o epóxidos en al caso del carfilzomib. Estos compuestos se unen gracias a sus grupos electrófilos a residuos tirosina del sitio catalítico del proteasoma impidiendo que lleve a cabo su actividad aminopeptidasa.
Por lo tanto, una pregunta lógica que surge es ¿Se puede emplear el proteasoma para degradar de forma selectiva proteínas sobreexpresadas o que estén teniendo una función aberrante? La respuesta es sí y la solución es el empleo de los PROTACs.
Aprovechando esta maquinaria, los PROTACs aproximan ligasas E3 con diferentes proteínas como receptores nucleares, factores de transcripción, proteínas estructurales, enzimas o proteínas reguladoras que de forma natural no se encontrarían próximas; favoreciendo así su degradación y corrigiendo la hiperactivación que puede ser la causa de diferentes condiciones patológicas o causantes de enfermedades (2).
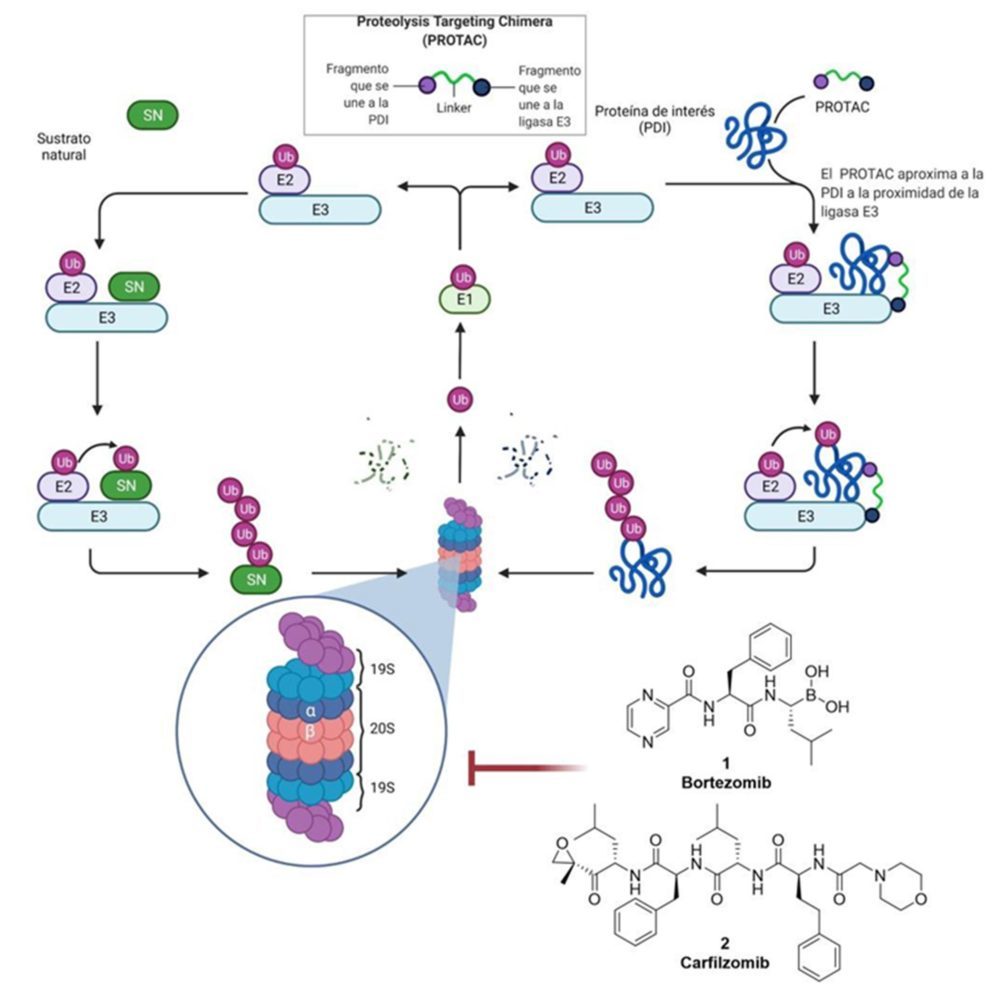
Figura 1: Funcionamiento del sistema UPS y como un PROTAC lo emplea para la degradación selectiva de proteínas de forma esquemática. Creado con BioRender.com.
La estrategia más empleada para la obtención de estos PROTACs se basa en el empleo de ligandos de una de estas ligasas bien establecidas y modificar la molécula que se une a la proteína de interés (PDI), así como el fragmento que une ambas subunidades. Este proceso de optimización generalmente da lugar a una molécula con capacidad para formar un complejo ternario y capaz de transferir las subunidades de ubiquitina a la proteína de interés. La primera vez que se mencionan estos compuestos bifuncionales es en el diseño de degradadores basados en β-TrCP por Crews y colaboradores (3).
2. PROTACs SEGÚN SU LIGASA E3
2.1 PROTAC basados en β-TrCP
La primera aplicación en la práctica de un PROTAC con capacidad para degradar una proteína fue descrita de nuevo por Crews y colaboradores en el que emplearon como ligasa E3 SCFβ-TrCP para la degradación de los receptores de andrógenos (AR) y de
estrógenos (ER). (4) β-TrCP es una proteína que contiene un dominio F-box que actúa como reclutador del resto de la ligasa E3 de la que forma parte formada por Skp1-Cul1- F-box (SCF). Dentro de la estructura de β-TrCP se encuentra un bolsillo formado por un dominio WD40 que reconoce como sustratos naturales proteínas que contienen secuencias fosforiladas en los residuos de serina con la secuencia genérica DpSGXXpS. (5) El dominio WD40 es el encargado de reconocer a la proteína IBk, el inhibidor natural de NF-kB, marcándola con ubiquitina y promoviendo su degradación, lo que finalmente induce la activación de NF-kB. La secuencia de IBk reconocida por β-TrCP es un decapéptido difosforilado en sus serinas DRHDSpGLDSpM. (6).
Empleando este péptido fosforilado y uniéndolo a través de un conector al fragmento capaz de reconocer los receptores de estrógenos y andrógenos, se obtuvieron las primeras quimeras capaces de degradar proteínas que no eran su sustrato natural dando lugar a Protac-2 (3) y Protac-3 (4).
Sin embargo, esta ligasa E3 no ha sido ampliamente empleada por las dificultades a la hora de buscar moléculas pequeñas capaces de reconocer a β-TrCP y poder generar así PROTACs que tengan unas buenas propiedades farmacocinéticas debido a la estructura peptídica del fragmento kB.
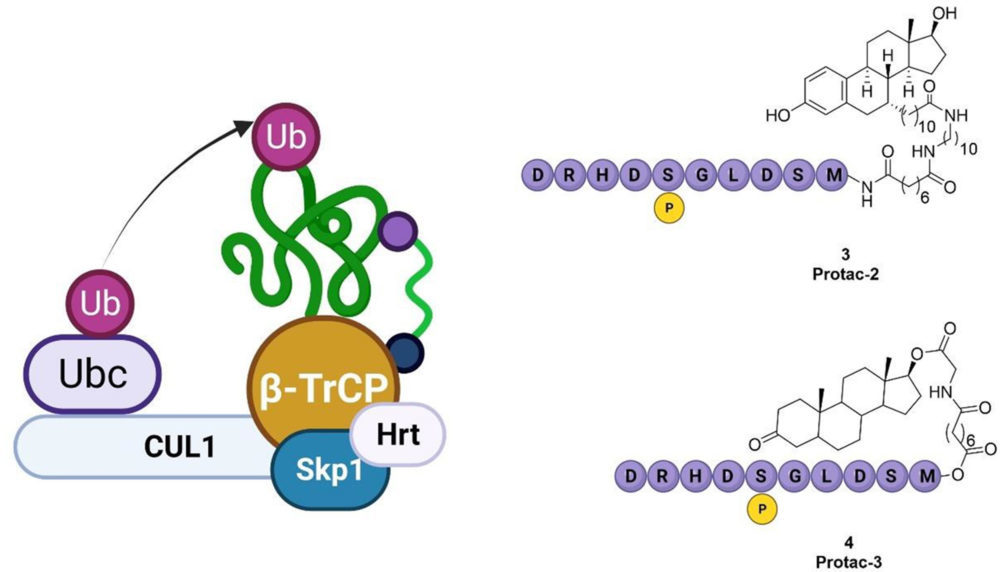
Figura 2. Esquema del complejo formado por β-TrCP y estructura de los PROTAC 2 y 3. Creado con BioRender.com.
2.2. PROTAC basados en VHL
El factor inducible por hipoxia 1-a (HIF1a) se encarga de diferentes funciones de regulación metabólica como respuesta a los niveles de oxígeno (7). En condiciones de normoxia se produce la hidroxilación selectiva de residuos de prolina que permiten su ubiquitinación y posterior degradación por el proteasoma, mientras que durante un proceso de hipoxia esta hidroxilación no se produce, permitiendo al factor ejercer su función (8). En 1999 se demostró la degradación el factor inducible por hipoxia 1-a por parte de la proteína supresora de tumores VHL (9) que es una de las subunidades del complejo E3 CRL2VHL formado por CUL2-RBX1-ElonginB-ElonginC-VHL (10).
La obtención de la estructura de rayos-X del complejo formado por pVHL con el extremo C-terminal de HIF1a encargado de la degradación dependiente de oxígeno, permitió identificar las interacciones clave del anillo de hidroxiprolina con los residuos de Ser111 e His151 y el posterior desarrollo de peptidomiméticos basados en esta secuencia (11, 12). Posteriormente, la obtención de potentes derivados como VH032 (5) y la publicación de la estructura cristalográfica unido a pVHL permitió seleccionar el grupo amida más próximo al extremo de la molécula como punto óptimo para la vectorización hacia el exterior de la molécula (13). Esto permitió la formación de una molécula quimérica por la fusión este fragmento a un conector con JQ1 en su extremo, dando lugar a MZ1 (5), el primer PROTAC basado en VHL. (13) El extremo al que está unido JQ1 se encarga de reconocer a proteínas bromodominio- y extraterminales (BET) BRD2, BRD3 y BRD4 relacionadas con procesos cancerosos y promoviendo su degradación VHL dependiente. En este caso se observó una inesperada selectividad por BRD4, que no existía en el ligando de partida de esta proteína (JQ1, 6), (13) recalcando la interesante posibilidad de usar estos compuestos para ganar selectividad entre subtipos de proteínas con grandes porcentajes de homología entre sus secuencias como se discute más adelante en el apartado 3.3.
Posteriormente en el mismo grupo de Ciulli se llevó a cabo una modificación estructural a través de la ciclación de la estructura de MZ1 (7), (14) lo que permitió la obtención de derivados macrocíclicos. Esta estrategia permite obtener un mejor perfil farmacocinético al reducir la penalización energética por reducción de los grados de libertad del compuesto (15) y se han descrito como estructuras privilegiadas a la hora de actuar como inhibidores de la interacción proteína-proteína (16).
El estudio sistemático de esta ligasa E3, así como la identificación de su estructura ha sido uno de los principales motivos por los que es uno de los sistemas más empleados en el desarrollo de PROTACs, dando lugar a una gran cantidad de ellos en los últimos años con capacidad para degradar un gran número de proteínas como receptores nucleares, enzimas y proteínas estructurales implicadas en varias patologías.
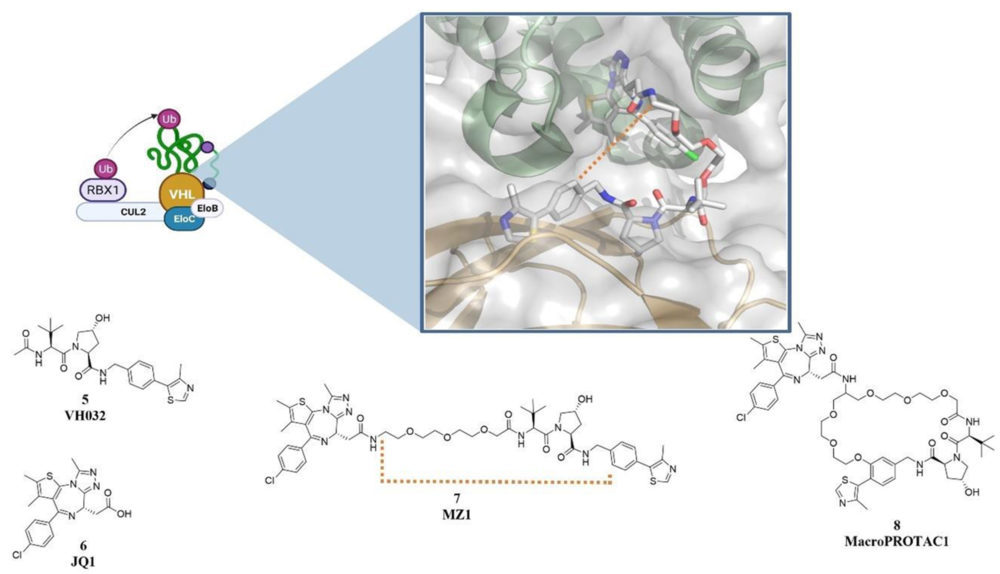
Figura 3: Estructura del complejo ligasa E3 formado por VHL. Detalle de la estructura cristalográfica entre VHL-BRD4-MZ1 PDB: 5T35 (17), Estructuras de JQ1, VH032, MZ1 y MacroPROTAC1. Creado con BioRender.com.
2.3 PROTAC basados en CRBN
La talidomida, empleada como sedante para el tratamiento de las molestias y el insomnio en embarazadas, fue retirada del mercado tras relacionarse con las malformaciones en 8000 a 12000 niños nacidos en la década de los 60. Años más tarde demostró ser un agente eficaz contra el mieloma múltiple, indicación para la que se sigue empleando como uso compasivo y su mecanismo de acción todavía no está completamente claro. En 2010, se describió por primera vez la unión de la talidomida a CRBN una subunidad de la ligasa E3 CRL4CRBN formada por CUL4-RBX1-DBB1- CRBN que explica, en parte, los efectos teratógenos de la talidomida.
La lenalidomida, un análogo estructural de la talidomida, se comporta como un pegamento molecular y mantiene unidos a CRBN con las proteínas de dedos de zinc de la familia ikaros 1 y 3 (IKZF1 y IKZF3) induciendo su degradación dependiente del proteasoma. Esto explica su actividad como agentes efectivos en el mieloma múltiple, ya que la sobreactuación de estas proteínas está relacionada con la división descontrolada de células linfáticas. (18) En 2014 se obtuvo la estructura cristalográfica del complejo DBB1-CRBN permitiendo observar la porción del ligando expuesto al exterior de la proteína (19) y que posteriormente aprovechando un proceso de diseño racional similar al empleado con VHL se obtuvieron PROTACs capaces de degradar BRD4 y FKBP tanto en células como en modelos con ratones (20).
Este reclutador de ligasas E3 junto con el VHL son los más ampliamente estudiados y empleados en el diseño de nuevos PROTACs. Sin embargo en el desarrollo clínico CRBN se encuentra un paso por delante, ya que es el sistema que emplea los dos PROTACs más avanzados en ensayos clínicos de fase II, ARV-110 contra el receptor de andrógenos en cáncer de próstata y ARV-471 en el receptor de estrógenos en cáncer de mama.
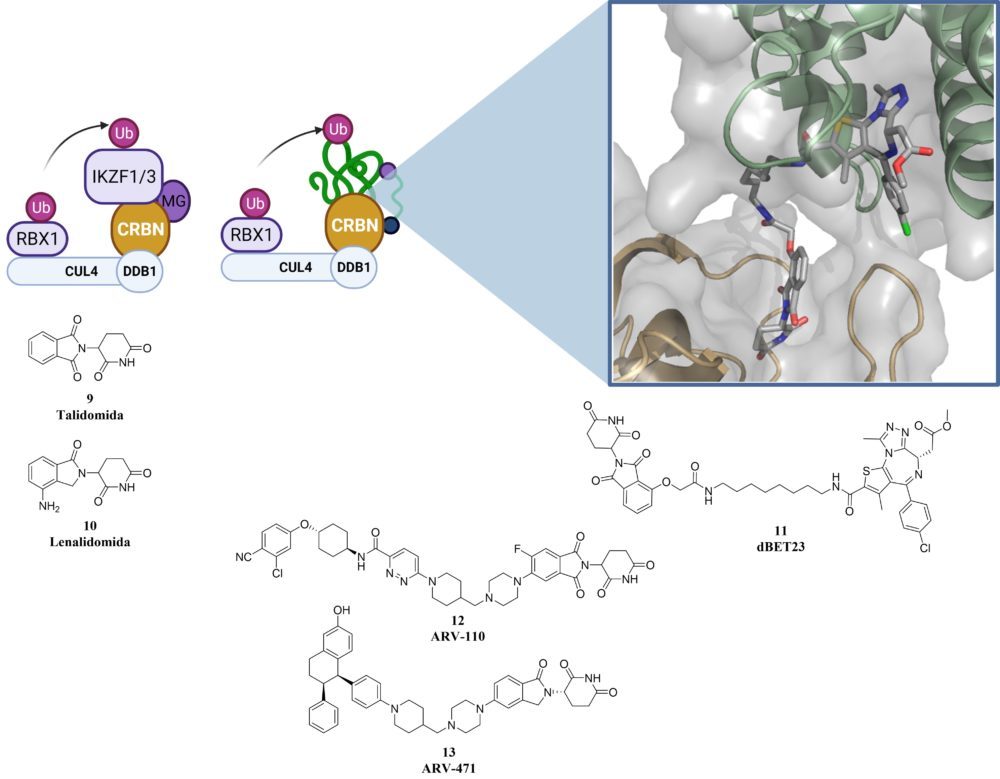
Figura 4: Estructura del complejo ligasa E3 formado por CRBN con la talidomida (MG) y IZKF1/3. Esquema del complejo formado por CRBN-PROTAC (dBET23)- con la proteína de interés (BRD4).
2.4. PROTACs basados en IAP
Las proteínas inhibidoras de la apoptosis (IAPs) fueron descritas por primera vez en baculovirus, (22) mientras que en mamíferos son reguladores negativos de la apoptosis por inhibición de las caspasas. La estructura de los IAPs esta y pueden estar conformadas por entre uno y tres dominios BIR, un dominio asociado a la ubiquitina (UBA) y un dominio de dedo de zinc RING dando lugar a una familia de 8 miembros, pero los más extensamente estudiados son IAP celulares 1 y 2 (cIAP1 y cIAP2) y IAP asociado al cromosoma X (XIAP) por encontrarse sobreexpresadas en algunos tipos de cáncer y estar asociadas a un peor pronóstico. (23) Esto se debe a que estas IAPs capaces de reconocer a caspasas activadas, uniéndose a ellas ubiquitinando las caspasas y a sí mismas lo que da lugar a su degradación por el proteasoma. Esta eliminación aberrante de caspasas genera una resistencia a la apoptosis lo que desencadena el proceso canceroso y del mal pronóstico de este. Por ese motivo la generación de inhibidores de IAPs ha sido una estrategia importante en el desarrollo de agentes contra los cánceres resistentes a la quimioterapia. Un ejemplo de estos agentes es la metilbestatina descrita por Sekine y colaboradores (24) capaz de incrementar la sensibilidad de las células a agentes antitumorales por una reducción de los niveles de cIAP1. (24) La degradación de cIAP1 se produce tras la unión de la metilbestatina, que produce la auto ubiquitinación por parte de una ligasa E2 que reconoce el dominio RING. El estudio de diferentes derivados de la bestatina dio lugar a una relación estructura-actividad en la que se observa como la modificación de su extremo carboxilo terminal por esteres con grupos voluminosos no afecta a su unión y, por lo tanto, ofrece un punto óptimo para poder generar moléculas bifuncionales capaces de aproximar a estos IAP proteínas que no serían reconocidas de forma natural para poder ubiquitinarlas y ser degradas por el proteasoma. (25) Estas moléculas quiméricas con capacidad de aproximar a las proteínas de interés a IAP se denominan SNIPERS (Specific and Non-genetic IAP-dependent Protein Erasers), (23) y son capaces de degradar diferentes proteínas como proteína de bromodominio 4 (BRD4), receptores de estrógenos (ER), receptores de andrógenos (AR) y la tirosina-quinasa BCR-ABL. (8) Todas las proteínas descritas anteriormente están involucradas en el cáncer de modo que es interesante como aproximación el uso de estos SNIPERS al eliminar a dos proteínas involucradas en la patología puede tener un efecto sinérgico.
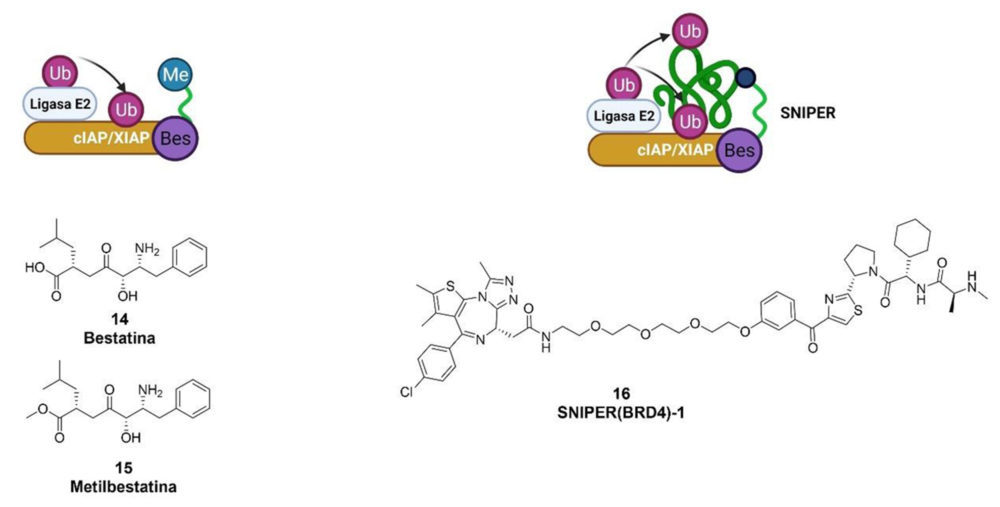
Figura 5. Esquema de ubiquitinación mediada por la metilbestatina y aplicación de un derivado para la obtención de un SNIPER. Creado con BioRender.com.
2.5 PROTACs basados en MDM2
La proteína MDM2 (murine double minute protein) es una proteína codificada por el oncogen mdm2 y que actúa como una ligasa E3 promoviendo la ubiquitanación tanto de MDM2 como de p53 y posterior degradación de todo el complejo MDM2-p53. Esta proteína se encarga de mantener la reparación del ADN y regula el ciclo celular y la apoptosis, por ese motivo se le conoce como “el guardían del genoma”. Además, p53 induce la expresión de MDM2 cuando los niveles de p53 son elevados, lo que hace que los niveles de ambos se mantengan estables a través de un ciclo de retroalimentación negativo por la degradación de p53 MDM2 dependiente. (26) Es importante destacar que una actividad elevada de MDM2 o una malfunción debida a mutaciones en p53 (presente en el 50% de los tumores) es un factor desencadenante de cáncer.
Derivados de tipo cis-imidazolina como la nutlina y la indasanutlina interfieren en ese proceso de reconocimiento impidiendo la degradación de p53 (27). Estos derivados se han empleado para generar moléculas bifuncionales capaces de aproximar proteínas a MDM2 promoviendo así su degradación como es el caso de los receptores de andrógenos (AR), (28) generando el primer PROTAC basado en MDM2 (PROTAC14), o BRD4 obteniendo PROTACs (A1874) (29) que tienen una doble actividad como agentes antitumorales al reducir los niveles de una proteína asociada con el proceso canceroso (AR y BRD4) además de incrementar los niveles de p53. Sin embargo, debido a la limitada capacidad de degradación que tienen los PROTACs basado en MDM2 unido al perfil farmacocinético complicado de sus ligandos hace que se prefieran otros sistemas para la obtención de PROTACs.
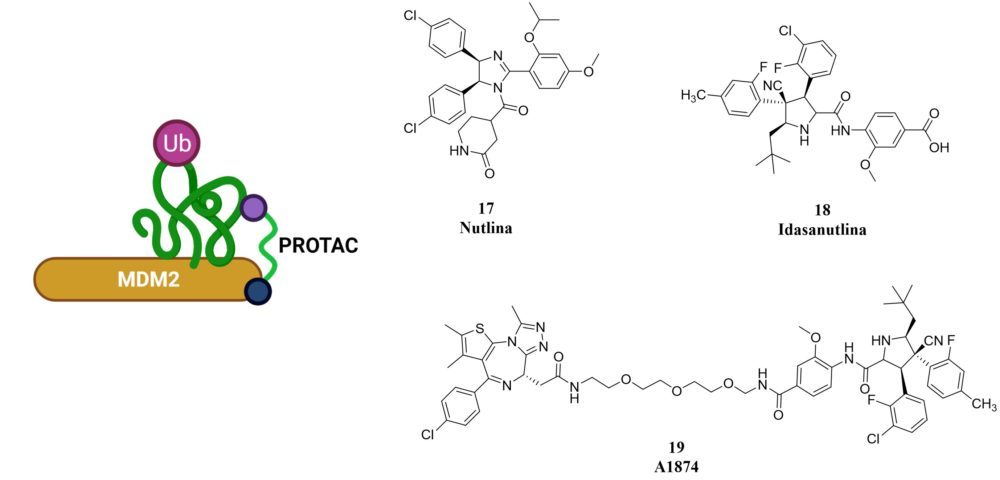
Figura 6. Esquema del funcionamiento de PROTACs basados en MDM2. Nutlina e idasanutlina como ligandos de MDM2 y el PROTAC A1874 basado en la idasanutlina para degradar BRD4. Creado con BioRender.com.
2.6 PROTACs basados en KEAP1
Un ejemplo de nuevos sustratos de ligasas E3 puede ser KEAP1, ya que esta expresada de forma ubicua y esto es debido en gran medida, a su papel dentro del sistema Keap1-Nrf2 como uno de los principales sistemas de defensa antioxidante celular. De forma constitutiva, Nrf2 es degradado por mediación de Keap1 impidiendo su acción. Sin embargo, en presencia de especies reactivas de oxígeno o de electrófilos promueven la liberación de Nrf2 que tras translocarse al núcleo produce un aumento en la expresión de proteínas citoprotectoras y antiinflamatorias.
No existe la estructura tridimensional completa cristalizada de Keap1 o de Nrf2; sin embargo, sí que existen fragmentos del complejo por separado y de otras proteínas con un elevado grado de homología. A través de estas estructuras tridimensionales, se ha podido obtener modelo próximo a la realidad (30) que puede permitir entender mejor la función de este sistema y relacionarlo con su estructura. En este modelo se pueden observar las diferentes proteínas que lo componen, como es el caso de la propia proteína Keap1, clasificada dentro de la familia BTB-Kelch. El dominio N-terminal BTB se encarga de la homo-dimerización de dos subunidades de Keap1 y contiene una región denominada B-box encargada de reconocer a Cul3. El dominio C terminal se denomina Kelch, y es el encargado de reconocer a dos secuencias específicas de Nrf2 (ETGE y DLG). La degradación de Nrf2 por parte del proteasoma ocurre tras la unión de ubiquitina por parte de una ligasa E2 que se encuentra unida a Cul3 gracias a una proteína accesoria denominada Rbx1.

Figura 7. A, estructura tridimensional del complejo Nrf2-Keap1(Kelch)-Keap1(BTB)-Cul1-Rbx1- Nedd8-E2-Ub; B, Dominio Kelch de Keap1 unido al dominio ETGE y evidenciados las 5 cavidades en colores P1, P2, P3, P4 y P5; C, Inhibidores de la interacción proteína-proteína con cada uno de los fragmentos resaltados de forma complementaria a su cavidad de unión; D, CDDO (magenta) unido covalentemente a la Cys151 de la región BTB de Kelch1 (gris) y el impedimento que provoca en la región en la que se une Cul3 en azul; E, Inhibidores covalentes que se unen a la región BTB de Keap1 con las grupos responsables de esta unión resaltados en amarillo.
Una característica muy importante de Keap1 es su elevada capacidad para reconocer otras proteínas para promover su degradación por parte de proteasoma. Debido a la relevancia de este sistema de defensa antioxidante y su intervención en diferentes enfermedades inflamatorias, neurodegenerativas o cáncer, se han desarrollado series de compuestos con capacidad para interferir con este sistema. Se han desarrollado inhibidores de este sistema que pueden categorizarse principalmente en inhibidores covalentes o en inhibidores de la interacción proteína-proteína.
2.6.a. Inhibidores covalentes reversibles e irreversibles
Como se ha discutido con anterioridad, Nrf2 se ve activado de forma fisiológica al incrementarse el estrés oxidativo en la célula, pero también lo puede hacer por causas exógenas como la exposición a ciertos electrófilos. Se han propuesto dos mecanismos mayoritarios por los que estos electrófilos pueden llevar a cabo su acción: (a) induciendo algún tipo de cambio conformacional en Keap1 impidiendo que reconozca a Nrf2 y disminuyensu degradación, o (b) tras unirse a alguna de las cisteínas en la región B-box de Keap1 impide el reconocimiento de esta región por Cul3 y finalmente no se forma el complejo de proteínas necesario para degradar Nrf2.
Estos inhibidores covalentes de Keap1 pueden ser de origen natural como la curcumina (20), el sulforafano (21) o diferentes derivados de quinona; de origen semisintético como los triterpenoides de cianoenona (22) como pueden ser la bradoloxona, metil bradoloxona, CDDO-etilamida y por ultimo los derivados sintéticos entre los que cabe destacar los esteres del ácido fumárico (23) como el MMF o el DMF (Tecfidera®, probado por la FDA para el tratamiento de la esclerosis múltiple).
Algunos de estos compuestos, se unen a la cisteína 151 del dominio BTB y en estos casos además de reducirse la degradación de Nrf2, producen una disminución llamativa de los niveles de Keap1.
2.6.b. Inhibidores de la interacción proteína-proteína
Los primeros inhibidores conocidos de esta interacción entre el dominio Kelch y la secuencia ETGE son péptidos que imitan a esta misma secuencia. Sin embargo, presentan una serie de limitaciones típicas de estos compuestos como son los problemas de estabilidad y su limitada capacidad para pasar por membranas a través de difusión pasiva, comprometiendo así su farmacocinética. Otra aproximación para interrumpir el reconocimiento del dominio Kelch de Keap1 por Nrf2 es el empleo moléculas pequeñas. Esto es posible gracias al tamaño asumible de la superficie del dominio Kelch (300-1000 Å2) y la clara definición de este sitio en un bolsillo en el que encaja perfectamente las secuencia ETGE de Nrf2 en forma de horquilla β. Este bolsillo a su vez puede verse dividido en cinco cavidades. Las regiones P1 y P2 se caracterizan por encontrarse cargadas positivamente por los residuos de arginina Arg415, Arg483 y Arg485 y que permiten la interacción con grupos de carácter ácido que mimeticen el comportamiento de los carboxilatos presentes en los residuos de Glu 79 y Glu82 en la secuencia ETGE de Nrf2. La zona central del bolsillo denominada P3 está formada por residuos de pequeño tamaño, lo que deja espacio importante para acomodar una gran diversidad de estructuras heterocíclicas. En sus proximidades se encuentran otras dos cavidades (P4 y P5) conformado por residuos de naturaleza alifática y en particular por residuos de tirosina. La definición tan clara de este sitio de unión ha permitido que en los últimos años un elevado número de compuestos hayan sido identificado y desarrollado para actuar sobre este dominio Kelch.
Un ejemplo ha sido la identificación de la tetrahydroisoquinolina 24 o la naftilsulfonamida 25 que han sido identificadas a través de campañas de cribado de alto rendimiento. La posterior caracterización estructural de estos compuestos en el bolsillo a través de cristalografía de rayos-x demostró como el compuesto 25a se localizaba en el entorno de P3, P4 y P5. Posteriormente a través de un proceso de optimización basado en la estructura se introdujeron dos grupos carboxilato que promovían la interacción con las cavidades P1 y P2, dando lugar al compuesto 25b. Sin embargo, la presencia de grupos carboxilo reduce drásticamente el paso a través de membranas y la presencia del naftilo como estructura central lo convierte en una molécula fácilmente metabolizable y capaz de interferir con otros fármacos fácilmente. Como solución a esta limitaciones siguió optimizando la estructura y la sustitución del naftilo por una isoquinolina y la sustitución de los ácidos carboxílicos por tetrazoles dio lugar a los derivados 25c y 25d respectivamente. Otros derivados han sido identificados como inhibidores de esta interacción (Figura 7).
Actualmente existen protacs basados en estos inhibidores ya conocidos de la vía Keap1- Nrf2. El primero de ellos desarrollado en 2018 se trata de un péptido de gran tamaño y que contiene la secuencia ETGE y capaz de reconocer la región PHF6 de TAU y eliminándola de forma efectiva. (31) Sin embargo, la utilidad de este PROTAC a nivel del SNC queda limitada por su baja capacidad para cruzar la barrera hematoencefálica. Recientemente en el año 2020, se ha descrito otro PROTAC (CDDO-JQ1, 27 )basado en la degradación a través de Keap1 pero en este caso a través de la unión a Keap1 por su región BTB empleando un derivado de la bradoloxona que se une covalentemente. (32) Esta aproximación consiguió eliminar la proteína de interés (BRD4) pero también produjo la eliminación de Keap1, de modo que este PROTAC no va a poder perpetuar su efecto en un tratamiento a largo plazo como puede ser el cáncer, enfermedad al que iría dirigido este PROTAC.
En general esta es una limitación que ocurriría con muchos de los inhibidores covalentes de Keap1 y que además puede tener repercusiones en otras rutas de señalización, debido a la implicación de esta en ellas como se ha comentado con anterioridad.
En contraposición, recientemente se ha descrito un PROTAC (MS83, 28) que emplea el compuesto 26 como fragmento encargado de reconocer a Keap1 por su dominio Kelch y enfocado como en el caso anterior a eliminar BRD4 (33). A través de esta aproximación se consiguió la degradación de la proteÍna diana (BRD4) sin reducir los niveles de Keap1 al contrario de lo que ocurría con los derivados que se unen de forma covalente a su región BTB.
Por todos estos motivos, la compresión profunda de la estructura de este complejo puede dar lugar a la obtención de compuestos con tamaños moderados para mantener unas buenas propiedades farmacocinéticas y su empleo en todo tipo de patologías como pueden sr las enfermedades neurodegenerativas, en el que el paso a través de la BHE es un factor muy limitante en el desarrollo de terapias efectivas.
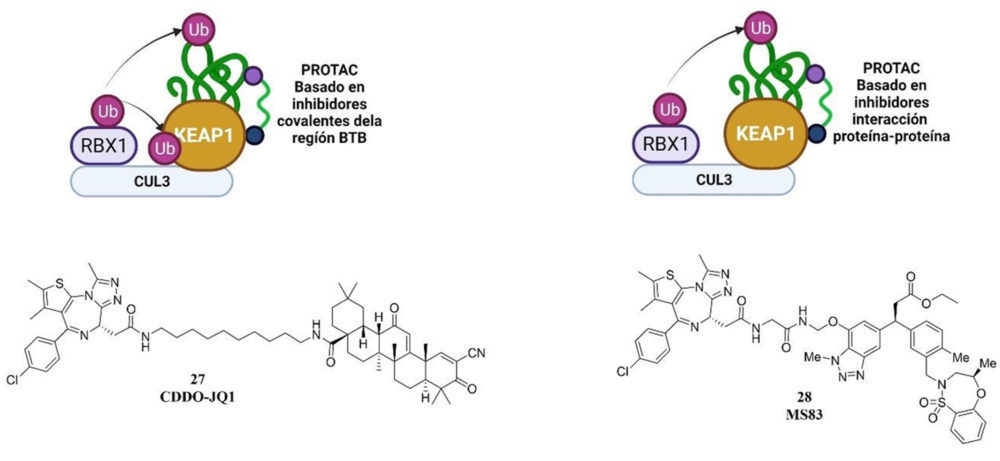
Figura 8: Esquema del complejo reclutado por Keap1 y la diferencia de ubiquitinación al emplear un inhibidor covalente como ligando (CDDO-JQ1) y un inhibidor IPP (26). Creado con BioRender.com.
2.7 PROTACs basados en DCAF15
Un ejemplo de estos nuevos sustratos de ligasas E3 puede ser DECAF15, que ha demostrado ser el mediador de la degradación de RBM39 gracias a la unión de estas dos proteínas por parte de diferentes arilsulfonamidas con carácter antitumoral como pueden ser el indisulam, tasisulam ó E7820. Estas arilsulfonamidas, actúan como «pegamentos moleculares» induciendo un cambio conformacional en la superficie de DECAF15 que permite el reconocimiento de RBM39. DCAF15 actúa como sustrato de una ligasa E3 que finalmente transfiere la ubiquitina a RBM39 y promueve su degradación por le proteasoma. La actividad antitumoral atribuida a esta degradación está relacionada con la propia actividad de RBM39 que se encarga del splicing de dos quinesinas fundamentales durante la fase G2/M KIF20A y KIF20B. De modo que al inducir la degradación RBM39 con las arylsulfonamidas, no se puede producir el procesamiento adecuado del ARNm que codifica para las quinesinas KIF20A y KIF20B funcionales impidiendo la división celular y dando lugar a su actividad antitumoral.
Gracias a la determinación de la estructura por cristalografía de rayos-X de DCAF15 unido a estas arylsulfonamidas, en concreto a E1820, (34) ha permitido el rediseño de estos pegamentos moleculares para poder vectorizar hacia el exterior de la proteína una cadena. Este nuevo punto de unión permite poder conectar fragmentos con capacidad para reconocer a otras proteínas, como ocurre en el caso de BRD4 (35) dado lugar a DP1 y a una nueva familia de PROTACs basados en DCAF15 con un perfil farmacocinético muy favorable.
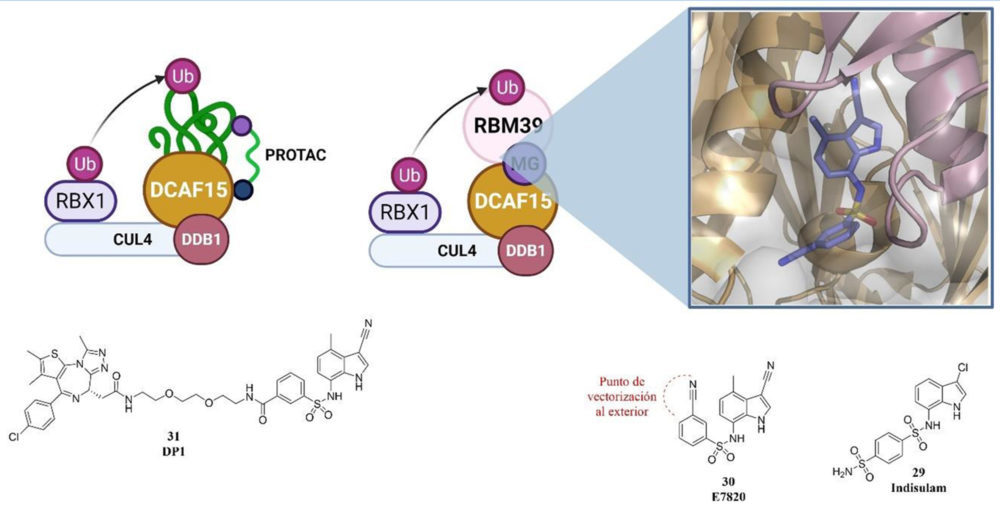
Figura 9. Esquema del funcionamiento de indisulam y E7820 como pegamentos moleculares MG. Detalle de la estructura cristalográfica del complejo DCAF15-RBM39-E7820 lo que permite observar la mejor posición para añadir un grupo que permite vectorizar un espaciador hacia el exterior para poder unirlo a una molécula con capacidad para reconocer otras proteínas. Estructura de DP1. Creado con BioRender.com.
3. FACTORES CLAVE EN EL DESARROLLO DE UN PROTAC
3.1. Expresión en el tejido en el que tiene que llevar a cabo su función
Uno de los factores clave a la hora de escoger una ligasa E3 es su expresión en el tejido en el que queremos actuar, ya que puede no expresarse de la misma forma en todos los tejidos. Este fenómeno puede ayudar a conseguir la degradación selectiva de una proteína en algún tejido concreto aprovechándose de este diferente perfil de expresión de ligasas E3. Por esta razón es conveniente el desarrollo de degradadores que recluten a diferentes ligasas E3 y con diferentes perfiles de distribución por el organismo tanto en condiciónse fisiológicas como patológicas añadiéndose así a las más comúnmente empleadas como CBRN y VHL para poder seleccionar la mejor opción. Por otro lado, si la proteína a degradar tiene una función fundamental se debería de emplear una ligasa E3 que se encuentra expresada solo o en mayor medida en el tejido o en la región en la que queremos actuar. Esto es particularmente útil en el caso de ligasas como las IAPs, MDM2 o DCAF15 que están sobreexpresadas en algunos tipos de cáncer.
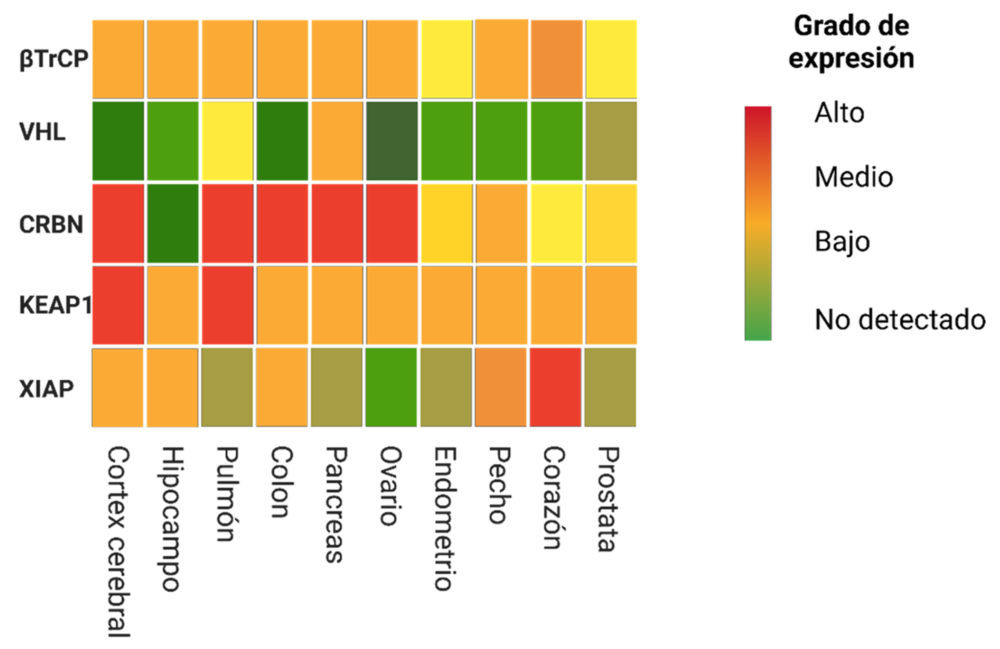
Figura 10. Expresión de Keap1 en los diferentes tejidos. Datos extraídos de https://www.proteinatlas.org. Creado con BioRender.com.
3.2. Propiedades farmacocinéticas, combatir su gran tamaño
El desarrollo de PROTACs como dos subunidades independientes unidas por un fragmento de tamaño variable, da lugar a la obtención de moléculas de gran tamaño, un elevado número de grados de libertad y una amplia presencia de grupos funcionales susceptibles de sufrir metabolismo, lo que a su vez puede acarrear problemas de estabilidad y toxicidad poco deseables. A pesar de estas limitaciones, algunos PROTACs han demostrado tener un perfil farmacocinético deseable y una amplia distribución en tejidos, tras procesos complejos de optimización. Una estrategia que permitiría, a pesar del gran tamaño de estos compuestos, mejorar el paso de estos compuestos por las barreras celulares, es la reducción de los grados de libertad por restricción conformacional al incluirlos en macrociclos. (15) Como alternativa a ello están surgiendo con fuerza los denominados pegamentos moleculares, estos se tratan de moléculas de menor tamaño y que pueden reconocer a ambas proteínas o que en su defecto, al reconocer una de las proteínas la convierten en una superficie complementaria a la de la otra, favoreciendo la interacción proteína-proteína y la degradación de la proteína diana al encontrarse en el entorno de la ligasa E3. El menor tamaño de estos pegamentos moleculares los convierte en una mejor aproximación para la degradación dirigida de proteínas al presentar “a priori” unas mejores propiedades farmacocinéticas.
3.3 Reconocimiento mutuo de ambas proteínas y degradación efectiva
Sin embargo, esto no siempre ocurre, ya sea por la falta de complementariedad entre las superficies de ambas estructuras que les impidan estar próximas en el espacio. Esta complementariedad pude medirse con la cooperatividad (α). Esta medida nos permite conocer si el complejo ternario se puede formar a partir de sus constantes de equilibrio. Cuando α< 0 se dice que la cooperatividad es negativa, esto significa que las fuerzas repulsivas entre ambas proteínas con tan grandes que aunque le PROTAC se una a ambas por separado, nunca se formará el complejo ternario. Cuando α > 0 se dice que la cooperatividad es positiva y por lo tanto que además de interacciona el PROTAC aproximando ambas proteínas espacialmente, existe complementariedad entre la superficie de ambas proteínas, dando lugar a un complejo ternario muy estable. Por otro lado, aunque se forme el complejo ternario, la transferencia de ubiquitina por parte de la ligasa E3 necesita encontrar un residuo sobre el que la ligasa E3 pueda unir la ubiquitina, de no ser así, la formación del complejo no dará lugar a una degradación efectiva (21).

Figura 11. Equilibrio entre los diferentes complejos binarios que dan lugar al complejo ternario, calculo de la cooperatividad (α) y significado de la misma. Creado con BioRender.com.
Este hecho hace necesario que nos planteemos el estudio sistemático de estas ligasas para caracterizar su capacidad de ser reprogramadas para reconocer nuevos sustratos y aumentar así el número de ligasas disponibles para la degradación de proteínas que no pueden ser modificadas por moléculas pequeñas con las técnicas de química médica tradicionales.
Además de las posibles soluciones aportadas ya en cada uno de estos apartados, el estudio sistemático de las ligasas E3 puede conducir a un mayor número de alternativas a elegir para la obtención de degradadores efectivos. Uno de los puntos clave a la hora de diseñar un nuevo PROTAC es la elección de la ligasa E3, que permite destruir una proteína que, debido a sus características topológicas, carece de sitios de unión adecuados para su inhibición con moléculas pequeñas. Se han descrito alrededor de 600 ligasas E3 en todo el proteoma. Sin embargo, solamente en torno al 1% de ellas se han estudiado como degradadores en PROTAC y solamente 2 (CBRN y VHL) son las que se emplean de forma habitual para dar lugar a la mayoría de PROTACs útiles.
4. OTRAS ESTRATEGIAS DE DEGRADACIÓN DE PROTEÍNAS
Tomando como modelo la estrategia de los PROTACs, se han desarrollado una serie de moléculas pequeñas denominadas ENDTACs/LYTACs y AUTACs (36,37) que aprovechan los lisosomas y la autofagia, respectivamente, para llevar a cabo la degradación de proteínas. Presentan la ventaja frente a los PROTACs de que también permiten la degradación de proteínas de membrana y extracelulares.
4.1. LYTAC (lysosome targeting chimera) technology
Los lisosomas degradan proteínas con dominios intracelulares así como proteínas extracelulares. La degradación de estas últimas está mediada por receptores de superficie de membrana (LTRs, lysosome-targeting receptors) que facilitan el transporte de dichas proteínas hasta los lisosomas. El receptor prototipo es el receptor de manosa-6-fosfato independiente de catión (CI-M6PR) que reconoce proteínas que contienen N-polisacáridos con un resto de manosa-6-fosfato (M6Pn) terminal.
El grupo de Bertozzi ha diseñado una serie de moléculas quiméricas pequeñas diseñadas para promover la degradación de proteínas extracelulares a través de la vía lisosomal, a las que han denominado LYTAcs (lysosome targeting chimera). (38) Un LYTAC consta de una molécula o un anticuerpo de la PDI que incorpora manosa-6-fosfonato (M6Pn), un oligopétido sintético, en los residuos de serina o lisina.
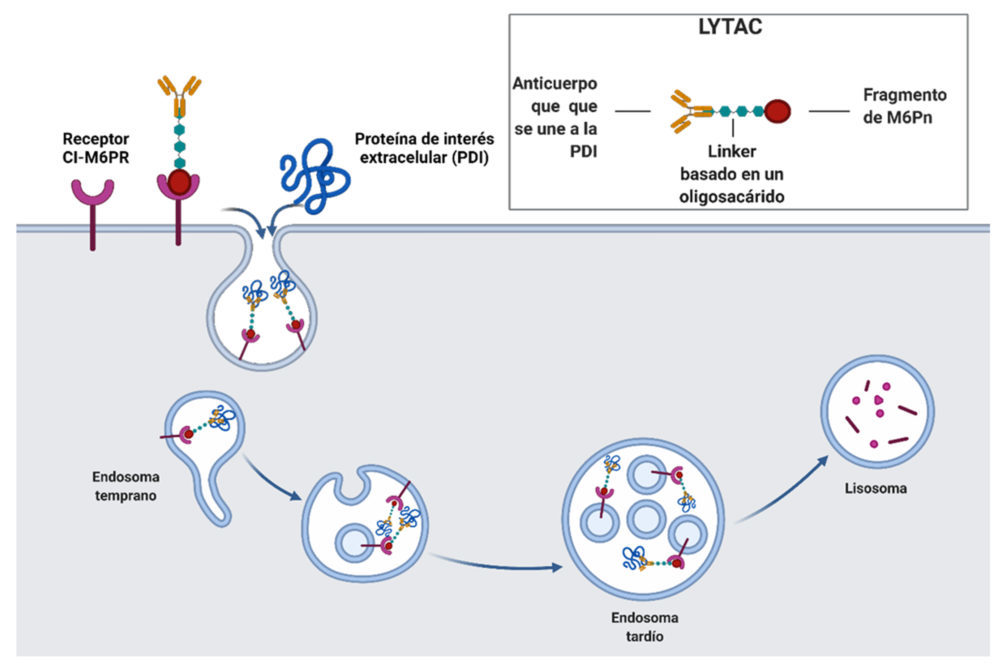
Figura 12. LYTACs formados por un anticuerpo conjugado a un glucopéptido capaz de unirse a CI- M6PR y internalizar proteínas secretadas o de membrana a un lisosoma. Creado con BioRender.com.
Los ensayos biológicos llevados a cabo confirmaron la efectividad de estos compuestos sobre sobre proteínas extracelulares como la estraptavidina o la apolipoproteína E4 (ApoE4) implicada en la enfermedad de Alzheimer. También demostraron que esta estrategia se puede aplicar a la degradación de receptores de membrana utilizando cetuximab, un anticuerpo monoclonal del receptor del factor de crecimiento epidérmico (EGFR), funcionalizado con (M6Pn). Se observó una degradación del receptor por encima del 70% y que se mantuvo durante tres días.
A pesar de los buenos resultados obtenidos, estos LYCTACs presentan algunos inconvenientes como son su alto peso molecular y la posibilidad de inducir respuestas inmunes debido a la presencia de anticuerpos o polipétidos en su estructura.
4.2. Macroautophagy Degradation Targeting Chimeras (MADTACS): AUTACs/ATTECs
La macroautofagia es el principal mecanismo de degradación celular. Puede llevar a cabo la degradación de proteínas, así como diferentes orgánulos celulares y agentes patógenos intracelulares. Es un proceso celular que tiene lugar en varios pasos y en el que las proteínas a degradar son englobadas en unas estructuras de doble membrana denominadas autofagosomas que contienen proteínas LC3 unidas a cadenas lipídicas. Las proteínas LC3 presentan homología estructural con la ubiquitina por lo que también se conocen como ubiquitin-like proteins. Contribuye a la formación del autofagosoma y a la selección de los sustratos para la autofagia.
Los receptores de la autofagia (SQSTM1, p62) reconocen proteínas poliubiquitinadas en el residuo de lisina 63 y las conducen a los autofagosomas para su eliminación (39).
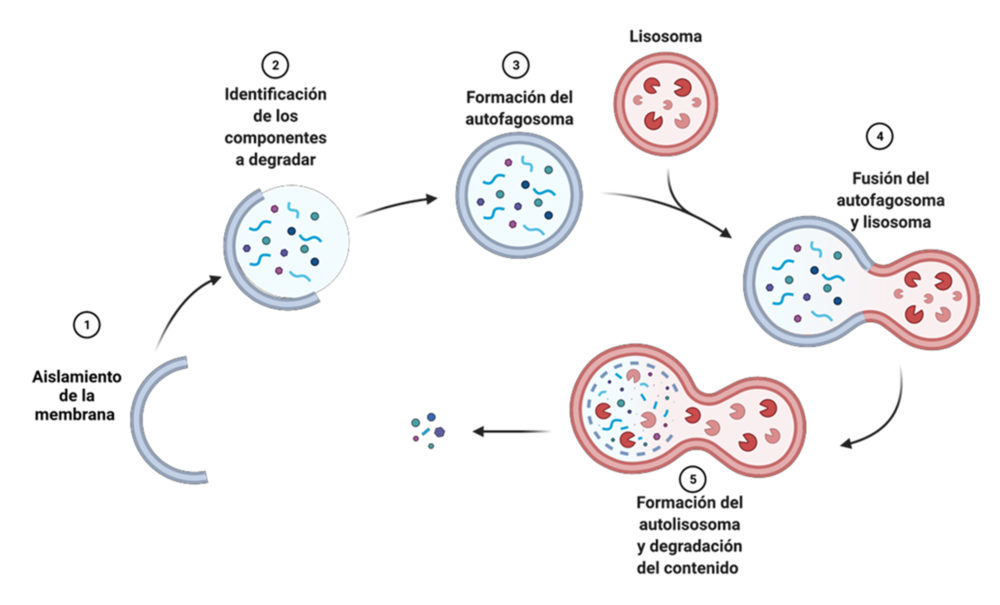
Figura 13. Esquema del mecanismo de la macroautofagia. Creado con BioRender.com.
En la eliminación de bacterias por macroaautofagia se ha visto que es importante un proceso de conjugación de cisteína con 8-nitro-cGMP (S-guanilación) que favorece la ubiquitinación de los residuos de lisina y la degradación de los microorganismos en el lisosoma. Un estudio llevado a cabo por Arimoto y col. (39) ha demostrado que la introducción de una marca de cGMP en la proteína a degradar es suficiente para inducir una autofagia selectiva. Este grupo ha desarrollado una serie de moléculas denominadas AUTACs (autophagy targeting chimeras) para la degradación selectiva de proteínas y orgánulos celulares por medio de la autofagia. Estas moléculas contienen en su estructura un derivado de guanina (p-fluorobencilguanina, FBnG) y un ligando específico para el sustrato a degradar unidos por un espaciador de polietilénglicol (PEG). Los AUCTACs 1y 2 fueron capaces de silenciar de manera selectiva las proteínas citosólicas metionina aminopeptidasa 2 (MetAP2) y la proteína de unión a FK506 (FKBP12), respectivamente, utilizando en el primero de los casos un ligando de unión covalente a la proteína y en el segundo, un ligando de unión no covalente.
También ensayaron un AUTAC para la degradación selectiva de BRD4, perteneciente a la familia de proteínas BET (bromodomain extraterminal proteins), por la importancia de su papel en el mantenimiento de los melanomas. Estas proteínas están localizadas en el núcleo y no es fácil su degradación por lisosomas que solo se encuentran en el citoplasma. La degradación de BRD4 utilizando una AUTAC basado en su inhibidor JQ-1 fue muy baja, probablemente debido a que solo puede tener lugar durante la división celular, momento en el que BRD4 es accesible en el citoplasma.
Este mismo grupo ha demostrado que los AUTACs que contienen FBnG pueden también utilizarse para la degradación de sustratos no proteícos, como el AUTAC4 que indujo la degradación de la mitocondria. Este tipo de AUTACs serían útiles para inducir la mitofagia de sistema mitocondriales disfuncionales.
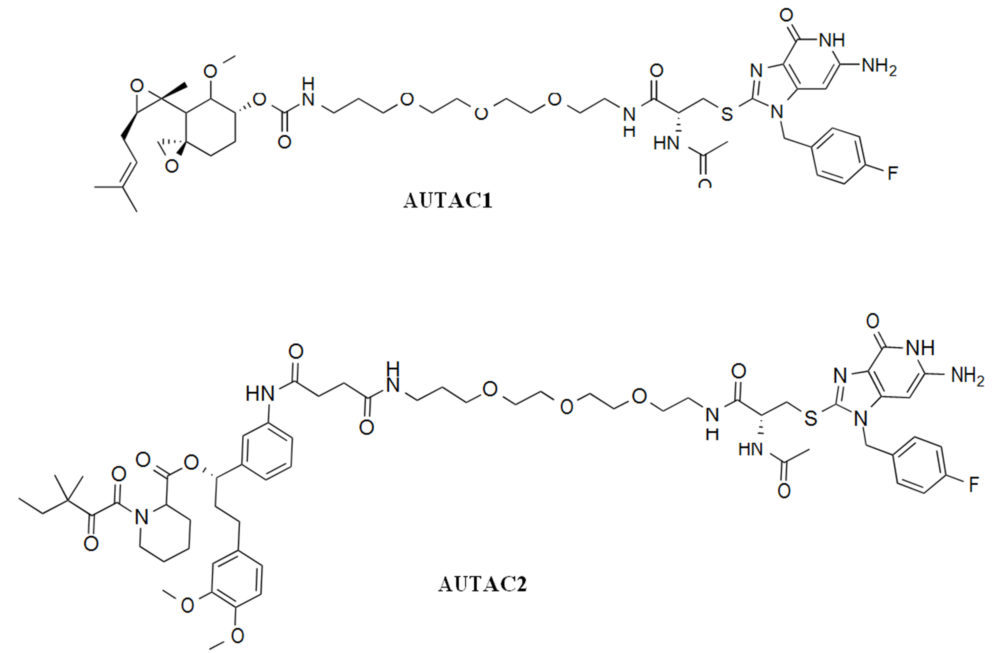

Figura 14. Estructuras de AUTAC1, 2 y 4.
Una variante de esta estrategia es la utilización de un único ligando capaz de unirse a la PDI y a la proteína LC3, clave en la formación del autofagosoma. Este tipo de compuestos ha sido desarrollado por el grupo de Lu y se denominan compuestos de unión al autofagosoma, ATTECs (autophagosome-tethering compound). (40) Los cuatro ATTEcs ensayados fueron capaces de promover la degradación selectiva por autofagia de la proteína huntingtina mutada sin afectar a la proteína natural. Estos resultados son esperanzadores y abren una nueva vía de investigación en la búsqueda de nuevos fármacos para el tratamiento de proteinopatías
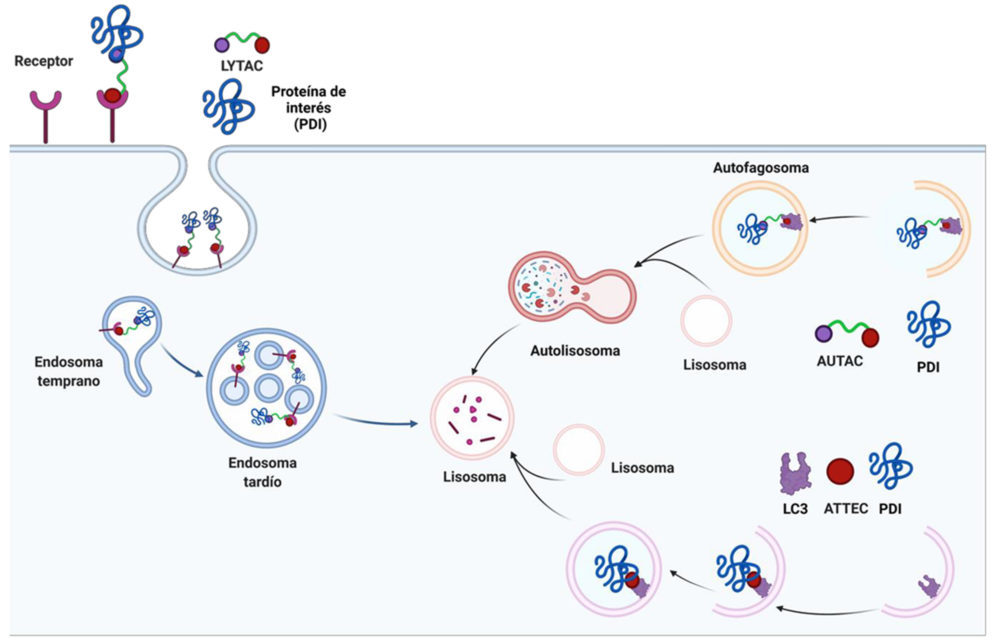
Figura 15. Resumen de otras estrategias de degradación de proteínas. Creado con BioRender.com.
5. REFERENCIAS
- Belcher BP, Ward CC, Nomura DK. Ligandability of E3 Ligases for Targeted Protein Degradation Applications. Biochemistry. 2021.
- Paiva SL, Crews CM, Targeted protein degradation: elements of PROTAC design. Curr Opin Chem. Biol. 2019;50:111-9.
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. PROTACs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98(15):8554-9.
- Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2(12):1350-8.
- Castillo-Quan, JI, Li L, Kinghorn KJ, Ivanov DK, Tain LS, Slack C, Kerr F, Nespital T, Thornton J, Hardy J, Bjedov I, Partridge L. Lithium promotes longevity through GSK-3/NRF2-dependent hormesis. Cell Rep . 2016;15(3):638-650.
- Kanarek N, London N, Schueler-Furman O, Ben-Neriah Y. Ubiquitination and degradation of the inhibitors of NF-κB. Cold Spring Harbor perspectives in biology, 2010, 2, a000166.
- Semenza GL, Wang GL. A Nuclear Factor Induced by Hypoxia via De Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol Cell Biol. 1992;12(12):5447-5454.
- Ishida T, Ciulli A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. SLAS DISCOVERY: Advancing the Science of Drug Discovery, 2021;26(4):484-502.
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399 (6733):271-5.
- Cardote TAF, Gadd MS, Ciulli A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure. 2017;25(6):901-911.e3.
- Van Molle I, Thomann A, Buckley DL. Dissecting Fragment-Based Lead Discovery at the von Hippel-Lindau Protein: Hypoxia Inducible Factor 1α Protein-Protein Interface. Chem Biol. 2012;19(10):1300-1312.
- Galdeano C, Gadd MS, Soares P, Scaffidi S, Van Molle I, Birced I, Hewitt S, Dias DM, Ciulli A. Structure-Guided Design and Optimization of Small Molecules Targeting the Protein-Protein Interaction Between the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with In Vitro Nanomolar Affinities. J Med Chem. 2014;57(20):8657-8663.
- Zengerle M, Chan K-H, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10(8):1770-7.
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514-521.
- Glas A, Wamhoff EC, Krgger DM, Rademacher C, Grossmann TN. Increased Conformational Flexibility of a Macrocycle–Receptor Complex Contributes to Reduced Dissociation Rates. Chem. Eur. J. 2017;23(64):16157-16161.
- Valeur E, Guret SM, Adihou H, Gopalakrishnan R, Lemurell M, Waldmann H, Grossmann TN, Plowright AT. New Modalities for Challenging Targets in Drug Discovery. Angew Chem Int Ed. 2017;56(35):10294-10323.
- Glas A, Wamhoff EC, Krgger DM, Rademacher C, Grossmann TN. Increased Conformational Flexibility of a Macrocycle–Receptor Complex Contributes to Reduced Dissociation Rates. Chem. Eur. J. 2017;23(64):16157-16161.
- Lu G, Middleton RE, Sun, H, Naniong M, Ott CJ, Mitsiades CS, Wong K-K., Breadner JE, Kaelin WG. The myeloma drug lenalidomide promotes the cereblon- dependent destruction of Ikaros proteins. Science. 2014;343(6168):305-9.
- Fischer ES, Böhm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith REJ, Harper JW, Jenkins JL, Thomä NH. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature, 2014;512(7512):49-53.
- Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Fischer ES. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14(7):706-714.
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514-521.
- Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67(4):2168-2174.
- Ma Z, Ji Y, Yu Y, Liang D. Specific non-genetic IAP-based protein erasers (SNIPERs) as a potential therapeutic strategy. Eur J Med Chem. 2021;216:113247.
- Sekine K, Takubo K, Kikuchi R, Nishimoto M, Kitagawa M, Abe F, Nishikawa K, Tsuruo T, Natio M. Small molecules destabilize cIAP1 by activating auto- ubiquitylation. J Biol Chem. 2008;283(14):8961-8.
- Ma Z, Ji Y, Yu Y, Liang D. Specific non-genetic IAP-based protein erasers (SNIPERs) as a potential therapeutic strategy. Eur J Med Chem. 2021;216:113247.
- Huun J, Gansmo LB.; Mannsåker B, Iversen GT, Sommerfelt-Pettersen J, Øvrebø JI, Knappskog S. The Functional Roles of the MDM2 Splice Variants P2-MDM2-10 and MDM2-∆ 5 in Breast Cancer Cells. Transl Oncol. 2017;10(5):806-817.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu E.A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844-8.
- Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted Intracellular Protein Degradation Induced by a Small Molecule: En Route to Chemical Proteomics. Bioorg Med Chem Lett. 2008;18(22):5904-8.
- Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79(1):251-262.
- Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015:88;101-7.
- Lu M, Liu T, Jiao Q, Ji J, Ta M, Liu Y, Jiang Z. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018;146:251-9.
- Tong B, Luo M, Xie Y, Spradlin JN, Tallarico JA, McKenna JM, Schirle M, Nomura DK. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci Rep. 2020;10(1):15543.
- Wei J, Meng F, Park KS, Yim H, Velez J, Kumar P, Wang L, Xie L, Chen H, Shen Y, Teichman E, Li D, Wang GG, Che X, Kaniskan HU, Jin J. Harnessing the E3 Ligase KEAP1 for Targeted Protein Degradation. J Am Chem Soc. 2021;143(37):15073-15083.
- Du X, Volkov OA, Czerwinski RM, Tan H, Huerta C, Morton ER, Rizzi JR, Wehn PM, Xu R, Nijhawan D, Wallace EM. Structural basis and kinetic pathway of RBM39 recruitment to DCAF15 by a sulfonamide molecular glue E7820. Structure. 2019;27(11):1625-1633.e3.
- Li L, Mi D, Pei H, Duan Q, Wang X, Zhou W, Jin J, Li D, Liu M, Chen Y. In Vivo Target Protein Degradation Induced by PROTACs Based on E3 Ligase DCAF15. Signal Transduct Target Ther. 2020;5(1):129.
- Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020; 41(7):464-74.
- Alabi SA, Crews CM. Major advances in the targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem. 2021;296:100647.
- Banik S, Pedram K, Wisnovsky S, Riley N, Bertozzi C. Lysosome Targeting Chimeras (LYTACs) for the Degradation of Secreted and Membrane Proteins. Nature. 2020;584:291-7.
- Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, Akaike T, Itto-Nakama K, Arimoto H. AUTACs: cargo-specific degraders using selective autophagy. Mol Cell. 2019;76:797-810.
- Li Z, Zhu C, Ding Y, Fei Y, Lu, B. ATTEC: a potential new approach to target proteinopathies. Autophagy 2020;16(1):185-7.
