(B) SANGRE Y SISTEMA HEMATOPOYÉTICO
FITUSIRAN (QFITLIA®) GENZYME (USA)
Indicación: Profilaxis de rutina para prevenir o reducir la frecuencia de episodios de sangrado en pacientes adultos y pediátricos de 12 años o más con hemofilia A o B, con o sin inhibidores del factor VIII o IX.
Tipo: Medicamento biológico constituido por un ácido ribonucleico de interferencia pequeño (siRNA) bicatenario, en el que la cadena sentido está conjugada con un complejo de N-acetilgalactosamina triantenaria (GalNAc). Autorizado (FDA) en Estados Unidos el 28 de marzo de 2025 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track); no disponible en la Unión Europea.
Mecanismo: Es un ARN pequeño que provoca la degradación del ARN mensajero (ARNm) de la proteína antitrombina (SERPINC1) mediante la interferencia con el ARNm, lo que reduce los niveles plasmáticos de antitrombina, como mecanismo para promover la síntesis de trombina y reequilibrar la hemostasia (y prevenir hemorragias) en pacientes con hemofilia A o B. La conjugación con GalNAc es una estrategia química utilizada para promover la captación del fármaco por el hígado.
Eficacia clínica: Dos ensayos clínicos multicéntricos, aleatorizados, que reclutaron a un total de 177 pacientes varones adultos y pediátricos con hemofilia A o hemofilia B. En los dos ensayos aleatorizados, los participantes recibieron una dosis fija de Qfitlia mensualmente o su tratamiento habitual a demanda (agentes de derivación o concentrados de factores de coagulación) según fuera necesario durante nueve meses. La variable principal de eficacia fue la tasa de sangrado anualizada. En los participantes con inhibidores que recibieron el régimen de dosificación basado en antitrombina de Qfitlia, hubo una reducción del 73% en la tasa de sangrado anualizada estimada en comparación con aquellos que recibieron tratamiento a demanda con agentes de derivación. En los participantes sin inhibidores que recibieron el régimen de dosificación basado en antitrombina de Qfitlia, hubo una reducción del 71%.
Eventos adversos: Los más comunes (≥10%) son infección viral (29%), nasofaringitis (26%) e infección bacteriana (11%).
GARADACIMAB (ANDENBRY®) CSL BEHRING (UE)
Indicación: Prevención rutinaria de las crisis recurrentes de angioedema hereditario (AEH) en pacientes adultos y adolescentes a partir de 12 años de edad.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal recombinante IgG4/lambda totalmente humano. Autorizado en la Unión Europea el 10 de febrero de 2025; no disponible en Estados Unidos.
Mecanismo: Anticuerpo que se une al dominio catalítico del factor XII activado (FXIIa y βFXIIa) e inhibe su actividad catalítica. La inhibición del FXIIa, el primer factor activado en el sistema de contacto, previene los ataques de angioedema hereditario (AEH) al bloquear la activación de precalicreína a calicreína y la generación de bradicinina, que se asocia con la inflamación y la hinchazón en los ataques de AEH.
Eficacia clínica: Un estudio de fase 3, multicéntrico, aleatorizado, doble ciego y controlado con placebo de grupos paralelos, en 64 pacientes mayores de 12 años (58 adultos y 6 pacientes pediátricos) que experimentaron al menos 2 ataques durante el período de preinclusión de hasta 2 meses. La variable primaria de eficacia fue el número normalizado (media ajustada a la tasa basal de ataques) de ataques de angioedema en el tiempo desde el día 1 hasta el final del período de tratamiento de 6 meses: 0,22 vs 2,07. Las principales variables secundarias fueron la reducción porcentual en el número medio de ataques de AEH normalizados en el tiempo (85,5%), el porcentaje de sujetos que estuvieron libres de ataques desde el día 1 hasta el final de los primeros 3 meses (71,8 vs 8,3%) y el porcentaje de sujetos con respuestas buenas o excelentes al SGART (evaluación global de la respuesta al tratamiento por parte de los sujetos) desde el día 1 hasta el final del período de tratamiento de 6 meses (82 vs 33%).
Eventos adversos: Los más comunes (≥1/100 a <1/10) son reacciones en el lugar de inyección (eritema, hematomas, prurito o urticaria), cefalea y dolor abdominal.
(H) PREPARACIONES HORMONALES SISTÉMICAS
TIRATRICOL (EMCITATE®) RARE THYROID THERAPEUTICS (UE)
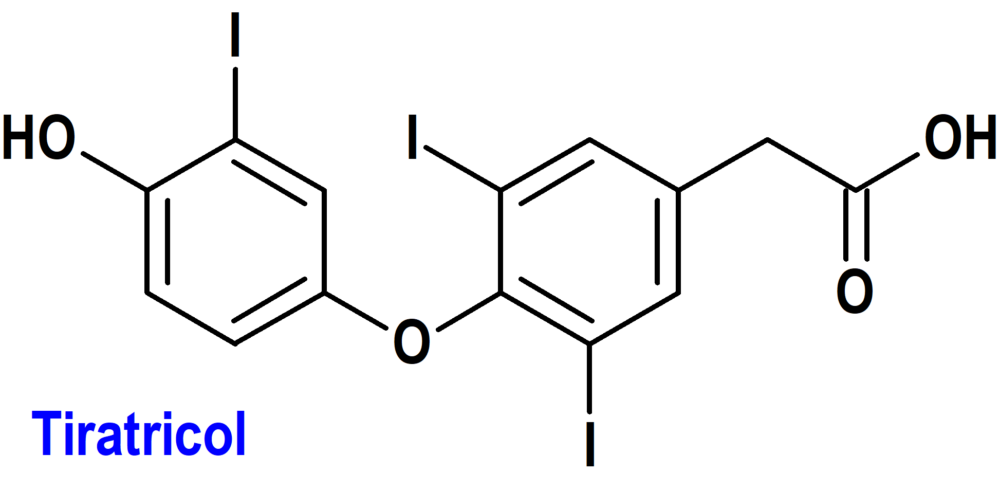
Indicación: Tratamiento de la tirotoxicosis periférica en pacientes con deficiencia del transportador 8 de monocarboxilato (MCT8) (síndrome de Allan-Herndon-Dudley) desde el nacimiento.
Tipo: Medicamento sintético estándar constituido por el ácido 2-[4-(4-hidroxi-3-iodofenoxi)-3,5-diiodofenil]acético. Autorizado en la Unión Europea el 12 de febrero de 2025 como medicamento huérfano (Orphan drug); no disponible en Estados Unidos.
Mecanismo: Es un metabolito circulante natural de la hormona tiroidea activa (T3), que presenta un alto grado de similitud estructural con esta hormona y sigue la misma vía de degradación posterior (desiodación y conjugación) y eliminación a través de la bilis y la orina. El tiratricol es biológicamente activo, se une con gran afinidad a los receptores de hormona tiroidea TRα y TRβ y ejerce efectos biológicos similares a los de la T3, aunque con una especificidad tisular diferente. El tiratricol puede entrar en células dependientes de MCT8 sin un transportador de MCT8 funcional, a diferencia de la T3 y la T4; por lo tanto, el tiratricol puede sustituir a la T3 en los tejidos dependientes de MCT8 y restablecer la actividad normal de las hormonas tiroideas en todos los tejidos.
Eficacia clínica: Un estudio multicéntrico, abierto y de un solo grupo en el que participaron 46 pacientes que fueron tratados durante un máximo de 12 meses. El tratamiento con tiratricol redujo la concentración sérica media de T3 de 4,97 nmol/l al inicio hasta 1,82 nmol/l a los 12 meses. A los 12 meses o en la última evaluación disponible, 25 de 45 pacientes (56 %) alcanzaron unos niveles séricos de T3 dentro del intervalo objetivo, 13 de 45 pacientes (29 %) presentaron unos niveles de T3 por debajo del intervalo objetivo y 7 de 45 pacientes (16 %) presentaron unos niveles séricos de T3 por encima del intervalo objetivo. Todos los pacientes (45 de 45; 100 %) mejoraron en al menos una de las variables de peso corporal, frecuencia cardíaca en reposo o presión arterial sistólica, y 31 de 45 (69 %) mejoraron en al menos dos de estas tres variables. En total, 39 de 45 pacientes (87 %) mejoraron en al menos una de las variables de puntuación Z del peso corporal en función de la edad con deficiencia de MCT8, puntuación Z de la frecuencia cardíaca en reposo o puntuación Z de la presión arterial sistólica, y 21 de 45 (47 %) mejoraron en al menos dos de estas tres variables. El número medio de extrasístoles auriculares (ESA) medidas por el ECG de 24 horas disminuyó desde 899,7 ESA/24 horas al inicio hasta 313,9 ESA/24 horas a los 12 meses. Las concentraciones de creatina cinasa aumentaron desde 108 U/l al inicio hasta 160,7 U/l a los 12 meses.
Eventos adversos: Los más comunes son hiperhidrosis (7%), diarrea (6%), irritabilidad (2%), ansiedad (2%) y pesadillas (2%).
(J) ANTIINFECCIOSOS SISTÉMICOS
GEPOTIDACINA (BLUJEPA®) GLAXOSMITHKLINE (USA)

Indicación: Tratamiento de pacientes mujeres adultas y pediátricas de 12 años de edad y mayores que pesen al menos 40 kilogramos (kg) con infecciones del tracto urinario (ITU) no complicadas causadas por los siguientes microorganismos susceptibles: Escherichia coli, Klebsiella pneumoniae, complejo Citrobacter freundii, Staphylococcus saprophyticus y Enterococcus faecalis.
Tipo: Medicamento sintético estándar constituido por la (3R)-3-[[4-(3,4-dihidro-2H-pirano[2,3-c]piridin-6-ilmetilamino)piperidin-1-il]metil]-1,4,7-triazatriciclo[6.3.1.04,12]dodeca-6,8(12),9-trieno-5,11-diona. Autorizado (FDA) en Estados Unidos el 25 de marzo de 2025 mediante revisión prioritaria (Priority Review); no disponible en la Unión Europea.
Mecanismo: Inhibe las topoisomerasas de tipo II, incluidas la topoisomerasa bacteriana II (ADN girasa) y la topoisomerasa IV, inhibiendo así la replicación del ADN bacteriano. Es el primero (cabeza de serie) de los antibacterianos con este mecanismo de acción.
Eficacia clínica: Dos ensayos multicéntricos, de grupos paralelos, doble ciego, con doble simulación y de no inferioridad. Ambos ensayos compararon gepotidacina con nitrofurantoína. La eficacia se evaluó como una combinación de curación clínica y respuesta microbiológica en la visita de prueba de curación (TOC) (días del estudio 10 al 13) en la población microbiológicamente sensible a la nitrofurantoína (micro-ITTS), que incluyó a todos los pacientes que recibieron al menos una dosis de la medicación del estudio, tenían al menos un uropatógeno calificado basalmente (≥10(5) unidades formadoras de colonias [UFC]/ml) y excluyeron a los pacientes con microorganismos no sensibles a la nitrofurantoína. La curación clínica se definió como la resolución de todos los signos y síntomas de cistitis aguda presentes al inicio del estudio y la ausencia de nuevos signos y síntomas sin que el paciente recibiera otros antimicrobianos sistémicos. La respuesta microbiológica se definió como la reducción de todos los uropatógenos considerados al inicio del estudio en ≥10(5) UFC/ml a <10(3) UFC/ml sin que el paciente recibiera otros antimicrobianos sistémicos. En el primer ensayo clínico (N=634), las tasas de respuesta combinada fueron del 52% (gepotidacina) vs. 47% (nitrofurantoína); la curación clínica se alcanzó en el 67/66% y la respuesta microbiológica fue del 73/67%. En el segundo ensayo clínico (N=567), las tasas de respuesta combinada fueron del 59/44%; la curación clínica se alcanzó en el 68/64% y la respuesta microbiológica fue del 73/58%.
Eventos adversos: Los más comunes (>5%) son diarrea (16%) y náusea (9%).
SIPAVIBART (KAVIGALE®) ASTRAZENECA (UE)
Indicación: Profilaxis previa a la exposición de COVID-19 en adultos y adolescentes a partir de 12 años de edad que pesen al menos 40 kg y que estén inmunodeprimidos debido a una enfermedad o que reciban tratamientos inmunosupresores.
Tipo: Medicamento biológico constituido por un anticuerpo basado en la inmunoglobulina (Ig) G1 humana recombinante. Autorizado en la Unión Europea el 20 de enero de 2025 mediante evaluación acelerada (Accelerated assessment); no disponible en Estados Unidos.
Mecanismo: Anticuerpo monoclonal IgG1 humano recombinante que proporciona inmunización pasiva al unirse al dominio de unión al receptor de la proteína de la espícula (spike protein receptor binding domain, RBD) de SARS-CoV-2. Sipavibart posee acción prolongada, con sustituciones de aminoácidos para aumentar la vida media del anticuerpo y para reducir la función efectora del anticuerpo y el posible riesgo de enfermedad incrementada dependiente de anticuerpos. Sipavibart se une al RBD de la proteína de la espícula RBD de SARS-CoV-2 (BA.2) bloqueando la unión RBD al receptor ACE2 humano. Esto da lugar a un bloqueo de la entrada del virus.
Eficacia clínica: Un ensayo clínico en fase 3, aleatorizado (1:1), doble ciego y controlado con comparador que estudia sipavibart para la profilaxis previa a la exposición a la COVID-19 en 1669 adultos inmunodeprimidos y adolescentes ≥ 12 años de edad, para recibir una dosis única de sipavibart 300 mg mediante inyección intramuscular y 1666 fueron aleatorizados para recibir un comparador (tixagevimab 300 mg + cilgavimab 300 mg o placebo). Los participantes recibieron una segunda dosis de sipavibart 300 mg o placebo 6 meses 12 después de la dosis inicial. Las dos variables primarias de eficacia comparando la eficacia de sipavibart con un comparador en la prevención de la COVID-19 sintomática causada por cualquier variante del SARS-CoV-2 hasta 181 días después de la última dosis, confirmada por RT-PCR (porcentaje de acontecimientos: 9,2 vs 12,7%; reducción del riesgo relativo del 29,9%) y las atribuibles a variantes coincidentes (variantes que no contienen la mutación F456L según los datos de secuenciación viral y que se espera que sean susceptibles a sipavibart) hasta 181 días después de la última dosis, también confirmada por RT-PCR (porcentaje de acontecimientos: 4,4 vs 6,6%; reducción del riesgo relativo del 35,3%).
Eventos adversos: En inyección intramuscular, el más común es la reacción en el lugar de la inyección (4,1%). Con perfusión intravenosa son la reacción en el lugar de la perfusión (1,9%) y reacciones relacionadas con la perfusión (1,9%).
ZAPOMERAN (KOSTAIVE®) ARCTURUS (UE)
Indicación: Inmunización activa destinada a prevenir la COVID-19 causada por el SARS-CoV-2 en personas de 18 años o más.
Tipo: Medicamento biológico constituido por UN ARN mensajero autoamplificado (ARNm-aa) de COVID-19 (encapsulado en nanopartículas lipídicas). Autorizado en la Unión Europea el 12 de febrero de 2025; no disponible en Estados Unidos.
Mecanismo: Es un ARNm autoamplificado que codifica la proteína espicular del SARS-CoV-2, que está diseñado para producir copias adicionales de ARNm dentro de las células huésped después de la inyección intramuscular, para lograr una expresión mejorada del antígeno de la proteína espicular. Esto da lugar a anticuerpos neutralizantes y respuestas inmunitarias celulares al antígeno espicular, lo que contribuye a la protección contra la COVID-19. El proceso de autoamplificación del ARNm es transitorio y no genera partículas infecciosas.
Eficacia clínica: un estudio clínico aleatorizado, controlado, con enmascaramiento del observador y multicéntrico realizado en participantes de 18 años y mayores en Vietnam en un momento en que la variante dominante era la delta. La eficacia se evaluó en el grupo de análisis por ITTm, que incluyó a 15.458 participantes, 7.762 en el grupo de Kostaive (zapomerán) y 7.696 en el grupo de placebo. La variable principal de eficacia consistió en el número y porcentaje de casos de COVID-19 virológicamente confirmados con inicio entre el día 36 (7 días después de la dosis 2) y el día 92, inclusive. Para los participantes sin evidencia de infección por SARS-CoV-2 previa (antes de 7 días después de la dosis 2), el número total de casos de COVID-19 fue de 200 en el grupo vacunado y de 440 en el placebo, lo que supone una eficacia global de la vacuna del 56,7%. En personas entre 18 y 60 años, los porcentajes de eficacia fueron del 49,8% para personas sanas y del 69,7% para personas en riesgo, mientras que para mayores de 60 años fue del 53,5%.
Eventos adversos: Los más comunes (≥10 %) son dolor en el lugar de la inyección (49%), sensibilidad en el lugar de la inyección (49%), fatiga (42%), dolor de cabeza (35 %), mialgia (30%), escalofríos (29%), artralgia (27%), mareos (20%) y pirexia (11%).
VACUNA CHIKUNGUNYA RECOMBINANTE (VIMKUNYA®) BABARIAN NORDIC (UE; USA)
Indicación: Prevención de la enfermedad causada por el virus Chikungunya (CHIKV) en personas de 12 años de edad y mayores.
Tipo: Medicamento biológico constituido por partículas similares a virus purificadas (VLP) que consisten en proteína de la cápside (C) de CHIKV y proteínas de envoltura E1 y E2, derivadas de la cepa CHIKV Senegal 37997. Las VLP se producen transfectando un plásmido de expresión que codifica la poliproteína estructural C-E3-E2-6K-E1 de CHIKV en HEK293 (una línea continua de células renales embrionarias humanas). Autorizado en la Unión Europea el 28 de febrero de 2025 mediante evaluación acelerada (Accelerated assessment); disponible en Estados Unidos desde el 14 de febrero de 2025.
Mecanismo: Inducción de respuestas inmunes específicas del CHIKV.
Eficacia clínica: Dos estudios en los que la evaluación de la eficacia se basó en la tasa de serorrespuesta y en la media geométrica del título de anticuerpos neutralizantes séricos. La tasa de serorrespuesta a los 183 días posteriores a la vacunación en el Estudio 1 (N=2.893) fue del 85,5 % con VIMKUNYA y del 1,5 % con placebo (diferencia de la tasa de serorrespuesta: 84,0 puntos porcentuales, pp). La tasa de serorespuesta a la vacunación en el Estudio 2 (N=372) fue del 75,5 vs 1,2% (diferencia de 74,3 pp).
Eventos adversos: Los más comunes en personas de 12 a 64 años de edad son dolor en el lugar de la inyección (24%), fatiga (20%), dolor de cabeza (18%) y mialgia (18%); en personas de 65 años de edad y mayores son mialgia (6,3%), fatiga (6,3%) y dolor en el lugar de la inyección (5,4%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
MIRDAMETINIB (GOMEKLI®) SPRINGWORKS (USA)

Indicación: Tratamiento de pacientes adultos y pediátricos de 2 años de edad o más con neurofibromatosis tipo 1 (NF1) que tienen neurofibromas plexiformes (PN) sintomáticos no susceptibles de resección completa.
Tipo: Medicamento sintético estándar constituido por la N-[(2R)-2,3-dihidroxipropoxi]-3,4-difluoro-2-[(2-fluoro-4-yodofenil)amino]benzamida. Autorizado en Estados Unidos el 11 de febrero de 2025 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no disponible en la Unión Europea.
Mecanismo: Inhibidor de las cinasas 1 y 2 de la proteína cinasa activada por mitógeno (MEK1/2). Las proteínas MEK1/2 son reguladores ascendentes de la vía de la cinasa relacionada con la señal extracelular (ERK). En un modelo de ratón de NF1, mirdametinib inhibió la fosforilación de ERK y redujo el volumen tumoral y la proliferación del neurofibroma.
Eficacia clínica: Un ensayo multicéntrico de un solo brazo en 114 pacientes ≥2 años de edad (58 adultos, 56 pacientes pediátricos) con NP asociada a NF1 sintomática e inoperable que causaba una morbilidad significativa. La principal variable de eficacia fue la tasa de respuesta global confirmada (ORR), definida como el porcentaje de pacientes con respuesta completa (desaparición del PN objetivo) o respuesta parcial (≥20% de reducción en el volumen del PN) con confirmación de las respuestas dentro de los 2 a 6 meses durante la fase de tratamiento de 24 ciclos: 41% en adultos y 52% en la cohorte pediátrica.
Eventos adversos: Los más comunes (>25%) en pacientes adultos son erupción cutánea, diarrea, náuseas, dolor musculoesquelético, vómitos y fatiga; la anomalía de laboratorio de grado 3 o 4 más frecuente (>2%) fue el aumento de la creatina fosfocinasa. En pacientes pediátricos, son erupción cutánea, diarrea, dolor musculoesquelético, dolor abdominal, vómitos, dolor de cabeza, paroniquia, disfunción ventricular izquierda y náuseas; las anomalías de laboratorio de grado 3 o 4 más frecuentes (>2%) fueron disminución del recuento de neutrófilos y aumento de la creatina fosfocinasa.
VIMSELTINIB (ROMVIMZA®) DECIPHERA (USA)

Indicación: Tratamiento de pacientes adultos con tumor tenosinovial de células gigantes (TGCT) sintomático en el que la resección quirúrgica potencialmente causará empeoramiento de la limitación funcional o morbilidad grave.
Tipo: Medicamento sintético estándar constituido por la 3-metil-5-[6-metil-5-[2-(1-metilpirazol-4-il)piridin-4-il]oxipiridin-2-il]-2-(propan-2-ilamino)pirimidin-4-ona. Autorizado en Estados Unidos el 14 de febrero de 2025 por vía rápida (Fast Track); no disponible en la Unión Europea (EMA).
Mecanismo: Inhibidor de la cinasa que inhibe el receptor del factor estimulante de colonias 1 (CSF1R). Vimseltinib inhibe la autofosforilación de CSF1R, la señalización inducida por la unión del ligando CSF1 y la proliferación de células que expresan CSF1R.
Eficacia clínica: Un ensayo doble ciego, multicéntrico, aleatorizado (2:1), controlado con placebo en 123 pacientes con TGCT para quienes la resección quirúrgica puede causar empeoramiento de la limitación funcional o morbilidad grave. Los pacientes fueron asignados aleatoriamente a placebo o vimseltinib durante 24 semanas, durante el período doble ciego (Parte 1). Durante el período abierto (Parte 2), los pacientes podrían continuar con vimseltinib y aquellos que recibían placebos podrían pasar a vimseltinib. La principal variable de eficacia fue la tasa de respuesta global (ORR) evaluada mediante una revisión radiológica independiente ciega en la semana 25 (40 vs. 0%). La duración media de la respuesta (DOR) no se alcanzó en el grupo de vimseltinib y, con base en 6 meses adicionales de seguimiento, 28 respondedores (85%) tuvieron una DOR≥6 meses y 19 (58%) tuvieron una DOR ≥9 meses.
Eventos adversos: Los más comunes (≥20%) son aumento de la aspartato aminotransferasa, edema periorbitario, fatiga, erupción cutánea, aumento del colesterol, edema periférico, edema facial, disminución de neutrófilos, disminución de leucocitos, prurito y aumento de la alanina aminotransferasa.
SERPLULIMAB (HETRONIFLY®) HENLIUS (UE)
Indicación: Tratamiento de primera línea, en combinación con carboplatino y etopósido, de pacientes adultos con cáncer de pulmón microcítico en estadio extendido (CPM-EE).
Tipo: Medicamento biológico constituido por un anticuerpo humanizado (IgG4/isotipo kappa con una alteración de la secuencia estabilizadora en la región bisagra). Autorizado en la Unión Europea el 3 de febrero de 2025 como medicamento huérfano (orphan drug); no disponible en Estados Unidos.
Mecanismo: Anticuerpo monoclonal humano de tipo IgG4 que se une al receptor de muerte programada 1 (PD-1) y bloquea su interacción con los ligandos PD-L1 y PD-L2. El receptor PD-1 es un regulador negativo de la actividad de los linfocitos T e implicado en el control de la respuesta inmunitaria de estos. El acoplamiento de PD-1 con los ligandos PD-L1 y PD-L2, que se expresan en las células presentadoras de antígenos y que podrían ser expresados por tumores u otras células en el microambiente tumoral, produce la inhibición de la proliferación de los linfocitos T y la secreción de citocinas. Serplulimab potencia las respuestas de los linfocitos T, incluyendo sus respuestas antitumorales, por medio del bloqueo de PD-1, evitando su unión a los ligandos PD-L1 y PD-L2.
Eficacia clínica: Un ensayo clínico de fase 3 multirregional, aleatorizado y doble ciego (serplulimab + carboplatino + etopósido frente a placebo + carboplatino + etopósido) sobre un conjunto de 585 pacientes. La variable primaria de la eficacia fue la supervivencia global (SG) entre los meses 25 y 33 después del inicio del ensayo clínico: 15,4 vs 10,9 meses, con un 37,5 vs 51,0% de pacientes con acontecimientos. Las variables secundarias de la eficacia fueron la supervivencia libre de progresión (SLP: 5,7 vs. 4,3 meses) y la tasa de respuesta objetiva (TRO: 67,4 vs 58,7%).
Eventos adversos: Los más comunes son neutropenia (83%), leucopenia (74%), anemia (73%), trombocitopenia (56%), alopecia (54%), náuseas (36%), hiperlipidemia (32%), apetito disminuido (28%), hipoproteinemia (25%) e hiponatremia (25%). Las reacciones de grado ≥3 más frecuentes son neutropenia (65%), leucopenia (33%), trombocitopenia (23%), anemia (20%), hiponatremia (10%) y linfopenia (5%).
CATUMAXOMAB (KORJUNY®) LINDIS (UE)
Indicación: Tratamiento intraperitoneal de la ascitis maligna en adultos con carcinomas positivos para la molécula de adhesión de células epiteliales (EpCAM), que no son candidatos a terapia sistémica contra el cáncer.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal IgG2 híbrido de rata-ratón. Autorizado en la Unión Europea el 10 de febrero de 2025 como medicamento huérfano (Orphan drug); no disponible en Estados Unidos.
Mecanismo: Anticuerpo monoclonal trifuncional dirigido específicamente contra la molécula de adhesión de las células epiteliales (EpCAM) y el antígeno CD3. El antígeno EpCAM se expresa en la mayoría de los cánceres, especialmente en los carcinomas. El CD3 se expresa en los linfocitos T maduros como componente del receptor de los linfocitos T. El tercer lugar de unión funcional en la región Fc del catumaxomab permite su interacción con las células inmunitarias accesorias a través de los receptores Fc-gamma. Debido a las propiedades de unión de catumaxomab, las células tumorales, las células T y las células inmunitarias accesorias se encuentran muy próximas; en consecuencia, se induce una inmunorreacción concertada frente a las células tumorales que incluye diferentes mecanismos de acción como la activación de los linfocitos T, la destrucción mediada por los linfocitos T a través del sistema perforina/granzima, la citotoxicidad celular dependiente de anticuerpos (ADCC), la citotoxicidad dependiente del complemento (CDC) y la fagocitosis. Esto provoca la destrucción de las células tumorales de la cavidad peritoneal, eliminando así una de las principales causas de la ascitis maligna.
Eficacia clínica: Estudio clínico de fase 2/3 pivotal, de dos grupos (paracentesis más catumoxamab o paracentesis sola), aleatorizado y abierto, en el que participaron 258 pacientes con ascitis maligna sintomática debido a carcinomas EpCAM positivos, de los cuales 170 fueron aleatorizados para el tratamiento de catumaxomab. La variable principal de la eficacia fue la supervivencia sin punción en el periodo de estudio principal aleatorizado y controlado, una variable compuesta definida como el tiempo hasta la primera necesidad de punción de ascitis terapéutica o la muerte, sea cual sea lo que ocurra primero: 48 vs. 11 días (pacientes con cáncer de ovarios) y 30 vs. 14 días (pacientes con cáncer no ovárico). La tasa de supervivencia fue del 27,5 vs 17,1% a los 6 meses y del 11,4 vs 2,6% a los 12 meses, siendo la mediana global de supervivencia de 72 y 71 días, respectivamente.
Eventos adversos: Los más comunes son pirexia (62%), dolor abdominal (42%), náuseas (41%) y vómitos (38%). Las reacciones adversas más graves son el síndrome de respuesta inflamatoria sistémica y la insuficiencia hepática.
VILOBELIMAB (GOHIBIC®) INFLARX (UE)
Indicación: Tratamiento de pacientes adultos con síndrome de dificultad respiratoria aguda (SDRA) inducido por el SARS-CoV2 que reciben corticosteroides sistémicos como parte del tratamiento estándar de elección y que reciben ventilación mecánica invasiva (MVI) (con o sin oxigenación por membrana extracorpórea (ECMO).
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal IgG4 quimérico humano/ratón. Autorizado en la Unión Europea el 14 de enero de 2025 en condiciones excepcionales (Exceptional circumstances); disponible en Estados Unidos desde el 4 de abril de 2023.
Mecanismo: Inhibidor específico del componente C5a del complemento humano soluble. El complemento puede ser activado directamente por el SARS-CoV-2, y los niveles elevados de C5a están asociados con la gravedad de la enfermedad y la mortalidad.
Eficacia clínica: Ensayo de fase 3, doble ciego, aleatorizado, controlado con placebo, multicéntrico y multinacional en 369 pacientes adultos (≥ 18 años) que requieren ventilación 6 mecánica invasiva (VMI) (con o sin oxigenación por membrana extracorpórea (ECMO). La variable principal de eficacia clínica fue la mortalidad por cualquier causa a los 28 días: 31,7 vs. 41,6%.
Eventos adversos: Los más comunes son neumonía (21,7 %), herpes simple (6,3 %), aspergilosis broncopulmonar (5,7 %) y sepsis (5,1 %).
DATOPOTAMAB DERUXTECAN (DATROWAY®) DAIICHI SANKYO (USA; UE)
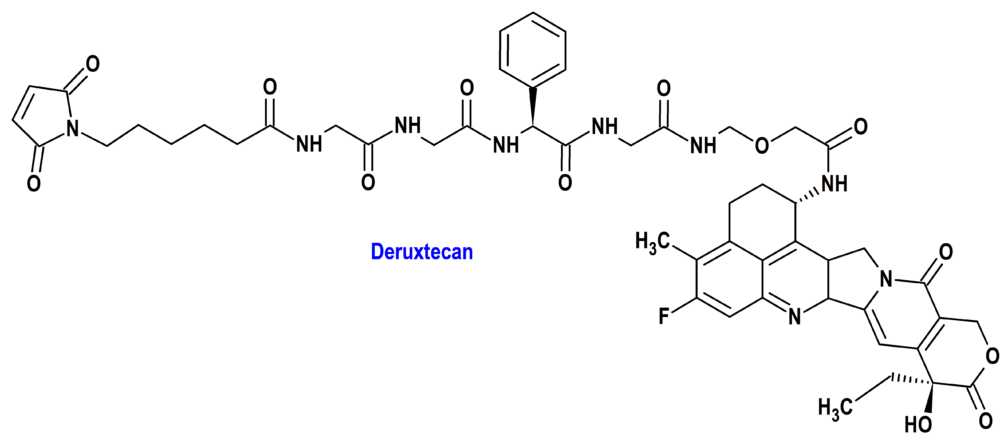
Indicación: Tratamiento de pacientes adultos con cáncer de mama irresecable o metastásico, positivo para el receptor hormonal (HR), negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2) (IHC 0, IHC 1+ o IHC 2+/ISH-), que hayan recibido terapia endocrina previa y quimioterapia para la enfermedad irresecable o metastásica.
Tipo: Medicamento biológico formado por un conjugado anticuerpo-fármaco (ADC) compuesto por un anticuerpo monoclonal humanizado anti-Trop2 IgG1, unido covalentemente a deruxtecan (Dxd), un inhibidor de la topoisomerasa I derivado del exatecan, mediante un enlazador tetrapeptídico maleimida escindible por proteasas. El deruxtecan es la 6-(2,5-dioxopirrol-1-il)-N-[2-[[2-[[(2S)-1-[[2-[[2-[[(10S,23S)-10-etil-18-fluoro-10-hidroxi-19-metil-5,9-dioxo-8-oxa-4,15-diazahexaciclo[14.7.1.02,14.04,13.06,11.020,24]tetracosa-1,6(11),12,14,16,18,20(24)-heptaen-23-il]amino]-2-oxoetoxi]metilamino]-2-oxoetil]amino]-1-oxo-3-fenilpropan-2-il]amino]-2-oxoetil]amino]-2-oxoetil]hexanamida. Autorizado (FDA) en Estados Unidos desde el 17 de enero de 2025; evaluado favorablemente en la Unión Europea (CHMP, EMA) el 30 de enero de 2025.
Mecanismo: Después de unirse a Trop2 (transductor de señal de calcio asociado a tumores-2) en las células, incluidas las células tumorales, datopotamab deruxtecan experimenta internalización y escisión intracelular del enlazador por enzimas lisosomales. Tras su liberación, el inhibidor de la topoisomerasa I (Dxd) permeable a la membrana nuclear provoca daño del ADN y muerte celular apoptótica. La topoisomerasa I está implicada en el desenrollamiento de las hebras de ADN, proceso previo a la replicación y transcripción del ADN. La enzima actúa uniéndose a regiones específicas de la cadena de ADN, rompiendo una de las hebras del ADN; por decirlo de otro modo, la topoisomerasa I libera la energía torsional en la hélice de ADN durante la replicación y transcripción, induciendo rupturas de cadena única. Posteriormente, la enzima vuelve a soldar la cadena tras haberla desenrollado. Determinados fármacos antineoplásicos, como el deruxtecan, se unen al complejo ADN-topoisomerasa I y lo estabilizan; aunque este fenómeno permite el primer paso de la acción enzimática, sin embargo, impide el segundo, la reconstrucción de la hebra de ADN y, con ello, queda paralizada la síntesis de nuevas moléculas de ADN. Las células neoplásicas parecen presentar niveles de actividad de topoisomerasa I superiores a los de las células normales.
Eficacia clínica: Ensayo multicéntrico, abierto y aleatorizado en 732 pacientes adultos con cáncer de mama irresecable o metastásico, positivo para el receptor hormonal (HR) y negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2) que habían experimentado progresión de la enfermedad, no habían sido considerados aptos para recibir terapia endocrina adicional y habían recibido una o dos líneas de quimioterapia previa, comparando datopotamab deruxtecan (n=365) con la quimioterapia elegida por el investigador (n=367; eribulina, 60%; capecitabina, 21%; vinorelbina, 10%; o gemcitabina, 9%). Las variables principales de eficacia fueron la supervivencia libre de progresión (SLP: mediana de 6,9 vs 4,9 meses; p bilateral <0,0001) y la supervivencia general (SG: mediana de 18,6 vs 18,3 meses, sin diferencia estadísticamente significativa). Los resultados de eficacia adicionales incluyeron la tasa de respuesta objetiva confirmada (TRG: 36 vs 23%) y la duración de la respuesta (DOR: mediana de 6,7 vs 5,7 meses).
Eventos adversos: Los más comunes (≥20%) son estomatitis, náuseas, fatiga, disminución de leucocitos, disminución de calcio, alopecia, disminución de linfocitos, disminución de hemoglobina, estreñimiento, disminución de neutrófilos, ojo seco, vómitos, aumento de ALT, queratitis, aumento de AST y aumento de fosfatasa alcalina.
TISOTUMAB VEDOTINA (TIVDAK®) PFIZER (USA; UE)

Indicación: Tratamiento del cáncer de cuello uterino recurrente o metastásico con progresión de la enfermedad durante o después de la quimioterapia.
Tipo: Medicamento biológico constituido por un conjugado de fármaco-anticuerpo (ADC) dirigido al factor tisular (TF) compuesto por un anticuerpo IgG1-kappa humano anti-TF conjugado con el agente disruptor de microtúbulos monometil auristatina E (MMAE) a través de un enlazador vc (valina-citrulina) escindible por proteasa. Autorizado (FDA) en Estados Unidos el 29 de abril de 2024, completando la autorización previa de forma acelerada (Accelerated Approval) llevada a cabo el 20 de septiembre de 2021; autorizado en la Unión Europea el 28 de marzo de 2025.
Mecanismo: Es un conjugado anticuerpo-fármaco dirigido al factor tisular (TF) presente en la superficie celular, donde actúa como iniciador primario de la cascada de coagulación sanguínea extrínseca. La monometil auristatina E (MMAE) es un agente disruptor de microtúbulos, que se encuentra unido al anticuerpo a través de un enlazador escindible por proteasa. La actividad anticancerígena de tisotumab vedotin se debe a la unión del conjugado (ADC) a las células cancerosas que expresan TF, seguida de la internalización del complejo ADC-TF y la liberación de MMAE a través de la escisión proteolítica. MMAE interrumpe la red de microtúbulos de células en división activa, lo que conduce a la detención del ciclo celular y la muerte celular apoptótica.
Eficacia clínica: Un ensayo abierto, controlado con fármaco activo (quimioterapia elegida por el investigador consistente en topotecán, vinorelbina, gemcitabina, irinotecán o pemetrexed), multicéntrico y aleatorizado en el que participaron 502 pacientes con cáncer de cuello uterino recurrente o metastásico que habían recibido uno o dos regímenes sistémicos previos, incluida quimioterapia con o sin bevacizumab y/o un agente anti-PD-(L)-1. La principal variable de eficacia fue la supervivencia general (SG): 11,5 meses (tisotumab vedotina) vs. 9,5 meses (quimioterapia electiva). Las variables de eficacia adicionales fueron la supervivencia libre de progresión (SLP: 4,2 vs. 2,9 meses) y la tasa de respuesta objetiva confirmada (TRF: 17,8 vs. 5,2%).
Eventos adversos: Los más comunes (≥25%), incluidas las anomalías de laboratorio, son disminución de la hemoglobina (45%), neuropatía periférica (39%), reacciones adversas conjuntivales (38%), náuseas (37%), fatiga (36%), aumento de la aspartato aminotransferasa (33%), epistaxis (33%), alopecia (31%), aumento de la alanina aminotransferasa (30%) y hemorragia (28%).
BELZUTIFAN (WELIREG®) MERCK, SHARP & DOHME (UE; USA)

Indicación: Tratamiento (en monoterapia) de pacientes adultos con carcinoma renal de células claras avanzado, que ha progresado después de recibir dos o más líneas de tratamiento que incluyeron un inhibidor de PD-(L)1 y al menos dos tratamientos dirigidos al factor de crecimiento del endotelio vascular (VEGF); y tratamiento (en monoterapia) de pacientes adultos con enfermedad de Von Hippel-Lindau, que requieren tratamiento para un carcinoma de células renales localizado (CCR), hemangioblastomas del sistema nervioso central (SNC) o tumores neuroendocrinos pancreáticos (pNET) asociados a la enfermedad, y para quienes los procedimientos localizados no son adecuados.
Tipo: Medicamento sintético estándar constituido por el 3[[(1S,2S,3R)-2,3-difluoro-2,3-dihidro-1-hidroxi-7-(metilsulfonil)-1H-inden-4-il]oxi]-5-fluorobenzonitrilo. Autorizado condicionalmente (Conditional marketing authorisation) en la Unión Europea el 12 de febrero de 2025; disponible en Estados Unidos desde el 13 de agosto de 2021.
Mecanismo: La proteína VHL (Von Hippel-Landau) es codificada por el gen del mismo nombre, un gen supresor tumoral que ayuda a controlar la división y la multiplicación de las células, así como otras funciones celulares importantes. Se ha encontrado este gen mutado en casi todos los pacientes con la enfermedad de Von Hippel-Lindau, de carácter hereditario que a veces causa cáncer de riñón y tumores en el encéfalo, la médula espinal, el ojo, el oído, la glándula suprarrenal, el páncreas u otras partes del cuerpo. El belzutifan es un inhibidor del factor de transcripción inducible por hipoxia 2 alfa (HIF-2α). Con niveles normales de oxígeno, la proteína VHL determina la degradación de HIF-2α. La alteración de la función de la proteína VHL provoca la acumulación de HIF-2α. En consecuencia, el HIF-2α se transloca al núcleo y regula la expresión de genes asociados a la proliferación celular, la angiogénesis y el crecimiento tumoral. Belzutifán se une al HIF-2α y, en condiciones de hipoxia o alteración de la función de la proteína VHL, bloquea la interacción HIF-2α-HIF-1β, lo que reduce la transcripción y la expresión de los genes diana del HIF-2α.
Eficacia clínica: Dos ensayos clínicos, uno para el cáncer de células renales claras y el otro para tumores asociados a la enfermedad de Von Hippel-Lindau (VHL). El primer ensayo clínico fue de Fase 3, abierto, aleatorizado y controlado con tratamiento activo para comparar belzutifán con everolimus en 746 pacientes con CCR de células claras no resecable, localmente avanzado o metastásico, que ha progresado después de administrar inhibidores de puntos de control PD-1/L1 y tratamientos dirigidos al receptor del VEGF en secuencia o en combinación. Las variables primarias de eficacia fueron la supervivencia libre de progresión (SLP, mediana de 4,6 con belzutifan vs 5,4 meses con everolimus) y la supervivencia global (SG, mediana de 21,8 vs 18,1 meses), mientras que las variables secundarias fueron la tasa de respuesta objetiva (TRO: 2,7 vs 0% completa y 21,4 vs 3,3% parcial) y la duración de la respuesta (DOR: no alcanzada con belzutifan vs mediana de 17,2 meses con everolimus). El segundo ensayo clínico fue de Fase 2, abierto, en 61 pacientes con enfermedad de VHL que tenían al menos un tumor sólido medible localizado en el riñón y que no requería cirugía inmediata, aunque también podían presentar otros tumores asociados a la enfermedad de VHL, como hemangioblastomas en el SNC y tumores neuroendocrinos pancreáticos. La variable primaria de eficacia para el tratamiento del CCR asociado a la enfermedad de VHL fue la TRO (11,5% completa y 55,7% parcial); otras variables de eficacia fueron la duración de la respuesta (no alcanzada, pero el 100% tuvo una duración ≥12 meses) y el tiempo hasta la respuesta (mediana de 11,1 meses).
Eventos adversos: Los más comunes (≥10%) son anemia, mareos, disnea, hipoxia, hemorragia, náusea y fatiga.
(N) SISTEMA NERVIOSO
SUZETRIGINA (JOURNAVX®) VERTEX (USA)
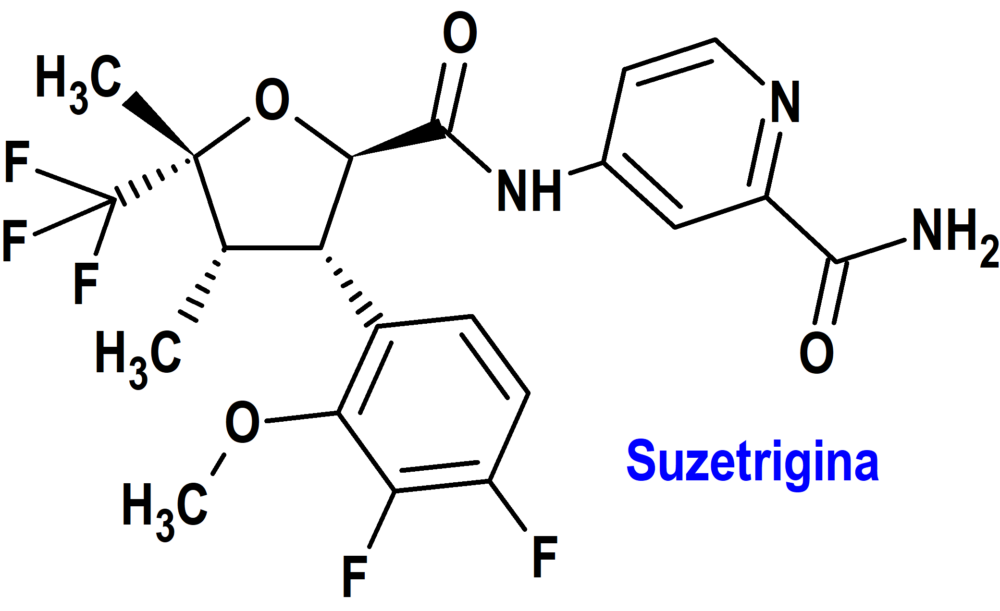
Indicación: Tratamiento del dolor agudo moderado a severo en adultos.
Tipo: Medicamento sintético estándar constituido por la 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-metoxifenil)-4,5-dimetil-5-(trifluorometil)oxolano-2-amido]piridina-2-carboxamida. Autorizado (FDA) en Estados Unidos el 30 de enero de 2025 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no disponible en la Unión Europea.
Mecanismo: Bloqueador selectivo del canal de sodio dependiente de voltaje NaV1.8 (pero no de otros canales de sodio dependientes de voltaje de la serie NaV1). NaV1.8 se expresa en neuronas sensoriales periféricas, incluidas las neuronas ganglionares de la raíz dorsal, donde su función es transmitir señales de dolor (potenciales de acción). Al inhibir selectivamente los canales NaV1.8, la suzetrigina inhibe la transmisión de señales de dolor a la médula espinal y al cerebro. M6-SUZ, un metabolito activo principal, es 3,7 veces menos potente que la suzetrigina como inhibidor de NaV1.8.
Eficacia clínica: Dos ensayos aleatorizados, doble ciego, controlados con placebo y con fármaco activo (combinación de hidrocodona y paracetamol, HP) sobre el dolor agudo, uno después de una abdominoplastia completa (ensayo 1) y el otro después de una bunionectomía (ensayo 2). En cada ensayo, se midió la intensidad del dolor utilizando una escala numérica de valoración del dolor (NPRS) de 11 puntos informada por el paciente, que va de 0 a 10, donde cero corresponde a ningún dolor y 10 corresponde al peor dolor imaginable. El ensayo 1 evaluó la eficacia de suzetrigina durante 48 horas en 1118 pacientes adultos con dolor agudo de moderado a intenso después de un procedimiento de abdominoplastia completa. La eficacia se evaluó mediante la suma ponderada en el tiempo de la diferencia de intensidad del dolor de 0 a 48 horas (SPID48), encontrándose una diferencia media de 48,4 vs placebo y de 6,6 vs HP; asimismo, el tiempo medio hasta el alivio significativo del dolor (definido como una reducción ≥ 2 puntos en NPRS) fue de 119 minutos para los pacientes del grupo suzetrigina y de 480 minutos para los pacientes del grupo placebo, mientras que el tiempo medio hasta el inicio del alivio perceptible del dolor (definido como una reducción ≥ 1 punto en NPRS) para los pacientes del grupo suzetrigina fue de 34 minutos. El ensayo 2 evaluó la eficacia de suzetrigina durante 48 horas en 1073 pacientes adultos con dolor agudo moderado a severo después de una bunionectomía; la diferencia media SPID48 fue de 29,3 vs placebo y de -20,2 vs HP; asimismo, el tiempo medio hasta el alivio significativo del dolor fue de 240 vs 480 minutos en el grupo placebo y el tiempo medio hasta la aparición de un alivio perceptible del dolor para los pacientes del grupo suzetrigina fue de 60 minutos.
Eventos adversos: Los más comunes (con mayor incidencia que el placebo) son prurito (2,1%), espasmos musculares (1,3%), elevación de la creatina fosfocinasa sérica (CPK, 1,1%) y erupción exantemática (1,1%).
(V) VARIOS
ANTÍGENOS DERIVADOS DE MYCOBACTERIUM
TUBERCULOSIS (rdESAT-6/rCFP-10) (SIILTIBCY®) SERUM LIFE SCIENCE (UE)
Indicación: Como ayuda diagnóstica para la detección de la infección por Mycobacterium tuberculosis, incluida la enfermedad, en adultos y niños de 28 días o más.
Tipo: Medicamento biológico constituido por un antígeno quimérico formado por las proteínas CFP10 y ESAT6. CFP10 es un antígeno de 10 kDa codificado por el gen esxB, mientras que ESAT6 es un antígeno temprano de 6 kDa y codificado por el gen esat-6. Autorizado en la Unión Europea el 13 de enero de 2025; no disponible en Estados Unidos.
Mecanismo: Antígeno quimérico formado por las proteínas CFP10 y ESAT6. CFP10 es un antígeno de 10 kDa codificado por el gen esxB; esta proteína juega un papel en la virulencia de la bacteria y no está presente en muchas de las micobacterias no tuberculosas (NTMs). ESAT6 es un antígeno temprano de 6 kDa y codificado por el gen esat-6; esta proteína también juega un papel en la virulencia de la bacteria formando un complejo de secreción 1:1 con la proteína CFP10. La presentación intradérmica de los antígenos al sistema inmunitario induce una reacción de hipersensibilidad de tipo retardado en las personas portadoras de la bacteria, que se manifiesta como una induración en el lugar de la inyección.
Eficacia clínica: El rendimiento diagnóstico de Siiltibcy (sensibilidad y especificidad) se comparó con dos productos destinados al diagnóstico de Mycobacterium tuberculosis: la prueba Quantiferon TB Gold In-Tube (QFT, una prueba in vitro) y el derivado purificado de la tuberculina (PPD RT23, una prueba utilizada intradérmicamente, como Siiltibcy). En comparación con QFT, Siiltibcy es más fácil de usar. En comparación con PPD RT23, Siiltibcy tiene una sensibilidad ligeramente inferior, pero la ventaja de una mayor especificidad en personas vacunadas con Bacillus Calmette-Guérin y da menos falsos positivos, ya que excluye mejor a las personas infectadas por micobacterias distintas de Mycobacterium tuberculosis.
Eventos adversos: Los más comunes son prurito, dolor y hematoma en el lugar de la inyección.
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE) Medicamentos Prioritarios
(Priority Medicines; PRIME): Es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): Son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes); no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca o el cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (lo que supone una prevalencia aproximada de 7,5/10.000 habitantes en la actualidad).
Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
Producto calificado para enfermedades infecciosas. (Qualified Infectious Disease Product): un medicamento antibacteriano o antifúngico para uso humano destinado a tratar infecciones graves o potencialmente mortales, incluidas aquellas causadas por un patógeno resistente a antibacterianos o antifúngicos, incluidos patógenos infecciosos nuevos o emergentes.
