Fármacos de nueva aprobación – Newly approved drugs:
(A) TRACTO ALIMENTARIO Y METABOLISMO: Diabetes mellitus: Insulina Icodec (Awiqli®; EMA). Colangitis biliar primaria: Elafibranor (Iqirvo®; FDA).(B) SANGRE Y SISTEMA HEMATOPOYÉTICO: Hemofilia B: Fidanacogene Elaparvovec (Beqvez®; FDA). (C) SISTEMA CARDIOVASCULAR: (D) DERMATOLOGÍA: Hiperhidrosis axilar primaria: Sofpironio (Sofdra®; FDA). (G) SISTEMA GENITOURINARIO Y HORMONAS SEXUALES: Vejiga hiperactiva: Vibegron (Obgemsa®; EMA). (J) ANTIINFECCIOSOS SISTÉMICOS: Infección intraabdominal: Aztreonam / Avibactam (Emblaveo®; EMA). Neumonía neumocócica: Vacuna neumococo polisacárida conjugada 22-valente (Capvaxine®; FDA). Neumonía por Virus Respiratorio Sincitial (VRS): vacuna VRS (mResvia®; EMA/FDA). (L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES: Cáncer de pulmón de células pequeñas: Tarlatamab (Imdelltra®; FDA). Cáncer de vejiga: Nogapendekina alfa inbakicept (Anktiva®; FDA). Glioma: Tovorafenib (Ojemda®; FDA). Síndrome WHIM: Mavorixafor (Xolremdi®; FDA). Hemoglobinuria nocturna paroxística: Danicopan (Voydeya®; EMA/FDA). Hemoglobinuria nocturna paroxística: Crovalimab (Piasky®; FDA). Síndromes mielodisplásicos: Imetelstat (Rytelo®; FDA). (R) SISTEMA RESPIRATORIO: EPOC: Ensifentrina (Ohtuvayre®; FDA). (V) VARIOS: Diagnóstico por imagen (fluorescencia): Pegulicianina (Lumisight®; FDA).
(A) TRACTO ALIMENTARIO Y METABOLISMO
Insulina Icodec (Awiqli®) Novo Nordisk (EMA, UE)
Indicación: Tratamiento de la diabetes mellitus en adultos.
Tipo: Medicamento biológico constituido por un análogo de la insulina humana, compuesto por dos cadenas peptídicas unidas por un puente disulfuro a las que se ha añadido una cadena lateral que contiene un diácido graso C20 para permitir una unión fuerte aunque reversible a la albúmina, así como tres sustituciones de aminoácidos que proporcionan estabilidad molecular y atenúan la unión y eliminación del receptor de insulina; todas estas modificaciones prolongan la semivida de eliminación de más de ocho días, lo que permite una única administración semanal. Autorizado en la Unión Europea (EMA) el 21 de mayo de 2024; no autorizado previamente en Estados Unidos (FDA).
Mecanismo: La insulina y sus análogos, como la insulina icodec, regulan el metabolismo de la glucosa. Reduciendo los niveles sanguíneos de glucosa mediante la activación de los receptores específicos de insulina para estimular la captación periférica de glucosa, especialmente por el músculo esquelético y la grasa, así como para inhibir la producción de glucosa hepática. La insulina también inhibe la lipolisis y la proteolisis e incrementa la síntesis de proteínas. La semvida de eliminación prolongada de la insulina icodec refleja un depósito de insulina icodec en la circulación y en el compartimento intersticial, desde donde se libera lenta y continuamente y se une específicamente al receptor de insulina. Cuando la insulina icodec se une al receptor de insulina humana, produce los mismos efectos farmacológicos que los de la insulina humana.
Eficacia clínica: Cinco ensayos clínicos de fase 3 internacionales, aleatorizados, con control activo, abiertos o ciegos y de grupos paralelos de 26 o 52 semanas de duración, en un total de 1.628 pacientes (1.338 con diabetes mellitus tipo 2 y 290 con diabetes mellitus tipo 1). El objetivo principal era demostrar el efecto sobre el control glucémico de la insulina icodec semanal en comparación con una insulina basal diaria (insulina degludec o insulina glargina) en la población diabética, comprándose el cambio (%) en la HbA1c. En diabetes mellitus tipo 2, el cambio medio en la HbA1c desde el inicio hasta el final del tratamiento fue de -1,6 puntos porcentuales con insulina icodec vs. -1,4 con insulina degludec, y de -1,2 vs. -1,2 con insulina glargina; el porcentaje de pacientes que presentaron unos niveles de HbA1c <7% fue del 53 vs. 43% con insulina degludec y del 26 vs. 25% con insulina glargina. En diabetes mellitus tipo 1, el cambio medio en la HbA1c desde el inicio hasta el final del tratamiento fue de -0,5 puntos porcentuales con insulina icodec vs. -0,5 con insulina degludec; el porcentaje de pacientes que presentaron unos niveles de HbA1c <7% fue del 9,6 vs. 16,7% con insulina degludec.
Eventos adversos: La más común (≥10%) es hipoglucemia ; son frecuentes (≥1%) las reacciones en el lugar de inyección y edema periférico.
Elafibranor (Iqirvo®) Ipsen (FDA, USA)
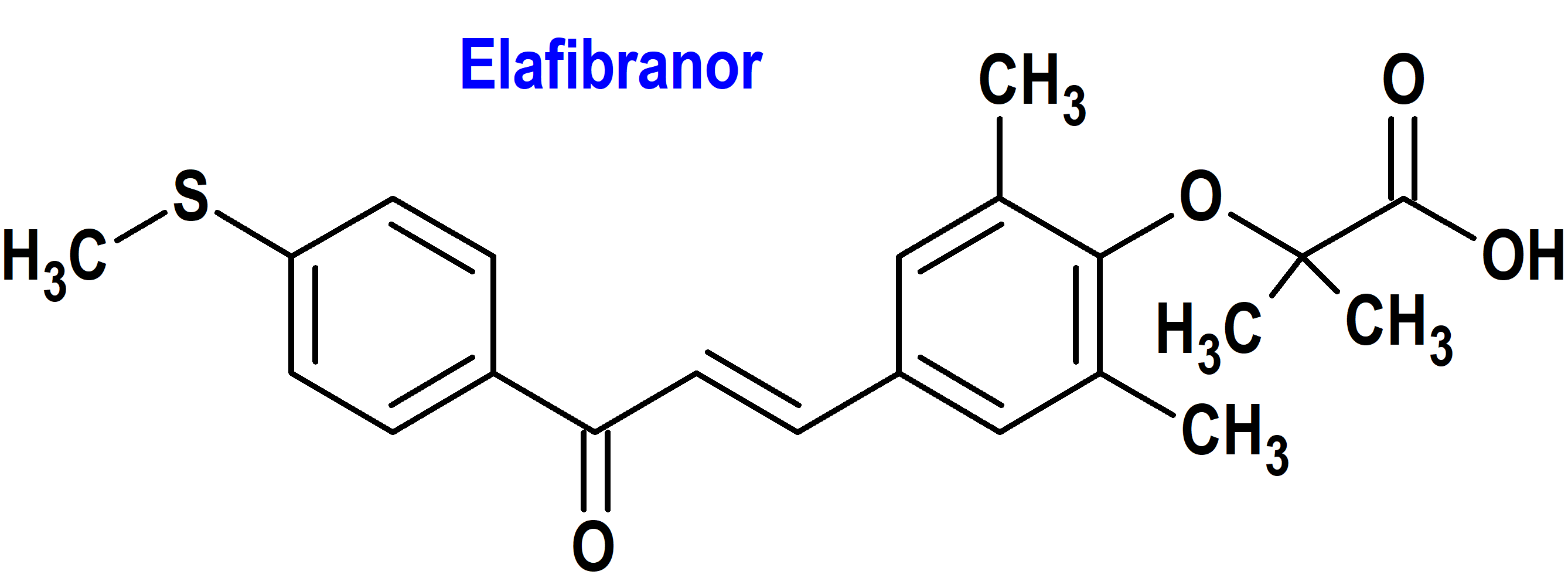
Indicación: Tratamiento de la colangitis biliar primaria (CBP) en combinación con ácido ursodesoxicólico (AUDC) en adultos que han tenido una respuesta inadecuada al AUDC, o como monoterapia en pacientes que no pueden tolerar el AUDC.
Tipo: Medicamento sintético estándar constituido por el ácido 2-(2,6-dimetil-4-{3-[4-(metilsulfanil)fenil]-3-oxoprop-1-en-1-il}fenoxi)-2-metilpropanoico. Autorizado en Estados Unidos (FDA) el 10 de junio de 2024 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Elafibranor y su principal metabolito activo, GFT1007, son agonistas del receptor activado por proliferador de peroxisomas (PPAR), los cuales activan PPAR-alfa, PPAR-gamma y PPAR-delta. Sin embargo, no se comprende bien el mecanismo por el cual elafibranor ejerce sus efectos terapéuticos en pacientes con colangitis biliar primaria, aunque la actividad farmacológica que podría ser relevante para los efectos terapéuticos incluye la inhibición de la síntesis de ácidos biliares mediante la activación de PPAR-alfa y PPAR-delta. La vía de señalización para PPAR-delta incluye la regulación negativa dependiente del factor de crecimiento de fibroblastos 21 (FGF21) del citocromo CYP7A1, la enzima clave para la síntesis de ácidos biliares a partir del colesterol.
Eficacia clínica: Un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo que incluyó a 161 adultos con CBP con respuesta inadecuada o intolerancia al AUDC. Los pacientes fueron tratados al menos 52 semanas. La variable principal de eficacia fue la respuesta bioquímica en la semana 52 (nivel de fosfatasa alcalina – FA – menor de 1,67 veces el límite superior normal – LSN – de 129 U/L para hombres y 104 U/L para mujeres; bilirrubina total menor o igual que el LSN de 1,20 mg/dL; y una disminución de la FA mayor o igual al 15 % desde el inicio): 51 vs 4%. Como variable secundaria se consideró la normalización de la FA (menor o igual al LSN) en la semana 52: 15 vs 0%.
Eventos adversos: Los más comunes (≥5%, con una diferencia de ≥3% con placebo) son dolor abdominal (11/6%), náusea (11/6%), vómitos (11/2%), artralgia (8/4%), estreñimiento (8/2%), mialgia (7/2%), reflujo gastroesofágico (6/2%), sequedad de boca (5/2%) y pérdida de peso (5/0%).
(B) SANGRE Y SISTEMA HEMATOPOYÉTICO
Fidanacogene Elaparvovec (Beqvez®) Pfizar (FDA, USA)
Indicación: Tratamiento de XXXX
Tipo: Medicamento de terapia avanzada (génica) basada en virus adenoasociados (AAV) que consiste en una cápside viral recombinante (AAVRh74var) derivada de un vector de serotipo de AAV (Rh74) de origen natural que contiene el transgén del factor IX de coagulación humano (FIX) modificado a una variante de actividad de factor IX altamente específica (FIX-R338L). Autorizado en Estados Unidos (FDA) el 25 de abril de 2024 como medicamento huérfano (Orphan drug), designado como terapia innovadora (Breakthrough Therapy) y como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Terapia génica para introducir en las células transducidas una copia funcional del gen del factor IX que codifica una variante de FIX de alta actividad (FIX-R338L, hFIX Padua). La cápside AAVRh74var es capaz de transducir hepatocitos, el sitio natural de síntesis del factor IX.
Eficacia clínica: Un estudio multinacional, prospectivo, abierto, de un solo brazo y en actualmente en curso, realizado en 45 pacientes varones adultos con hemofilia B de moderada a grave (actividad del factor IX ≤2 UI/dL). Los pacientes recibieron una única infusión intravenosa del medicamento y entraron en un período de seguimiento (PS) de 6 años, de los que 41 completaron al menos 15 meses, siendo la mediana de los 45 pacientes tratados fue de 2,0 años (rango: 0,4 a 3,2 años) desde el momento de la infusión. La variable primaria de eficacia de eficacia fue una prueba de no inferioridad (NI) de la tasa de hemorragia anualizada (THA) durante el período de evaluación de eficacia (PEE), semana 12 (día 82) hasta el corte de datos después del tratamiento, en comparación con la la THA inicial durante el período inicial, de 3,0 sangrados/año. Las diferencias entre el valor inicial (período inicial prospectivo) versus el período de evaluación de eficacia posterior (a partir de 12 semanas) fueron una mediana de THA (sangrados/año) de 1,3 frente a 0,0; THA media derivada del modelo [sangrados/año] 4,5 frente a 2,5; (%) de pacientes sin sangrado 29 vs. 60%; número total de hemorragias observadas 225 frente a 98; número de hemorragias espontáneas observadas (proporción del total de hemorragias) 70 frente a 61%; número de hemorragias articulares observadas (proporción del total de hemorragias): 82 frente a 72 %.
Eventos adversos: El más común (>10%) consiste en aumento de alanina transaminasa (ALT) y de aspartato transaminasa (AST).
(D) DERMATOLOGÍA
Sofpironio (Sofdra®) Botanix (FDA, USA)
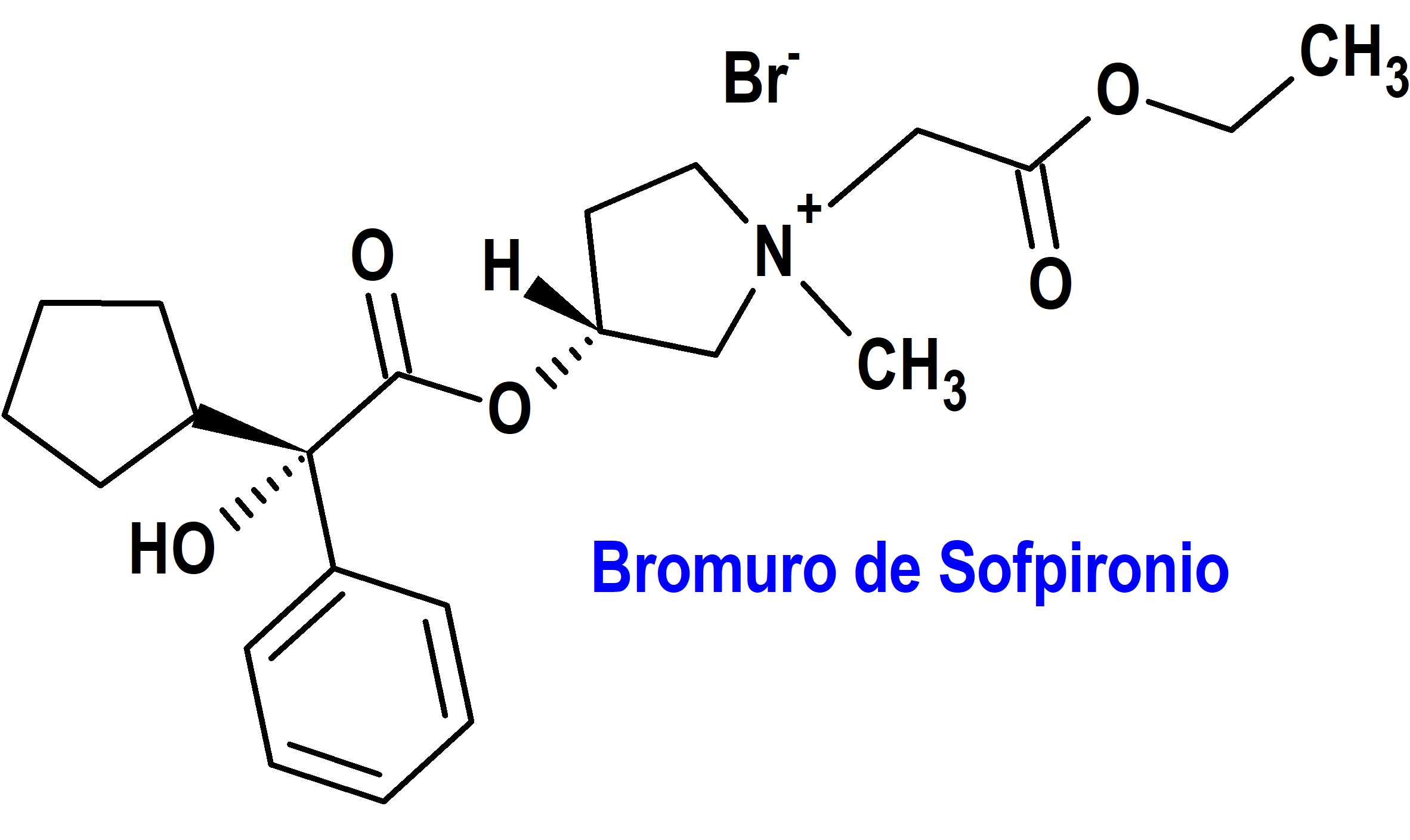
Indicación: Tratamiento de la hiperhidrosis axilar primaria en adultos y pacientes pediátricos de 9 años de edad y mayores.
Tipo: Medicamento sintético estándar constituido por el bromuro de 3’(R)-[2(R)ciclopentilfenilhidroxi-acetoxi]-1’-metil-1’-etoxicarbonilmetil-pirrolidinio. Autorizado en Estados Unidos (FDA) el 18 de junio de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor competitivo de los receptores de acetilcolina que se encuentran en ciertos tejidos periféricos, incluidas las glándulas sudoríparas. El sofpironio reduce indirectamente la tasa de sudoración al prevenir la estimulación de estos receptores.
Eficacia clínica: Dos ensayos multicéntricos, aleatorizados y controlados con placebo incluyendo a un total de 701 sujetos de 9 o más años con hiperhidrosis axilar primaria, con síntomas durante al menos 6 meses de duración, produciendo al menos 50 mg de sudor en cada axila con un total combinado de al menos 150 mg durante un período de 5 minutos y tener una puntuación ≥3 en la escala HDSM-Ax7 (medida de la gravedad de la enfermedad de hiperhidrosis: puntuación en la escala axilar de 7 ítems). La variable primaria de eficacia fue la tasa de pacientes que experimentaron una mejora ≥2 puntos en la puntuación de la escala HDSM-Ax-7a desde el inicio hasta el día 43: 49 vs. 28% (estudio 1) y 64 vs. 48% (estudio 2).
Eventos adversos: Los más comunes (≥2%) son boca seca (14%), visión borrosa (9%), midriasis (7%) y retención urinaria (2%).
(G) SISTEMA GENITOURINARIO Y HORMONAS SEXUALES
Vibegron (Obgemsa®) Pierre Fabre (EMA, UE)
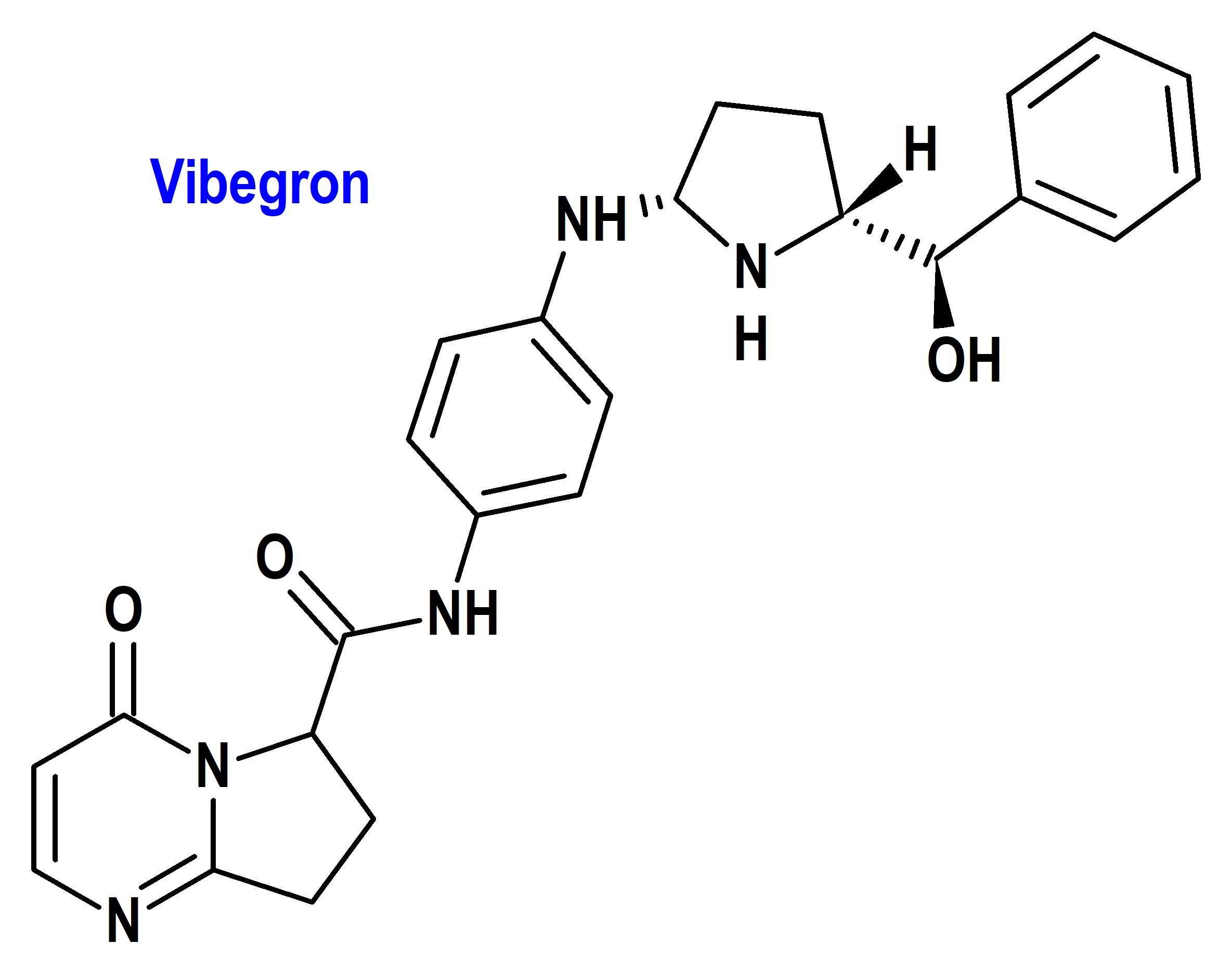
Indicación: Tratamiento sintomático de pacientes adultos con síndrome de vejiga hiperactiva.
Tipo: Medicamento sintético estándar constituido por la (6S)-N-[4-[[(2S,5R)-5-[(R)-hidroxi(fenil)metil]pirrolidin-2-il]metil]fenil]-4-oxo-7,8-dihidro-6H-pirrolo[1,2-a]pirimidina-6-carboxamida. Autorizado en la Unión Europea (EMA) el 27 de junio de 2024; autorizado previamente en Estados Unidos (FDA) el 23 de diciembre de 2020.
Mecanismo: Agonista selectivo y potente del receptor adrenérgico beta-3 humano sobre β1-AR y β2-AR. La activación del receptor adrenérgico beta-3 localizado en el músculo detrusor de la vejiga aumenta la capacidad vesical al relajar el músculo liso detrusor durante el llenado de la vejiga.
Eficacia clínica: Ensayo clínico de fase 3, doble ciego, aleatorizado, controlado con placebo y con un ensayo activo (tolterodina), de 12 semanas de duración, en 1.518 pacientes con vejiga hiperactiva (VH) con síntomas de urgencia y frecuencia urinaria con o sin incontinencia urinaria de urgencia (IUU). Las variables co-primarias fueron el cambio con respecto al valor basal en el número medio diario de micciones (-1,8 con vibegron vs. -1,6 con tolterodina y -1,3 con placebo) y el número medio diario de episodios de IUU en la semana 12 (-2,0/-1,8/-1,4).
Eventos adversos: Los más comunes son infección urinaria (6,6%), cefalea (5,0%), diarrea (3,1%) y náuseas (3,0%). La frecuencia de reacciones adversas al medicamento que condujeron a la interrupción del tratamiento es del 0,9%.
(J) ANTIINFECCIOSOS SISTÉMICOS
Aztreonam / Avibactam (Emblaveo®) Pfizer (EMA, UE)
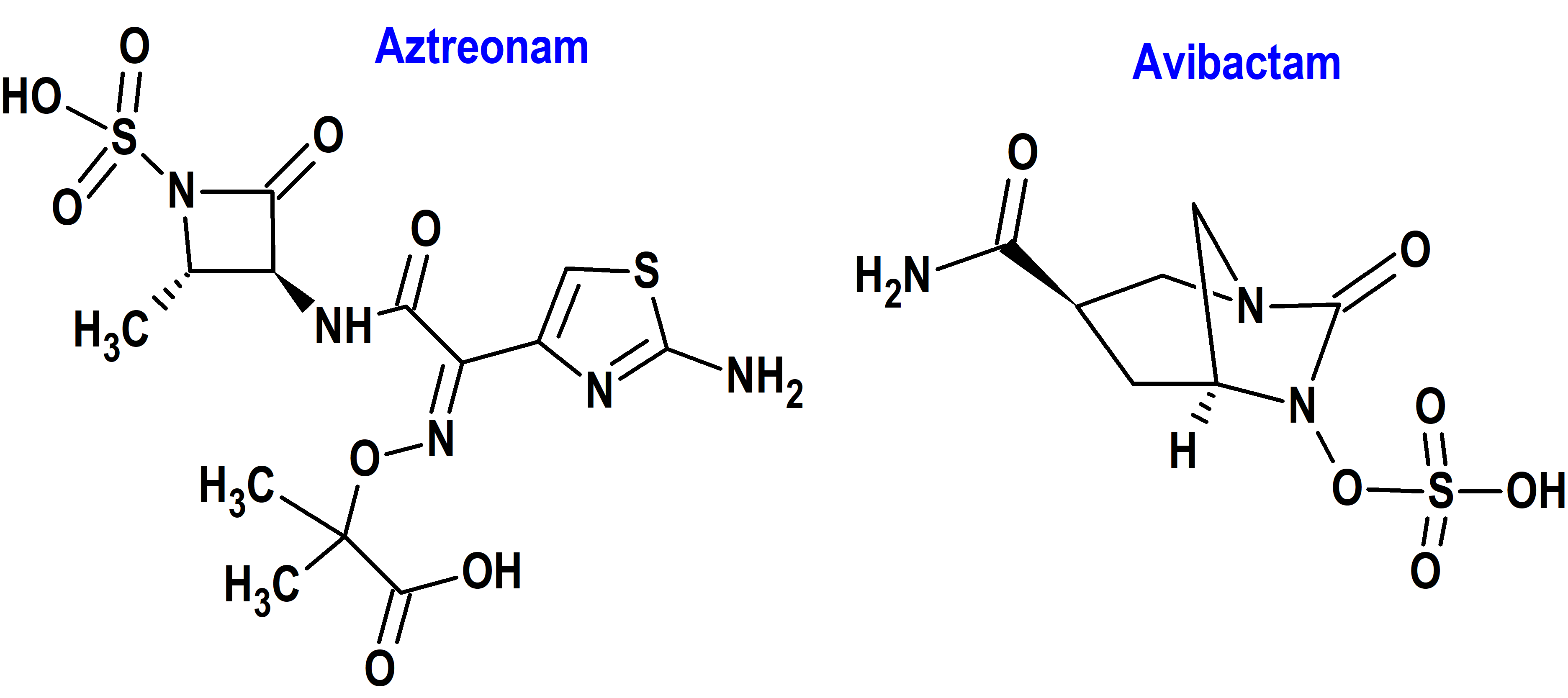
Indicación: Tratamiento en pacientes adultos de infección intraabdominal complicada (IIAc), neumonía adquirida en el hospital (NAH), incluyendo neumonía asociada a ventilación mecánica (NAV) e infección del tracto urinario complicada (ITUc), incluyendo pielonefritis.
Tipo: Medicamento sintético estándar constituido por una combinación de un antibiótico monobactámico (aztreonam: 3-[2-(2-azanioil-1,3-tiazol-4-il)-2-(1-hidroxi-2-metil-1-oxo-propan-2-il)oxiimino-acetil]amino-2-methil-4-oxo-azetidin-1-sulfonato) y un inhibidor de betalactamasas (avibactam: [1R,2S,5R]-7-oxo-6-sulfooxi-1,6-diazabiciclo[3.2.1]octano-2-carboxamida. Autorizado en la Unión Europea (EMA) el 22 de abril de 2024 ; no autorizado previamente en Estados Unidos (FDA). Tanto aztreonam como avibactam ya estaban previamente autorizados en la Unión Europea, pero no formando esta combinación; en concreto, el avibactam había sido autorizado tanto en la UE (2016) como en Estados Unidos (2015) en combinación con ceftazidima.
Mecanismo: Aztreonam es un antibiótico monobactámico que, como el resto de betalactámico, inhibe la síntesis de peptidoglicano de la pared celular bacteriana mediante la unión a las proteínas de unión a penicilinas (PBP), lo que conduce a la lisis y muerte de la célula bacteriana. Aztreonam es generalmente estable a la hidrólisis por las enzimas de clase B (metalo-β-lactamasas). Por su parte, avibactam es un inhibidor de β-lactamasas no betalactámico que actúa mediante la formación de un enlace covalente con la enzima que es estable a la hidrólisis; inhibe las β-lactamasas de clase A y C de Ambler y algunas enzimas de clase D, incluyendo las β-lactamasas de amplio espectro (BLEEs), las carbapenemasas de Klebsiella pneumoniae (KPC) y OXA-48, y las enzimas AmpC; no inhibe las enzimas de clase B y no es capaz de inhibir muchas enzimas de clase D.
Eficacia clínica: Los estudios muestran que se espera que el medicamento sea eficaz en el tratamiento de infecciones para las que ya se utiliza aztreonam, así como infecciones debidas a organismos gramnegativos aeróbicos. Los datos microbiológicos indican que aztreonam en combinación con avibactam también puede tener una utilidad importante en infecciones causadas por enterobacterias productoras de metalo-β-lactamasa.
Eventos adversos: Los más comunes son anemia (6,9%), diarrea (6,2%), y elevación de la alanina aminotransferasa (ALT) (6,2%) y de la aspartato aminotransferasa (AST) (5,2%).
Vacuna neumococo polisacárida conjugada 22-valente (Capvaxine®) Merck Sharp Dohme (FDA, USA)
Indicación: Inmunización activa para la prevención de enfermedades invasivas causadas por Streptococcus pneumoniae serotipos 3, 6A, 7F, 8, 9N, 10A, 11A, 12F, 15A, 15B, 15C, 16F, 17F, 19A, 20A, 22F, 23A, 23B, 24F, 31, 33F y 35B en personas de 18 años en adelante. Inmunización activa para la prevención de la neumonía causada por S. pneumoniae serotipos 3, 6A, 7F, 8, 9N, 10A, 11A, 12F, 15A, 15C, 16F, 17F, 19A, 20A, 22F, 23A, 23B, 24F, 31, 33F y 35B en personas mayores de 18 años.
Tipo: Medicamento biológico constituido por polisacáridos capsulares purificados de S. pneumoniae serotipos 3, 6A, 7F, 8, 9N, 10A, 11A, 12F, 15A, 15B (des-O-acetilado antes de la conjugación), 16F, 17F, 19A, 20A, 22F, 23A, 23B, 24F, 31, 33F y 35B conjugados individualmente con la proteína transportadora CRM197 (un mutante no tóxico de la toxina diftérica originada de Corynebacterium diphtheriae C7, expresada de forma recombinante en Pseudomonas fluorescens). Autorizado en Estados Unidos (FDA) el 17 de junio de 2024 de forma acelerada (Accelerated Approval); no autorizado aún en la Unión Europea (EMA).
Mecanismo: La protección contra la enfermedad neumocócica invasiva se confiere principalmente mediante la destrucción opsonofagocítica de S. pneumoniae. La vacuna induce actividad opsonofagocítica (OPA) contra 22 serotipos de S. pneumoniae. El polisacárido des-O-acetilado del serotipo 15B tiene una estructura molecular similar al polisacárido del serotipo 15C e induce OPA al serotipo 15C y también induce OPA con reacción cruzada contra el serotipo 15B.
Eficacia clínica: Cuatro estudios de inmunogenicidad evaluada mediante respuestas de actividad opsonofagocítica (OPA) específicas del serotipo un mes después de la vacunación. Los criterios de valoración principales de inmunogenicidad incluyeron los títulos medios geométricos (GMT) de OPA y la proporción de individuos que lograron un aumento ≥4 veces en las respuestas de OPA desde la prevacunación hasta 1 mes después de la vacunación. La eficacia de Capvaxive® en personas de 18 años o más para la prevención de enfermedades invasivas causadas por S. pneumoniae se demostró basándose en la inmunogenicidad comparativa con una vacuna neumocócica autorizada (Prevnar 20®), donde la tasa de respuesta observada con Capvaxive®fue superior a la de Prevnar 20® para los serotipos específicos de Capvaxive®.
Eventos adversos: Los más comunes (>10%) en personas de 18 a 49 años de edad son dolor en el lugar de la inyección (73%), fatiga (36%), dolor de cabeza (28%), mialgia (16% ), eritema en el lugar de la inyección (14%) e hinchazón en el lugar de la inyección (13%).
Vacuna Virus Respiratorio Sincitial (mResvia®) Moderna (EMA, UE; FDA, USA)
Indicación: Inmunización activa para la prevención de la enfermedad del tracto respiratorio inferior causada por el virus respiratorio sincitial (VRS) en personas de 60 años de edad y mayores.
Tipo: Medicamento sintético biológico constituido por un ARNm modificado con nucleósidos que codifica la glicoproteína F del VRS estabilizada en la conformación de prefusión (proteína pre-F). Autorizada en Estados Unidos (FDA) el 31 de mayo de 2024; no autorizada aún en la Unión Europea, pero aprobada por el CHMP (EMA) el 27 de junio de 2024.
Mecanismo: Induce una respuesta inmune contra la proteína pre-F del RSV.
Eficacia clínica: Estudio clínico aleatorizado, controlado con placebo, ciego al observador y basado en casos en personas de 60 años o más con o sin afecciones médicas subyacentes después de recibir una dosis única de MRESVIA. El estudio se lleva a cabo en 22 países e incluye participantes de las regiones de América del Norte/Europa, América Central/Latina, África y Asia/Pacífico y está diseñado para seguir a los participantes durante hasta 24 meses después de la vacunación. La población de análisis de eficacia primaria (Conjunto de eficacia por protocolo) incluyó 35 064 participantes que recibieron MRESVIA (n = 17 561) o placebo (n = 17 503). La variable principal de eficacia fue la prevención de un primer episodio de enfermedad del tracto respiratorio inferior por VRS con ≥2 signos/síntomas (15 vs. 70 casos; eficacia del 79%) o ≥3 signos/síntomas (5 vs. 26 casos; eficacia del 81%) comenzando 14 días después de la vacunación.
Eventos adversos: Los más comunes (≥10%) son dolor en el lugar de la inyección (56%), fatiga (31%), dolor de cabeza (27%), mialgia (26%), artralgia (22%), hinchazón o sensibilidad en la axila (15%) y escalofríos (12%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
Tarlatamab (Imdelltra®) Amgen (FDA, USA)
Indicación: Tratamiento de pacientes adultos con cáncer de pulmón de células pequeñas en estadio extenso (SCLC) con progresión de la enfermedad durante o después de la quimioterapia basada en platino.
Tipo: Medicamento sintético biológico recombinante constituido por un acoplador de células T CD3 biespecífico dirigido al ligando tipo delta 3 (DLL3) y al CD3 expresados en la superficie de las células T, formado por 982 aminoácidos y un peso molecular 105 kDa. Autorizado en Estados Unidos (FDA) el 16 de mayo de 2024 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Es un acoplador biespecífico de células T que se une a DLL3 expresado en la superficie de las células, incluidas las células tumorales, y a CD3 expresado en la superficie de las células, provocando la activación de las células T, la liberación de citocinas inflamatorias y la lisis de las células que expresan DLL3, el cual que se expresa de manera aberrante en la mayoría de los cánceres pulmonares de células pequeñas.
Eficacia clínica: Un estudio abierto, multicéntrico y de múltiples cohortes, realizado en 99 pacientes con cáncer de pulmón de células pequeñas en estadio extenso y en recaída/refractario con progresión de la enfermedad después de quimioterapia basada en platino excluyéndose a los pacientes con metástasis cerebrales sintomáticas, enfermedad pulmonar intersticial o neumonitis no infecciosa e inmunodeficiencia activa. Los pacientes recibieron tarlatamab hasta la progresión de la enfermedad o toxicidad inaceptable. Las principales variables de eficacia fueron la tasa de respuesta general (ORR: 40% global, 52% en pacientes con SCLC resistente al platino y 31% con SCLC sensible al platino) y la duración de la respuesta (DOR: mediana de 9,7 meses).
Eventos adversos: Los más comunes (>20%) fueron síndrome de liberación de citocinas (SLC), fatiga, pirexia, disgeusia, disminución del apetito, dolor musculoesquelético y estreñimiento, anemia y náuseas. Las anomalías de laboratorio de grado 3 o 4 más comunes (≥5%) fueron disminución de linfocitos, disminución de sodio, aumento de ácido úrico, disminución de neutrófilos totales, disminución de hemoglobina, aumento del tiempo de tromboplastina parcial activada y disminución de potasio.
Nogapendekina Alfa Inbakicept (Anktiva®) Altor (FDA, USA)
Indicación: Tratamiento, en combinación con Bacillus Calmette-Guérin (BCG), de pacientes adultos con cáncer de vejiga no músculo-invasivo (NMIBC) que no responde a BCG con carcinoma in situ (CIS) con o sin tumores papilares.
Tipo: Medicamento biológico constituido por un complejo soluble que consiste en nogapendekina alfa (una variante humana de IL-15N72D, 114 aminoácidos) unida a inbakicept [un dominio sushi dimérico de IL-15Rα humano (65 aminoácidos)/proteína de fusión IgG1 Fc humana (232 aminoácidos)]. Cada complejo nogapendekina alfa inbakicept completamente ensamblado consta de una molécula de inbakicept y dos de nogapendekina alfa. Cada componente de IL-15N72D está unido a uno de los dominios sushi de IL-15Rα. El peso molecular del complejo nogapendekin alfa inbakicept desglucosilado es 92,1 kDa y los de las subunidades desglucosiladas individuales son 66,6 kDa (dímero) y 12,8 kDa para el dominio Fc de IL-15RαSu/IgG1 y el dominio IL-15N72D, respectivamente. Autorizado en Estados Unidos (FDA) el 22 de abril de 2024 y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Nogapendekin alfa inbakicept es un agonista del receptor de IL-15. La IL-15 envía señales a través de un receptor heterotrimérico que está compuesto por la subunidad común de la cadena gamma (γc), la subunidad de la cadena beta (βc) y la subunidad alfa específica de IL-15, el receptor α de IL-15. La IL-15 es transpresentada por el receptor α de IL-15 al receptor compartido de IL-2/IL-15 (βc y γc) en la superficie de las células T CD4+ y CD8+ y de las células NK. La unión de nogapendequina alfa inbakicept a su receptor da como resultado la proliferación y activación de células NK, CD8+ y T de memoria sin proliferación de células Treg inmunosupresoras.
Eficacia clínica: Un ensayo multicéntrico de un solo grupo de 77 pacientes con NMIBC de alto riesgo y que no responden al BCG con CIS con o sin enfermedad papilar Ta/T1 después de una resección transuretral. Los pacientes recibieron inducción de nogapendekin alfa inbakicept mediante instilación intravesical con BCG seguida de terapia de mantenimiento durante hasta 37 meses. El estado del tumor se evaluó con cistoscopia y citología de orina cada 3 meses durante un máximo de 2 años y se requirió una biopsia (aleatoria o dirigida por cistoscopia) dentro de los primeros 6 meses después del inicio del tratamiento. Las principales medidas de resultados de eficacia fueron la tasa de respuesta completa (RC), definida como cistoscopia negativa (62%) y la duración de la respuesta completa, con el 58% por ciento de los pacientes ≥ 12 meses y el 40% ≥ 24 meses.
Eventos adversos: Los más comunes (≥15%) son aumento de creatinina, disuria, hematuria, frecuencia urinaria, urgencia miccional, infección del tracto urinario, aumento de potasio, dolor musculoesquelético, escalofríos y pirexia.
Tovorafenib (Ojemda®) One Day (FDA, USA)
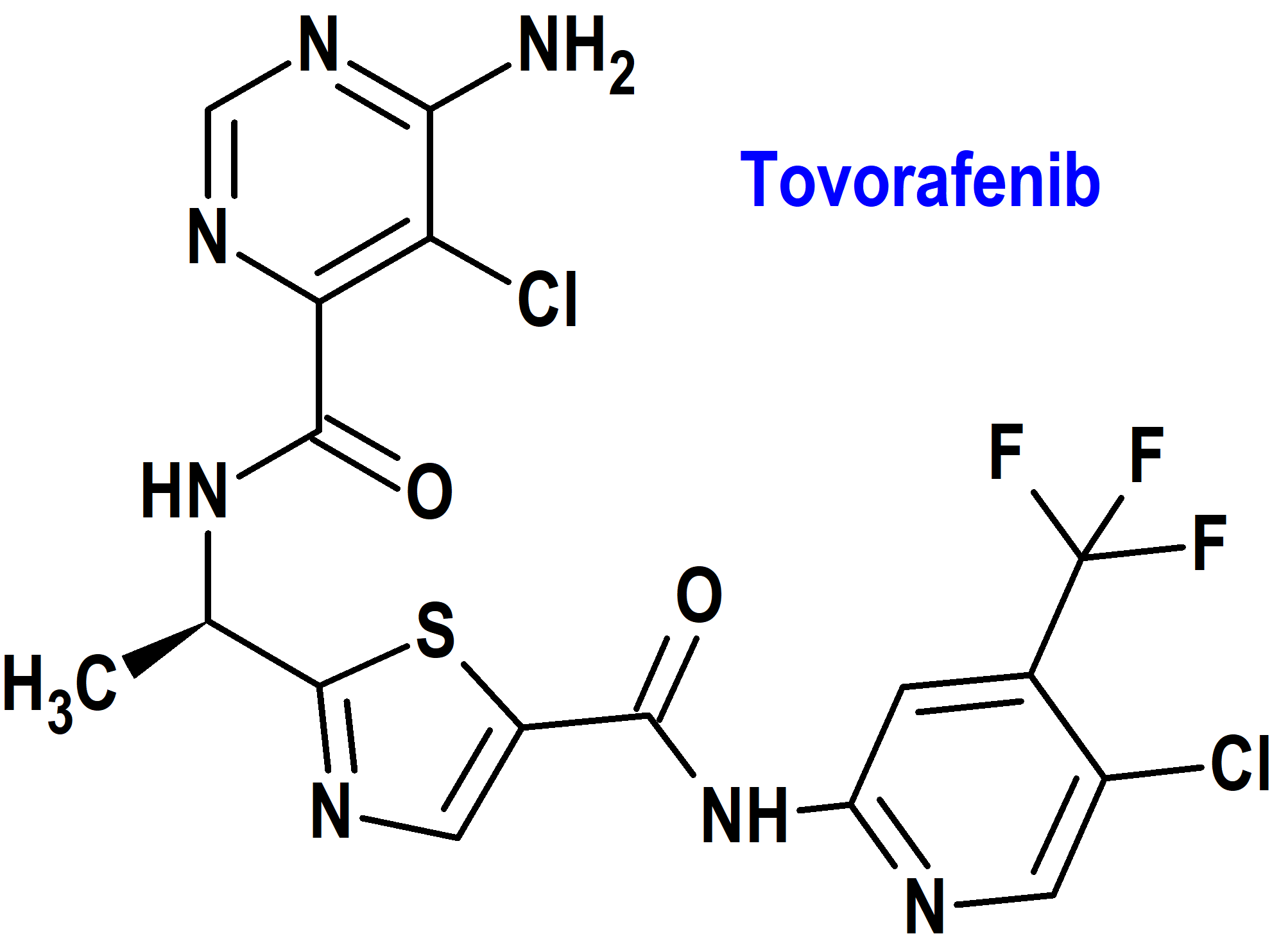
Indicación: Tratamiento de pacientes de 6 meses de edad y mayores con glioma pediátrico de bajo grado (LGG) en recaída o refractario que alberga una fusión o reordenamiento BRAF, o una mutación BRAF V600.
Tipo: Medicamento sintético estándar constituido por la 6-amino-5-cloro-N-[(1R)-1-(5-{[5-cloro-4-(trifluorometil)piridin2-il]carbamoil}-1,3-tiazol-2-il)etil]pirimidina-4-carboxamida. Autorizado en Estados Unidos (FDA) el 23 de abril de 2024 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD) y designado como terapia innovadora (Breakthrough Therapy) ; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de cinasa RAF tipo II de las cinasas BRAF V600E mutante, BRAF de tipo salvaje y CRAF de tipo salvaje.
Eficacia clínica: Un ensayo multicéntrico, abierto y de un solo brazo en 76 pacientes con LGG pediátrico en recaída o refractario que alberga una alteración activadora de BRAF y que habían recibido al menos un línea de terapia sistémica previa. Se excluyeron los pacientes con tumores que albergaban alteraciones moleculares activadoras adicionales (p. ej., mutaciones IDH1/2, mutaciones FGFR) o con un diagnóstico conocido o sospechado de neurofibromatosis tipo 1. La variable primaria de eficacia fue la tasa de respuesta general (51%), definida como la proporción de pacientes con respuesta completa (CR, 0%), respuesta parcial (PR, 37%) o respuesta menor (MR, 14%) mediante una revisión central independiente ciega basada en la respuesta. Adicionalmente, se determinó la mediana de la duración de la respuesta: 13,8 meses (85%≥6 meses y 23% ≥12 meses).
Eventos adversos: Los más comunes (≥30%) son sarpullido, cambios de color de cabello, fatiga, infección viral, vómitos, dolor de cabeza, hemorragia, pirexia, piel seca, estreñimiento, náuseas, dermatitis acneiforme e infección del tracto respiratorio superior.
Mavorixafor (Xolremdi®) X4 (FDA, USA)
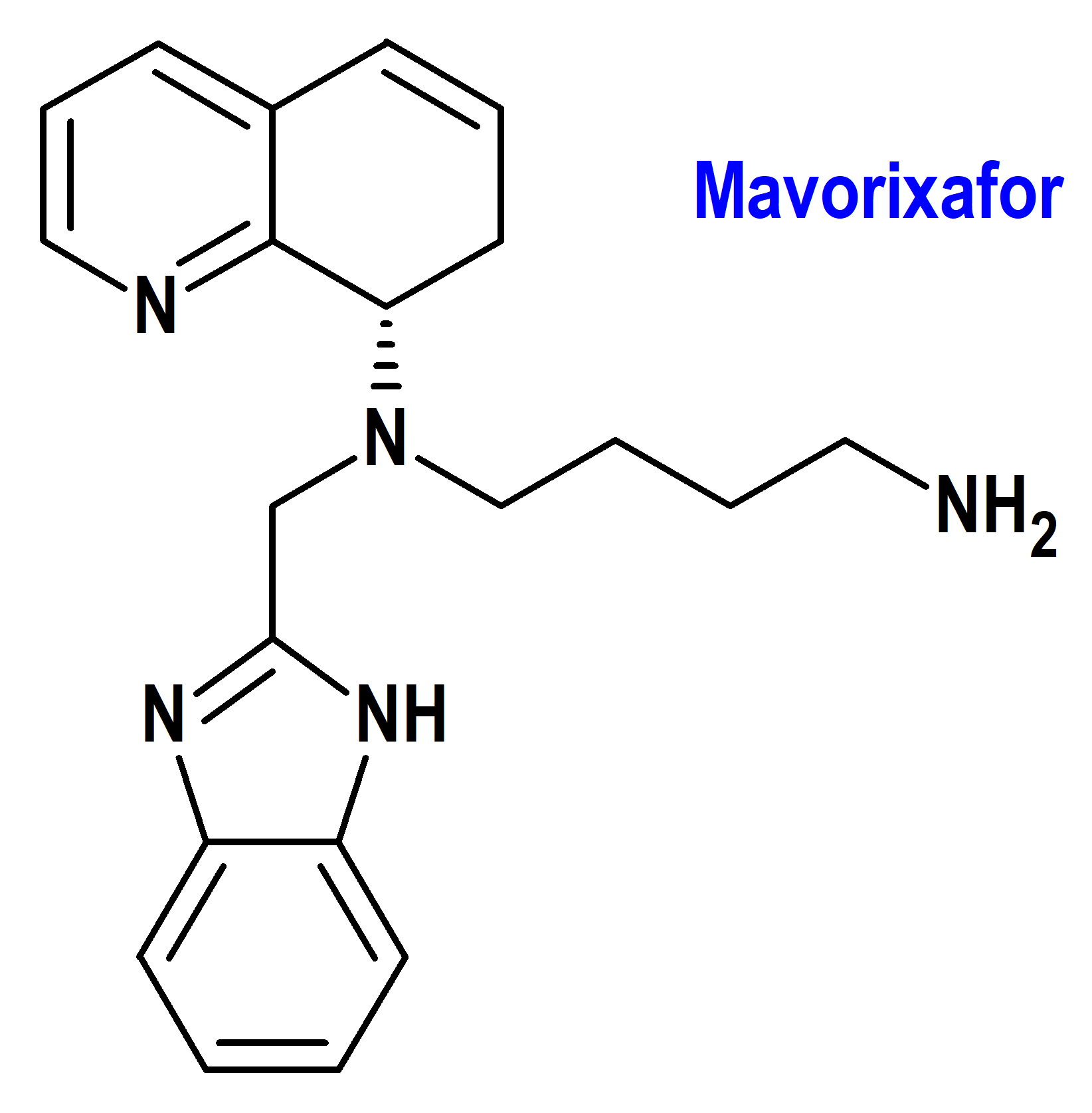
Indicación: En pacientes de 12 años de edad y mayores con síndrome WHIM (verrugas, hipogammaglobulinemia, infecciones y mielocatexis) para aumentar el número de neutrófilos y linfocitos maduros circulantes.
Tipo: Medicamento sintético estándar constituido por la N1-(1H-bencimidazol-2-ilmetil)-N1-[(8S)-5,6,7,8-tetrahidroquinolin-8-il]butano-1,4-diamina. Autorizado en Estados Unidos (FDA) el 26 de abril de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Antagonista del receptor de quimiocina CXC 4 (CXCR4) que bloquea la unión del ligando CXCR4, factor-1α derivado del estroma (SDF-1α)/ligando de quimiocina CXC 12 (CXCL12). SDF-1/CXCR4 desempeña un papel en el tráfico y la localización de leucocitos hacia y desde el compartimento de la médula ósea. Las mutaciones de ganancia de función en el gen del receptor CXCR4 que ocurren en pacientes con síndrome WHIM conducen a una mayor capacidad de respuesta a CXCL12 y a la retención de leucocitos en la médula ósea. Mavorixafor inhibe la respuesta a CXCL12 en las variantes CXCR4 tanto de tipo salvaje como mutadas asociadas con el síndrome WHIM. El tratamiento con mavorixafor produce una mayor movilización de neutrófilos y linfocitos desde la médula ósea hacia la circulación periférica. El síndrome WHIM es una enfermedad rara (prevalencia <1/1.000.000) inmunitaria congénita autosómica dominante caracterizada por la retención anómala de los neutrófilos maduros en la médula ósea (mielocatexis) con hipogammaglobulinemia ocasional, asociado a un aumento del riesgo de infecciones bacterianas y a lesiones inducidas por la susceptibilidad al virus del papiloma humano (HPV) (verrugas cutáneas, displasia genital y carcinoma mucoso invasivo).
Eficacia clínica: En ensayo aleatorizado, doble ciego y controlado con placebo de 52 semanas de duración en el que participaron 31 adolescentes y adultos con síndrome WHIM. Mavorixafor mejoró los recuentos absolutos de neutrófilos y de linfocitos, evaluados durante un período de 24 horas cuatro veces a lo largo del estudio. Los recuentos absolutos de neutrófilos por debajo de 500 células/μl y los recuentos absolutos de linfocitos por debajo de 1000 células/μl se asocian con un mayor riesgo de infecciones. El período de tiempo promedio durante 24 horas en que los recuentos estuvieron por encima de estos niveles fue significativamente mayor con mavorixafor en comparación con placebo (15,0 vs 2,8 horas para los recuentos absolutos de neutrófilos; 15,8 vs 4,6 horas para los recuentos absolutos de linfocitos). Mavorixafor redujo la puntuación de infección en un 40 % durante el período de tratamiento en comparación con placebo; sin embargo, no mejoró las verrugas. Eventos adversos: Los más comunes son trombocitopenia (21%), pitiriasis (14%), erupción cutánea (14%), rinitis (14%), epistaxis (14%), vómitos (14%) y mareos (14%).
Danicopan (Voydeya®) Alexion (EMA, UE; FDA, USA)
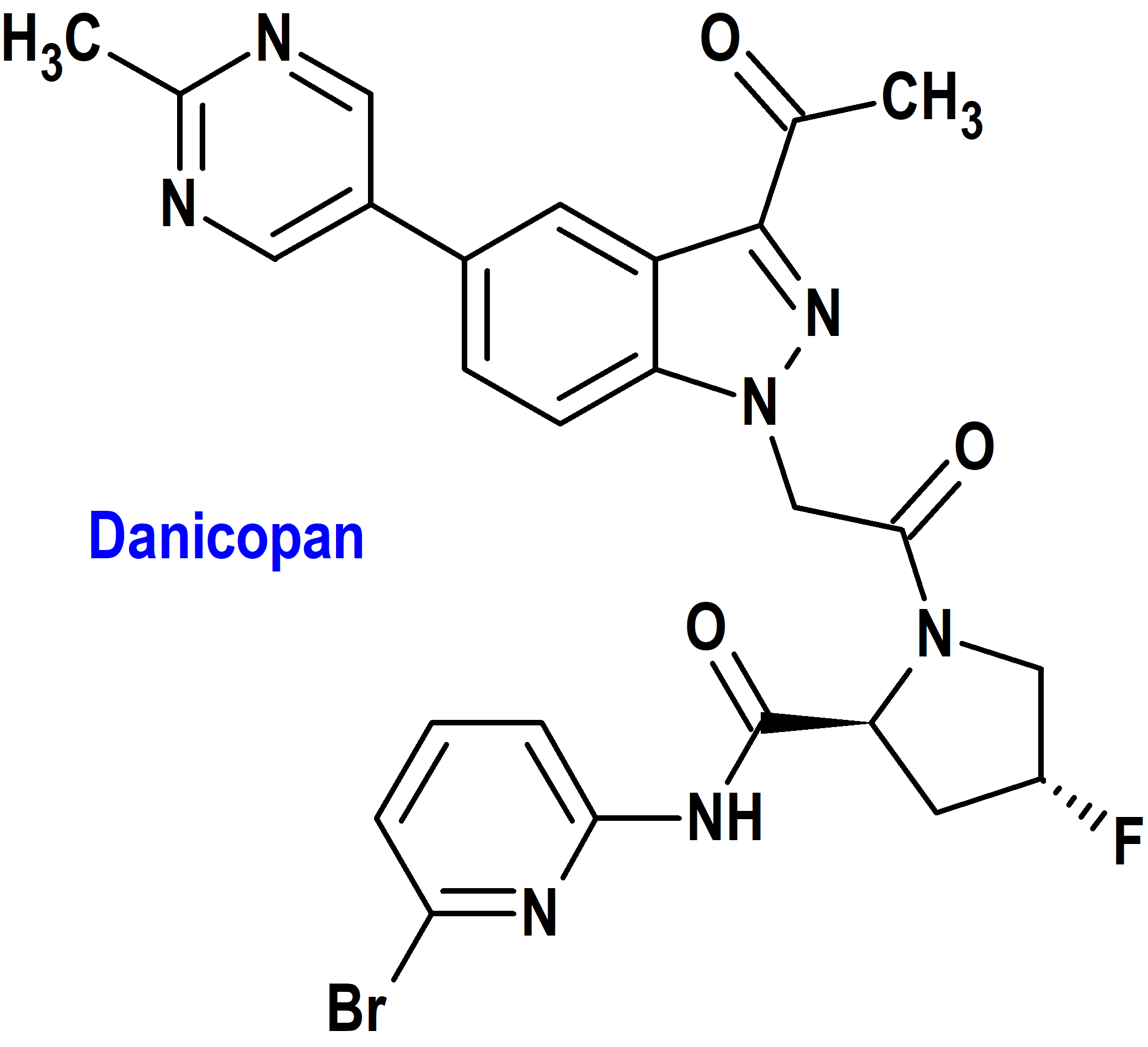
Indicación: Tratamiento, de forma complementaria a ravulizumab o eculizumab, de pacientes adultos con hemoglobinuria paroxística nocturna (HPN) que presentan anemia hemolítica residual.
Tipo: Medicamento sintético estándar constituido por la (2S,4R)-1-[2-[3-acetil-5-(2-metilpirimidin-5-il)indazol-1-il]acetil]-N-(6-bromopiridin-2-il)-4- fluoropirrolidina-2-carboxamida. Autorizado en la Unión Europea (EMA) el 19 de abril de 2024, como medicamento huérfano (Orphan drug); autorizado en Estados Unidos (FDA) el 29 de marzo de 2024.
Mecanismo: Se une de manera reversible al factor D (FD) del complemento y actúa como inhibidor selectivo de la función del FD. Al inhibir el FD, danicopán bloquea selectivamente la activación de la vía alternativa del complemento, lo que impide la producción de múltiples efectores, incluidos los fragmentos C3, después de la activación de la vía alternativa. Las otras dos vías del complemento (clásica y de la lectina) siguen estando activas. El efecto inhibidor de danicopán sobre la activación de la vía alternativa inhibe el depósito de fragmentos C3 en los glóbulos rojos de la HPN. Este depósito es la causa principal de la hemólisis extravascular, que puede ser clínicamente significativa en un pequeño subgrupo de pacientes con HPN que reciben un inhibidor de C5. El mantenimiento de la inhibición de C5 controla las consecuencias fisiopatológicas potencialmente mortales de la activación del complemento terminal que subyacen a la HPN.
Eficacia clínica: Estudio de fase 3, de varias regiones, aleatorizado, con doble enmascaramiento y comparativo con placebo en 86 pacientes con HPN que habían sido tratados con una dosis estable de ravulizumab o eculizumab durante al menos los 6 meses previos, y que tenían anemia (hemoglobina [Hgb] ≤9,5 g/dl) con un recuento absoluto de reticulocitos ≥120 × 10(9) /l, con o sin transfusiones. La variable principal de eficacia fue el cambio en el nivel de Hgb desde el inicio hasta la semana 12, que fue de 2,81 vs 0,46 g/dl (diferencia de 2,35 g/dl favorable a danicopan vs. placebo). También se observó una mejoría estadísticamente significativa con danicopán en comparación con placebo en las 4 variables secundarias analizadas: proporción de pacientes con aumento de la Hgb de ≥2 g/dl en ausencia de transfusiones (54,4 vs. 0%), proporción de pacientes con evitación de transfusiones (79,9 vs. 27,6%), cambio en la puntuación FACIT-Fatiga (8,10 vs. 2,38) y cambio en el recuento absoluto de reticulocitos (−92,6 vs. -0,9).
Eventos adversos: Los más comunes son pirexia (25%), cefalea (20%), niveles de enzimas hepáticas elevados (12 %) y dolor en las extremidades (12%).
Crovalimab (Piasky®) Genentech (FDA, USA)
Indicación: Tratamiento de pacientes adultos y pediátricos de 13 años y mayores con hemoglobinuria paroxística nocturna (HPN) y un peso corporal de al menos 40 kg.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal humanizado de origen recombinante basado en una estructura de IgG1 humana que consta de dos cadenas pesadas (451 aminoácidos cada una) y dos cadenas ligeras (217 aminoácidos cada una), con un peso molecular de 145 kDa. Autorizado en Estados Unidos (FDA) el 20 de junio de 2024 como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea (EMA), pero aprobado por el CHMP (EMA) el 27 de junio de 2024.
Mecanismo: Se une específicamente con alta afinidad a la proteína del complemento C5, inhibiendo su escisión en C5a y C5b, previniendo la formación del complejo de ataque a la membrana (MAC). Inhibe la hemólisis intravascular mediada por el complemento terminal en pacientes con HPN.
Eficacia clínica: Estudio de no inferioridad, abierto y controlado con tratamiento activo (eculizumab) sobre 204 pacientes (peso corporal ≥ 40 kg) con HPN no tratados previamente con un inhibidor del complemento. Las variables de eficacia clínica principales fueron la tasa de control de la hemólisis, medida por la proporción media de pacientes con LDH ≤ 1,5 x límite normal superior desde la semana 5 a la semana 25: (79,3% con crovalimab vs. 79,0% con eculizumab); y la proporción de pacientes que lograron evitar las transfusiones, definida como pacientes que no recibieron transfusiones de concentrados de glóbulos rojos desde el inicio hasta la semana 25 (65,7 vs. 68,1%).
Eventos adversos: Los más comunes (≥5%) son reacción relacionada con la infusión (16%), infección del tracto respiratorio (13%), infección viral (11%), hiperuricemia (8%), dolor de cabeza (8%) y diarrea (7%).
Imetelstat (Rytelo®) Geron (FDA, USA)
Indicación: Tratamiento de pacientes adultos con síndromes mielodisplásicos de riesgo bajo a intermedio con anemia dependiente de transfusiones que requieren 4 o más unidades de glóbulos rojos durante 8 semanas y que no han respondido o han perdido la respuesta o no son elegibles para recibir agentes estimulantes de la eritropoyesis.
Tipo: Medicamento sintético estándar constituido por un oligonucleótido de fórmula d(3’-amino-3’-desoxi-P-tio) (T-AG-G-G-T-T-A-G-A-C-A-A), fosforotioato de 5’-[O-[2-hidroxi-3-(hexadecanoilamino)propilo]] . Autorizado en Estados Unidos (FDA) el 6 de junio de 2024 como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea (EMA). Mecanismo: Imetelstat es un oligonucleótido inhibidor de la telomerasa humana que se une a la región del componente de ARN de la telomerasa humana (hTR), inhibe la actividad enzimática de la telomerasa y previene la unión de los telómeros. Se ha informado de un aumento de la actividad de la telomerasa y de la expresión del ARN de la transcriptasa inversa de la telomerasa humana en los síndromes mielodisplásicos y en cuadros con células madre y progenitoras malignas. El imetelstat reduce la longitud de los telómeros, así como la proliferación de células madre y progenitoras malignas, e induce su muerte celular apoptótica.
Eficacia clínica: Un ensayo multicéntrico aleatorizado, doble ciego y controlado con placebo en 178 pacientes con síndromes mielodisplásicos, que recibieron imetelstat o placebo en ciclos de tratamiento de 28 días hasta la progresión de la enfermedad o una toxicidad inaceptable. La eficacia se estableció después de una mediana de tiempo de seguimiento de 19,5 meses en el grupo de imetelstat y 17,5 meses en el grupo de placebo según la proporción de pacientes que alcanzaron una independencia transfusional de glóbulos rojos (RBC-TI), definida como la ausencia de necesidad de transfusiones de glóbulos rojos durante cualquier período consecutivo de 8 semanas y durante cualquier período consecutivo de 24 semanas, respectivamente, desde la aleatorización hasta el inicio de la terapia anticancerosa posterior (si corresponde). La tasa de RBC-TI de ≥8 semanas fue del 39,8 % con imetelstat vs. 15 % con placebo. La tasa de RBC-TI de ≥24 semanas fue del 28 vs. 3,3 %.
Eventos adversos: Los más comunes (≥10%, con una diferencia >5% con placebo) son anomalías de laboratorio (disminución de plaquetas, de glóbulos blancos y de neutrófilos, y aumento de aspartato aminotransferasa, de fosfatasa alcalina, de alanina aminotransferasa), fatiga, prolongación del tiempo de tromboplastina, artralgia/mialgia, infecciones por COVID-19 y dolor de cabeza.
(R) SISTEMA RESPIRATORIO
Ensifentrina (Ohtuvayre®) Verona (FDA, USA)
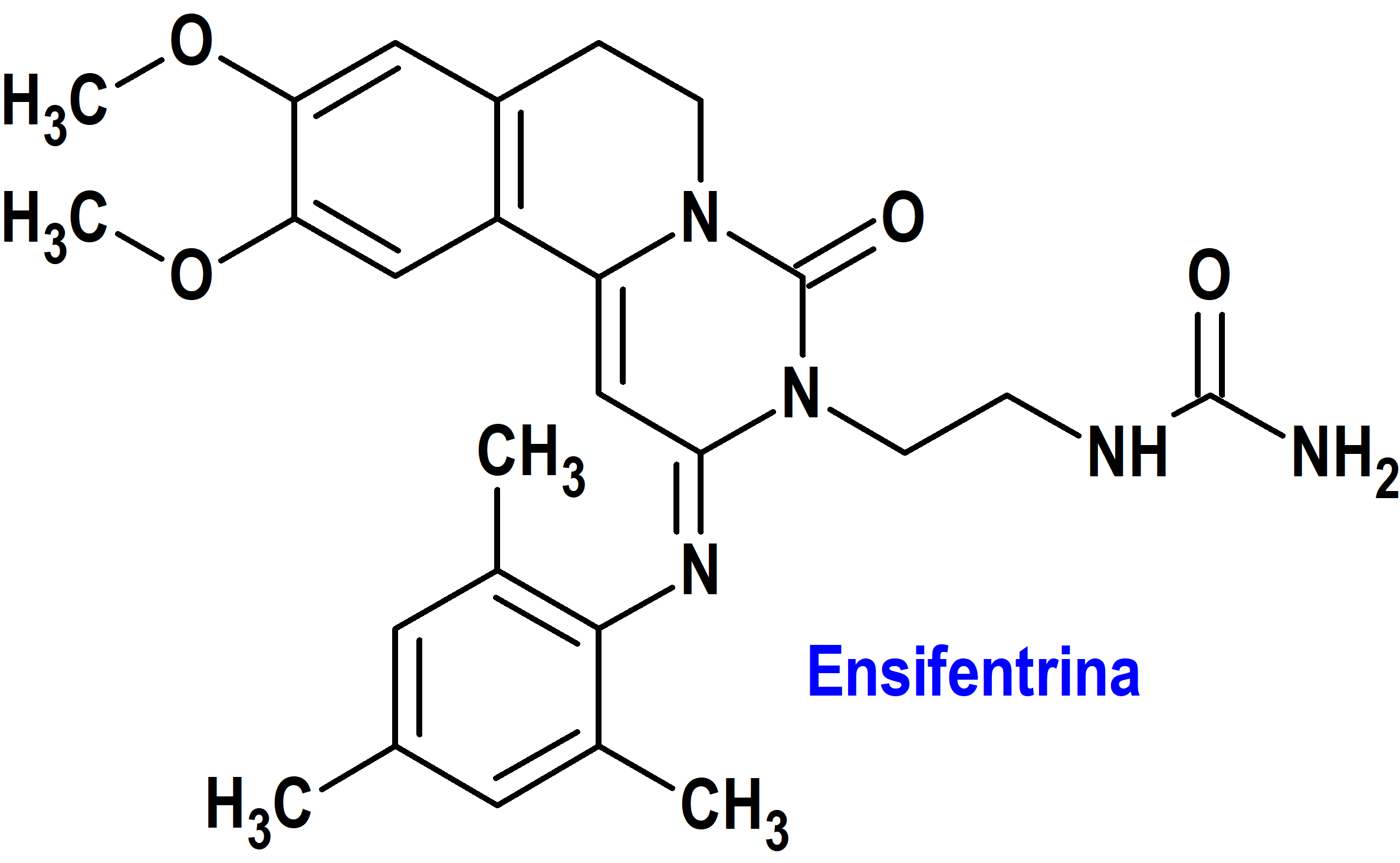
Indicación: Tratamiento de mantenimiento de la enfermedad pulmonar obstructiva crónica (EPOC) en pacientes adultos.
Tipo: Medicamento sintético estándar constituido por la N-(2-{(2E)-9,10-dimetoxi-4-oxo-2-[(2,4,6-trimetilfenil)imino]-6,7-dihidro-2H-pirimido[6,1-a]isoquinolin3(4H)-il}etil)urea. Autorizado en Estados Unidos (FDA) el 26 de junio de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de las enzimas PDE3 y PDE4. La PDE3 hidroliza principalmente la molécula de segundo mensajero monofosfato de adenosina cíclico (AMPc), pero también es capaz de hidrolizar monofosfato de guanosina cíclico (GMPc). La PDE4 hidroliza únicamente el AMPc. La inhibición de PDE3 y PDE4 da como resultado la acumulación de niveles intracelulares de AMPc y/o GMPc, lo que produce diversos efectos de señalización posteriores. La inhibición dual de PDE3 y PDE4 da lugar a efectos broncodilatadores y antiinflamatorios, con mejoras significativas en la función pulmonar, síntomas, calidad de vida y frecuencia de exacerbaciones en pacientes sintomáticos con EPOC.
Eficacia clínica: Dos ensayos clínicos aleatorizados, doble ciego, controlados con placebo y de grupos paralelos de 24 semanas de duración. Los dos ensayos reclutaron a un total de 1553 adultos con EPOC de moderada a grave. El criterio de valoración principal para ambos fue el cambio medio desde el valor inicial en el FEV1 después de la dosis en la semana 12 (+61 vs. -26 ml y +48 vs. -46 ml).
Eventos adversos: Los más comunes son dolor de espalda (1,8 ensifentrina vs. 1,0% placebo), hipertensión (1,7 vs. 0,9%), infección del tracto urinario (1,3 vs. 1,0%), diarrea (1,0 vs. 0,7%). La proporción de pacientes que interrumpieron el tratamiento debido a reacciones adversas fue del 7,6 % con ensifentrina vs. 8,2 % con placebo.
(V) VARIOS
Pegulicianina (Lumisight®) Lumicell (FDA, USA)
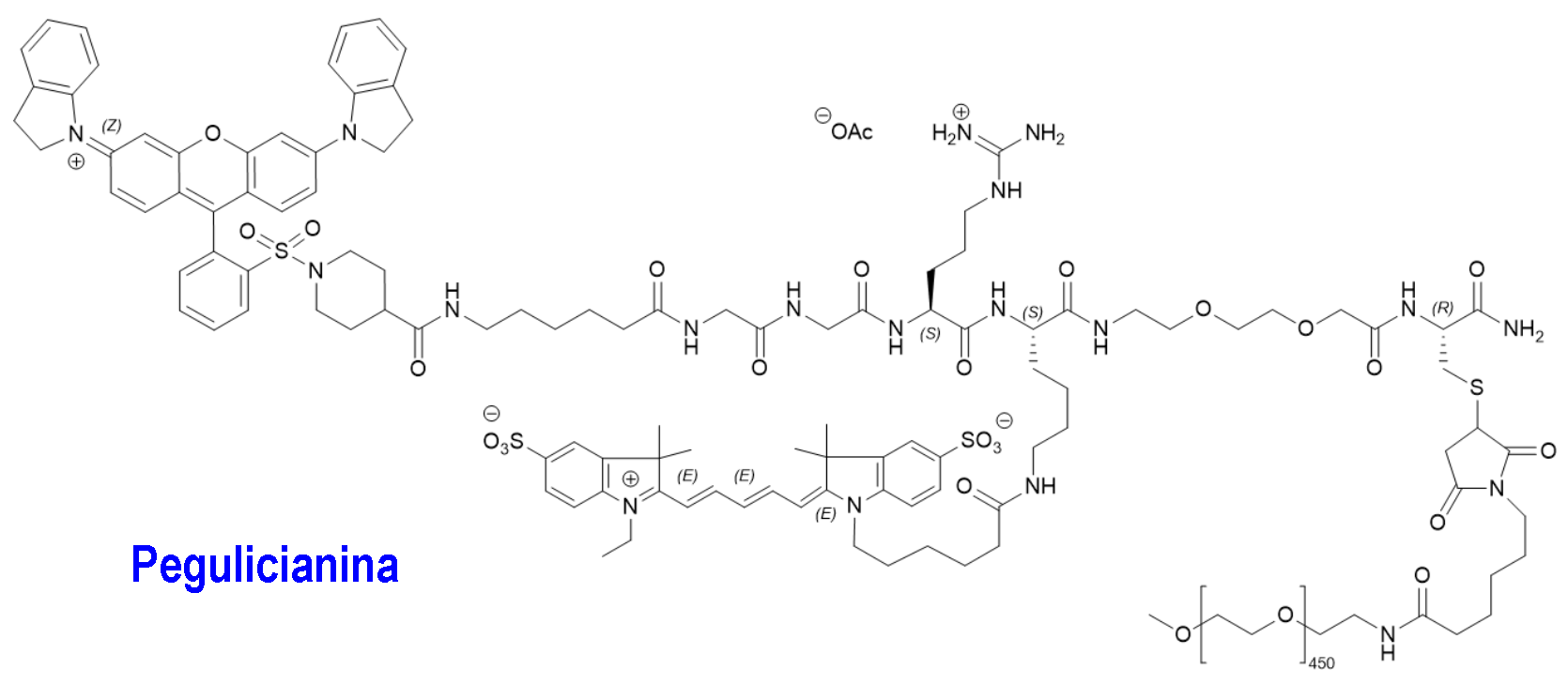
Indicación: Realización de imágenes de fluorescencia en adultos con cáncer de mama como complemento para la detección intraoperatoria de tejido canceroso dentro de la cavidad de resección luego de la extracción de la muestra primaria durante la cirugía de lumpectomía.
Tipo: Medicamento sintético estándar constituido por la N-[6-(1-{2-[3,6-bis(2,3-dihidro-1H-indol-1-il)xantilio-9-il]benceno-1-sulfonil}piperidina-4- carboxamido)hexanoil]glicilglicil-L-arginil-N6-(6-{2-[(1E,3E,5Z)-5-(1-etil-3,3-dimetil-5-sulfonato-1,3-dihidro-2H-indol-2-ilideno)penta-1,3-dien-1-il]-3,3- dimetil-5-sulfonato-3H-indol-1-io-1-il}hexanoil)-L-lisil-[2-(2-aminoetoxi)etoxi]acetil-S-[(3RS)-1-{6-[αmetilpoli(oxietileno)-ω-amino]-6-oxohexil}-2,5- dioxopirrolidin-3-il]-L-cisteinamida. La pegulicianina forma parte de un sistema guiado por fluorescencia (FGS) que comprende, además de este agente generador de imágenes fluorescentes activable, un dispositivo portátil y un algoritmo de detección de tumores específico. Autorizado en Estados Unidos (FDA) el 17 de abril de 2024 por vía rápida (Fast Track) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: La pegulicianina es un profármaco que es ópticamente inactivo cuando está intacto y produce una señal fluorescente después de que las catepsinas y las metaloproteasas de matriz (MMP) escinden su cadena peptídica. Los niveles de estas enzimas son más altos dentro y alrededor de las células tumorales y asociadas a tumores que en las células normales. Esta escisión enzimática da como resultado el “fragmento 2” y el “fragmento 3”, que son metabolitos ópticamente activos que emiten fluorescencia, así como el “fragmento 1” que contiene el extintor de fluorescencia que mantiene la molécula intacta ópticamente inactiva. El “fragmento 2” y el “fragmento 3” absorben luz en la región de luz visible con un pico de absorción a 650 nm y fluorescen con un pico de emisión a 675 nm.
Eficacia clínica: Un ensayo clínico aleatorizado, multicéntrico y controlado entre 406 pacientes adultos con cáncer de mama invasivo confirmado, carcinoma ductal in situ (CDIS) o ambos, que recibieron LUMISIGHT 2 a 6 horas antes de la obtención de imágenes con Lumicell DVS, de los 357 pacientes se sometieron a imágenes guiadas por LUMISIGHT después de completar el procedimiento estándar de lumpectomía. El rendimiento diagnóstico mostró una sensibilidad del 49,1% y una especificidad del 86,5%.
Eventos adversos: Los más comunes son hipersensibilidad (1,4%, incluida anafilaxia) y cromaturia (85%).
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE)
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
Producto Calificado para Enfermedades Infecciosas. (Qualified Infectious Disease Product): un medicamento antibacteriano o antifúngico para uso humano destinado a tratar infecciones graves o potencialmente mortales, incluidas aquellas causadas por un patógeno resistente a antibacterianos o antifúngicos, incluidos patógenos infecciosos nuevos o emergentes.
