(A) TRACTO ALIMENTARIO Y METABOLISMO: Colitis ulcerosa: Etrasimod (Velsipity®; EMA/FDA). Hiperoxaluria primaria: Nedosiran (Rivfloza®; FDA). Deficiencia de arginasa-1: Pegzilarginasa (Loargys®; EMA). Polineuropatía de la amiloidosis hereditaria mediada por transtiretina: Eplontersen (Wainua®; FDA). (B) SANGRE Y SISTEMA HEMATOPOYÉTICO: Anemia de células falciformes (drepanocítica) y beta talasemia: Exagamglogene Autotemcel (Casgevy®; EMA/FDA). Anemia de células falciformes (drepanocítica): Lovotibeglogene Autotemcel (Lyfgenia®; FDA). Púrpura trombótica trombocitopénica: ADAMTS13 (Adzynma®; FDA). Profilaxis antiinfecciosa en cateterismo venoso: Taurolidina/Heparina (Defencath®; FDA). (D) DERMATOLOGÍA: Dermatitis atópica: Lebrikizumab (Ebglyss®; EMA). (H) PREPARACIONES HORMONALES SISTÉMICAS: Hipoparatiroidismo: Palopegteriparatida (Yorvipath®; EMA). (J) ANTIINFECCIOSOS SISTÉMICOS: Gripe A : Vacuna gripe A zoonótica (H5N1) (Zoonotic Influenza Vaccine Seqirus®; EMA). Virus Chikungunya: Vacuna Virus Chikungunya viva (Ixchiq®; FDA). (L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES: Leucemia mieloide aguda: Decitabina/Cedazuridina (Inaqovi®; EMA). Carcinoma nasofaríngeo: Toripalimab (Loqtorzi®; FDA). Cáncer colorrectal: Fruquintinib (Fruzaqla®; FDA). Cáncer de mama: Capivasertib (Truqap®; FDA). Cáncer de pulmón: Repotrectinib (Augtyro®; FDA). Tumores desmoides:
Nirogacestat (Ogsiveo®; FDA). Miastenia grave: Zilucoplan (Zilbrysq®; EMA/FDA). Neutropenia: Efbemelanograstim (Ryzneuta®; FDA). Hemoglobinuria nocturna paroxística: Iptacopan (Fabhalta®; FDA).(M) SISTEMA MÚSCULO-ESQUELÉTICO: Distrofia muscular de Duchenne: Vamorolona (Agamre®; EMA/FDA). (N) SISTEMA NERVIOSO: Depresión: Gepirona (Exxua®; FDA). (V) VARIOS: Hiperfosfatemia en insuficiencia renal: Tenapanor (Xphozah®; FDA).
(A) TRACTO ALIMENTARIO Y METABOLISMO
Etrasimod (Velsipity®) Pfizer (FDA, USA; EMA, UE)
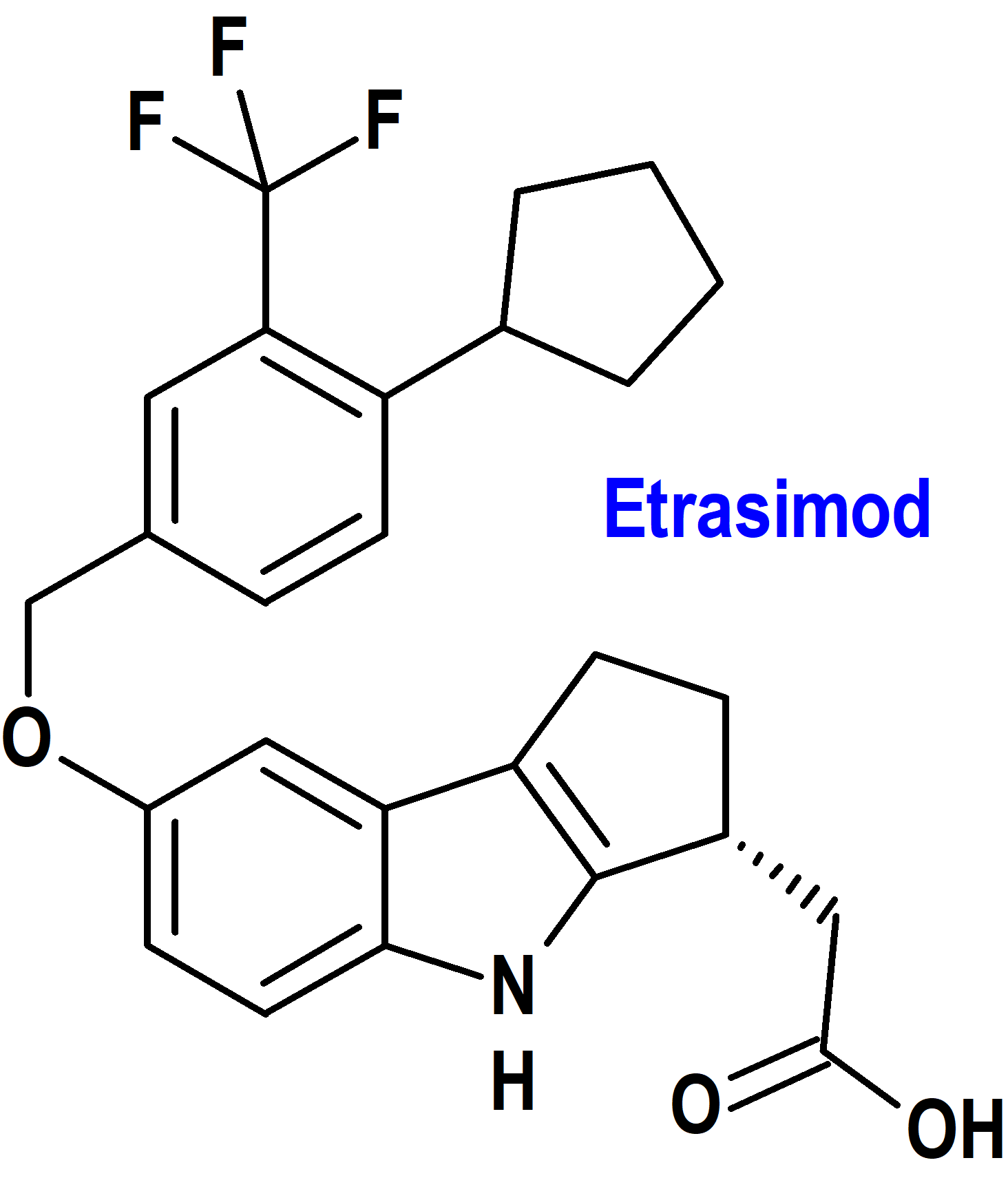
Indicación: Tratamiento de la colitis ulcerosa activa de moderada a grave en adultos.
Tipo: Medicamento sintético estándar constituido por L-arginina, (3R)-7-[[4-ciclopentil-3-(trifluoroetil)fenil]metoxi]-1,2,3,4-tetrahidrociclopent[b]indol-3-acetato. Autorizado en Estados Unidos (FDA) el 12-10-2023; evaluado favorablemente en la Unión Europea (EMA) el 14-12-2023.
Mecanismo: Modulador del receptor de esfingosina 1-fosfato (S1P) que se une con alta afinidad a los subtipos 1, 4 y 5 del receptor S1P. Bloquea parcial y reversiblemente la capacidad de los linfocitos para salir de los órganos linfoides, reduciendo el número de linfocitos en la sangre periférica. Los efectos terapéuticos parecen deberse a la reducción de la migración de linfocitos hacia el intestino.
Eficacia clínica: Dos estudios clínicos aleatorizados, doble ciego y controlados con placebo en sujetos adultos con colitis ulcerosa activa de moderada a grave que tuvieron una respuesta inadecuada, pérdida de respuesta o intolerancia a una o más opciones de tratamiento. El primero tuvo una duración de 52 semanas y el segundo de 12. En el primer estudio, la eficacia se evaluó en 408 sujetos adultos con una mMS (puntuación de Mayo modificada) inicial de 5 a 9 (mediana de 7) y los criterios de valoración coprimarios fueron la proporción de sujetos logrando la remisión clínica en la semana 12 (27 vs. 7%) y en la semana 52 (32 vs. 7%). En el segundo estudio, la eficacia se evaluó en 333 sujetos adultos con similares características a los anteriores, siendo el criterio de valoración principal la proporción de sujetos que lograron la remisión clínica en la Semana 12 (26 vs. 15%).
Eventos adversos: Los más comunes son cefalea (9%), aumento de los valores enzimáticos hepáticos (6%) y mareos (5%).
Nedosiran (Rivfloza®) Novo Nordisk (FDA, USA)
Indicación: Indicado para reducir los niveles de oxalato urinario en niños de 9 años de edad y mayores y adultos con hiperoxaluria primaria tipo 1 (PH1) y función renal relativamente conservada (eGFR ≥30 ml/min/1,73 m2).
Tipo: Medicamento biológico constituido por un pequeño ARN de interferencia (siRNA) bicatenario con cuatro residuos de N-acetil-D-galactosamina (GalNAc) unidos covalentemente. Autorizado en Estados Unidos (FDA) el 29-9-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Tras su administración subcutánea, los azúcares conjugados con GalNAc se unen a los receptores de asialoglicoproteína (ASGPR) permitiendo la incorporación de nedosiran a los hepatocitos, donde reduce los niveles de lactato deshidrogenasa hepática (LDH) mediante la degradación del ácido ribonucleico mensajero (ARNm) de LDHA mediante la interferencia del ARN. La reducción de la LDH hepática mediante nedosiran reduce la producción de oxalato en el hígado, reduciendo así la carga de oxalato.
Eficacia clínica: Un ensayo aleatorizado, doble ciego y controlado con placebo, en 35 pacientes de 6 años o más con PH1 una TFGe ≥30 ml/min/1,73 m², con una mediana de edad de 20 años (9-46). La variable principal de eficacia fue el área bajo la curva, desde los días 90 a 180, del cambio porcentual desde el valor inicial en la excreción urinaria de oxalato de 24 horas (AUC Uox 24 horas). La media de mínimos cuadrados (LS) del AUC Uox en 24 horas fue -3486 en el grupo de nedosiran en comparación con +1490 en el de placebo. El cambio porcentual medio LS desde el valor inicial en la excreción urinaria de oxalato de 24 horas, promediado durante los días 90, 120, 150 y 180, fue -37% en el grupo de nedosiran y +12% en el de placebo (diferencia de 49 puntos porcentuales).
Eventos adversos: Los más comunes son reacciones en el lugar de la inyección (39%): eritema, dolor, hematomas y erupción cutánea; en general, fueron leves y no condujeron a la interrupción del tratamiento.
Pegzilarginasa (Loargys®) Immedica (EMA, UE)
Indicación: Tratamiento la deficiencia de arginasa 1 (ARG1-D), también conocida como hiperargininemia, en adultos, adolescentes y niños a partir de 2 años de edad.
Tipo: Medicamento biológico constituido por arginasa 1 recombinante humana sustituida por cobalto, producida en células de Escherichia coli, conjugada covalentemente con metoxipolietilenglicol (mPEG). Autorizado por la Unión Europea (EMA) el 15-12-2023 como medicamento huérfano (Orphan drug); no autorizado previamente en Estados Unidos (FDA).
Mecanismo: Arginasa 1 humana recombinante sustituida por cobalto conjugada con portadores de mPEG de 5 kDa en un grado de sustitución de 6-12 moles de mPEG por mol de proteína. El portador de mPEG reduce el aclaramiento de la pegzilarginasa, lo que provoca como resultado una semivida prolongada, manteniendo al mismo tiempo las funciones de la enzima.
Pegzilarginasa se emplea para sustituir la actividad deficiente de la enzima arginasa 1 humana en pacientes con ARG1D, una enfermedad metabólica hereditaria caracterizada por la deficiencia de la enzima arginasa 1 y asociada a la elevación persistente de la arginina plasmática que conduce a las manifestaciones de la enfermedad y a la progresión de los síntomas clínicos.
Eficacia clínica: Un ensayo multicéntrico, doble ciego y controlado con placebo que incluyó a 32 sujetos pediátricos y adultos de 2 a 29 años con ARG1-D. La variable principal de eficacia midió la reducción con respecto al nivel basal de la arginina plasmática en los sujetos tratados con pegzilarginasa en comparación con placebo a la semana 24 (76,7% vs. 0%). La proporción de sujetos que alcanzaron los niveles diana de arginina recomendados por las guías (< 200 µM) fue del 90.5% vs. 0%. Las variables secundarias clave para evaluar la movilidad funcional fueron la medición de la función motora gruesa, ítem E de la GMFM (Gross Motor Function Measure; E: caminar, correr, saltar): [4,2 vs -0,4] y la prueba de marcha de 2 minutos (2MWT) [7,4 vs. 1,9 m]. Durante el periodo de extensión abierto, los sujetos que habían recibido previamente pegzilarginasa demostraron mejorías sostenidas en los niveles plasmáticos de arginina, las puntuaciones GMFM-E y 2 MWt. Los pacientes aleatorizados inicialmente a placebo y tratados con pegzilarginasa en el periodo de extensión abierto mostraron también reducciones similares con respecto al valor basal de los niveles plasmáticos medios de arginina.
Eventos adversos: El más común es la hipersensibilidad (12,5 %).
Eplontersen (Wainua®) Ionis (FDA, USA)
Indicación: Tratamiento de la polineuropatía de la amiloidosis hereditaria mediada por transtiretina (TTR) en adultos.
Tipo: Medicamento sintético estándar constituido por un oligonucleótido antisentido (ASO) unido covalentemente a un ligando que contiene tres residuos de N-acetil galactosamina (GalNAc) para permitir la entrega del ASO a los hepatocitos. Autorizado en Estados Unidos (FDA) el 21-12-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Provoca la degradación del ARNm de TTR mutante y de tipo salvaje mediante la unión al ARNm de TTR, lo que da como resultado una reducción de la proteína TTR sérica y los depósitos de proteína TTR en los tejidos.
Eficacia clínica: Un ensayo clínico multicéntrico, aleatorizado y abierto en pacientes adultos con polineuropatía causada por amiloidosis hereditaria mediada por transtiretina. Los pacientes fueron aleatorizados en una proporción de 6:1 para recibir 45 mg de eplontersen una vez cada 4 semanas (N=144) o 284 mg de inotersen una vez por semana (N=24), respectivamente, como inyecciones subcutáneas. El noventa y siete por ciento de los pacientes tratados con eplontersen y el 83% de los pacientes tratados con inotersen completaron al menos 35 semanas del tratamiento asignado. Las evaluaciones de eficacia se basaron en una comparación del grupo eplontersen con un grupo de placebo externo (N=60) en otro estudio compuesto por una población comparable de pacientes adultos. Los criterios de valoración de eficacia fueron el cambio desde el inicio hasta la semana 35 en la puntuación compuesta de la Escala de deterioro de neuropatía modificada+7 (mNIS+7) [0,2 vs. 9,2] y el cambio desde el inicio hasta la semana 35 en la puntuación total de la calidad de vida de Norfolk-Neuropatía diabética (QoL-DN) [-3,1 vs. 8,7] .
Eventos adversos: Los más comunes son disminución de vitamina A (15%), vómitos (9%), proteinuria (8%), reacciones en el lugar de la inyección (7%) y visión borrosa (6%).
(B) SANGRE Y SISTEMA HEMATOPOYÉTICO
Exagamglogene Autotemcel (Casgevy®) Vertex (FDA, USA; EMA, UE)
Indicación: Tratamiento de la anemia de células falciformes (drepanocítica) grave en pacientes de 12 años de edad y mayores con crisis vasooclusivas (COV) recurrentes para quienes es apropiado un trasplante de células madre hematopoyéticas y un antígeno leucocitario humano (HLA). Tratamiento de la b-talasemia dependiente de transfusiones en pacientes de 12 años de edad y mayores para quienes el trasplante de células madre hematopoyéticas (HSC) es apropiado y no lo es un donante de HSC emparentado con antígeno leucocitario humano (HLA). La segunda indicación solo corresponde a la Unión Europea (EMA).
Tipo: Medicamento de terapia avanzada (génica), constituido por células madre hematopoyéticas CD34+ autólogas editadas mediante tecnología CRISPR/Cas9 en la región potenciadora específica de eritroides del gen BCL11A para reducir la expresión de BCL11A en células del linaje eritroide, lo que conduce a una mayor producción de proteína de hemoglobina fetal (HbF). Autorizado en Estados Unidos (FDA) el 8-12-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); evaluado favorablemente en la Unión Europea (EMA) el 14-12-2023, con revisión prioritaria (Priority Medicines; PRIME), como medicamento huérfano (Orphan drug) y de forma condicional (Conditional marketing authorisation).
Mecanismo: Las células CD34+ editadas se injertan en la médula ósea del paciente y se diferencian en células del linaje eritroide con expresión reducida de BCL11A. La expresión reducida de BCL11A da como resultado un aumento en la expresión de g-globina y la producción de proteína HbF en células eritroides. En pacientes con anemia falciforme grave, la expresión de HbF reduce la concentración de hemoglobina S intracelular (HbS), evitando que los glóbulos rojos se hagan falciformes y abordando la causa subyacente de la enfermedad, eliminando así las crisis vasooclusivas recurrentes y aborda la hemoglobina A ausente en pacientes con b-talasemia.
Eficacia clínica: Un ensayo multicéntrico de un solo grupo todavía en curso que evalúa la seguridad y eficacia de una dosis única de CASGEVY en pacientes adultos y adolescentes con anemia de células falciformes. Los pacientes elegibles se sometieron a movilización y aféresis para recolectar células madre CD34+ para la fabricación de CASGEVY, seguido de acondicionamiento mieloablativo e infusión de CASGEVY. Luego se realizó un seguimiento de los pacientes en el ensayo 1 durante 24 meses después de la infusión de CASGEVY. Los pacientes que completen o abandonen el ensayo 1 fueron alentados a inscribirse en el ensayo 2, un ensayo de seguimiento a largo plazo en curso para un seguimiento adicional durante un total de 15 años después de la infusión de CASGEVY. En el momento del análisis intermedio, un total de 63 pacientes se inscribieron en el ensayo, de los cuales 58 (92%) comenzaron la movilización. Un total de 44 (76%) pacientes recibieron una infusión de CASGEVY y formaron el conjunto de análisis completo (FAS). Treinta y un pacientes de la FAS (70%) tuvieron un seguimiento adecuado para permitir la evaluación del criterio de valoración principal de eficacia y formaron el conjunto primario de eficacia. La variable primaria de eficacia fue la proporción de respondedores (VF12: pacientes que no experimentaron ninguna crisis vaso-oclusiva grave durante al menos 12 meses consecutivos dentro de los primeros 24 meses después de la infusión de CASGEVY en el ensayo 1. La evaluación de VF12 comenzó 60 días después de la última transfusión de glóbulos rojos para apoyo postrasplante o manejo de la ECF. La mediana del tiempo transcurrido hasta la última transfusión de glóbulos rojos fue de 19 días después de la infusión de CASGEVY para los pacientes del grupo primario de eficacia y la tasa de respuesta de VF12 fue del 93,5 %. Los pacientes que respondieron a VF12 no experimentaron crisis graves durante el período de evaluación, con una duración media de 22,2 meses en el momento del análisis intermedio.
Eventos adversos: Los más comunes son mucositis (87%), neutropenia febril (48%), reducción del apetito (41%), dolor muscoloesquelético (14%), dolor abdominal, colelitiasis y prurito (todos con el 11%).
Lovotibeglogene Autotemcel (Lyfgenia®) Bluebird Bio (FDA, USA)
Indicación: Tratamiento de pacientes de 12 años de edad o mayores con anemia de células falciformes y antecedentes de eventos vaso-oclusivos.
Tipo: Medicamento de terapia avanzada (génica), constituido por células CD34+ autólogas de pacientes con anemia de células falciformes que contienen células madre hematopoyéticas (HSC) transducidas con BB305 LVV que codifica b(A-T87Q)-globina, suspendidas en una solución de criopreservación. Autorizado en Estados Unidos (FDA) el 8-12-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); no autorizado aún en la Unión Europea (EMA).
Mecanismo: El medicamento añade copias funcionales de un gen de b(A)-globina modificado (treonina [T] reemplazada por glutamina [Q] en la posición 87, T87Q: b(A-T87Q)-globina) en las células madre hematopoyéticas (HSC) de los pacientes mediante la transducción de células CD34+ autólogas con BB305 LVV. Después de la infusión de LYFGENIA, las HSC CD34+ transducidas se injertan en la médula ósea y se diferencian para producir glóbulos rojos que contienen b(A-T87Q)-globina biológicamente activa que se combinarán con a-globina para producir hemoglobina funcional que contiene b(A-T87Q)-globina (HbAT87Q). HbAT87Q tiene una afinidad de unión al oxígeno y una curva de disociación de la hemoglobina del oxígeno similares a la HbA natural, reduce los niveles intracelulares y totales de hemoglobina S (HbS) y está diseñado para inhibir estéricamente la polimerización de HbS, limitando así la formación de células falciformes de los glóbulos rojos.
Eficacia clínica: Un estudio de Fase 1/2 multicéntrico, abierto, de un solo brazo, de 24 meses de duración y continuó en un estudio de seguimiento a largo plazo. 43 sujetos se sometieron a aféresis después de la movilización con plerixafor, de los cuales 36 pacientes recibieron acondicionamiento mieloablativo con busulfano. La población de trasplantes para los resultados de eficacia incluyó pacientes con antecedentes de al menos 4 eventos vaso-oclusivos en los 24 meses anteriores al consentimiento informado. Los resultados de eficacia fueron la resolución completa de los eventos vaso-oclusivos (88%) y de los eventos graves (94%) entre 6 y 18 meses después de la infusión de LYFGENIA.
Eventos adversos: Los más comunes son estomatitis, niveles bajos de plaquetas y de glóbulos blancos y rojos, y neutropenia febril.
ADAMTS13 (Adzynma®) Takeda (FDA, USA)
Indicación: Tratamiento de reemplazo enzimático profiláctico o a demanda en pacientes adultos y pediátricos con púrpura trombocitopénica trombótica congénita (cTTP).
Tipo: Medicamento biológico constituido por es una forma recombinante humana bivariante la desintegrina y metaloproteinasa con motivos de trombospondina 13 (ADAMTS13). Autorizado en Estados Unidos (FDA) el 9-11-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea (EMA).
Mecanismo: ADAMTS13 es una metaloproteasa (zinc) plasmática que regula la actividad del factor von Willebrand (VWF) al escindir multímeros de VWF grandes y ultragrandes en unidades más pequeñas y, por lo tanto, reduciendo las propiedades de unión a plaquetas del VWF y su propensión a formar microtrombos.
Eficacia clínica: Un estudio global, prospectivo, aleatorizado, con control activo, abierto, multicéntrico, cruzado de dos períodos seguido de un período de continuación de un solo brazo que evaluó la eficacia y seguridad de la terapia profiláctica y a demanda con el fármaco en comparación con las terapias basadas en plasma en 46 pacientes que fueron aleatorizados para recibir 6 meses de tratamiento con ADZYNMA o terapias basadas en plasma (Período 1), luego pasaron al otro tratamiento durante 6 meses (Período 2). Treinta y cinco pacientes ingresaron al período de un solo brazo de 6 meses con ADZYNMA (Período 3). La eficacia del tratamiento profiláctico cse demostró con base en la incidencia de eventos de TTP agudos y subagudos definidos por el protocolo y la tasa anualizada de manifestaciones de la púrpura trombocitopénica trombótica: eventos agudos (0 con ADZYNMA vs. 0,05 con terapias basadas en plasma), eventos subagudos (0 vs. 4), eventos trombocitopénicos (2,0 vs. 4,44) y eventos microangiopáticos hemolíticos (0,38 vs. 1,47).
Eventos adversos: Los más comunes son cefalea (31,3%), diarrea (16,7%), migraña (14,6), dolor abdominal (12,5%), náuseas (12,5%), infección del tracto respiratorio superior (12,5%), mareos (10,4%) y vómitos (10,4%).
Taurolidina/Heparina (Defencath®) Cormedix (FDA, USA)
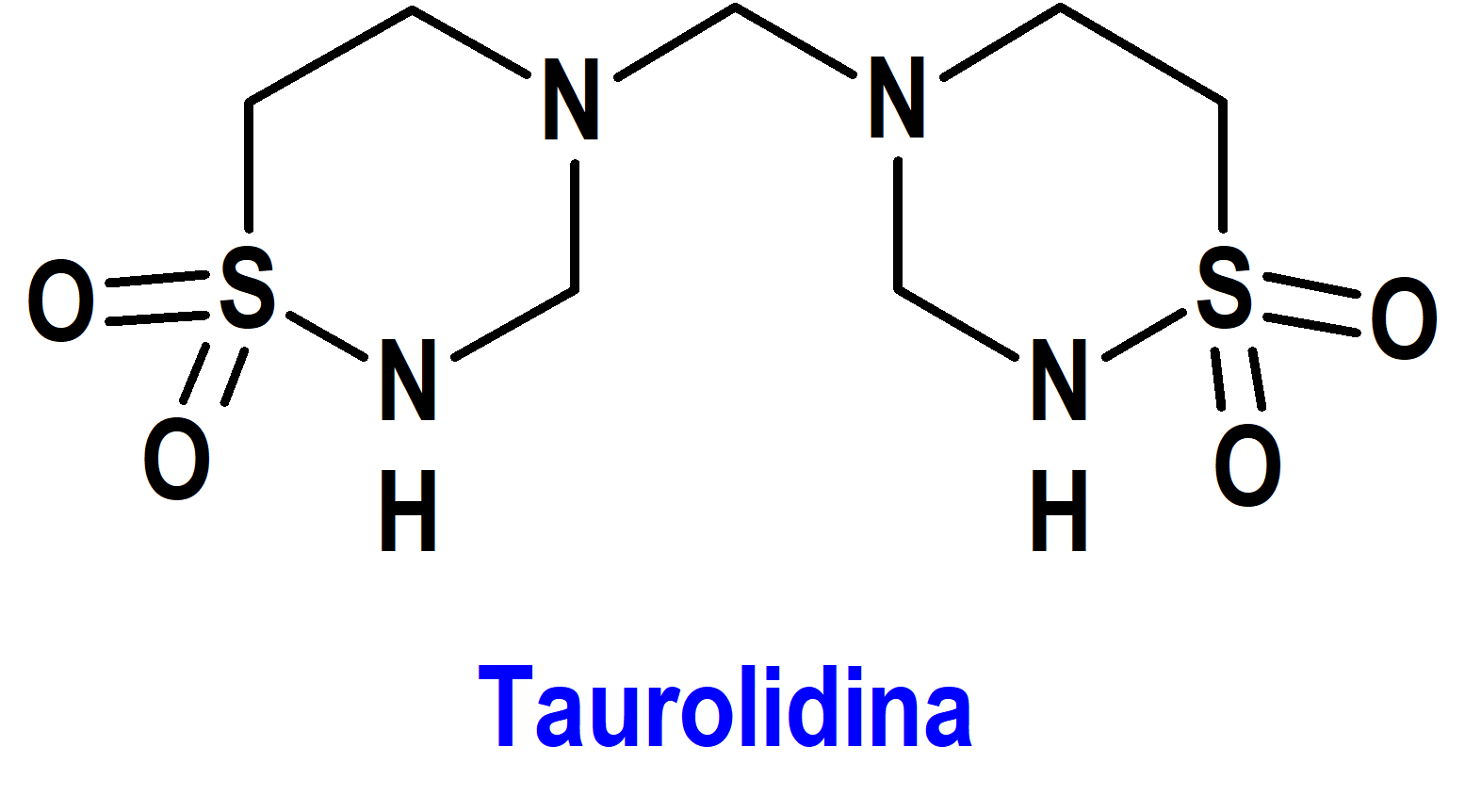
Indicación: Para reducir la incidencia de infecciones del torrente sanguíneo relacionadas con catéteres en pacientes adultos con insuficiencia renal que reciben hemodiálisis crónica a través de un catéter venoso central.
Tipo: Medicamento estándar constituido por un antimicrobiano sintético, la taurolidina [4,4’-metilenbis(1,2,4-tiadiazinano)-1,1,1’,1’-tetraóxido], y heparina sódica, un anticoagulante. Autorizado en Estados Unidos (FDA) el 15-11-2023 mediante revisión prioritaria (Priority Review) y por vía rápida (Fast Track); no autorizado aún en la Unión Europea (EMA).
Mecanismo: La taurolidina es un fármaco bactericida. Su mecanismo de acción es inespecífico; daña las paredes celulares microbianas e inhibe la adherencia de los microorganismos a las superficies biológicas. Se ha demostrado que es activo contra la mayoría de los aislados de bacterias Gram positivas (Staphylococcus aureus, incluidas las resistentes a meticilina, St.
Epidermidis, Enterococcus faecalis), Gram negativos (Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Serratia marcescens) y Hongos (Candida albicans, C. glabrata). La heparina es un agente anticoagulante que actúa sobre la antitrombina III para inducir un cambio conformacional que mejora notablemente la actividad serina proteasa de la antitrombina III, inhibiendo así los factores de coagulación activados implicados en la secuencia de coagulación, particularmente Xa y IIa.
Eficacia clínica: Se evaluó un total de 806 pacientes con insuficiencia renal que recibían hemodiálisis crónica en un ensayo multicéntrico, aleatorizado, doble ciego y controlado con tratamiento activo, siendo aleatorizados para recibir DEFENCATH o heparina sola como solución de sellado o bloqueo del catéter. La variable principal de eficacia fue la tasa de eventos de infección del torrente sanguíneo relacionadas con catéteres por 1000 días-catéter: 2,3% ( DEFENCATH) vs. 8,0% (heparina sola).
Eventos adversos: Los más comunes son mal funcionamiento del catéter de hemodiálisis (17%), hemorragia/sangrado (7%), náuseas (6%), vómitos (7%) y mareos (6%).
(D) DERMATOLOGÍA
Lebrikizumab (Ebglyss®) Almirall (EMA, UE)
Indicación: Tratamiento de la dermatitis atópica de moderada a grave en adultos y adolescentes a partir de 12 años con un peso corporal de al menos 40 kg que sean candidatos a una terapia sistémica.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal tipo IgG4. Autorizado por la Unión Europea (EMA) el 16-11-2023; no autorizado previamente en Estados Unidos (FDA).
Mecanismo: Inmunoglobulina monoclonal (IgG4) que se une con alta afinidad a la interleucina (IL)-13 e inhibe selectivamente su señalización a través del receptor heterodímero IL-4 alfa (IL-4Ra) / IL-13 alfa 1 (IL-13Ra1), de modo que inhibe los efectos resultantes de la IL-13. La inhibición de la señalización de la IL-13 parece ser beneficiosa en enfermedades en las que la IL-13 es un factor contribuyente clave en la patogenia de la enfermedad. Lebrikizumab no impide la unión de la IL-13 al receptor alfa 2 de la IL-13 (IL-13Ra2 o receptor decoy), lo cual permite la internalización de la IL-13 en la célula. Lebrikizumab reduce las concentraciones séricas de algunos mediadores inflamatorios de tipo 2, como la periostina, las inmunoglobulinas E (IgE) totales, ligandos de tipo quimiocina CC (CCL)17 (quimiocina del timo y regulada por activación [TARC]), CCL18 (quimiocina pulmonar y regulada por activación [PARC]) y CCL13 (proteína quimiotáctica de monocitos-4 [MCP-4]).
Eficacia clínica: Tres estudios clínicos aleatorizados, doble ciego y controlados con placebo en 1.062 adultos y adolescentes (de 12 a 17 años de edad y con un peso ≥40 kg) con dermatitis atópica de moderada a grave y que habían presentado previamente una respuesta inadecuada al tratamiento tópico o tenían una contraindicación médica para recibirlos. Las variables coprimarias de eficacia fueron el porcentaje de pacientes con una puntuación de la Evaluación global del investigador (IGA) de 0 o 1 (“aclaramiento completo” o “aclaramiento casi completo” de las lesiones, respectivamente), con una reducción ≥2 puntos con respecto al inicio (33,2-43,1% con lebrikizumab vs. 10,8-12,7% con placebo), y el porcentaje de pacientes que lograron al menos una reducción del 75% en el índice de extensión y gravedad del eccema (EASI) con respecto al inicio hasta la semana 16 (EASI 75: 52,1-58,8 con lebrikizumab vs. 16,2-18,1% con placebo). Las variables secundarias fueron el porcentaje de pacientes que lograron al menos una reducción del 90 % en el EASI (EASI 90: 30,7-7-38,1% vs. 9,0-9,5%) y el porcentaje de pacientes con al menos una mejora de 4 puntos con respecto al inicio en la Escala de valoración numérica (NRS) del prurito (39,8-45,9% vs. 11,5-13,0%).
Eventos adversos: Los más comunes son conjuntivitis, ojo seco y reacción en el lugar de la inyección (1-10%).
(H) PREPARACIONES HORMONALES SISTÉMICAS
Palopegteriparatida (Yorvipath®) Ascendis (EMA, UE)
Indicación: Tratamiento sustitutivo de la hormona paratiroidea (PTH) indicado para el tratamiento de adultos con hipoparatiroidismo crónico.
Tipo: Medicamento biológico constituido por un profármaco compuesto de una fracción (secuencia 1-34, del total de 84 aminoácidos) de la hormona paratiroidea (PTH), conjugada con un portador de metoxipolietilenglicol (mPEG) mediante un conector químico. Autorizado por la Unión Europea (EMA) el 17-11-2023 como medicamento huérfano (Orphan drug); no autorizado previamente en Estados Unidos (FDA).
Mecanismo: Profármaco conteniendo la fracción (1-34) de PTH. Ésta y su metabolito principal, PTH(1-33), tienen una afinidad y activación similar sobre el receptor 1 de la PTH (PTH1R), similar a la PTH endógena. En condiciones fisiológicas, la PTH se escinde de palopegteriparatida de forma controlada para proporcionar una exposición sistémica continua de PTH activa. La hormona paratiroidea endógena (PTH) es segregada por las glándulas paratiroides como un polipéptido de 84 aminoácidos, ejerciendo su acción mediante receptores específicos (PTH1R) de superficie celular que están expresados, entre otros tejidos, en hueso, riñón y tejido nervioso. La activación de PTH1R estimula la renovación ósea, aumenta la reabsorción renal de calcio y la excreción de fosfato y facilita la síntesis de la vitamina D activa.
Eficacia clínica: Ensayo clínico de fase 3 controlado con placebo, doble ciego de 26 semanas, como tratamiento sustitutivo de la PTH en 82 pacientes adultos con hipoparatiroidismo. La variable primaria de eficacia combinada se definió como la proporción de pacientes en la semana 26 que lograron niveles de calcio en suero dentro de los límites normales (2,07 a 2,64 mmol/l [8,3 a 10,6 mg/dl]), independencia del tratamiento convencional definido como la no necesidad de vitamina D activa y ≤600 mg/día de aporte suplementario de calcio, y sin aumento en el tratamiento del estudio prescrito en 4 semanas antes de la semana 26. La respuesta completa a todos los parámetros se alcanzó en el 78,7% (palopegteriparatida) vs. 4,8% (placebo), y la respuesta individualizada a cada parámetro fue del 80,3 vs. 47,6% (calcio en suero dentro de los límites normales), 98,4 vs. 23,8% (sin necesidad de vitamina D), 93,4 vs. 57,1% (sin aumento en el tratamiento).
Eventos adversos: Los más comunes son cefalea, parestesia, náuseas, reacciones en la zona de inyección y fatiga(≥10%).
(J) ANTIINFECCIOSOS SISTÉMICOS
Vacuna Gripe A Zoonótica (H5N1) (Zoonotic Influenza Vaccine Seqirus®) Seqirus (EMA, UE)
Indicación: Inmunización activa frente a los virus A de la gripe del subtipo H5N1.
Tipo: Medicamento biológico constituido por antígenos de superficie de virus de la gripe (hemaglutinina y neuraminidasa) de la cepa similar a A/turkey/Turkey/1/2005 (H5N1) (NIBRG-23) (clado 2.2.1); contiene como adyuvante MF59C.1 (escualeno, polisorbato 80, trioleato de sorbitán, citrato de sodio y ácido cítrico). Autorizado por la Unión Europea (EMA) el 6-10-2023; autorizado previamente en Estados Unidos (FDA) el 29-10-2021.
Mecanismo: Respuesta inmunitaria en forma de anticuerpos específicos frente al virus de la gripe A (H5N1).
Eficacia clínica: Dos ensayos clínicos de fase 3. En el primero 2.566 adultos sanos recibieron dos dosis de la vacuna contra la gripe zoonótica (H5N1) (A/Vietnam/1194/2004), administradas con un intervalo de tres semanas. En el segundo se incluyó a 194 sujetos adultos que recibieron dos dosis de la vacuna contra la gripe zoonótica (H5N1) (A/turkey/Turkey/1/2005) administradas con tres semanas de diferencia. La tasa de seroprotección (área de SRH, anticuerpos anti-HA ≥25 mm²) del 91% con ambos tipos de vacunas, que fue del 82% (también con las dos vacunas) en personas mayores de 60 años. La persistencia de anticuerpos tras la primovacunación en personas de mayores se redujo del 1/2 a 1/5 de los niveles posvacunación en el día 202 respecto a los títulos obtenidos el día 43 tras la finalización de la pauta de primovacunación. Hasta un 50% de las personas mayores inmunizadas con la vacuna contra la gripe zoonótica (H5N1) mantuvieron seroprotección a los seis meses.
Eventos adversos: Los más comunes (≥10%) son cefalea, mialgia e hinchazón, induración o enrojecimiento en el punto de la inyección.
Vacuna Chikungunya Viva (Ixchiq®) Valneva (FDA, USA)
Indicación: Prevención de la enfermedad causada por el virus chikungunya (CHIKV) en personas de 18 años y mayores que tienen un mayor riesgo de exposición al CHIKV.
Tipo: Medicamento biológico constituido por virus chikungunya vivos y atenuados (generados mediante genética inversa a partir del clon LR2006 OPY1 de la cepa LRCHIKV de La Reunión); el virus atenuado tiene una deleción en la proteína no estructural 3, que codifica un componente del complejo viral de replicasa, y se replica con menos eficiencia que el CHIKV de tipo salvaje .
Autorizado en Estados Unidos (FDA) el 9-11-2023 de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Activa la respuesta inmunitaria específicas frente al CHIKV.
Eficacia clínica: La eficacia de la vacuna contra la enfermedad causada por CHIKV se basó en una evaluación de la serorespuesta definida como un nivel de anticuerpos neutralizantes anti-CHIKV por encima de un umbral (título mPRNT50 ≥150), derivado de un modelo de primates no humanos. Según éste, la tasa de serorespuesta fue del 99% (vacuna) y 0% (placebo) a los 28 días, y del 96% y 0% a los 180 días.
Eventos adversos: Los más comunes (con una diferencia de al menos dos puntos porcentuales sobre el placebo) son cefalea (28,5% con la vacuna y 12,7% con placebo), mialgia (23,9/7,4%), artralgia (17,2/4,9%), fiebre (13,5/0,9%), náusea (11,2/5,6%), reacciones locales de hipersensibilidad (10,6/8,1%) y dolor (6,2/3,7%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
Decitabina/Cedazuridina/(Inaqovi®) Otsuka (EMA, UE)
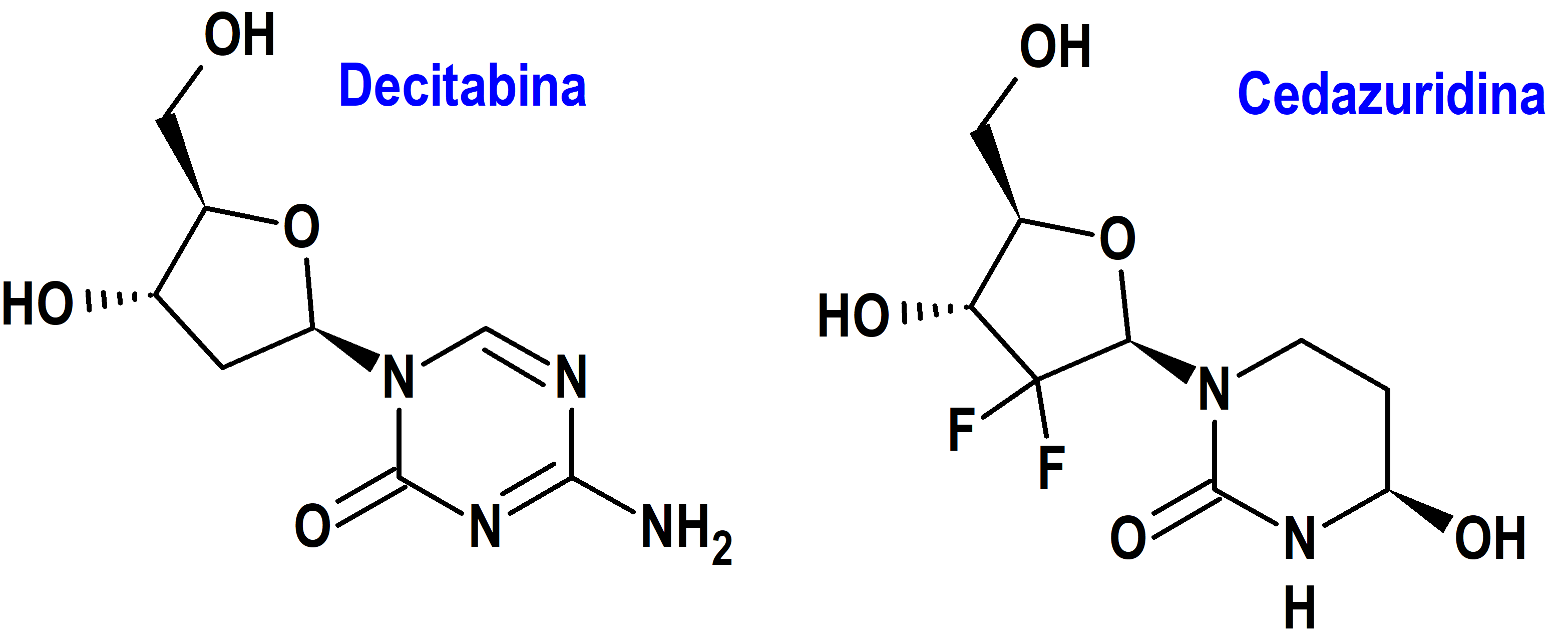
Indicación: Tratamiento en monoterapia de pacientes adultos con leucemia mieloide aguda (LMA) de nuevo diagnóstico que no son candidatos a la quimioterapia de inducción estándar.
Tipo: Medicamento sintético estándar compuesto por una combinación de dos nucleósidos. Un antineoplásico, la decitabina [4-amino-1-[(2R,4S,5R)-4-hidroxi-5(hidroximetil)oxolan-2-il]-1,3,5-triazin-2(1H)-ona], y un inhibidor del metabolismo hepático de la decitabina, la cedazuridina [(4R)-1-[(2R,4R,5R)-3,3-difluoro-4-hidroxi-5-(hidroximetil)oxolan-2-il]-4-hidroxi-1,3-diazinan-2-ona]. La combinación fue autorizada por la Unión Europea (EMA) el 15-9-2023 como medicamento huérfano (Orphan drug), aunque la decitabina había sido autorizada previamente (20-9-2012) por la EMA como monofármaco para la misma indicación; la combinación fue autorizada previamente en Estados Unidos (FDA) el 7-7-2020.
Mecanismo: La decitabina es un inhibidor metabólico de nucleósidos que actúa tras su fosforilación y la incorporación directa en el ADN. Asimismo, inhibe la ADN metiltransferasa, lo que provoca la hipometilación del ADN y la diferenciación celular o apoptosis. La hipometilación inducida por decitabina en células neoplásicas puede restaurar la función normal de los genes que son críticos para el control de la diferenciación y proliferación celular. En las células que se dividen rápidamente, la citotoxicidad de la decitabina también se puede atribuir a la formación de aductos covalentes entre la ADN metiltransferasa y la decitabina incorporada al ADN. La citidina desaminasa (CDA) es una enzima responsable de la degradación de los nucleósidos de citidina, incluida la decitabina. Los altos niveles de CDA en el tubo gastrointestinal y el hígado degradan rápidamente estos nucleósidos e impiden o limitan su biodisponibilidad oral. La cedazuridina inhibe la CDA y, con ello, su administración oral conjunta con decitabina aumenta la exposición sistémica de la decitabina a través de la inhibición del efecto de primer paso de la decitabina en el intestino y el hígado por la CDA.
Eficacia clínica: Dos ensayos cruzados, aleatorizados y abiertos, totalizando 213 pacientes adultos. El primer ensayo (n=80) demostró una tasa de respuesta completa (RC) del 18% con una duración media de 8,7 meses (rango: 1,1, 18,2). Entre 41 pacientes que dependían de transfusiones de glóbulos rojos y/o plaquetas al inicio del estudio, el 49% se volvieron independientes de estas transfusiones y entre aquellos incialmente independientes de las transfusiones de glóbulos rojos y plaquetas al inicio del estudio, el 64% permanecieron independientes de las transfusiones. El segundo estudio mostró una respuesta completa del 21 %, con una mediana de duración de 7,5 meses. Entre los pacientes que dependían de transfusiones de glóbulos rojos y/o plaquetas al inicio del estudio, el 53% se volvieron independientes de las transfusiones y entre los pacientes inicialmente independientes de las transfusiones el 63% permaneció independiente de éstas.
Eventos adversos: Los más comunes (≥20%) son fatiga, estreñimiento, hemorragia, mialgia, mucositis, artralgia, náuseas, disnea, diarrea, erupción cutánea, mareos, neutropenia febril, edema, dolor de cabeza, tos, disminución del apetito, tracto respiratorio superior. Infección, neumonía y aumento de transaminasas. Las anomalías de laboratorio de grado 3 o 4 más comunes (≥50%) son disminución de leucocitos, disminución del recuento de plaquetas, disminución del recuento de neutrófilos y disminución de la hemoglobina. Se produjeron muertes durante el tratamiento en el 24% de los pacientes, siendo las causas más frecuentes neumonía (8%), sepsis (3%) y hemorragia en el sistema nervioso central en el contexto de trombocitopenia (3%). Se produjeron interrupciones permanentes del tratamiento en el 14% e interrupciones temporales y reducciones de dosis en el 48%.
Toripalimab (Loqtorzi®) Choerus (FDA, USA)
Indicación: En combinación con cisplatino y gemcitabina, para el tratamiento de primera línea de adultos con carcinoma nasofaríngeo localmente avanzado metastásico o recurrente.
Tipo: Medicamento biológico constituido por una inmunoglobulina kappa humanizada G4 (IgG4). Autorizado en Estados Unidos (FDA) el 27-10-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo bloqueador del receptor de muerte programada 1 (PD-1). La unión de los ligandos PD-1, PD-L1 y PD-L2, al receptor PD-1 que se encuentra en las células T, inhibe la proliferación de células T y la producción de citocinas. En algunos tumores se produce una regulación positiva de los ligandos PD-1 y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmune activa de las células T de los tumores. Toripalima se une al receptor PD-1 y bloquea su interacción con PD-L1 y PD-L2, liberando la inhibición de la respuesta inmune mediada por la vía PD-1, incluida la respuesta inmune antitumoral.
Eficacia clínica: Ensayo aleatorizado, multicéntrico, de una sola región, doble ciego y controlado con placebo en 289 pacientes con carcinoma nasofaríngeo localmente avanzado metastásico o recurrente que no habían recibido quimioterapia sistémica previa para la enfermedad recurrente o metastásica. El tratamiento con toripalimab o placebo continuó hasta la progresión de la enfermedad, toxicidad inaceptable o un máximo de 2 años. Las evaluaciones del tumor se realizaron cada 6 semanas durante los primeros 12 meses y cada 9 semanas posteriormente. La principal medida de resultado de eficacia fue la supervivencia libre de progresión evaluada por el Comité de Revisión Independiente Cegado (BIRC) según RECIST v1.1. (11,7 vs. 8,0 meses). Las medidas de resultado de eficacia adicionales incluyen la tasa de respuesta general (77%, incluyendo 19% con respuesta completa vs. 66% con 11% de respuesta completa) y la supervivencia general evaluadas por BIRC a los cuatro años (61 vs. 47%).
Eventos adversos: Los más comunes (≥20%) son: náuseas (71%), vómitos (68%), disminución del apetito (55%), estreñimiento (39%), hipotiroidismo (38%), erupción cutánea (36%), pirexia. (32%), diarrea (31%), neuropatía periférica (30%), tos (26%), dolor musculoesquelético (25%), infección de las vías respiratorias superiores (23%), insomnio (23%), mareos (21%) y malestar (21%). Las anomalías de laboratorio de Grado 3 o 4 más comunes (≥2%) fueron: disminución de neutrófilos (58%), disminución de linfocitos (57%), disminución de hemoglobina (50%), disminución de plaquetas (33%), disminución de potasio (10%), disminución de sodio (9%), aumento de alanina aminotransferasa (6%), aumento o disminución de magnesio (4,2% cada uno), disminución de calcio (3,5%), aumento de aspartato aminotransferasa (2,7%) y aumento de bilirrubina (2,1%).
Fruquintinib (Fruzaqla®) Takeda (FDA, USA)
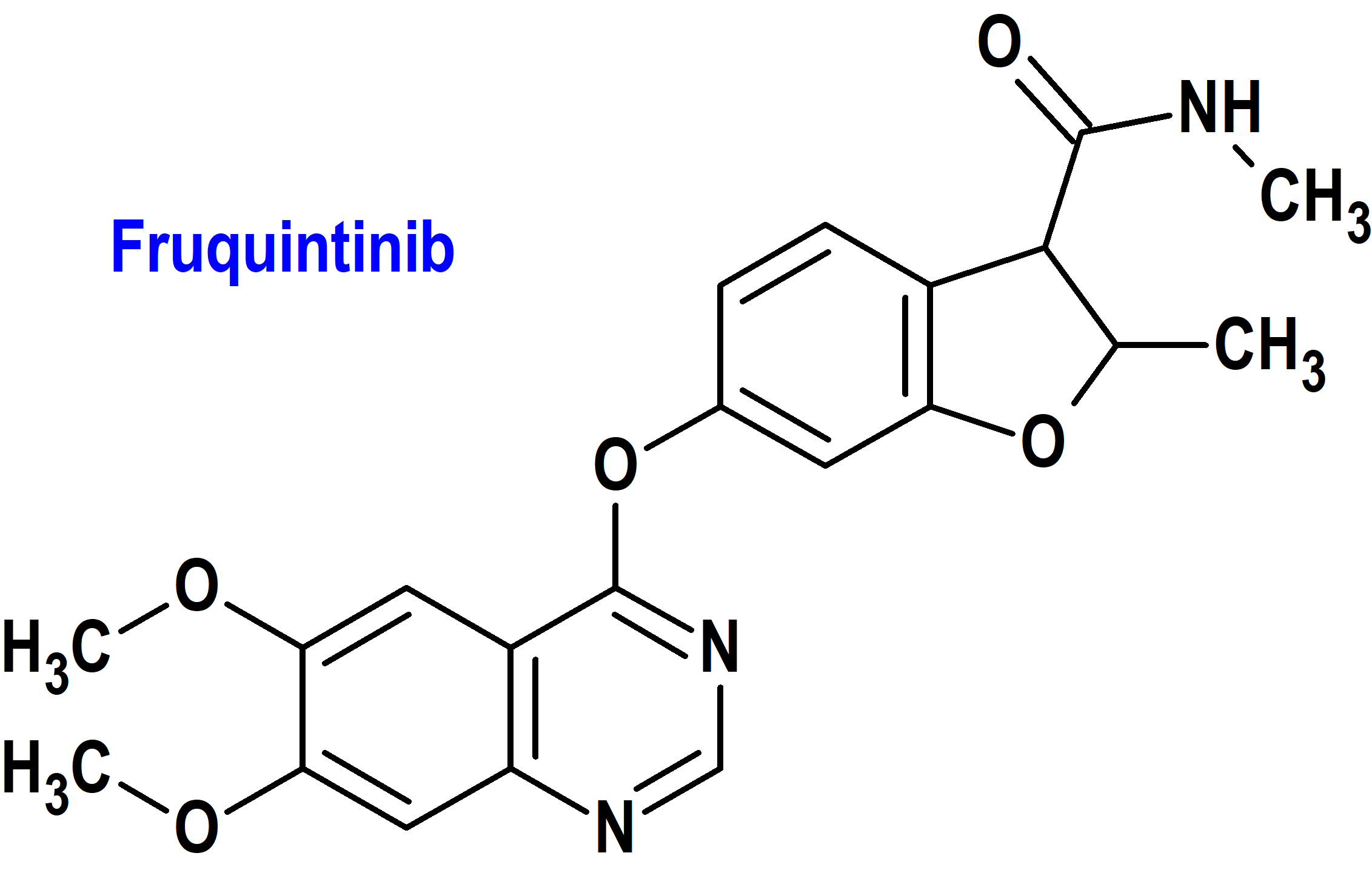
Indicación: Tratamiento pacientes adultos con cáncer colorrectal metastásico que han sido tratados previamente con quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, una terapia anti-VEGF y, si RAS de tipo salvaje y médicamente apropiado, una terapia anti-EGFR.
Tipo: Medicamento sintético estándar constituido por la 6-[(6,7-dimetoxiquinazolin-4-il)oxi]-N,2-dimetil-1-benzofuran-3-carboxamida. Autorizado en Estados Unidos (FDA) el 8-11-2023 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la cinasa de los receptores del factor de crecimiento endotelial vascular (VEGFR) -1, -2 y -3, inhibiendo la proliferación de células endoteliales y la formación de túbulos mediada por VEGF.
Eficacia clínica: Dos ensayos clínicos multicéntricos, aleatorizados, doble ciego y controlados con placebo, que evaluaron a un total de 1107 pacientes con cáncer colorrectal metastásico que tuvieron progresión de la enfermedad durante o después de una quimioterapia previa. La variable principal de eficacia en ambos ensayos fue la supervivencia global: mediana de 7,4 meses (fruquintinib) vs. 4,8 (placebo) en el primer estudio y 9,3 vs. 6,6 en el segundo.
Eventos adversos: Los más comunes (≥20%) son hipertensión, eritrodisestesia palmo-plantar, proteinuria, disfonía, dolor abdominal, diarrea y astenia.
Capivasertib (Truqap®) AstraZeneca (FDA, USA)
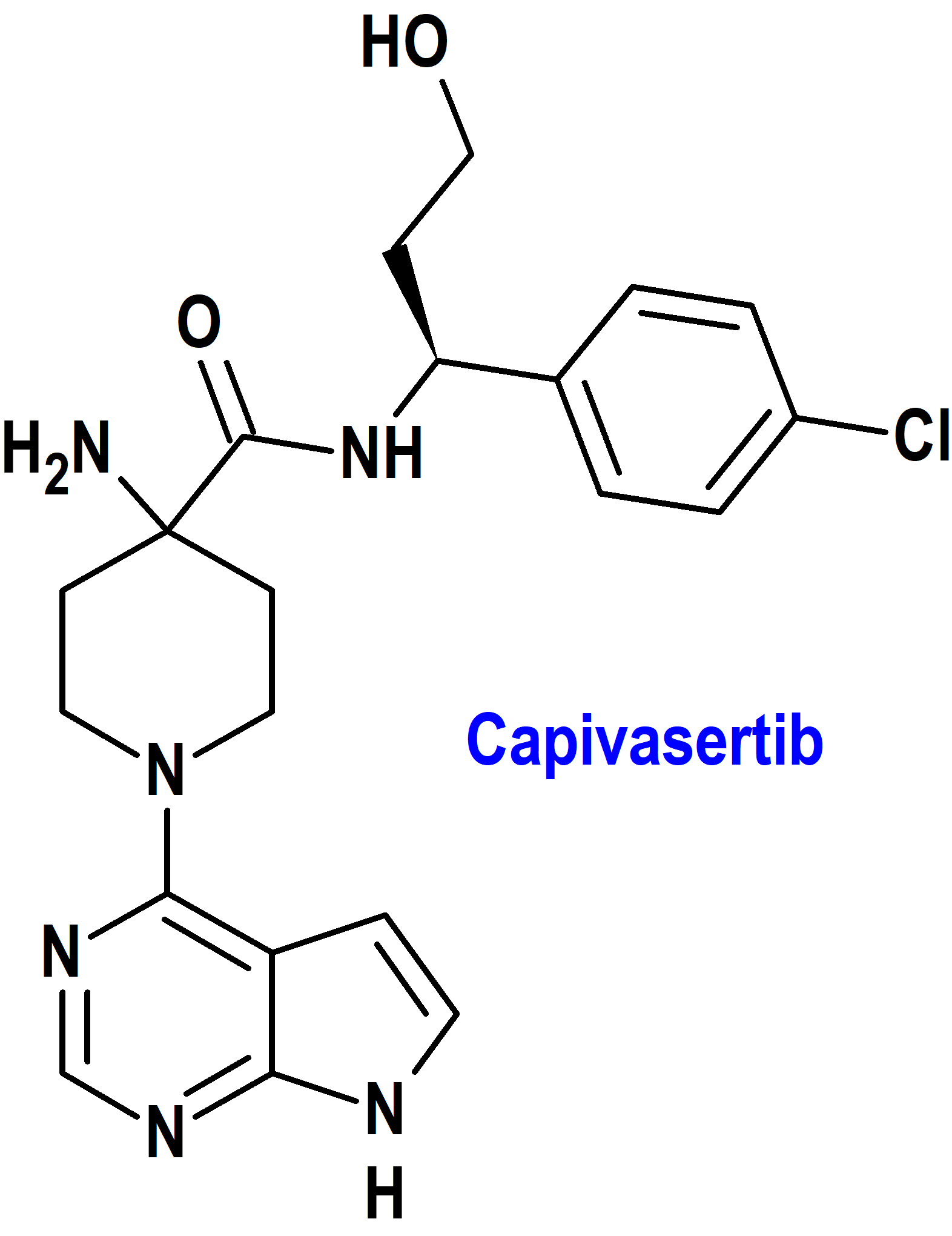
Indicación: Tratamiento en combinación con fulvestrant de pacientes adultos con cáncer de mama localmente avanzado o metastásico con receptor hormonal (HR) positivo y receptor 2 del factor de crecimiento epidérmico humano (HER2) negativo con una o más alteraciones de PIK3CA/AKT1/PTEN después de la progresión con al menos un régimen endocrino en el entorno metastásico o recurrencia en o dentro de los 12 meses posteriores a completar la terapia adyuvante.
Tipo: Medicamento sintético estándar constituido por la 4-amino-N-[(1S)-1-(4-clorofenil)-3-hidroxipropil]-1-(7H-pirrolo[2,3-d]pirimidin-4-il)-4-piperidinacarboxamida. Autorizado en Estados Unidos (FDA) el 16-11-2023 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de las 3 isoformas de la serina/treonina cinasa AKT (AKT1, AKT2 y AKT3) e inhibe la fosforilación de sustratos de AKT posteriores. La activación de AKT en tumores es el resultado de la activación de vías de señalización ascendentes, mutaciones en AKT1, pérdida de la función del homólogo de fosfatasa y tensina (PTEN) y mutaciones en la subunidad catalítica alfa de la fosfatidilinositol 3-cinasa (PIK3CA). In vitro, capivasertib reduce el crecimiento de líneas celulares de cáncer de mama, incluidas aquellas con mutaciones relevantes de PIK3CA o AKT1 o alteración de PTEN.
Eficacia clínica: Ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo en 708 pacientes adultos con cáncer de mama HR positivo localmente avanzado (inoperable) o metastásico y HER2 negativo, de los cuales 289 pacientes tenían tumores con alteraciones elegibles de PIK3CA/AKT1/PTEN. La variable principal de eficacia fue la supervivencia libre de progresión en la población de pacientes cuyos tumores tienen alteraciones de PIK3CA/AKT1/PTEN: mediana de 7,3 meses (capivasertib) vs. 3,1 meses (placebo). Las variables secundarias de eficacia fueron la supervivencia global a los 12 meses (24,5 vs. 12,7% ), la tasa de respuesta objetiva (26 vs. 8%) y la duración de la respuesta (medianas de 10,2 vs. 8,6 meses).
Eventos adversos: Los más comunes son diarrea (77%), reacción adversa cutánea (56%), disminución de linfocitos (49%), disminución de hemoglobina (47%), fatiga (38%), aumento de glucosa en ayunas (37%), náuseas y disminución de leucocitos (35% cada uno), aumento de triglicéridos (30%), estomatitis (25%), disminución de neutrófilos (25%) y vómitos (21%). Se produjeron reacciones adversas graves en el 18% de los pacientes y fue necesario interrumpir permanentemente el tratamiento en el 10%; un 1,3% experimentaron eventos adversos mortales.
Repotrectinib (Augtyro®) Bristol Myers Squibb (FDA, USA)
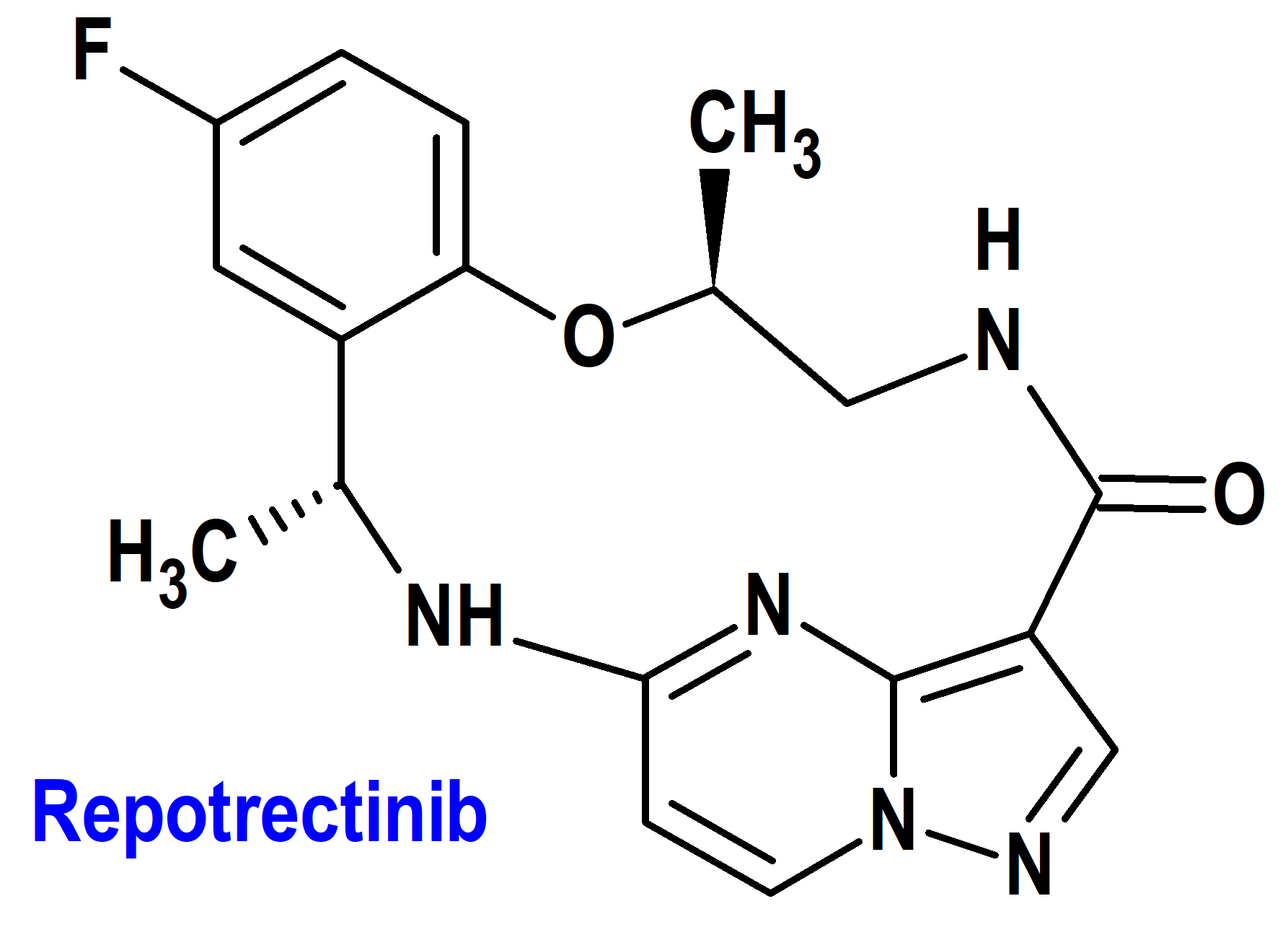
Indicación: Tratamiento de pacientes adultos con cáncer de pulmón de células no pequeñas localmente avanzado o metastásico positivo para ROS1.
Tipo: Medicamento sintético estándar constituido por la (3R,11S)-6-fluoro-3,11-dimetil-10-oxa-2,13,17,18,21-pentaazatetraciclo[13.5.2.04,9.018,22]docosa-1(21),4,6,8,15(22),16,19-heptaen-14-ona. Autorizado en Estados Unidos (FDA) el 15-11-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor del protooncogén ROS1 (tirosina-proteína cinasa ROS1) y de las tirosina cinasas del receptor de tropomiosina (TRK) TRKA, TRKB y TRKC. Las proteínas de fusión que incluyen dominios ROS1 pueden impulsar el potencial tumorigénico mediante la hiperactivación de vías de señalización posteriores que conducen a una proliferación celular sin restricciones.
Eficacia clínica: Ensayo clínico multicéntrico, de un solo brazo, abierto y de múltiples cohortes en 127 pacientes (71 pacientes sin tratamiento previo con ROS1 TKI que recibieron hasta 1 línea previa de quimioterapia y/o inmunoterapia basada en platino y 56 pacientes que recibieron 1 ROS1 TKI previo sin quimioterapia o inmunoterapia previa basada en platino). Las variables principales de eficacia fueron la tasa de respuesta general confirmada completa (6% en pacientes naïve vs. 5% en pretratados) y parcial (73 vs. 32%) y la mediana de la duración de la respuesta: 34,1 vs. 14,8 meses.
Eventos adversos: Los más comunes son mareos (64%), disgeusia (50%), neuropatía periférica (47%), estreñimiento (37%), disnea (30%), ataxia (29%), fatiga (29%), trastornos cognitivos (23%) y náuseas (20%).
Nirogacestat (Ogsiveo®) Springworks (FDA, USA)
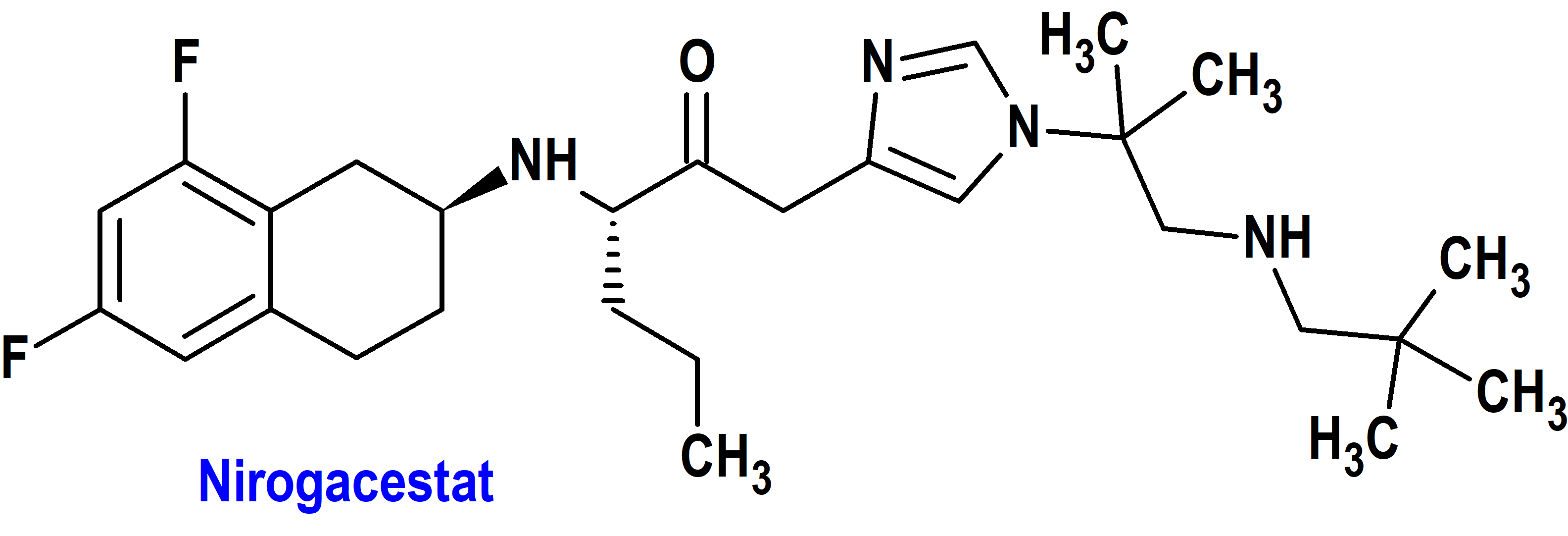
Indicación: Para pacientes adultos con tumores desmoides progresivos que requieren tratamiento sistémico.
Tipo: Medicamento sintético estándar constituido por la (S)-2-(((S)-6,8-difluoro-1,2,3,4-tetrahidronaftalen-2-il)amino)-N-(1-(2-metil-1-(neopentilamino)propan-2-il)-1H imidazol-4-il)pentanamida. Autorizado en Estados Unidos (FDA) el 27-11-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la gamma secretasa que bloquea la activación proteolítica del receptor Notch. Cuando este receptor Notch se encuentra desregulado, puede activar vías que contribuyen al crecimiento de tumores desmoides. Este tipo de tumores, que suelen ser benignos, están constituidos por tejidos blandos que se forman en el tejido fibroso (conjuntivo), localizándose más habitualmente en brazos, piernas o abdomen, aunque también pueden presentarse en la cabeza y el cuello. La vía de señalización Notch desempeña un papel clave para regular el destino celular, crecimiento, proliferación y la muerte celular programada durante el desarrollo y especialmente en procesos proliferativos de la neurogénesis y mantenimiento de nichos de célula madre.
Eficacia clínica: Un ensayo internacional, multicéntrico, aleatorizado, doble ciego y controlado con placebo en 142 pacientes adultos con tumores desmoides progresivos no susceptibles de cirugía. La variable clínica principal de eficacia fue la tasa de supervivencia libre de progresión (porcentaje de pacientes sin eventos radiográficos o clínicos: 83 vs. 49%) y la variable secundaria fue la tasa de respuesta objetiva (41%; 7% completa y 34% parcial vs. 8%; 0% completa y 8% parcial).
Eventos adversos: Los más comunes (>15%) son diarrea, toxicidad ovárica, erupción cutánea, náuseas, fatiga, estomatitis, dolor de cabeza, dolor abdominal, tos, alopecia, infección del tracto respiratorio superior y disnea.
Zilucoplan (Zilbrysq®) UCB (FDA, USA; EMA, UE)
Indicación: Tratamiento de la miastenia grave generalizada en pacientes adultos con anticuerpos anti-receptor de acetilcolina (AChR) positivos.
Tipo: Medicamento sintético estándar constituido por un péptido macrocíclico formado por 15 aminoácidos, en el que los aminoácidos Lys (1) y Asp (6) forman un anillo lactámico. Autorizado en Estados Unidos (FDA) el 17-10-2023 como medicamento huérfano (Orphan drug); autorizado en la Unión Europea (EMA) el 1-12-2023.
Mecanismo: Zilucoplan inhibe los efectos de la proteína del complemento C5 a través de un mecanismo de acción doble. Se une específicamente a C5, inhibiendo así su escisión por parte de la C5 convertasa en C5a y C5b, lo que da lugar a una disminución del ensamblaje y la actividad citolítica del complejo de ataque a la membrana (MAC). Además, al unirse a la fracción C5b de C5, zilucoplán impide estéricamente la unión de C5b a C6, lo que impide el posterior ensamblaje y la actividad del MAC, en caso de que se forme C5b. Un 80% de los pacientes con miastenia grave tienen anticuerpos positivos contra el receptor de acetilcolina (AchR), en los que la unión de autoanticuerpos antiAChR al AChR da como resultado una activación incontrolada e inapropiada de la vía clásica del complemento. El complejo inmunológico formado por el complejo autoanticuerpo-antígeno activa el componente C1 de la vía clásica del complemento. Esto conduce a una serie de pasos de escisión enzimática, que culminan en la escisión del componente 5 del complemento (C5) en C5a y C5b y el depósito del complejo de ataque a la membrana citolítica (C5b-9, MAC) en la membrana postsináptica de la unión neuromuscular y lesión posterior de la placa terminal neuromuscular, lo que conduce a una falla de la transmisión neuromuscular.
Eficacia clínica: Un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo de 12 semanas de duración en 174 pacientes. El criterio de valoración principal de eficacia fue una comparación del cambio desde el inicio entre los grupos de tratamiento en la puntuación total de la escala de actividades de la vida diaria específicas de miastenia gravis (MGADL) después de doce semanas de tratamiento. La MG-ADL evalúa el impacto de la gMG en las funciones diarias de 8 signos o síntomas que normalmente se ven afectados en la miastenia grave, con una puntuación total que varía de 0 a 24, y las puntuaciones más altas indican mayor deterioro. Los resultados mostraron reducciones de la escala MG-ADL de 4,4 puntos con zilucoplan y de 2,3 con placebo.
Eventos adversos: Los más comunes son reacciones en el lugar de la inyección (29% vs. 16% con placebo), infecciones del tracto respiratorio superior (14/7%), diarrea (11/2%), infección del tracto urinario (8/5%), náuseas o vómitos (8/7%), Aumento de lipasa (7/0%) y de amilasa (5/1%).
Efbemalenograstim (Ryzneuta®) Evive (FDA, USA)
Indicación: Para disminuir la incidencia de infección, manifestada por neutropenia febril, en pacientes adultos con neoplasias malignas no mieloides que reciben fármacos anticancerosos mielosupresores asociados con una incidencia clínicamente significativa de neutropenia febril.
Tipo: Medicamento biológico constituido por una proteína de fusión recombinante de 413 aminoácidos que consta de G-CSF (un factor de crecimiento de leucocitos) humano, un conector de 16 aminoácidos y la porción Fc de la IgG2 humana. Autorizado en Estados Unidos (FDA) el 16-11-2023; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Factor estimulante de colonias que actúa sobre las células hematopoyéticas uniéndose a receptores específicos de la superficie celular, estimulando la proliferación, diferenciación, compromiso y activación funcional de las células terminales.
Eficacia clínica: Dos estudios aleatorios y controlados. El primero fue un estudio aleatorizado, doble ciego y controlado con placebo que empleó doxorrubicina y docetaxes para el tratamiento de cáncer de mama. En este estudio, 122 pacientes fueron aleatorizados para recibir una única inyección subcutánea de efbemalenograstim o placebo el día 2 del ciclo 1 de quimioterapia. La variable principal de eficacia fue la duración media de la neutropenia grave (Grado 4) en el ciclo 1: 1,4 vs. 4,3 días; asimismo, la incidencia de neutropenia febril fue del 4,8 vs. 25,6%. El segundo fue un estudio aleatorizado y controlado con tratamiento activo (pegfilgrastim) que empleó docetaxel y ciclofosfamida para el tratamiento del cáncer de mama no metastásico en 393 pacientes. La media de días de neutropenia grave (Grado 4) fue la misma (0,6 días) con ambos tratamientos.
Eventos adversos: Los más comunes (≥10%) son fatiga, dolor óseo, náuseas, anemia y trombocitopenia.
Iptacopan (Fabhalta®) Novartis (FDA, USA)
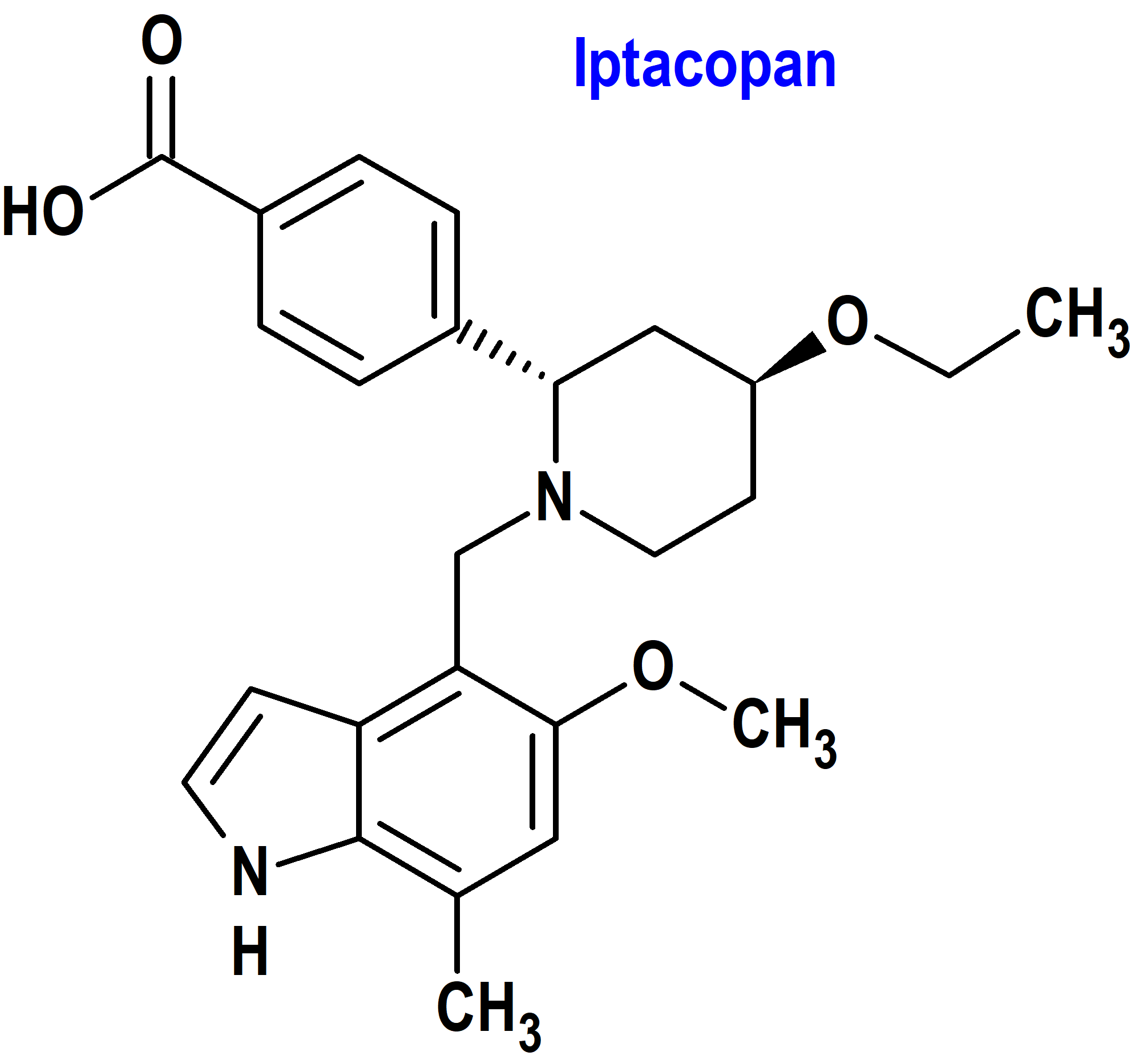
Indicación: Tratamiento de adultos con hemoglobinuria paroxística nocturna (HPN).
Tipo: Medicamento sintético estándar, constituido por la (2S,4S)-2-(4-carboxifenil)-4-etoxi-1-[(5-metoxi-7-metil-1H-indol-4-il)metil]piperidina. Autorizado en Estados Unidos (FDA) el 5-12-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Iptacopan se une al factor B de la vía alternativa del complemento y regula la escisión de C3, la generación de efectores posteriores y la amplificación de la vía terminal. En la hemoglobinuria paroxística nocturna, la hemólisis intravascular está mediada por el complejo de ataque a la membrana, mientras que la hemólisis extravascular se ve facilitada por la opsonización de C3b. Iptacopan actúa sobre la vía alternativa de la cascada del complemento para controlar tanto la hemólisis extravascular como la Hiv intravascular.
Eficacia clínica: Un estudio multicéntrico, abierto, de 24 semanas, controlado con comparador activo (eculizumab o ravulizumab) en 97 adultos con HPN y anemia residual (hemoglobina <10 g/dL) a pesar del tratamiento previo con un régimen estable de tratamiento anti-C5 (eculizumab o ravulizumab) durante al menos 6 meses antes de la aleatorización. Las variables clínicas principales fueron el porcentaje de pacientes con aumento sostenido de los niveles de hemoglobina ≥2 g/dL desde el inicio en ausencia de transfusiones (82,3% con iptacopan vs. 0% con eculizumab o raviluzimab) y el de pacientes con nivel sostenido de hemoglobina ≥12 g/dL en ausencia de transfusiones (67,7 vs. 0%). Como variable secundaria se determinó el porcentaje de pacientes que evitaron la transfusión (95,2 vs. 45,7%).
Eventos adversos: Los más comunes (≥10%) son dolor de cabeza (19%), nasofaringitis (16%), diarrea (15%), dolor abdominal (15%), infección bacteriana (11%), infección viral (10%) y náuseas (10%).
(M) SISTEMA MÚSCULO-ESQUELÉTICO
Vamorolona (Agamree®) Santhera (EMA, UE; FDA, USA)
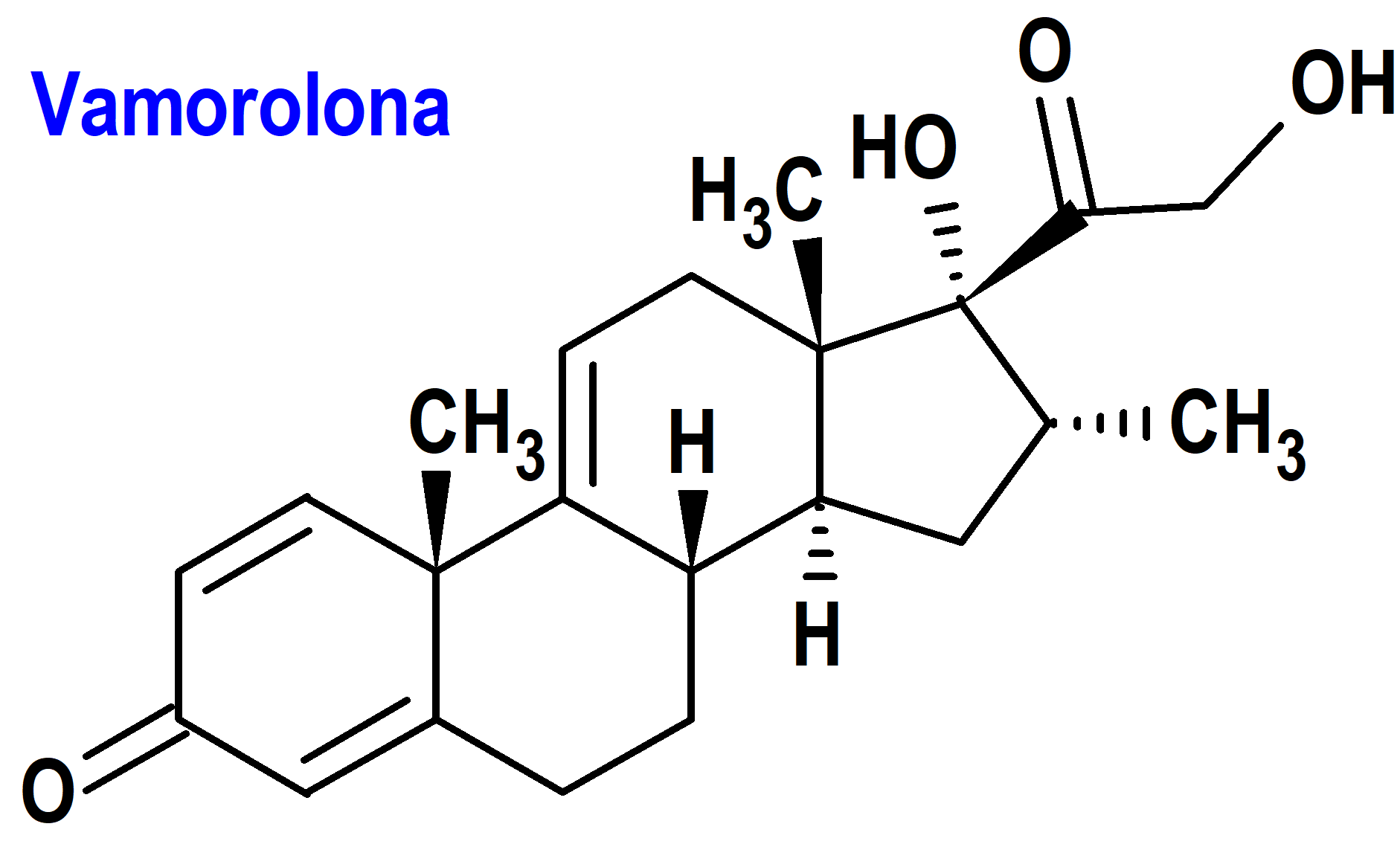
Indicación: Tratamiento de la distrofia muscular de Duchenne (DMD) en pacientes de 2 años de edad y mayores.
Tipo: Medicamento sintético estándar constituido por la 17a21-dihidroxi-16a-metil-pregna-1,4,9(11)-trieno-3,20-diona. Autorizado en Estados Unidos (FDA) el 26-10-2023 como medicamento huérfano (Orphan drug); autorizado en la Unión Europea (EMA) el 14-12-2023.
Mecanismo: Corticosteroide que actúa a través del receptor de glucocorticoides, produciendo efectos antiinflamatorios e inmunosupresores. Se desconoce el mecanismo preciso por el cual la vamorolona ejerce su efecto en pacientes con DMD.
Eficacia clínica: Un estudio multinacional, multicéntrico, aleatorizado, doble ciego, de grupos paralelos, controlado con placebo y activo, de 24 semanas de duración, sobre 121 pacientes masculinos con DMD. El criterio de valoración principal fue el cambio desde el valor inicial hasta la semana 24 en la velocidad de la prueba de tiempo para ponerse de pie (TTSTAND), una medida de la función muscular que mide el tiempo necesario para que el paciente se ponga de pie hasta una posición erguida desde una posición supina (piso): +0,048 (vamorolona) vs. -0,012 (placebo). Los criterios de valoración secundarios consistieron en el cambio desde el inicio hasta la semana 24 en la prueba de caminata de 6 minutos (6MWT, mide la distancia que un paciente puede caminar sobre una superficie plana y dura en un período de 6 minutos): +29 vs. -14 m; y el tiempo para correr/caminar 10 metros ( TTRW, mide el tiempo que tarda un paciente en correr o caminar 10 metros): 0,258 vs. 0,014 m/s.
Eventos adversos: Los más comunes son signos cushingoides (7% con la dosis de 2 mg/kg/d; 29% con la dosis de 6 mg/kg/d; 0% con placebo), trastornos psiquiátricos (comportamiento anormal, agresión, agitación, ansiedad, irritabilidad, estado de ánimo alterado, trastornos del sueño y estereotipos: 7/21/14%), vómitos (17/ 14/7%), aumento de peso (0/11/3%), deficiencia de vitamina D (7/11/0%), tos (10/7/3%), dolor de cabeza (7/7/3%), diarrea (3/7/3%), aumento del apetito (3/7/3%) y rinitis (3/7/3%).
(N) SISTEMA NERVIOSO
Gepirona (Exxua®) Fabre Kramer (FDA, USA)
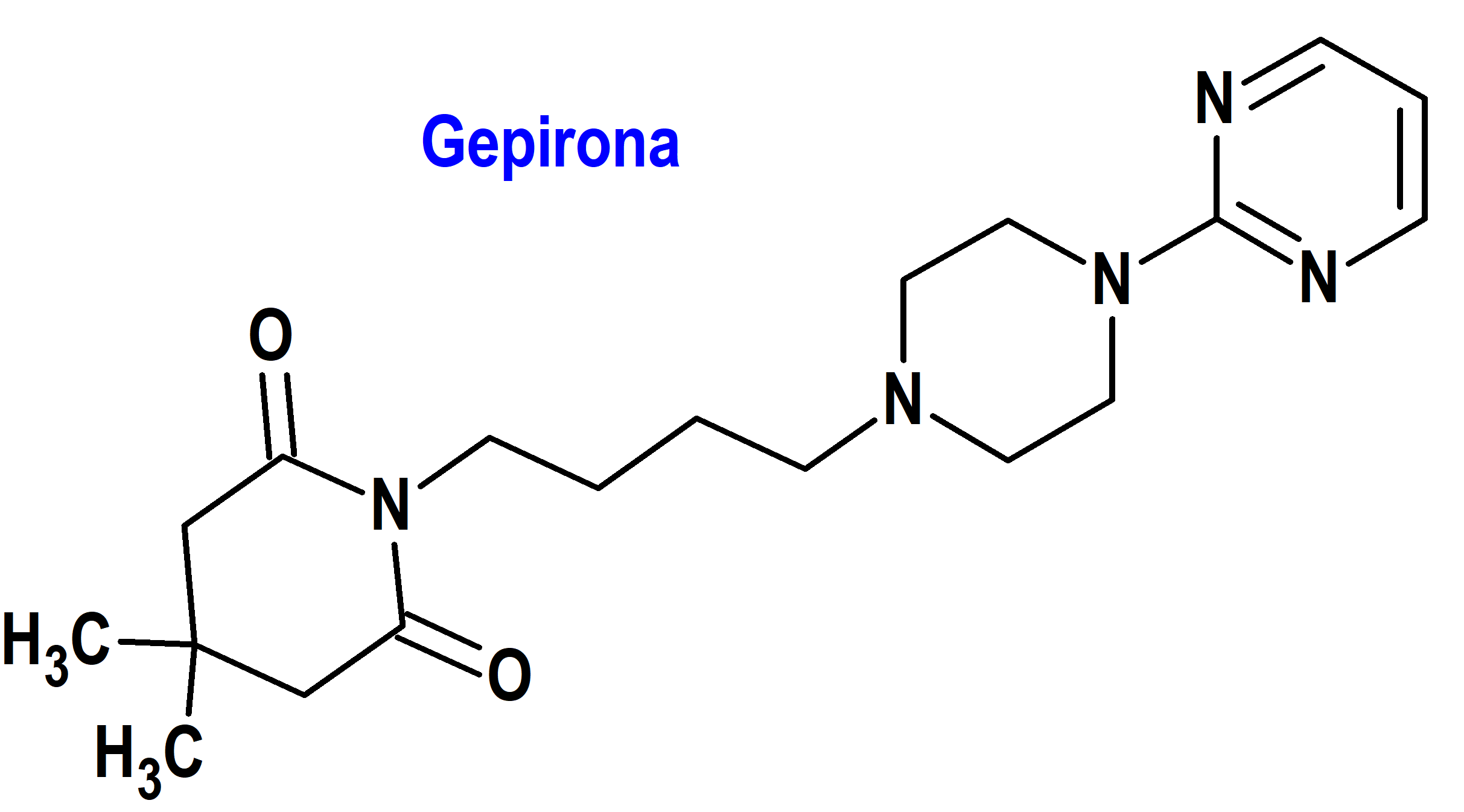
Indicación: Tratamiento del trastorno depresivo mayor (TDM) en adultos.
Tipo: Medicamento sintético estándar constituido por la 4,4-dimetil-1-[4-[4-(2-pirimidinil)-1-piperazinil]butil]-2,6-piperidinadiona. Autorizado en Estados Unidos (FDA) el 22-9-2023; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Modulación de la actividad serotoninérgica en el SNC a través de la actividad agonista selectiva en los receptores 5HT1A. Su metabolito 3’-OH-gepirona contribuye significativamente a la acción de la gepirona.
Eficacia clínica: Dos estudios de ocho semanas, aleatorizados, doble ciego, controlados con placebo y de dosis flexible en un total de 442 adultos (de 18 a 69 años) que cumplen con los criterios del Manual Diagnóstico y Estadístico de Trastornos Mentales (DSM-IV) para el TDM. La variable principal de eficacia en ambos estudios fue el cambio desde el inicio en la puntuación total de la Escala de Calificación de Depresión de Hamilton (HAMD-17) en la Semana 8 en comparación con el placebo, registrándose una reducción estadísticamente significativa del 40 vs 30% y del 43 vs. 33%, respectivamente.
Eventos adversos: Los más comunes (≥5 % y, al menos, doble de incidencia que el placebo) son mareos (49/10%), náusea (35/13%), insomnio (14/5%), dolor abdominal (7/3%) y dispepsia (6/2%).
(V) VARIOS
Tenapanor (Xphozah®) Ardelyx (FDA, USA)
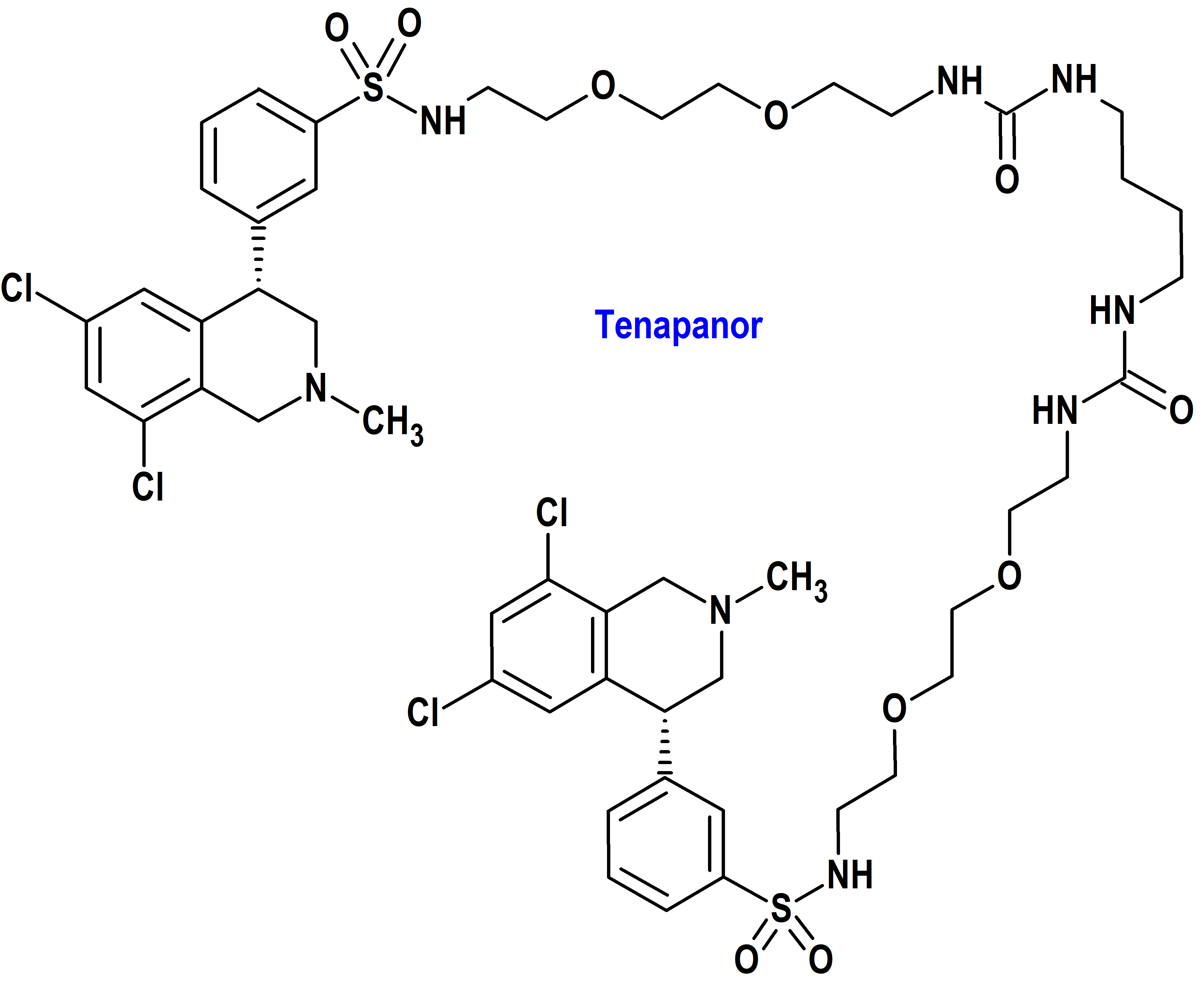
Indicación: Para reducir el fósforo sérico en adultos con enfermedad renal crónica (ERC) en diálisis como terapia complementaria en pacientes que tienen una respuesta inadecuada a los quelantes de fosfato o que son intolerantes a cualquier dosis de terapia con quelantes de fosfato.
Tipo: Medicamento sintético estándar constituido por un derivado de la 12,15-dioxa-2,7,9-triazaheptadecanamida. Autorizado en Estados Unidos (FDA) el 17-10-2023; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de acción local que se dirige al intercambiador 3 de sodio/hidrógeno (NHE3), un antiportador expresado en la superficie apical del epitelio del intestino delgado y el colon. La inhibición de NHE3 por tenapanor da como resultado una reducción de la absorción de sodio y una disminución de la absorción de fosfato al reducir la permeabilidad del fosfato a través de la vía paracelular.
Eficacia clínica: Tres estudios clínicos. El primero incluyó un período de tratamiento abierto, aleatorizado y controlado con activo de 26 semanas, seguido de un período de retiro aleatorizado, ciego y controlado con placebo de 12 semanas, sobre un total de 564 pacientes; durante la fase de retirada aleatoria, la concentración de fósforo aumentó en el grupo de placebo en 0,7 mg/dL en relación con los pacientes que permanecieron con tenapanor. El segundo estudio incluyó un período doble ciego, aleatorizado de 8 semanas que evaluó tres regímenes de dosificación de tenapanor (3 mg dos veces al día, 10 mg dos veces al día o un régimen de titulación). A este período le siguió una fase de retirada aleatoria controlada con placebo de 4 semanas, durante la cual los pacientes fueron reasignados 1:1 a su tratamiento actual con tenapanor o a placebo, en 219 pacientes; durante la fase de retirada aleatoria, la concentración de fósforo aumentó en el grupo grupo placebo en 0,7 mg/dL en relación con los pacientes que permanecieron con tenapanor. El tercero fue un estudio aleatorizado, de grupos paralelos, doble ciego y controlado con placebo que evaluó el efecto de tenapanor sobre el cambio en el fósforo sérico cuando se usó como terapia complementaria en pacientes en tratamiento estable con quelantes de fosfato con fósforo sérico mayor o igual a 5,5 mg/dL, en un total de 236 pacientes; durante el período de 4 semanas, el fósforo sérico disminuyó en 0,7 mg/dL en el grupo de tenapanor en comparación con el placebo.
Eventos adversos: La diarrea (43-53% de los pacientes) es la única reacción adversa reportada en al menos el 5% de los pacientes tratados.
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE)
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
