1. INTRODUCCIÓN
1.1 Etiología y fisiopatología de la esquizofrenia y su tratamiento con fármacos antipsicóticos
La esquizofrenia es un trastorno psiquiátrico grave y crónico que afecta a 21 millones de personas en todo el mundo. Esta enfermedad se manifiesta generalmente en el final de la adolescencia/principio de la edad adulta y está asociada a síntomas positivos que reflejan un exceso y distorsión de las funciones normales, resultando en delirios, alucinaciones y dificultad para pensar y concentrarse; y/o a síntomas negativos relacionados con el deterioro de funciones cognitivas normales, entre los que se incluyen apatía, abulia, alogia y anhedonia (1, 2). A menudo estos síntomas están asociados con episodios psicóticos que alteran la estabilidad y calidad de vida de las personas, lo que les impide una vida normal en sociedad. Afortunadamente, las intervenciones farmacológicas existentes hoy en día son altamente eficaces en suprimir la sintomatología y restablecer así una vida satisfactoria permitiendo la integración de las personas con esquizofrenia en la sociedad (3). Según las directrices actuales, los fármacos antipsicóticos son la primera línea de tratamiento de la esquizofrenia (4, 5). Dado que la interrupción del tratamiento está asociada con un riesgo exponencial de recaída en comparación con la terapia de mantenimiento, las personas afectadas generalmente continúan el mismo tratamiento que fue efectivo durante la fase aguda de la enfermedad, siempre y cuando el fármaco sea bien tolerado.
Desde la introducción de la clorpromazina en 1952, los fármacos antipsicóticos de primera generación (FAPG) han cambiado drásticamente la atención psiquiátrica, permitiendo que muchas personas con desórdenes mentales graves y debilitantes (esquizofrenia, trastorno bipolar y agitación aguda, entre otras afecciones) se reintegren en la sociedad. Los FAPG contrarrestan predominantemente los síntomas positivos de la esquizofrenia a través de mecanismos que aún se desconocen. La hipótesis más aceptada sobre la acción de los FAPG se relaciona con la teoría dopaminérgica de la esquizofrenia, que postula que la sintomatología positiva es causada por el aumento de la liberación subcortical de dopamina y la posterior sobreactivación de los receptores de dopamina-2 (D2) (6, 7), probablemente derivado de alteraciones en la vía cortical a través del núcleo accumbens. Por el contrario, los síntomas negativos parecen ser causados por una señalización dopaminérgica atenuada debida a la reducción de la actividad del receptor de dopamina-1 (D1) en la corteza prefrontal y núcleo caudado (7-9). Las alteraciones en la expresión y actividad de los receptores de dopamina-3 (D3) también se han asociado con los síntomas negativos de la esquizofrenia (10). Algunos estudios han corroborado la teoría dopaminérgica de la esquizofrenia, mostrando que los antagonistas D2 como los FAPG actúan sobre diferentes vías dopaminérgicas (mesolímbica, mesocortical, nigroestriatal y tuberoinfundibular) para controlar la sintomatología de la esquizofrenia. Por el contrario, los agonistas D2, incluidas la l-3,4-dihidroxifenilalanina (L-DOPA), la cocaína y las anfetaminas, desencadenan efectos psicomiméticos en individuos que no padecen esquizofrenia per se. Sin embargo, los FAPG se asocian con efectos secundarios extrapiramidales como discinesia y distonía (11). En un intento por contrarrestar estos efectos, en la década de 1980 se investigaron una variedad de nuevos fármacos, dando lugar a la aprobación de los denominados fármacos antipsicóticos de segunda generación (FASG) o atípicos, que son actualmente los medicamentos de elección para el tratamiento de las personas con esquizofrenia y otros trastornos psicóticos (4). Más allá de la interacción con los receptores dopaminérgicos al igual que los FAPG, los FASG también tienen la capacidad de bloquear los receptores serotoninérgicos (5-HT2A y 5- HT2C), con una mayor afinidad que la de los receptores D2 (8, 11), sugiriendo que las alteraciones en las vías serotoninérgicas también podrían desempeñar un papel relevante en el desarrollo de la esquizofrenia. Además es importante destacar que los efectos secundarios neurológicos asociados con los FAPG no son tan evidentes en individuos tratados con los FASG, contrarrestando así los síntomas positivos y negativos de la enfermedad (4).
Sin embargo, las observaciones clínicas han revelado una variedad de alteraciones metabólicas en una proporción relevante de individuos tratados con FASG, como son un aumento anormal de peso corporal, hiperglucemia y dislipidemia (12-14). Estos efectos secundarios (Tabla 1) sugieren que el uso de FASG podría tener efectos directos y/o indirectos en diferentes tejidos y sistemas periféricos, incluidos los depósitos de grasa, el hígado y las células inmunes.
Tabla 1. Lista de antipsicóticos de primera y segunda generación y la correspondiente incidencia de efectos secundarios.
1.2 El tejido adiposo: un órgano clave para el control del balance energético y la señalización endocrina
El aumento anormal de peso corporal es un efecto secundario sustancial de la terapia con FASG (11) y se asocia con un aumento de los depósitos de grasa (3, 14, 15). El tejido adiposo es un órgano altamente especializado que regula la homeostasis energética y el metabolismo, y en los mamíferos se compone principalmente de tres tipos de adipocitos, blancos, marrones y beige, que tienen orígenes, morfologías, distribución de gotas de lípidos, redes mitocondriales y patrones de expresión génica distintos entre sí (16).
Funcionalmente, el tejido adiposo blanco (TAB) es el almacén predominante del excedente de energía del organismo en forma de triglicéridos (TG), y contiene adipocitos con una gran gota lipídica unilocular y pocas mitocondrias. El TAB representa del 5 al 50 % del peso corporal total en humanos e incluye depósitos viscerales (dentro de la cavidad abdominal) y subcutáneos (debajo de la piel), con importantes diferencias ontogenéticas y metabólicas entre ambos (16). En un estado de equilibrio energético positivo, el TAB se expande mediante hipertrofia de las células adiposas existentes, así como mediante hiperplasia, con formación de novo de adipocitos. Más allá de su papel clásico como unidad de almacenamiento y liberación de energía, el TAB también funciona para proteger otros órganos (hígado y músculos) de la toxicidad asociada a los lípidos (lipotoxicidad) (17), además de tener un papel clave clave en la señalización endocrina (18).
El tejido adiposo marrón o pardo (TAM) es mucho menos abundante que el TAB y se localiza principalmente en las regiones interescapulares subcutáneas. El TAM se caracteriza por la presencia de pequeños adipocitos con gotas lipídicas multioculares que inicialmente se percibieron como células de linaje similar al músculo esquelético, pero más recientemente se ha sugerido su origen adipocitario (19, 20). El TAM es abundante en mitocondrias enriquecidas en la proteína desacoplante-1 o termogenina (UCP-1) localizada en la membrana interna, que funciona para disipar energía química en forma de calor al desacoplar la oxidación de substratos, fundamentalmente ácidos grasos, de la síntesis de ATP (21-23). En los seres humanos, anteriormente se creía que el TAM existía sólo en los neonatos, pero recientemente se ha identificado en adultos en la zona inferior del cuello (24-26), donde sus funciones y características se encuentran actualmente bajo estudio exhaustivo.
Los adipocitos beige están presentes principalmente en depósitos blancos subcutáneos y en cantidades restringidas de TAB visceral (27). Los adipocitos beige se consideran células adiposas termogénicas de tipo marrón con una morfología de gotas lipídicas multiloculares y expresión de UCP-1, y se desarrollan a partir del proceso llamado en inglés beiging o browning del TAB. Este fenómeno de diferenciación es inducido por el estrés del frío o por agonistas que mimetizan este efecto, como son los agonistas de los receptores adrenérgicos b3 (28, 29). A pesar de que el origen de los adipocitos beige no se conoce completamente, se ha descrito que pueden derivar de un linaje progenitor beige (19, 27, 30), pero también que pueden transdiferenciarse a partir de adipocitos blancos maduros (31, 32), o incluso diferenciarse a partir de células de otros orígenes (16).
Como órgano endocrino, el tejido adiposo participa activamente en los procesos inflamatorios produciendo y secretando una amplia variedad de péptidos bioactivos, incluidas citoquinas y las denominadas adipoquinas, como la leptina y la adiponectina. Estos péptidos tienen acciones tanto locales como a distancia y están relacionadas con la modulación del metabolismo de lípidos y glucosa, pero también con el equilibrio energético (16).
Dada la evidente desregulación metabólica en algunos individuos tratados con FASG, es importante profundizar en el estudio de los efectos de estos fármacos sobre los adipocitos para descifrar los mecanismos moleculares subyacentes a estas disfunciones asociadas al tratamiento con estos fármacos. Por todo ello, en esta revisión se examinarán los avances recientes en la investigación sobre los efectos de los FASG en los mecanismos de diferenciación y función de los adipocitos.
2. EFECTO DE LOS FÁRMACOS ANTIPSICÓTICOS DE SEGUNDA GENERACIÓN SOBRE EL TEJIDO ADIPOSO BLANCO
El TAB se organiza en depósitos discretos (principalmente viscerales y subcutáneos) que se asocian de manera diferencial con la resistencia a la insulina y el riesgo de síndrome metabólico. Si bien la acumulación de TAB visceral se considera perjudicial debido a la inflamación sistémica que promueve, el TAB subcutáneo parece proteger contra el desarrollo de enfermedades metabólicas en ratones y humanos (33, 34). En este sentido, se ha descrito que las personas con esquizofrenia tienen significativamente más TAB visceral que los individuos sanos (15), pudiendo esto explicar su mayor susceptibilidad a desarrollar resistencia a la insulina y disfunciones metabólicas más graves como la diabetes mellitus tipo 2 (T2D) (35). Como se ha mencionado anteriormente, la terapia con FASG induce con frecuencia un aumento de la grasa subcutánea y visceral (15), y, como se detallará a continuación también altera los mecanismos de diferenciación y respuesta a diferentes estímulos fisiológicos.
2.1. Efecto de los fármacos antipsicóticos de segunda generación sobre la diferenciación de los adipocitos blancos.
Una posible explicación del aumento de peso asociado al tratamiento con los FASG reside en su capacidad de modular la diferenciación de los preadipocitos a adipocitos maduros. En relación con la diferenciación de adipocitos, destacan la familia de las proteínas reguladoras de elementos de unión a esteroles (del inglés, Sterol regulatory element-binding proteins o SREBP), constituida por las isoformas SREBP-1a y -1c (codificadas por el gen SREBF1) y SREBP-2 (codificada por SREBF2) (36), que son factores de transcripción que desempeñan un importante papel regulador de este proceso. Yang et al. mostraron que el tratamiento de preadipocitos murinos 3T3-L1 diferenciados con el FASG olanzapina (usado a las concentraciones de 10 o 50 μM durante 24 h) aumentó la expresión de SREBP-1c y, por consiguiente, de la enzima lipogénica ácido graso sintasa (del inglés, Fatty acid synthase, FAS), lo que resultó en una acumulación de TG (36). Estos autores también encontraron que el SREBP-1c, pero no la proteína alfa potenciadora de unión a CCAAT (del inglés, CCAAT/enhancer-binding protein-a, C/EBP-a), estaba sobreexpresada y activada en los adipocitos 3T3-L1 estimulados con olanzapina. De manera similar, la olanzapina (usada a 100 µM durante 3 h) aumentó la lipogénesis y redujo la lipólisis (37). Asimismo, la clozapina (usada a 10 µM) indujo la expresión de la estearoil-CoA desaturasa-1 (del inglés, Stearoyl- CoA desaturase, SCD1) y de SREBP-1c tanto en las etapas más tempranas (día 3) como tardías (día 7) de la diferenciación de células madre humanas derivadas del tejido adiposo (del inglés, adipose-derived stem cells, ASC) (38). En general, estos estudios muestran que algunos FASG aumentan la expresión de SREBP-1c y de sus dianas lipogénicas a diferentes niveles. De hecho, se ha sugerido que los efectos de los FASG en la formación de gotas lipídicas (clozapina> olanzapina>risperidona) dependen de la translocación nuclear de SREBP-1c y la posterior modulación de la adipogénesis (38). Además, en la línea celular hepática humana HepG2, el tratamiento con olanzapina (10 y 50 µM durante 24 h) aumenta la actividad de SREBP-1c de manera dosis dependiente (39). Sin embargo, estos efectos no son tan evidentes en otras líneas celulares hepáticas tratadas con olanzapina, haloperidol o mirtazapina (usados a 25 µM durante 24 h), como han reportado Raeder y colaboradores (40). Dados los resultados anteriormente mencionados en adipocitos, se podría especular que, además de actuar en el sistema nervioso central (SNC) provocando un aumento de peso y diferentes alteraciones lipídicas (41,42), los FASG también median un efecto celular autónomo al aumentar la lipogénesis directamente en los adipocitos.
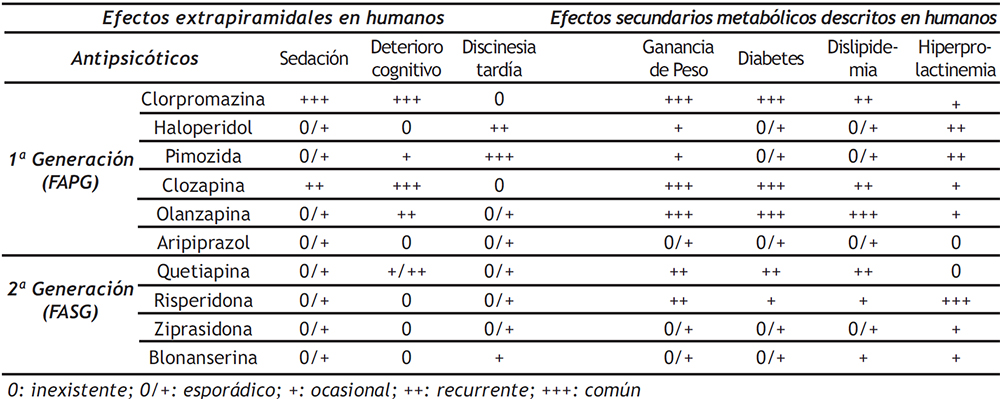
Las proteínas de la familia del gen inducido por la insulina (del inglés, Insulin-induced gene, Insig) son importantes reguladores negativos de la función de SREBP-1c y la lipogénesis. Las Insigs son proteínas chaperonas de las proteínas SREBP que forman un complejo con SREBP y la proteína activadora de escisión de SREBP (del inglés, SREBP cleavage- activating protein, SCAP) en el retículo endoplásmico (RE). Cuando los niveles de esteroles son bajos, las proteínas SREBP se activan mediante escisión proteolítica y el dominio N- terminal es liberado del RE y trasladado al núcleo, donde actúa como factor de transcripción para múltiples genes de biosíntesis de lípidos (43). La familia Insig se compone de dos isoformas: Insig-1, una diana de las SREBP nucleares cuyos niveles de ARNm están directamente asociados con su presencia nuclear, e Insig-2, que está regulada negativamente por la insulina de manera independiente de las proteínas SREBP (36). En condiciones de exceso de esteroles, Insig-2 puede retener el complejo SCAP/SREBP en el RE (44). Es interesante destacar que Chen et al. describieron una relación entre la biosíntesis de lípidos inducida por SREBP-1c, el bloqueo de Insig-2 y el aumento de peso asociado al tratamiento con los FASG, mostrando que la clozapina (usada a 10 µM) suprime significativamente la expresión del gen INSIG2 durante la adipogénesis de las ASC humanas, tanto en la fase temprana como en la fase tardía. Además, después del tratamiento, los niveles de INSIG2 se correlacionaron negativamente con la expresión de SREBP-1c y otros genes de biosíntesis de lípidos. Además, es de destacar que los autores encontraron que, en el tercer día de diferenciación, todos los FASG probados (clozapina (10 µM), olanzapina (1 µM) y risperidona (0,4 µM)) disminuyeron la expresión de INSIG2; sin embargo, sólo la clozapina suprimió significativamente INSIG2 en el día 7 de diferenciación y, en consecuencia, la actividad de SREBP-1c aumentó con el tratamiento (38). Asimismo, aunque el tratamiento con olanzapina no mantuvo la supresión de la expresión de INSIG2 en el último día de diferenciación, sí que aumentó la expresión de SREBP-1c. Por el contrario, la sobreexpresión de INSIG2 durante la diferenciación de las ASC humanas resultó en una bajada de los niveles SREBP-1c, inhibiendo así la biosíntesis de lípidos inducida por los FASG (38). De esta manera, este estudio implica a Insig-2 en la patogénesis de las disfunciones metabólicas desarrolladas por los individuos en tratamiento con los FASG. A pesar de que las interacciones entre las dos isoformas de las proteínas Insig están relacionadas con el síndrome metabólico inducido por los FASG, se ha descrito que los niveles de Insig-1 no se correlacionan con estos efectos secundarios metabólicos per se (45) (Figura 1A).
En las células de mamíferos, el receptor activado por el proliferador de peroxisomas gamma (del inglés, peroxisome proliferator-activated receptor gamma, PPAR-γ) es fundamental no sólo para la diferenciación adipogénica, sino también para mantener el fenotipo del adipocito maduro (46, 47) y, junto con las C/EBP, se considera una proteína esencial para la regulación de la adipogénesis (48). Sertie et al. han descrito que los FASG, particularmente clozapina (20-30 µM) y olanzapina (40-100 µM), aumentan la expresión temprana (hasta día 3) de C/EBPb y de PPARγ2, y tardía (día 14) de PPAR-γ y de la lipoproteína lipasa (LPL) durante la diferenciación adipogénica de las ASC humanas (49). En línea con estos resultados, Hemmrich et al. diferenciaron ASC humanas en presencia de clozapina (5 y 50 µM) durante los primeros 5 días y luego en su ausencia durante 9 días adicionales (14 días de diferenciación en total), encontrando una mayor actividad de la enzima glicerol 3-fosfatasa deshidrogenasa, otro marcador clave de la diferenciación adipogénica. También encontraron que la clozapina aumentó el porcentaje de células diferenciadas en comparación con los cultivos no tratados (50). Estos resultados proporcionan una explicación plausible de porqué el tratamiento con clozapina está asociado con un mayor riesgo de aumento de peso en comparación con otros FASG (51, 52). Del mismo modo, se evidencia que otros fármacos, en particular la pimozida (10 µM), también promovieron la adipogénesis de los preadipocitos 3T3-L1 hasta el día 6 de la diferenciación al inhibir la proteína de unión de ácidos grasos 4 (del inglés, Fatty acid-binding protein 4, FABP4) y aumentar los niveles proteicos de PPAR- γ (53). De manera similar, el tratamiento de ASC humanas durante 3 días con clozapina, olanzapina, quetiapina o risperidona (todas utilizadas a 250 nM) aumentó la producción total de lípidos, un efecto bloqueado por la adición de un inhibidor de la proteína quinasa C (del inglés, protein kinase C, PKC)-b (51). A nivel molecular, este estudio muestra que el tratamiento de las ASC con los FASG, particularmente clozapina, promueve la translocación de PKC-β a la membrana plasmática y su posterior activación. Es importante destacar que los FASG también estimulan la adipogénesis de células madre derivadas del músculo esquelético, sugiriendo que el aumento de peso asociado con los FASG también implica la adquisición de características adipogénicas en otras poblaciones de células madre (54).
El aumento de peso corporal asociado con el tratamiento con FASG también podría deberse a alteraciones en el almacenamiento de lípidos en los adipocitos (hipertrofia). Por ejemplo, la diferenciación de ASC de rata en presencia de clozapina (10 µM) aumenta la formación de gotas lipídicas, mientras que la adición de olanzapina (1 µM) o risperidona (0,4 µM) induce sólo un ligero aumento en el día 7 de diferenciación (38). Asimismo, en los adipocitos diferenciados de células madre mesenquimales (MSC) humanas, el tratamiento con olanzapina (5-100 µM) durante 12 días aumenta la acumulación de lípidos de manera dosis- dependiente (52). Estudios proteómicos adicionales revelaron que la olanzapina regula positivamente los niveles de perilipina-4 (PLIN4) y de otras enzimas relacionadas con el metabolismo de los lípidos, incluida la FAS. De hecho, las MSC diferenciadas en presencia de olanzapina mostraron gotas lipídicas más grandes recubiertas de PLIN1, 2 y 4. Sin embargo, solo los niveles proteicos de PLIN2 aumentaron por la olanzapina en las etapas tempranas de la diferenciación y disminuyeron a partir del día 12, mientras que PLIN4 y PLIN1 aumentan en etapas posteriores. Si bien se sabe que las proteínas PLIN están reguladas por PPAR-γ, los autores de este estudio no encontraron alteraciones en sus niveles de ARNm por la olanzapina. Sin embargo, los niveles de ARNm de C/EBP-a disminuyeron en los días 9 a 16 en presencia de olanzapina, lo que podría resultar en una regulación negativa de su proteína diana, la translocasa de ácido graso/CD36, en las últimas etapas de diferenciación (55). No obstante, la relevancia de esta regulación negativa es controvertida, ya que se ha encontrado que la eliminación de CD36 en ratones confiere protección frente al aumento de tejido adiposo inducido por una dieta rica en grasas (56). Es de destacar que el tratamiento con este FAPG no alteró otros genes que codifican proteínas relevantes del metabolismo de los lípidos como SREBP-1c, FAS, LPL, leptina y adiponectina. El estudio mencionado anteriormente también demostró que la olanzapina aumentó la expresión de la triglicérido lipasa del tejido adiposo (del inglés, adipose triglyceride lipase, ATGL). En los adipocitos, ATGL se localiza en gotas lipídicas grandes positivas para PLIN1 y se libera tras la fosforilación de PLIN1, por lo que los autores postulan que la expresión de ATGL aumenta en las MSC tratadas con olanzapina de manera concomitante con la acumulación de gotas lipídicas (Figura 1A). Además, los cambios en la expresión génica y los niveles de proteínas de los miembros de la familia PLIN mediados por la olanzapina podrían sugerir un posible mecanismo para el aumento de la adiposidad en pacientes sometidos a tratamiento con este fármaco (55).
La dislipidemia es otro efecto secundario metabólico asociado a la terapia con FASG y está directamente relacionada con la capacidad de los adipocitos para almacenar ácidos grasos libres. En este sentido, un estudio con adipocitos humanos demostró que la clozapina, olanzapina, quetiapina o risperidona (usadas a 50 o 100 µM) no inhibieron el transporte de ácidos grasos, concluyendo que probablemente este no sea el mecanismo responsable de la dislipidemia observada en individuos tratados con FASG (57). A pesar de todos estos hallazgos, se requieren más estudios para comprender mejor el impacto de los FASG en los eventos tempranos y tardíos de la adipogénesis, así como en la hipertrofia de los adipocitos.
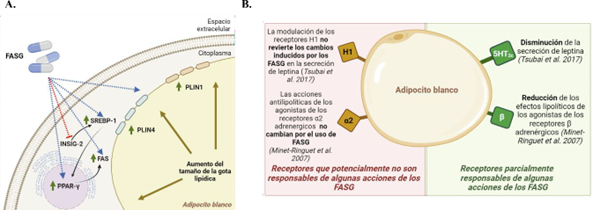
Figura 1. Acciones de los FASG en los adipocitos blancos. A. Esquema de los efectos moleculares mediados por los FASG en los adipocitos blancos, particularmente enfocados en las proteínas relacionadas con el metabolismo de lípidos y el aumento de las gotas lipídicas. B. Tipos de receptores a través de los cuales los FASG pueden actuar sobre los adipocitos blancos
2.2. Los fármacos antipsicóticos de segunda generación modifican la función endocrina de los adipocitos blancos
El TAB tiene un papel esencial en el metabolismo y la inflamación sistémica (inmunometabolismo) (16, 58), produciendo y liberando una gran cantidad de biomoléculas con funciones paracrinas, autocrinas y endocrinas asociadas con el equilibrio entre el gasto energético y la ingesta, la regulación de la homeostasis de lípidos y glucosa, así como con procesos inflamatorios (16).
La adiponectina es la adipoquina más abundante, y una de las más relevantes secretadas por los adipocitos maduros. Los niveles de esta adipoquina se encuentran alterados en la obesidad, la resistencia a la insulina y el síndrome metabólico (59). La adiponectina modula vías relacionadas con el metabolismo de carbohidratos y lípidos, y también procesos vasculares, debido a sus propiedades antiinflamatorias, antiaterogénicas y sensibilizadoras a la insulina (60). Varios estudios han investigado la respuesta de la adiponectina a los FASG. Las ASC humanas diferenciadas en presencia de clozapina (100 ng/ml), quetiapina (50 ng/ml) o aripiprazol (100 ng/ml) mostraron una expresión elevada de adiponectina (61). De manera similar, el tratamiento con olanzapina (10 µM) durante la diferenciación aumentó la expresión de adiponectina también en células 3T3-L1 (39). En otro estudio se encontró que el tratamiento corto (2 días en adipocitos maduros) o largo (10 días durante la diferenciación) con clozapina (10–30 µM) en adipocitos 3T3-L1 disminuía la secreción de adiponectina sin alterar sus niveles de ARNm (62). Asimismo, la exposición de 10 días a blonanserina (0,01– 0,1 µM) también redujo los niveles de ARNm de adiponectina. Por lo tanto, este efecto opuesto sobre la expresión/secreción de adiponectina resulta intrigante y puede depender del origen de los adipocitos o de la sensibilidad de los sistemas celulares a los FASG.
A diferencia de la adiponectina, la leptina es una adipoquina secretada por los adipocitos blancos que se relaciona positivamente con la cantidad de grasa corporal, modulando la ingesta de alimentos y el gasto energético (63); en consecuencia, la leptina suele estar elevada en el suero de pacientes obesos (64). Tsubai et al. reportaron que la exposición de los adipocitos 3T3-L1 a clozapina (10–30 µM) durante 2 o 10 días disminuyó los niveles de expresión génica de leptina, así como su secreción al medio de cultivo (62). Además, este trabajo demostró el papel de los receptores serotoninérgicos 5-HT2c e histaminérgicos H1 en el impacto de la clozapina en la expresión y secreción de leptina, ya que se expresan de manera ubicua en el cerebro y los tejidos periféricos (65, 66) y desempeñan un papel importante en alteraciones/trastornos de los patrones alimentarios que pueden conducir a un metabolismo lipídico desregulado (67-69). Los autores utilizaron histamina (agonista del receptor H1) y difenhidramina (antagonista del receptor H1), así como serotonina (agonista del receptor 5-HT2c) y SB242084 (antagonista del receptor 5-HT2c).
El tratamiento a corto plazo de las células 3T3-L1 con histamina o difenhidramina no alteró la secreción de leptina, lo que sugiere que este proceso no está mediado por los receptores H1. Sin embargo, el tratamiento con serotonina disminuyó la secreción de leptina en comparación con las células control, aunque no se encontró ningún efecto sinérgico o aditivo cuando se combinó con clozapina (30 µM). Curiosamente, el SB242084 también disminuyó la secreción de leptina en los adipocitos3T3-L1, disminuyendo de forma dosis-dependiente aún más la secreción de leptina cuando se combinó con clozapina (30 y 50 µM). Sin embargo, esta relación parece ser más compleja porque la serotonina no logró revertir este efecto al aumentar la secreción de leptina (Figura 1B) (62). Por el contrario, el tratamiento de ASC humanas con varios FASG aumentó la expresión de leptina (61). Por consiguiente, se requieren más estudios para comprender completamente los mecanismos que regulan las interacciones entre los FASG y los receptores serotoninérgicos en la hipertrofia de los adipocitos y la secreción de adipoquinas.
Los adipocitos secretan citoquinas con propiedades proinflamatorias que son los principales impulsores de la inflamación crónica de bajo grado asociada a la obesidad y a sus consecuentes anomalías metabólicas (70). En el estudio antes mencionado de Sarvari et al. (61), el tratamiento in vitro de ASC humanas con olanzapina, ziprasidona, clozapina, quetiapina, aripiprazol, risperidona o haloperidol indujo la expresión del factor de transcripción nuclear κB (del inglés, nuclear factor κB, NF-κB), un regulador clave de la respuesta inflamatoria, y un aumento en la expresión de las citoquinas proinflamatorias, como el factor de necrosis tumoral (del inglés, tumor necrosis factor, TNF)-α, interleuquina (IL)-1β, IL-8 y la proteína quimioatrayente de monocitos 1 (del inglés, Monocyte chemoattractant protein-1, MCP-1), en paralelo a la secreción de IL-8 y MCP-1 al medio de cultivo. Estos resultados sugieren que los tratamientos antipsicóticos podrían “sensibilizar” o “predisponer” para el desarrollo de un estado proinflamatorio crónico de bajo grado. De hecho, la exposición de los adipocitos 3T3-L1 a clozapina (30 µM) durante 10 días aumentó la expresión de MCP-1 e IL-6 (62). Este aumento en la producción de moléculas proinflamatorias a su vez promueve la resistencia a la insulina y exacerba la disfunción metabólica (71, 72). Es de destacar que la adiponectina puede bloquear la producción local de citoquinas proinflamatorias al antagonizar los receptores tipo toll (del inglés, toll like receptor, TLR) (73) e inhibir a NF-κB (74, 75). Por lo tanto, la hipoadiponectinemia asociada con la hiperleptinemia tras los tratamientos con FASG se acompaña de una mayor expresión y secreción de citoquinas proinflamatorias (61, 62). El patrón alterado de secreción de citoquinas proinflamatorias en los adipocitos y la afinidad promiscua de los FASG por varios receptores, incluidos los receptores histaminérgicos H1, serotoninérgicos 5-HT2c y adrenérgicos (35), son probablemente la causa del desarrollo de resistencia a la insulina y T2D.
2.3. Los fármacos antipsicóticos de segunda generación modulan la sensibilidad a la insulina y el transporte de glucosa en los adipocitos blancos
Además del control del balance energético del organismo, el TAB también regula el metabolismo glucídico y lipídico (16, 58, 76). El transporte de glucosa resultante de la activación de la señalización de la insulina es muy relevante porque su metabolismo proporciona tanto ácidos grasos como glicerol para la síntesis de TG (77, 78). Vestri et al. compararon los efectos de clozapina, olanzapina, risperidona y quetiapina (1–500 µM) con los FAPG butirofenona y trifluoperazina relativos al transporte de glucosa inducido por insulina, así como la lipogénesis/lipólisis en células 3T3-L1 y en adipocitos primarios de rata. En ambos sistemas, la olanzapina y la clozapina usadas a concentraciones bajas (5 µM) redujeron fuertemente el transporte de glucosa inducido por la insulina, mientras que los FAPG no alteraron esta respuesta. Además, todos los antipsicóticos (testados a 100 µM) aumentaron la tasa de oxidación de glucosa basal e inducida por insulina (37). Curiosamente, otro estudio demostró que las células 3T3-L1 tratadas con haloperidol, quetiapina o clozapina a la concentración de 10 µM presentaron una captación de glucosa reducida sin alteraciones en la sensibilidad a la insulina o en la activación del mediador de la señalización de la insulina Akt/proteína quinasa B, pero esto no ocurría a la concentración de 1 µM (79). En esta línea, Robinson et al. encontraron que concentraciones más bajas de olanzapina (7-350 nM) no alteraron el transporte de glucosa basal o inducido por insulina en las células 3T3-L1 (80). Estos resultados sugieren que los efectos de los FASG sobre el transporte de glucosa dependen de la dosis. Además, los FAPG no afectaron a la lipólisis en respuesta al isoproterenol o a la captación de glucosa en respuesta a la insulina, lo que podría explicar por qué las terapias convencionales están menos asociadas con efectos secundarios metabólicos (37).
2.4. El tejido adiposo blanco y los efectos de los fármacos antipsicóticos en modelos animales: alteraciones en el metabolismo lipídico
Los modelos animales son una herramienta traslacional de gran utilidad para descifrar las bases moleculares de los efectos secundarios metabólicos asociados con los FASG. Varios estudios han descrito que la clozapina y la olanzapina son responsables del mayor aumento de peso en roedores asociado con déficits en el metabolismo glucídico y lipídico (81-84). Por ejemplo, Yang et al. describieron que ratas hembra Sprague-Dawley tratadas con olanzapina (a la dosis de 2 mg/kg dos veces al día mediante sonda gástrica) durante 2 semanas mostraron un aumento significativo del peso corporal total que se debía principalmente al aumento de peso del hígado y TAB (85). Además, estos animales desarrollaron hiperlipidemia, hiperglucemia, hiperinsulinemia, resistencia a la insulina y presentaron niveles séricos elevados de IL-6 en paralelo con una depleción de cromo en los tejidos. Curiosamente, la suplementación diaria de cromo durante el tratamiento con olanzapina produjo un fenotipo más leve, y la suplementación con AICAR, un activador de la proteína quinasa activada por AMP (del inglés, adenosine monophosphate-activated protein kinase, AMPK), mejoró la hiperglucemia e hiperlipidemia inducidas por olanzapina, lo que sugiere que la baja actividad de cromo y AMPK está relacionada con la disfunción metabólica inducida por olanzapina (85). Curiosamente, las ratas hembras son más susceptibles que las ratas macho a los efectos metabólicos nocivos inducidos por los tratamientos con FASG (81, 86). Albaugh et al. describieron que un régimen de dosificación creciente de olanzapina mezclada con masa de galletas (4 mg/kg del día 0 al 6; 8 mg/kg del día 7 al 20 y 12 mg/kg del día 21 al 29) aumenta el peso corporal de ratas hembras Wistar y Sprague-Dawley, pero no de ratas macho (81). Pouzet y colaboradores reportaron previamente resultados comparables en ratas hembra Wistar tratadas con 5 y 20 mg/kg de olanzapina mediante administración oral durante 21 días (86). Además, Goudie et al. demostraron que ratas hembra Wistar inyectadas por vía intraperitoneal con olanzapina (4 mg/kg) durante 19 días aumentaron su peso corporal (83). Es de destacar que se ha descrito que en estos modelos el aumento de peso es muy rápido al inicio del tratamiento y es reversible una vez finalizado el mismo (83, 86). En este sentido, Albaugh et al. evidenciaron la asociación entre el aumento de peso corporal y la hiperfagia a partir de las primeras 24 horas de tratamiento en ratas hembra que recibieron olanzapina administrada por vía oral incluida en masa de galletas (81). En el mismo estudio, la administración de olanzapina aumentó de los niveles de leptina, así como de la adiposidad, induciendo una resistencia leve a la insulina a partir del día 12 de tratamiento, lo que sugiere que el aumento de peso inducido por la olanzapina podría ser un efecto secundario de la hiperfagia relacionada con la resistencia a la leptina (hiperleptinemia). Por el contrario, en un estudio realizado en ratas Sprague-Dawley hembra a las que se les inyectó olanzapina (100 mg/kg) por vía intramuscular durante 14 días en un tratamiento de 4 inyecciones, Horska et al. describieron un aumento de peso corporal paralelo a una elevación significativa en la cantidad de TAB visceral, pero sin hiperfagia (87). Además, conjuntamente con los aumentos tanto en la adiposidad como en el tamaño de los adipocitos, Tan et al. encontraron que el tratamiento con olanzapina en el agua de bebida (entre 0,003 mg/ml y 0,03 mg/ml) provocaba alteraciones morfológicas en el TAB subcutáneo de ratas Wistar hembra, un efecto relacionado con el aumento del número de células adiposas indiferenciadas en este depósito que fue detectado al tercer día de tratamiento de forma dependiente de la dosis, pero independientemente del aumento de peso corporal (87, 88).
A diferencia de lo que ocurre en las hembras tratadas con FASG, el aumento de peso corporal es más difícil de reproducir en machos. Minet-Ringuet et al. demostraron que ratas Sprague- Dawley macho alimentadas con una dieta suplementada con FASG durante 5 semanas (1 mg/kg de haloperidol u olanzapina o 10 mg/kg de ziprasidona) no ganaron peso corporal pero sí mostraron un aumento de la adiposidad en los depósitos subcutáneos y viscerales (89). Un análisis más profundo de los adipocitos aislados de estos animales tratados no mostró alteraciones en la actividad lipolítica basal; sin embargo, la olanzapina redujo los efectos lipolíticos desencadenados por el agonista b-adrenérgico isoproterenol o por el inhibidor de la fosfodiesterasa, 3-isobutil-1-metilxantina (IBMX).
Además, las acciones antilipolíticas de la insulina, así como las del análogo de adenosina feniliso-propiladenosina o del agonista adrenérgico a2 UK14304 no se alteraron en los adipocitos primarios tratados con olanzapina. En general, los resultados de este estudio apuntan a una reducción de la sensibilidad de los receptores b-adrenérgicos por parte de la olanzapina, lo que podría explicar su impacto negativo sobre el metabolismo de los lípidos. Sin embargo, el hecho de que la lipólisis inducida por el IBMX también se afecta sugiere que los mecanismos moleculares podrían ser más complejos que el bloqueo de los receptores b-adrenérgicos por los FASG (89). Por otro lado, la olanzapina, el haloperidol y la ziprasidona son antagonistas moderados de los receptores adrenérgicos a1 y a2 (90), pero dado que los receptores adrenérgicos a1 no participan en la lipólisis en ratas (91), es poco probable que estén implicados en el mecanismo de acción de estos fármacos. Además, en el estudio de Minet-Ringuet et al., el hecho de que el tratamiento con FASG no lograra contrarrestar la inhibición de la lipólisis inducida por agonistas de los receptores adrenérgicos a2 (89) descarta a estos receptores en el mecanismo por el cual los FASG interrumpen la movilización de lípidos.
La homeostasis lipídica se sustenta en el equilibrio entre la lipogénesis y la lipólisis. Curiosamente, las ratas Sprague-Dawley macho tratadas con olanzapina presentan una expresión aumentada de la FAS y una expresión disminuida de la lipasa sensible a hormonas (del inglés, hormone-sensitive lipase, HSL) en los adipocitos, favoreciendo así la síntesis de lípidos frente a la lipólisis, y proporcionando una posible explicación al aumento de los depósitos adiposos en ratas (89). Por el contrario, Victoriano et al. encontraron que ratas Sprague-Dawley macho tratadas con olanzapina (2 mg/kg) o haloperidol (1 mg/kg) suplementado en la dieta durante 46 días no mostraron cambios en los niveles de marcadores lipogénicos (FAS y acetil-CoA carboxilasa (ACC)) y de enzimas lipolíticas (LPL y HSL) en el TAB (92). Claramente, se necesitan más estudios preclínicos para dilucidarlas bases moleculares del impacto de los FASG en el metabolismo lipídico, incluida la susceptibilidad intrínseca del sexo, para poder trasladar estos resultados a los humanos.
2.4.1. Efecto de los fármacos antipsicóticos de segunda generación sobre la expresión y secreción de hormonas/citoquinas en el TAB
Como se ha mencionado anteriormente, el tejido adiposo actualmente se reconoce como un modulador clave en el desarrollo de la inflamación sistémica y, en consecuencia, de la resistencia a la insulina y la disfunción metabólica (93, 94). Aunque los mecanismos a través de los cuales los FASG influyen en la inflamación sistémica y/o local no estén claramente definidos, se ha planteado la hipótesis de que el tejido adiposo es responsable de la respuesta inflamatoria inducida por los FASG, que a su vez sería la causa principal de las disfunciones metabólicas asociadas con las terapias antipsicóticas (93). Así, Li y sus colaboradores encontraron un aumento en los niveles de citoquinas proinflamatorias circulantes TNF-α, IL- 6, IL-8 e IL-1β, que se correlacionaban con mayores niveles de expresión de génica en el TAB de ratones Balb/c y de ratas Sprague-Dawley hembras tratadas por vía intraperitoneal con olanzapina (10 mg/kg) durante 8 semanas (95). De manera similar, la administración de olanzapina (10 mg/kg) mediante bombas osmóticas en ratas Sprague-Dawley macho aumenta los niveles de inmunotinción de IL-6 y F4/80 en muestras de TAB, lo que se correlacionó positivamente con los niveles de proteína translocadora (del inglés, translocator protein, TSPO) – una diana para radiomarcadores que potencialmente indican microgliosis en estudios clínicos de neuroimagen – mientras que no se encontraron cambios en los niveles de IL-1β, IL-4, IL-5, IFN-γ o TNF-α (93). La olanzapina administrada por vía oral a ratas Sprague-Dawley hembra (1 mg/kg, 3 veces al día) provocó un aumento en la infiltración de monocitos en el TAB, paralelamente a un aumento del peso corporal y del tamaño de los adipocitos (96). Además, los autores encontraron una alta correlación entre el tamaño de los adipocitos y la infiltración de macrófagos en el TAB, que se acompañó de un aumento en la expresión de TNF-α, IL-1β e IL-6. Curiosamente, la respuesta inflamatoria en el TAB de los animales tratados con FASG también podría tener un componente dependiente del sexo. Por ejemplo, Davey et al. investigaron los efectos de la olanzapina administrada por vía intraperitoneal (2 o 4 mg/kg, dos veces al día) en ratas Sprague-Dawley macho y hembra, encontrando que las hembras ganaron más peso corporal que los machos, un efecto asociado a la hiperfagia (97). Además, las hembras, pero no los machos, mostraron un incremento de la expresión de la IL-6 en el TAB, así como niveles plasmáticos elevados de IL-8 e IL-1β. Sin embargo, tanto la adiposidad como la infiltración de macrófagos aumentaron con el tratamiento con olanzapina en ambos sexos, junto con alteraciones en la microbiota intestinal (97). El estudio mencionado de Victoriano et al. realizado en ratas macho Sprague-Dawley tratadas con olanzapina o haloperidol suplementados en la dieta, también describió inflamación en el TAB, manifestada por los altos niveles de expresión de TNF-α y de infiltración de células inflamatorias positivas para el marcador de superficieCD68 (92). Dado que el tratamiento con FASG conduce a la sobreexpresión de NF-κB en las ASC humanas (61), y que la relación entre la inflamación y la resistencia a la insulina está bien documentada (98), un cambio hacia un perfil proinflamatorio podría explicar algunas de las disfunciones metabólicas descritas en pacientes en tratamiento con FASG que ganan peso. En consecuencia, el factor de transcripción NF-κB podría ser una potencial diana terapéutica para prevenir y/o mitigar la resistencia a la insulina inducida por olanzapina debido a la inflamación del TAB (95).
Es importante mencionar que la acción de los FASG en el SNC también podría desempeñar un papel relevante en los patrones de apetito e ingesta de alimentos, ya que la olanzapina podría activar la hormona concentradora de melanina (del inglés, Melanin-concentrating hormone, MCH) (sistema de iniciación de la alimentación) en el hipotálamo lateral que tiene proyecciones hacia el núcleo accumbens (42), aumentando la ingesta de alimentos. En lo que respecta a los efectos mediados por el SNC de los FASG que resultan en un perfil proinflamatorio, Guesdon et al. describieron que la administración de olanzapina (1 mg/kg, 13 días) a través de mini-bombas implantadas en ratas Wistar macho, aumentó moderadamente los niveles de expresión génica del receptor de la hormona concentradora de melanina en la capa del núcleo accumbens (41). Con el tiempo, esto podría provocar una hiperfagia y, en consecuencia, un aumento del TAB y, en consecuencia, el reclutamiento de células inmunes, desencadenando así procesos inflamatorios. Sin embargo, como se mencionó anteriormente, la conexión entre los efectos periféricos y centrales durante el tratamiento con los FASG no está totalmente esclarecida y necesita ser investigada en estudios futuros.
2.4.2. Alteraciones in vivo de la respuesta a la insulina en el TAB por los fármacos antipsicóticos de segunda generación
Varios estudios en modelos animales han reportado una influencia directa de los FASG en la respuesta del TAB a la insulina. En esta línea, Cui et al. encontraron que el aumento de peso corporal observado en ratones hembra C57Bl/6 tratadas con olanzapina (3 mg/kg/día) suplementada en la dieta durante 2, 4 u 8 semanas se asociaba con hiperinsulinemia y resistencia a la insulina (99). Estos hallazgos fueron corroborados por Coccurello et al. en el mismo modelo animal (100), y también por Hou et al. en ratones hembra C57Bl/6 que recibieron olanzapina (6 mg/kg) mediante sonda oral durante 7 semanas (101). Calevro et al. plantearon la hipótesis de que la resistencia a la insulina en estos animales tratados está estrechamente relacionada con la inflamación, ya que la olanzapina aumenta la infiltración de macrófagos y la expresión de citoquinas proinflamatorias en el TAB, particularmente IL- 6 (93). Además, el efecto de los FASG sobre la vía de señalización de Wnt parece ser relevante para las alteraciones del metabolismo de la glucosa y la sensibilidad a la insulina en el TAB. En este sentido, Li y colaboradores encontraron que, además de un aumento en los niveles de insulina durante el ayuno, la expresión de TFC7L2, un efector clave de la vía Wnt, aumentaba en el TAB, hígado y músculo esquelético de ratones machos C57Bl/6 tratados con olanzapina (4 mg/kg/día) por vía oral durante 8 semanas. El tratamiento combinado con metformina, un fármaco anti-hiperglucémico, bloqueó eficazmente los cambios en los niveles plasmáticos de insulina y en la expresión de TFC7L2, sugiriendo una posible correlación entre la resistencia a la insulina inducida por olanzapina y TCF7L2 (102). Además, a nivel molecular, una de las la etapas finales de la cascada de señalización de la insulina en el tejido adiposo es la translocación del transportador de glucosa-4 (GLUT4) a la membrana plasmática. Sin embargo, los estudios de clamp euglucémico-hiperglucémico realizados en ratas Sprague-Dawley macho tratadas con olanzapina mezclada en masa de galletas con un régimen de dosis creciente (día 0-6: 4 mg/kg/día; día 7-13: 8 mg/kg/día y día 14-29: 12 mg/kg/día) no revelaron alteraciones en la captación de glucosa en el TAB, aunque sí que estaba alterada en el músculo esquelético (103).
Además de la relación directa entre la resistencia a la insulina y los FASG, un estudio en ratones C57Bl/6 machos obesos demostró que el tratamiento con olanzapina (2 mg/kg/día) mediante sonda oral una vez al día durante 4 semanas, aumentaba la sensibilidad a la insulina al reducir los niveles plasmáticos de glucosa e insulina, un efecto relacionado con la autofagia por la potenciación de la función lisosomal en los adipocitos (104). Por lo tanto, futuras investigaciones proporcionarán nuevos conocimientos para comprender completamente el efecto de estas terapias in vivo con el fin de desentrañar su relevancia clínica.
2.5. Efectos directos e indirectos de los fármacos antipsicóticos de segunda generación en el TAB humano
Los estudios en humanos son indispensables para trasladar los resultados sobre los mecanismos de acción de los FASG identificados en modelos celulares o animales, ya que estos últimos no recapitulan completamente la enfermedad en las personas. Con respecto al efecto de los FASG en la expresión de adipoquinas y la resistencia a la insulina, se sabe que los niveles de la adipoquina proinflamatoria resistina están elevados en individuos con esquizofrenia tratados con clozapina (105, 106), y se correlacionan con los niveles circulantes de IL-1Ra, TNF-α y proteína C reactiva (107). De acuerdo con estos resultados, Sapra y colaboradores compararon el peso corporal y varios indicadores inflamatorios entre un grupo de 8 hombres no diabéticos con esquizofrenia que habían estado bajo tratamiento con SGA (independientemente del agente elegido) durante al menos 6 meses, y hombres sanos con edad e índice de masa corporal (IMC) comparables y encontraron que los niveles plasmáticos de adiponectina eran más bajos y los niveles de proteína C reactiva eran más altos en los primeros después de un ayuno nocturno, lo que se asociaba con una mayor resistencia a la insulina (108). De manera similar, en un estudio prospectivo, abierto y unicéntrico con 113 individuos tratados ya sea con risperidona (54 individuo scon una dosis promedio de 4.4 mg/día) u olanzapina (59 individuos con una dosis promedio de 17.4 mg/día), el aumento de peso y la prevalencia del síndrome metabólico fueron significativamente mayores en el grupo de olanzapina que en el grupo de risperidona. Además, mientras que los niveles de adiponectina aumentaron significativamente con el tiempo en el grupo de risperidona, disminuyeron significativamente en el grupo de olanzapina. Por el contrario, no se encontraron diferencias significativas entre grupos en los niveles de glucosa e insulina en ayunas, , lo que sugiere que el efecto de la olanzapina en los niveles de adiponectina precede a las disfunciones en el metabolismo sistémico de la glucosa (109). En un estudio similar, Richards et al. (110) examinaron los efectos de la olanzapina y otros FASG sobre los niveles de adiponectina en personas con esquizofrenia versus controles sanos comparables, y encontraron hipoadiponectinemia asociada a olanzapina con una disminución específica de las formas de la proteína de alto peso molecular. Sin embargo, en este estudio no se encontraron alteraciones en la expresión de adiponectina o en la composición de multímeros en adipocitos primarios aislados de TAB subcutáneo y tratados ex vivo con olanzapina (10 ng/ml) hasta 7 días. Estos resultados apuntan a la idea de que las alteraciones en la expresión y secreción de adiponectina podrían ocurrir progresivamente.
Más allá de los efectos específicos de los FASG sobre los parámetros metabólicos, también se deben considerar los cambios específicos de la enfermedad. En un estudio realizado con 9 individuos no diabéticos sin medicación y controles emparejados, Cohn et al. evidenciaron una resistencia a la insulina relacionada con la esquizofrenia con una compensación inadecuada en la secreción de insulina (111). Sin embargo, esto no se asoció con una pérdida significativa de los niveles de adiponectina, como sí se ha descrito en otro estudio en individuos con esquizofrenia sin medicación (112). Se conoce que los niveles de adiponectina pueden verse influenciados por el sexo, ya que la testosterona modula su expresión, así como la secreción de multímeros in vitro (113). Por tanto, los efectos indirectos y directos de los fármacos, los fenómenos inflamatorios y/o la hipoadiponectinemia también pueden considerarse mecanismos potenciales para las alteraciones metabólicas inducidas por los FASG (108). Asimismo, los niveles plasmáticos de la proteína B fijadora de calcio (del inglés, S100 calcium binding protein b, S100B) aumentan en mujeres, pero no en hombres, tratados con clozapina (125 a 900 mg/día) y se correlacionan positivamente con el IMC, lo que señala una posible relación entre esta proteína y el aumento de adiposidad (114). Cabe mencionar que a pesar de que se ha postulado que los astrocitos son las únicas células que expresan y secretan S100B, también se ha demostrado que los adipocitos secretan S100B en un proceso que está controlado negativamente por la insulina (115). S100B inhibe la adenilato ciclasa activando los receptores D2 y potenciando las quinasas reguladas por señales extracelulares (del inglés, extracellular-signal-regulated kinase, ERK) en los astrocitos (116) y, en consecuencia, apuntando al complejo receptor S100B/D2 como una potencial diana molecular de los FASG (114).
Los voluntarios sanos son fundamentales en estudios de ensayos clínicos porque normalmente no han estado expuestos a los tratamientos farmacológicos evaluados y pueden proporcionar información sobre los efectos del tratamiento en sí mismo, independientemente de la patología. En voluntarios sanos, una dosis única de olanzapina (10 mg) eleva los niveles de glucosa en ayunas en las primeras 4,25 horas después de la administración, sin alterar el peso corporal, y también disminuye los niveles de cortisol y ácidos grasos libres en suero (117). Dado que el cortisol activa la LPL en el hígado y la ATGL en los adipocitos (118), su disminución podría explicar la reducción de los niveles de ácidos grasos libres. También se encontró una disminución en las concentraciones de ácidos grasos libres en ayunas y posprandiales en voluntarios sanos que recibieron olanzapina (10 mg/día) durante 8 días, lo que se asoció con un aumento en los niveles nocturnos de adiponectina independientemente de las alteraciones en el IMC (119). Además, el tratamiento durante 15 días con olanzapina (10 mg/día) en hombres sanos elevó los niveles de adiponectina, leptina y TNF-a, y disminuyó los de grelina, sin encontrarse una correlación con cambios en la adiposidad (120). En general, estos estudios ilustran el efecto directo de los FASG en el organismo independientemente de las alteraciones en el peso corporal o la adiposidad (117).
Debido a que la esquizofrenia puede manifestarse en la edad adulta temprana, algunos estudios se han centrado en cohortes de individuos más jóvenes. Un estudio en personas de 6 a 18 años sin tratamiento previo con FASG diagnosticados con trastornos de conducta disruptiva y tratados aleatoriamente con aripiprazol, olanzapina o risperidona (comúnmente utilizado en niños) durante 12 semanas mostró que la olanzapina conduce a un mayor aumento de peso (4,12%) en comparación con el aripiprazol (1,66%) o la risperidona (1,18%), asociado con un aumento de la grasa subcutánea (119). Además, los individuos tratados con olanzapina o aripiprazol mostraron una reducción en la captación de glucosa estimulada por la insulina (29,34% y 30,26%, respectivamente), lo que indica una situación de resistencia a la insulina. El aumento del peso corporal y la reducción de la sensibilidad a la insulina en las primeras 12 semanas de tratamiento podrían ser responsables de la futura morbilidad y mortalidad cardiometabólica asociada con el tratamiento con FASG en la población joven (121).
La reciente introducción de tratamientos combinatorios ha mejorado algunos de los efectos secundarios metabólicos inducidos por los FASG. Por ejemplo, en una cohorte de 30 individuos con esquizofrenia bajo tratamiento estable con olanzapina, Taveira et al. testaron el efecto de la coadministración de naproxeno, un antagonista de los receptores opioides. Esta terapia combinatoria disminuyó la masa grasa corporal y aumentó la masa libre de grasa (masa magra), lo que confiere protección frente al aumento de peso. Aunque este estudio no prueba que el aumento de peso asociado a la olanzapina es inducido por la activación del sistema opioide ya que por sí solo este sistema tiene la capacidad de mejorar el control del peso corporal, sugiere que el bloqueo de los receptores opioides podría contrarrestar las alteraciones del metabolismo lipídico asociadas al tratamiento con los FASG (122).
El impacto molecular de la terapia con FASG en el tejido adiposo humano aún no se conoce en profundidad y, de hecho, se han publicado varios estudios contradictorios. Una posible explicación de estas discrepancias es la variabilidad en los grupos control utilizados en los estudios clínicos (edad, IMC, sexo, tabaquismo, entre otros parámetros). Además, el efecto de los FASG es claramente diferente entre voluntarios sanos y personas con esquizofrenia u otros trastornos psicóticos, por lo que es posible que omitan alteraciones específicas de la enfermedad en estudios con sujetos sanos. Es importante destacar que las alteraciones en la adiposidad y el aumento del IMC observados en individuos sometidos a tratamientos con FASG están estrechamente relacionadas con la comorbilidad cardiometabólica y la mortalidad. En consecuencia, se requieren estudios adicionales para comprender el impacto directo (o indirecto) de estos agentes en el tejido adiposo humano en lo que respecta a la diferenciación, la expresión génica, las vías metabólicas y los patrones secretores.
3. EFECTO DE LOS FÁRMACOS ANTIPSICÓTICOS DE SEGUNDA GENERACIÓN EN EL TEJIDO ADIPOSO MARRÓN
El TAM es el principal modulador de la termogénesis adaptativa que funciona para mantener la temperatura corporal en ambientes fríos. A diferencia del TAB, tal como se ha señalado anteriormente, los adipocitos marrones expresan la UCP-1 que disipa energía en forma de calor (21-23). Sin embargo, a pesar de sus diferentes funciones, la diferenciación de los adipocitos blancos y marrones está controlada por cascadas de transcripción similares (123), aunque la adipogénesis del TAM requiere la expresión de factores de transcripción adicionales como la proteína de zinc PRDM16, que forma un complejo con C/EBP- b y activa PPAR-γ2, lo que resulta en la expresión de marcadores específicos del TAM como UCP-1, coactivador gamma 1 alfa del receptor activado por proliferador de peroxisomas (del inglés, peroxisome proliferator-activated receptor-γ coactivator-1a, PGC-1α) y Cidea (124).
3.1.El impacto de los fármacos antipsicóticos de segunda generación en la termogénesis del TAM
Durante muchos años se reconoció que TAM era funcionalmente importante para la aclimatación al frío en pequeños mamíferos, como ratones y ratas, y en humanos recién nacidos. Estudios recientes que utilizan tomografía por emisión de positrones con 18F- fluorodesoxiglucosa combinada con tomografía computarizada han demostrado la existencia de áreas de TAM metabólicamente activo en humanos adultos (24, 125), que está controlado funcionalmente tanto por catecolaminas como por insulina (126). Mientras que la diferenciación de los adipocitos marrones aumenta el gasto energético y contribuye a la reducción del peso corporal (127), la disminución de la adipogénesis de los adipocitos marrones está relacionada con la obesidad y la resistencia a la insulina (128).
En un intento por descifrar la contribución de los adipocitos marrones en el aumento de peso inducido por los FASG, Oh et al. diferenciaron una línea celular de adipocitos marrones murinos en presencia de clozapina (40 µM), quetiapina (30 µM) o ziprasidona (10 µM). En el día 8 de diferenciación, la clozapina inhibió casi por completo la expresión de PRDM16, UCP-1 y Cidea. La quetiapina también redujo la expresión de estos genes, pero su efecto fue menos potente. Además, el tratamiento con ziprasidona inhibió la expresión de PPAR-γ al inicio, pero no al final del proceso de diferenciación. Estos hallazgos se confirmaron con la tinción de lípidos con Oil Red-O, que mostró una inhibición casi completa de la adipogénesis marrón con clozapina, una inhibición moderada con quetiapina y ausencia de inhibición con ziprasidona (129). Paralelamente al programa termogénico, es bien conocido en roedores que los adipocitos marrones se diferencian al final de la vida fetal a través de un programa adipogénico relacionado con la síntesis de lípidos y la expresión de enzimas lipogénicas, lo que resulta en un fenotipo de gotas de grasa multiloculares (130). En el estudio mencionado anteriormente, la clozapina redujo los niveles de expresión de los genes que codifican las proteínas ACC, SCD1, GLUT4, la proteína de adipocitos 2 (AP2) y CD36, pero no FAS. En contraste con la respuesta de la clozapina, la quetiapina aumentó los niveles de ACC y FAS, mientras que el tratamiento con ziprasidona no moduló la expresión de los genes lipogénicos (129) (Figura 2A). Es importante mencionar que los efectos de los FASG en la diferenciación de los adipocitos marrones se correlacionan con su capacidad para inducir el aumento de peso en personas (clozapina> quetiapina> ziprasidona). En general, estos resultados apoyan la hipótesis de que la inhibición de la adipogénesis del TAM puede ser un mecanismo por el cual los FASG inducen un aumento de peso como efecto secundario (129). En contraste con estos hallazgos, Ota et al. encontraron que ratas macho Sprague-Dawley tratadas por vía subcutánea con risperidona (0,1 mg/kg/día) durante 21 días presentaron hipotermia sin cambios en los programas de expresión génica relacionados con adipogénesis, lipogénesis o termogénesis en el TAM (131).
Como se mencionó anteriormente, se cree que la hiperfagia es la principal causa del aumento de peso inducido por los tratamientos de corta duración con los FASG. Sin embargo, en el tratamiento a largo plazo, cuando se normaliza la ingesta de alimentos, es probable que el aumento del peso corporal sea consecuencia de una reducción en el gasto energético debido a la disminución de la termogénesis y la actividad locomotora. Zhang y sus colaboradores encontraron en ratas hembra Sprague-Dawley que un tratamiento de 34 días con olanzapina (1 mg/kg, 3 veces al día) mezclada con masa de galletas reduce significativamente la temperatura del TAM analizada entre 45 y 150 minutos después del tratamiento. Esta disminución se asoció con una reducción de los niveles de UCP-1 y PGC-1a y una menor abundancia de adipocitos marrones, lo que sugiere que la olanzapina induce alteraciones morfológicas del TAM, así como de su función termogénica (132). Además de estos estudios, cabe señalar que recientemente se ha identificado un vínculo directo entre la hiperfagia y la termogénesis del TAM. Tras la ingesta, un aumento en la circulación de la hormona intestinal secretina activa la termogénesis del TAM al unirse a su receptor en los adipocitos marrones, estimulando así la lipólisis que se detecta en el cerebro y promueve la saciedad (133). Queda por dilucidar si este mecanismo regulador se ve afectado por los tratamientos con los FASG.
La demostración del metabolismo activo del TAM en humanos adultos (24-26, 125, 134) ha conducido a la hipótesis de que la producción de calor en el TAM podría contribuir a la hipertermia emocional. De hecho, recientemente se ha demostrado que el estrés psicológico leve activa la termogénesis del TAM en humanos adultos (135). En relación con este aspecto, Blessing et al. realizaron un estudio en ratas macho Sprague-Dawley tratadas por vía subcutánea o intraperitoneal (catéter) con el FAPG clorpromazina (0,1–5 mg/kg) o con los FASG clozapina (30 µg–2 mg/kg) o risperidona (6,25 µg–1 mg /kg) y expuestos a una rata desconocida (intrusa). El objetivo del estudio era comprender el efecto de los agentes antipsicóticos sobre la hipertermia emocional, que activa la termogénesis del TAM y la constricción de la arteria de la cola. Todos los antipsicóticos probados redujeron significativamente la termogénesis del TAM provocada por la rata intrusa, y la vasocontricción de la cola de la arteria, disminuyendo la hipertermia emocional de una manera dependiente de la dosis (136). Además, todas las dosis necesarias para provocar un efecto sobre la capacidad termogénica del TAM fueron inferiores a las que afectan a los parámetros cardiovasculares inducidos por el estrés en campo abierto (137), la respuesta de sobresalto acústico (138) o la inhibición previa al pulso (139-141), lo que indica una mayor sensibilidad del TAM a estos fármacos. Aunque el antagonista D2 clorpromazina podría inhibir las acciones termorreguladoras de manera similar a la clozapina o la risperidona, esto no significa necesariamente que el mecanismo de acción dependa del bloqueo del receptor D2, como lo demuestra un estudio previo con el FAPG haloperidol, otro un potente antagonista D2, que no logró mostrar un efecto agudo sobre la temperatura corporal en reposo cuando se administró por vía subcutánea a ratas Sprague-Dawley macho en una dosis única (hasta 3 mg/kg) (142). Blessing et al. también analizaron el potente y selectivo antagonista D2 racloprida y descubrieron que no reducía la termogénesis del TAM provocada por la rata intrusa (136). Por el contrario, la acción hipotérmica del agonista D2 apomorfina fue contrarrestada por el haloperidol de manera dosis dependiente (142). De manera similar, una dosis baja del antagonista D2 espiperona atenuó la hipotermia inducida por apomorfina, así como la inhibición de la termogénesis del TAM mediada por el quinpirol (agonista D2). Además, la espiperona disminuyó la vasoconstricción de la arteria de la cola inducida por la clozapina (143). Todos estos hallazgos sugieren que quizás la estimulación, y no el bloqueo, de los receptores de dopamina D2 en el SNC reduce la temperatura corporal (136, 143). Además, los estudios farmacológicos demostraron que los agonistas de 5-HT1A y los antagonistas de 5-HT2A reducen la termogénesis del TAM y la vasoconstricción cutánea, contribuyendo así a la hipotermia (144, 145), y también que los agonistas de la señalización adrenérgica a2 reducen la termogénesis TAM (146). Por el contrario, la activación de los receptores H1 en las neuronas productoras de histamina aumenta la temperatura corporal en paralelo a la estimulación del sistema de excitación ascendente (147). Debido a su cinética de unión, la clozapina y la risperidona actúan a través de todas las familias de receptores antes mencionadas, lo que permite hipotetizar que la influencia de estos FASG en la función termogénica del TAM puede ser consecuencia de interacciones sinérgicas entre ellos (136) (Figura 2B). Estas observaciones son aún preliminares y se necesitan más investigaciones para comprender cómo los FASG impactan en la termogénesis del TAM.
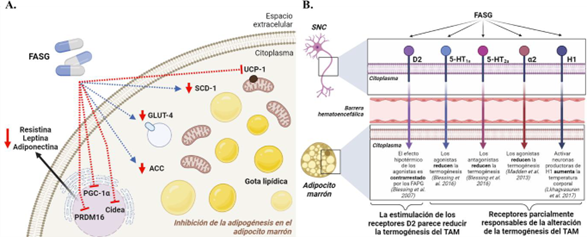
Figura 2. Efecto de los FASG en la adipogénesis de los adipocitos marrones y en la función termogénica del TAM. A. Esquema de los efectos celulares moleculares autónomos de los FASG en los adipocitos marrones, centrándose en sus efectos en las proteínas relacionadas con el metabolismo lipídico y la termogénesis, así como en la inhibición de la adipogénesis marrón. B. Receptores en el sistema nervioso central (SNC) a través de los cuales actúan los FASG
La activación del TAM puede ser crítica para las diferencias de género encontradas en el aumento de peso tras el tratamiento con FASG. Esta hipótesis fue probada por Ferno et al. en ratas macho Sprague-Dawley tratadas con dos inyecciones intramusculares de olanzapina disponible comercialmente (ZypAdheras®, 100 mg/kg) en los días 0 y 9, y sacrificadas el día 17 después de la primera inyección (148). Los autores utilizaron la dosis que previamente habían utilizado para inducir hiperfagia en ratas hembra Sprague-Dawley (149). También un segundo grupo de animales alimentados con una dieta rica en grasas fueron tratados del mismo modo. Los autores encontraron que la olanzapina aumenta transitoriamente la ingesta de alimentos en los días posteriores a su administración, con un efecto prolongado en el grupo alimentado con una dieta estándar. Sin embargo, a pesar de la inducción de hiperfagia temporal, los grupos tratados con olanzapina (independientemente de la dieta) presentaron una disminución en el peso corporal, lo que sugiere una reducción de la eficiencia alimenticia. Sin embargo, el tratamiento no logró modificar los niveles de expresión génica de UCP-1 y de PGC-1a en el TAM de los grupos experimentales, lo que probablemente excluye un efecto mediado por un aumento de la actividad del TAM. Los autores también trataron otra cohorte de animales con una única administración a dosis más elevadas de la misma formulación de olanzapina (150, 200 o 250 mg/kg) y los sacrificaron 13 días después de la inyección. Este tratamiento agudo también provocó un aumento transitorio de la ingesta acompañado de una disminución en el aumento de peso acumulado que, a diferencia del tratamiento anterior, podría explicarse por un incremento de la expresión de UCP-1 y PGC- 1a en el TAM de los animales tratados con olanzapina, indicando un aumento en el gasto energético (148). Estos resultados son contrarios a los reportados en hembras (149, 150). Por ejemplo, Skrede et al. describieron que el tratamiento de ratas hembra Sprague-Dawley con olanzapina (6 mg/kg) dos veces al día mediante sonda oral durante 13 días disminuyó tanto la UCP-1 como la PGC-1a en el TAM y provocó un aumento de peso mayor. Es de destacar que cuando los animales fueron tratados con aripiprazol en la mismas condiciones experimentales, únicamente disminuyó la expresión de PGC-1a (151). En otro estudio (150), ratas hembra Sprague-Dawley tratadas por vía oral con olanzapina (6 mg/kg/día) durante 24 días mostraron un aumento transitorio en la ingesta de alimentos conjuntamente con un mayor aumento del peso corporal a lo largo del tratamiento, en comparación con las ratas no tratadas.
Del mismo modo, en ratas con alimentación controlada que recibieron olanzapina, el peso corporal aumentó a lo largo del tratamiento en comparación con las ratas tratadas con el vehículo, en paralelo a una disminución del gasto energético, una reducción en la temperatura del TAM y una disminución en la expresión de la UCP-1. Curiosamente, se encontró un aumento en la proteína FOS en las neuronas de la médula espinal que proyectan en el tronco cerebral e hipotálamo, sugiriendo su activación excitatoria, y se observó también que algunas de estas neuronas, específicamente las ubicadas en la región perifornical del hipotálamo lateral, eran positivas para la orexina A (150), un neuropeptido clave en las neuronas del hipotálamo lateral dirigidas al TAM (152). En un estudio anterior, la activación de estas neuronas tras el tratamiento con olanzapina se asoció con hiperfagia, probablemente debido a que sus proyecciones se dirigen a las áreas asociadas con el hambre en la corteza cerebral (153). Además, las mismas neuronas orexina A-positivas en la región perifornical del hipotálamo proyectan sus axones a regiones relacionadas con la ingesta de alimentos y la termogénesis (154), lo que corrobora que la activación de estas neuronas por la olanzapina proporciona una posible explicación de los cambios mediados por los FASG tanto en la ingesta como en el gasto energético, provocando así al aumento de peso. Otro posible factor relacionado con la disminución de la termogénesis por parte de los FASG es la estimulación de diferentes subregiones del hipotálamo lateral que pueden inducir respuestas inhibidoras, limitando la actividad del nervio simpático (155), aunque esto aún no se ha analizado en el TAM. En este escenario, la respuesta a la olanzapina mediaría acciones inhibidoras a nivel de la médula espinal; sin embargo, se necesitan más estudios para comprender completamente la participación del eje hipotálamo-TAM en los efectos secundarios inducidos por los FASG (150). Además, se ha demostrado que S100B y su chaperona calsyntenin 3b influyen positivamente en la inervación simpática del TAM, mientras que su deficiencia predispone a los ratones a la obesidad inducida por la dieta (156). Como se mencionó anteriormente, el S100B aumenta en el plasma de mujeres bajo tratamiento con clozapina (114); sin embargo, cabe mencionar que una elevación de S100B circulante no significa necesariamente un efecto positivo en la inervación funcional del TAM, ya que esta adipoquina puede activar células inmunes innatas mediante la interacción con receptores para productos finales de glicación avanzada (del inglés, Receptor for Advanced Glycation Endproducts, RAGE) (157), lo que podría resultar en una sensibilidad defectuosa a la insulina y una función termogénica alterada en el TAM (158). En este sentido, se requieren más estudios para dilucidar el efecto de la elevación plasmática de S100B en el tratamiento con los FASG.
Sin embargo, otros estudios describen diferentes efectos de los FASG sobre la expresión de los genes termogénicos del TAM. El tratamiento de ratones hembra C57Bl/6J con risperidona durante 3 semanas (4 mg/kg/día, vía intraperitoneal) aumenta la ingesta y el peso corporal, asociada a una reducción de la actividad locomotora en los primeros 2 días después de la primera inyección, sin producir alteraciones en la temperatura corporal central en este período de tiempo (159). Sin embargo, al final de la tercera semana, la temperatura corporal de los animales que recibieron risperidona aumentó, y esto se asoció con una menor actividad locomotora durante la fase de oscuridad. Además, la risperidona elevó la expresión de UCP- 1 en el TAM y UCP-3 en el músculo gastrocnemio, mientras que el ARNm que codifica la orexina A disminuyó en el hipotálamo. Estos resultados sugieren que el aumento de peso inducido por la risperidona en ratones es una consecuencia de una mayor ingesta y una actividad reducida, mientras que la mayor temperatura corporal puede ser el resultado de la termogénesis inducida por la dieta y un aumento de UCP-1 y UCP-3 junto con una reducción en la expresión hipotalámica de la orexina A (159). Por el contrario, utilizando ratas macho Sprague-Dawley tratadas con olanzapina (1 mg/kg/día) suplementada en la dieta durante 6 semanas, Minet-Ringuet et al. no detectaron alteraciones en la termogénesis mitocondrial en el TAM de los animales tratados con olanzapina. Incluso en presencia de guanosina difosfato, que inhibe la UCP-1, las tasas respiratorias a diferentes potenciales de membrana no mostraron alteraciones en la conductancia de protones después del tratamiento (160). Teniendo en cuenta los resultados aparentemente contradictorios en algunos de estos estudios, no podemos descartar que estos resultados puedan estar relacionados, al menos en parte, con las diferentes afinidades de unión, dosis y/o rutas de administración de los FASG evaluados.
Por otra parte, se han probado tratamientos combinatorios para contrarrestar los efectos secundarios de los FASG en la función del TAM. En concreto, y dado que se identificó el antagonismo de estos fármacos con los receptores histaminérgicos como uno de los principales contribuyentes a las alteraciones del peso corporal, se probó betahistina, agonista del receptor H1 y antagonista del receptor H3, en ratas hembra Sprague-Dawley como cotratamiento con la olanzapina (3 mg/kg/día en masa de galletas) durante 3 semanas después de 23 días de tratamiento con olanzapina y posteriormente seguido de 19 días de lavado (161). El contratamiento con betahistina redujo en un (45 % la ganancia de peso corporal inducida por olanzapina y contrarrestó los aumentos inducidos por la olanzapina del receptor H1 y de la fosforilación de AMPKa en el hipotálamo, así como la disminución de UCP-1 y PGC-1a en el TAM. Es importante destacar que este trabajo condujo a la hipótesis de que la activación de AMPK inducida por olanzapina en el hipotálamo media los desequilibrios de peso al disminuir la termogénesis del TAM a través del receptor hipotalámico H1 y la vía de AMPK (161). Estos resultados concuerdan con los efectos observados tras la activación específica de la AMPK hipotalámica en el núcleo ventromedial del hipotálamo, que contrarresta el efecto central de la hormona tiroidea triyodotironina en la activación del TAM (162). Otro cotratamiento testado es el antagonista b-adrenérgico no selectivo propranolol, que mitigó las alteraciones inducidas por risperidona (0.75 mg/kg/día, sonda gástrica durante 4 semanas) en el TAM en hembras C57BL/6 J al aumentar los niveles de UCP-1 y PGC-1a (163).
3.2. El impacto de los fármacos antipsicóticos de segunda generación en la expresión de citoquinas en el TAM
Más allá del impacto de los FASG en la adipogénesis, lipogénesis y función del TAM, el estudio antes mencionado de Oh et al. (129) demostró que la diferenciación de preadipocitos marrones murinos en presencia de clozapina también modula la expresión de adipoquinas al reducir la expresión de resistina, leptina y adiponectina. Se observó el mismo resultado para los niveles de resistina y leptina en las células tratadas con quetiapina, mientras que la ziprasidona no alteró la expresión de adipoquinas. Nuevamente, el efecto de estos FASG sobre la expresión de leptina refleja su capacidad para inducir un aumento de peso (129). Es interesante destacar que la síntesis y secreción de leptina está modulada por la insulina (164) y se ha reportado que el tratamiento con FASG interfiere con la señalización de la insulina (165), aunque esto aún no se ha descrito particularmente en el TAM. De esta manera se podría plantear que el impacto de los FASG en la respuesta a la insulina en el TAM podría favorecer la reducción de los niveles de leptina (129). Además, también se debe considerar el papel de la UCP-1 en el aumento del efecto anorexigénico de la leptina (166), lo que sugiere que, en sinergia con la inhibición de la expresión de la leptina, la clozapina también reduce las acciones de la leptina mediante la inhibición de la expresión de la UCP-1 (129). Independientemente de estos estudios, algunos trabajos sugieren que la clozapina aumenta los niveles séricos de esta adipoquina en individuos con esquizofrenia (167). De interés, el trabajo de Zhang et al. en ratas tratadas con olanzapina mencionado anteriormente mostró una mayor infiltración de macrófagos en el TAM, que era acompañada de un incremento de los niveles de las citoquinas inflamatorias TNF-a, IL-1b e IL-6, lo que sugiere que la olanzapina puede desencadenar una inflamación periférica. Además, estas citoquinas inflamatorias también están incrementadas en el hipotálamo (96), un efecto posiblemente relacionado con una disminución de la termogénesis, como se ha descrito en otros contextos (168).
4. EFECTO DE LOS FÁRMACOS ANTIPSICÓTICOS DE SEGUNDA GENERACIÓN SOBRE EL TEJIDO ADIPOSO BEIGE
El tejido adiposo beige se encuentra dentro de los depósitos blancos, sobre todo en los depósitos subcutáneos. Al igual que los adipocitos del TAM, los adipocitos beige se caracterizan por tener un alto número de mitocondrias y por su capacidad para disipar energía mediante la termogénesis debido a los considerables niveles de UCP-1 (16). Kristof et al. demostraron que el tratamiento ex vivo de ASC humanas aisladas de voluntarios sanos de 20 a 65 años (IMC <29,9) con clozapina (100 ng/ml) durante la diferenciación aumentó 10 veces los niveles de expresión de UCP-1, apuntando a un proceso de beiging/pardeamiento de estos precursores (169). Este aumento de los niveles de ARNm de UCP-1 se asoció con una mayor expresión de otros genes marcadores de beiging, incluido TBX1 (T-box transcription factor 1). De hecho, el tratamiento con clozapina de las ASC en diferenciación resultó en un aumento de 1,5 veces en el número de células beige. Este patrón de expresión génica relacionada con el pardeamiento se reguló positivamente incluso cuando las células fueron tratadas únicamente en los últimos 2 o 4 días del proceso de diferenciación, lo que sugiere que la clozapina puede promover la transición de adipocitos blancos a beige en las últimas etapas de diferenciación. Sin embargo, a pesar del aumento del beiging, las células no activaron la termogénesis basal. Independientemente de estos efectos, la clozapina añadida durante 12 h a adipocitos ASC ya diferenciados no alteró el perfil de expresión de genes marcadores de beiging. Esto sugiere que existe una etapa crítica del proceso de diferenciación en la que la clozapina compromete el paso de los progenitores de adipocitos mesenquimales a adipocitos beige.
Además, las gotas lipídicas encontradas en las células diferenciadas tratadas con clozapina eran más pequeñas y presentaban una distribución multiocular, lo que se asoció con niveles elevados de ADN mitocondrial, pero con una sensibilidad disminuida a la estimulación con AMPc (169). En este sentido, se ha encontrado una señalización defectuosa de AMPc en el cerebro de humanos (170) y ratones (171) bajo tratamiento con FASG. Por el contrario, y como se mencionó anteriormente, el tratamiento de los adipocitos blancos con olanzapina durante la diferenciación aumenta la acumulación de lípidos. Además, la diferenciación de ASC humanas en presencia de olanzapina, ziprasidona, risperidona, quetiapina, haloperidol (todos utilizados a 50 ng/ml) o aripiprazol (100 ng/ml) no alteró la expresión de UCP-1 (169). Es interesante destacar que se ha descrito que las elevaciones de los niveles periféricos de serotonina en ratones obesos inhiben el beiging, así como la sensibilidad de las células beige y marrones al estímulo termogénico de una manera celular autónoma (172, 173). Además, los niveles elevados de serotonina periférica (174) y los polimorfismos en el gen que codifica el enzima triptófano hidroxilasa 1, que cataliza el paso limitante de la producción de serotonina fuera del SNC, se asocian con la obesidad (175). En este sentido, Kristof et al. demostraron la importancia de la señalización de la serotonina al tratar conjuntamente los adipocitos con serotonina exógena y clozapina durante la diferenciación. Estos investigadores encontraron una reducción parcial del beiging causado por la menor expresión de genes relacionados con este proceso. Es interesante destacar que se encontró que el receptor serotoninérgico HTR2A estaba inducido en los adipocitos tratados con clozapina (169), y que recientemente se ha descrito que la activación de HTR2A conduce a una señalización mediada por Gq capaz de suprimir el beiging en ratones y humanos (176). De hecho, la expresión de la proteína Gq se correlaciona negativamente con los niveles de UCP-1 en el TAB en ratones. Teniendo en cuenta todos estos estudios, y considerando que la clozapina antagoniza varios receptores serotoninérgicos con alta afinidad específica por HTR2A (177), la alteración en la señalización Gq inducida por la serotonina puede, al menos en parte, explicar los resultados no esperados de Kristof y colaboradores. Es interesante destacar que la clozapina es un agonista bien conocido de los receptores H4 (178,179) que se expresan abundantemente en los adipocitos. En un estudio reciente en el que estos receptores fueron inactivados en el TAB subcutáneo se observó una reducción en el pardeamiento y la lipólisis inducidos por el frío. Por el contrario, cuando se inyectó 4-metil histamina, un agonista selectivo del receptor H4 en el TAB subcutáneo, el pardeamiento aumentó mediante la activación de las vías de señalización mediadas por las quinasas p38MAPK y ERK1/2, junto con una aceleración de la tasa metabólica y la tolerancia a la hipotermia (180). Teniendo en cuenta estos resultados, el aumento del pardeamiento en las ASC tratadas con clozapina descrito por Kristof et al. podría ser una consecuencia, al menos en parte, de la activación de los receptores H4. Sin embargo, se necesitan más estudios para comprender el impacto preciso de los FASG en los adipocitos beige y el proceso de beiging. En este sentido, el estudio de Kristof et al. (169) es un excelente punto de partida. Además, destaca vías de señalización específicas que podrían afectar la diferenciación de los adipocitos por la clozapina, con especial importancia a la señalización serotoninérgica y b-adrenérgica, apuntando a un posible enfoque combinando ambos abordajes para mejorar los efectos adversos de estos fármacos en los adipocitos beige.
5. CONCLUSIONES
El estudio de los depósitos de tejido adiposo como tejidos metabólicamente activos y órganos endocrinos que coordinan las acciones periféricas de la insulina, la inflamación y el gasto energético es un campo relativamente novedoso. En los últimos años, se están realizando muchos esfuerzos para comprender la relevancia de los tejidos adiposos como desencadenantes de alteraciones metabólicas y/o posibles objetivos terapéuticos para diversas patologías metabólicas, incluidas las asociadas con el tratamiento de los FASG. En esta revisión, hemos intentado ilustrar la modulación del destino celular de los adipocitos y sus funciones metabólicas y endocrinas mediadas por los FASG, tanto en sistemas celulares como en estudios preclínicos y clínicos. Estos fármacos pueden modular la diferenciación de los adipocitos blancos, lo que resultada en una mayor acumulación de lípidos y una adiposidad que se relaciona con las manifestaciones clínicas de los individuos bajo terapia con FASG de una manera aparentemente dependiente del sexo. Además, los FASG exacerban los procesos inflamatorios crónicos de bajo grado del TAB al aumentar la expresión y secreción de citoquinas proinflamatorias y el reclutamiento de células inmunes, lo que se asocia con patrones secretores alterados de adipoquinas que se correlacionan a su vez con la obesidad, la resistencia a la insulina y la hiperfagia. Estos procesos son claramente los principales contribuyentes a las disfunciones metabólicas asociadas a la terapia con FASG. Además, el tratamiento con estos fármacos afecta la adipogénesis marrón y la homeostasis del TAM a través de su capacidad para modular el programa de diferenciación termogénica mediante el control de la expresión de UCP-1, también de manera dependiente del sexo. De manera similar, los FASG, en particular la clozapina, alteran el beiging de los adipocitos blancos. Hasta la fecha, sólo un estudio ha examinado este depósito adiposo en particular, pero podría ser un factor crucial en los efectos secundarios durante los tratamientos con FASG. Otro tema importante pero menos estudiado es cómo los FASG afectan el eje SNC-tejido adiposo. Las salidas centrales de este eje son los principales coordinadores de la función y la homeostasis del tejido adiposo periférico al suministrar neurotransmisores y, en este sentido, las alteraciones en los circuitos neuronales que controlan el gasto energético o la ingesta de alimentos potencian las alteraciones periféricas en sinergia con los efectos autónomos celulares específicos de los FASG. Finalmente, los déficits metabólicos en individuos bajo tratamiento con FASG podrían deberse a una intercomunicación defectuosa entre la grasa y el hígado/músculo esquelético, que son tejidos clave para la lipogénesis de novo o la captación de glucosa, respectivamente.
En conclusión, a pesar de los grandes avances logrados en nuestra comprensión de las alteraciones metabólicas de los tratamientos con los FASG, se requieren más investigaciones para abordar completamente el impacto de estos fármacos en los diferentes depósitos de tejido adiposo del organismo y el interactoma entre ellos ya sea por moléculas inflamatorias, adipoquinas y/u otros compuestos activadores o inhibidores, así como las conexiones entre el SNC y el tejido adiposo.
LISTA DE ABREVIATURAS
ACS, células madre derivadas del tejido adiposo (del inglés, adipose-derived stem cells); AMPK, proteína quinasa activada por AMP (del inglés, adenosine monophosphate-activated protein kinase); ATGL, triglicérido lipasa del tejido adiposo (del inglés, adipose triglyceride lipase); Insig, gen inducido por la insulina (del inglés, Insulin-induced gene); FAPG, fármacos antipsicóticos de primera generación; FAS, ácido graso sintasa (del inglés, Fatty acid synthase); FASG, fármacos antipsicóticos de segunda generación; HSL, lipasa sensible a hormonas (del inglés, hormone-sensitive lipase); IBMX, 3-isobutil-1-metilxantina; MCP- 1, proteína quimioatrayente de monocitos 1 (del inglés, Monocyte chemoattractant protein- 1); MSC, células madre mesenquimales; PLIN, perilipina; PGC-1a, coactivador gamma 1 alfa del receptor activado por proliferador de peroxisomas (del inglés, peroxisome proliferator-activated receptor-γ coactivator-1a); PPAR-γ, receptor activado por el proliferador de peroxisomas gamma (del inglés, peroxisome proliferator-activated receptor gamma); SCAP, proteína activadora de escisión de SREBP (del inglés, SREBP cleavage- activating protein); SNC, sistema nervioso central; SREBP, proteínas reguladoras de elementos de unión a esteroles (del inglés, Sterol regulatory element-binding proteins); T2D, diabetes mellitus tipo 2; TAB, tejido adiposo blanco; TAM, tejido adiposo marrón; TG, triglicéridos; UCP-1, Proteína desacomplante 1 (del inglés, Uncoupling protein 1).
6. REFERENCIAS
- Rosen, W.G., et al., Positive and negative symptoms in schizophrenia. Psychiatry Res, 1984. 13(4): p. 277-84.
- Carra, G., et al., Positive and negative symptoms in schizophrenia: A longitudinal analysis using latent variable structural equation modelling. Schizophr Res, 2019. 204: p. 58-64, DOI: 10.1016/j.schres.2018.08.018.
- Fleischhacker, W.W., et al., Schizophrenia—time to commit to policy change.
- Schizophr Bull, 2014. 40 Suppl 3: p. S165-94, DOI: 10.1093/schbul/sbu006.
- Patel, K.R., et al., Schizophrenia: overview and treatment options. P T, 2014. 39(9):p. 638-45.
- Remington, G., et al., Guidelines for the Pharmacotherapy of Schizophrenia in Adults. Can J Psychiatry, 2017. 62(9): p. 604-616, DOI: 10.1177/0706743717720448.
- Brisch, R., et al., The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Front Psychiatry, 2014. 5: p. 47, DOI: 10.3389/fpsyt.2014.00047.
- Shen, L.H., M.H. Liao, and Y.C. Tseng, Recent advances in imaging of dopaminergic neurons for evaluation of neuropsychiatric disorders. J Biomed Biotechnol, 2012. 2012: p. 259349, DOI: 10.1155/2012/259349.
- Howes, O.D. and R.M. Murray, Schizophrenia: an integrated sociodevelopmental- cognitive model. Lancet, 2014. 383(9929): p. 1677-1687, DOI: 10.1016/S0140- 6736(13)62036-X.
- O’Donnell, P. and A.A. Grace, Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull, 1998. 24(2): p. 267-83, DOI: 10.1093/oxfordjournals.schbul.a033325.
- Simpson, E.H., et al., Selective overexpression of dopamine D3 receptors in the striatum disrupts motivation but not cognition. Biol Psychiatry, 2014. 76(10): p. 823- 31, DOI: 10.1016/j.biopsych.2013.11.023.
- Lally, J. and J.H. MacCabe, Antipsychotic medication in schizophrenia: a review. Br Med Bull, 2015. 114(1): p. 169-79, DOI: 10.1093/bmb/ldv017.
- Cohen, D., et al., Hyperglycemia and diabetes in patients with schizophrenia or schizoaffective disorders. Diabetes Care, 2006. 29(4): p. 786-91.
- Casey, D.E., Dyslipidemia and atypical antipsychotic drugs. J Clin Psychiatry, 2004. 65 Suppl 18: p. 27-35.
- Kurzthaler, I. and W.W. Fleischhacker, The clinical implications of weight gain in schizophrenia. J Clin Psychiatry, 2001. 62 Suppl 7: p. 32-7.
- Manu, P., et al., Weight gain and obesity in schizophrenia: epidemiology, pathobiology, and management. Acta Psychiatr Scand, 2015. 132(2): p. 97-108, DOI: 10.1111/acps.12445.
- Luo, L. and M. Liu, Adipose tissue in control of metabolism. J Endocrinol, 2016. 231(3): p. R77-R99, DOI: 10.1530/JOE-16-0211.
- Tan, C.Y. and A. Vidal-Puig, Adipose tissue expandability: the metabolic problems of obesity may arise from the inability to become more obese. Biochem Soc Trans, 2008. 36(Pt 5): p. 935-40, DOI: 10.1042/BST0360935.
- Kajimura, S., Adipose tissue in 2016: Advances in the understanding of adipose tissue biology. Nat Rev Endocrinol, 2017. 13(2): p. 69-70, DOI: 10.1038/nrendo.2016.211.
- Seale, P., et al., PRDM16 controls a brown fat/skeletal muscle switch. Nature, 2008.454(7207): p. 961-7, DOI: 10.1038/nature07182.
- Lepper, C. and C.M. Fan, Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis, 2010. 48(7): p. 424-36, DOI: 10.1002/dvg.20630.
- Aherne, W. and D. Hull, Brown adipose tissue and heat production in the newborn infant. J Pathol Bacteriol, 1966. 91(1): p. 223-34, DOI: 10.1002/path.1700910126.
- Cannon, B. and J. Nedergaard, Brown adipose tissue: function and physiological significance. Physiol Rev, 2004. 84(1): p. 277-359, DOI: 10.1152/physrev.00015.2003.
- Fedorenko, A., P.V. Lishko, and Y. Kirichok, Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell, 2012. 151(2): p. 400-13, DOI: 10.1016/j.cell.2012.09.010.
- Nedergaard, J., T. Bengtsson, and B. Cannon, Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab, 2007. 293(2): p. E444-52, DOI: 10.1152/ajpendo.00691.2006.
- Cypess, A.M., et al., Identification and importance of brown adipose tissue in adult humans. N Engl J Med, 2009. 360(15): p. 1509-17, DOI: 10.1056/NEJMoa0810780.
- Zingaretti, M.C., et al., The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J, 2009. 23(9): p. 3113-20, DOI: 10.1096/fj.09-133546.
- Wu, J., et al., Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell, 2012. 150(2): p. 366-76, DOI: 10.1016/j.cell.2012.05.016.
- Young, P., J.R. Arch, and M. Ashwell, Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett, 1984. 167(1): p. 10-4.
- Harms, M. and P. Seale, Brown and beige fat: development, function and therapeutic potential. Nat Med, 2013. 19(10): p. 1252-63, DOI: 10.1038/nm.3361.
- Petrovic, N., et al., Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem, 2010. 285(10): p. 7153-64, DOI: 10.1074/jbc.M109.053942.
- Himms-Hagen, J., et al., Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol, 2000. 279(3): p. C670-81, DOI: 10.1152/ajpcell.2000.279.3.C670.
- Barbatelli, G., et al., The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab, 2010. 298(6): p. E1244-53, DOI: 10.1152/ajpendo.00600.2009.
- Foster, M.T., et al., Subcutaneous Adipose Tissue Transplantation in Diet-Induced Obese Mice Attenuates Metabolic Dysregulation While Removal Exacerbates It. Physiol Rep, 2013. 1(2), DOI: 10.1002/phy2.15.
- Smith, U. and B.B. Kahn, Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J Intern Med, 2016. 280(5): p. 465-475, DOI: 10.1111/joim.12540.
- Starrenburg, F.C. and J.P. Bogers, How can antipsychotics cause Diabetes Mellitus? Insights based on receptor-binding profiles, humoral factors and transporter proteins. Eur Psychiatry, 2009. 24(3): p. 164-70, DOI: 10.1016/j.eurpsy.2009.01.001.
- Ye, J. and R.A. DeBose-Boyd, Regulation of cholesterol and fatty acid synthesis.Cold Spring Harb Perspect Biol, 2011. 3(7), DOI: 10.1101/cshperspect.a004754.
- Vestri, H.S., et al., Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology, 2007. 32(4): p. 765-72, DOI: 10.1038/sj.npp.1301142.
- Chen, C.C., et al., Overexpression of Insig-2 inhibits atypical antipsychotic-induced adipogenic differentiation and lipid biosynthesis in adipose-derived stem cells. Sci Rep, 2017. 7(1): p. 10901, DOI: 10.1038/s41598-017-11323-9.
- Yang, L.H., et al., Olanzapine induces SREBP-1-related adipogenesis in 3T3-L1 cells. Pharmacol Res, 2007. 56(3): p. 202-8, DOI: 10.1016/j.phrs.2007.05.007.
- Raeder, M.B., et al., SREBP activation by antipsychotic- and antidepressant-drugs in cultured human liver cells: relevance for metabolic side-effects? Mol Cell Biochem, 2006. 289(1-2): p. 167-73, DOI: 10.1007/s11010-006-9160-4.
- Guesdon, B., R.G. Denis, and D. Richard, Additive effects of olanzapine and melanin- concentrating hormone agonism on energy balance. Behav Brain Res, 2010. 207(1): p. 14-20, DOI: 10.1016/j.bbr.2009.09.032.
- Georgescu, D., et al., The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced- swim performance. J Neurosci, 2005. 25(11): p. 2933-40, DOI: 10.1523/JNEUROSCI.1714-04.2005.
- Walker, A.K., et al., A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell, 2011. 147(4): p. 840-52, DOI: 10.1016/j.cell.2011.09.045.
- Yabe, D., M.S. Brown, and J.L. Goldstein, Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc Natl Acad Sci U S A, 2002. 99(20): p. 12753-8, DOI: 10.1073/pnas.162488899.
- Liou, Y.J., et al., Gene-gene interactions of the INSIG1 and INSIG2 in metabolic syndrome in schizophrenic patients treated with atypical antipsychotics. Pharmacogenomics J, 2012. 12(1): p. 54-61, DOI: 10.1038/tpj.2010.74.
- Imai, T., et al., Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci U S A, 2004. 101(13): p. 4543-7, DOI: 10.1073/pnas.0400356101.
- Tontonoz, P. and B.M. Spiegelman, Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem, 2008. 77: p. 289-312, DOI: 10.1146/annurev.biochem.77.061307.091829.
- Moseti, D., A. Regassa, and W.K. Kim, Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int J Mol Sci, 2016. 17(1), DOI: 10.3390/ijms17010124.
- Sertie, A.L., et al., Effects of antipsychotics with different weight gain liabilities on human in vitro models of adipose tissue differentiation and metabolism. Prog Neuropsychopharmacol Biol Psychiatry, 2011. 35(8): p. 1884-90, DOI: 10.1016/j.pnpbp.2011.07.017.
- Hemmrich, K., et al., Clozapine enhances differentiation of adipocyte progenitor cells. Mol Psychiatry, 2006. 11(11): p. 980-1, DOI: 10.1038/sj.mp.4001892.
- Newcomer, J.W., Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs, 2005. 19 Suppl 1: p. 1-93, DOI: 10.2165/00023210-200519001-00001.
- Allison, D.B., et al., Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry, 1999. 156(11): p. 1686-96, DOI: 10.1176/ajp.156.11.1686.
- Wang, Y., et al., Pimozide, a novel fatty acid binding protein 4 inhibitor, promotes adipogenesis of 3T3-L1 cells by activating PPARgamma. ACS Chem Neurosci, 2015. 6(2): p. 211-8, DOI: 10.1021/cn5002107.
- Pavan, C., et al., Weight gain related to treatment with atypical antipsychotics is due to activation of PKC-beta. Pharmacogenomics J, 2010. 10(5): p. 408-17, DOI: 10.1038/tpj.2009.67.
- Nimura, S., et al., Olanzapine promotes the accumulation of lipid droplets and the expression of multiple perilipins in human adipocytes. Biochem Biophys Res Commun, 2015. 467(4): p. 906-12, DOI: 10.1016/j.bbrc.2015.10.045.
- Qiao, L., et al., Transcriptional regulation of fatty acid translocase/CD36 expression by CCAAT/enhancer-binding protein alpha. J Biol Chem, 2008. 283(14): p. 8788-95, DOI: 10.1074/jbc.M800055200.
- Saini, N., et al., Fatty acid transport protein-2 inhibitor Grassofermata/CB5 protects cells against lipid accumulation and toxicity. Biochem Biophys Res Commun, 2015. 465(3): p. 534-41, DOI: 10.1016/j.bbrc.2015.08.055.
- Hotamisligil, G.S., Inflammation, metaflammation and immunometabolic disorders. Nature, 2017. 542(7640): p. 177-185, DOI: 10.1038/nature21363.
- Yamauchi, T. and T. Kadowaki, Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond), 2008. 32 Suppl 7: p. S13-8, DOI: 10.1038/ijo.2008.233.
- Trujillo, M.E. and P.E. Scherer, Adiponectin—journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med, 2005. 257(2): p. 167- 75, DOI: 10.1111/j.1365-2796.2004.01426.x.
- Sarvari, A.K., et al., Atypical antipsychotics induce both proinflammatory and adipogenic gene expression in human adipocytes in vitro. Biochem Biophys Res Commun, 2014. 450(4): p. 1383-9, DOI: 10.1016/j.bbrc.2014.07.005.
- Tsubai, T., et al., Effects of clozapine on adipokine secretions/productions and lipid droplets in 3T3-L1 adipocytes. J Pharmacol Sci, 2017. 133(2): p. 79-87, DOI: 10.1016/j.jphs.2017.01.004.
- Friedman, J.M. and J.L. Halaas, Leptin and the regulation of body weight in mammals. Nature, 1998. 395(6704): p. 763-70, DOI: 10.1038/27376.
- Heymsfield, S.B., et al., Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA, 1999. 282(16): p. 1568-75.
- Heisler, L.K., et al., Activation of central melanocortin pathways by fenfluramine. Science, 2002. 297(5581): p. 609-11, DOI: 10.1126/science.1072327.
- Yamashita, M., et al., Expression cloning of a cDNA encoding the bovine histamine H1 receptor. Proc Natl Acad Sci U S A, 1991. 88(24): p. 11515-9, DOI: 10.1073/pnas.88.24.11515.
- He, M., C. Deng, and X.F. Huang, The role of hypothalamic H1 receptor antagonism in antipsychotic-induced weight gain. CNS Drugs, 2013. 27(6): p. 423-34, DOI: 10.1007/s40263-013-0062-1.
- Tecott, L.H., et al., Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature, 1995. 374(6522): p. 542-6, DOI: 10.1038/374542a0.
- Sakata, T., H. Yoshimatsu, and M. Kurokawa, Hypothalamic neuronal histamine: implications of its homeostatic control of energy metabolism. Nutrition, 1997. 13(5):p. 403-11.
- Makki, K., P. Froguel, and I. Wolowczuk, Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm, 2013. 2013: p. 139239, DOI: 10.1155/2013/139239.
- Ryan, A.S., et al., Plasma adiponectin and leptin levels, body composition, and glucose utilization in adult women with wide ranges of age and obesity. Diabetes Care, 2003. 26(8): p. 2383-8.
- Sugai, T., et al., Dysregulation of adipocytokines related to second-generation antipsychotics in normal fasting glucose patients with schizophrenia. J Clin Psychopharmacol, 2012. 32(3): p. 390-3, DOI: 10.1097/JCP.0b013e3182524393.
- Fantuzzi, G., Adiponectin in inflammatory and immune-mediated diseases. Cytokine, 2013. 64(1): p. 1-10, DOI: 10.1016/j.cyto.2013.06.317.
- Robinson, K., J. Prins, and B. Venkatesh, Clinical review: adiponectin biology and its role in inflammation and critical illness. Crit Care, 2011. 15(2): p. 221, DOI: 10.1186/cc10021.
- Lira, F.S., et al., Both adiponectin and interleukin-10 inhibit LPS-induced activation of the NF-kappaB pathway in 3T3-L1 adipocytes. Cytokine, 2012. 57(1): p. 98-106, DOI: 10.1016/j.cyto.2011.10.001.
- Gastaldelli, A., M. Gaggini, and R.A. DeFronzo, Role of Adipose Tissue Insulin Resistance in the Natural History of Type 2 Diabetes: Results From the San Antonio Metabolism Study. Diabetes, 2017. 66(4): p. 815-822, DOI: 10.2337/db16-1167.
- Harvey, P.D. and C.R. Bowie, Ziprasidone: efficacy, tolerability, and emerging data on wide-ranging effectiveness. Expert Opin Pharmacother, 2005. 6(2): p. 337-46, DOI: 10.1517/14656566.6.2.337.
- Hunter, S.J. and W.T. Garvey, Insulin action and insulin resistance: diseases involving defects in insulin receptors, signal transduction, and the glucose transport effector system. Am J Med, 1998. 105(4): p. 331-45.
- Smith, G.C., et al., Atypical antipsychotic drugs induce derangements in glucose homeostasis by acutely increasing glucagon secretion and hepatic glucose output in the rat. Diabetologia, 2008. 51(12): p. 2309-
- Robinson, K.A., S.Z. Yacoub Wasef, and M.G. Buse, At therapeutic concentrations, olanzapine does not affect basal or insulin-stimulated glucose transport in 3T3-L1 adipocytes. Prog Neuropsychopharmacol Biol Psychiatry, 2006. 30(1): p. 93-8, DOI: 10.1016/j.pnpbp.2005.06.008.
- Albaugh, V.L., et al., Hormonal and metabolic effects of olanzapine and clozapine related to body weight in rodents. Obesity (Silver Spring), 2006. 14(1): p. 36-51, DOI: 10.1038/oby.2006.6.
- Zugno, A.I., et al., Energy metabolism, leptin, and biochemical parameters are altered in rats subjected to the chronic administration of olanzapine. Braz J Psychiatry, 2012. 34(2): p. 168-75.
- Goudie, A.J., J.A. Smith, and J.C. Halford, Characterization of olanzapine-induced weight gain in rats. J Psychopharmacol, 2002. 16(4): p. 291-6, DOI: 10.1177/026988110201600402.
- Volpato, A.M., A.I. Zugno, and J. Quevedo, Recent evidence and potential mechanisms underlying weight gain and insulin resistance due to atypical antipsychotics. Braz J Psychiatry, 2013. 35(3): p. 295-304, DOI: 10.1590/1516-4446- 2012-1052.
- Yang, C.P., et al., Olanzapine Induced Dysmetabolic Changes Involving Tissue Chromium Mobilization in Female Rats. Int J Mol Sci, 2019. 20(3), DOI: 10.3390/ijms20030640.
- Pouzet, B., et al., Chronic treatment with antipsychotics in rats as a model for antipsychotic-induced weight gain in human. Pharmacol Biochem Behav, 2003. 75(1): p. 133-40.
- Horska, K., et al., Olanzapine-depot administration induces time-dependent changes in adipose tissue endocrine function in rats. Psychoneuroendocrinology, 2016. 73: p. 177-185, DOI: 10.1016/j.psyneuen.2016.07.218.
- Tan, W., H. Fan, and P.H. Yu, Induction of subcutaneous adipose proliferation by olanzapine in rodents. Prog Neuropsychopharmacol Biol Psychiatry, 2010. 34(6): p. 1098-103, DOI: 10.1016/j.pnpbp.2010.06.002.
- Minet-Ringuet, J., et al., Alterations of lipid metabolism and gene expression in rat adipocytes during chronic olanzapine treatment. Mol Psychiatry, 2007. 12(6): p. 562- 71, DOI: 10.1038/sj.mp.4001948.
- Duncan, G.E., S. Zorn, and J.A. Lieberman, Mechanisms of typical and atypical antipsychotic drug action in relation to dopamine and NMDA receptor hypofunction hypotheses of schizophrenia. Mol Psychiatry, 1999. 4(5): p. 418-28.
- Lafontan, M., et al., Adrenergic receptors and fat cells: differential recruitment by physiological amines and homologous regulation. Obes Res, 1995. 3 Suppl 4: p. 507S-514S.
- Victoriano, M., et al., Olanzapine-induced accumulation of adipose tissue is associated with an inflammatory state. Brain Res, 2010. 1350: p. 167-75, DOI: 10.1016/j.brainres.2010.05.060.
- Calevro, A., et al., Effects of chronic antipsychotic drug exposure on the expression of Translocator Protein and inflammatory markers in rat adipose tissue. Psychoneuroendocrinology, 2018. 95:p.28-33, DOI: 10.1016/j.psyneuen.2018.05.021.
- Rotter, V., I. Nagaev, and U. Smith, Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem, 2003. 278(46): p. 45777-84, DOI: 10.1074/jbc.M301977200.
- Li, H., et al., Chronic olanzapine administration causes metabolic syndrome through inflammatory cytokines in rodent models of insulin resistance. Sci Rep, 2019. 9(1):p. 1582, DOI: 10.1038/s41598-018-36930-y.
- Zhang, Q., et al., Effects of olanzapine on the elevation of macrophage infiltration and pro-inflammatory cytokine expression in female rats. J Psychopharmacol, 2014. 28(12): p. 1161-9, DOI: 10.1177/0269881114555250.
- Davey, K.J., et al., Gender-dependent consequences of chronic olanzapine in the rat: effects on body weight, inflammatory, metabolic and microbiota parameters. Psychopharmacology (Berl), 2012. 221(1): p. 155-69, DOI: 10.1007/s00213-011-2555-2.
- Shoelson, S.E., J. Lee, and A.B. Goldfine, Inflammation and insulin resistance. J Clin Invest, 2006. 116(7): p. 1793-801, DOI: 10.1172/JCI29069.
- Cui, D., et al., Macrophage migration inhibitory factor mediates metabolic dysfunction induced by atypical antipsychotic therapy. J Clin Invest, 2018. 128(11): p. 4997-5007, DOI: 10.1172/JCI93090.
- Coccurello, R., et al., Olanzapine (LY170053, 2-methyl-4-(4-methyl-1-piperazinyl)- 10H-thieno(2,3-b)(1,5) benzodiazepine), but not the novel atypical antipsychotic ST2472 (9-piperazin-1-ylpyrrolo(2,1-b)(1,3)benzothiazepine), chronic administration induces weight gain, hyperphagia, and metabolic dysregulation in mice. J Pharmacol Exp Ther, 2008. 326(3): p. 905-11, DOI: 10.1124/jpet.108.137240.
- Hou, P.H., et al., Long-term administration of olanzapine induces adiposity and increases hepatic fatty acid desaturation protein in female C57BL/6J mice. Iran J Basic Med Sci, 2018. 21(5): p. 495-501, DOI: 10.22038/IJBMS.2018.22759.5780.
- Li, R., et al., The Wnt Signaling Pathway Effector TCF7L2 Mediates Olanzapine- Induced Weight Gain and Insulin Resistance. Front Pharmacol, 2018. 9: p. 379, DOI: 10.3389/fphar.2018.00379.
- Albaugh, V.L., et al., Olanzapine promotes fat accumulation in male rats by decreasing physical activity, repartitioning energy and increasing adipose tissue lipogenesis while impairing lipolysis. Mol Psychiatry, 2011. 16(5): p. 569-81, DOI: 10.1038/mp.2010.33.
- Zhang, X., et al., Regulation of obesity-associated metabolic disturbance by the antipsychotic drug olanzapine: Role of the autophagy-lysosome pathway. Biochem Pharmacol, 2018. 158: p. 114-125, DOI: 10.1016/j.bcp.2018.10.001.
- Kunnari, A.M., et al., The expression of human resistin in different leucocyte lineages is modulated by LPS and TNFalpha. Regul Pept, 2009. 157(1-3): p. 57-63, DOI: 10.1016/j.regpep.2009.05.002.
- Schwartz, D.R. and M.A. Lazar, Human resistin: found in translation from mouse to man. Trends Endocrinol Metab, 2011. 22(7): p. 259-65, DOI: 10.1016/j.tem.2011.03.005.
Klemettila, J.P., et al., Resistin as an inflammatory marker in patients with schizophrenia treated with clozapine. Nord J Psychiatry, 2017. 71(2): p. 89-95, DOI: 10.1080/08039488.2016.1230649. - Sapra, M., et al., Adiposity – independent hypoadiponectinemia as a potential marker of insulin resistance and inflammation in schizophrenia patients treated with second generation antipsychotics. Schizophr Res, 2016. 174(1-3): p. 132-136, DOI: 10.1016/j.schres.2016.04.051.
- Wampers, M., et al., Differential effects of olanzapine and risperidone on plasma adiponectin levels over time: results from a 3-month prospective open-label study. Eur Neuropsychopharmacol, 2012. 22(1): p. 17-26, DOI: 10.1016/j.euroneuro.2011.03.010.
- Richards, A.A., et al., Olanzapine treatment is associated with reduced high molecular weight adiponectin in serum: a potential mechanism for olanzapine- induced insulin resistance in patients with schizophrenia. J Clin Psychopharmacol, 2006. 26(3): p. 232-7, DOI: 10.1097/01.jcp.0000218404.64619.52.
- Cohn, T.A., et al., Insulin resistance and adiponectin levels in drug-free patients with schizophrenia: A preliminary report. Can J Psychiatry, 2006. 51(6): p. 382-6, DOI: 10.1177/070674370605100608.
- van Nimwegen, L.J., et al., Hepatic insulin resistance in antipsychotic naive schizophrenic patients: stable isotope studies of glucose metabolism. J Clin Endocrinol Metab, 2008. 93(2): p. 572-7, DOI: 10.1210/jc.2007-1167.
- Xu, A., et al., Testosterone selectively reduces the high molecular weight form of adiponectin by inhibiting its secretion from adipocytes. J Biol Chem, 2005. 280(18):p. 18073-80, DOI: 10.1074/jbc.M414231200.
- O’Connell, K., J. Thakore, and K.K. Dev, Levels of S100B are raised in female patients with schizophrenia. BMC Psychiatry, 2013. 13: p. 146, DOI: 10.1186/1471- 244X-13-146.
- Steiner, J., et al., S100B Serum Levels in Schizophrenia Are Presumably Related to Visceral Obesity and Insulin Resistance. Cardiovasc Psychiatry Neurol, 2010. 2010:p. 480707, DOI: 10.1155/2010/480707.
- Liu, Y., D.C. Buck, and K.A. Neve, Novel interaction of the dopamine D2 receptor and the Ca2+ binding protein S100B: role in D2 receptor function. Mol Pharmacol, 2008. 74(2): p. 371-8, DOI: 10.1124/mol.108.044925.
- Hahn, M.K., et al., Acute effects of single-dose olanzapine on metabolic, endocrine, and inflammatory markers in healthy controls. J Clin Psychopharmacol, 2013. 33(6):p. 740-6, DOI: 10.1097/JCP.0b013e31829e8333.
- Samra, J.S., et al., Effects of physiological hypercortisolemia on the regulation of lipolysis in subcutaneous adipose tissue. J Clin Endocrinol Metab, 1998. 83(2): p. 626-31, DOI: 10.1210/jcem.83.2.4547.
- Vidarsdottir, S., et al., Orally disintegrating and oral standard olanzapine tablets similarly elevate the homeostasis model assessment of insulin resistance index and plasma triglyceride levels in 12 healthy men: a randomized crossover study. J Clin Psychiatry, 2010. 71(9): p. 1205-11, DOI: 10.4088/JCP.08m04654yel.
- Fountaine, R.J., et al., Increased food intake and energy expenditure following administration of olanzapine to healthy men. Obesity (Silver Spring), 2010. 18(8): p. 1646-51, DOI: 10.1038/oby.2010.6.
- Nicol, G.E., et al., Metabolic Effects of Antipsychotics on Adiposity and Insulin Sensitivity in Youths: A Randomized Clinical Trial. JAMA Psychiatry, 2018. 75(8):p.788-796,DOI: 10.1001/jamapsychiatry.2018.1088.
- Taveira, T.H., et al., The effect of naltrexone on body fat mass in olanzapine-treated schizophrenic or schizoaffective patients: a randomized double-blind placebo- controlled pilot study. J Psychopharmacol, 2014. 28(4): p. 395-400, DOI: 10.1177/0269881113509904.
- Kajimura, S., P. Seale, and B.M. Spiegelman, Transcriptional control of brown fat development. Cell Metab, 2010. 11(4): p. 257-62, DOI: 10.1016/j.cmet.2010.03.005.
- Kajimura, S., et al., Initiation of myoblast to brown fat switch by a PRDM16-C/EBP- beta transcriptional complex. Nature, 2009. 460(7259): p. 1154-8, DOI: 10.1038/nature08262.
- van Marken Lichtenbelt, W.D., et al., Cold-activated brown adipose tissue in healthy men. N Engl J Med, 2009. 360(15): p. 1500-8, DOI: 10.1056/NEJMoa0808718.
- Orava, J., et al., Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab, 2011. 14(2): p. 272-9, DOI: 10.1016/j.cmet.2011.06.012.
- Tseng, Y.H., et al., New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature, 2008. 454(7207): p. 1000-4, DOI: 10.1038/nature07221.
- Yang, X., S. Enerback, and U. Smith, Reduced expression of FOXC2 and brown adipogenic genes in human subjects with insulin resistance. Obes Res, 2003. 11(10):p. 1182-91, DOI: 10.1038/oby.2003.163.
- Oh, J.E., et al., Inhibition of mouse brown adipocyte differentiation by second- generation antipsychotics. Exp Mol Med, 2012. 44(9): p. 545-53, DOI: 10.3858/emm.2012.44.9.062.
- Valverde, A.M. and M. Benito, The brown adipose cell: a unique model for understanding the molecular mechanism of insulin resistance. Mini Rev Med Chem, 2005. 5(3): p. 269-78.
- Ota, M., et al., Resistance to excessive bodyweight gain in risperidone-injected rats. Clin Exp Pharmacol Physiol, 2005. 32(4): p. 279-87, DOI: 10.1111/j.1440- 1681.2005.04184.x.
- Zhang, Q., et al., Olanzapine reduced brown adipose tissue thermogenesis and locomotor activity in female rats. Prog Neuropsychopharmacol Biol Psychiatry, 2014. 51: p. 172-80, DOI: 10.1016/j.pnpbp.2014.02.003.
- Li, Y., et al., Secretin-Activated Brown Fat Mediates Prandial Thermogenesis to Induce Satiation. Cell, 2018. 175(6): p. 1561-1574 e12, DOI: 10.1016/j.cell.2018.10.016.
- Virtanen, K.A., et al., Functional brown adipose tissue in healthy adults. N Engl J Med, 2009. 360(15): p. 1518-25, DOI: 10.1056/NEJMoa0808949.
- Robinson, L.J., et al., Brown adipose tissue activation as measured by infrared thermography by mild anticipatory psychological stress in lean healthy females. Exp Physiol, 2016. 101(4): p. 549-57, DOI: 10.1113/EP085642.
- Blessing, W.W., et al., Clozapine, chlorpromazine and risperidone dose-dependently reduce emotional hyperthermia, a biological marker of salience. Psychopharmacology (Berl), 2017. 234(21): p. 3259-3269, DOI: 10.1007/s00213- 017-4710-x.
- van den Buuse, M., Acute effects of antipsychotic drugs on cardiovascular responses to stress. Eur J Pharmacol, 2003. 464(1): p. 55-62.
- Swerdlow, N.R., et al., Effects of spiperone, raclopride, SCH 23390 and clozapine on apomorphine inhibition of sensorimotor gating of the startle response in the rat. J Pharmacol Exp Ther, 1991. 256(2): p. 530-6.
- Geyer, M.A., et al., Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl), 2001. 156(2-3): p. 117-54.
- Kollias, J. and R.W. Bullard, The Influence of Chlorpromazine on Physical and Chemical Mechanisms of Temperature Regulation in the Rat. J Pharmacol Exp Ther, 1964. 145: p. 373-81.
- Swerdlow, N.R., et al., Seroquel, clozapine and chlorpromazine restore sensorimotor gating in ketamine-treated rats. Psychopharmacology (Berl), 1998. 140(1): p. 75-80.
- Chipkin, R.E., Effects of D1 and D2 antagonists on basal and apomorphine decreased body temperature in mice and rats. Pharmacol Biochem Behav, 1988. 30(3): p. 683-6.
- Blessing, W.W. and Y. Ootsuka, Activation of dopamine D2 receptors in the CNS inhibits sympathetic cutaneous vasomotor alerting responses (SCVARs), contributing to clozapine’s SCVAR-inhibiting action. Prog Neuropsychopharmacol Biol Psychiatry, 2007. 31(2): p. 328-36, DOI: 10.1016/j.pnpbp.2006.09.005.
- Morrison, S.F. and C.J. Madden, Central nervous system regulation of brown adipose tissue. Compr Physiol, 2014. 4(4): p. 1677-713, DOI: 10.1002/cphy.c140013.
- Blessing, W., R. McAllen, and M. McKinley, Control of the Cutaneous Circulation by the Central Nervous System. Compr Physiol, 2016. 6(3): p. 1161-97, DOI: 10.1002/cphy.c150034.
- Madden, C.J., et al., alpha2 Adrenergic receptor-mediated inhibition of thermogenesis. J Neurosci, 2013. 33(5): p. 2017-28, DOI: 10.1523/JNEUROSCI.4701-12.2013.
- Lkhagvasuren, B. and T. Oka, The histaminergic system is involved in psychological stress-induced hyperthermia in rats. Physiol Rep, 2017. 5(8), DOI: 10.14814/phy2.13204.
- Ferno, J., et al., Olanzapine depot exposure in male rats: Dose-dependent lipogenic effects without concomitant weight gain. Eur Neuropsychopharmacol, 2015. 25(6): p. 923-32, DOI: 10.1016/j.euroneuro.2015.03.002.
- Skrede, S., et al., Olanzapine depot formulation in rat: a step forward in modelling antipsychotic-induced metabolic adverse effects. Int J Neuropsychopharmacol, 2014. 17(1): p. 91-104, DOI: 10.1017/S1461145713000862.
- Stefanidis, A., et al., The role of thermogenesis in antipsychotic drug-induced weight gain. Obesity (Silver Spring), 2009. 17(1): p. 16-24, DOI: 10.1038/oby.2008.468.
- Skrede, S., et al., Olanzapine, but not aripiprazole, weight-independently elevates serum triglycerides and activates lipogenic gene expression in female rats. Int J Neuropsychopharmacol, 2012.15(2):p.163-79, DOI: 10.1017/S1461145711001271.
- Oldfield, B.J., et al., The neurochemical characterisation of hypothalamic pathways projecting polysynaptically to brown adipose tissue in the rat. Neuroscience, 2002. 110(3): p. 515-26.
- Fadel, J., M. Bubser, and A.Y. Deutch, Differential activation of orexin neurons by antipsychotic drugs associated with weight gain. J Neurosci, 2002. 22(15): p. 6742- 6, DOI: 20026632.
- Oldfield, B.J., et al., Lateral hypothalamic ‘command neurons’ with axonal projections to regions involved in both feeding and thermogenesis. Eur J Neurosci, 2007. 25(8): p. 2404-12, DOI: 10.1111/j.1460-9568.2007.05429.x.
- Allen, G.V. and D.F. Cechetto, Functional and anatomical organization of cardiovascular pressor and depressor sites in the lateral hypothalamic area: I. Descending projections. J Comp Neurol, 1992. 315(3): p. 313-32, DOI: 10.1002/cne.903150307.
- Zeng, X., et al., Innervation of thermogenic adipose tissue via a calsyntenin 3beta- S100b axis. Nature, 2019. 569(7755): p. 229-235, DOI: 10.1038/s41586-019-1156-9.
- Donato, R., S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol, 2001. 33(7): p. 637-68, DOI: 10.1016/s1357-2725(01)00046-2.
- Villarroya, F., et al., Inflammation of brown/beige adipose tissues in obesity and metabolic disease. J Intern Med, 2018. 284(5): p. 492-504, DOI: 10.1111/joim.12803.
- Cope, M.B., et al., Risperidone alters food intake, core body temperature, and locomotor activity in mice. Physiol Behav, 2009. 96(3): p. 457-63, DOI: 10.1016/j.physbeh.2008.11.011.
- Minet-Ringuet, J., et al., Long term treatment with olanzapine mixed with the food in male rats induces body fat deposition with no increase in body weight and no thermogenic alteration. Appetite, 2006. 46(3): p. 254-62, DOI: 10.1016/j.appet.2006.01.008.
- Lian, J., et al., Preventing olanzapine-induced weight gain using betahistine: a study in a rat model with chronic olanzapine treatment. PLoS One, 2014. 9(8): p. e104160, DOI: 10.1371/journal.pone.0104160.
- Martinez-Sanchez, N., et al., Hypothalamic AMPK-ER Stress-JNK1 Axis Mediates the Central Actions of Thyroid Hormones on Energy Balance. Cell Metab, 2017. 26(1): p. 212-229 e12, DOI: 10.1016/j.cmet.2017.06.014.
- Motyl, K.J., et al., Propranolol Attenuates Risperidone-Induced Trabecular Bone Loss in Female Mice. Endocrinology, 2015. 156(7): p. 2374-83, DOI: 10.1210/en.2015-1099.
- Saladin, R., et al., Transient increase in obese gene expression after food intake or insulin administration. Nature, 1995. 377(6549): p. 527-9, DOI: 10.1038/377527a0.
- Cui, J., et al., Macrophage migration inhibitory factor promotes cardiac stem cell proliferation and endothelial differentiation through the activation of the PI3K/Akt/mTOR and AMPK pathways. Int J Mol Med, 2016. 37(5): p. 1299-309, DOI: 10.3892/ijmm.2016.2542.
- Okamatsu-Ogura, Y., et al., Possible involvement of uncoupling protein 1 in appetite control by leptin. Exp Biol Med (Maywood), 2011. 236(11): p. 1274-81, DOI: 10.1258/ebm.2011.011143.
- Monteleone, P., et al., Pronounced early increase in circulating leptin predicts a lower weight gain during clozapine treatment. J Clin Psychopharmacol, 2002. 22(4):p. 424-6.
- Arruda, A.P., et al., Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology, 2011. 152(4): p. 1314-26, DOI: 10.1210/en.2010-0659.
- Kristof, E., et al., Clozapine modifies the differentiation program of human adipocytes inducing browning. Transl Psychiatry, 2016. 6(11): p. e963, DOI: 10.1038/tp.2016.230.
- Elman, I., et al., Effects of risperidone on the peripheral noradrenegic system in patients with schizophrenia: a comparison with clozapine and placebo. Neuropsychopharmacology, 2002. 27(2): p. 293-300, DOI: 10.1016/S0893- 133X(02)00314-7.
- Dwivedi, Y., H.S. Rizavi, and G.N. Pandey, Differential effects of haloperidol and clozapine on ((3)H)cAMP binding, protein kinase A (PKA) activity, and mRNA and protein expression of selective regulatory and catalytic subunit isoforms of PKA in rat brain. J Pharmacol Exp Ther, 2002. 301(1): p. 197-209, DOI: 10.1124/jpet.301.1.197.
- Crane, J.D., et al., Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat Med, 2015. 21(2): p. 166-72, DOI: 10.1038/nm.3766.
- Oh, C.M., et al., Regulation of systemic energy homeostasis by serotonin in adipose tissues. Nat Commun, 2015. 6: p. 6794, DOI: 10.1038/ncomms7794.
- Kim, H.J., et al., Metabolomic analysis of livers and serum from high-fat diet induced obese mice. J Proteome Res, 2011. 10(2): p. 722-31, DOI: 10.1021/pr100892r.
- Kwak, S.H., et al., Association of variations in TPH1 and HTR2B with gestational weight gain and measures of obesity. Obesity (Silver Spring), 2012. 20(1): p. 233-8, DOI: 10.1038/oby.2011.253.
- Klepac, K., et al., The Gq signalling pathway inhibits brown and beige adipose tissue. Nat Commun, 2016. 7: p. 10895, DOI: 10.1038/ncomms10895.
- Correll, C.U., From receptor pharmacology to improved outcomes: individualising the selection, dosing, and switching of antipsychotics. Eur Psychiatry, 2010. 25 Suppl 2: p. S12-21, DOI: 10.1016/S0924-9338(10)71701-6.
- Buckland, K.F., T.J. Williams, and D.M. Conroy, Histamine induces cytoskeletal changes in human eosinophils via the H(4) receptor. Br J Pharmacol, 2003. 140(6):p. 1117-27, DOI: 10.1038/sj.bjp.0705530.
- Lim, H.D., et al., Evaluation of histamine H1-, H2-, and H3-receptor ligands at the human histamine H4 receptor: identification of 4-methylhistamine as the first potent and selective H4 receptor agonist. J Pharmacol Exp Ther, 2005. 314(3): p. 1310-21, DOI: 10.1124/jpet.105.087965.
- Zhao, Y.X., et al., Stimulation of histamine H4 receptor participates in cold-induced browning of subcutaneous white adipose tissue. Am J Physiol Endocrinol Metab, 2019, DOI: 10.1152/ajpendo.00131.2019.
