1. INTRODUCCIÓN
La infección por el virus de la Hepatitis C (VHC) supone un importante problema de salud pública con 170 millones de personas infectadas de manera crónica en todo el mundo (1). De hecho, la infección por el VHC se ha convertido en la principal causa de enfermedad hepática crónica relacionada con el desarrollo de otras enfermedades hepáticas tales como la fibrosis, cirrosis o carcinoma hepatocelular (CHC), y ha sido la principal causa de trasplante hepático durante los últimos veinte años (2).
Curiosamente, la infección crónica por el VHC está asociada con la resistencia a la insulina y diabetes tipo 2 (3, 4). De hecho, la prevalencia de resistencia a la insulina y diabetes tipo 2 es significativamente mayor en los pacientes infectados por el VHC en comparación con individuos no infectados o que padecen otras enfermedades hepáticas crónicas (5, 6). Además, varios estudios han demostrado que la resistencia a la insulina hepática asociada al VHC puede afectar negativamente la respuesta al tratamiento con interferón (7-9).
A nivel experimental, existen evidencias que indican que el VHC interfiere de manera directa en la ruta de señalización de la insulina. De hecho, se ha detectado una reducción en la expresión hepática de mediadores clave en el inicio de esta cascada de señalización, como es el caso de los sustratos del receptor de insulina (IRS), IRS1 e IRS2, en pacientes con infección crónica por VHC, en ratones transgénicos que expresan VHC, así como en un modelo celular experimental de infección por VHC (10, 11). Los mecanismos moleculares implicados en la deficiente expresión de IRS1 e IRS2 son múltiples, como la inducción de supresores de la señalización de citocinas (SOCS) (11-13) o la activación de diferentes quinasas que promueven la fosforilación de residuos de serina de IRS1, inhibiendo la fosforilación en residuos tirosina, imprescindible para la activación del mismo, y promoviendo su degradación (14). Asimismo, el VHC induce la expresión de moduladores negativos de la vía de señalización de la insulina como la proteína tirosina fosfatasa 1B (PTP1B), el homólogo de fosfatasa y tensina (PTEN) y la proteína fosfatasa 2A (PP2A) (15). En conjunto, todos estos mecanismos impiden que la activación de AKT, un efector clave en la acción de la insulina en el hígado, que conduce a la resistencia a la insulina (10, 14).
En los últimos años, el descubrimiento de tratamientos antivirales basados en los nuevos y altamente eficaces agentes antivirales directos se ha convertido en un avance revolucionario en el tratamiento de la infección por VHC. En comparación con el tratamiento estándar utilizado anteriormente (terapia con interferón pegilado más ribavirina), los tratamientos basados en estos nuevos antivirales muestran una actividad pangenotípica, una mayor tolerabilidad y mínimos efectos adversos asociados a la medicación, y logran una mayor respuesta viral sostenida, casi del 100%, con tratamientos más cortos y sencillos en pacientes infectados por el VHC (16-18). Entre ellos, sofosbuvir (SOF), un nucleótido análogo del inhibidor de la polimerasa NS5B del VHC, fue aprobado por la Administración de Alimentos y Medicamentos de Estados Unidos (FDA) para el tratamiento de la infección crónica por VHC en combinación con otros agentes antivirales en diciembre de 2013 (19, 20). Curiosamente, la terapia antiviral con agentes antivirales directos, incluido el sofosbuvir, parece mejorar notablemente la sensibilidad a la insulina periférica en pacientes infectados por VHC (21); sin embargo, se desconocen los mecanismos moleculares involucrados en la relación entre la erradicación del VHC y la recuperación en la respuesta a la insulina inducida por estos antivirales.
Por todo lo anterior, el principal objetivo de este estudio fue determinar los efectos del sofosbuvir sobre la resistencia a la insulina inducida por el VHC, tanto en pacientes tratados con regímenes basados en este antiviral como en hepatocitos que expresan un replicón completo de VHC tratados in vitro con dicho antiviral.
2. MATERIAL Y MÉTODOS
2.1. Estudio clínico
El estudio clínico se realizó de acuerdo con los principios de la Declaración de Helsinki y la Conferencia Internacional de Armonización de Buenas Prácticas Clínicas. La inclusión de los pacientes en el estudio así como la obtención de las muestras biológicas de los mismos se llevó a cabo tras la firma de un consentimiento informado específico aprobado por el Comité Ético de Investigación Clínica del Hospital Universitario Virgen del Rocío (Sevilla, España). Los datos personales y el material biológico de todos los pacientes se tratarán con confidencialidad y seguridad, de acuerdo con la normativa basada en la Ley 15/1999 y en la Ley 14/2007 sobre Investigación Biomédica.
Este estudio retrospectivo incluyó 42 pacientes con enfermedad hepática crónica asociada a VHC reclutados de manera aleatoria y consecutiva en el Servicio de Digestivo del Hospital Universitario Virgen del Rocío (Sevilla, España) entre enero de 2015 y febrero de 2017. Los criterios de inclusión fueron los siguientes: mayores de 18 años de edad, presencia de resistencia a la insulina (índice de resistencia a la insulina, HOMA > 2,5) al inicio del estudio, y tratamiento antiviral completado. Se incluyeron pacientes con cualquier estadio de fibrosis hepática (de F0 a F4), independientemente del genotipo viral.
Los pacientes fueron tratados con diferentes regímenes antivirales basados en sofosbuvir (SOF) según la elección del médico y recibieron SOF/daclatasvir (DAC)/ribavirina (RVB) (n=7), SOF/Ledipasvir (LED)/RBV (n=12); SOF/DAC (n=5); SOF/LED (n=16) o SOF/Simeprevir (SIM) (n=2). La duración del tratamiento osciló entre 12 semanas (n=35) y 24 semanas (n=7).
La presencia de fibrosis hepática se evaluó mediante elastografía transitoria no invasiva (Fibroscan® 402, Echosens, París, Francia) y los pacientes se estratificaron siguiendo la siguiente escala: F0-F1, < 7 kPa, ausencia de fibrosis o fibrosis leve; F2, 7-9,4 kPa, fibrosis moderada; F3, 9,5-13 kPa, fibrosis avanzada; F4, > 13 kPa, cirrosis.
Los parámetros demográficos, antropométricos, virológicos y bioquímicos se registraron de la siguiente manera: edad, sexo, índice de masa corporal (IMC), cuantificación de ARN del VHC, genotipo del VHC, historia previa del tratamiento antiviral, y pruebas generales de laboratorio (aspartato aminotransferasa -AST-, alanina aminotransferasa -ALT-, gammaglutamiltransferasa -GGT-, fosfatasa alcalina -ALP-, albúmina, bilirrubina, creatinina, glucosa, insulina, hemoglobina glicosilada -HBa1c-, colesterol total, colesterol unido a lipoproteínas de baja densidad -LDL-, colesterol unido a lipoproteínas de alta densidad -HDL-, apolipoproteína B -ApoB- y triglicéridos -TG-). El índice de resistencia a la insulina HOMA se calculó aplicando la siguiente fórmula: insulina en ayunas (μU/mL) × glucosa en ayunas (mg/dL) / 405. El índice de fibrosis FIB4, método no invasivo que predice la presencia de fibrosis hepática, se calculó aplicando la siguiente fórmula: edad (años) × AST (U/L) / [plaquetas (109/L) × ALT/2 (U/L)].
Para el análisis de estas variables virológicas, bioquímicas y metabólicas se recogieron muestras de sangre al inicio del estudio (basal, B), al final del tratamiento (FdT) y un año después del final del tratamiento (final del seguimiento, FdS).
2.2. Cultivo celular
Para el estudio experimental, se utilizó la línea celular de hepatoma humano Huh7. Las células Huh7 y sus derivados se cultivaron en medio DMEM (medio Eagle modificado por Dulbecco) enriquecido (suero fetal bovino 10%, L-glutamina 2 mM y antibióticos: gentamicina 50 μg/mL, penicilina 100 U/mL y 100 estreptomicina μg/mL), a 37ºC en una atmósfera de CO2 al 5%. Las células Huh7 que expresan un replicón completo de VHC de genotipo 1b (Con1; número de acceso a la base de datos EMBL AJ238799) se establecieron como se ha descrito previamente (22). Brevemente, las construcciones pI389/Core-3’/5.1 y pI377/NS3-3’ se linealizaron con ScaI y se usaron como secuencia molde para la síntesis de ARN utilizando la polimerasa ARN T7 (Promega, Madison, WI). Para la transfección mediante electroporación de 107 células Huh7 se utilizaron 20 μg del ARN sintetizado, y 24 horas más tarde se realizó la selección antibiótica de las células transfectadas añadiendo gentamicina B1 (500 μg/ml, G418). Durante 4 semanas tras la transfección, el medio de cultivo suplementado con gentamicina B1 se reemplazó 2 veces por semana, y, finalmente, se aislaron las colonias resistentes que son las que expresan un replicón completo de VHC.
2.3. Tratamiento de las células con el fármaco antiviral
El sofosbuvir (SOF, proporcionado amablemente por Gilead Science Inc., EE. UU.) se reconstituyó en DMSO a una concentración de 10 mM. Las células portadoras del replicón del VHC se trataron con sofosbuvir a 10 μM durante 9 días en ausencia de G418 para evitar la muerte de las células una vez eliminado el replicón. Cada 3 días el medio de cultivo se sustituyó por medio fresco que contenía sofosbuvir o vehículo (DMSO).
2.4. Análisis de la activación de la cascada de señalización de insulina
Para evaluar los efectos de sofosbuvir sobre la señalización de la insulina, se retiró el suero del medio a las células portadoras del replicón del VHC tratadas con sofosbuvir o DMSO antes de la estimulación con insulina (1 o 10 nM, 10 minutos).
2.5. Análisis de proteínas por inmunofluorescencia
Las células se cultivaron sobre un cubreobjetos de vidrio de 12 mm de diámetro. Al final del experimento, las células se fijaron con paraformaldehído al 4% en solución salina tamponada con fosfato (PBS) durante 10 minutos a temperatura ambiente, y se permeabilizaron con NP-40 al 0,1% en PBS durante 10 minutos a temperatura ambiente. A continuación, las células se bloquearon con TNB (Tris-HCl 0,1 M, NaCl 0,15 M, reactivo de bloqueo al 0,5% -Boehringer Mannheim GmbH, Mannheim, Alemania-) durante 30 minutos a 37ºC y se incubaron con los correspondientes anticuerpos primarios diluidos en TNB durante 1 hora a 37ºC: anti-VHC core (clon C7-50, Affinity BioReagents, Goleen, CO) o anti-VHC NS5A (Virostat, Portland, ME). Después de lavar con NP-40 al 0,1% en PBS, las células se incubaron con el anticuerpo secundario IgG de cabra anti-ratón Alexa 488 (Molecular Probes, Inc., Eugene, OR) durante 20 minutos a 37ºC, se contratiñeron con DAPI (Pierce, Rockford, IL) y se montaron en el medio de montaje DakoCytomation (DAKO A / S, Glostrup, Dinamarca). Las preparaciones se examinaron con un microscopio confocal Leica TCS-SP5 (Leica Microsystems, Heidelberg, Alemania).
2.6. Preparación de extractos de proteínas para inmunoprecipitación
Al final del experimento, las células se lisaron con tampón RIPA (Tris-HCl 50 mM, pH 7,4, Triton X-100 al 1%, SDS al 0,2%, EDTA 1 mM, PMSF 1 mM y 5 μg/mL de leupeptina) durante 10
minutos en hielo para obtener los lisados celulares totales. Después de la determinación del contenido de proteína con el reactivo de Bradford, se inmunoprecipitó una cantidad igual de proteína (500 μg) con anticuerpo anti-IRS1 (sc-8038, Santa Cruz Biotechnology Inc., Heidelberg, Alemania). Los complejos inmunes se recogieron en perlas de sefarosa (GE17-0120-01, Merck KGaA, Darmstadt, Alemania), se calentaron a 95ºC durante 5 minutos en tampón de carga Laemmli (Tris 100 mM pH 7,6, glicerol al 10% (v/v), SDS al 6% (p/v), azul de bromofenol al 0,2% (p/v), β mercaeptoetanol 2 mM) y se sometieron al análisis de proteínas por Western blot.
2.7. Preparación de extractos de proteínas totales
Al final del experimento, después de lavar con PBS, las células se lisaron en la placa con 100 μL de tampón de carga Laemmli, se calentaron a 95ºC durante 5 minutos y se sometieron al análisis de proteínas por Western blot.
2.8. Análisis de proteínas por Western blot
Las proteínas se separaron en un gel de poliacrilamida-SDS y se transfirieron a membranas de nitrocelulosa (Bio-Rad Inc., Hercules, CA, EE.UU.) que se incubaron en solución de bloqueo (leche desnatada al 5% en solución salina tamponada con Tris -TBS- o BSA al 3% en TBS) durante 1 hora a temperatura ambiente. A continuación, las membranas se incubaron durante la noche a 4ºC con los correspondientes anticuerpos primarios diluidos en TBS-T (0,1% Tween-20 en TBS) con leche desnatada o BSA al 1%: anti-VHC core (clon C7-50) de Affinity BioReagents; anti-HCV NS5A de Virostat; anti-IRS1 (06-248) de Merk-Millipore (Darmstadt, Alemania); anti-p53 (sc-126), anti-pTyr1146 IR (sc-81499), anti-IR (sc-711), anti-fosfoserina (4A3, sc-81516), anti-pSer473 AKT (sc-7985-R) y anti-AKT (sc-5298) de Santa Cruz Biotechnology; anti-pSer256 FoxO1 (9461) y anti-pSer9 GSK3β (9336) de Cell Signaling Technology (Boston, MA, EE.UU.); anti-βactina (A-5441) de Sigma-Aldrich Inc. (Madrid, España).
Posteriormente, las membranas se lavaron tres veces en TBS-T y se incubaron con IgG de cabra anti-ratón o anti-conejo marcada con peroxidasa (Pierce, Rockford, IL) diluida en TBS-T durante 45 minutos a temperatura ambiente. Después de tres lavados en TBS-T, se visualizó el anticuerpo unido a la membrana con el sustrato quimioluminiscente SuperSignal West Pico (Pierce). El análisis densitométrico de las bandas de proteína se realizó utilizando el software ImageJ Biological Image Analysis (NIH, Bethesda, MD, EE. UU.). Los resultados de la cuantificación de los blots de todos los experimentos se expresaron en porcentaje de estimulación con insulina en relación con la condición de DMSO (100%), o en veces de cambio en relación a la condición DMSO (1).
2.9. Análisis de expresión génica mediante PCR cuantitativa en tiempo real (RT-qPCR)
El ARN total se extrajo con Reactivo TRI (Vitro, Sevilla, España). Seguidamente, el ADNc se obtuvo a partir de 1 μg de ARN mediante transcripción inversa (kit del sistema de transcripción inversa, Promega Inc., Madison, WI, EE. UU.). La RT-qPCR se llevó a cabo en un sistema detector de secuencias de PCR en tiempo real StepOnePlus ™(Thermo Fisher Scientific Inc., Madrid, España) utilizando un kit SYBR Green (Promega Inc.) y cebadores aleatorios d(N)6 que se compraron a Metabion (Planegg, Alemania):
5’-TACGTCCTCTTCCCCATCTG-3’ y 5’-TCCCTGGTCCAGTCTCACAA-3’ para glucosa 6 fosfatasa, 5’-CCAGGCAGTGAGGGAGTTTCT-3’ y 5’-ACTGTGTCTCTTTGCTCTTGG-3’ para fosfoenolpiruvato carboxiquinasa, 5’-CACCTGCTGGAAACCACACTT-3’ y 5’-AAGGTGAACCGGGACAGAGA-3’ para PTP1B, 5’-AAGACATTATGACACCGCCAAA-3’ y 5’-GTTCTAGCTGTGGTGGGTTATGG-3’ para PTEN, 5’-GAGCACTTGATCGCCTACAAGA-3’ y 5’-ACCACGGTCATCTGGATCTGA-3’ para PP2A. El gen 36B4 se utilizó como gen constitutivo normalizador. Los resultados se expresaron en veces de cambio en relación a la condición DMSO (1).
2.10. Análisis del contenido celular de glucógeno
Después retirar el suero del medio durante la noche, las células se lavaron dos veces con PBS, se resuspendieron con 200 μL de H2Od y se incubaron en hielo durante 10 minutos. A continuación, las muestras se hirvieron a 100ºC durante 10 minutos para inactivar las enzimas y se centrifugaron a 13.000 rpm durante 10 minutos más. El contenido de glucógeno en los sobrenadantes se midió con el kit de glucógeno Glycogen Assay Kit II (ab169558, Abcam, Cambridge, Reino Unido). Los resultados se expresaron en μg de glucógeno normalizados por la proteína total (mg de proteína).
2.11. Tinción de PAS (Periodic acid-Schiff)
Las células se cultivaron sobre un cubreobjetos de vidrio de 12 mm de diámetro. Tras retirar el suero del medio durante la noche, las células se lavaron dos veces con PBS y se fijaron con paraformaldehído al 4% durante 10 minutos. Para la tinción de PAS se añadió una solución de ácido peryódico al 0.5% (Sigma-Aldrich Inc. Madrid, España) durante 5 minutos y, a continuación, se tiñeron con el reactivo de Schiff (Sigma-Aldrich Inc. Madrid, España) durante 15 minutos. Por último, se realizó la contratinción con hematoxilina durante 2 minutos y se montaron con el medio de montaje Permount (Thermo Fisher Scientific Inc.). Las preparaciones se examinaron con un microscopio óptico Nikon Eclipse E400 (Nikon, Japón). El análisis de las imágenes se realizó utilizando el software ImageJ Biological Image Analysis (NIH). Los resultados de la cuantificación de las imágenes de todos los experimentos se expresaron en unidades arbitrarias (u.a.).
2.12. Ensayo de producción de glucosa
Las células se incubaron durante la noche en DMEM sin suero y sin glucosa suplementado con piruvato de sodio 2 mM. Luego, se recogió el medio de cultivo y se midió el contenido de glucosa usando el kit de detección colorimétrica de glucosa. (Ref.15880712, Invitrogen, Thermo Fisher Scientific Inc.). Los resultados se expresaron en mg/dL de glucosa normalizados por la proteína total (mg de proteína).
2.13. Análisis estadístico
Se utilizó la prueba de Shapiro-Wilk para verificar la normalidad de la distribución de datos. Los datos obtenidos del estudio clínico se presentaron como media ± desviación estándar (SD). Las características basales de los pacientes se presentaron como frecuencia y porcentaje. Las variables se compararon entre los grupos de pacientes (basal -B-, final del tratamiento -FdT- y final del seguimiento -FdS-) mediante el test de Wilcoxon de pares equiparados. Los datos experimentales obtenidos del estudio in vitro se presentaron como media ± error estándar de la media (SEM) y se compararon utilizando el test t para datos no apareados. Todos los análisis estadísticos se realizaron utilizando el software IBM SPSS Statistics 24.0 (SPSS Inc., IBM, Armonk, NY, EE. UU.) y el software GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, EE. UU.) con contraste bilateral, considerando estadísticamente significativo un p valor < 0,05.
3. RESULTADOS
3.1. Características demográficas y basales de los pacientes
La población de estudio incluyó a 42 pacientes resistentes a la insulina (índice de resistencia a la insulina, HOMA > 2.5) antes de comenzar el tratamiento antiviral (29 hombres (69%), media de edad 53.5 años). El índice de masa corporal (IMC) medio fue de 28.5 kg/m2. El examen mediante elastografía transitoria (TE) reveló solo 5 pacientes con F0-F1 y 7 pacientes con fibrosis moderada (F2), mientras que 30 pacientes tenían fibrosis avanzada o cirrosis (8 casos con F3 y 22 casos con F4) (Tabla 1).
Tabla 1. Características demográficas, antropométricas, virológicas y bioquímicas de la población de estudio. Los datos se muestran como media ± SD.

Los pacientes fueron tratados según la elección del médico con regímenes terapéuticos basados en sofosbuvir (SOF). Los antivirales de acción directa utilizados para el tratamiento fueron: SOF / LED (± RBV) 66.7% (n=28/42), SOF / DAC (± RBV) 28.6% (n=12/42) y SOF / SIM (± RBV) 4.7 % (n=2/42). Al final del tratamiento, el 100% de los pacientes eliminó el ARN del VHC, consiguiendo una respuesta virológica sostenida en todos los pacientes. Como era de esperar, se detectó una disminución significativa de los marcadores séricos de daño hepático, como ALT, AST, GGT y bilirrubina total en los pacientes al final del tratamiento que se mantuvo un año después (Tabla 2). Además, el índice predictivo de fibrosis hepática FIB4 se redujo a lo largo de todo el estudio (p<0.0001) (Tabla 2).
Tabla 2. Los regímenes antivirales basados en sofosbuvir producen una disminución significativa de la concentración sérica de marcadores de daño hepático en los pacientes con VHC

Los datos se muestran como media ± SD. B: basal; FdT: fin del tratamiento; FdS: final del seguimiento
3.2. Cambios en la resistencia a la insulina sistémica y el metabolismo de la glucosa
Los datos sobre los cambios en los niveles de glucosa e insulina en suero de los pacientes incluidos en el estudio se muestran en la Tabla 3. La glucosa en ayunas fue menor al final del tratamiento con respecto a los valores basales (p=0.0687), alcanzando una disminución significativa en la visita de seguimiento al año (p=0.0156). No se observaron cambios en los niveles de insulina tras el tratamiento (p=0.1113); sin embargo, se logró una diferencia estadísticamente significativa respecto al nivel basal un año después del final del tratamiento (p<0.001). Además, se observó una bajada significativa en el índice de resistencia a la insulina HOMA desde el final del tratamiento (p=0.0048), siendo más pronunciada un año después (p<0.001), en comparación con los datos iniciales. En cuanto a los niveles de HbA1c, no hubo diferencia a lo largo del estudio.
Tabla 3. Los regímenes antivirales basados en sofosbuvir mejoran el estado de resistencia a la insulina de los pacientes con VHC

Los datos se muestran como media ± SD. B: basal; FdT: fin del tratamiento; FdS: final del seguimiento
3.3. Variaciones en marcadores lipídicos
A continuación, se analizó el perfil lipídico de la población de estudio, encontrando un aumento significativo en el contenido de colesterol total, LDL y ApoB mantenido en el tiempo después del tratamiento (p <0.005), mientras que no se observaron diferencias en los valores de TG y HDL (Tabla 4).
Tabla 4. Los regímenes antivirales basados en sofosbuvir modifican el perfil lipídico de los pacientes con VHC.
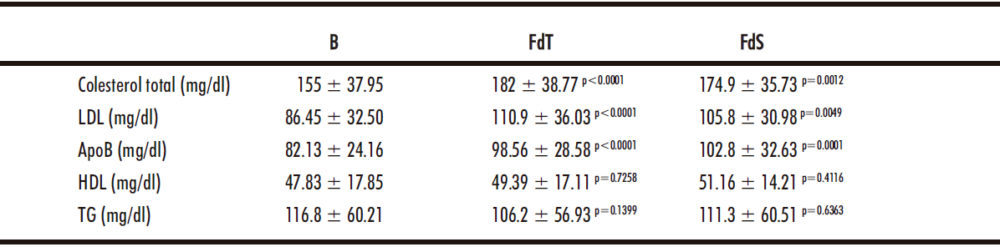
Los datos se muestran como media ± SD. B: basal; FdT: fin del tratamiento; FdS: final del seguimiento.
3.4. El tratamiento con sofosbuvir consigue la eliminación del VHC en hepatocitos que expresan un replicón del VHC de manera estable
Con el fin de explorar los posibles mecanismos moleculares implicados en los efectos de sofosbuvir sobre la mejora de la resistencia a la insulina observada en los pacientes, realizamos un modelo experimental in vitro: células Huh7 que expresan un replicón del VHC de manera estable tratadas con sofosbuvir para la eliminación del virus y, posteriormente, estimuladas con diferentes dosis de insulina.
Tras 9 días de tratamiento con sofosbuvir (10 μM), no se detectó la expresión de las proteína virales core y NS5A ni por inmunofluorescencia (Figura 1A) ni por análisis por Western blot (Figura 1B) en los hepatocitos que expresan un replicón del VHC en comparación con los que recibieron el vehículo (DMSO), lo que indica que mediante este protocolo de tratamiento con sofosbuvir se consigue la eliminación del VHC de manera eficaz.
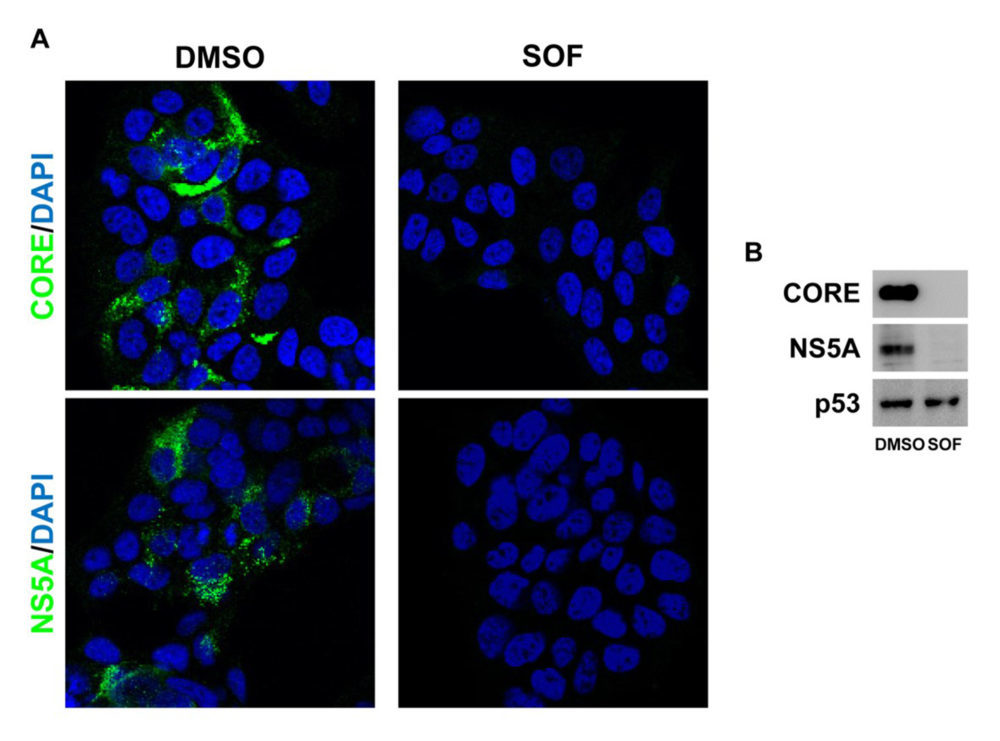
Figura 1. La eliminación del VHC se consigue tras el tratamiento con sofosbuvir en hepatocitos que expresan replicones del virus. Análisis por inmunofluorescencia (A) y Western blot (B) de la expresión de las proteínas virales core y NS5A de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o sofosbuvir (SOF). p53 se utilizó como control de carga. (n=3 experimentos independientes realizados por duplicado).
3.5. Efectos del sofosbuvir sobre la respuesta deficiente a la insulina inducida por el VHC
A continuación, las células tratadas con DMSO o sofosbuvir se incubaron con insulina (1 o 10 nM) durante 10 minutos. Como se muestra en la Figura 2, se observa un ligero aumento de la fosforilación en tirosina del receptor de la insulina (IR) tras la inducción con insulina a 10 nM en los hepatocitos con VHC tratados con sofosbuvir en comparación con aquellos tratados con DMSO. Además, la fosforilación de AKT en respuesta a todas las dosis de insulina mejoró considerablemente en las células con VHC tratadas con el antiviral.
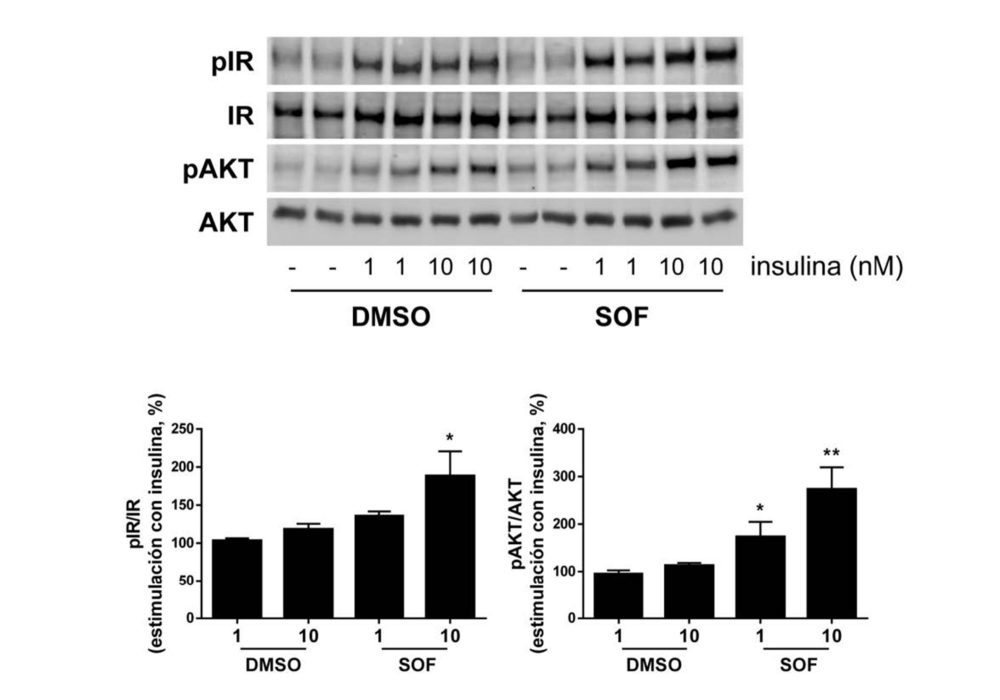
Figura 2. El tratamiento con sofosbuvir mejora la respuesta a la insulina en las células Huh7 con VHC. Análisis por Western blot de la fosforilación del receptor de insulina (pIR) y de la AKT de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o sofosbuvir (SOF) y, posteriormente, se estimularon con insulina (1 o 10 nM) durante 10 min. Los datos se presentan como media ± SEM. *p <0.05 y **p <0.01, SOF vs DMSO (n=4 experimentos independientes realizados por duplicado).
Con respecto a los sustratos de AKT, el tratamiento con sofosbuvir supuso un aumento en la fosforilación tanto de FoxO1 como de GSK3β tras la estimulación con insulina (Figura 3).
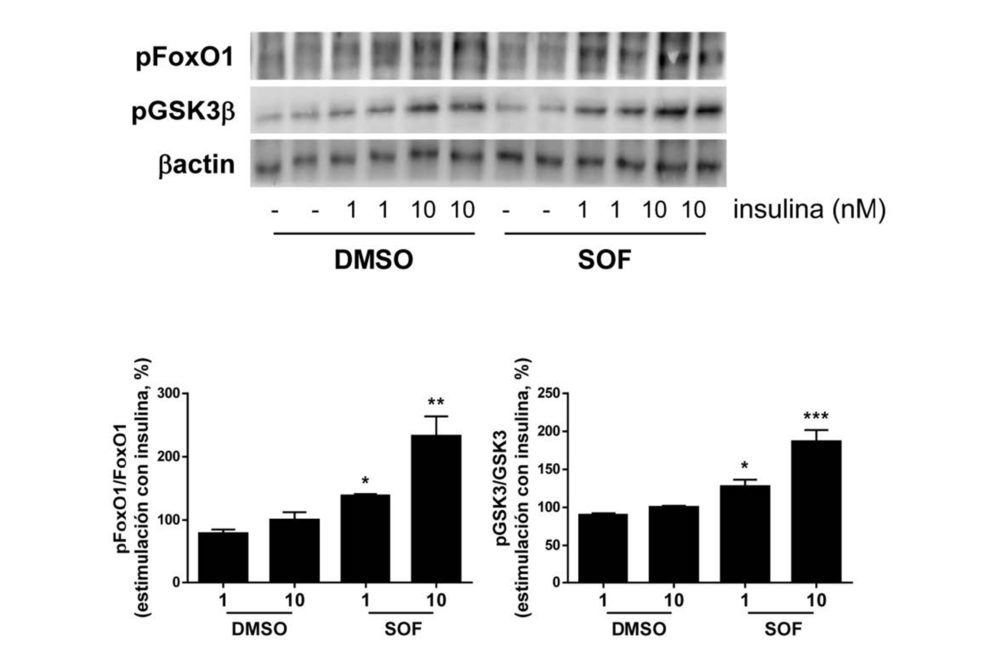
Figura 3. El tratamiento con sofosbuvir mejora la respuesta a la insulina en los hepatocitos con VHC. Análisis por Western blot de la fosforilación de FoxO1 y de GSK3 de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o sofosbuvir (SOF) y, posteriormente, se estimularon con insulina (1 o 10 nM) durante 10 min. β actina se utilizó como control de carga. Los datos se presentan como media ± SEM. *p<0.05, **p<0.01 y ***p<0.005, SOF vs. DMSO (n=3 experimentos independientes realizados por duplicado).
Está ampliamente establecido que AKT fosforila al factor de transcripción FoxO1, impidiendo así su entrada en el núcleo y, en consecuencia, la transcripción de genes gluconeogénicos como glucosa 6 fosfatasa (G6PC) y fosfoenolpiruvato carboxiquinasa (PCK1) (23). Así que la modulación negativa de la señalización de insulina mediada por AKT que induce el VHC tiene como resultado la regulación positiva de G6PC y PCK1, lo que desencadena en un aumento en la producción de glucosa hepática; estos eventos provocados por el virus se revirtieron tras el tratamiento con sofosbuvir (Figura 4A y 4B).
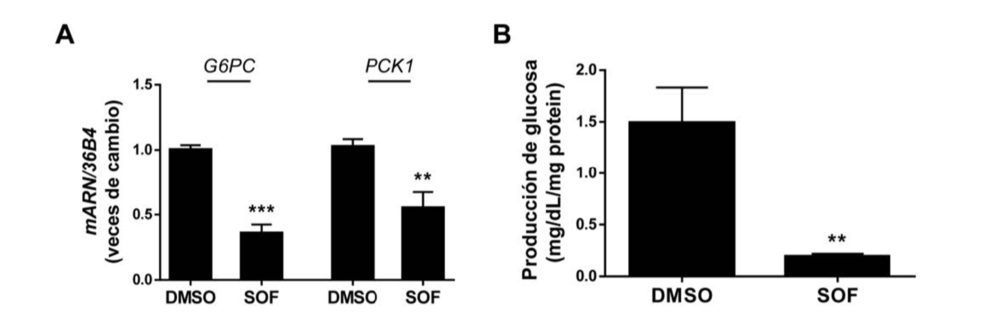
Figura 4. El tratamiento con sofosbuvir reduce la gluconeogénesis exacerbada por el VHC. Análisis por RT-qPCR de los niveles de ARNm de G6PC y PCK1 (A) y de la producción de glucosa (B) de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o tratamiento con SOF. Los datos se presentan como media ± SEM. **p<0.01 y ***p<0.005, SOF vs. DMSO (n=3 experimentos independientes realizados por duplicado).
Por otro lado, la AKT induce la fosforilación de GSK3, inactivando su actividad quinasa, lo que posteriormente conduce a una activación de la glucógeno sintasa (GS), favoreciendo así la síntesis de glucógeno (24). En este sentido, la síntesis de glucógeno se vio impedida en las células con VHC y, tras el tratamiento con sofosbuvir, los niveles de glucógeno intracelular se incrementaron significativamente en comparación con las células tratadas con DMSO, como consecuencia directa de la recuperación de la señalización de AKT (Figura 5).
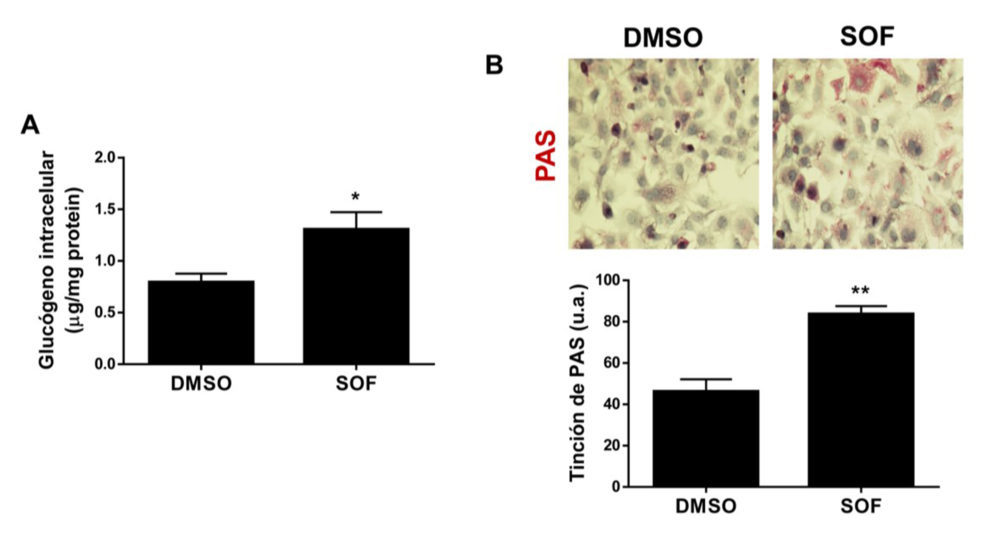
Figura 5. El tratamiento con sofosbuvir favorece la síntesis de glucógeno en los hepatocitos con VHC. Análisis del contenido de glucógeno intracelular por colorimetría (A) y por tinción de PAS (B) de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o tratamiento con sofosbuvir (SOF). Los datos se presentan como media ± SEM. *p<0.05 y **p<0.01, SOF vs. DMSO (n=3 experimentos independientes realizados por duplicado).
3.6. El traramiento con sofosbuvir recupera la expresión de proteína de IRS1 en hepatocitos con VHC
Lo siguiente fue dilucidar los mecanismos moleculares subyacentes a los efectos del sofosbuvir sobre la mejora de la respuesta a la insulina en los hepatocitos con VHC. Mientras que los niveles proteicos del receptor de la insulina se mantuvieron sin cambios (Figura 2), los de IRS1 aumentaron en las células tratadas con sofosbuvir (Figura 6A).
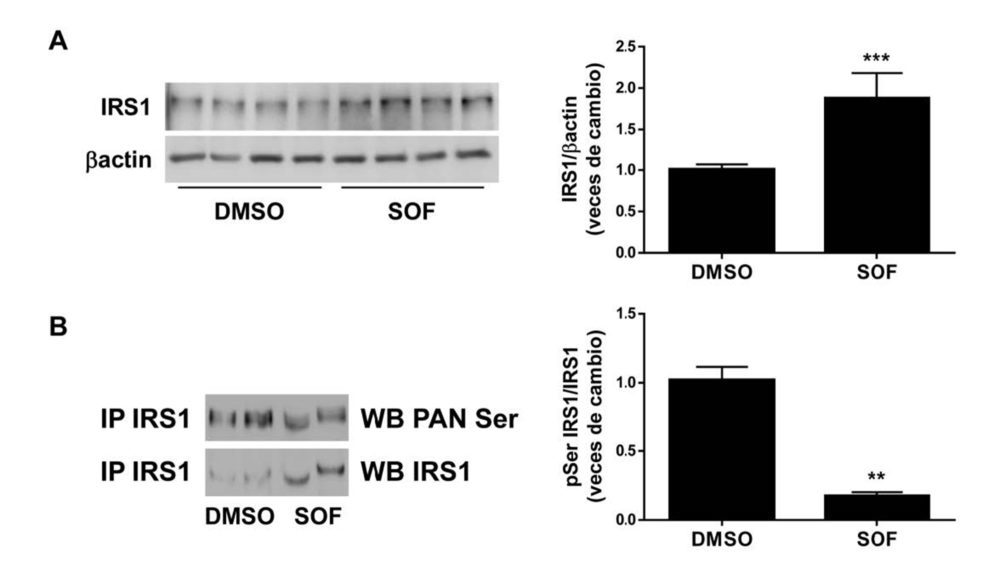
Figura 6. El sofosbuvir induce un aumento del contenido proteico de IRS1 paralelo a una reducción en su fosforilación en residuos serina en hepatocitos con VHC. Análisis por Western blot de la expresión de IRS1 (A) y de su fosforilación en residuos serina (B) de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o sofosbuvir (SOF). β actina se utilizó como control de carga. Los datos se presentan como media ± SEM. **p<0,01 y ***p<0,005, SOF vs. DMSO (n=3-4 experimentos independientes realizados por duplicado/triplicado).
Puesto que es un hecho consolidado que el VHC altera la señalización de la insulina a través de la fosforilación en residuos serina de IRS1 (25), se analizaron los niveles de esta fosforilación, encontrando una atenuación de la misma en las células con VHC tratadas con sofosbuvir en comparación con las tratadas con DMSO (Figura 6B), lo que explicaría la restitución de la acción de la insulina, sobre todo a nivel de AKT, tras el tratamiento con el antiviral.
Finalmente, evaluamos la posibilidad de que los cambios en la expresión de moduladores negativos de la señalización de la insulina también pudieran explicar la sensibilización a la insulina inducida por sofosbuvir en los hepatocitos con VHC. De acuerdo con esta hipótesis, medimos la expresión de PTP1B, PTEN y PP2A en los hepatocitos que expresan el VHC tratados con DMSO o sofosbuvir. Como se muestra en la Figura 7, los niveles de ARNm de todas estas fosfatasas (PTPN1, PTEN y PPP2CA, respectivamente) permanecieron inalterados.
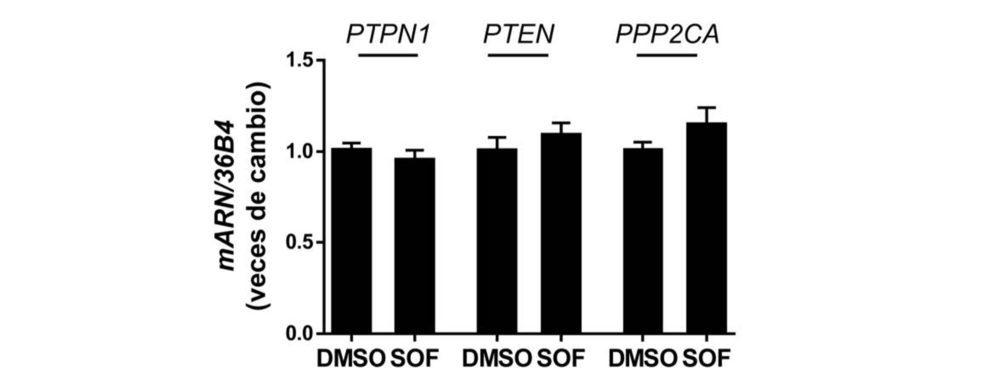
Figura 7. El sofosbuvir no altera la expresión de moduladores negativos de la señalización de insulina en células con VHC. Análisis por RT-qPCR de los niveles de ARNm de PTPN1, PTEN y PPP2CA de las células Huh7 con VHC que fueron tratadas durante 9 días con vehículo (DMSO) o tratamiento con SOF. Los datos se presentan como media ± SEM (n=3 experimentos independientes realizados por duplicado).
4. DISCUSIÓN
El presente estudio traslacional ha demostrado que el tratamiento con sofosbuvir mejora la resistencia sistémica a la insulina en pacientes con VHC y, por primera vez, se ha revelado qué mecanismos moleculares están implicados en los efectos de este antiviral sobre la respuesta insulínica alterada inducida por el VHC en los hepatocitos.
Una de las principales preocupaciones en el campo de la atención sanitaria es el gran número de personas afectadas por la infección crónica por el VHC y sus complicaciones asociadas. En este sentido, la infección crónica por el VHC está asociada con la aparición de resistencia a la insulina, y se ha descrito un aumento en el riesgo de padecer diabetes tipo 2 en pacientes infectados por el VHC (4, 5). De hecho, el desarrollo de estas complicaciones metabólicas relacionadas con el VHC afecta la calidad de la salud de los pacientes y supone una carga importante para la atención médica. En los últimos años, la llegada de terapias anti-VHC basadas en antivirales de acción directa se ha convertido en un avance revolucionario en el tratamiento de la infección por VHC. La infección crónica por VHC parece estar disminuyendo con la llegada de estos nuevos tratamientos. Este hecho se manifiesta en una menor carga de pacientes con cirrosis e insuficiencia hepática crónica entre aquellos añadidos a lista de espera y los nuevos receptores de trasplantes.
Los resultados obtenidos en el estudio clínico mostraron que los pacientes con VHC resistentes a la insulina tratados con regímenes terapéuticos basados en sofosbuvir fueron respondedores al tratamiento, independientemente del genotipo o del grado de fibrosis hepática. Como era de esperar, se detectó una disminución significativa de la concentración de marcadores séricos de daño hepático -AST, ALT, GGT y bilirrubina- en los pacientes al final del tratamiento que se mantuvo un año después, en concordancia con la consecución de una respuesta viral sostenida, en paralelo a una reducción del índice predictivo de fibrosis hepática FIB4 a lo largo del estudio. Además, se encontró que los tratamientos basados en sofosbuvir parecen mejorar la homeostasis glucídica de los pacientes con VHC. En particular, se observó una reducción significativa del índice HOMA post-tratamiento, que se mantuvo un año después. Estos hallazgos concuerdan con datos previos que mostraron que los regímenes de antivirales de acción directa libres de interferón mejoran la resistencia sistémica a la insulina y los niveles de glucosa en sangre en pacientes que eliminaron el VHC (21, 26-31). Con respecto al hígado, sólo un estudio ha demostrado que la erradicación viral conseguida con estos nuevos tratamientos mejora la sensibilidad a la insulina hepática en pacientes con VHC que se sometieron a un clamp euglucémico-hiperinsulinémico (32).
A nivel molecular, este estudio también ha demostrado que el sofosbuvir mejora la resistencia a la insulina hepática inducida por la infección por el VHC. En este sentido, es bien sabido que el VHC interfiere en los primeros pasos de la cascada de señalización de la insulina, específicamente mediante la reducción de la expresión de las proteínas IRS, IRS1 “and” IRS2 (10, 11). La inactivación de estas proteínas “embarcadero” por diferentes mecanismos, como la degradación mediada por el proteasoma, se ha asociado en gran medida con la resistencia a la insulina (33). Es importante destacar que en este estudio el VHC provocó una reducción del contenido de proteínan de IRS1 en los hepatocitos, lo cual concuerda con estudios previos realizados en diferentes sistemas celulares para el estudio del VHC (12, 34), y el tratamiento con sofosbuvir fue capaz de equilibrar los bajos niveles de esta proteína IRS1 en células con VHC .
De hecho, se observó que los niveles de fosforilación en residuos serina de IRS1 disminuyeron en las células con VHC tratadas con sofosbuvir en comparación con las células con VHC no tratadas, lo que podría explicar la recuperación del contenido de proteína total de IRS1 después del tratamiento con este antiviral. En este sentido, está bien establecido que la proteína VHC-core induce la fosforilación en residuos serina de IRS1 bloqueando la fosforilación en tirosina e induciendo la degradación de IRS1 por el proteasoma (10, 14, 34). Puesto que IRS1 es una molécula crítica involucrada en la transducción de la señal de insulina desde el receptor de la insulina, su degradación altera la activación de AKT, lo que conduce a la resistencia a la insulina. En este estudio, se encontró que el tratamiento con sofosbuvir aumentó considerablemente la fosforilación de AKT en respuesta a la insulina en hepatocitos con VHC en comparación con la condición de DMSO, revirtiendo el estado de resistencia a la insulina y, en consecuencia, la expresión elevada de genes gluconeogénicos, el aumento de la producción de glucosa y la deficiencia en la síntesis de glucógeno, todos ellos eventos inducidos por el VHC. Otros estudios también han demostrado que el tratamiento de células infectadas con VHC con interferón modifica parcialmente los marcadores indirectos de resistencia a la insulina, como la inducción de la gluconeogénesis (35, 36). Sin embargo, este es el primer estudio que muestra una reversión en el bloqueo de la vía de señalización IR / IRS1 / AKT en respuesta a la insulina en células con VHC curadas, que se refleja en una disminución de la gluconeogénesis y una recuperación de la síntesis de glucógeno, otorgando al sofosbuvir un papel clave en la regulación de esta vía y explicando sus efectos metabólicos sobre la señalización de la insulina.
Dado que se ha descrito que el VHC también puede modificar la expresión de moduladores negativos de la señalización de la insulina como son las fosfatasas PTP1B, PTEN y PP2A, se evaluó la posibilidad de que cambios en su expresión pudieran explicar la sensibilización a la insulina inducida por sofosbuvir en hepatocitos que expresan el VHC; sin embargo, el antiviral no moduló sus niveles de expresión génica en las células con VHC.
Por otro lado, el VHC es también capaz alterar el metabolismo de los lípidos, ya que varios estudios han documentado hipocolesterolemia e hipolipidemia, particularmente niveles más bajos de colesterol total, LDL y ApoB, el principal componente proteico de LDL y VLDL, en pacientes con infección crónica por VHC (37-39). Aunque nuestro estudio no fue diseñado para aclarar los efectos del VHC sobre el metabolismo de lípidos, ha demostrado que la eliminación exitosa del VHC resultó en un restablecimiento de los niveles circulantes de colesterol total, LDL y ApoB, de acuerdo con las observaciones en pacientes que lograron una respuesta viral sostenida tras el tratamiento con antivirales de acción directa o con interferón (40, 41).
5. CONCLUSIONES
Tomados en conjunto, los resultados derivados de este estudio traslacional indican que el sofosbuvir mejora la respuesta a la insulina alterada por la infección del VHC. De hecho, este estudio es especialmente relevante ya que es el primero que revela los mecanismos moleculares implicados en la sensibilización a la insulina hepática inducida por el tratamiento con sofosbuvir, que implica la recuperación de los niveles de proteína IRS1 como evento clave de sus efectos.
Conflicto de intereses
Los autores declaran que NO tienen conflicto de intereses y agradecen el apoyo financiero de la subvención CP14/00181 del Instituto de Salud Carlos III con financiada con fondos FEDER.
6. REFERENCIAS
- WHO. Guidelines for the screening, care and treatment of persons with chronic hepatitis C infection. Guidelines for the Care and Treatment of Persons Diagnosed with Chronic Hepatitis C Virus Infection. WHO Guidelines Approved by the Guidelines Review Committee. Geneva2018.
- Goldberg D, Ditah IC, Saeian K, Lalehzari M, Aronsohn A, Gorospe EC, et al. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology. 2017;152(5):1090-9 e1.
- Desbois AC, Cacoub P. Diabetes mellitus, insulin resistance and hepatitis C virus infection: A contemporary review. World journal of gastroenterology. 2017;23(9):1697-711.
- Gastaldi G, Goossens N, Clement S, Negro F. Current level of evidence on causal association between hepatitis C virus and type 2 diabetes: A review. Journal of advanced research. 2017;8(2):149-59.
- Wang CC, Cheng PN, Kao JH. Systematic review: chronic viral hepatitis and metabolic derangement. Alimentary pharmacology & therapeutics. 2020;51(2):216-30.
- White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. Journal of hepatology. 2008;49(5):831-44.
- Cacoub P, Carrat F, Bedossa P, Lambert J, Penaranda G, Pol S, et al. Insulin resistance impairs sustained virological response rate to pegylated interferon plus ribavirin in HIV-hepatitis C virus-coinfected patients: HOMAVIC-ANRS HC02 Study. Antiviral therapy. 2009;14(6):839-45.
- El-Zayadi AR, Anis M. Hepatitis C virus induced insulin resistance impairs response to anti viral therapy. World journal of gastroenterology. 2012;18(3):212-24.
- Romero-Gomez M, Del Mar Viloria M, Andrade RJ, Salmeron J, Diago M, Fernandez-Rodriguez CM, et al. Insulin resistance impairs sustained response rate to peginterferon plus ribavirin in chronic hepatitis C patients. Gastroenterology. 2005;128(3):636-41.
- Garcia-Monzon C, Lo Iacono O, Mayoral R, Gonzalez-Rodriguez A, Miquilena-Colina ME, Lozano-Rodriguez T, et al. Hepatic insulin resistance is associated with increased apoptosis and fibrogenesis in nonalcoholic steatohepatitis and chronic hepatitis C. Journal of hepatology. 2011;54(1):142-52.
- Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. The American journal of pathology. 2004;165(5):1499-508.
- Pazienza V, Clement S, Pugnale P, Conzelman S, Foti M, Mangia A, et al. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45(5):1164-71.
- Ahmed QL, Manzoor S, Tariq M, Khalid M, Ashraf W, Parvaiz F, et al. Hepatitis C virus infection in vitro triggers endoplasmic reticulum stress and downregulates insulin receptor substrates 1 and 2 through upregulation of cytokine signaling suppressor 3. Acta virologica. 2014;58(3):238-44.
- Banerjee S, Saito K, Ait-Goughoulte M, Meyer K, Ray RB, Ray R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. Journal of virology. 2008;82(6):2606-12.
- Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126(1):263-77.
- Pecoraro V, Banzi R, Cariani E, Chester J, Villa E, D’Amico R, et al. New Direct-Acting Antivirals for the Treatment of Patients With Hepatitis C Virus Infection: A Systematic Review of Randomized Controlled Trials. Journal of clinical and experimental hepatology. 2019;9(4):522-38.
- Shah N, Pierce T, Kowdley KV. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert opinion on investigational drugs. 2013;22(9):1107-21.
- Umar M, Akhter TS. New Direct Acting Antiviral Agents for the Treatment of Hepatitis C: 2016 and Beyond. Journal of the College of Physicians and Surgeons–Pakistan : JCPSP. 2016;26(10):843-50.
- Herbst DA, Jr., Reddy KR. Sofosbuvir, a nucleotide polymerase inhibitor, for the treatment of chronic hepatitis C virus infection. Expert opinion on investigational drugs. 2013;22(4):527-36.
- Rodriguez-Torres M, Lawitz E, Kowdley KV, Nelson DR, Dejesus E, McHutchison JG, et al. Sofosbuvir (GS-7977) plus peginterferon/ribavirin in treatment-naive patients with HCV genotype 1: a randomized, 28-day, dose-ranging trial. Journal of hepatology. 2013;58(4):663-8.
- Ribaldone DG, Sacco M, Saracco GM. The Effect of Viral Clearance Achieved by Direct-Acting Antiviral Agents on Hepatitis C Virus Positive Patients with Type 2 Diabetes Mellitus: A Word of Caution after the Initial Enthusiasm. Journal of clinical medicine. 2020;9(2).
- Benedicto I, Molina-Jimenez F, Barreiro O, Maldonado-Rodriguez A, Prieto J, Moreno-Otero R, et al. Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology. 2008;48(4):1044-53.
- Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. The Journal of biological chemistry. 2006;281(15):10105-17.
- Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes research and clinical practice. 2007;77 Suppl 1:S49-57.
- Douglas MW, George J. Molecular mechanisms of insulin resistance in chronic hepatitis C. World journal of gastroenterology. 2009;15(35):4356-64.
- Fabrizio C, Procopio A, Scudeller L, Dell’Acqua R, Bruno G, Milano E, et al. HCV and diabetes: towards a ‘sustained’ glycaemic improvement after treatment with DAAs? Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2017;23(5):342-3.
- Gastaldi G, Gomes D, Schneiter P, Montet X, Tappy L, Clement S, et al. Treatment with direct-acting antivirals improves peripheral insulin sensitivity in non-diabetic, lean chronic hepatitis C patients. PloS one. 2019;14(6):e0217751.
- Huang JF, Huang CF, Yeh ML, Dai CY, Hsieh MH, Yang JF, et al. The outcomes of glucose abnormalities in chronic hepatitis C patients receiving interferon-free direct antiviral agents. The Kaohsiung journal of medical sciences. 2017;33(11):567-71.
- Hum J, Jou JH, Green PK, Berry K, Lundblad J, Hettinger BD, et al. Improvement in Glycemic Control of Type 2 Diabetes After Successful Treatment of Hepatitis C Virus. Diabetes care. 2017;40(9):1173-80.
- Pavone P, Tieghi T, d’Ettorre G, Lichtner M, Marocco R, Mezzaroma I, et al. Rapid decline of fasting glucose in HCV diabetic patients treated with direct-acting antiviral agents. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2016;22(5):462 e1-3.
- Ciancio A, Bosio R, Bo S, Pellegrini M, Sacco M, Vogliotti E, et al. Significant improvement of glycemic control in diabetic patients with HCV infection responding to direct-acting antiviral agents. Journal of medical virology. 2018;90(2):320-7.
- Lim TR, Hazlehurst JM, Oprescu AI, Armstrong MJ, Abdullah SF, Davies NP, et al. Hepatitis C virus infection is associated with hepatic and adipose tissue insulin resistance that improves after viral cure. Clinical endocrinology. 2019;90(3):440-8.
- Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012;55(10):2565-82.
- Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. Journal of virology. 2012;86(11):6315-22.
- Deng L, Shoji I, Ogawa W, Kaneda S, Soga T, Jiang DP, et al. Hepatitis C virus infection promotes hepatic gluconeogenesis through an NS5A-mediated, FoxO1-dependent pathway. Journal of virology. 2011;85(17):8556-68.
- Shlomai A, Rechtman MM, Burdelova EO, Zilberberg A, Hoffman S, Solar I, et al. The metabolic regulator PGC-1alpha links hepatitis C virus infection to hepatic insulin resistance. Journal of hepatology. 2012;57(4):867-73.
- Jung HJ, Kim YS, Kim SG, Lee YN, Jeong SW, Jang JY, et al. The impact of pegylated interferon and ribavirin combination treatment on lipid metabolism and insulin resistance in chronic hepatitis C patients. Clinical and molecular hepatology. 2014;20(1):38-46.
- Lacerda GS, Medeiros T, Rosario NFD, Peralta RHS, Cabral-Castro MJ, Esberard EBC, et al. Exploring lipid and apolipoprotein levels in chronic hepatitis C patients according to their response to antiviral treatment. Clinical biochemistry. 2018;60:17-23.
- Sun HY, Cheng PN, Tseng CY, Tsai WJ, Chiu YC, Young KC. Favouring modulation of circulating lipoproteins and lipid loading capacity by direct antiviral agents grazoprevir/elbasvir or ledipasvir/sofosbuvir treatment against chronic HCV infection. Gut. 2018;67(7):1342-50.
- Corey KE, Kane E, Munroe C, Barlow LL, Zheng H, Chung RT. Hepatitis C virus infection and its clearance alter circulating lipids: implications for long-term follow-up. Hepatology. 2009;50(4):1030-7.
- El Sagheer G, Soliman E, Ahmad A, Hamdy L. Study of changes in lipid profile and insulin resistance in Egyptian patients with chronic hepatitis C genotype 4 in the era of DAAs. The Libyan journal of medicine. 2018;13(1):1435124.
