1. INTRODUCCIÓN
1.1. Los comienzos de la investigación. Objetivos
En 2013, nuestro Grupo de Terapias Avanzadas de la Universidad Complutense de Madrid estaba centrado en el desarrollo de protocolos de terapia génica no viral para la hemofilia B. Estos protocolos, sin embargo, eran muy poco eficaces y las posibilidades de traslación clínica eran muy bajas. Este hecho coincidió con el conocimiento de una paciente, Celia, de Jaén, a través de la Asociación Andaluza de Hemofilia. Esta paciente presentaba y presenta un déficit grave de factor V de la coagulación que ni siquiera estaba bien diagnosticada ni mucho menos estudiada en su causa mutacional. Gracias a la colaboración de la presidenta de aquella asociación de pacientes en aquel momento, y por el reto que representaba para el coordinador del Grupo de Terapias Avanzadas de la Universidad Complutense de Madrid el abordar una enfermedad ultra rara como es el déficit de factor V, que no tenía, ni tiene, un tratamiento específico, hizo que, en 2014, se cambiara el rumbo de las investigaciones. En 2015, se organiza, gracias a las iniciativas de micro mecenazgo de los padres de la paciente, un primer evento de crowdfunding para el proyecto de factor V. En noviembre de ese mismo año se registraba el proyecto en Orphanet. En 2017 se recibe la primera ayuda de financiación por una empresa farmacéutica y el proyecto se inicia poniendo, esta vez sí, no como en ocasiones anteriores, cara y la máxima relevancia a un trabajo de investigación. Los desafíos y la responsabilidad adquiridos eran muchos porque nada se había investigado sobre los tratamientos para este déficit de la coagulación, pero a cambio se recibía un alto reconocimiento y apoyo incondicional de la familia de Celia y de la sociedad que apoyaba el proyecto.
El éxito del proyecto por la alta transversalidad de los objetivos requería inevitablemente la colaboración de distintos grupos e instituciones de investigación de forma multidisciplinar, como así ha sido. Más tarde y todo canalizado a través de una joven asociación creada por los padres de la paciente para este fin, la Asociación para la Investigación y Cura del Déficit de Factor V, Una Esperanza para Celia, con iniciativas de micro mecenazgo o financiación colectiva (crowdfunding), así como la financiación recibida de algunas empresas farmacéuticas y de la propia Universidad Complutense de Madrid con contratos predoctorales, se inició el ambicioso proyecto de investigación para lograr la “cura” del déficit de factor V.
El objetivo general del proyecto era establecer protocolos de terapias avanzadas, génica o celular para el tratamiento de esta patología. De este objetivo general derivaron los objetivos específicos. Así, el primero de ellos era establecer la causa mutacional de este déficit congénito en el gen F5 de la paciente y su segregación parental; después como para cualquier otro tipo de enfermedad se precisaba contar con modelos experimentales de patología tanto para estudios in vitro en cultivos celulares (modelo celular) como en modelo animal vivo, que presentara deficiencia de factor V y que se pudiera utilizar para estudios in vivo; era también necesario estandarizar las medidas de la actividad funcional de factor V en ratón que hasta ese momento nada se había descrito, y por último, como objetivo final, era demostrar la prueba de concepto de la utilidad de una terapia génica, en nuestro caso con vectores lentivirales, para la corrección del defecto genético en el gen F5.
1.2. Las técnicas clásicas y biotecnológicas que fueron imprescindibles
En el momento presente, con el fin de abordar un proyecto de este tipo, muy complejo y costoso, es fundamental la colaboración entre distintos centros y grupos de investigación con el fin de lograr una sinergia tanto en ideas y conocimientos específicos como en infraestructura y medios en general. Esta colaboración multidisciplinar ha permitido disponer de las últimas y más novedosas técnicas, muchas de ellas del área de la biotecnología.
Se han utilizado técnicas clásicas mejoradas como las propiamente dichas de obtención de muestras, tanto de líquidos fisiológicos como sangre y orina, o a partir de procedimientos quirúrgicos como la obtención de órganos y tejidos. Técnicas otras relacionadas con la preservación de las muestras en cadena de frio o fijación celular, su procesamiento y fraccionamiento y su adecuación a las exigencias de las diversas técnicas de evaluación. Técnicas analíticas en el campo de la bioquímica como las determinaciones enzimáticas o de otros parámetros bioquímicos; también aquellas relacionadas con la Biología Molecular o de ADN recombinante, como la obtención de ácidos nucleicos, su purificación y su amplificación mediante la reacción en cadena de la polimerasa, el estudio de expresión de ARNm, la subclonación en plásmidos, la purificación de vectores virales, la preparación de virus recombinantes mediante el empaquetado de fragmentos de ADN, o estudios de integración viral en órganos y tejidos. También la proteómica ha sido un campo de gran utilidad para nuestros objetivos que nos han permitido el estudio de proteínas en su estructura primaria, secundaria y terciaria.
Por otro lado, los métodos de cultivos celulares nos han permitido llevar a cabo estudios in vitro y la reducción del número de animales de experimentación. También para evaluar los efectos tras la administración de un vector viral en un organismo se han realizado estudios anatomopatológicos mediante análisis histopatológicos y de inmunohistoquímica, utilizando complejos medios iconográficos con el empleo de microscopia óptica y de fluorescencia de alta resolución.
No menos importantes han sido los estudios in silico que han permitido llegar muy lejos antes de iniciar los experimentos reales reduciendo también así el numero de animales de experimentación. Para ello se han utilizado bases de datos de mutaciones humanas y repositorios científicos; bases de datos genéticos y de genoma de ratones; herramientas de alineamiento de secuencias múltiples; herramientas de predicción de efectos de variantes; algoritmos para predecir el impacto funcional de mutaciones; servidores web de modelado de homología 3D de proteínas; análisis de impacto de mutaciones en la estabilidad de proteínas; software de visualización de gráficos moleculares y repositorios para visualización online de modelos 3D.
Por las características particulares de este proyecto que está estrechamente relacionado con la coagulación sanguínea, se han utilizado análisis hematimétricos desde los parámetros típicamente de rutina de hemograma hasta los estudios específicos de coagulación como ha sido la cuantificación del tiempo de protrombina (TP), del tiempo de tromboplastina parcial activada (TTPa) o la medida de la actividad y niveles de factor V.
Pero realmente las técnicas que condicionan el éxito de objetivos en terapias avanzadas han sido tres las imprescindibles, todas ellas biotecnológicas por excedencia. Así, la Secuenciación de Nueva Generación (NGS) que ofrece la secuenciación rápida y precisa de grandes secuencias de material genético, analizando múltiples fragmentos de ADN simultáneamente, lo que le da un alto nivel de detalle, precisión y velocidad. Esto nos ha permitido la detección de mutaciones en el gen F5 y el genotipado del genoma de animales. También las más modernas técnicas de reproducción asistida como ha sido la microinyección en cigotos que nos ha permitido la obtención de modelos animales con una determinada mutación en el gen F5. Y, por último, y la técnica estrella, la edición génica mediante el sistema CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats o Repeticiones Palindrómicas Cortas Agrupadas y Regularmente Espaciadas), que permite realizar modificaciones muy precisas en el ADN. Ha posibilitado la inducción de mutaciones concretas en el genoma, la recuperación y corrección de mutaciones y la producción de modelos animales transgénicos con mutaciones en el gen F5, mediante la microinyección de los componentes del sistema CRISPR en el propio cigoto para inducir la mutación.
1.3. La bioética y la significación estadística como condiciones sine qua non para la investigación
La ética en investigación es una condición sine qua non para desarrollar cualquier labor investigadora tanto con la utilización de muestras humanas como en el uso de animales de experimentación. Así en relación con muestras humanas se han respetado en todo momento las directrices de la Declaración de Helsinki y los experimentos se aprobaron siempre por los comités de ética de investigación en los centros hospitalarios y de investigación en los que se obtenían muestras o se realizaba la experimentación. Se obtuvieron los consentimientos informados de todos los pacientes participantes en el estudio en el momento de la toma de muestras de sangre con fines clínicos. Estos estudios se aprobaron por el Comité de Ética Médica del Hospital Universitario de Jaén y del Hospital Santa Creu i Sant Pau de Barcelona. Los donantes de sangre aceptaron participar mediante consentimiento informado por escrito.
Los estudios con animales de experimentación se llevaron a cabo de acuerdo con el Real Decreto 118/2021 por el que se modifica el Real Decreto 53/2013 y la Directiva de la UE 2010/63/EU. El estudio experimental fue aprobado por el Comité de Ética de la Facultad de Veterinaria de la Universidad Complutense de Madrid, el Comité de Ética para Experimentación Animal de la Universidad Complutense y los comités de ética del Instituto Nacional de Investigación y Tecnología Agraria (INIA) y de la Comunidad de Madrid (PROEX 258.1/21; PROEX 358.4/21; PROEX 040/17). Para los procedimientos de terapia génica lentiviral, que se llevaron a cabo en el Animalario del Hospital Nacional de Parapléjicos de Toledo, con un nivel de contención II, se obtuvo la aprobación del Comité de Ética y del Órgano Habilitador del Hospital Nacional de Parapléjicos de Toledo y la Comunidad de Castilla La Mancha. (Proyecto de experimentación 10-2023) Durante todo el transcurso de los experimentos con animales de experimentación siempre se consideró el principio de las 3Rs (Reducción, Reemplazo y Refinamiento) propuesto en 1959 por William Russel y Rex Burch (1). Además, se garantizó el bienestar animal en toda su extensión desde el mantenimiento de su salud física hasta su salud comportamental, así como la reducción o eliminación total del dolor y la angustia de los animales, en base a las reglas mínimas de las libertades.
En cuanto a la significación estadística, ésta da veracidad a los resultados a los que les infiere una correlación contrastada con la realidad. El análisis estadístico se realizó con los paquetes Statgraphics Centurion XVII (Statgraphics Technologies Inc, The Plains, VA, EE.UU.) y SPSS 27, v9.4 (SAS Institute, Cary, NC, EE.UU.), considerándose significativos los resultados cuando p < 0,05. Las curvas de calibración se generaron en Excel (Microsoft Office 365, Microsoft, Albuquerque, NM EE.UU.), y los gráficos se diseñaron con GraphPad Prism 8 (GraphPad Software, La Jolla, CA, EE.UU.). En el caso de la obtención de un modelo celular deficiente en factor V mediante edición génica, y su corrección, se empleó la prueba t de Student para el análisis estadístico. En la estandarización de la medida de actividad de factor V en Mus musculus, se usaron Kolmogorov-Smirnov para evaluar la normalidad y t de Student para diferencias entre sexos. En la obtención de un modelo animal patológico en ratón deficiente en factor V mediante edición génica y técnicas de reproducción asistida, la normalidad se evaluó con Shapiro-Wilk y la homogeneidad con Levene; se aplicó la prueba t de Student para diferencias entre los grupos, salvaje (WT), heterocigoto (Hz) y homocigoto (Hm). Para la prueba de concepto de terapia génica lentiviral para la corrección in vitro e in vivo de la mutación en el gen F5, para la expresión transitoria en HepG2 y HEK-293T, se emplearon ANOVA (ajuste de Tukey) y Kruskal-Wallis (ajuste de Dunn), y para comparaciones entre 48 y 72 horas, la prueba de Wilcoxon. El análisis en HepG2 mutadas a distintas dosis incluyó ANOVA (ajustes de Tukey y Games-Howell) y prueba t para muestras no pareadas entre WT y mutadas a dosis 0. Las comparaciones intragrupo en el experimento in vivo entre semanas 8 y 11 se realizaron con Wilcoxon y entre grupos independientes con Kruskal-Wallis (ajuste de Dunn). Las integraciones del vector en tejido hepático se evaluaron con Mann-Whitney, y para la gradación histológica se emplearon Mann-Whitney (WT vs Hm) y ANOVA o Kruskal-Wallis según correspondiera.
1.4. Hemostasia, el proceso homeostático por excelencia
La hemostasia es un proceso fisiológico que se encuentra presente en todos los vertebrados y que actúa a modo de respuesta fisiológica frente a una lesión, traumatismo o daño vascular. En este sistema se encuentran implicados multitud de componentes entre los que destacan los factores de coagulación, el fibrinógeno, las proteínas asociadas a la coagulación como la proteína C, S y Z, las plaquetas, el factor von Willebrand (FvW) y las células endoteliales que interactúan en una serie de reacciones en cadena para dar lugar al coágulo de fibrina (2). Estos componentes llegan a formar el trombo y de igual manera reestablecen la fluidez de la sangre mediante el proceso de fibrinolisis (3). La hemostasia primaria es aquella que ofrece la primera respuesta del organismo frente a la lesión vascular y endotelial, centrándose en una acción rápida en la que intervienen las plaquetas y el endotelio vascular dañado en la que el objetivo es el de la formación del trombo blanco o primario (2,4). La función de este coágulo es principalmente mecánica sellando la lesión del vaso y previniendo la pérdida continua de sangre mientras se activan mecanismos de coagulación más duraderos que corresponden a la hemostasia secundaria. Esta hemostasia secundaria o también llamada cascada de la coagulación tiene como objetivo la formación de la fibrina por la conversión de fibrinógeno soluble tras una serie de reacciones pro enzimáticas formando el trombo rojo o coágulo de fibrina. A su vez todos estos procesos constan de varias vías, donde encontramos la vía del factor tisular (FT) o vía extrínseca, la vía de amplificación o intrínseca y la vía común (5). La vía extrínseca es la que primero reacciona y se desencadena tras el daño vascular, entrando los componentes de la sangre en contacto con las células endoteliales. Estas células endoteliales secretarán una glicoproteína integral transmembrana, el FT, encargado de iniciar la cascada de la coagulación. Este FT entrará en contacto con el factor VII activado (FVIIa) formando el llamado complejo iniciador. Este complejo transformará al factor IX (FIX) en activado (6). Por otro lado, se encuentra la vía intrínseca, encargada de generar la trombina. Esta vía comienza con la activación del factor XII (FXII), que cuando entra en contacto con una superficie cargada negativamente, producto de un daño vascular, hace que este factor se active. El FXIIa se encargará de transformar la precalicreína (Pre-K), a la cual va unida el quininógeno de alto peso molecular (HMWK; high molecular weight kininogen), en calicreína (Kal), la cual también activará más FXII a FXIIa. Tras esto el FXIIa activará al factor XI (FXI) y este a su vez al factor IX (FIX), que se unirá al factor VIII activado (FVIIIa) que lleva asociado el FvW para evitar su degradación. Este FVIII ha sido activado previamente por la trombina para formar el complejo tenasa. Este complejo tenasa está formado por la unión del FIXa con el FVIIIa junto con calcio (Ca2+) y fosfolípidos (FL), que activarán al factor X (FX) (7). Tras esto, el FXa se unirá al factor V activado (FVa) para formar el complejo protrombinasa, que transforma la protrombina en trombina en presencia de FL y Ca2+. Tras esto, la trombina activará al fibrinógeno que formará polímeros de fibrina que serán estabilizados por el factor XIII activado (FXIIIa) uniéndose mediante enlaces covalentes a los residuos de glutamina y lisina de las fibras de fibrina. Este FXIII se activa previamente por la trombina en presencia de Ca2+ adquiriendo capacidad transglutaminasa. Esta fibrina va a interactuar con el trombo plaquetario y formará el coágulo final que ocluirá la lesión vascular (8) (Figura 1).
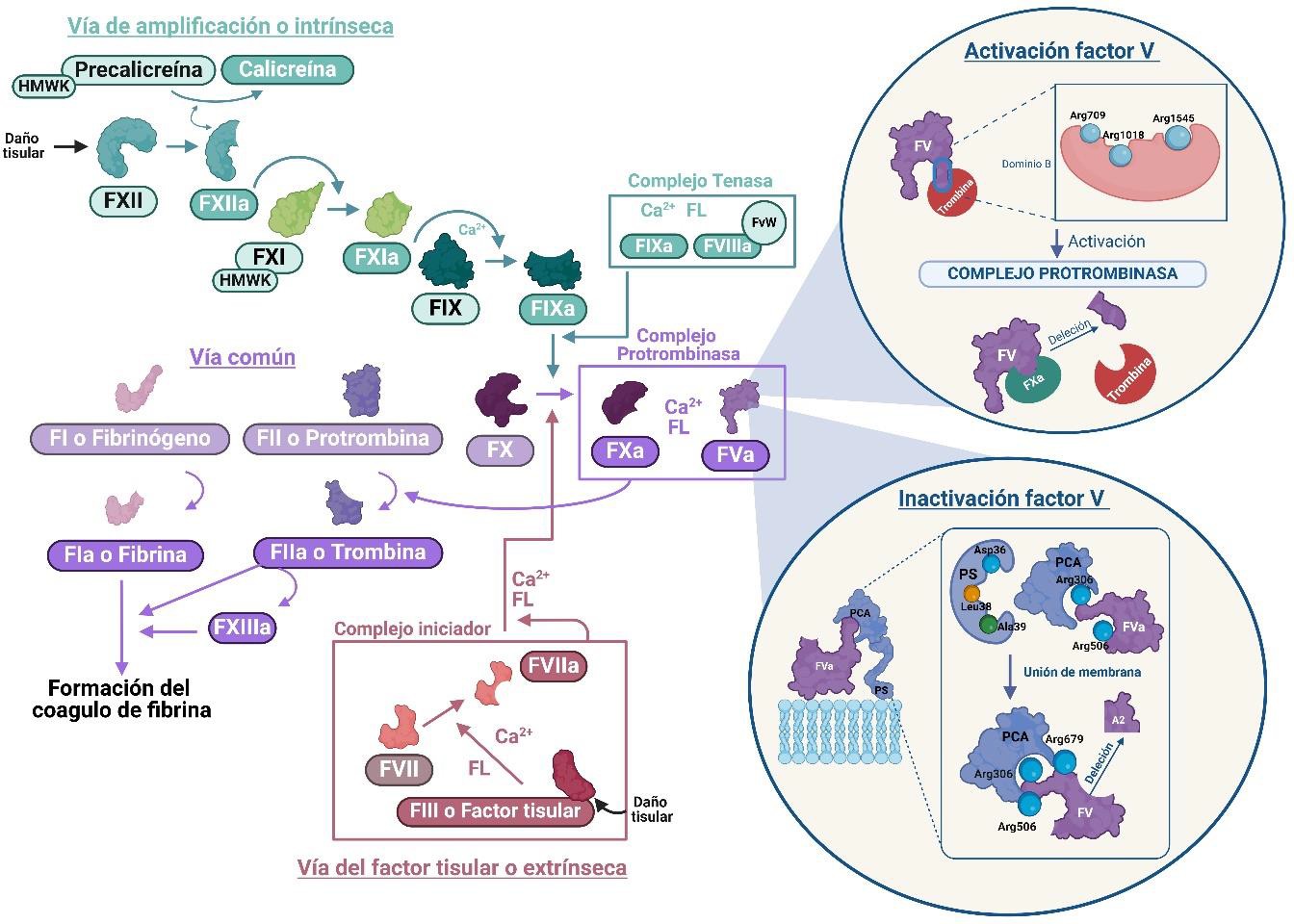
Figura 1. Hemostasia y factor V (HMWK, quininógeno de alto peso molecular; F, factor de la coagulación; FL, fosfolípidos; FvW, factor von Willebrand; PCA, proteína C activada; PS, fosfatidilserina)
La hemostasia se considera un proceso homeostático por excelencia ya que la restauración de la circulación sanguínea tras una lesión se basa en un equilibrio muy fino entre coagulación (formación de trombos) y la fibrinolisis, proceso mediante el cual la fibrina y el coágulo son degradados por la plasmina generando componentes y productos de degradación que posteriormente son eliminados a través del hígado. Este equilibrio de procesos es crucial para la prevención de la formación de trombos que puedan ocluir los vasos sanguíneos y causar complicaciones isquémicas (9). Pero en ocasiones este fino equilibrio de fuerzas se altera en situaciones de determinadas patologías relacionadas con la coagulación. Algunas afectan indirectamente a la hemostasia ya que modifican el equilibrio hemostático a través de mecanismos que alteran la función endotelial, la respuesta inflamatoria, el metabolismo de los lípidos o la función renal y hepática (10). Las enfermedades hepáticas, como la cirrosis, también tienen un impacto significativo ya que el hígado es el principal productor de factores de la coagulación (10). Las patologías autoinmunes, como el lupus eritematoso sistémico, afectan también a la hemostasia a través de la producción de anticuerpos antifosfolípidos, que elevan el riesgo de trombosis y complicaciones obstétricas. Hay otras, las coagulopatías congénitas, que afectan directamente a la coagulación. Estas pueden presentarse por diferentes causas, ya sea por un exceso de coagulación (trombofilias), como la enfermedad de Leiden, o por una hipocoagulabilidad (trastornos prohemorrágicos), lo que da lugar a problemas clínicos significativos que en muchas ocasiones producen enfermedades raras como la hemofilia A y B y otras ultra raras como la deficiencia de FV (11). En este caso pueden afectar tanto a la hemostasia primaria, que involucra principalmente a las plaquetas y al FvW (12), como a la hemostasia secundaria, que incluye los factores de la coagulación responsables de la formación de fibrina. Los pacientes con coagulopatías por defecto, a menudo, experimentan sangrados prolongados o espontáneos, hematomas extensos y hemorragias articulares (hemartros) o musculares, e incluso hemorragias en órganos internos comprometiendo su vida, lo que puede afectar significativamente a su calidad de vida y llevar a complicaciones crónicas. Las hemofilias tanto la A como la B son los trastornos de la hemostasia secundaria más conocidos que, aunque son de baja prevalencia y se consideran enfermedades raras, disponen de mayores avances en su tratamiento. Se caracterizan por ser trastornos hereditarios ligados al cromosoma X, ya que los genes afectados se encuentran en este cromosoma (gen F8 y gen F9 respectivamente) (13). Los avances en factores recombinantes con vidas medias prolongadas (14), y el desarrollo de los llamados fármacos miméticos (15) ofrecen para estas dos enfermedades un óptimo tratamiento paliativo seguro y eficaz. Existen otras deficiencias menos frecuentes y conocidas de otros factores de la coagulación que de igual forma siguen caracterizándose como enfermedades raras o ultra raras, como es el déficit de factor V, que no gozan de ningún tratamiento específico (11).
1.5. Enfermedades raras y ultra raras
Las llamadas enfermedades raras o poco frecuentes son aquellas que presentan una prevalencia menor de 1 por cada 2000 personas. Este tipo de enfermedades son producidas en su mayor parte por causas genéticas hereditarias (alrededor de un 80%), aunque también las hay adquiridas o derivadas de procesos neoplásicos o autoinmunes. Estas enfermedades aparecen en un 70% en la infancia; cerca del 95% carecen de tratamientos aprobados; el tiempo medio para un diagnóstico preciso es de 4 a 8 años; cerca del 30% de los niños con una enfermedad rara fallecen antes de los 5 años; el 50% no tienen un método de diagnóstico, y además, son la causa del 25% de las enfermedades crónicas conocidas (16). Se ha propuesto también el concepto de enfermedades ultra raras, que son aquellas que presentan una prevalencia de 1 por cada 50000. El diagnóstico de las enfermedades raras representa un gran desafío en la práctica médica debido a su baja prevalencia, a la alta diversidad de manifestaciones clínicas y a la falta de conocimientos etiopatogénicos. En muchos casos el diagnóstico puede demorarse muchos años, dificultando la aplicación de un tratamiento adecuado, si lo hubiere, para los pacientes (16). Las pruebas genéticas juegan un papel crucial en el diagnóstico de muchas enfermedades raras, especialmente aquellas de origen hereditario. El avance en la secuenciación de nueva generación (17) y en la inteligencia artificial (18) ha permitido un acceso más amplio a diagnósticos genéticos, facilitando la identificación de mutaciones responsables de estas patologías. Esto permite también un diagnóstico precoz que es crucial para mejorar el pronóstico y la calidad de vida de los pacientes con enfermedades raras. En algunos casos, el diagnóstico neonatal y la implementación de programas de cribado pueden identificar enfermedades antes de la aparición de los síntomas, lo que permite intervenciones tempranas. El tratamiento de las enfermedades raras presenta desafíos únicos debido a la diversidad de estas patologías y la limitada disponibilidad de terapias específicas. Se sabe que un 95% de todas estas enfermedades no presenta un tratamiento específico. Dado que cada enfermedad rara afecta a un pequeño número de personas, la investigación farmacéutica y el desarrollo de tratamientos específicos suelen estar limitados, lo que se traduce en un número reducido de opciones terapéuticas aprobadas (19). Así que la realidad es que la mayoría de los pacientes con enfermedades raras dependen de tratamientos paliativos que se centran en aliviar los síntomas y mejorar la calidad de vida, más que en curar la enfermedad subyacente. En cuanto a los tratamientos específicos, los medicamentos huérfanos juegan un papel crucial. Estos son medicamentos desarrollados específicamente para tratar enfermedades raras y que, debido a la poca rentabilidad económica para las empresas farmacéuticas, reciben incentivos regulatorios y financieros para fomentar su investigación y desarrollo (20). Las nuevas terapias avanzadas representan áreas prometedoras en el tratamiento de enfermedades raras, especialmente aquellas de origen genético, con potencial de corregir la mutación genética subyacente y, en algunos casos, ofrecer una curación. Las enfermedades raras no impactan solamente en los individuos que las padecen, sino que también tienen un efecto profundo en las familias y en la sociedad en su conjunto. Desde el punto de vista social, estas enfermedades generan grandes retos en términos de atención médica, inclusión, y comprensión por parte de la comunidad. Las personas con enfermedades raras a menudo se enfrentan a estigmatización y al aislamiento social debido a la falta de conocimiento y escasa visibilidad de su problemática (16). Desde una perspectiva económica, las enfermedades raras imponen una carga financiera considerable tanto para las familias como para los sistemas de salud, principalmente en aspectos de financiación pública, suponiendo una gran carga económica. Los costes asociados a la atención médica especializada y a los tratamientos y los medicamentos huérfanos, suelen ser elevados, lo que genera dificultades económicas para las familias afectadas (21). A nivel político, el reconocimiento de las enfermedades raras y la creación de políticas públicas para su manejo son esenciales para garantizar que los pacientes reciban la atención y el apoyo que precisan. Esto incluye la implementación de programas nacionales de atención, de incentivos económicos a las familias, la promoción de la investigación, y el desarrollo de registros de enfermedades raras para mejorar la recogida de datos y la planificación sanitaria, así como la implantación de leyes que beneficien a este tipo de pacientes. Las organizaciones de pacientes también desempeñan un papel crucial en abogar por los derechos de los pacientes y en la creación de redes de apoyo. Es importante destacar el papel de la comunidad y las organizaciones no gubernamentales en el apoyo a las personas con enfermedades raras. Estas organizaciones, a menudo lideradas por pacientes y sus familias, proporcionan recursos, información, y un sentido de comunidad que es esencial para afrontar los desafíos que se plantean al vivir el día a día con una enfermedad rara. Además, impulsan la investigación y el desarrollo de tratamientos, y luchan por políticas que mejoren la calidad de vida de los afectados.
1.6. Déficit de factor V de la coagulación
El FV es una proteína que actúa en la hemostasia secundaria, concretamente en la vía común. Esta proteína junto con el FX, previamente ambos activados, forman el complejo protrombinasa para que en presencia de Ca2+ y FL de membrana se unan a la protrombina y produzcan trombina (2). Este factor de la coagulación se codifica por el gen F5 que se encuentra en el brazo largo del cromosoma 1 en la posición q23. Fue descrito por primera vez en 1986 y está conformado por 26 intrones y 25 exones, siendo el exón 13 el de mayor longitud y menos conservado entre especies. El gen tiene 80 kb y el ARN mensajero, que codifica la proteína, 6,8 kb dando lugar a una proteína de un peso molecular de 330 kDa. Esta proteína se caracteriza por tener diferentes regiones y aminoácidos de interés entre los que destacan los anclajes a trombina, proteína C, sitios de N-glicosilación (FV1 y FV2), sitios de glicosilación, cisteínas, regiones ricas en tirosina sulfato, regiones donde se produce fosforilación de serina, región de inactivación del FVa, región procoagulante, región anticoagulante, de unión al FX, inactivación de la plasmina (Pn), unión a multimerina 1 y repeticiones en tándem en el dominio B (11). El FV se produce en un 80% en los hepatocitos y en un 20% en los megacariocitos (precursores de las plaquetas) (11) y está formado por unos 2224 aminoácidos en el ser humano y compuesto por una cadena pesada (dominio A1 y A2), un gran dominio B que posteriormente se escinde al activarse la proteína y una cadena ligera (dominio A3, C1 y C2) (Figura 1).
Circula libre en el plasma de forma inactiva, que para activarse requiere que previamente se active el propio sistema de la hemostasia, es decir que haya un daño vascular y una respuesta por parte del organismo (8,11). Para sufrir esta activación deben mediar proteasas como la trombina, el FXa o la plasmina. Aunque estas 3 moléculas pueden activar el FV, la molécula que lo hace con mayor eficacia y en mayor cantidad es la trombina, que se une a los residuos de arginina 709, 1018 y 1545. El FVa resultante está compuesto por una cadena pesada de 105 kDa y una cadena ligera de 71 o 74 kDa, que están conectadas entre sí por interacciones hidrofóbicas y un único ion Ca2+. Tras esto el FXa se unirá a las argininas 348, 709, 1045 y 1745 formando el complejo protrombinasa. Desde un punto de vista filogenético, respecto al FV en otras especies, no se encuentran diferencias en la función de la hemostasia secundaria. Sin embargo, las proteínas de cada especie distan en longitud, siendo por ejemplo en el ratón de 2184 aminoácidos (22). Entre especies existen dominios y regiones muy conservadas salvo el dominio B, que se escinde y es el menos conservado. Así mismo, se han reportado diferencias de funcionalidad con respecto al ser humano en otras especies como el ratón o el conejo, presentando en estos casos hasta una tasa de coagulación de 10 veces superior al ser humano (23). La deficiencia de FV se produce principalmente por etiología genética. Se trata de una enfermedad genética poco común con una herencia autosómica recesiva, causada por mutaciones en el gen F5 (11,24). Hasta la fecha, se han descrito más de 250 mutaciones en este gen, responsables de la enfermedad. La deficiencia de FV se clasifica como una enfermedad ultra rara, con una incidencia estimada de 1 a 9 casos por cada millón de nacidos vivos (11). Este déficit congénito puede presentarse en dos formas: la deficiencia tipo I, caracterizada por niveles muy bajos o indetectables tanto de antígeno como de actividad funcional del factor V, y la deficiencia tipo II, donde los niveles de antígeno son normales o levemente reducidos, pero la actividad funcional del factor V está muy disminuida. La mayoría de los pacientes presenta la deficiencia tipo I, siendo la forma más común de esta condición (11). Las mutaciones que causan la deficiencia de FV son diversas. Incluyen mutaciones missense, nonsense, inserciones, deleciones pequeñas, mutaciones splice y frameshifts, entre otras (11). La mayoría de las mutaciones presentes son de tipo nonsense y se concentran sobre todo en el dominio B de la proteína, mientras que las mutaciones missense se distribuyen principalmente en los dominios A y C, dando lugar, a veces, a las formas más leves de la enfermedad (11). La gran variedad de mutaciones en el gen F5 significa que los casos de deficiencia de FV son únicos, asociados a una familia o región geográfica específica (25). En algunas ocasiones, los pacientes presentan mutaciones combinadas, lo que se conoce como heterocigotos compuestos, y que pueden mostrar una variedad de síntomas dependiendo de la naturaleza y localización de las mutaciones (11). Las manifestaciones clínicas de esta patología van desde condiciones asintomáticas hasta hemorragias potencialmente fatales. Los pacientes pueden presentar hemorragias (sangrados en mucosas como epistaxis, menorragia y sangrado en la cavidad oral) espontáneas o sangrados exacerbados debido a traumatismos o procedimientos quirúrgicos. Las hemorragias más severas suelen ocurrir en órganos internos como el tracto digestivo o el sistema nervioso central, clasificándose la severidad según los niveles de FV circulante en el plasma. Un fenotipo leve se considera cuando los niveles son superiores al 10%, moderado cuando están entre el 1% y el 10%, y severo cuando están por debajo del 1% o son indetectables (11). Como la mayoría de las mutaciones que causan esta deficiencia congénita son recesivas, los individuos deben ser homocigotos o heterocigotos compuestos para manifestar un fenotipo patológico. Los heterocigotos para una de estas mutaciones presentan niveles plasmáticos de FV inferiores a los normales o son normales, pero generalmente no muestran síntomas ni problemas de coagulación, lo que correspondería a un fenotipo leve (25). El diagnóstico de la deficiencia de FV se puede realizar a través de un enfoque clínico y de laboratorio comenzando con pruebas rutinarias de coagulación como el TP y el TTPa cuyo alargamiento sería un indicador de una posible deficiencia. La confirmación de la sospecha se realizaría mediante estudio específico de los niveles de FV mediante métodos coagulométricos y estudios moleculares para identificar mutaciones en el gen F5. El tratamiento ―siempre paliativo― para esta enfermedad se basa en la administración de plasma fresco congelado (PFC) (26), o de algunos productos como el Octaplas®, que es un preparado intravenoso caracterizado por una combinación preestablecida cuantitativa y cualitativamente optimizada para distintos factores de la coagulación incluyendo el FV (27). La administración repetida de PFC puede provocar efectos secundarios como la lesión pulmonar aguda, hipervolemia, y a veces generación de inhibidores o procesos anafilácticos. Los antifibrinolíticos, como el ácido tranexámico o el ácido β-aminocaproico, pueden administrarse por vía oral o intravenosa para controlar hemorragias, especialmente en mucosas, y antes de una intervención quirúrgica invasiva. A las pacientes con sangrado menstrual abundante se les puede administrar terapia de sustitución con estrógenos o progesterona (28). En el año 2014, von Drygalski y colaboradores (29) consiguieron preparar un FV recombinante humano al que denominaron SUPERFVa. Este factor resiste la inactivación mediada por la proteína C activada (PCA), por lo que aumenta su vida media en plasma y muestra además una seguridad, eficacia y estabilidad óptimas, una baja inmunogenicidad, ausencia de efectos trombogénicos y un perfil farmacocinético óptimo. En el momento presente este factor no se encuentra en ningún tipo de ensayo clínico. Respecto a intentos mediante terapias avanzadas no se ha descrito todavía un protocolo potencialmente esperanzador, tan solo primeros abordajes con tímidos resultados con células madre pluripotentes inducidas (iPSCs) (30) o mediante la utilización de vectores adenoasociados que expresan FVa (31).
1.7. Modelos experimentales patológicos de enfermedad
1.7.1. Los modelos in vitro con líneas celulares
Este tipo de modelos se emplean para predecir respuestas clínicas in vitro para determinadas enfermedades. Un ejemplo relevante es el uso de líneas celulares linfoblastoides transformadas por el virus de Epstein-Barr, que se han utilizado ampliamente para analizar el efecto de fármacos frente a variaciones genéticas. Este tipo de modelos se ha empleado durante décadas para estudiar el comportamiento celular, la toxicidad de fármacos y las interacciones entre células y compuestos químicos. Para mejorar estos modelos en 2D, se han desarrollado modelos in vitro como tejidos tridimensionales u órganos en microchip siendo estos últimos los más usados (32). A pesar de los avances, los modelos in vitro aún presentan limitaciones. Por ejemplo, no siempre pueden replicar con precisión el microambiente de un organismo vivo, especialmente en estudios que requieren interacciones a nivel sistémico. Así mismo, la variabilidad en los resultados entre diferentes laboratorios, su precio en el mercado y de fabricación y la dificultad para reproducir algunos modelos más complejos, hacen que todavía sea necesario validarlos con otros enfoques, como estudios in vivo en animales o en humanos (33).
1.7.2. Los modelos animales
Un modelo animal de enfermedad es una especie animal no humana que presenta analogías patológicas con una enfermedad que se produce en humanos o en otros animales. Esto ha sido posible gracias al gran desarrollo biotecnológico desde que se creara el primer modelo animal en embriones de ratón insertando ADN exógeno mediante recombinación homóloga, hasta nuestros días en que, por ejemplo, la edición génica con el sistema CRISPR está haciendo posible la modificación del genoma de los animales. Los modelos animales se pueden clasificar, por semejanza a los seres humanos con respecto a la patología o situación que se quiera emular en: homólogos, isomórficos o parciales. Los modelos homólogos, son aquellos que presentan síntomas y procesos idénticos al ser humano; los isomórficos presentan síntomas similares, pero la causa es diferente entre el modelo animal y el humano, y los modelos animales parciales no imitan el proceso completo de estudio, sino que se usan para estudiar algunos aspectos o posibles tratamientos de dicho proceso patológico (34). Los modelos de enfermedad inducidos o experimentales son aquellos a los que se les provoca la condición a emular del ser humano de manera experimental (manipulación quirúrgica, administración de dietas modificadas, administración de sustancias biológicamente activas, cambios etiológicos, modificación genética). El uso de modelos animales se ha convertido en una herramienta fundamental para comprender y estudiar numerosos procesos biológicos tanto en humanos como en animales. Estos modelos han sido clave en la caracterización de distintos tipos de cáncer, enfermedades cardiovasculares, Parkinson, así como en el desarrollo de nuevos fármacos (35). En algunas ocasiones, la elección del modelo animal adecuado puede ser un desafío, ya que se requiere definir con precisión los objetivos y limitaciones de la investigación y basarse en los protocolos del estudio para su elección. Se busca establecer homologías entre el animal y el ser humano y delimitar las características del fenómeno biológico a estudiar, con el fin de obtener resultados fiables. Los vertebrados, en especial los mamíferos, y de ellos los roedores, son los animales más utilizados en la investigación biomédica. Cualquier especie puede ser susceptible de uso en investigación, aunque las especies más comunes son animales pequeños como ratas, ratones, cobayas y conejos, debido a su tamaño reducido, alta tasa de reproducción, rápida madurez sexual y facilidad de manejo (36). La transgénesis, o la introducción de material genético exógeno en el genoma de un organismo, ha sido y es el método casi de elección para generar modelos animales. Este proceso permite la creación de organismos que expresan genes modificados, lo que es muy valioso para estudiar la función de genes específicos y el desarrollo de enfermedades, así como el efecto de las nuevas terapias avanzadas como la terapia génica o celular en fases preclínicas de investigación. Más recientemente, las herramientas de edición genética como CRISPR/Cas han revolucionado la capacidad de generar modelos animales con mutaciones precisas y específicas, lo que ha mejorado enormemente la precisión y la eficacia en la creación de modelos animales. El ratón es el modelo más ampliamente utilizado debido a su similitud genética con los humanos y la facilidad con la que pueden ser modificados genéticamente. Los ratones ofrecen varias características ventajosas que, combinadas con su mantenimiento relativamente barato, los han convertido en importantes organismos modelo en la investigación de la hemostasia y la trombosis.
1.7.3. Modelos animales para la deficiencia de factor V
En el caso del déficit de FV, los modelos animales que se han desarrollado se caracterizan por ser no viables, lo que significa que no son susceptibles de ensayo de procedimientos ni estudios de una enfermedad ni de un tratamiento. En 1996 Cui y colaboradores (37) obtuvieron el primer modelo deficiente en FV mediante la técnica de recombinación homóloga. Lo que se observó es que aproximadamente la mitad de los individuos homocigotos se reabsorbían en torno al día E9-10 del periodo embrionario, posiblemente debido a una anomalía en el saco vitelino. El resto de los animales homocigotos que llegaron a término, murieron debido a una hemorragia masiva en las 2 horas posteriores al nacimiento. Se postula que esto es debido a la falta de trombina, relacionada con receptores PAR que son fundamentales para el desarrollo embrionario. En aquellos ratones con anomalías en estos receptores se producen lesiones vasculares en el saco vitelino y defectos en las paredes de grandes vasos. Años más tarde, el mismo grupo de investigación estudió los requerimientos del FV en la embriogénesis y en la hemostasia en ratones, generando líneas de ratones transgénicos que expresaban un minigén del FV bajo el control de los promotores específicos de tejido de albúmina (Malb) o del factor 4 plaquetario de rata (Rpf4). A pesar del bajo nivel de expresión del transgén de FV, se observó la recuperación del fenotipo letal. Sin embargo, la recuperación parecía ser incompleta, con una pérdida continuada de más de la mitad de los ratones esperados durante la embriogénesis temprana. Se concluyó que la presencia de FV en niveles inferiores al 0,1% es suficiente para permitir la supervivencia postnatal (38). Recientemente, Weyand y colaboradores (39) obtuvieron el primer modelo con déficit de FV mediante la técnica CRISPR/Cas9 en pez cebra, cuya hemostasia presenta sorprendentes similitudes con la hemostasia en humanos, generando un modelo KO deficiente mediante la deleción de 49 pares de bases en el exón 4 del gen F5. Aunque la embriogénesis de los individuos homocigotos no sufrió ninguna alteración, estos individuos sí experimentaron hemorragias fatales en la edad adulta.
1.8. Edición génica con CRISPR. La “herramienta”
Las posibilidades de transgénesis han evolucionado significativamente, desde los primeros métodos de inyección pronuclear hasta las modernas técnicas de edición de genes que permiten una manipulación genética precisa (40). Además, las técnicas de transgénesis han permitido la creación de modelos animales con genes informadores (reporter genes), que facilitan el estudio de la expresión génica y la actividad de proteínas en tiempo real en un organismo vivo. El sistema CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats o Repeticiones Palindrómicas Cortas Agrupadas y Regularmente Espaciadas), en particular, ha revolucionado el campo de la transgénesis al permitir la edición génica dirigida y eficiente en una amplia gama de organismos. Esta tecnología ha hecho posible la generación de modelos animales que contienen mutaciones específicas, lo que es esencial para el estudio de enfermedades genéticas y el desarrollo de nuevas terapias (41). Inicialmente descubierto en bacterias, CRISPR es un sistema inmune adaptativo que permite a los microorganismos defenderse contra virus invasores, conocidos como bacteriófagos. Funciona en combinación con proteínas asociadas, siendo Cas9 (CRISPR-associated protein 9) la más utilizada en biotecnología para la edición genética (42). El avance clave en la aplicación de CRISPR en biotecnología se produjo en 2012, cuando Jennifer Doudna y Emmanuelle Charpentier demostraron que el sistema CRISPR/Cas9 podía ser reprogramado para cortar ADN en secuencias específicas en organismos eucariotas, incluyendo células humanas. Este descubrimiento abrió la puerta a la edición génica dirigida, permitiendo realizar modificaciones precisas en el genoma de una manera más rápida, eficiente y económica que con tecnologías anteriores como las nucleasas de dedo de zinc (ZFNs) y las nucleasas TALEN (42). Cas9 es una endonucleasa, lo que significa que puede cortar las cadenas de ADN en un sitio específico. Esta especificidad es dirigida por un ARN guía que se empareja con la secuencia diana en el ADN, permitiendo a Cas9 realizar un corte preciso en el genoma, concretamente en la secuencia PAM (protospacer adjacent motif; motivo adyacente al protoespaciador), que se trata de una secuencia de nucleótidos complementaria al ARN guía que va a indicar a la proteína Cas dónde “cortar” (43). Este corte desencadena mecanismos de reparación del ADN en la célula, que pueden ser aprovechados para introducir mutaciones o corregir secuencias defectuosas. El sistema CRISPR/Cas es altamente modular, lo que significa que puede ser adaptado a diferentes propósitos de edición génica. Al cambiar la secuencia del ARN guía, es posible dirigir la proteína Cas a diferentes partes del genoma, permitiendo la edición simultánea de múltiples genes. La unión de extremos no homólogos (Non- Homologous End Joining, NHEJ) es uno de los principales mecanismos de reparación del ADN que se activa tras el corte inducido por Cas en el genoma o para realizar reparaciones de errores que ocurren de forma normal en células que no se encuentran en división (44). La Reparación Dirigida por Homología (Homology-Directed Repair, HDR) es otro mecanismo de reparación del ADN que se activa después de un corte de la doble hebra inducido por Cas o en células en división. A diferencia de NHEJ, HDR utiliza una secuencia de plantilla homóloga como referencia para reparar la rotura, lo que permite la corrección precisa de mutaciones o la inserción de secuencias específicas en el genoma. Sin embargo, HDR es menos eficiente que NHEJ y solo ocurre durante ciertas fases del ciclo celular, lo que limita su uso en algunas aplicaciones de edición génica 45). CRISPR/Cas ha encontrado una amplia gama de aplicaciones en biología y medicina, desde la creación de modelos animales hasta la terapia génica y la biotecnología agrícola. En la investigación biomédica, CRISPR se utiliza para generar modelos animales que replican enfermedades humanas, lo que permite estudiar la función de genes específicos y probar nuevas terapias. Sin embargo, a pesar de su gran potencial, la terapia génica basada en CRISPR/Cas aún presenta limitaciones, incluidas aquellas relacionadas con la seguridad, la especificidad y los posibles efectos fuera del objetivo (efectos off-target), que deben resolverse (46).
1.9. Nuevas terapias avanzadas
Las terapias avanzadas han revolucionado el campo de la medicina, proporcionando nuevas estrategias terapéuticas que van más allá de las terapias convencionales. Éstas incluyen la terapia génica, la terapia celular y la medicina regenerativa, dirigidas a abordar enfermedades complejas que carecen de tratamientos efectivos. Se basan en genes o células (advanced therapy medicinal products, ATMPs), que han abierto la puerta a tratamientos personalizados, ajustados a las necesidades individuales de los pacientes, y abriendo el paradigma hacia una medicina individualizada (47). La terapia celular implica el uso de células vivas para tratar enfermedades o reparar tejidos dañados, que han sido mínimamente manipuladas o procesadas ex vivo mediante propagación, expansión, selección, tratamiento farmacológico o alguna otra modificación de sus características biológicas. Conlleva el trasplante de células de un donante (terapia alogénica) o del propio paciente (terapia autóloga), o incluso en algunos casos de forma xenogénica (de otra especie). Así, las células madre mesenquimales (MSCs), han mostrado un gran potencial en la regeneración tisular y en la modulación del sistema inmunológico (48). En el ámbito de las coagulopatías, las células iPSCs muestran interesantes posibilidades para corregir deficiencias en factores de la coagulación, abriendo nuevas posibilidades terapéuticas para enfermedades como la hemofilia (49). La terapia génica se centra en la modificación o transferencia del material genético de las células para corregir mutaciones que causan enfermedades. Esta tecnología se ha expandido rápidamente en los últimos años desde que comenzara su investigación en la década de los 90´ para el trastorno de la inmunodeficiencia combinada severa, en donde se utilizó una copia funcional del gen que codifica la enzima adenosina desaminasa. Su utilidad potencial se contempla para enfermedades monogénicas como la hemofilia, así como en otras enfermedades complejas como el cáncer y trastornos neurodegenerativos. La terapia génica puede ser administrada ex vivo cuando, las células del paciente se modifican fuera del organismo y luego se reimplantan, o in vivo, donde los vectores se administran directamente al paciente (50). Los avances en vectores virales y no virales han permitido mejoras en la transferencia y eficacia del material genético. Sin embargo, la seguridad sigue siendo una preocupación clave, especialmente en lo que respecta a la inmunogenicidad y el riesgo de mutagénesis insercional cuando se utilizan vectores virales. El objetivo final de la terapia génica para las enfermedades genéticas es su “curación” a largo plazo mediante la expresión sostenida del gen transferido en niveles terapéuticos. Aún así, la terapia génica viral ha emergido como una herramienta potente y versátil para el tratamiento de una amplia gama de enfermedades genéticas, cardiovasculares, infecciosas y el cáncer. Esta técnica se basa en la utilización de virus modificados genéticamente e inactivados que actúan como vectores para introducir material genético terapéutico en las células del paciente, con el objetivo de corregir mutaciones genéticas, reprogramar funciones celulares o inducir la muerte de células patológicas. Los virus, debido a su capacidad natural para infectar células y transferir su genoma, son vectores ideales para la terapia génica. Sin embargo, el diseño de estos vectores requiere de una cuidadosa modificación para asegurar que sean seguros y eficaces, evitando efectos adversos. Los virus adenoasociados (AAV) y los lentivirales (LVV) son los que en la actualidad ofrecen un mayor potencial terapéutico, aunque se deben solucionar algunos inconvenientes como la capacidad de integración en el genoma del huésped y el perfil inmunogénico (51). El éxito de la terapia génica viral depende en gran medida de la selección adecuada del promotor que regula la expresión del transgén, la ruta de administración (local o sistémica) y la evasión de la respuesta inmune del huésped (52). Los vectores lentivirales de nueva generación han sido diseñados para minimizar riesgos, eliminando genes virales innecesarios y utilizando promotores que limitan la expresión del transgén a las células diana (53). Los lentivirus, debido a su naturaleza, presentan una baja inmunogenicidad y toxicidad, lo que significa que no provocan una respuesta inmune exacerbada por parte del huésped, permitiendo una expresión más estable y prolongada del gen terapéutico. La producción de vectores lentivirales implica la utilización de células empaquetadoras, comúnmente derivadas de la línea celular HEK 293-T, que se transfectan transitoriamente con plásmidos que codifican las proteínas virales necesarias para la producción del vector, junto con el transgén de interés. Después de la producción, el vector es purificado para eliminar cualquier contaminante y asegurar la calidad del producto final. Este proceso debe cumplir con las buenas prácticas de fabricación (GMP; Good Manufacturing Practices) para garantizar la seguridad y eficacia del vector en aplicaciones clínicas (54).
2. ESTUDIO MUTACIONAL CAUSAL DEL DÉFICIT DE FACTOR V
El primer objetivo específico planteado, fue definir el diagnóstico de nuestra paciente, en cuanto a la causa mutacional mediante técnicas de Biología Molecular y en cuanto a su fenotipo por medida de los niveles de factor V en plasma. También llegar a establecer la segregación parental para explicar su situación clínica (Figura 2). Los datos de la anamnesis de la paciente indicaban una alteración en la coagulación sanguínea lo que hacía pensar como causa más probable una mutación en un gen de algún factor de la coagulación. Se realizó el estudio mediante secuenciación masiva del genoma de la paciente y de sus progenitores. Esto llevó a su publicación, destacando el hallazgo de una nueva mutación todavía no descrita en ese momento en el gen F5 (25).
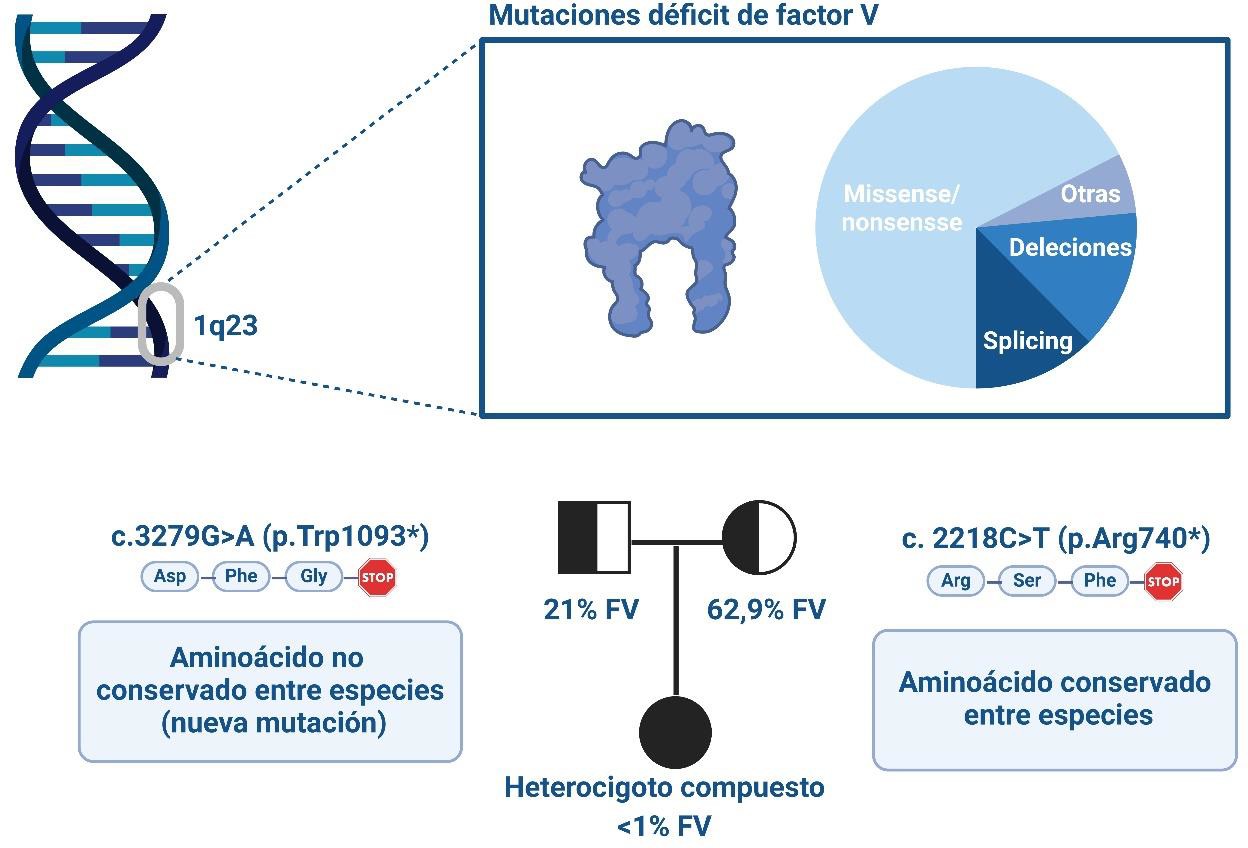
Figura 2. Estudio mutacional causal del déficit de factor V.
Los resultados mostraron un fenotipo grave por debajo del 1% de FV circulante en la paciente que se correspondía con los graves episodios de sangrado que había sufrido, y unos niveles moderados en sus progenitores que eran por decirlo así portadores asintomáticos de la enfermedad. Otra de las conclusiones del estudio fue el alto nivel de heterogeneidad mutacional del gen F5, mutaciones sin sentido, mutaciones con cambio de marco, cambios sin sentido, variantes de secuencias sinónimas y cambios intrónicos. Además, el estudio también incluyó un análisis filogenético comparado entre el FV y el FVIII dado su alto grado de homología genética y que comparten rutas biogenéticas y de la cascada de coagulación. En cuanto a la segregación familiar, la madre de la paciente era heterocigota para el gen F5 con la presencia de la mutación c.2218C>T, p.Arg740* ya descrita anteriormente en la literatura científica (55), y con niveles plasmáticos de FV del 62,9%, indicativo de un fenotipo leve sin síntomas clínicos dignos de mención. El padre, por su parte era heterocigoto para la mutación c.3279G>A, p.Trp1093*, que produce un codon de parada prematuro. Esta mutación, antes no descrita, que fue la gran novedad del estudio, producía en el padre unos niveles plasmáticos de FV del 21%, también sin síntomas significativos. La paciente presenta dos mutaciones diferentes heredadas de sus padres por lo que se trata de una heterocigosis compuesta que da lugar a una patología grave por estar afectados los dos alelos. Estas situaciones que se deben a la consanguinidad, en este caso debido a una procedencia de poblaciones muy próximas de los progenitores (en Andalucía se da todavía una cierta consanguinidad). Este hecho se produce en algunas regiones del Mundo de forma más frecuente, debido a una endogamia muy alta, por ejemplo, en países del norte de África, el África subsahariana, el mundo árabe, Asia y especialmente India y Pakistán, donde la cultura favorece los matrimonios consanguíneos por razones sociales, económicas o políticas.
La gran heterogeneidad de las mutaciones que afectan al gen F5, justifica la realización de un cribado mutacional completo del gen para obtener un diagnóstico molecular preciso de la enfermedad. Esto explica la alta correlación con un gran número de efectos muy variables a nivel funcional y molecular.
3. ESTANDARIZACIÓN, EN RATÓN, DE LA MEDIDA DE FACTOR V Y DE TIEMPOS DE COAGULACIÓN
El segundo gran objetivo específico se planteó a raíz de que nuestra intención era preparar en un tiempo razonablemente corto, como así fue después, un modelo patológico en ratón deficiente en FV, donde tendríamos que evaluar a la postre los niveles funcionales de esta proteína de la coagulación. Pero, aquí surgió el inconveniente, porque la especie Mus musculus, que se utilizaría para obtener el modelo, se caracteriza por niveles extraordinariamente elevados de este factor y tiempos de coagulación muy cortos con respecto a humanos (56), lo que dificulta la realización de medidas precisas. Por ello, era indispensable una caracterización detallada de tales parámetros (23).
Así, en el marco de este estudio se diseñó un método para determinar y normalizar con precisión los niveles de factor V, del tiempo de protrombina y del tiempo de tromboplastina parcial activada en Mus musculus (Figura 3). Estos parámetros se evaluaron en una muestra de 66 animales sanos, utilizando un coagulómetro semiautomatizado y reactivos, todo ello utilizado en el diagnóstico humano, y además en un intento de determinar la hora del día más adecuada para las extracciones, ya que en este tipo de animales el ciclo circadiano puede influir enormemente en los resultados, como así se comprobó.
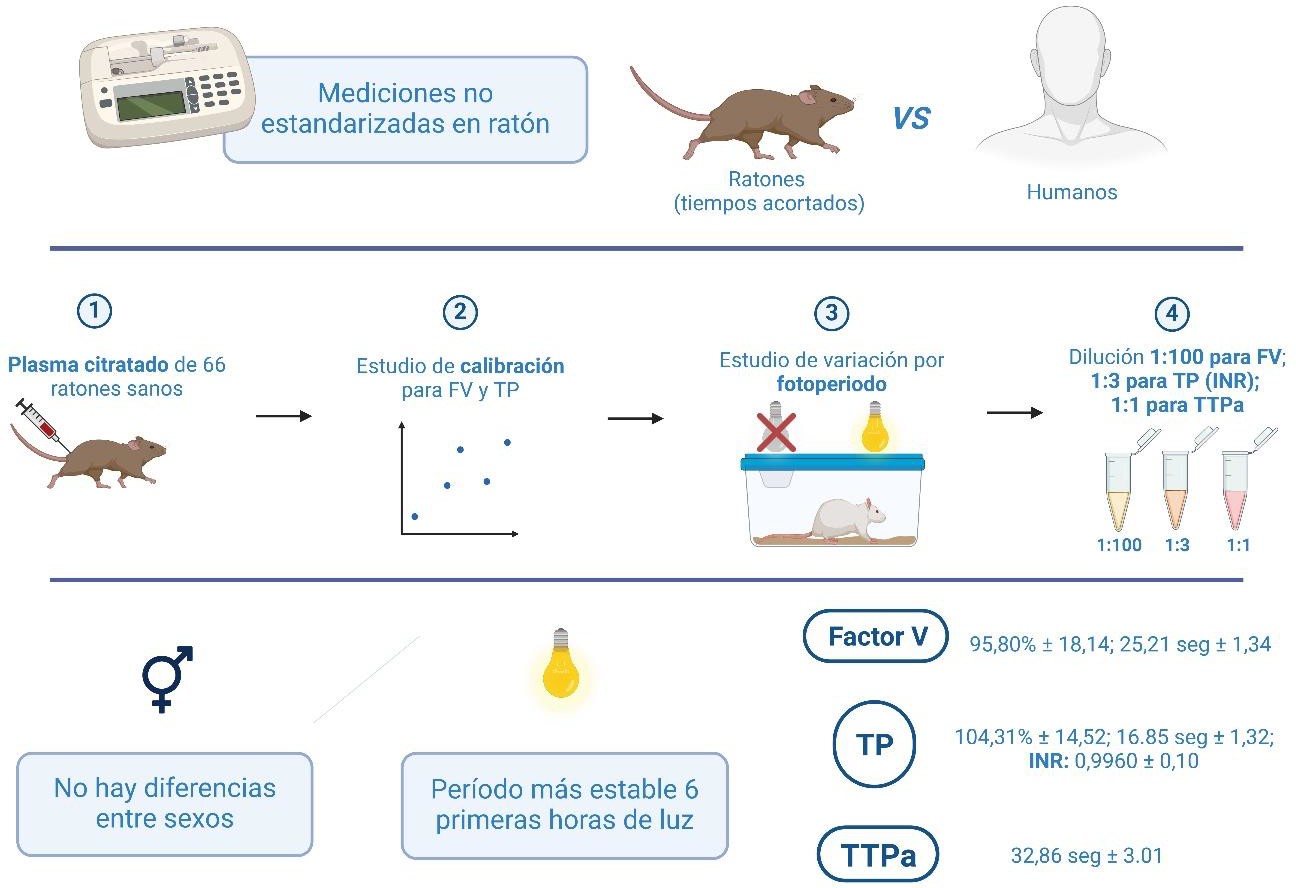
Figura 3. Estandarización, en ratón, de la medida de factor V y de tiempos de coagulación (FV, factor V; INR, Índice Internacional Normalizado; TP, tiempo de protrombina; TTPa, tiempo de tromboplastina parcial activada).
Se diseñó un protocolo específico para ratones, capaz de conseguir correcciones en las muestras a diluciones de 1:100 para factor V y de 1:3 para el tiempo de protrombina. El objetivo era “suavizar” las curvas de calibración, que presentan pendientes muy pronunciadas y rangos de medición muy estrechos entre un punto de calibración y otro. Se comprobó que el periodo más estable y óptimo para la extracción de las muestras de sangre era el comprendido en las primeras 6 horas del periodo luminoso. No se observaron diferencias clínicas entre sexos y se establecieron los intervalos de referencia para factor V (95,80% ± 18,14; 25,21 seg ± 1,34), para el tiempo de protrombina (104,31% ± 14,52; 16,85 seg ± 1,32) y para el tiempo de tromboplastina parcial activada, dilución 1:1, (32,86 seg ± 3,01). También se utilizó el INR (Índice Internacional Normalizado) como herramienta de estandarización para el modelo de ratón´(0,996 ± 0,10).
Además, siguiendo la práctica habitual de la clínica en humanos, y contrariamente a informes anteriores de la literatura, se establecieron los porcentajes y los segundos como unidades de medida del nivel de FV en ratones. La estrategia planteada en este estudio puede ayudar a establecer intervalos de referencia y a detectar pequeñas diferencias entre individuos sanos y enfermos en el contexto de la biomedicina veterinaria o humana, en la medicina transfusional o en los modelos patológicos utilizados para las coagulopatías congénitas, como puede ser el déficit de FV.
4. OBTENCIÓN DE UN MODELO CELULAR DEFICIENTE EN FACTOR V MEDIANTE EDICIÓN GÉNICA, Y SU CORRECCIÓN
Como ya se ha comentado más arriba, para el establecimiento de nuevos fármacos y estrategias como es el caso de las nuevas terapias avanzadas, se precisa ineludiblemente la utilización de modelos patológicos tanto para estudios iniciales in vitro como los posteriores in vivo. Para el primer abordaje de los estudios in vitro utilizando cultivos celulares, en el caso específico de la enfermedad que padece nuestra paciente, déficit severo de FV, debido a sus características clínicas de riesgo de sangrado, no podíamos contemplar la posibilidad de la obtención de células por biopsia hepática para su posterior cultivo. Por esta razón, una investigación, en general, debe adaptarse a las peculiaridades concretas de la enfermedad, por lo que se decidió preparar un modelo celular deficiente en FV.
La «herramienta» más adecuada, como también se ha explicado más arriba, era la edición génica mediada por CRISPR/Cas9, por homología en células HepG2 (Figura 4). Esta línea celular humana se caracteriza por estar inmortalizada, por su robustez, durabilidad, rápida proliferación y potencial para generar colonias a partir de una sola célula. Además, expresa FV de forma constitutiva, como lo hace el hígado de forma fisiológica (57).
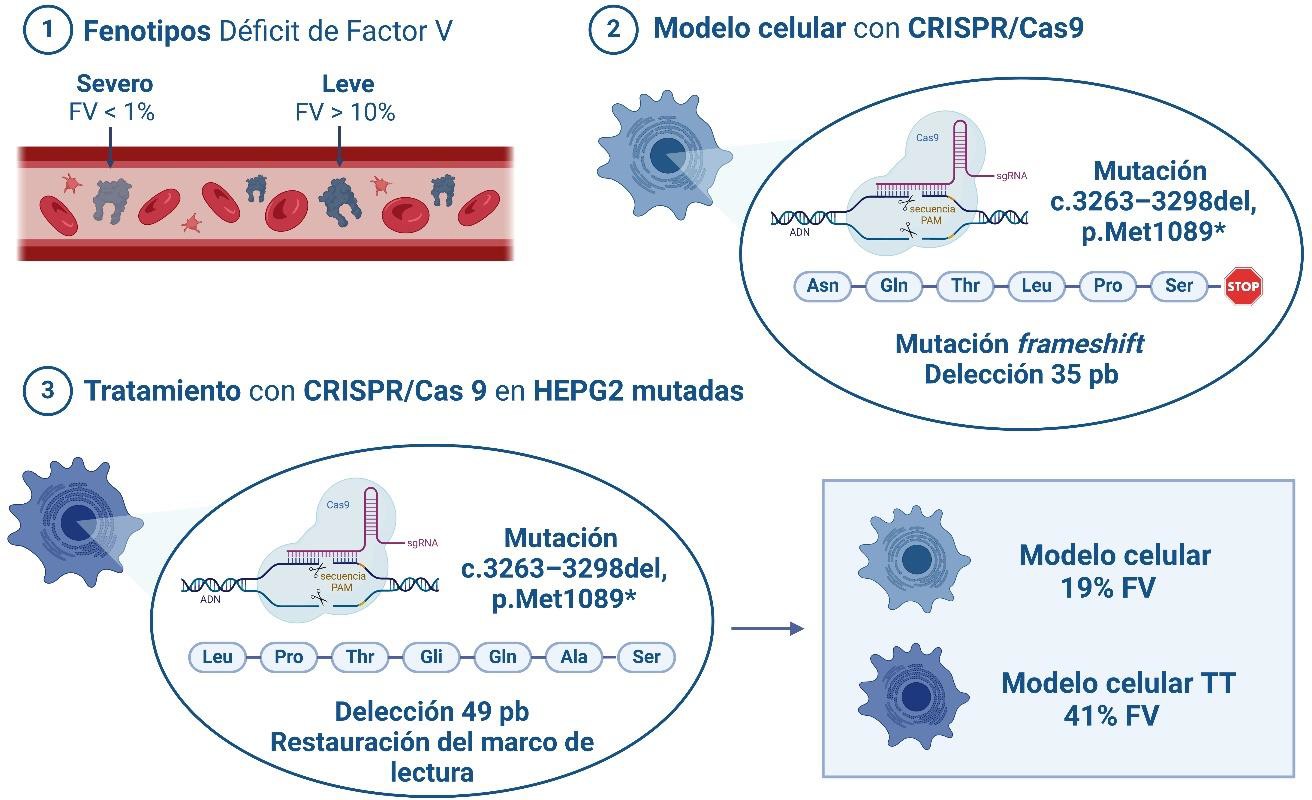
Figura 4. Obtención de un modelo celular deficiente en factor V mediante edición génica, y su corrección (Cas9, proteína asociada a CRISPR; CRISPR, repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas; del, deleción; FV, factor V; pb, pares de bases; PAM, motivo adyacente al protoespaciador; sgRNA, ARN guía único; TT, modelo celular tratado).
Así (58), se generó un modelo celular deficiente en FV mediante edición génica CRISPR/Cas9 produciendo una mutación similar a la que presentaba la paciente estudiada (c.3279G>A, p.Trp1093*) (25). Este modelo celular mutado presentaba, aproximadamente, un 19% de funcionalidad de FV con respecto a las células nativas.
Se propuso además y así se demostró, una nueva terapia avanzada basada en edición génica para la corrección de la mutación. Pasos adicionales de deleción de precisión por unión homóloga CRISPR/Cas9 permitieron corregir el 41% de las células mutadas en el gen F5, produciéndose una proteína funcional. El análisis de los genes fuera de diana (off-target) no mostró ninguna modificación en las secuencias genéticas derivadas de edición inespecífica al menos en los genes estudiados. Este método de corrección mediante edición génica se protegió mediante patente española (59) con cobertura internacional.
Teniendo en cuenta las concentraciones plasmáticas correspondientes a los diferentes niveles de gravedad de la deficiencia de factor V, se puede argumentar que la corrección lograda en este estudio podría, en condiciones ideales, ser suficiente para convertir un fenotipo grave en uno leve asintomático.
5. OBTENCIÓN DE UN MODELO ANIMAL PATOLÓGICO EN RATÓN DEFICIENTE EN FACTOR V MEDIANTE EDICIÓN GÉNICA Y MICROINYECCIÓN DIRECTA EN CIGOTOS
Una vez establecido el modelo celular con deficiencia en FV, se inició el desarrollo de un modelo animal patológico en ratón, utilizando la tecnología CRISPR/Cas9 y microinyección directa en cigotos, que sirviera para los posteriores estudios in vivo de prueba de protocolos de terapia génica (Figura 5). Debía de ser un modelo viable, como así se demostró en nuestros estudios, resultado éste que daba la máxima originalidad al modelo porque los previamente descritos nunca lo fueron y en consecuencia no tenían utilidad en estudios farmacológicos in vivo. Nuestro modelo se protegió mediante patente española (60) con cobertura internacional.
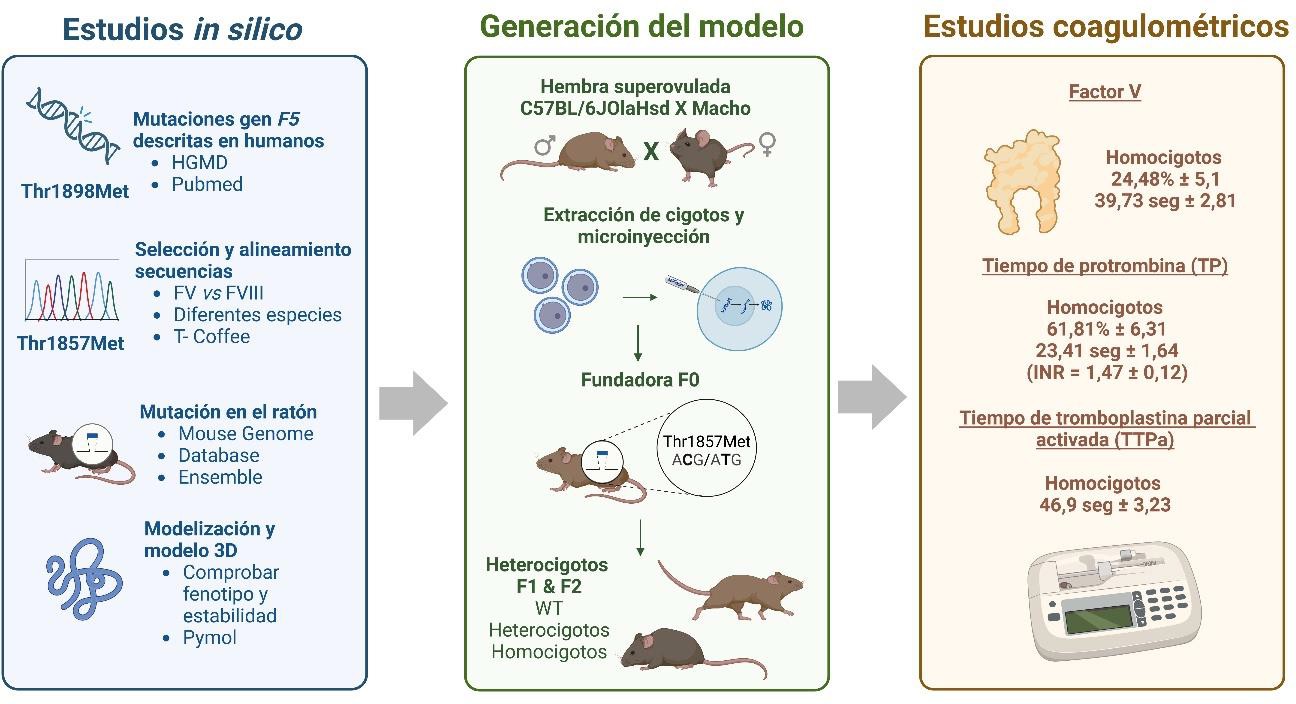
Figura 5. Obtención de un modelo animal patológico en ratón deficiente en factor V mediante edición génica y microinyección directa en cigotos (F, generación filial; FV, factor V; FVIII, factor VIII; HGMD, Human Gene Mutation Database; INR, Índice Internacional Normalizado; TP, tiempo de protrombina; TTPa, tiempo de tromboplastina parcial activada; WT, tipo salvaje).
Sobre la base de hallazgos previos que mostraban que concentraciones muy bajas de FV daban lugar a modelos animales no viables, se tomó la decisión de generar un modelo de fenotipo leve (aproximadamente del 25% de FV con respecto al de referencia), replicando una mutación humana que da lugar a un fenotipo leve de la enfermedad en una región conservada del FV de mamífero. La caracterización del fenotipo se realizó mediante ensayos de coagulometría para determinar la funcionalidad de la proteína, el tiempo de protrombina y el tiempo de tromboplastina parcial activada.
Se realizó un estudio previo in silico para seleccionar una mutación causante de un fenotipo leve de la enfermedad en humanos (Thr1898Met) (61). Dicha mutación se replicó en ratones (Thr1857Met) mediante edición génica dirigida por homología mediada por CRISPR y por microinyección directa, en el citoplasma de cigotos, de los componentes necesarios del sistema CRISPR/Cas9. Tras los distintos cruces necesarios, los individuos homocigotos obtenidos y tras su genotipado, fueron sometidos a ensayos de coagulometría, incluyendo niveles de FV, tiempo de protrombina y tiempo de tromboplastina parcial activada.
Los estudios in silico ya sugerían que la mutación desestabilizaba la estructura del FV (activado sin el dominio B) tanto en las variantes de ratón como humanas, lo que daba lugar a un fenotipo leve de la enfermedad.
Se observó herencia mendeliana en la descendencia y no se produjeron signos espontáneos por alteración de la coagulación sanguínea, ni muertes prematuras o disfunciones gestacionales. Los niveles de FV en los animales homocigotos fueron de 24,48% ± 5,1 y 39,73 seg ± 2,81; el TP un 61,81% ± 6,31 y 23,41 seg ± 1,64 (INR = 1,47 ± 0,12); y el TTPa de 46,9 seg ± 3,23. Estos datos demostraban un modelo viable en ratón deficiente en FV por una mutación sin sentido inducida en el gen F5, con un fenotipo leve de la enfermedad, similar al observado en humanos.
6. PRUEBA DE CONCEPTO DE TERAPIA GÉNICA LENTIVIRAL PARA LA CORRECCIÓN IN VITRO E IN VIVO DE LA MUTACIÓN EN EL GEN F5
Y con todos estos resultados previos, llegaba el momento de demostrar la prueba de concepto de que un protocolo de terapia génica viral utilizando vectores lentivirales, podía ser capaz de revertir un fenotipo patológico de una deficiencia de factor V.
La elección del vector viral es una de las cuestiones más difíciles y a la vez más cruciales porque hay que intentar el equilibrio entre la eficacia y los efectos adversos. En la actualidad, son los vectores lentivirales y adenoasociados los que más se utilizan en aplicaciones clínicas. Pero se ha de saber que los vectores adenoasociados aunque se integran mínimamente en el genoma, son altamente inmunogénicos y desencadenan una respuesta inmune muy fuerte que finaliza en hepatotoxicidad. Los vectores lentivirales por el contrario no son inmunogénicos. La elección, a parte de tener en cuenta estas características propias del vector, se debe basar en las condiciones clínicas del paciente receptor. En nuestro caso, nuestra paciente tiene una muy alta predisposición a sufrir graves efectos anafilácticos con plasma fresco congelado y otros productos sanguíneos. Por esta razón nuestra elección del vector se dirigió sin lugar a duda hacia los vectores lentivirales. Esta estrategia con vector lentiviral, permite la integración del gen terapéutico en las células del huésped, proporcionando, teóricamente, una expresión sostenida de la proteína FV.
La prueba de concepto para el protocolo de terapia génica lentiviral (Figura 6), se llevó a cabo, en primer lugar, a través de estudios in vitro en células HepG2 nativas y mutadas en el gen F5 como se describió más arriba en el modelo celular de patología (Figura 4). Respecto al constructo del vector recombinante se partió de la base del éxito de los constructos de moléculas de FVIII (62), y se elaboró un transgén de FV humano activado en el cual se eliminó la secuencia codificante del dominio B (función solamente estructural de la proteína), permitiendo reducir el tamaño del empaquetamiento en el vector y pudiendo mejorar las titulaciones del vector y la infectividad. Además, de esta forma, el transgén utilizado sintetiza, tras integrarse, una proteína FV directamente activada y funcional que pueda intervenir en la cascada de la coagulación.
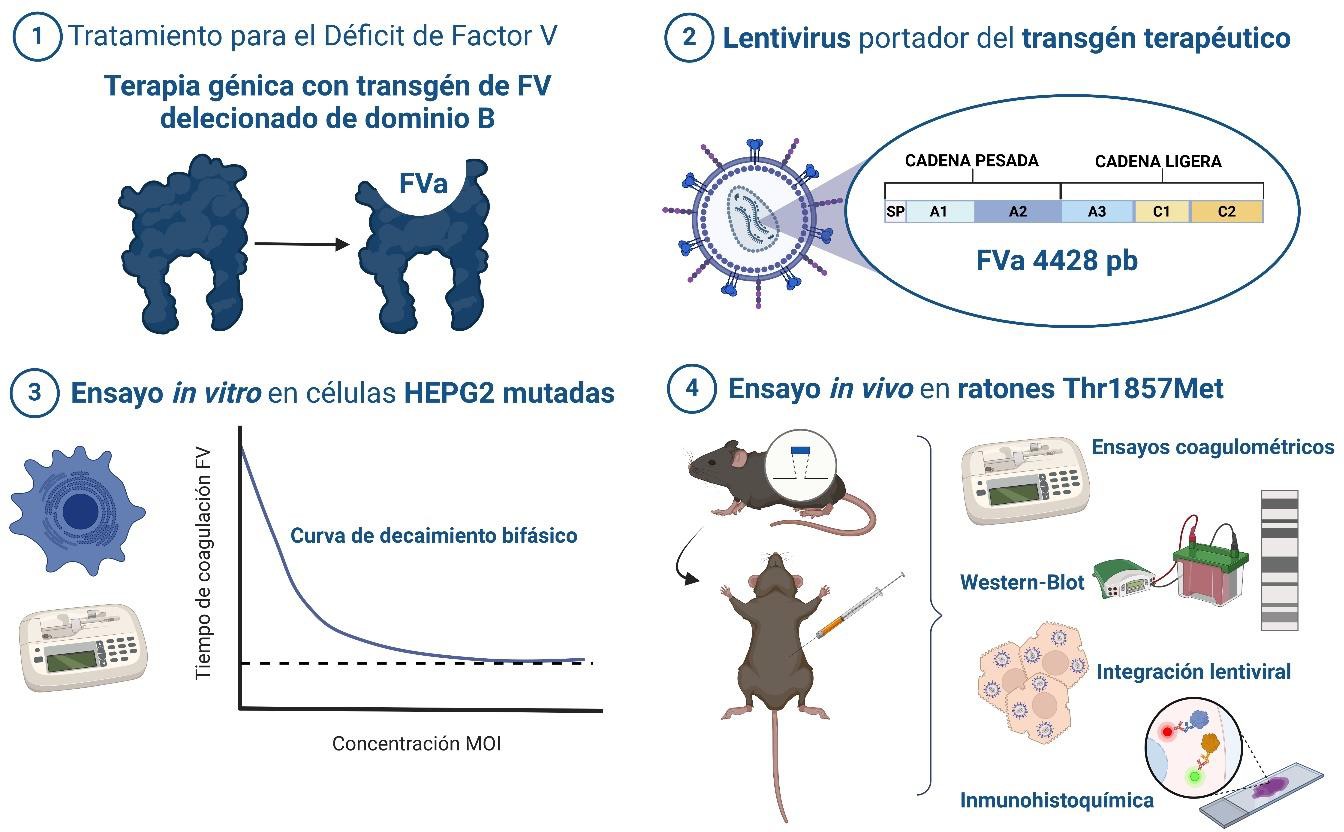
Figura 6. Prueba de concepto de terapia génica lentiviral para la corrección in vitro e in vivo de la mutación en el gen F5 (FVa, factor V activado; FV, factor V; MOI, dosis del virus o multiplicidad de infección; pb, pares de bases; SP, péptido señal).
Los resultados del experimento de transducción in vitro con el vector lentiviral en células HepG2 mutadas en el gen F5, demostraron una relación clara y proporcional entre la dosis del vector y la actividad de FV. Por un lado, los valores obtenidos respecto de las células WT (nativas) y mutadas sin transfectar con el vector lentiviral, se ajustaban a los descritos más arriba (Figura 4). A medida que se incrementa la dosis del vector, se observa una reducción progresiva en los tiempos de coagulación y un aumento significativo en la actividad de FV, lo que sugiere que el vector recombinante con el transgén de FV restaura la funcionalidad del FV en las células mutadas. Sin embargo, no se observaron aumentos pronunciados entre las dosis más altas (MOI 5 vs MOI 10) en términos de tiempos de coagulación y actividad porcentual, lo que sugiere que puede haber un límite máximo en la capacidad de las células para procesar o expresar FV a partir del vector lentiviral.
En los estudios in vivo, respecto a los valores de los parámetros coagulométricos en animales WT, Hz y Hm sin tratamiento, los resultados obtenidos coinciden con los descritos en el modelo animal patológico (Figura 5), lo que refuerza la consistencia y reproducibilidad de los datos. Esta estabilidad en los parámetros coagulantes de referencia permite una base sólida para comparar los efectos del tratamiento. En los animales Hm tratados se observó un aumento significativo en los niveles de FV, y una disminución del TP y del TTPa, lo cual sugiere una mejora en la función coagulante en respuesta a la terapia, evidenciando un efecto positivo del vector lentiviral. Además, la proteína FV humana se detectó mediante Western blot tanto en el plasma control de origen humano como en el plasma de los animales tratados con vector lentiviral.
Se analizó, además, la integración del vector lentiviral en los tejidos hepáticos de los ratones mediante la cuantificación del número de copias del vector por célula (VCN), utilizando qPCR. En todas las muestras, se observaron valores detectables de VCN salvo en los controles, confirmando la integración del vector lentiviral en el genoma de los hepatocitos, lo que concuerda con la llegada del vector que porta promotor hepático. Estos resultados de integración hepática coinciden con estudios previos con vectores lentivirales en modelos de ratón con hemofilia B (63).
También se llevó a cabo un estudio exhaustivo ―a modo de prueba preclínica en animal― de las lesiones histopatológicas. En animales WT y Hm sin tratar, no se observaron lesiones tisulares ni en órganos. Sin embargo, en los animales tratados con el vector lentiviral, sí se observaron diferencias significativas en la gravedad de las lesiones, particularmente en el tejido pulmonar, mientras que en otros órganos solo se encontraron alteraciones ocasionales y sin relevancia estadística. Las lesiones pulmonares severas, en los animales tratados, podrían deberse a una respuesta inflamatoria localizada inducida por el tratamiento lentiviral, favorecida por eventos de micro tromboembolismos o acumulación de fibrina en la microvasculatura pulmonar. La acumulación de fibrina también podría estar estimulando la inflamación peribronquiolar y perivascular, actuando como un potente desencadenante para la migración de células inflamatorias, lo que siempre agrava el daño histológico en el pulmón. De igual forma, podría existir un período inicial de actividad procoagulante derivado de la elevada cantidad de FV introducido, que podría estar generando un estado de hipercoagulabilidad temporal en los animales tratados.
Respecto a los estudios de inmunohistoquímica, en los órganos que presentaron inmunorreacción positiva para FV en animales WT, hubo descenso de ésta en animales Hm sin tratar; la inmunorreacción se volvió a recuperar en los animales Hm tratados con lentivirus, en la mayoría de los tejidos sin diferencia entre las dosis. Por otro lado se analizó la trombina, que es una molécula esencial en la cascada de la coagulación, siendo su producción dependiente de la acción del FV, por lo que su expresión va a estar influenciada directamente por la de FV (64). Al igual que en FV, se encuentran también diferencias de inmunorreacción para trombina en pulmón y en cerebro, lo cual confirmaría la relación entre ambas proteínas. La trombina, además, es conocida por su capacidad para inducir respuestas inflamatorias a nivel celular y en el pulmón, mediante la producción de citoquinas, como la IL-8 en células epiteliales, que estimulan el reclutamiento de células inflamatorias adicionales, exacerbando la inflamación local.
Por último, se analizó la α-actina de músculo liso que es una proteína estructural esencial para la contracción de las células en ese tejido y que desempeña un papel clave en el mantenimiento de la integridad y funcionalidad de los vasos sanguíneos y órganos internos. Esta proteína podría tener un papel en la generación de sangrados espontáneos, ya que el soporte de las células musculares lisas marcado por α-actina (como en la vena porta o en la arteria hepática) puede ser insuficiente para compensar la debilidad del coágulo, dejando a las estructuras vasculares más vulnerables a fugas y sangrados. El FV y la trombina están estrechamente relacionados con la α-actina en contextos de remodelación tisular e inflamación. Así el análisis de α-actina por inmunohistoquímica mostró diferencias cualitativas, en los órganos estudiados, entre los grupos de animales control y tratados. Esta inmunorreacción se caracterizaba por una tinción marrón en el citoplasma de las fibras musculares lisas de arterias y arteriolas musculares. En el bazo, los ratones WT mostraron una fuerte expresión de α-actina en la cápsula del bazo, compuesta por músculo liso, y en los senos venosos de la zona marginal y arteriolas peniciladas de la pulpa blanca. Los ratones Hm tratados con suero salino presentaron una reducción significativa de la expresión de α-actina en estas áreas. En los ratones tratados con lentivirus, tanto a dosis baja como alta, se observó un aumento en la expresión de α-actina en el hígado, en la cápsula del bazo y en los senos venosos, con niveles de expresión comparables entre una y otra dosis del vector.
7. CONCLUSIONES Y EXPECTATIVAS
Ya han pasado siete años desde que comenzara este proyecto de investigación. Una revisión, como es esta, retrospectiva y actualizada en un contexto científico propio del Grupo de Terapias Avanzadas de la Universidad Complutense de Madrid, se escribe en pocas páginas, sin embargo, el trasfondo acumula años y esfuerzo que casi no se alcanza a vislumbrar en un artículo, pero que han estado ahí. Los primeros momentos fueron de incertidumbre porque el objetivo era ambicioso y los medios escasos. No obstante, la ilusión de unos padres y de la propia paciente por conseguir un ansiado tratamiento, animaba a iniciar y continuar. El momento no tenía ya marcha atrás. El aunar fuerzas e ilusiones envolvían y suavizaban las inquietudes. La colaboración de otros grupos de investigación e investigadores que ahora forman parte de este equipo que se presentó con ilusión a este prestigioso premio de la Real Academia Nacional de Farmacia, hicieron posible la consecución de objetivos. Algunas empresas farmacéuticas y la propia Universidad Complutense de Madrid confiaron también en el proyecto y todos y todas, en conjunto, hemos llegado hasta aquí. El sentirse arropado por una asociación de pacientes como es la Asociación para la Investigación y Cura del Déficit de Factor V, Una Esperanza para Celia, creada para este fin en concreto, potencia más, si cabe, la vocación investigadora.
Aún así, ¿qué significa llegar hasta aquí? Significa haber abierto camino a una enfermedad ultra rara que a casi nadie interesa, pero que tiene cara y alma en la paciente y en sus padres; significa dignificar a una enfermedad minoritaria y concienciar acerca de la necesidad de investigar las enfermedades olvidadas.
No se trata tan solo de desbrozar el camino sino también de construir a lo largo de la senda. Así, se ha diagnosticado a la paciente y se ha establecido el hilo conductor de la transmisión hereditaria de su déficit de factor V; se ha entrado en el mundo de la biología celular y animal, y se han interferido los mecanismos genéticos para conseguir modificar el genoma de unas células y de un animal para ser utilizados como modelos experimentales. Y, en fin, se ha demostrado mediante una prueba de concepto que el protocolo que se proponía de terapia génica lentiviral, puede ser una primera “llama” de esperanza para la curación de esa patología.
Se es consciente de que son los primeros pasos, como también de que sin ellos no se hace camino. Los estudios in vitro e in vivo han demostrado que el vector lentiviral, portador de parte del gen F5, es capaz de restaurar de manera eficiente los niveles de FV en un modelo celular y en un modelo animal de patología deficiente en esa proteína. Esto sugiere que esta aproximación tiene potencial para su aplicación preclínica en animales y posterior clínica en humanos.
Ahora, con las bases establecidas de los protocolos, la mirada como expectativa se orientará en definir la dosificación óptima y afianzar los parámetros de seguridad farmacológica en fase preclínica. También se avanzará en otras líneas ya abiertas por el grupo, basadas en terapia celular para conseguir el microtrasplante de hepatocitos sanos en el modelo animal patológico para conseguir revertir el fenotipo.
La labor investigadora, en general, se basa en ir aportando y colocando las piezas de un puzle para que quienes vengan detrás puedan ir acercándose cada vez más a la meta final. Esto, no es de la noche a la mañana, pero dejar la semilla hace atisbar mejor el objetivo.
Agradecimientos
En las primeras etapas de la investigación fue la Asociación Andaluza de Hemofilia la que financió parte de la actividad científica (ASANHEMO FV2016–20). También participó en la financiación la empresa farmacéutica Octapharma S.A (OCPH2019-20). Posteriormente, y a través de una colaboración científica con la Fundación Jiménez Diaz y el Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) de Madrid, la financiación vino del Instituto de Salud Carlos III, de fondos para el Desarrollo Regional Europeo (RETICS RD 16/0011/00011 y RD 16/0011/0013), de la Dirección General de Investigación de la Comunidad de Madrid (AvanCell-CM; Ref. S2017/BMD-3692) y de la Agencia Estatal de Investigación (PID2020- 119637RB-I00). En los últimos tres años de investigación la financiación se recibió de la Universidad Complutense de Madrid a través de contratos predoctorales (CT63/19-CT64/19) y de la Asociación para la Investigación y Cura del Déficit de Factor V, Una Esperanza para Celia (ASDEFAV/2021-24). También, a través de la colaboración con el Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), los fondos aplicados fueron del Ministerio de Ciencia, Innovación y Universidades (PID2020-117501RB-I00). Agradecimiento especial a Benedicto Jerónimo, supervisor del animalario de la Facultad de Veterinaria de la Universidad Complutense de Madrid donde se llevaron a cabo los procedimientos, por preservar tan seriamente la seguridad y el bienestar de los animales. También agradecer al Centro de Asistencia a la Investigación, Unidad de Genómica, de la Universidad Complutense de Madrid, y a los doctores Enrique Páramo y Mónica Carballo, responsables del Servicio de Animalario y Cirugía Experimental del Hospital Nacional de Parapléjicos de Toledo, por su profesionalidad y facilidades ofrecidas para llevar a cabo todos los experimentos de terapia génica lentiviral. Agradecer, por último, a la Fundación Inocente, la ayuda concedida (FII2024-151) para el desarrollo de este proyecto.
8. REFERENCIAS
- Russel WMS, Burch RL. The Principles of Humane Experimental Technique. Med J Aust 1960; 1: 500-500. https://doi.org/10.5694/j.1326-5377.1960.tb73127.x.
- Versteeg HH, Heemskerk JWM, Levi M, et al. New fundamentals in hemostasis. Physiol Rev 2013; 93: 327-58. https://doi.org/10.1152/physrev.00016.2011.
- Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev 2015; 29: 17-24. https://doi.org/10.1016/j.blre.2014.09.003.
- De Pablo-Moreno JA, Serrano LJ, Revuelta L, et al. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. Int J Mol Sci 2022; 23: 8283. https://doi.org/10.3390/ijms23158283.
- Monagle P, Massicotte P. Developmental haemostasis: Secondary haemostasis. Semin Fetal Neonatal Med 2011; 16: 294-300. https://doi.org/10.1016/j.siny.2011.07.007.
- Grover SP, Mackman N. Intrinsic Pathway of Coagulation and Thrombosis: Insights From Animal Models. Arterioscler Thromb Vasc Biol 2019; 39: 331-8. https://doi.org/10.1161/ATVBAHA.118.312130.
- Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth 2014; 58: 515-23. https://doi.org/10.4103/0019-5049.144643.
- De Pablo-Moreno JA, Miguel-Batuecas A, De Sancha M, et al. The Magic of Proteases: From a Procoagulant and Anticoagulant Factor V to an Equitable Treatment of Its Inherited Deficiency. Int J Mol Sci 2023; 24: 6243. https://doi.org/10.3390/ijms24076243.
- Mukhopadhyay S, Johnson TA, Duru N, et al. Fibrinolysis and Inflammation in Venous Thrombus Resolution. Front Immunol 2019; 10: 1348. https://doi.org/10.3389/fimmu.2019.01348.
- McMurry HS, Jou J, Shatzel J. The hemostatic and thrombotic complications of liver disease. Eur J Haematol 2021; 107: 383-92. https://doi.org/10.1111/ejh.13688.
- Tabibian S, Shiravand Y, Shams M, et al. A Comprehensive Overview of Coagulation Factor V and Congenital Factor V Deficiency. Semin Thromb Hemost 2019; 45: 523-43. https://doi.org/10.1055/s-0039-1687906.
- Weyand AC, Flood VH. Von Willebrand Disease: Current Status of Diagnosis and Management. Hematol Oncol Clin North Am 2021; 35: 1085-101. https://doi.org/10.1016/j.hoc.2021.07.004.
- Castaman G, Matino D. Hemophilia A and B: molecular and clinical similarities and differences. Haematologica 2019; 104: 1702-9. https://doi.org/10.3324/haematol.2019.221093.
- Malec L, Peyvandi F, Chan AKC, et al. Efanesoctocog Alfa Prophylaxis for Children with Severe Hemophilia A. N Engl J Med 2024; 391: 235-46. https://doi.org/10.1056/NEJMoa2312611.
- Callaghan MU, Negrier C, Paz-Priel I, et al. Long-term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1-4 studies. Blood 2021; 137: 2231-42. https://doi.org/10.1182/blood.2020009217.
- The Lancet Global Health. The landscape for rare diseases in 2024. Lancet Glob Health 2024; 12: e341. https://doi.org/10.1016/S2214-109X(24)00056-1.
Vinkšel M, Writzl K, Maver A, et al. Improving diagnostics of rare genetic diseases with NGS approaches. J Community Genet 2021; 12: 247-56. https://doi.org/10.1007/s12687-020-00500-5. - Choon YW, Choon YF, Nasarudin NA, et al. Artificial intelligence and database for NGS-based diagnosis in rare disease. Front Genet 2024; 14: 1258083. https://doi.org/10.3389/fgene.2023.1258083.
- Lopes-Júnior LC, Ferraz VEF, Lima RAG, et al. Health Policies for Rare Disease Patients: A Scoping Review. Int J Environ Res Public Health 2022; 19: 15174. https://doi.org/10.3390/ijerph192215174.
- Bannister JB. Regulating Rare Disease: Safely Facilitating Access to Orphan Drugs. Fordham Law Rev 2018; 86: 1889- 921. PMID: 29993206.
- Buckle N, Doyle O, Kodate N, et al. The economic impact of living with a rare disease for children and their families: a scoping review protocol. HRB Open Res 2024; 6: 41. https://doi.org/10.12688/hrbopenres.13765.2.
- Yang TL, Cui J, Rehumtulla A, et al. The Structure and Function of Murine Factor V and Its Inactivation by Protein C. Blood 1998; 91: 4593-9. PMID: 9616155.
- De Pablo-Moreno JA, Liras A, Revuelta L. Standardization of Coagulation Factor V Reference Intervals, Prothrombin Time, and Activated Partial Thromboplastin Time in Mice for Use in Factor V Deficiency Pathological Models. Front Vet Sci 2022; 9: 846216. https://doi.org/10.3389/fvets.2022.846216.
- Tabibian S, Dorgalaleh A, Camire RM. Congenital Factor V Deficiency. En: Dorgalaleh A, Ed. Congenital Bleeding Disorders. Cham: Springer International Publishing 2018; pp. 201-18. Disponible en: (http://link.springer.com/10.1007/978-3-319-76723-9_8).
- Bernal S, Pelaez I, Alias L, et al. High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. Int J Mol Sci 2021; 22: 9705. https://doi.org/10.3390/ijms22189705.
- Jain S, Acharya SS. Management of rare coagulation disorders in 2018. Transfus Apher Sci 2018; 57: 705-12. https://doi.org/10.1016/j.transci.2018.10.009.
- Heger A, Svae TE, Neisser-Svae A, et al. Biochemical quality of the pharmaceutically licensed plasma OctaplasLG ® after implementation of a novel prion protein (PrP Sc) removal technology and reduction of the solvent/detergent (S/D) process time. Vox Sang 2009; 97: 219-25. https://doi.org/10.1111/j.1423-0410.2009.01190.x.
- Naderi M, Tabibian S, Shamsizadeh M, et al. Miscarriage and recurrent miscarriage in patients with congenital factor V deficiency: a report of six cases in Iran. Int J Hematol 2016; 103: 673-5. https://doi.org/10.1007/s12185-016-1981-7.
- von Drygalski A, Bhat V, Gale AJ, et al. An Engineered Factor Va Prevents Bleeding Induced by Anticoagulant wt Activated Protein C. Doering CB, editor. PLoS ONE 2014; 9: e104304. https://doi.org/10.1371/journal.pone.0104304.
- Nakamura T, Morishige S, Ozawa H, et al. Successful correction of factor V deficiency of patient‐derived iPSCs by CRISPR/Cas9‐mediated gene editing. Haemophilia 2020; 26: 826-33. https://doi.org/10.1111/hae.14104.
- Sun J, Chen X, Chai Z, et al. Adeno-associated virus-mediated expression of activated factor V (FVa) for hemophilia phenotypic correction. Front Med (Lausanne) 2022; 9: 880763. https://doi.org/10.3389/fmed.2022.880763.
- Ma C, Peng Y, Li H, et al. Organ-on-a-Chip: A New Paradigm for Drug Development. Trends Pharmacol Sci 2021; 42: 119-33. https://doi.org/10.1016/j.tips.2020.11.009.
- Cacciamali A, Villa R, Dotti S. 3D Cell Cultures: Evolution of an Ancient Tool for New Applications. Front Physiol 2022; 13: 836480. https://doi.org/10.3389/fphys.2022.836480.
- Tkacs NC, Thompson HJ. From Bedside to Bench and Back Again: Research Issues in Animal Models of Human Disease. Biol Res Nurs 2006; 8: 78-88. https://doi.org/10.1177/1099800406289717.
- Domínguez-Oliva A, Hernández-Ávalos I, Martínez-Burnes J, et al. The Importance of Animal Models in Biomedical Research: Current Insights and Applications. Animals (Basel) 2023; 13: 1223. https://doi.org/10.3390/ani13071223.
- Kiani AK, Pheby D, Henehan G, et al. Ethical considerations regarding animal experimentation. J Prev Med Hyg 2022; 63: E255-E266. https://doi.org/10.15167/2421-4248/jpmh2022.63.2S3.2768.
- Cui J, O’Shea KS, Purkayastha A, et al. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature 1996; 384: 66-8. https://doi.org/10.1038/384066a0.
- Yang T, Cui J, Taylor J, et al. Rescue of Fatal Neonatal Hemorrhage in Factor V Deficient Mice by Low Level Transgene Expression. Thromb Haemost 2000; 83: 70-7. PMID: 10669158.
- Weyand AC, Grzegorski SJ, Rost MS, et al. Analysis of factor V in zebrafish demonstrates minimal levels needed for early hemostasis. Blood Adv 2019; 3: 1670-80. https://doi.org/10.1182/bloodadvances.2018029066.
- Shakweer WME, Krivoruchko AY, Dessouki ShM, et al. A review of transgenic animal techniques and their applications. J Genet Eng Biotechnol 2023; 21: 55. https://doi.org/10.1186/s43141-023-00502-z.
- Birling MC, Herault Y, Pavlovic G. Modeling human disease in rodents by CRISPR/Cas9 genome editing. Mamm Genome 2017; 28: 291-301. https://doi.org/10.1007/s00335-017-9703-x.
- Xu Y, Li Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput Struct Biotechnol J 2020; 18: 2401-15. https://doi.org/10.1016/j.csbj.2020.08.031.
- Mojica FJM, Montoliu L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol 2016; 24: 811-20. https://doi.org/10.1016/j.tim.2016.06.005.
- Stinson BM, Loparo JJ. Repair of DNA Double-Strand Breaks by the Nonhomologous End Joining Pathway. Annu Rev Biochem 2021; 90: 137-64. PMID: 33556282.
- Riesenberg S, Kanis P, Macak D, et al. Efficient high-precision homology-directed repair-dependent genome editing by HDRobust. Nat Methods 2023; 20: 1388-99. https://doi.org/10.1038/s41592-023-01949-1.
- Lopes R, Prasad MK. Beyond the promise: evaluating and mitigating off-target effects in CRISPR gene editing for safer therapeutics. Front Bioeng Biotechnol 2024; 11: 1339189. https://doi.org/10.3389/fbioe.2023.1339189.
- Ho D, Quake SR, McCabe ERB, et al. Enabling Technologies for Personalized and Precision Medicine. Trends Biotechnol 2020; 38: 497-518. https://doi.org/10.1016/j.tibtech.2019.12.021.
- Gao K, Kumar P, Cortez-Toledo E, et al. Potential long-term treatment of hemophilia A by neonatal co-transplantation of cord blood-derived endothelial colony-forming cells and placental mesenchymal stromal cells. Stem Cell Res Ther 2019; 10: 34. PMID: 30670078.
- Olgasi C, Talmon M, Merlin S, et al. Patient-Specific iPSC-Derived Endothelial Cells Provide Long-Term Phenotypic Correction of Hemophilia A. Stem Cell Reports 2018; 11: 1391-406. https://doi.org/10.1016/j.stemcr.2018.10.012.
- High KA, Roncarolo MG. Gene Therapy. N Engl J Med 2019; 381: 455-64. https://doi.org/10.1056/NEJMra1706910.
- Li X, Le Y, Zhang Z, et al. Viral Vector-Based Gene Therapy. Int J Mol Sci 2023; 24: 7736. https://doi.org/10.3390/ijms24097736.
- Naso MF, Tomkowicz B, Perry WL, et al. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017; 31: 317-34. https://doi.org/10.1007/s40259-017-0234-5.
- Cornetta K, Duffy L, Turtle CJ, et al. Absence of Replication-Competent Lentivirus in the Clinic: Analysis of Infused T Cell Products. Mol Ther 2018; 26: 280-8. https://doi.org/10.1016/j.ymthe.2017.09.008.
- Ausubel LJ, Hall C, Sharma A, et al. Production of CGMP-Grade Lentiviral Vectors. Bioprocess Int 2012; 10: 32-43. PMCID: PMC3374843.
- Lunghi B, Castoldi E, Mingozzi F, et al. A Novel Factor V Null Mutation Detected in a Thrombophilic Patient with Pseudo-Homozygous APC Resistance and in an Asymptomatic Unrelated Subject. Blood 1998; 92: 1463-64. PMID: 9694743.
- Yang TL, Pipe SW, Yang A, et al. Biosynthetic origin and functional significance of murine platelet factor V. Blood 2003; 102: 2851-5. https://doi.org/10.1182/blood-2003-04-1224.
- Aden DP, Fogel A, Plotkin S, et al. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979; 282: 615-6. https://doi.org/10.1038/282615a0.
- Serrano LJ, Garcia-Arranz M, De Pablo-Moreno JA, et al. Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations. Int J Mol Sci 2022; 23: 5802. https://doi.org/10.3390/ijms23105802.
- Liras Martín A, Serrano Ramos LJ, Bernal Noguera S, inventores; Universidad Complutense de Madrid y Fundació Institut de Recerca de L’hospital de La Santa Creu I Sant Pau, assignees. Método in vitro para recuperar la expresión del gen F5 que codifica el factor V de la coagulación. Spanish patent ES2785323B2. 2020.
- Liras Martín A, De Pablo Moreno JA, Revuelta Rueda L, Miguel Batuecas A, Bermejo Alvarez P, González Brusi L, inventores; Universidad Complutense de Madrid y Consejo Superior de
- Investigaciones Científicas, assignees. Mouse model, deficient in factor V. Spanish patent ES2948817B2. 2024.
- Delev D, Pavlova A, Heinz S, et al. Factor 5 mutation profile in German patients with homozygous and heterozygous factor V deficiency. Haemophilia 2009; 15: 1143-53. https://doi.org/10.1111/j.1365-2516.2009.02048.x.
- Miao HZ, Sirachainan N, Palmer L, et al. Bioengineering of coagulation factor VIII for improved secretion. Blood 2004; 103: 3412-9. https://doi.org/10.1182/blood-2003-10-3591.
- Follenzi A, Battaglia M, Lombardo A, et al. Targeting lentiviral vector expression to hepatocytes limits transgene- specific immune response and establishes long-term expression of human antihemophilic factor IX in mice. Blood 2004; 103: 3700-9. https://doi.org/10.1182/blood-2003-09-3217.
- Barcellona D, Marongiu F. The hemostatic system. 1st Part. J Pediatr Neonatal Individ Med 2020; 9: e090106. https://doi.org/10.7363/090106.
