1. INTRODUCCIÓN
Los últimos treinta años se han caracterizado por una mejora de los estándares de calidad de vida en los países industrializados. En determinados aspectos, ello tiene consecuencias negativas en el medio ambiente. De ello surge la motivación y la búsqueda de metodologías respetuosas con el medioambiente (“environmentally-friendly”) (1,2). En el campo de la química analítica, se han ido introduciendo metodologías, procedimientos y técnicas cuya finalidad es reducir el impacto medioambiental que de los distintos métodos analíticos se derivan (3). Desde el control de calidad de materias primas y productos terminados en la industria farmacéutica, las determinaciones analíticas en laboratorios de control oficiales (Agencias Estatales y Europeas, de control de medicamentos, alimentos, contaminantes ambientales…) hasta en la rutina de los laboratorios de análisis clínicos, se ha intentado buscar métodos calificados como “verdes” o “sostenibles”, que tiendan a disminuir los residuos generados en el proceso analítico. En los últimos años distintas publicaciones científicas de revisión en el campo de la química analítica sostenible (4,5,6) analizan cómo las metodologías que, desarrolladas para un formato que podríamos denominar convencional, se han transformado en metodologías sostenibles. Así, una gran parte de los procedimientos de pre-tratamiento analítico se ha transformado en sus equivalentes “micro” o miniaturizados (7,8). En los últimos años se han ido introduciendo diferentes formas de evaluar la sostenibilidad de los métodos analíticos (9) como: NEMI (National Environmental Method Index), GAPI (Green Analytical Procedure Index), AMGS (Analytical Method Greenness Score), y AGREE (Analytical greenness), entre otros. La reducción del número y del tamaño de la muestra, la elección de las técnicas de muestreo directo tratando de evitar aquellas que impliquen un pre-tratamiento, la miniaturización en las medidas, las determinaciones analíticas “in situ”, así como la utilización de disolventes y reactivos “renovables” y “reutilizables”, la disminución de residuos generados, o bien, el descenso del consumo de energía, son algunas de las variables a considerar para que una técnica analítica pueda ser calificada como sostenible.
Pocas técnicas instrumentales han alcanzado tanto grado de madurez y aplicación para los análisis de rutina como la cromatografía de líquidos de alta eficacia (HPLC). Ello se debe en parte a la gran diversidad de las aplicaciones de esta técnica en aspectos tan significativos como el análisis y purificación de fármacos, la determinación de parámetros de interés en clínica y toxicología o el análisis y la cuantificación de numerosos compuestos de interés biológico, nutricional y medioambiental. Hoy en día, la cromatografía de líquidos de alta eficacia (HPLC) es la técnica analítica más empleada rutinariamente en la industria farmacéutica. Aunque la cantidad de residuos de disolventes orgánicos generada por un equipo de HPLC utilizado a pleno rendimiento no es demasiado grande (aprox. 0,5-1 L/día), el efecto acumulativo de cientos de miles de equipos de HPLC de todo el mundo hace que el volumen de residuos generados a nivel global sea preocupante (10). Por ello, cada vez son mayores los esfuerzos por hacer de la cromatografía de líquidos una técnica analítica “verde” (11) dentro de lo que se denomina química sostenible. En el caso de la cromatografía, estos esfuerzos se basan en reducir el volumen de los disolventes de desecho, remplazar los disolventes no respetuosos con el medio ambiente por otros que sí lo son (agua y alcoholes (12)), y reciclar (13) los disolventes a través de procesos respetuosos con el medio ambiente. Por todo ello, los profesionales que trabajamos en cromatografía y sabemos de su gran eficacia y versatilidad como técnica analítica debemos preguntarnos: ¿Cómo podemos convertir la cromatografía de líquidos en una metodología más respetuosa con el medio ambiente? La respuesta más sencilla a esta pregunta se basa en el diseño y desarrollo de metodologías en las que el volumen de los residuos generados sea mínimo y, además, éstos sean respetuosos con el entorno. Si nuestra muestra a analizar contiene compuestos contaminantes, se tratará de tomar medidas para evitar que contamine en gran escala a los residuos generados en el análisis. Esta idea se esquematiza en la Figura 1.

Figura 1. Tipos de residuos generados en cromatografía en función de la naturaleza de la muestra y de los disolventes empleados en la fase móvil. El color rojo simboliza la situación desfavorable, y el color verde indica un resultado favorable desde el punto de vista medioambiental.
“A priori”, en cromatografía de líquidos, la forma más sencilla de reducir el consumo de disolventes y residuos, es reducir el tiempo de análisis, lo que a su vez conlleva un importante ahorro energético. En equipos convencionales de HPLC se puede lograr reducir el tiempo de análisis y por tanto el consumo energético:
- Reduciendo la longitud de la columna (por ejemplo 50 mm frente a 150 mm convencionales)
- Reduciendo el diámetro interno de la columna (por ejemplo 2,1 mm frente a 4,6 mm convencionales)(10, 14).
- Reduciendo el tamaño de las partículas que forman la fase estacionaria (por ejemplo 3 μm frente a 5 μm)
La modalidad denominada UHPLC (“Ultra-High Pressure Liquid Chromatography”), proporciona separaciones rápidas y eficaces con un ahorro de hasta un 80% en el consumo de disolventes, dado que el tiempo de análisis puede reducirse a la décima parte en comparación con los sistemas convencionales de HPLC. De esta manera son posibles separaciones rápidas sin sacrificar la resolución, gracias a la utilización de columnas empaquetadas con partículas de diámetro inferior a 2 μm. Sin embargo, la reducción del tamaño de partícula conlleva el aumento en la presión en la columna así como el calentamiento por fricción debido a las presiones elevadas. Además, se hace necesario el empleo de bombas capaces de trabajar a presiones más elevadas (max. 1300-1500 bares, UHPLC) que las utilizadas en separaciones convencionales (máx. 400-600 bares, HPLC). Así, mientras que columnas empaquetadas con partículas totalmente porosas con tamaños de partícula de 3-5 μm dan separaciones robustas independientemente del equipo instrumental de HPLC empleado, sin embargo, la elección del equipo instrumental para UHPLC trabajando con partículas totalmente porosas con diámetro inferior a 2 mm resulta determinante (15). Por todo ello hay que considerar: a) los volúmenes extra columna, e intentar reducirlos o minimizarlos, b) las características técnicas de las bombas y los inyectores, y c) la importancia de los hornos para columnas en la eficacia y selectividad de las separaciones. La Figura 2 muestra una comparación de las características de las columnas rellenas con partículas superficialmente porosas.
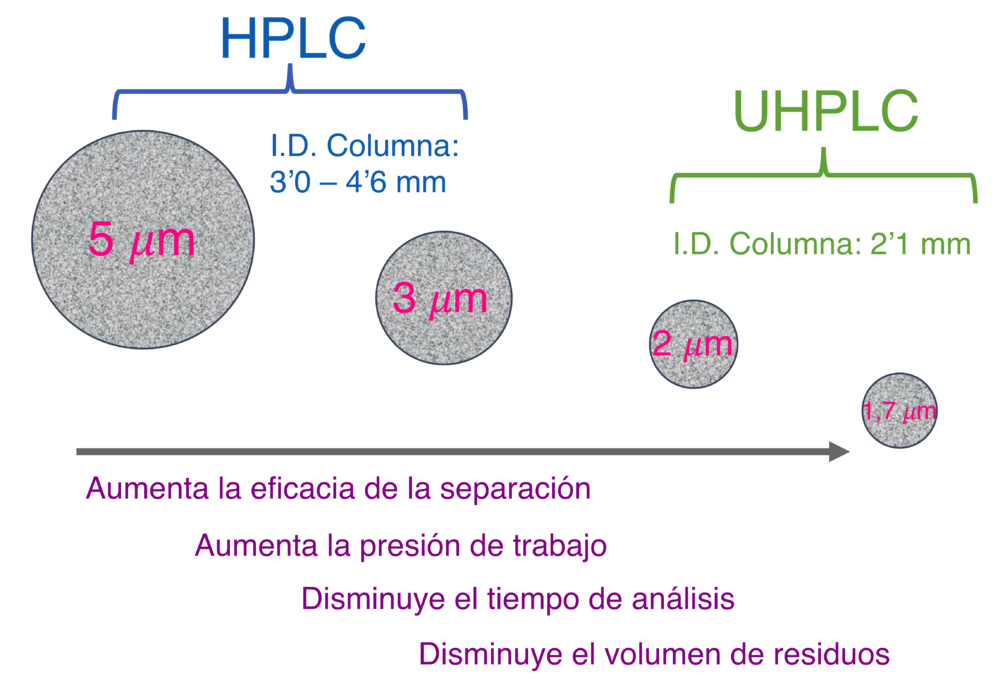
Figura 2. Comparación de las características de las separaciones en cromatografía de líquidos (HPLC y UHPLC) según el tamaño de las partículas de sílice totalmente porosa empleado en las columnas analíticas.
Una alternativa respetuosa con el medio ambiente y cuyas condiciones de trabajo no implican presiones tan elevadas como el UHPLC, es la utilización de columnas empaquetadas con partículas de sílice superficialmente porosa, tipo “core-shell”. Estas partículas están constituidas por un núcleo duro de sílice fundida rodeado de una serie de capas de sílice porosa que les confieren características idóneas desde el punto de vista cromatográfico.
Las fases estacionarias de núcleo de sílice fundido (16,17, 18) se conocían desde mediados de los años 90 del siglo pasado, pero se empezaron a comercializar en 2006. Están disponibles en una variedad química considerable de rellenos con un gran potencial en la resolución de problemas analíticos (19), y, además, presentan significativas ventajas con respecto a las partículas totalmente porosas; así, partículas superficialmente porosas de 5 μm poseen la eficacia de las separaciones comparable a la de partículas totalmente porosas de 2 μm permitiendo trabajar a presiones bajas similares a las presiones de trabajo en HPLC pero con eficacias semejantes a las de UHPLC, lo que conlleva una disminución en el tiempo de análisis de 3-10 veces y un ahorro en el consumo de energía y disolventes que varía entre el 50-80 % según las condiciones de separación. Además, a diferencia de las fases estacionarias de partículas sub-2 μm totalmente porosas utilizadas en UHPLC, las fases estacionarias de partículas superficialmente porosas pueden utilizarse en equipos de HPLC convencionales y sólo requieren adaptaciones sencillas como la reducción en el diámetro interno de las conexiones para reducir el volumen muerto, ya que volúmenes muertos significativos contribuyen a la disminución de la eficacia. Presentan la desventaja de que su capacidad de carga es sensiblemente menor por lo que hay que inyectar disoluciones suficientemente diluidas. En la Figura 3 se muestran dos cromatogramas correspondientes a la separación de los dos mismos compuestos en una fase superficialmente porosa y totalmente porosa. Se puede apreciar que la retención de los analitos en las fases “core-shell” es significativamente menor como se ilustra con las flechas, y ello se traduce en menores tiempos de retención con eficacias mayores.
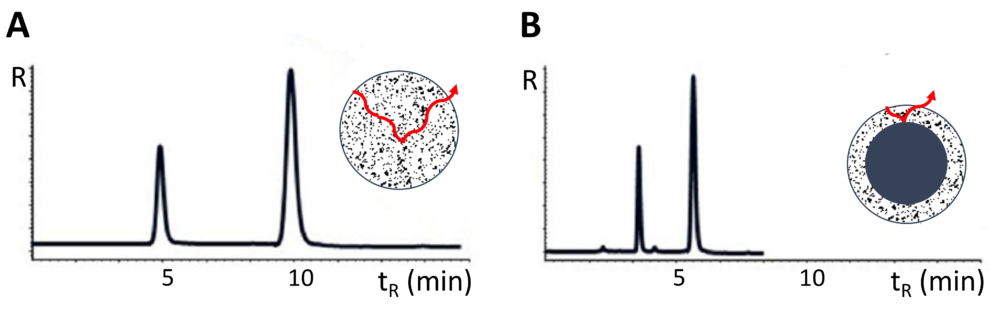
Figura 3. Cromatogramas obtenidos en las separaciones de dos analitos en fases estacionarias de partículas de sílice totalmente porosa (A) y partículas de sílice superficialmente porosa, “core-shell” (B). R: respuesta del detector, tR: tiempo de retención
Asimismo, se pueden lograr separaciones sostenibles utilizando fases estacionarias convencionales y modificando las fases móviles mediante la utilización de disolventes respetuosos con el medio ambiente, así como el empleo de agua a temperaturas elevadas y en condiciones de temperatura sub-crítica (20). Estas alternativas han sido revisadas detalladamente (4). La sustitución de acetonitrilo por etanol y otros disolventes eco-compatibles ha sido exhaustivamente revisada y aplicada al análisis de fármacos (21). Por ejemplo, la separación isocrática de rosuvastatina utilizando fases estacionarias de octilsilano y como fase móvil etanol:metanol:acetato de etilo (6:3:1) altamente eco-compatible proporciona una buena sensibilidad y exactitud para la determinación de este principio activo en formas farmacéuticas (22).
El empleo de aditivos en las fases móviles fundamentalmente tensioactivos (23, 24), ciclodextrinas (25, 26) o líquidos iónicos (27, 28) permite la utilización de fases móviles mayoritariamente acuosas con columnas y equipos de HPLC convencionales, lo que convierte las separaciones en metodologías sostenibles.
Los líquidos iónicos (ILs) lo constituyen un amplio y complejo grupo de compuestos, sales líquidas a temperatura inferior a 100ºC. La singulares propiedades que poseen se deben a que son compuestos completamente iónicos semejantes a las sales clásicas como el NaCl, por ello difieren significativamente de los líquidos moleculares (disolventes en general). Tienen muy baja volatilidad, buena estabilidad térmica, buena conductividad electrolítica, amplio intervalo de viscosidades, buena miscibilidad y además no son inflamables (29). Están constituidos por un catión orgánico (alquilpiridinio, alquilpirrolidino, alquilimidazolinio, alquilfosfonio, alquilamonio) y un contraión que suele ser inorgánico, desde cloruro a tetrafluoroborato. Los ILs poseen un gran potencial de aplicación en las distintas técnicas analíticas; en cromatografía de líquidos, puesto que son miscibles con una gran variedad de disolventes utilizados en HPLC, tanto agua como disolventes orgánicos. Así se ha descrito la significación de la adsorción de estos líquidos iónicos sobre los grupos silanol (30) de las fases estacionarias que no ha sido recubiertos de forma efectiva en el proceso de “end-capping” lo que conlleva a un significativo incremento de la eficacia de la separación. Incrementan la selectividad de las separaciones debido a la interacción con los analitos y generan separaciones más eficaces debido a su adsorción sobre las fases estacionarias. Reducen la retención de los analitos básicos sobre las fases estacionarias al mismo tiempo que favorecen la simetría de los picos e incrementan la resolución de numerosos compuestos (31, 32).
En el presente artículo se describe la separación de antitumorales inhibidores de topoisomerasas utilizando fases estacionarias superficialmente porosas y se compara con la separación en fases estacionarias convencionales utilizando líquidos iónicos como aditivos en las fases móviles y se valoran las ventajas y limitaciones no solamente desde las condiciones analíticas de la separación sino también desde el punto de vista de la sostenibilidad.
2. MATERIALES Y MÉTODOS
2.1. Instrumentación y reactivos
Para la verificación de la concentración de las disoluciones patrón de los antitumorales a separar se ha empleado un espectrofotómetro de absorción UV-Vis de doble haz Kontron Uvikon 810P y un espectrofotómetro de absorción UV-VIS Cary 60 de Agilent controlado mediante el software Cary WinUV8, siempre se han utilizando cubetas de cuarzo de 1 cm de paso óptico. Se utilizó un pH-metro Crison Micro-pH 2001 calibrado diariamente con disoluciones reguladoras de pH 4,00 y 7,00, para la preparación de las disoluciones tampón empleadas en las separaciones.
En las separaciones cromatográficas se ha utilizado un cromatógrafo de líquidos HPLC de la firma Merck-Hitachi provisto de una bomba de gradiente cuaternario (L-7100), un sistema de termostatización por efecto Peltier para columnas (Horno L-2300), el cual se programó para mantener la temperatura a 25 ºC para todos las separaciones. El equipo posee un detector de fluorescencia (L-7485). Como condiciones de detección se utilizó λex= 370 nm y λem= 440 nm ya que a estas longitudes de onda tanto la camptotecina como la luotonina A se excitan y emiten. El control del sistema cromatográfico fue realizado por ordenador a través del software HPLC System Manager, versión 4.1. En los experimentos con columnas de sílice totalmente porosa se emplearon capilares de acero inoxidable de 1000 × 0,2 mm (longitud × diámetro interno) del inyector a la columna y de PTFE de 450 × 0,2 mm (longitud × diámetro interno) de la columna al detector. En estas condiciones experimentales, se empleó un inyector Rheodyne 7725i con bucle de 20 μL. En el caso de las columnas de sílice superficialmente porosa se utilizaron capilares Viper (Dionex) de 750 × 0,13 mm (longitud × diámetro interno) y de 350 × 0,13 mm (longitud × diámetro interno) como conexiones del inyector a la columna y de la columna al detector respectivamente, con el fin de reducir el volumen muerto y minimizar el ensanchamiento de los picos. En estas condiciones experimentales, se empleó un inyector Rheodyne 7725i con bucle de 5 μL con la finalidad de adaptarlo a la capacidad de carga de las columnas de sílice superficialmente porosa. Las columnas empleadas fueron: a) columna analítica para elución en fase inversa Luna C18 (sílice totalmente porosa, 5 μm, 150 x 3 mm; tamaño de partícula, longitud × diámetro interna) de Phenomenex; y b) columna analítica para elución en fase inversa Kinetex C18 (sílice superficialmente porosa, 2,6 μm, 50 x 3 mm; tamaño de partícula, longitud × diámetro interno) de Phenomenex, equipada con una pre-columna de seguridad también C18 de Phenomenex.
Todos los disolventes utilizados fueron calidad espectroscopía o calidad cromatografía. El agua ultrapura se obtuvo gracias al equipo Milli-Q Direct 8 de Millipore. Los antitumorales camptotecina y luotonina A fueron suministrados por Sigma. Los líquidos iónicos cloruro de 1-butil-3-metil-imidazolinio y cloruro de 1-butil-1-metil-pirrolidinio también se adquirieron en Sigma-Merck.
Las fases móviles se filtraron con ayuda de vacío, a través de filtros de membrana de poliamida de tamaño de poro de 0,45 mm y 47 mm de diámetro (GPH, Waters) en el caso de las columnas Luna C18, y filtros de membrana de nylon de tamaño de poro de 0,22 mm y 47 mm de diámetro (Phenomenex) en el caso de las columnas Kinetex C18. En ambos casos seguidamente se aplicó sonicación durante 30 min para facilitar su desgasificación.
2.2 Preparación de patrones, líquidos iónicos y disoluciones tampón
Se prepararon disoluciones patrón de camptotecina y luotonina A, pesando una cantidad apropiada y disolviéndola en dimetilsulfóxido en concentración 1,0 x 10-3 M. Se verificó el valor de la concentración de estas disoluciones mediante la determinación espectrofotométrica de disoluciones 100 veces diluidas en etanol o acetonitrilo. Se determinó exactamente el valor de la concentración a través de la absorbancia medida en los máximos de absorción y teniendo en cuenta los valores de las absortividades molares, para la camptotecina (λmax= 368 nm, ε= 27530) (33) y la luotonina A (λmax= 342 nm, ε= 16218) (34). Estas disoluciones se alicuotaron y se mantuvieron a -20 ºC. Las disoluciones de camptotecina y luotonina A se prepararon independientemente en concentración 1,0 x 10-7 M en las fases móviles, según las condiciones experimentales a estudiar y seguidamente se filtraron a través de filtros de muestras de 45 mm de tamaño de poro. Las disoluciones de camptotecina se mantuvieron en baño de hielo para reducir al mínimo la formación del carboxilato de la camptotecina.
Se prepararon disoluciones acuosas de 1-butil-3-metil-imidazolinio, y cloruro de 1-butil-1-metil-pirrolidinio en concentración 0,5 M (500 mM) y a partir de estas se ensayaron las concentraciones de 10, 20, 30, 40 y 50 mM en tampón acético/acetato de amonio 10 mM (pH = 3,0) como parte acuosa integrante de la fase móvil.
La separaciones se realizaron en elución isocrática, acetonitrilo:agua; 40:60 y 35:65; v:v utilizando un caudal de 0,5 mL/min en el caso de las columnas de sílice totalmente porosa y con una fase móvil acetonitrilo:tampón acetato; 35:65; v:v utilizando un caudal de 0,5 mL/min en las columnas de sílice superficialmente porosa.
Para evaluar la eficacia de la separación se determinó el número de platos teóricos, siguiendo el criterio de la Farmacopea Europea.
3. RESULTADOS Y DISCUSION
Los resultados previos de nuestro grupo de investigación en las separaciones cromatográficas de antitumorales inhibidores de topoisomerasas permiten la separación de camptotecina y un grupo de análogos de luotonina A (14, 34). Además hemos podido demostrar la eficacia en la estabilización de la camptotecina mediante cromatografía de líquidos cuantificando las formas farmacológicamente activas de camptotecina (lactona) y la forma carente de actividad (carboxilato) gracias a la determinación de ambas por RP-HPLC y empleando la luotonina A como patrón interno. En este trabajo hemos puesto de manifiesto el incremento de la actividad antitumoral de ambos productos naturales (camptotecina y luotonina A) mediante la vehiculización de los mismos en forma de complejos de inclusión con ciclodextrinas (35).
En el presente trabajo se describe la separación de camptotecina en columnas de sílice modificada y empaquetadas con partículas de núcleo de sílice superficialmente porosas y se compara su comportamiento cromatográfico con las columnas convencionales de sílice totalmente porosa. Para ello se ha empleado acetonitrilo:agua como fase móvil. En la Figura 4 se muestra la comparación de los cromatogramas obtenidos en la separación de camptotecina según se utilicen fases estacionarias totalmente porosas o superficialmente porosas (“core-shell”).,
Como se aprecia en la Figura 4 al pasar de una fase estacionaria convencional a una fase superficialmente porosa, utilizando análogas condiciones de separación, el tiempo de retención de la camptotecina se reduce entre 3,5 y 4 veces con respecto a las fases estacionarias convencionales y sin que se sacrifique la eficacia de la separación. La reducción del tiempo de análisis significa un considerable ahorro de tiempo y de disolventes orgánicos, logrando unas separaciones cromatográficas sostenibles al economizar en la generación de residuos y en el consumo energético.
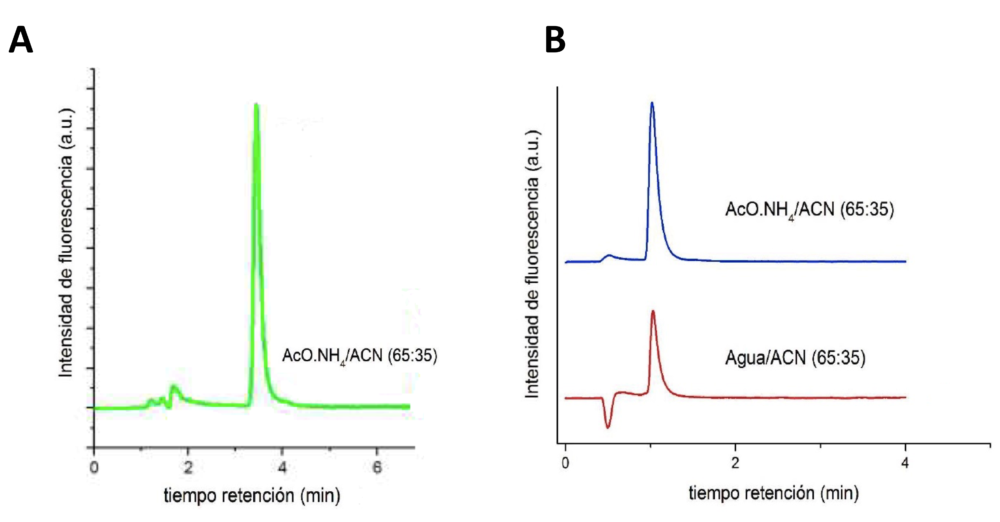
Figura 4. Cromatogramas obtenidos en las separaciones de camptotecina en fases estacionarias C18 de partículas de sílice totalmente porosa (A) y partículas de sílice superficialmente porosa, “core-shell” (B). Caudal 0,5 mL/min. Fase móvil agua o tampón acuoso de acetato amónico 0,15 M (pH 6,5) y acetonitrilo (ACN) en las proporciones indicadas.
Las separaciones de compuestos básicos (por ejemplo, alcaloides) por HPLC continúan presentado ciertas dificultades, como asimetría de los picos cromatográficos, mostrando “colas” debido a la adsorción de los compuestos básicos sobre los grupos silanol libres de las sílices químicamente modificadas. Algunos modificadores de las fases móviles mejoran la eficacia de las separaciones debido a su adsorción sobre las fases estacionarias. Los líquidos iónicos se encuentran en el grupo de compuestos que pueden “bloquear” los grupos silanol. Estas interacciones con la fase estacionaria y también con los analitos en las fases móviles conllevan modificaciones en los tiempos de retención de los solutos(36, 37). En la Figura 5 puede apreciarse como en el caso de los líquidos iónicos utilizados, tanto el derivado imidazolinio como el pirrolidinio producen un aumento en el valor del tiempo de retención de la camptotecina y de la luotonina A. Además a medida que aumenta la concentración de las sales de imidazolinio y prirrolidinio el incremento en el tiempo de retención (tR) es mayor. El aumento en el tiempo de retención de estos solutos en presencia de líquidos iónicos, no tiene los efectos buscados respecto a la sostenibilidad de las fases móviles, ya que implica un incremento en el consumo de fase móvil.
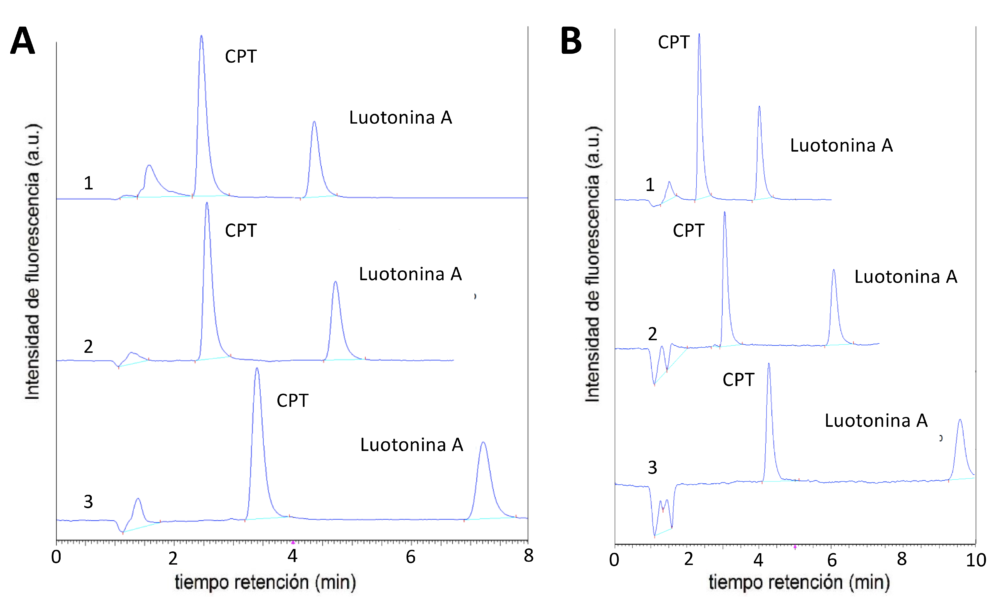
Figura 5. Cromatogramas obtenidos en las separaciones de camptotecina y luotoninina A en fases estacionarias C18 de partículas de sílice totalmente porosa y empleando líquidos iónicos, ILs, como aditivos en las fases móviles (A) cloruro de 1-butil-3-metil-imidazolinio, y (B) cloruro de 1-butil-1-metil-pirrolidinio. Caudal. 0,5 mL/min. Fases móviles: (1) agua : acetonitrilo; 60:40; v:v; (2) tampón acuoso de acetato amónico 10 mM (pH 3,0): acetonitrilo; 60:40; v:v conteniendo IL en concentración 20 mM; (3) tampón acuoso de acetato amónico 10 mM (pH 3,0): acetonitrilo; 60:40; v:v conteniendo IL en concentración 30 mM
La presencia de líquidos iónicos en la fase móvil conlleva una mejora significativa en la eficacia de las separaciones, evaluada como número de platos teóricos (N), así al aumentar la concentración de ILs en la fase móvil el número de platos teóricos se duplica tanto para la camptotecina como para la luotonina A. Teniendo en cuenta esta mejora en la eficacia que se muestra en la Figura 6, se podría incrementar la proporción de disolvente orgánico en la fase móvil para lograr tiempos de retención más cortos, sin embargo, ello supone un incremento en la proporción de disolvente orgánico lo cual nos aleja de nuevo del objetivo de diseñar separaciones cromatográficas sostenibles. Así, estas condiciones de separación con una proporción de disolvente acuoso ligeramente superior al empleado con las fases estacionaras tipo “core-shell”, nos mantiene dentro de los objetivos de sostenibilidad buscados. En las separaciones en las que se emplean ILs el papel más relevante en la separación lo ejerce el catión orgánico. En este caso se demuestra que esto es así, ya que los dos ILs estudiados presentan como anión cloruro, por tanto, las diferencias observadas en el tiempo de retención de la camptotecina y la luotonina A se deben a las interacciones del catión del IL en la fase móvil. Ambos cationes favorecen la retención de los analitos en la fase estacionaria aumentando también el factor de selectividad de la pareja de analitos (Figura 6).
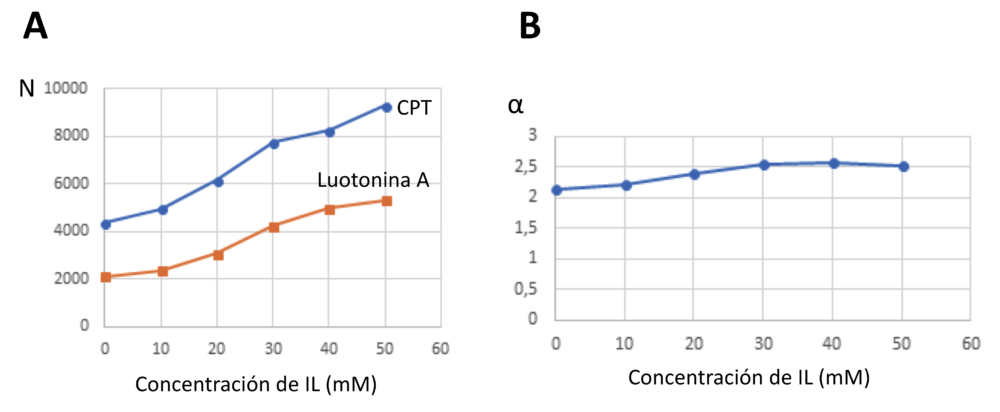
Figura 6. Comportamiento de la eficacia y de la selectividad de las separaciones empleando cloruro de 1-butil-3-metil-imidazolinio (IL) como aditivo en la fase móvil. (A) Variación de la eficacia, evaluada como número de platos teóricos N, en función de la concentración de IL en la fase móvil; y (B) Variación de la selectividad (α) en función de la concentración de IL en la fase móvil
En conclusión de las dos alternativas propuestas para mejorar la sostenibilidad de las separaciones por cromatografía de líquidos, el empleo de fases estacionarias de partículas superficialmente porosas reduce en casi un 50% el tiempo de análisis, el consumo de disolventes y los residuos generados. El empleo de líquidos iónicos como aditivos en las fases móviles mejora la eficacia de la separación pero no logra los objetivos de sostenibilidad de las separaciones. La combinación de fases estacionarias de sílice superficialmente porosa y fases móviles que incluyan líquidos iónicos como aditivos constituye un reto atractivo que podrá aunar una buena eficacia cromatográfica con un reducido consumo de disolventes y energía.
Agradecimientos
Los autores agradecen la financiación al Ministerio de Ciencia e Innovación a través de los proyectos CTQ2009-11312/BQU y RTI2018-097662-B-I00; así como a la Universidad Complutense de Madrid, Grupos de Investigación GR35/10-A-920234. FABL y FMSS agradecen la financiación de las Instituciones a las que pertenecen de sus respectivos países (Chile y Argentina) para su estancia en España.
5. REFERENCIAS
- Bennett GD. “Green Chemistry as an Expression of Environmental Ethics”. En Sharma SK, Mudhoo A. (Eds.) Green Chemistry for Environmental Sustainability, Boca Raton: CRC Press 2011.
- Gałuszka A, Migaszewski Z, Namieśnik J. The 12 Principles of Green Analytical Chemistry and the Significance Mnemonic of Green Analytical Practices. Trac-Trend Anal Chem 2013; 50: 78-84.
- Keith LH, Gron LU, Young JL. Green analytical methodologies. Chem Rev 2007; 107: 2695-2708.
- Olives AI, González-Ruiz V, Martín MA. Sustainable and Eco-Friendly Alternatives for Liquid Chromatographic Analysis. ACS Sustainable Chem. Eng 2017; 5: 5618-5634.
- Koel, M. Do we need Green analytical chemistry? Green Chem 2016; 18, 923−931.
- Armenta S, Garrigues S, de la Guardia M. Green Analytical Chemistry. Trac-Trend Anal Chem 2008; 27: 497-511.
- Mahugo-Santana C, Sosa-Ferrera Z, Torres-Padrón ME, Santana-Rodríguez JJ. Application of new approaches to liquid-phase microextraction for the determination of emerging pollutants. Trac-Trend Anal Chem 2011; 30: 732-748.
- Tobiszewski M, Mechlinska A, Zygmunt B, Namiesnik J. Green analytical chemistry in sample preparation for determination of trace organic pollutants. Trac-Trend Anal Chem 2009; 28: 943-951.
- Kannaiah KP, Sugumaran A, Chanduluru HK, Rathinam S. Environmental impact of greenness assessment tools in liquid chromatography–A review. Microchem J 2021;170: 106685- 106703.
- Welch CJ, Brkovic T, Schafer W, Gong X. Performance to burn? Re-evaluating the choice of acetonitrile as the platform solvent for analytical HPLC. Green Chem 2009; 11: 1232-1238.
- Welch CJ, Wu N, Biba M, Hartman R, Brkovic T, Gong X, Helmy R, Schafer W, Cuff J, Pirzada Z, Zhou L. Greening analytical chromatography. Trac-Trend Anal Chem 2010; 29: 667-680.
- Kerton FM. Alternative Solvents for Green Chemistry. Cambridge: RSC 2009.
- Garrigues S, Armenta S, de la Guardia M. Green strategies for decontamination of analytical wastes. Trac-Trend Anal Chem 2010; 29: 592-601.
- González-Ruiz V, Olives AI, Martín MA. Challenging core-shell stationary phases with the separation of closely related anti-cancer compounds: performance studies and application to drug quantitation in cell cultures with multi-well plate clean-up. J Chromatogr A 2014; 1364: 83-95.
- De Vos J, Broeckhoven K, Eeltink S. Advances in Ultrahigh-Pressure Liquid Chromatography Technology and System Design. Anal Chem 2016; 88: 262-278.
- González-Ruiz V, Olives AI, Martín MA. Core-shell particles lead the way to renewing high-performance liquid chromatography. Trac-Trend Anal Chem 2015; 64: 17-28.
- Tanaka N. Core−Shell, Ultrasmall Particles, Monoliths, and Other Support Materials in High-Performance Liquid Chromatography. Anal Chem 2016; 88: 279-298.
- Gumustas M, Zalewski P, Ozkan SA, Uslu B. The History of the Core–Shell Particles and Applications in Active Pharmaceutical Ingredients Via Liquid Chromatography. Chromatographia 2019; 82: 17-48.
- Ahmed A, Skinley K, Herodotou S, Zhang H. Core–shell microspheres with porous nanostructured shells for liquid chromatography. J Sep Sci 2018; 41: 99-124.
- Ordoñez, EY, Rodil R, Quintana JB, Cela R. Determination of artificial sweeteners in beverages with green mobile phases and high temperature liquid chromatography–tandem mass spectrometry. Food Chem 2015; 169: 162-168.
- Yabré M, Ferey L, Somé IT, GaudinK. Greening Reversed-Phase Liquid Chromatography Methods Using Alternative Solvents for Pharmaceutical Analysis, Molecules 2018, 23, 1065-1090.
- Haq N, Shakeel F, Alanazi F, Alshora FA, Ibrahim MA. Development and validation of a green RP-HPLC method for the analysis of rosuvastatin: a step towards making liquid chromatography environmentally benign. Green Process Synth 2018; 7: 160-169.
- García-Alvarez-Coque MC, Ruiz Ángel MJ, Peris-García E. Liquid chromatography| Micellar Liquid Chromatography. Academic Press, 2019.
- García-Alvarez-Coque MC, Ruiz Ángel MJ, Cardá Broch S. Micellar liquid chromatography: fundamentals. In Anderson JL, Berthod A, Pino V, Stalcup AM. Analytical Separation Science. Vol. 2. Wiley-VCH, 2015.
- González-Ruiz V, León AG, Olives AI, Martín MA, Menéndez JC. Eco-friendly liquid chromatographic separations based on the use of cyclodextrins as mobile phase additives. Green Chem., 2011, 13, 115-126.
- González-Ruiz V, Olives AI, Martín MA. SPE/RP-HPLC using C1 columns: an environmentally friendly alternative to conventional reverse-phase separations for quantitation of beta-carboline alkaloids in human serum samples. Anal Bioanal Chem 2011; 400: 395-401.
- García-Alvarez-Coque MC, Ruiz Ángel MJ, Berthod A, Cardá Broch S. On the use of ionic liquids as mobile phase additives in high-performance liquid chromatography. A review. Anal Chim Acta 2015; 883: 1-21.
- Peris-García E, García-Alvarez-Coque MC, Carda Broch S, Ruiz Ángel MJ. Effect of buffer nature and concentration on the chromatographicperformance of basic compounds in the absence and presence of1-hexyl-3-methylimidazolium chloride. J Chromatogr A 2019; 1602: 397-408.
- Sun P, Armstrong DW. Ionic liquids in analytical chemistry. Anal Chim Acta 2010; 661: 1-16.
- Buszewska-Forajta M, Markuszewski MJ, Kaliszan R. Free silanols and ionic liquids as their suppressors in liquid chromatography. J Chromatogr A 2018; 1559: 17-43.
- Ali I, Suhail M, Sanagi MM, Sanagi HY. Ionic Liquids in HPLC and CE: A Hope for Future. Crit Rev Anal Chem 2017; 47: 332-339.
- Ubeda-Torres MT, Ortiz-Bolsico C, García-Álvarez-Coque MC, Ruiz-Angel MJ. Gaining insight in the behaviour of imidazolium-based ionic liquids asadditives in reversed-phase liquid chromatography for the analysis of basic compounds. J Chromatogr A 2015; 1380: 96-103.
- Di Nunzio M, Cohen B, Douhal A. Structural photodynamics of camptothecin, an anticancer drug in aqueous solutions. J Phys Chem A 2011; 115: 5094-5104.
- Gonáez-Ruiz V, Mussardo P, Corda E, Girotti S, Olives AI, Martín MA. Liquid chromatographic analysis of the anticancer alkaloid luotonin A and some new derivatives in human serum samples. J Sep Sci 2010; 33: 2086-2093.
- González-Ruiz V, Cores A, Martín-Cámara O, Orellana K, Cervera-Carrascón V, Michalska P, Olives AI, León R, Martín MA, Menéndez JC. Enhanced Stability and Bioactivity of Natural Anticancer Topoisomerase I Inhibitors through Cyclodextrin Complexation. Pharmaceutics 2021; 13: 1609-1618.
- Berthod A, Ruiz-Angel MJ, Carda-Broch S. Ionic liquids in separation techniques. J Chromatogr A, 2008; 1184: 6-18.
- Han D, Row KH. Recent applications of ionic liquids in separation technology. Molecules, 2010; 15: 2405-2426.
