1. INTRODUCCIÓN
Síndrome metabólico (SM) se puede definir como un conjunto de trastornos metabólicos asociados al desarrollo de enfermedad cardiovascular aterosclerótica (1). La prevalencia de SM depende del aumento de peso y la edad (2).
Debido a la gran variedad de factores de riesgo implicados en el desarrollo de síndrome metabólico, el tratamiento tiene varios objetivos como (3,4):
- Promover estilos de vida saludables.
- Disminuir la obesidad visceral.
- Controlar las patologías asociadas que se relacionan con un mayor riesgo cardiovascular.
Existen varias alternativas para el tratamiento de síndrome metabólico que incluyen, en primer lugar, cambios en el estilo de vida, enfocados a disminuir de peso y aumentar la actividad física mediante dieta y ejercicio (5,6). Sin embargo, en múltiples ocasiones es fundamental la combinación con fármacos insulinosecretores, hipolipemiantes y antihipertensivos (7).
Hasta el momento, no existe un tratamiento farmacológico único para el síndrome metabólico, se trata cada componente por separado mediante terapia combinada con el objetivo de disminuir el desarrollo de eventos cardiovasculares (8,9).
En definitiva, son múltiples las patologías implicadas en el desarrollo de síndrome metabólico y, con ello, una gran variedad de fármacos que es necesario administrar, sobre todo, en aquellos pacientes que no respondan de forma adecuada al tratamiento dietético y tengan un mayor riesgo de enfermedad cardiovascular aterosclerótica (10).
2. OBJETIVO
El desarrollo de esta investigación tiene como principal objetivo el tratamiento de pacientes con síndrome metabólico (SM) mediante el desarrollo de cápsulas duras que contienen minicomprimidos de los siguientes grupos farmacológicos: una estatina, un fármaco insulinosecretor y un antagonista del receptor de angiotensina II (ARA II o sartan). De este modo, con la administración de una única dosis se engloba el tratamiento de cada una de las patologías asociadas al desarrollo de SM, disminuyendo el número de administraciones, mejorando la adherencia al tratamiento y facilitando la deglución en aquellos pacientes en los que se vea más limitada. Los fármacos seleccionados fueron: rosuvastatina cálcica (ROS), repaglinida (REP) y olmesartan medoximil (OLM).
3. MATERIALES Y MÉTODOS
La metodología utilizada en esta investigación se describe en dos fases, en la fase 1 (Figura 1) se estudia la caracterización de los principios activos, compatibilidad con los excipientes seleccionados y capacidad de la mezcla principio activo – excipiente para ser utilizada en compresión directa. Por el contrario, en la fase 2 (Figura 2) se estudian las características tecnológicas farmacéuticas de los minicomprimidos fabricados.
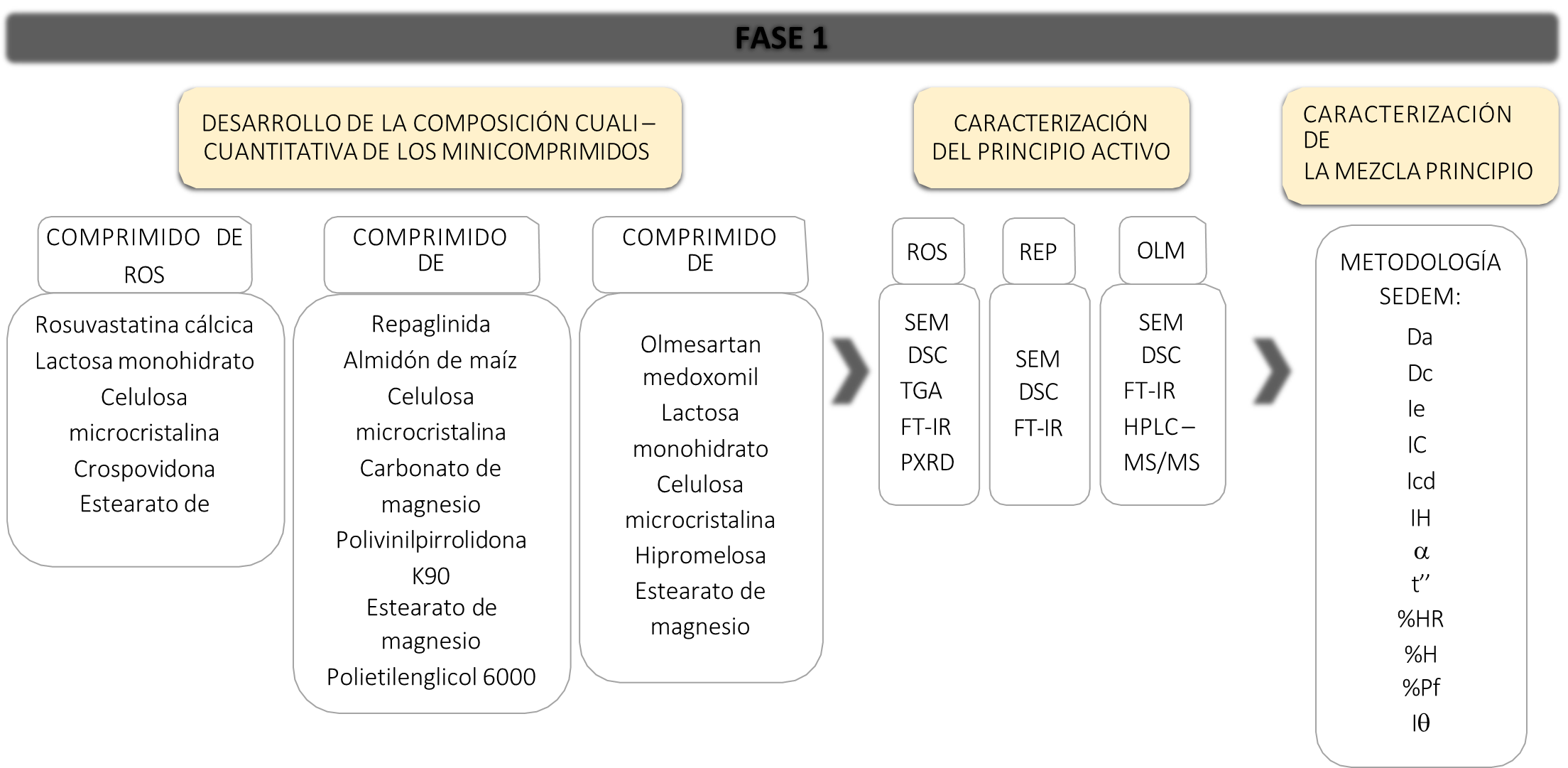
Figura 1. Desarrollo de la fórmula y caracterización de los principios activos y excipientes (Fase 1).
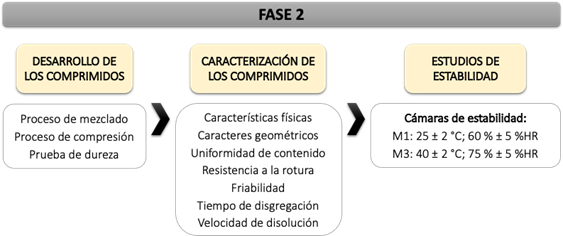
Figura 2. Desarrollo y caracterización de los comprimidos.
4. RESULTADOS Y DISCUSIÓN
4.1. Resultados minicomprimidos de rosuvastatina
El desarrollo de los comprimidos de ROS comenzó con la búsqueda de las formulaciones de ROS 20 mg comercializadas y preparadas por compresión directa. Debido al elevado número de medicamentos comercializados se realizó una búsqueda más selectiva, en primer lugar, en la base de datos CIMA (Centro de Información de Medicamentos en Línea de la Agencia Española de Medicamentos y Productos Sanitarios) y en diferentes tipos de libros oficiales (11). Crestor® 20 mg comprimidos recubiertos y Provisacor® 20 mg comprimidos recubiertos son los comprimidos ya comercializados de referencia de laboratorios Astrazeneca Farmacéutica Spain S.A. (12,13).
Una vez determinados los excipientes más utilizados y teniendo en cuenta las propiedades fisicoquímicas y biofarmacéuticas del principio activo, se definieron los excipientes ideales para el desarrollo galénico de los minicomprimidos de ROS (Tabla 1).
Tabla 1. Medicamentos con mayor (>3) número de extensiones de indicaciones aprobadas por el CHMP 2021-2024.
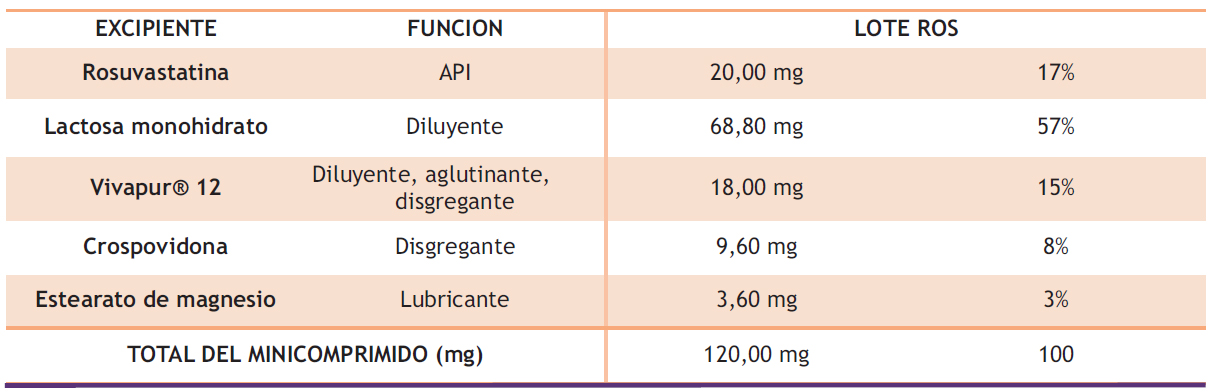
4.1.1. Caracterización del principio activo rosuvastatina cálcica.
Calorimetría diferencial de barrido (DSC):
La ROS es un fármaco de clase II según el Sistema de Clasificación Biofarmacéutica, con baja solubilidad en agua y alta permeabilidad (14). Diferentes factores influyen en la solubilidad de un fármaco como la forma, el tamaño y la distribución de las partículas, el área superficial, la porosidad y la morfología superficial o polimorfismo.
La ROS la podemos encontrar como sólido amorfo, estabilidad satisfactoria y baja tendencia a la cristalización, o utilizar este estado amorfo para fabricar formas sólidas cristalinas. Se han descrito varias formas cristalinas de dicho fármaco: A, B, B1, C M, R, S, TW1, que podrían mejorar su biodisponibilidad (15). Generalmente, el estado amorfo presenta una conformación aleatoria de las moléculas y es termodinámicamente inestable, a diferencia del estado cristalino con orden tridimensional y termodinámicamente estable. Sin embargo, se pueden encontrar formas amorfas más estables que sus cristales, como es el caso de la ROS, el estado amorfo puede tener mayor solubilidad que los cristales y, por tanto, mayores tasas de disolución y mejores características de compresión (15).
En la presente investigación, el termograma DSC de la ROS mostró un primer pico endotérmico localizado a Tonset = 170,15 ºC (ΔF = 19,80 J/g), precedido por un proceso endotérmico a Tonset = 72,29 ºC correspondiente a las pérdidas de agua (Figura 3). Según los resultados obtenidos, se utilizó la forma amorfa de ROS, ya que, el análisis entálpico diferencial reveló temperaturas similares a las publicadas en la bibliografía (15).
En la Figura 3 se detallan los termogramas de ROS y de los excipientes utilizados para la elaboración del comprimido (lactosa monohidrato, celulosa microcristalina, crospovidona y estearato de magnesio). Se pueden diferenciar dos estados físicos de lactosa, cristalino y amorfo. El primero se corresponde con un estado sólido, termodinámicamente estable cuyas moléculas se encuentran dispuestas formando una red regular, en cambio, el estado amorfo es termodinámicamente inestable e higroscópico. Los formas cristalina y amorfa de la lactosa presentan diferentes características relacionadas con distintos tamaños y formas de partícula, densidad, propiedades fisicoquímicas, estabilidad química, solubilidad en agua, higroscopicidad, propiedades de flujo y compactabilidad. En función del grado de cristalización se pueden encontrar diferentes formas de lactosa cristalina:α-lactosa anhidra estable, α-lactosa anhidra inestable, β-lactosa anhidra, cristales compuestos de α/β-lactosa y α-lactosa monohidrato (16). En el termograma DSC de α-lactosa monohidrato se observan varios picos endotérmicos. Los dos primeros a Tonset = 144,44 ºC y a Tonset = 158,40 ºC son consecuencia de la pérdida de agua. El tercer pico (Tonset = 214,45 ºC) podría tratarse de la anomerización de α-lactosa a β-lactosa debido a la presencia de agua residual. El último pico (Tonset = 228,38 ºC) es el resultado de la fusión del cristal de lactosa y su posterior descomposición (Figura 3).
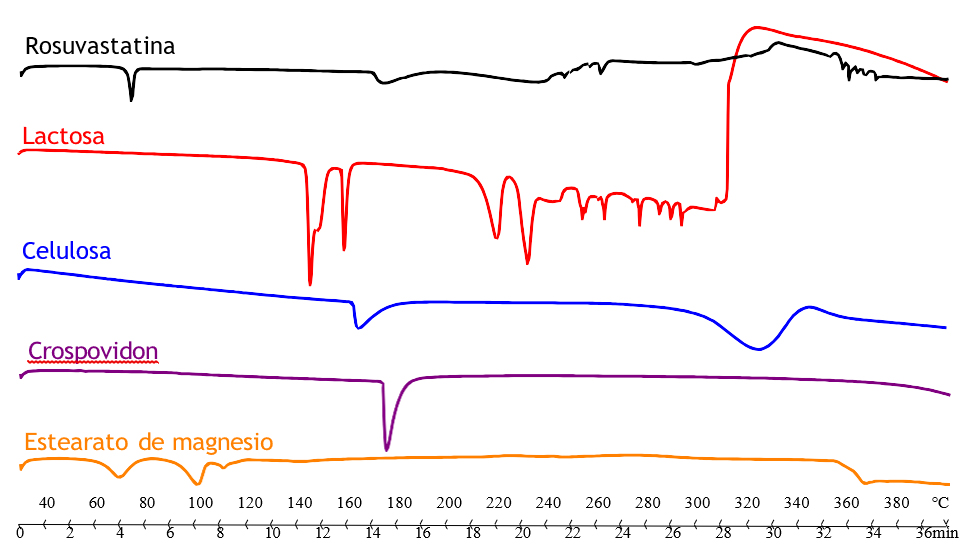
Figura 3. Termograma DSC de ROS y excipientes seleccionados.
La celulosa microcristalina (Vivapur 12®) evidenció un primer pico endotérmico (Tonset = 162,86 ºC) que se atribuye a la hidrólisis ácida de la celulosa cristalina. Sin embargo, la cristalización se consigue al aumentar el tiempo de hidrólisis, continuando el calentamiento hasta los 301,46 ºC (Figura 3). En el análisis térmico de la crospovidona (PVPP) se observa un único pico endotérmico (Tonset = 173,91 ºC) correspondiente a su punto de fusión, tal y como describe en la literatura, la PVPP presenta una fase de transición vítrea (Tg) cerca de los 200 ºC (17) (Figura 3). El estearato de magnesio muestra dos primeros picos correspondientes a la evaporación de agua, a 61,72 ºC y 93,02ºC, seguidos de un tercer pico a Tonset = 107,97 ºC que se debe a la fusión del palmitato de magnesio (Figura 3), ya que en su composición aparece ácido esteárico y ácido palmítico (esta impureza se encuentra habitualmente en los lotes comerciales).
Termogravimetría (TGA):
El termograma DSC de ROS evidenció un pico endotérmico a Tonset = 72,29 ºC correspondiente a las pérdidas de agua del principio activo. El contenido de agua (equivalente a dos moléculas de agua) se determinó mediante análisis termogravimétrico. La Figura 4 exterioriza la pérdida de agua de la ROS a 100,00 ºC, aproximadamente, con una variación de peso del 3,7 %. A continuación, el fármaco muestra estabilidad hasta los 200,00 ºC, donde se observa la descomposición del fármaco con una pérdida de masa máxima del 42,0 %. Estos resultados son similares a los descritos en la bibliografía (15,18).
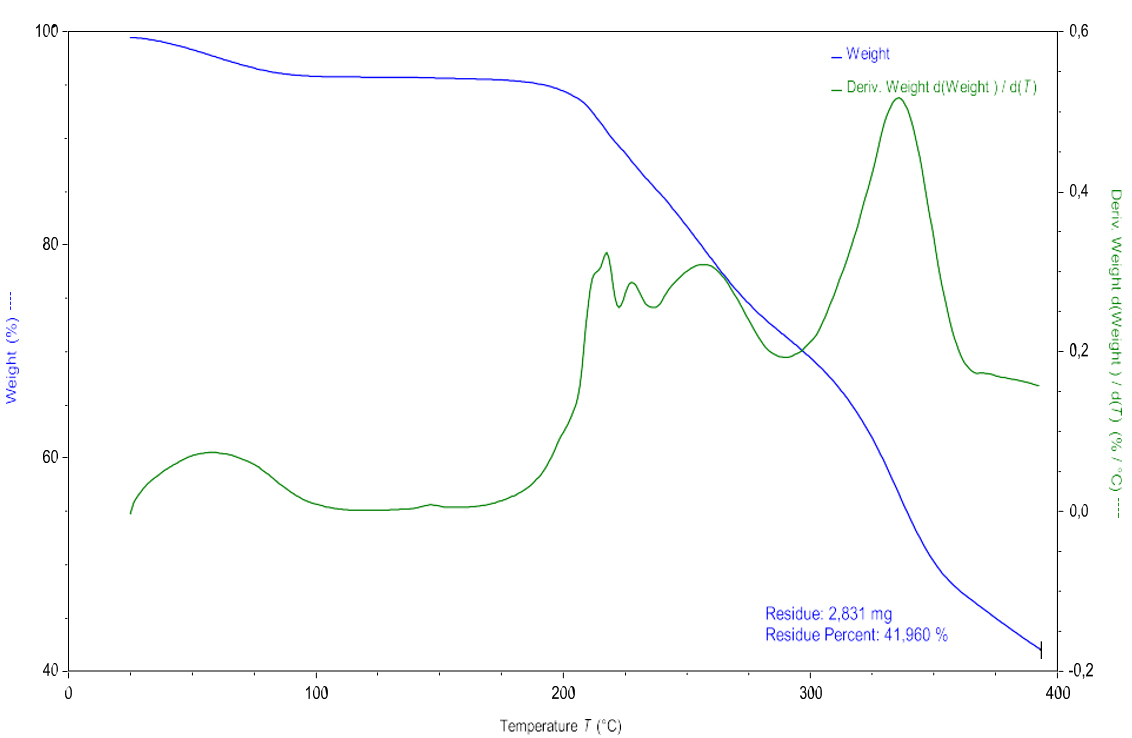
Figura 4. Análisis termogravimétrico de ROS.
Espectrofotometría infrarroja con transformada de Fourier (FT-IR):
La espectrofotometría infrarroja con transformada de Fourier (FT-IR) se utiliza principalmente para determinar posibles interacciones físicas o químicas entre principio activo y excipiente. En el espectro IR de ROS (Figura 5) se aprecian las principales bandas con los movimientos de los grupos funcionales de la molécula a 3376, 2932, 1437, 1336, 1155 y 1230 cm-1 que se confirman en la literatura (19). Estos picos son representativos de la presencia de aminas cíclicas (3376 cm-1), de la tensión C-H olefínica de la cadena lateral del heptano (2932 cm-1) y de la tensión C=O del grupo carboxilo (1437 cm-1). Las bandas a 1336 cm-1 y 1155 cm-1 se deben a las vibraciones de tensión simétricas y asimétricas por la presencia del grupo sulfóxido. El pico a 1230 cm-1 es característico de la tensión C-F del anillo aromático. Una banda ancha en la región de 3731 – 3376 cm-1 es una indicación de la presencia del grupo hidroxilo.
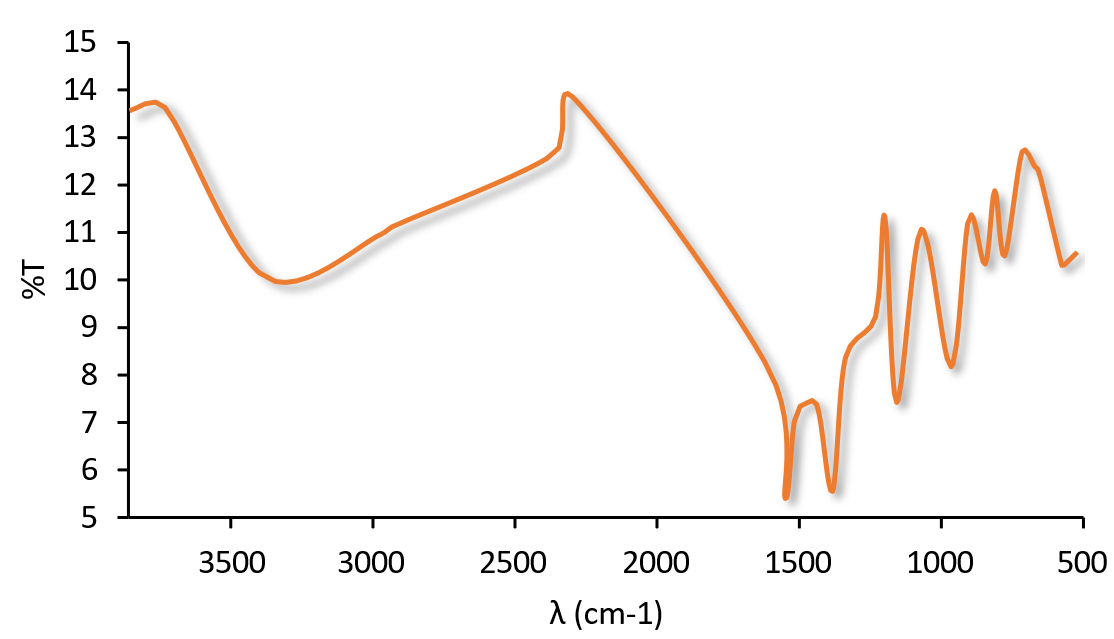
Figura 5. Espectro IR de ROS.
Difracción de rayos X (PXRD):
Las muestras analizadas por difracción de rayos X fueron: ROS pura, mezcla de ROS y excipientes, pulverización de los minicomprimidos en estudio y pulverización de comprimidos comercializados utilizados como parámetro de comparación. Los respectivos resultados de PXRD se ilustran en la Figura 6.
El patrón de difracción de la ROS pura indica la naturaleza amorfa del fármaco, apoyando así los datos de los resultados del DSC. A su vez, observamos que los excipientes utilizados son muy cristalinos, tanto la mezcla de fármaco y excipientes como la pulverización de los comprimidos de ROS mostraron nuevos picos que indican su naturaleza cristalina.
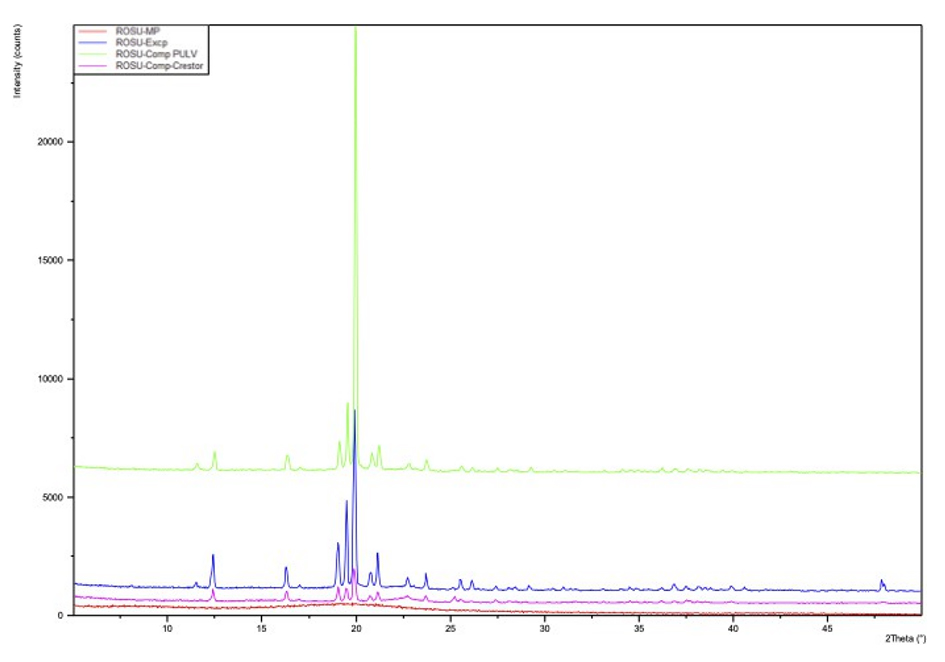
Figura 6. PXRD ROS pura (rojo), PXRD comprimidos comercializados (rosa), PXRD mezcla fármaco-excipientes (azul), PXRD minicomprimidos en estudio (verde).
4.1.2. Caracterización de la mezcla principio activo-excipiente
La idoneidad de la mezcla de ROS y los excipientes seleccionados (lactosa monohidrato, celulosa microcristalina, crospovidona y estearato de magnesio) para ser utilizada en compresión directa, pudo conocerse mediante el método del diagrama SeDeM. Los parámetros farmacotécnicos, densidad aparente (rDa) y densidad compactada (rDc) (parámetro dimensional); porosidad entre partículas (rIe), índice de Carr (IC) e índice de cohesión (rIcd) (parámetro de compresibilidad); índice de Hausner (IH), ángulo de reposo (rα) y tiempo de deslizamiento (rt’’) (parámetro de fluidez); humedad relativa (r%HR) e higroscopicidad (r%H) (parámetro de lubrificación/estabilidad); y, por último, tamaño de partícula < 50 μm (r%Pf) e índice de homogeneidad (rIØ) (parámetro de lubrificación/dosificación), se determinaron experimentalmente y se procesaron matemáticamente para expresarlos en una representación gráfica en forma de diagrama SeDeM (Figura 7). Los resultados de este método coinciden con los estudios de otros investigadores (20–23).
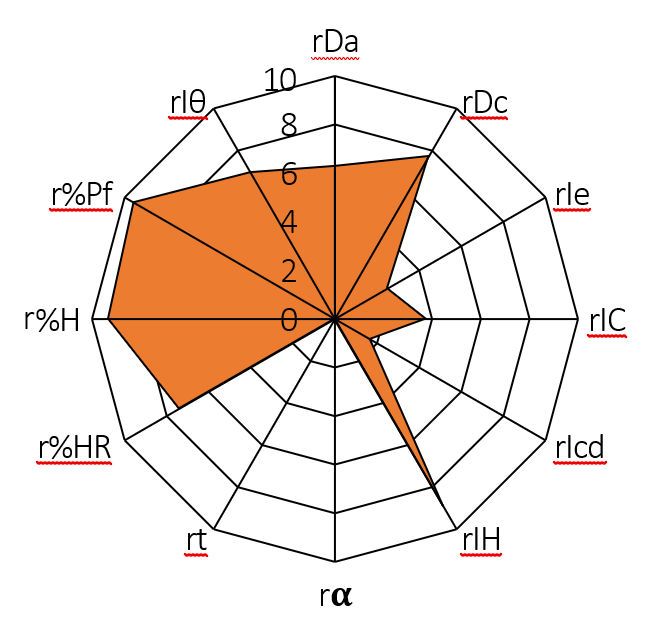
Figura 7. Diagrama SeDeM de la mezcla de ROS-excipientes
Según los resultados obtenidos de los diferentes parámetros y pruebas que conforman el sistema SeDeM, la mezcla de ROS, lactosa monohidrato, celulosa microcristalina, crospovidona y estearato de magnesio se considera idónea para ser utilizada en formulaciones de compresión directa.
Los índices de aceptación proporcionan valores por encima del valor mínimo esperado, índice paramétrico 0,58 (≥ 0,5), índice de perfil paramétrico 5,33 (≥ 5) e índice de buena compresión 5,08 (≥ 5). Del mismo modo, los valores de densidad aparente (0,63 g/mL) y densidad compactada (0,77 g/mL) se sitúan por encima de 0,50 g/mL. A pesar de la higroscopicidad de la ROS, es de gran importancia destacar que la mezcla con los excipientes seleccionados proporciona un bajo porcentaje de higroscopicidad (1,36 %), así como, valores de humedad relativa que facilitan la capacidad de compresión de la mezcla entre 2 – 3 % (2,61 %). Al mismo tiempo, hay que asegurar desde homogeneidad en la distribución de los tamaños de partículas de la mezcla (0,01) hasta el menor porcentaje de partículas inferior a 50 μm (2,31 %), obteniendo en ambos casos valores aceptables para su uso en compresión directa.
4.1.3. Características tecnológicas farmacéuticas de los comprimidos de ROS
En la Tabla 2 se describen las características tecnológicas farmacéuticas de los comprimidos de ROS 20 mg obtenidos por compresión directa (Lote ROS), que deben cumplir con los requisitos establecidos según la farmacopea, garantizando que la calidad de los minicomprimidos sea la esperada. A su vez, se realizó una comparación con el medicamento comercializado de referencia Crestor® 20 mg.
Tabla 2. Características tecnológicas farmacéuticas del Lote ROS.
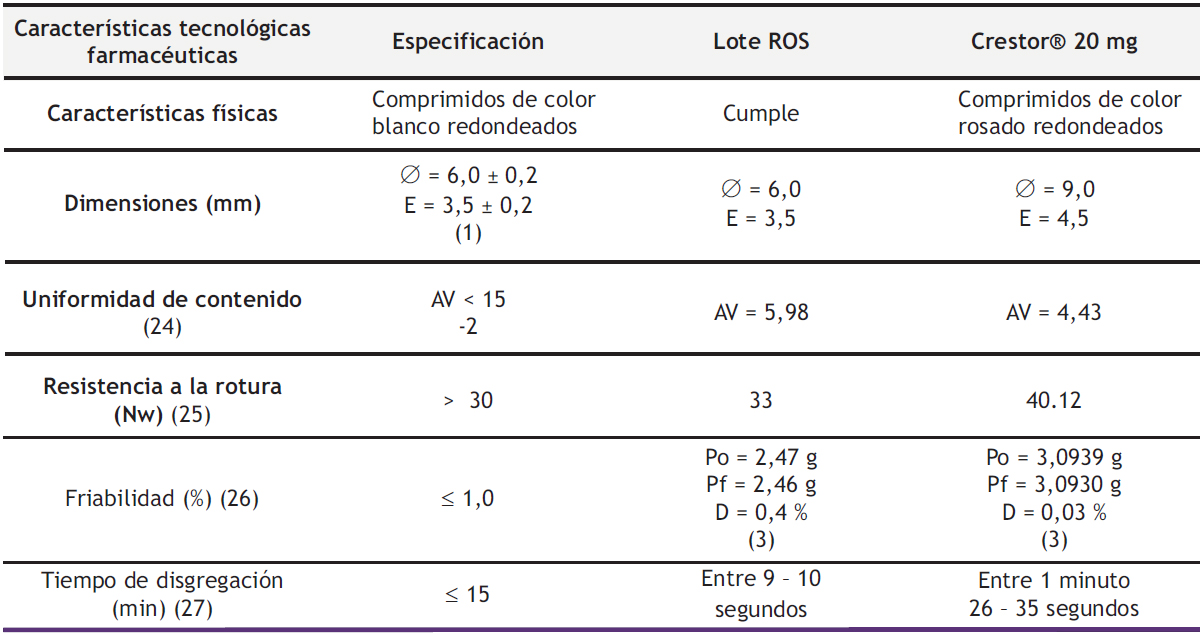
(1) Diámetro (Ø) medio de 10 comprimidos. Espesor (E) medio de 10 comprimidos. (2) Valor de aceptación (AV). (3) Po = peso inicial de 20 comprimidos; Pf = peso final de 20 comprimidos; D = desviación de 20 comprimidos.
Los minicomprimidos de ROS 20 mg (Lote ROS) presentan un aspecto visual blanco brillante con un grosor fino de 3,5 mm, debido al punzón utilizado de 6,0 mm, y una resistencia a la rotura de 33 Nw. El valor de aceptación (5,98) en la prueba de uniformidad de contenido está dentro de los límites permitidos según farmacopea, con un AV inferior a 15. La prueba de friabilidad cumple con las especificaciones de la RFE 2.9.7. (26) con una desviación de 0,4 %. A su vez, se observa una rápida disgregación de los comprimidos con un tiempo inferior a 10 segundos.
Estudio de velocidad de disolución:
El ensayo de velocidad de disolución se realizó con el objetivo de conocer la cinética de disolución de los minicomprimidos de ROS y como complemento al ensayo de disgregación, para ello, se comparó uno de los medicamentos de referencia, Crestor® 20 mg comprimidos recubiertos, y los minicomprimidos de ROS 20 mg (Lote ROS) de acuerdo con las especificaciones de la real farmacopea española (28).
Ambas formulaciones presentan un perfil de disolución similar (Tabla 3). La incorporación de superdisgregantes en la formulación como es la crospovidona, proporciona mayor absorción de agua y aumento del área superficial, lo que conlleva a tiempo de disgregación más cortos y, a su vez, mayor velocidad de disolución.
Tabla 3. Comparación del ensayo de disolución del Crestor® y de los minicomprimidos de ROS.
![]()
4.2. Resultados minicomprimidos de repaglinida
El desarrollo de los comprimidos de repaglinida (REP) comenzó con el estudio de las formulaciones de REP 2 mg comercializadas y preparadas por compresión directa, para ello se realizó una búsqueda en la base de datos CIMA. Los comprimidos ya comercializados de referencia para el desarrollo de la formulación de REP fueron: Prandin® 2 mg comprimidos y Novonorm® 2 mg comprimidos, ambos de laboratorios Novo Nordisk Pharma S.A. (29,30).
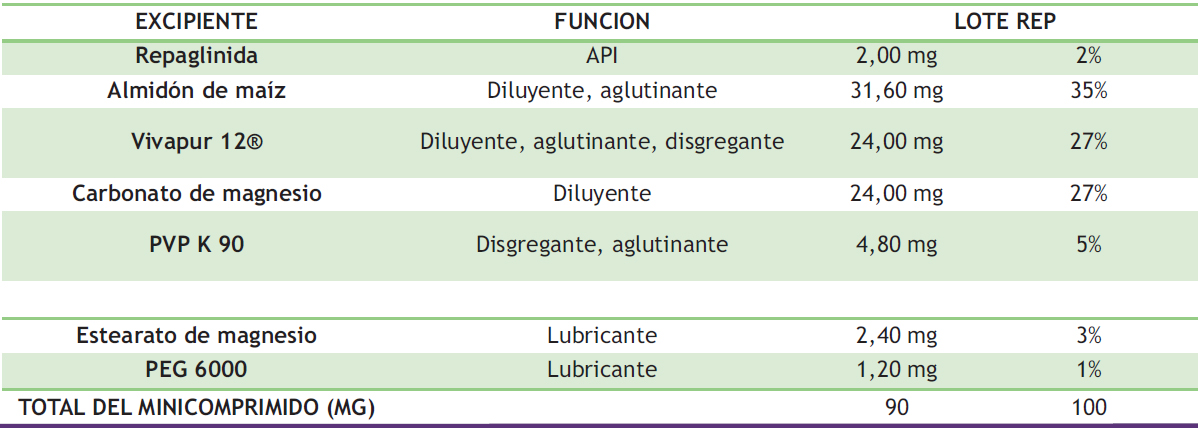
A partir de las formulaciones de referencias se seleccionaron los excipientes para el desarrollo de los comprimidos de REP según la características fisicoquímicas y biofarmacéuticas del principio activo. Los excipientes seleccionados según su funcionalidad y características tecnológicas para su uso en compresión directa se describen en la Tabla 4.
Tabla 4. Composición cualitativa y cuantitativa de minicomprimidos de REP
4.2.1. Caracterización del principio activo repaglinida
Calorimetría diferencial de barrido (DSC):
La REP es un fármaco de clase II según el Sistema de Clasificación Biofarmacéutica, caracterizado por una baja solubilidad en agua y alta lipofilia, en consecuencia, presenta una biodisponibilidad baja (31). Por ello es de gran importancia que, tanto los excipientes seleccionados como la forma farmacéutica mejoren la velocidad de disolución del principio activo y, con ello, su biodisponibilidad oral. La REP se puede encontrar como sólido cristalino o utilizar este estado cristalino para la modificación intencionada a sólido amorfo (32).
En la presente investigación la REP mostró un único pico endotérmico a Tonset = 133,31°C (ΔF = 93,15 J/g) indicando su naturaleza cristalina, estos resultados son similares a los publicados en la bibliografía (33, 34). En la Figura 8 se describen los termogramas de la REP y cada uno de los excipientes seleccionados (celulosa microcristalina, almidón, povidona K90, estearato de magnesio, carbonato de magnesio, polietilenglicol 6000).
El análisis entálpico diferencial (DSC) de la celulosa microcristalina mostró un primer pico endotérmico a Tonset = 162,86 °C, correspondiente a las pérdidas de agua por hidrólisis ácida de la celulosa cristalina, al continuar con el calentamiento se consigue la cristalización a Tonset = 301,46 ºC (Figura 8). En el análisis térmico del almidón observamos un único pico endotérmico correspondiente con su punto de fusión (Tonset = 152,42 ºC), no se observó ningún pico por debajo de 100 ºC por lo que se trata de un almidón libre de agua (Figura 8). Para analizar el pico de fusión, los resultados fueron comparados con los de la literatura (35). En nuestro caso, la entalpía de fusión no es muy elevada, esto indica menor contenido de amilopectina. A su vez, el pico observado es estrecho indicando una estructura homogénea (36). La povidona K90 evidenció un único pico endotérmico (Tonset = 166,87 ºC) correspondiente con su punto de fusión, tal y como ocurre con la PVPP, la PVP K90 presenta una fase de transición vítrea (Tg) cerca de los 200ºC (Figura 8). En el análisis térmico del estearato de magnesio se observan dos primeros picos relacionados con la pérdida de agua 61,72 ºC y 93,02 ºC. Al continuar con el calentamiento, se evidencia un tercer pico endotérmico (Tonset = 107,97 °C) relacionado con la fusión del palmitato de magnesio (Figura 8). La descomposición del carbonato de magnesio tuvo lugar en un proceso de dos pasos. El primero se observó a los 230 ºC, relacionado con la pérdida de agua, a diferencia del segundo a Tonset = 313,13 ºC, se corresponde con la pérdida de CO2 relacionado, por lo tanto, con la descomposición del carbonato de magnesio. Estos resultados están en concordancia con estudios previos (37) (Figura 8). Finalmente, el polietilenglicol es un polímero semicristalino con un único pico endotérmico (Tonset = 58,47 ºC) correspondiente con su punto de fusión.
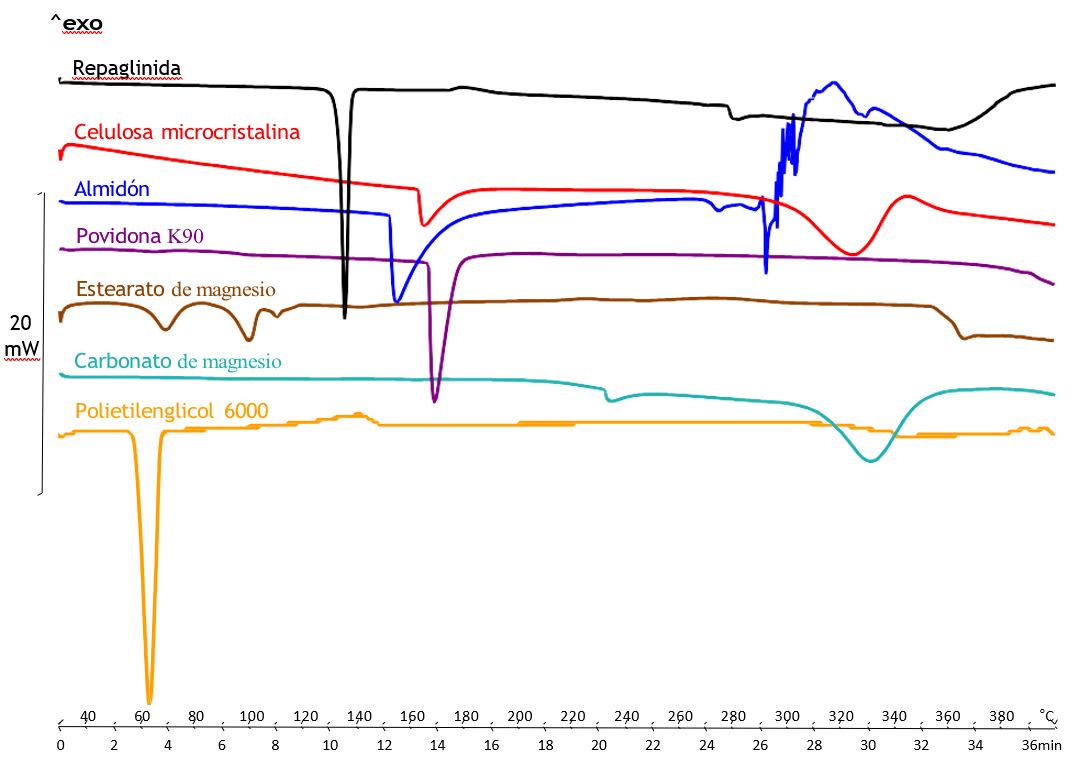
Figura 8. Termograma DSC de REP y excipientes seleccionados.
Espectrofotometría infrarroja con transformada de Fourier (FT-IR):
En el espectro infrarrojo de REP pura (Figura 9) se observaron picos de absorción característicos del principio activo, confirmando la presencia de ciertos grupos funcionales. Los resultados están en línea con la bibliografía consultada (38,39). El espectro mostró picos representativos de las vibraciones de estiramiento de diferentes grupos funcionales: N-H (3308 cm-1), C-H (2934 cm-1) y C=O (1687 cm-1). Las señales a 1637 cm-1 y 1568 cm-1 se deben a la presencia de N-H y C=C, respectivamente, del anillo aromático. Finalmente, se observaron cuatro picos para la tensión C-O: 1449 cm-1, 1429 cm-1, 1217 cm-1 y 1041 cm-1.
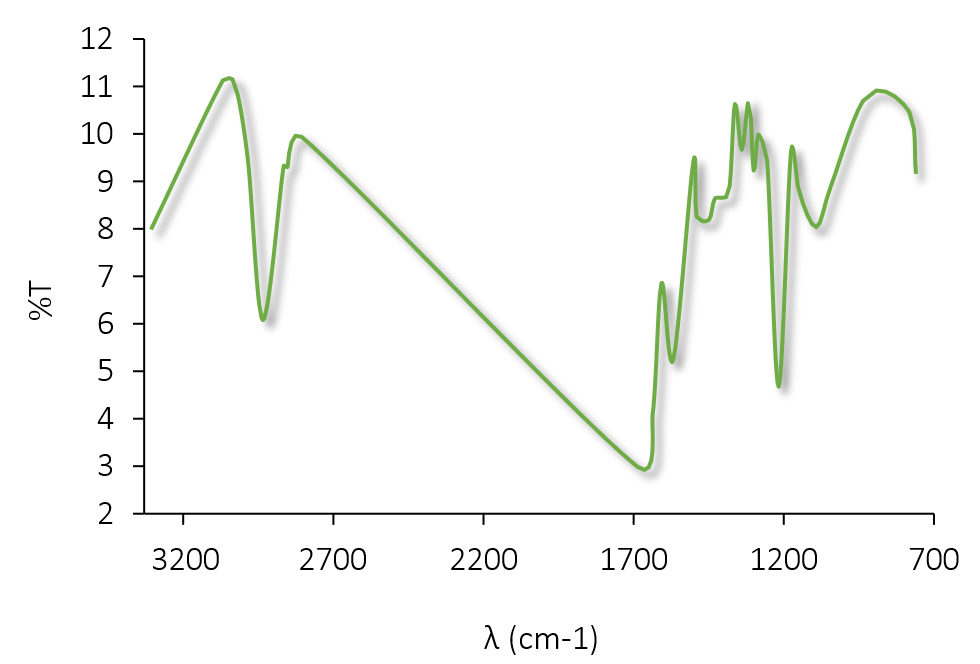
Figura 9. Espectro IR de REP
4.2.2. Caracterización de la mezcla principio activo – excipientes
La metodología galénica SeDeM se utilizó para determinar la idoneidad de la mezcla de REP y los distintos excipientes seleccionados (almidón de maíz, celulosa microcristalina, carbonato de magnesio, polivinilpirrolidona K 90, estearato de magnesio y polietilenglicol 6000) en cuanto a su disposición para ser utilizada en compresión directa. Se determinaron experimentalmente los siguientes parámetros farmacotécnicos: parámetro dimensional, densidad aparente (rDa) y densidad compactada (rDc); parámetro de compresibilidad, porosidad entre partículas (rIe), índice de Carr (rIC) e índice de Cohesión (rIcd); parámetro de fluidez, índice de Hausner (rIH), ángulo de reposo (rα) y tiempo de deslizamiento (rt); parámetro de lubricidad/estabilidad, humedad relativa (r%HR) e higroscopicidad (r%H); y, por último, parámetro de lubricidad/dosificación, tamaño de las partículas < 50 μm (r%Pf) e índice de homogeneidad (rIθ), dichos parámetros fueron procesados matemáticamente para construir el diagrama de SeDeM (Figura 10).
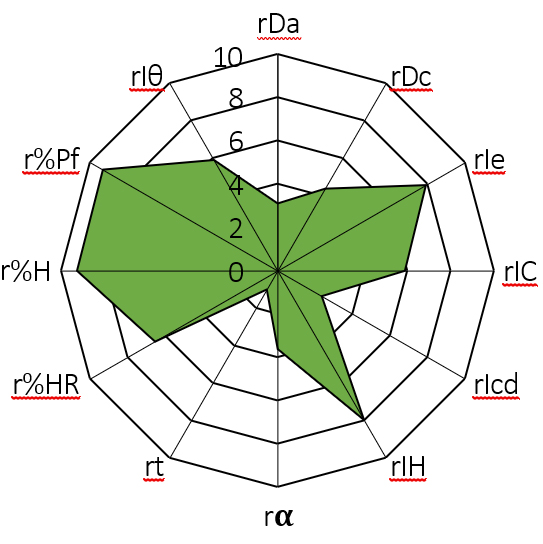
Figura 10. Diagrama SeDeM de la mezcla REP-excipientes.
Los parámetros farmacotécnicos calculados y su posterior representación gráfica en el diagrama de SeDeM demostraron que la mezcla de REP y excipientes seleccionados es idónea para ser utilizada en compresión directa.
El índice de buena compresión e índice de perfil paramétrico proporcionaron valores superiores a 5,00 (5,06 y 5,32, respectivamente). Del mismo modo, el índice paramétrico proporcionó un valor de 0,50 (≥0,5). Los resultados de los índices de aceptación demostraron que la mezcla es adecuada para su uso en compresión directa.
El porcentaje de REP en el total de la fórmula es muy bajo, 2 mg/dosis, por lo que es de gran importancia asegurar homogeneidad del principio activo durante el proceso de fabricación. En consecuencia, los resultados de índice de homogeneidad (0,012) superior a 0,01 y el bajo porcentaje de partículas de tamaño inferior a 50 μm (3,40 %) demostraron desde homogeneidad en la distribución de los tamaños de partícula hasta buenas propiedades reológicas y, con ello, homogeneidad de la REP.
Finalmente, la mezcla proporcionó porcentajes de higroscopicidad y humedad relativa elevados (4,15 % y 5,42 %, respectivamente), a pesar de ello y teniendo en cuenta los valores finales de índices de aceptación, los excipientes utilizados se consideraron adecuados para ser utilizados en compresión directa.
4.2.3. Características tecnológicas farmacéuticas de los comprimidos de REP
En la Tabla 5 se describen las características tecnológicas farmacéuticas: características físicas, dimensiones, uniformidad de contenido, resistencia a la rotura, friabilidad y tiempo de disgregación de los comprimidos de REP 2 mg obtenidos por compresión directa (Lote REP). Con el objetivo de garantizar la calidad de los comprimidos, se deben de cumplir las especificaciones establecidas según farmacopea. Del mismo modo, se realizó una comparación con el medicamento comercializado de referencia Prandin® 2 mg.
Tabla 5. Características tecnológicas farmacéuticas del Lote REP.
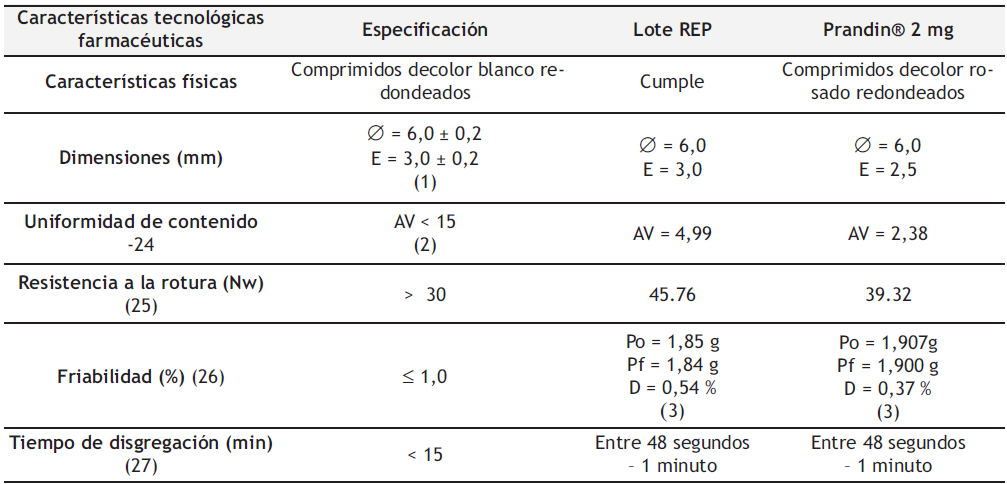
Los minicomprimidos de REP 2 mg (Lote REP) cumplen con las especificaciones de características físicas y dimensiones establecidas. Se obtuvieron comprimidos de color blanco redondeados, con un espesor fino de 3,5 mm y un diámetro de 6,0 mm, debido al punzón utilizado de 6,0 mm.
Debido al bajo peso de los comprimidos, 90 mg, fue crucial garantizar la uniformidad de contenido en el lote desarrollado, obteniendo un valor de aceptación inferior a 15 (4,99). A su vez, observamos que los comprimidos cumplen con las especificaciones de porcentaje de pérdida de peso y resistencia a la rotura según farmacopea (25,26), con valores de 0,54 % y 45,76 Nw, respectivamente. Por último, se observó la rápida disgregación de los comprimidos, con un tiempo de disgregación medio inferior a un minuto.
Los resultados obtenidos corroboran la idoneidad de los excipientes seleccionados para el desarrollo de los minicomprimidos de REP.
Estudio de velocidad de disolución:
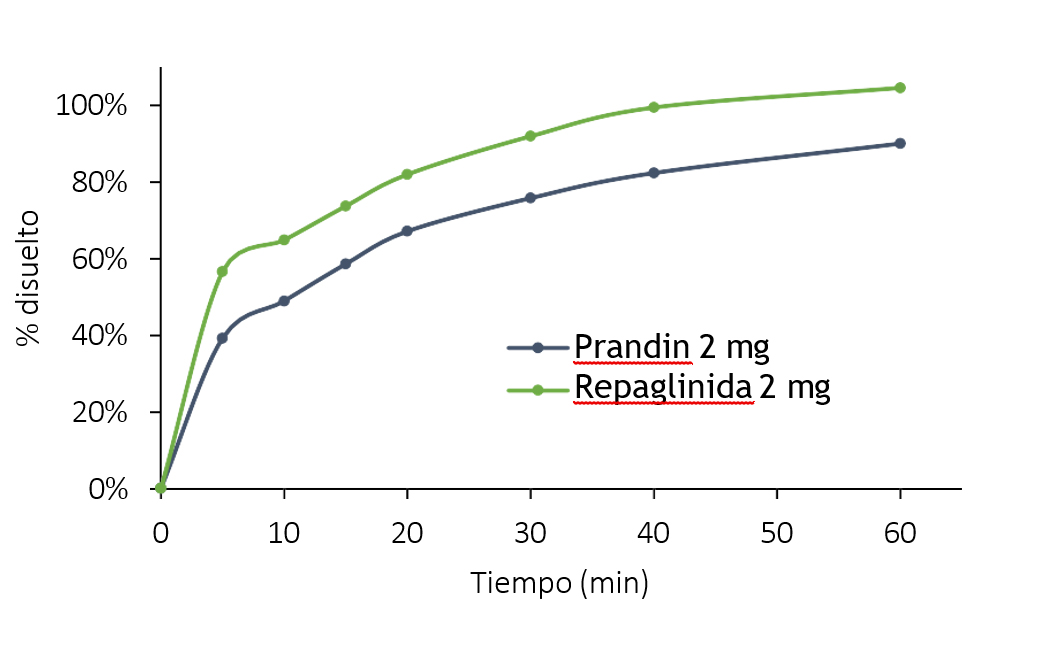
El estudio de velocidad de disolución se realizó con el objetivo de conocer la cinética de disolución de los minicomprimidos de REP y como complemento al ensayo de disgregación.
En la Tabla 6 se observa el perfil de disolución de ambas formulaciones. Los minicomprimidos de REP presentaron mayor velocidad de disolución desde el inicio. La incorporación de polietilenglicol 6000 en la formulación aumenta la humectabilidad y dispersión del principio activo, en consecuencia, favorece la solubilidad y, con ello, la velocidad de disolución del mismo (40).
Tabla 6. Comparación del ensayo de disolución de Prandin® y de los minicomprimidos de REP.
![]()
4.3. Resultados minicomprimidos de olmesartan
El desarrollo de los comprimidos de olmesartan medoxomil (OLM) comenzó con el estudio de los medicamentos comercializados con 20 mg de olmesartan preparados por compresión directa y teniendo en cuenta las características biofarmacéuticas y fisicoquímicas del principio activo. Ixia® 20 mg comprimidos recubiertos es el OLM de referencia ya comercializado de laboratorios Menarini S.A. (41).
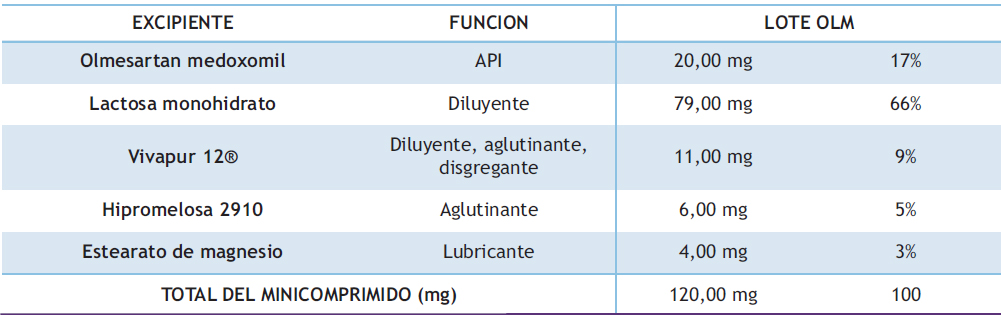
La selección de los excipientes en la fase de formulación es crucial. Para ello, se tuvo en cuenta desde la funcionalidad hasta las características tecnológicas de los excipientes para su uso en compresión directa (42). Los excipientes seleccionados y la fórmula definitiva se describen en la Tabla 7.
Tabla 7. Composición cualitativa y cuantitativa de minicomprimidos de OLM.
4.3.1. Caracterización del principio activo olmesartan medoxomil
Calorimetría diferencial de barrido (DSC):
OLM es un fármaco de naturaleza hidrofóbica (43), capaz de formar grandes agregados en la superficie del medio en el que se encuentre, reduciéndose la superficie real del tamaño de partícula de éste y, por tanto, su solubilidad. Para mejorar la solubilidad del OLM se han utilizado diferentes estrategias (44,45). Desde la combinación del fármaco con complejos hidrofílicos (44) hasta la reducción del tamaño de partícula a nanocristales (45), que lograron mejorar tanto los perfiles de solubilidad como los de disolución y biodisponibilidad.
El análisis entálpico diferencial (DSC) de OLM mostró un único pico endotérmico localizado a Tonset = 181,54 ºC ΔF = 91,97 J/g) correspondiente con su fusión y el cual es característico de su naturaleza cristalina (43). Los termogramas de OLM y cada uno de los excipientes seleccionados (lactosa monohidrato, celulosa microcristalina, hipromelosa y estearato de magnesio) se detallan en la Figura 11.
Figura 11. Termograma DSC de OLM y excipientes seleccionados.
En el estudio DSC de α-lactosa monohidrato se observan varios picos endotérmicos. Los dos primeros son consecuencia de la pérdida de agua: el primero (Tonset = 144,44 ºC) se corresponde con la pérdida de agua superficial y el segundo (Tonset = 158,40 ºC), por su parte, se asocia a la pérdida de agua de cristalización (46), ya que, de acuerdo con la literatura, α-lactosa monohidrato libera su agua de cristalización por encima de 150°C. El tercer pico (Tonset = 214,45 ºC) podría tratarse de la anomerización de α-lactosa a β- lactosa debido a la presencia de agua residual. El último pico (Tonset = 228,38 ºC) es el resultado de la fusión del cristal de lactosa y su posterior descomposición (Figura 11).
En el análisis térmico de la celulosa microcristalina se observó un primer pico endotérmico (Tonset = 162,86 ºC) que se atribuye a la evaporación de agua (hidrólisis ácida de la celulosa cristalina) (47). Sin embargo, la cristalización se consigue al aumentar el tiempo de hidrólisis continuando el calentamiento hasta los 301,46 ºC (Figura 11).
La hidroxipropilmetilcelulosa es un polímero amorfo que presenta un único pico endotérmico correspondiente a la temperatura de transición vítrea (Tg) a 180,70 °C (Figura 11) (48). Finalmente, en el estearato de magnesio se evidenciaron dos primeros picos correspondientes a la evaporación de agua a 61,72 °C y 93,02 °C, seguidos de un tercer pico a Tonset = 107,97°C que se debe a la fusión del palmitato de magnesio, ya que en su composición aparece ácido esteárico y ácido palmítico.
Espectrofotometría infrarroja con transformada de Fourier (FT-IR):
El espectro infrarrojo de OLM (Figura 12) reveló picos de absorción característicos como los publicados anteriormente asegurando la presencia de ciertos grupos funcionales (49). Se observó un pico característico a 3293 cm-1 debido a las vibraciones de estiramiento N-H, dos picos agudos a 1832 cm-1 y 1708 cm-1 característicos del grupo carbonilo (C=O). Las señales 1553 cm-1, 1532 cm-1 y 1474 cm-1 se deben al estiramiento C=C del aromático. Además, el espectro mostró seis picos para la tensión C-O, 1302 cm-1, 1227 cm-1, 1169 cm-1, 1136 cm-1, 1089 cm-1, 1054 cm-1.
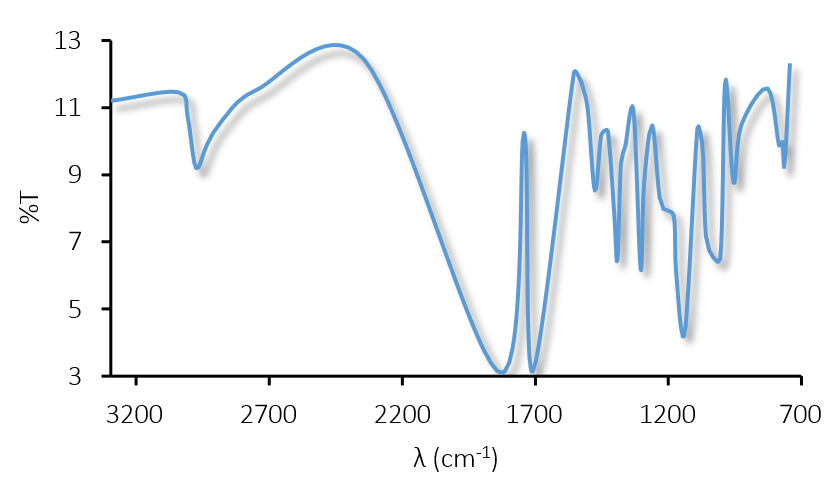
Figura 12. Espectro IR de OLM.
4.3.2. Caracterización de la mezcla principio activo-excipiente
La idoneidad de la mezcla de OLM y los distintos excipientes seleccionados (lactosa monohidrato, celulosa microcristalina, hipromelosa y estearato de magnesio) para ser utilizada en compresión directa se conoció mediante el diagrama de SeDeM. Los parámetros farmacotécnicos determinados experimentalmente fueron: parámetro dimensional, densidad aparente (rDa) y densidad compactada (rDc); parámetro de compresibilidad, porosidad entre partículas (rIe), índice de Carr (rIC) e índice de Cohesión (rIcd); parámetro de fluidez, índice de Hausner (rIH), ángulo de reposo (rα) y tiempo de deslizamiento (rt); parámetro de lubricidad/estabilidad, humedad relativa (r%HR) e higroscopicidad (r%H); y, por último, parámetro de lubricidad/dosificación, tamaño de las partículas < 50 μm (r%Pf) e índice de homogeneidad (rIθ). Los resultados fueron procesados matemáticamente para su posterior representación gráfica en forma de diagrama de SeDeM (Figura 13). Según los resultados obtenidos de los diferentes parámetros y pruebas que conforman el sistema SeDeM, la mezcla de OLM, lactosa monohidrato, celulosa microcristalina, hipromelosa y estearato de magnesio se consideró idónea para ser utilizada en formulaciones de compresión directa. Los índices de aceptación proporcionan resultados > 5 para el índice de perfil paramétrico (5, 37) e índice de buena compresión (5, 12), así como, resultados superiores a 0,5 para el índice paramétrico (0,58) indicando que la mezcla es adecuada para compresión directa. A su vez, los valores de densidad aparente y densidad compactada se encuentran próximos a 0,5 g/mL (Da = 0,45 g/mL) y por encima de este valor (Dc = 0,65 g/mL).
Es de gran importancia destacar los resultados implicados en el factor de incidencia de lubrificación/estabilidad con valores por encima de 5. La mezcla demostró desde excelentes cualidades reológicas hasta buena estabilidad como consecuencia del bajo porcentaje de higroscopicidad (0,92 %) y humedad relativa (1,32 %). Del mismo modo, la mezcla proporcionó excelente fluidez y compresión gracias también al bajo porcentaje de partículas inferior a 50 μm (5, 21 %).
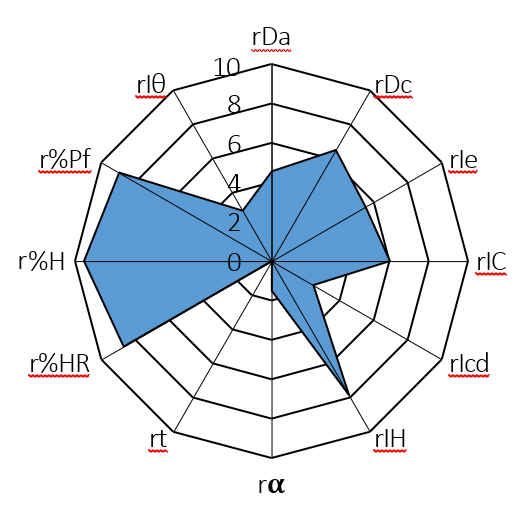
Figura 13. Diagrama SeDeM de la mezcla de OLM-excipientes.
4.3.3. Características tecnológicas farmacéuticas de los comprimidos de OLM
En la Tabla 8 se describen las características tecnológicas farmacéuticas de los comprimidos de OLM 20 mg obtenidos por compresión directa (Lote OLM). Se determinaron características físicas, dimensiones, uniformidad de contenido, resistencia a la rotura, friabilidad y tiempo de disgregación. Del mismo modo, se realizó una comparación de los resultados obtenidos con el medicamento comercializado de referencia Ixia® 20 mg. Para garantizar la calidad de los comprimidos se deben de cumplir con las especificaciones establecidas en farmacopea.
Tabla 8. Características tecnológicas farmacéuticas del Lote OLM.
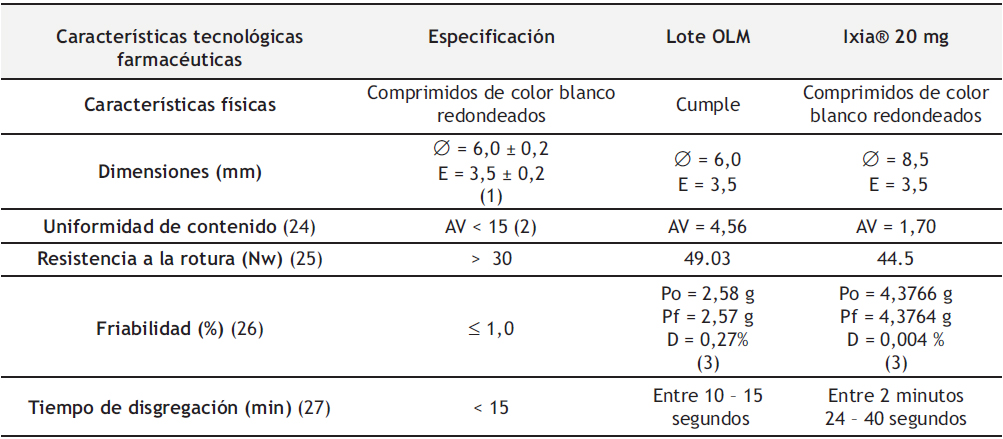
Los minicomprimidos obtenidos de OLM 20 mg (Lote OLM) tenían un aspecto visual blanco brillante. Debido al punzón utilizado de 6,0 mm, presentaron un diámetro de 6,0 mm y un espesor fino de 3,5 mm.
Se determinó que el Lote OLM cumple con las especificaciones de uniformidad de contenido según farmacopea, con un valor de aceptación inferior a 15 (4,56). Del mismo modo, la RFE 2.9.7 (26) establece que los comprimidos no recubiertos deben tener un porcentaje de pérdida de peso inferior al 1 %, los minicomprimidos de OLM cumplen con las especificaciones con una desviación del 0,27 %. Por otra parte, la fuerza aplicada durante el proceso de compresión resultó idónea, los comprimidos obtenidos presentaron una resistencia a la rotura de 49,03 Nw. Finalmente, hay que destacar la disgregación de los comprimidos inferior a 20 segundos, con un perfil de disgregación más rápido que los comprimidos de referencia.
Estudio de velocidad de disolución.
El estudio de velocidad de disolución se realizó con el objetivo de conocer la cinética de disolución de los minicomprimidos de OLM y como complemento al ensayo de disgregación.
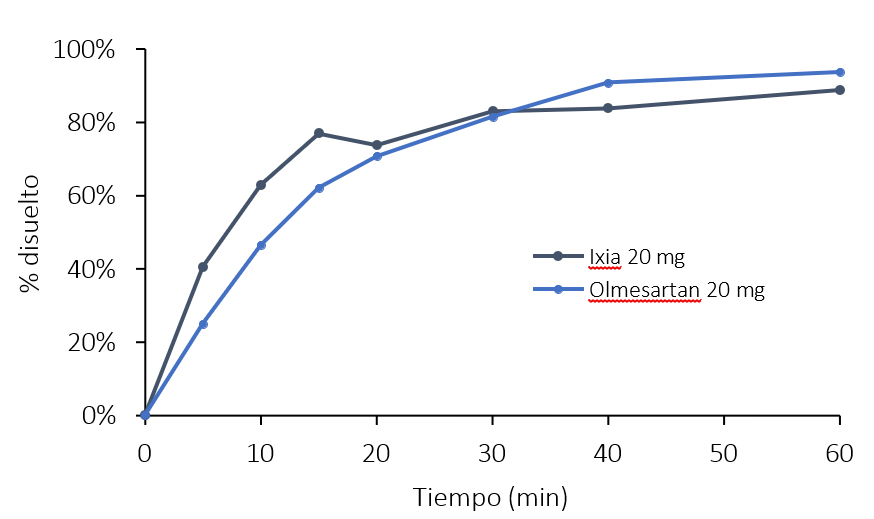
En la Tabla 9 se observó el perfil de disolución comparativo de los minicomprimidos de OLM y del medicamento de referencia Ixia®, con un perfil de disolución similar. Al inicio, Ixia® presenta una mayor liberación del principio activo, igualándose a los 20 minutos. A partir de este momento, los minicomprimidos de OLM aumentan su velocidad de disolución. Finalmente, a los 60 minutos se observó un 94 % del fármaco disuelto para los minicomprimidos de olmesartan frente a un 89 % para los comprimidos de Ixia®.
Tabla 9. Comparación del ensayo de disolución del Ixia® y de los minicomprimidos de OLM.
![]()
4.3.4. Determinación y cuantificación de N-nitrosodimetilamina
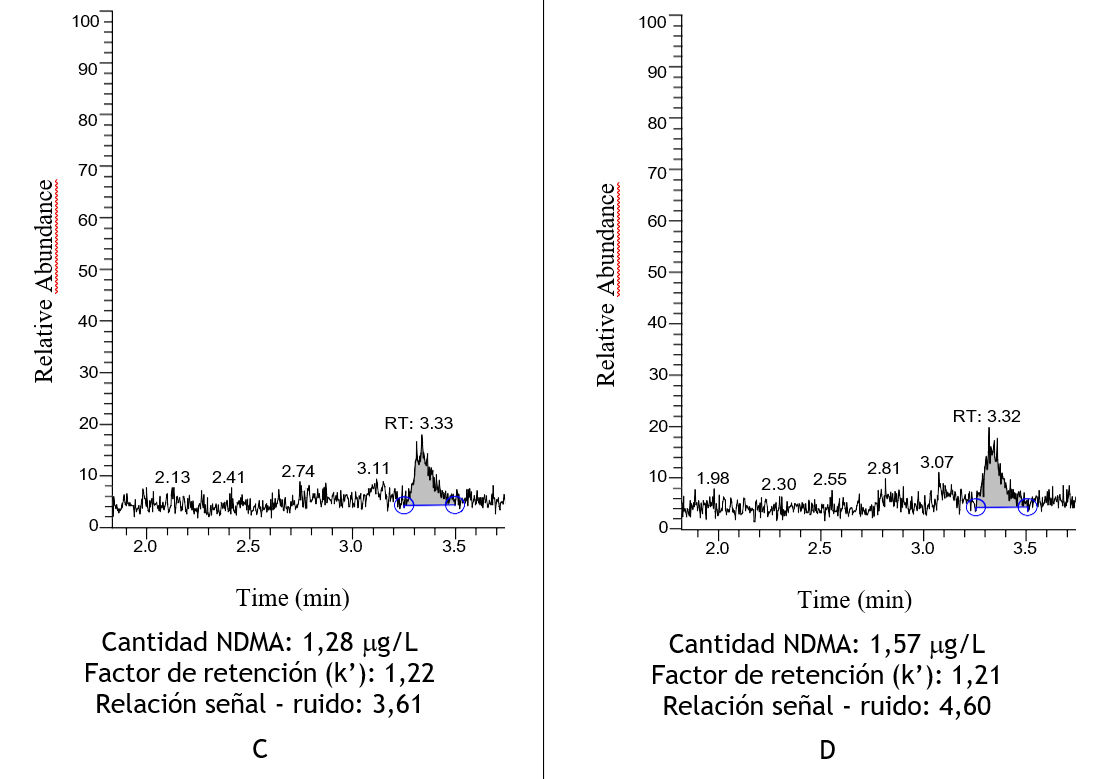
La detección, separación y cuantificación de NDMA se realizó en el principio activo de OLM y en comprimidos fabricados, para detectar posibles cambios en el proceso de fabricación. Así como en comprimidos fabricados con un año de diferencia. Por último, se analizaron los comprimidos comercializados.
Se detectó NDMA a una concentración de 0,26 μg/L en el API de OLM. En los comprimidos fabricados en septiembre de 2019 se detectó NDMA con una concentración de 0,24 μg/L y de 1,28 μg/L en los comprimidos fabricados en octubre de 2020. Por último, se analizaron comprimidos comercializados de OLM (Ixia®) y se detectó una concentración de NDMA de 1,57 μg/L. El resultado de las cuatro muestras se presenta en la Tabla 10. Las concentraciones de NDMA detectadas en todas las muestras estaban por debajo de los límites permitidos (50). A su vez, se observó que no existen variaciones significativas en la concentración de NDMA durante el proceso y el tiempo de fabricación.
Tabla 10. A. Determinación de N-Nitrosodimetilamina en el API de OLM; B. Determinación de N-Nitrosodimetilamina en los comprimidos de olmesartan fabricados en septiembre de 2019. C. Determinación de N-Nitrosodimetilamina en comprimidos de OLM fabricados en octubre de 2020. D. Determinación de N-Nitrosodimetilamina en comprimidos comercializados (Ixia®).
4.4. Estudio de estabilidad
Los resultados de humedad relativa del estudio de estabilidad de los comprimidos incorporados en la cápsula y en su material de acondicionamiento primario se describen en la tabla 1.
En los comprimidos a tiempo 3 meses de ROS no se observó aumento de humedad en comparación con los comprimidos a tiempo inicial, a su vez, no se observaron diferencias entre los comprimidos almacenados en el módulo 1 y módulo 3. En cuanto a los comprimidos almacenados a tiempo 6 meses, se observó un pequeño aumento de humedad (1 % aproximadamente), sin diferencias significativas entre módulos.
Los comprimidos de REP siguieron un patrón muy similar a los de rosuvastatina, sin diferencias entre comprimidos a tiempo inicial y tiempo 3 meses ni entre módulos. Sin embargo, en los comprimidos almacenados a tiempo 6 meses se observaron aumentos de humedad del 1 % y 2 % en los módulos 1 y 3, respectivamente.
Tabla 11. Porcentaje de humedad de los comprimidos a T0, T1 y T2 tras estudio de estabilidad.
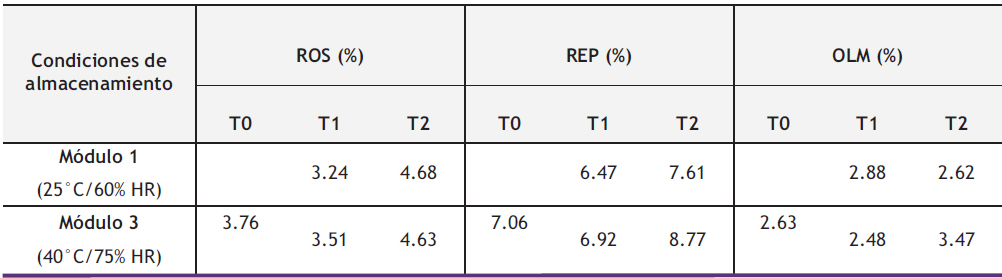
Finalmente, los comprimidos de OLM tuvieron comportamientos muy similares en cuanto a tiempo de almacenamiento y entre módulos. En el módulo 1 no se observaron diferencias significativas a tiempo inicial, tiempo 3 meses y tiempo 6 meses. En cuanto al módulo 3, no se observó aumento de humedad a tiempo inicial y tiempo 3 meses, sin embargo, a tiempo 6 meses hubo un pequeño aumento de humedad inferior al 1 %.
Por otra parte, se determinaron las características organolépticas (aspecto y color) de los comprimidos almacenados en los módulos 1 y 3 durante el período de estabilidad establecido, los resultados se observan en la tabla 11.
Los comprimidos a tiempo inicial sirvieron de referencia para su comparación entre módulos y tiempo de almacenamiento. Tras su análisis visual no se observaron diferencias de color ni aspecto con los comprimidos almacenados a 3 y 6 meses, T1 y T2 respectivamente, ni entre módulos. Los comprimidos seguían manteniendo la coloración blanca y los bordes redondeados sin estar deteriorados en ningún caso.
Tabla 12. Cápsulas con los minicomprimidos a T0, T1 y T2.
5. CONCLUSIONES
Los resultados obtenidos tras el desarrollo de la presente investigación permiten establecer las siguientes conclusiones. Las conclusiones resultaron de aplicación general en el diseño y desarrollo de formulación de medicamentos.
La búsqueda bibliográfica exhaustiva permitió seleccionar los principios activos idóneos para el tratamiento del síndrome metabólico, considerando desde su seguridad, eficacia y rapidez de acción hasta la relevancia de estos fármacos relacionada con el elevado consumo de estos medicamentos y, por consiguiente, con la elevada prevalencia de SM por encima de los 60 años.
La investigación permitió seleccionar los excipientes adecuados para el desarrollo de los comprimidos preparados por compresión directa y elegir las marcas comerciales que sirvieron de comparación en los estudios llevados a cabo. La incorporación de los tres minicomprimidos en una cápsula disminuye el número de administraciones, mejorando la adherencia al tratamiento.
La caracterización fisicoquímica de los principios activos y la compatibilidad principio activo – excipiente mediante análisis SEM, DSC, FT-IR y PXRD han asegurado una adecuada adaptación en las diferentes etapas tecnológicas, manteniendo siempre la calidad como objetivo primordial.
En la misma dinámica, la impureza genotóxica NDMA se detectó, separó y cuantificó con éxito mediante HPLC-MS/MS. Se observó que el tiempo y el proceso de fabricación del fármaco no influyeron en el desarrollo de impurezas. En todos los análisis, la presencia de NDMA en olmesartan se determinó en concentraciones por debajo de los límites permitidos.
Según el sistema de experto de pre-formulación SeDeM para la evaluación de las características reológicas y granulométricas, las formulaciones seleccionadas poseen propiedades adecuadas para su uso en compresión directa, los excipientes seleccionados, y sus proporciones, así como el método de fabricación utilizado en el desarrollo de los minicomprimidos, son favorables y rentables.
Los comprimidos desarrollados cumplieron con las especificaciones, características físicas, dimensiones, uniformidad del contenido, resistencia a la rotura, friabilidad y tiempo de desintegración, establecidas en la Real Farmacopea Española.
A su vez, en el estudio comparativo de velocidad de disolución con su equivalente comercializado los comprimidos fabricados demostraron idoneidad del proceso de producción, con iguales y mejores perfiles de disolución.
6. REFERENCIAS
- Grundy SM. Metabolic syndrome update. Trends Cardiovasc Med. 2016;26(4):364-73.
- Cordero A, Alegría E, León M. Prevalencia de síndrome metabólico. Rev Esp Cardiol Supl. 2005;5(4):11D-15D.
- Maiz A. El síndrome metabólico y riesgo cardiovascular. Bol Esc Med. 2005;30(1):25-30.
- Cequier Á, Arrarte V, Campuzano R, Castro A, Cordero A, Fernández Olmo MR, et al. Tratamiento hipolipemiante en los pacientes con enfermedad cardiovascular de riesgo muy elevado. Documento de consenso SEC sobre las indicaciones de los iPCSK9 en la práctica clínica. REC CardioClinics. enero de 2021;56(1):39-48.
- Fernández-Travieso JC. Síndrome metabólico y riesgo cardiovascular. Rev CENIC Cienc Biológicas. 2016;47(2):106-19.
- Rodríguez Perón JM, Rodríguez Izquierdo MM. Síndrome metabólico y su correlación con ecuaciones de predicción del riesgo global de enfermedad cardiovascular. Rev Cuba Med Gen Integral. 2022;38(3).
- Lahsen MR. Síndrome metabólico y diabetes. Revista Médica Clínica Las Condes. 2014;25(1):47-52.
- Carvajal Carvajal C. Síndrome metabólico: definiciones, epidemiología, etiología, componentes y tratamiento. Med Leg Costa Rica. 2017;34(1):175-93.
- Standl E. Tratamientos actuales y futuros del síndrome. El Síndr Metabólico. 2006;51:31-4.
- Barrios V, Escobar C, Calderón A, Llisterri JL, Alegría E, Muñiz J, et al. Adecuación del tratamiento de los pacientes hipertensos con síndrome metabólico Adequacy of the treatment of hypertensive patients with metabolic syndrome. Med Clínica. 2007;128(17):647-51.
- Davies P. Oral Solid Dosage Forms. In: Pharmaceutical Preformulation and Formulation. 2nd ed. New York, USA: Informa Healthcare; 2009.
- Ficha técnica Provisacor 20 mg comprimidos recubiertos con película. [Internet]. Disponible en: https://cima.aemps.es/cima/dochtml/ft/70241/FT_70241.html
- Ficha técnica Crestor 20 mg comprimidos recubiertos con película. [Internet]. Disponible en: https://cima.aemps.es/cima/publico/detalle.html?nregistro=70244
- Rohini P, Pavani A, Raja Reddy R. Formulation, and evaluation of orally disintegrating tablets of rosuvastatin. Int J Pharm Sci Rev Res. 2014;24(1):209-14.
- Ângelo ML, Ruela ALM, Ferreira ACM, Ramos MV de F, Montanari CM, Silva LM da, et al. Evaluating the discriminatory power of a dissolution assay for rosuvastatin calcium capsules: Solid-state properties and dissolution media. Braz J Pharm Sci. 2019;55:e17520.
- Vázquez Díaz S. Puesta a punto de metodologías analíticas de lactosa en bases lácteas [Trabajo de Fin de Máster]. Universidad de Oviedo; 2017.
- Marini A, Berbenni V, Pegoretti M, Bruni G, Cofrancesco P, Sinistri C, et al. Drug-excipient compatibility studies by physico-chemical techniques. J Therm Anal Calorim. 2003;73(2):547-61.
- Belozerova NM, Bilski P, Jarek M, Jenczyk J, Kichanov SE, Kozlenko DP, et al. Exploring the molecular reorientations in amorphous rosuvastatin calcium. RSC Adv. 2020;10(55):33585-94.
- Kishore CRP, Mohan GVK. Structural identification and estimation of Rosuvastatin calcium related impurities in Rosuvastatin calcium tablet dosage form. Anal Chem Res. 2017;12:17-27.
- Suñe Negre J, Roig Carreras M, Fuster García R, Hernández Pérez C, Ruhí Roura R, García Montoya E, et al. Nueva metodología de preformulación galénica para la caracterización de sustancias en relación a su viabilidad para la compresión: Diagrama SeDeM. Cienc Tecnol Pharm. 2005;125-36.
- Suñe Negre J, Perezlozano P, Minarro M, Roig M, Fuster R, Hernandez C, et al. Application of the SeDeM Diagram and a new mathematical equation in the design of direct compression tablet formulation. Eur J Pharm Biopharm. 2008;69(3):1029-39.
- Suñé Neģre JM, Roiģ Carreras M, García RF, Montoya EG, Lozano PP, Aģuilar JE, et al. SeDeM Diagram: an expert system for preformation, characterization and optimization of tablets obtained by direct compression. En: Formulation Tools for Pharmaceutical Development [Internet]. Elsevier; 2013 [citado 26 de diciembre de 2021]. p. 109-35. Disponible en: https://linkinghub.elsevier.com/retrieve/pii/B9781907568992500050
- Aguilar-Díaz JE, García-Montoya E, Pérez-Lozano P, Suñé-Negre JM, Miñarro M, Ticó JR. SeDeM expert system a new innovator tool to develop pharmaceutical forms. Drug Dev Ind Pharm. 2014;40(2):222-36.
- European Pharmacopoeia (2005) 2.9.40. Uniformity of dosage units. content uniformity. [Internet]. [citado 28 de junio de 2022]. Disponible en: http://www.uspbpep. com/ep50/2.9.40%20Uniformity%20of%20dosage%20units.pdf.
- Real Farmacopea Española (2008) 2.9.8. Resistencia de los comprimidos a la rotura. [Internet]. [citado 22 de abril de 2020]. Disponible en: https://extranet.boe.es/farmacopea/doc.php?id=20908
- Real Farmacopea Española (2010) 2.9.7. Friabilidad de los comprimidos no recubiertos. [Internet]. [citado 22 de abril de 2020]. Disponible en: https://extranet.boe.es/farmacopea/doc.php?id=20907
- Real Farmacopea Española (2011) 2.9.1. Disgregación de comprimidos y cápsulas. [Internet]. [citado 24 de abril de 2020]. Disponible en: https://extranet.boe.es/farmacopea/doc.php?id=20901
- Real Farmacopea Española (2012) 2.9.3. Ensayo de disolución de las formas farmacéuticas sólidas. [Internet]. [citado 24 de abril de 2020]. Disponible en: https://extranet.boe.es/farmacopea/doc.php?id=20903
- Ficha técnica Prandin 2 mg comprimidos. [Internet]. Disponible en: https://cima.aemps.es/cima/pdfs/ft/00162016/FT_00162016.pdf
- Ficha técnica Novonorm 2 mg comprimidos. [Internet]. Disponible en: https://cima.aemps.es/cima/dochtml/ft/98076019/FT_98076019.html
- Rao MEB, Swain S, Patra CN, Sruti J, Patra S. Development and in vitro evaluation of floating multiparticulate system of repaglinide. FABAD J Pharm Sci. 2011;36(2):75-92.
- Chadha R, Bhandari S, Arora P, Chhikara R. Characterization, quantification and stability of differently prepared amorphous forms of some oral hypoglycaemic agents. Pharm Dev Technol. 2013;18(2):504-14.
- Aramă C, Nicolescu C, Nedelcu A, Monciu CM. Synthesis and characterization of the inclusion complex between repaglinide and sulfobutylether-β-cyclodextrin (Captisol®). J Incl Phenom Macrocycl Chem. 2011;70(3-4):421-8.
- Su M, Xia Y, Shen Y, Heng W, Wei Y, Zhang L, et al. A novel drug–drug coamorphous system without molecular interactions: improve the physicochemical properties of tadalafil and repaglinide. RSC Adv. 2020;10(1):565-83.
- Prieto Chacón EM. Estudio de las transiciones térmicas del almidón y el almidón termoplástico mediante análisis térmicos (DSC y TGA). Uniandes; 2008.
- Medeiros ACD, Correia LP, Simões MO da S, Macêdo RO. Technological quality determination of pharmaceutical disintegrant by DSC cooling and DSC photovisual. J Therm Anal Calorim. 2007;88(2):311-5.
- Mahon D, Claudio G, Eames P. An Experimental Study of the Decomposition and Carbonation of Magnesium Carbonate for Medium Temperature Thermochemical Energy Storage. Energies. 2021;14(5):1316.
- Awasthi R, Kulkarni GT, Ramana MV, de Jesus Andreoli Pinto T, Kikuchi IS, Molim Ghisleni DD, et al. Dual crosslinked pectin–alginate network as sustained release hydrophilic matrix for repaglinide. Int J Biol Macromol. 2017;97:721-32.
- Yang XD, Li WS, Tian YJ, Liu CG, Gao DH, Ma HL. Dissolution rate enhancement of repaglinide by solid dispersion. Trop J Pharm Res. 8 de junio de 2016;15(6):1123.
- Rautray RK, Biswal B, Patra RK, Mallik S. Research Article Dissolution Rate Enhancement of Ganciclovir by Solid Dispersions with Polyethylene Glycol 6000. Res J Pharm Life Sci Vol. 2021;2(1):44-54.
- Ficha técnica Ixia 20 mg comprimidos recubiertos con película. [Internet]. Disponible en: https://cima.aemps.es/cima/dochtml/ft/65499/FT_65499.html
- Royce A, Ruegger C, Mecadon M, Karnachi A, Valazza S. Scale-up of the compaction and tableting process. En: Pharmaceutical Process Scale-Up. CRC Press; 2005. p. 405-42.
- Jain S, Patel K, Arora S, Reddy VA, Dora CP. Formulation, optimization, and in vitro–in vivo evaluation of olmesartan medoxomil nanocrystals. Drug Deliv Transl Res. 2017;7(2):292-303.
- Abd-El Bary A, D. Louis SS. Olmesartan medoxomil surface solid dispersion- based orodispersible tablets: formulation and in vitro characterization. J Drug Deliv Sci Technol. 2014;24(6):665-72.
- Chai R, Gao H, Ma Z, Guo M, Fu Q, Liu H, et al. In Vitro and In Vivo Evaluation of Olmesartan Medoxomil Microcrystals and Nanocrystals: Preparation, Characterization, and Pharmacokinetic Comparison in Beagle Dogs. Curr Drug Deliv. 2019;16(6):500-10.
- Listiohadi Y, Hourigan JA, Sleigh RW, Steele RJ. Thermal analysis of amorphous lactose and α-lactose monohydrate. Dairy Sci Technol. 2009;89(1):43-67.
- Aprilia NS, Davoudpour Y, Zulqarnain W, Khalil HA, Hazwan CCM, Hossain M, et al. Physicochemical characterization of microcrystalline cellulose extracted from kenaf bast. BioResources. 2016;11(2):3875-89.
- Gómez-Carracedo A, Alvarez-Lorenzo C, Gómez-Amoza J, Concheiro A. Chemical structure and glass transition temperature of non-ionic cellulose ethers. J Therm Anal Calorim. 2003;73(2):587-96.
- Nasr A, Gardouh A, Ghorab M. Novel Solid Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) for Oral Delivery of Olmesartan Medoxomil: Design, Formulation, Pharmacokinetic and Bioavailability Evaluation. Pharmaceutics. 2016;8(3):20.
- FDA updates and press announcements on angiotensin II receptor blocker (ARB) recalls (valsartan, losartan, and irbesartan): interim limits for NDMA, NDEA, and NMBA in angiotensin II receptor blockers (ARBs). [Internet]. Disponible en: https://www.fda.gov/drugs/drug-safety-and-availability
