1. INTRODUCCIÓN
1.1. Las enfermedades neurodegenerativas
Las enfermedades neurodegenerativas constituyen un grupo heterogéneo de patologías caracterizadas por el deterioro funcional y estructural del sistema nervioso central y/o periférico. Dentro de este grupo destacan, por su impacto sanitario y socioeconómico, las enfermedades de Alzheimer, Parkinson y Huntington, así como la esclerosis lateral amiotrófica.
Aunque bajo esta denominación se incluyen patologías de índole muy diversa, todas comparten una serie de características: tienen un origen multifactorial, un desarrollo progresivo y una relación íntima con el envejecimiento. Todas las áreas del sistema nervioso pueden verse afectadas por un proceso neurodegenerativo, siendo específicos de cada enfermedad el foco originario del daño celular y su patrón de extensión anatómica, que se correlacionan directamente con las manifestaciones clínicas (1).
El principal problema que envuelve a las enfermedades neurodegenerativas es el desconocimiento que aún reina sobre su origen y los mecanismos patológicos subyacentes que condicionan su progresión, aunque los enormes esfuerzos consagrados a su estudio en los últimos años hayan permitido realizar ciertos avances. Esta falta de conocimiento tiene unas consecuencias nefastas, pues implica que se carezca de métodos fiables para realizar un diagnóstico temprano de estas patologías, y que los pocos tratamientos farmacológicos disponibles sirvan únicamente para paliar los síntomas, sin una capacidad real para detener o revertir su progreso clínico.
El hecho de que estas enfermedades estén estrechamente relacionadas con el envejecimiento agrava considerablemente el alcance del problema, pues los avances médicos del último siglo han impulsado un notable aumento de la esperanza de vida a nivel mundial que conlleva un aumento paralelo de la incidencia de las patologías neurodegenerativas, con los consecuentes costes sanitarios y socioeconómicos. Están más que justificados, pues, los enormes esfuerzos que está invirtiendo la comunidad científica en la comprensión de estas enfermedades.
1.1.a. Epidemiología y etiología generales
En este apartado reside, como se ha señalado anteriormente, el reto que suponen las enfermedades neurodegenerativas. La esperanza de vida se ha doblado desde los inicios del siglo XX, situándose en unos 72 años a nivel mundial y en torno a los 80 años en las regiones con mayor nivel socioeconómico (2). La consecuencia inevitable de esta buena noticia es el aumento de la incidencia de todas aquellas patologías relacionadas con el envejecimiento. Así, el cáncer y las enfermedades cardiovasculares se han erigido como las primeras causas de muerte, mientras que la incidencia de las enfermedades neurodegenerativas se está disparando, un incremento que se agudizará a medida que la esperanza de vida en los países en vías de desarrollo se aproxime a la de los países más avanzados.
Como en ocasiones una cifra vale más que mil palabras, un vistazo a la evolución epidemiológica de las principales enfermedades neurodegenerativas bastará para poner en valor el reto a enfrentar. La asociación Alzheimer´s Disease International señaló, en un informe del año 2019, que los 20 millones de enfermos de demencia que se estimaban en el año 1990 a nivel mundial se han convertido en unos 50 millones en la actualidad, y serán aproximadamente 150 millones en el año 2050. Los costes asociados suponen cifras mareantes: se estima que los gastos sanitarios derivados de las demencias solo en Estados Unidos rondan el billón de dólares, e incluso dicho informe asevera que el coste global de la demencia sería la decimoctava economía mundial si fuese un país (3). El mismo panorama se extiende a otras enfermedades neurodegenerativas: el número de enfermos de Parkinson en 2050 se quintuplicará respecto al existente en el año 1990, y se prevé que la incidencia de la esclerosis lateral amiotrófica aumente un 70 % al cabo de las dos próximas décadas (4,5).
Es importante insistir en que buena parte de este incremento se producirá en los países en vías de desarrollo, que no cuentan con sistemas sanitarios y de cuidados capaces de absorber la consiguiente demanda de servicios, de modo que la búsqueda de nuevos métodos diagnósticos tempranos y nuevas terapias capaces de modificar el curso de estas enfermedades se ha convertido en una necesidad acuciante.
El principal obstáculo al que hay que hacer frente para alcanzar este objetivo tiene que ver con las enormes lagunas de conocimiento que existen sobre el funcionamiento del sistema nervioso, en general, y del origen de las enfermedades neurodegenerativas, en particular. Se sabe que estas enfermedades tienen una etiología multifactorial, en la que multitud de mecanismos celulares y fenómenos fisiopatológicos se interconectan en el origen y el desarrollo del proceso neurodegenerativo, pero persisten numerosas incógnitas sobre los actores de este proceso y la manera en que se relacionan entre sí. Hay que imaginar el estado actual del conocimiento sobre la neurodegeneración como un mapa vial en el que faltasen numerosas carreteras y poblaciones: hasta que no se arroje luz sobre las partes del mapa que faltan es imposible conocer las causas íntimas de la enfermedad y dilucidar a qué niveles se puede actuar para prevenirla o curarla.
Se puede considerar que la degeneración del sistema nervioso, como la del resto del organismo, es una consecuencia ineludible del envejecimiento. (Figura 1) Serían otros factores como los genéticos y los ambientales los que determinen cuándo, cómo y con qué intensidad se manifiesta este deterioro estructural y funcional (6).
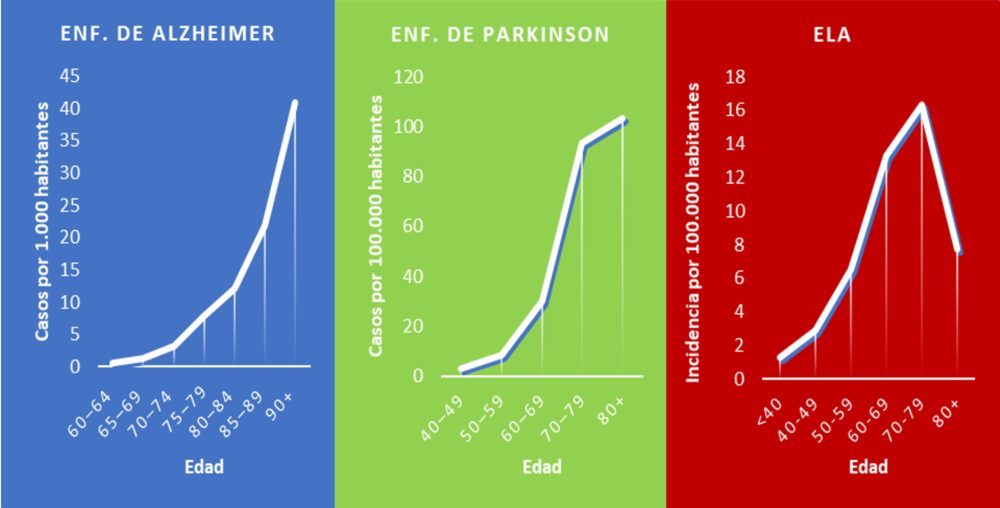
Figura 1: Relación de las principales patologías neurodegenerativas con el envejecimiento. Prevalencia de la enfermedad de Alzheimer en Europa (azul), de la enfermedad de Parkinson en el mundo (verde) e incidencia de ELA en Europa (rojo) desagregada por grupos de edad. (7–10)
En las fases últimas de la vida se constata una acumulación progresiva de alteraciones genéticas y proteicas que determinan múltiples perturbaciones de la fisiología celular a nivel del metabolismo, la homeostasis y las señales de supervivencia/muerte celular. Este fenómeno tiene un impacto aún más marcado a nivel del sistema nervioso, ya que el hecho de que las neuronas sean células postmitóticas implica que la capacidad de regeneración estructural y funcional del tejido nervioso frente a una situación de deterioro sea muy limitada. De ahí que explorar las causas primigenias de la enfermedad, y no sus manifestaciones posteriores, sea de imperiosa necesidad si se desea encontrar estrategias terapéuticas eficaces.
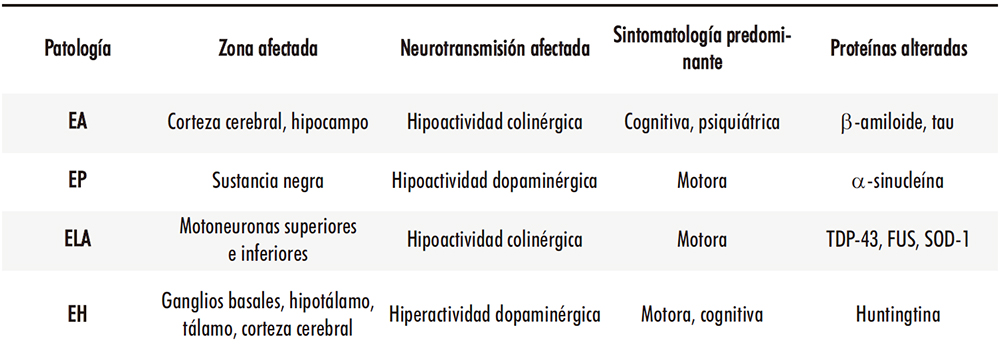
Decíamos antes que ciertos factores genéticos pueden influir decisivamente en el desarrollo de las patologías neurodegenerativas. (Tabla 1) El ejemplo más evidente es el de la enfermedad de Huntington, ligada a una mutación del gen que codifica para la proteína huntingtina y que se considera una enfermedad autosómica dominante en el 90 % de los casos, mientras que el porcentaje restante se debe a mutaciones espontáneas de dicho gen (11). En el caso de otras enfermedades como las de Alzheimer, Parkinson o la ELA, el porcentaje de casos que se pueden asociar a un defecto genético hereditario es considerablemente menor, si bien ciertas alteraciones genéticas hereditarias o adquiridas pueden participar de forma más o menos significativa en el origen del proceso patológico. A veces, determinados polimorfismos genéticos pueden modificar el riesgo de sufrir la enfermedad: uno de los casos más estudiados es el de la apolipoproteína E, cuyo alelo ε4 triplica el riesgo de padecer la enfermedad de Alzheimer, mientras que el alelo ε2 podría comportarse para sus portadores como un factor de protección (12).
Tabla 1: Origen de las enfermedades neurodegenerativas más comunes. Afectaciones más comunes a nivel proteico y genético.
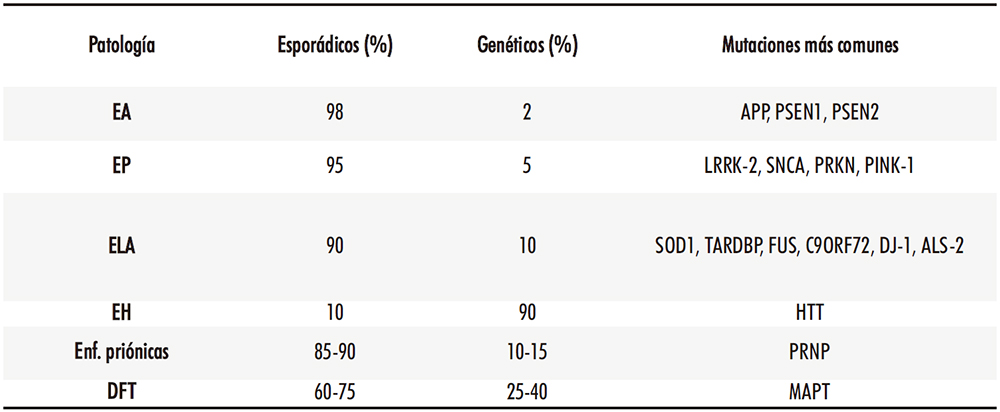
Tabla realizada con datos de (7–9,13,14)
Se ha estudiado ampliamente el papel de muchos factores ambientales en relación con las principales enfermedades neurodegenerativas, si bien la variedad en las conclusiones invita a leer los resultados con cautela. No en vano, es difícil llevar a cabo estudios consistentes en este tipo de pacientes por los largos periodos de latencia de estas enfermedades, la ausencia de criterios diagnósticos sólidos y la complejidad de los casos clínicos, en los que es frecuente que se solapen varias patologías.
Así, se ha relacionado la exposición a metales, pesticidas, disolventes y contaminantes ambientales con un mayor riesgo de desarrollar alguna enfermedad neurodegenerativa, aunque la claridad de la posible relación causa-efecto en estudios in vitro o in vivo no se ha reproducido en los estudios de exposición en humanos (15–17). En cuanto al sexo, está bien establecido que los hombres tienen más riesgo de padecer la enfermedad de Parkinson que las mujeres, y se sugiere que podría ocurrir lo contrario en la enfermedad de Alzheimer, aunque en este caso la evidencia es menos sólida (18). Se han relacionado ciertas patologías como la diabetes mellitus tipo II, las enfermedades cardiovasculares y la obesidad con un mayor riesgo de desarrollar demencia; también un historial de traumatismos craneoencefálicos, insomnio crónico, o determinadas infecciones víricas o bacterianas podrían aumentar el riesgo de padecer posteriormente alguna de las enfermedades neurodegenerativas más comunes. Finalmente, el estilo de vida también parece influir en la aparición del proceso neurodegenerativo: el hábito tabáquico podría ser un factor de protección frente a la enfermedad de Parkinson pero de riesgo frente a la enfermedad de Alzheimer y la ELA, un consumo moderado de alcohol podría ser protector frente a un aumento del riesgo si este es excesivo, y el ejercicio físico moderado y la dieta mediterránea podrían otorgar un cierto grado de protección. Hay que destacar también que una mayor formación académica e intelectual parecen constituir un factor de protección frente a las demencias (19,20).
Las enfermedades neurodegenerativas son, por tanto, el fruto de un deterioro inherente al envejecimiento que se ve agravado por la concurrencia de ciertos factores genéticos y ambientales, de modo que este declive funcional y estructural traspasa el umbral considerado como patológico. A nivel fisiopatológico, distintos factores como el estrés oxidativo, la alteración de las proteostasis y la neuroinflamación se solapan y retroalimentan abocando a las neuronas a una situación de fracaso fisiológico y funcional caracterizada por una homeostasis general y del calcio alteradas, disfunción mitocondrial, fallos en el transporte axonal y, finalmente, la muerte celular.
1.1.b. Evolución y diagnóstico de las enfermedades neurodegenerativas
Mucho de lo que sabemos sobre las enfermedades neurodegenerativas se ha descubierto a través del desarrollo de modelos in vitro e in vivo que tratan de imitar sus aspectos fisiopatológicos o clínicos más relevantes. No obstante, el grado de aproximación de estos modelos a la realidad queda en entredicho si se comparan los resultados de numerosos fármacos experimentales en estos modelos con los que arrojan en los ensayos clínicos. Queda claro, por tanto, que aún hay mucho trecho por recorrer a la hora de entender la naturaleza real de los distintos procesos neurodegenerativos, y uno de los campos donde se han depositado más esperanzas es la búsqueda de biomarcadores. El hallazgo de biomarcadores fiables de la neurodegeneración permitiría extraer información muy útil sobre el origen de la misma, así como seguir su evolución y elegir las dianas terapéuticas más adecuadas para circunstancia. Todo ello conllevaría la posibilidad de ajustar el inicio del tratamiento al del proceso patológico subyacente sin esperar a la aparición de las manifestaciones clínicas, momento en el que el daño tisular y el deterioro funcional suelen ser ya irreversibles (21).
El enfoque tradicional para predecir la aparición de una enfermedad neurodegenerativa se basaba en la búsqueda de determinadas mutaciones que se asociaban a la misma. No obstante, como ya se comentó anteriormente, los casos clínicos suelen ser de origen esporádico, e incluso muchas de las mutaciones que se relacionan con estas patologías no constituyen una causa única o necesaria, de modo que esta aproximación resulta poco adecuada. Hoy en día se consagran muchos estudios a la búsqueda de biomarcadores bioquímicos, de mayor utilidad en el diagnóstico y monitorización de estas enfermedades.
Para explicar los avances en la búsqueda de biomarcadores y cómo éstos han permitido extender considerablemente la comprensión del desarrollo del proceso degenerativo, tomaremos como ejemplo el caso de la enfermedad de Alzheimer, por ser de largo la patología más estudiada. Los niveles de proteína tau, tau fosforilada y péptido Aβ42 en líquido cefalorraquídeo (LCR) podrían ser suficientemente específicos y selectivos para el diagnóstico adecuado de la enfermedad, mientras que la ratio Aβ42/p-tau parece ofrecer un buen valor predictivo para seguir la enfermedad desde el estadio prepatológico hasta la fase de deterioro cognitivo severo. Otras técnicas que se están estudiando son la tomografía de emisión de positrones (PET) para la detección de depósitos de péptido β-amiloide, que parece tener un buen valor predictivo y diagnóstico, y la detección de la captación cerebral de 18F-fluorodeoxiglucosa mediante PET, pues su captación reducida es un signo de hipometabolismo que muestra una correlación predictiva y diagnóstica aceptable con la enfermedad (22).
El estudio de estos biomarcadores ha permitido seguir la evolución de la enfermedad de Alzheimer con una fiabilidad hasta ahora desconocida, constatándose que el verdadero inicio del proceso fisiopatológico puede tener lugar entre cinco y diez años antes que la aparición de las manifestaciones clínicas. Las primeras alteraciones observables serían el aumento de los niveles del péptido Aβ42 y la ratio Aβ42/p-tau en líquido cefalorraquídeo, así como la presencia de depósitos cerebrales de Aβ42. Años más tarde, se podría comenzar a observar el deterioro funcional a través de la detección de 18F-fluorodeoxiglucosa por PET o la resonancia magnética nuclear (RMN) de imagen funcional en las áreas cerebrales afectadas. Poco después aumentan los niveles de proteína tau en LCR, un hallazgo que se suele considerar un signo inespecífico de daño neuronal. Tras otro periodo de duración variable, que puede durar años, la disfunción cerebral se puede constatar mediante RMN de imagen volumétrica de los territorios cerebrales afectados y mediante pruebas funcionales que permiten evaluar el estatus cognitivo del paciente. Esta fase, que se conoce como periodo de deterioro cognitivo leve, anunciaría la próxima instauración de la demencia típica de la enfermedad de Alzheimer. (24,25) Como se refleja en la Figura 2, la evolución de todos estos parámetros no sería de carácter lineal, sino más bien se suele representar como funciones sigmoideas cuya pendiente reviste una importante variedad interindividual, que en ocasiones puede incluso afectar al orden de aparición de las alteraciones descritas.
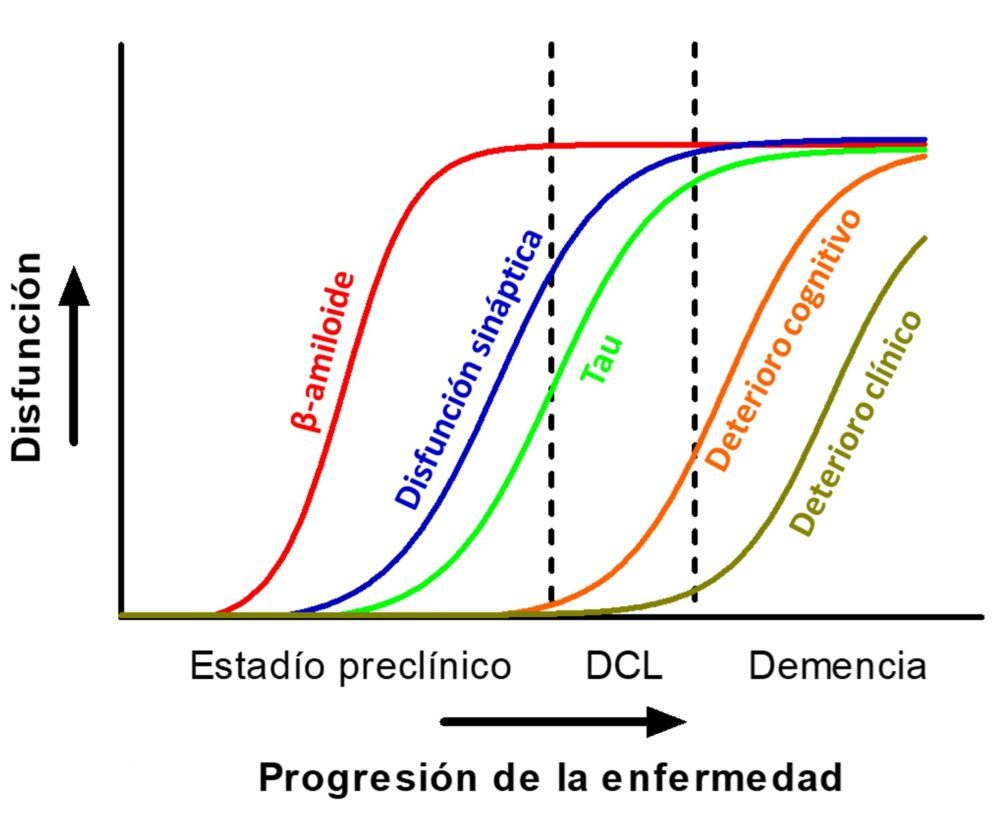
Figura 2:. Progresión temporal de los marcadores fisiopatológicos y bioquímicos de la EA en relación con la situación clínica. Adaptado de (23).
A pesar de estos avances, el diagnóstico de las enfermedades neurodegenerativas en la práctica clínica habitual se sigue basando en la sintomatología del paciente, pues los métodos en desarrollo aún carecen de la practicidad, la fiabilidad y el bajo coste necesarios para su aplicación a gran escala. Procede continuar, por tanto, con la búsqueda de métodos diagnósticos sensibles, específicos, económicos y no invasivos que permitan una detección temprana en la práctica clínica, ampliando la ventana temporal en la que se pueden poner en marcha las estrategias terapéuticas.
1.1.c Tratamiento de las enfermedades neurodegenerativas
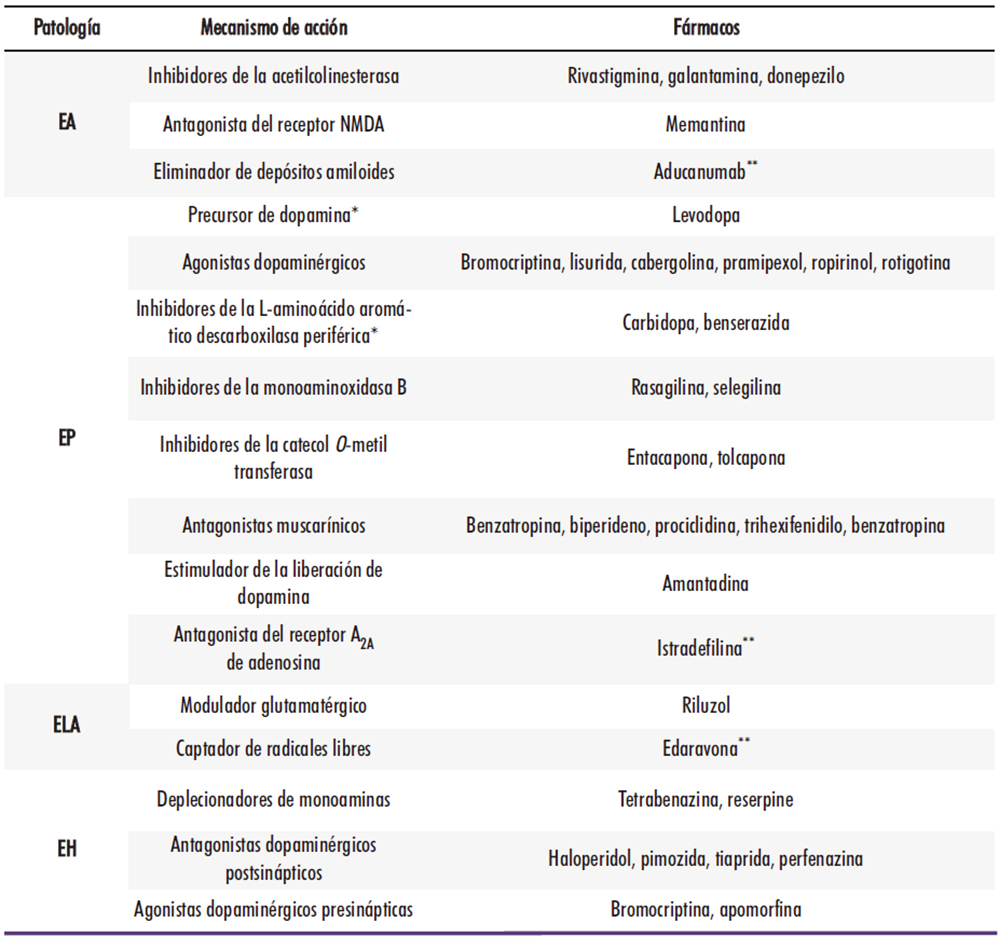
El gran problema a nivel práctico de las enfermedades neurodegenerativas, que supone un futuro oscuro para los pacientes que son diagnosticados de alguna y para sus allegados, es la carencia de tratamientos realmente efectivos. Se dispone para la mayoría de estas patologías de diversos fármacos que tienen una cierta capacidad para aliviar los síntomas o retrasar ligeramente el progreso de la enfermedad, pero ninguno puede realmente ejercer un impacto decisivo, deteniendo o revirtiendo su desarrollo. En la Tabla 2 se resumen los tratamientos disponibles hoy en día para las principales enfermedades neurodegenerativas, aunque posteriormente se describirán con más detalle patología por patología:
Tabla 2: Fármacos actualmente disponibles para el tratamiento de las principales enfermedades neurodegenerativas. (26) * Se administran conjuntamente. ** No aprobado aún por la Agencia Europea del Medicamento (sí por FDA y/o Japón
¿Por qué los enormes esfuerzos consagrados al tratamiento de estas enfermedades han dado tan poco fruto? La respuesta está, además de en la falta de métodos para el diagnóstico temprano, en el desconocimiento que envuelve el origen de estas patologías. Hasta hace poco, los tratamientos se han basado eminentemente en la corrección de determinadas anomalías que se observaban en el transcurso de la enfermedad. Así, la hipótesis colinérgica por la que la enfermedad de Alzheimer se relaciona con un agotamiento de la neurotransmisión colinérgica llevó al empleo de inhibidores de la acetilcolinesterasa como terapia de primera línea. En el caso de la enfermedad de Parkinson, donde se constata un deterioro de la neurotransmisión dopaminérgica a nivel de los ganglios basales, las terapias de reemplazo o restitución dopaminérgicas siguen constituyendo la estrategia más extendida. El problema de estas terapias es que se enfocan hacia la compensación de un daño que ya se ha producido, y no hacia las verdaderas causas que han motivado la alteración de esos procesos de neurotransmisión. Aunque hoy en día se están explorando una gran cantidad de dianas terapéuticas novedosas, en muchos casos se siguen dando palos de ciego: baste mencionar que entre la aprobación de la memantina para el tratamiento de la enfermedad de Alzheimer en 2003 y la aprobación del aducanumab en 2021, unas 300 moléculas alcanzaron la fase clínica de desarrollo sin reproducir los resultados prometedores que habían exhibido en los ensayos previos (27).
Este fracaso a la hora de trasladar las moléculas potencialmente activas de la fase preclínica de desarrollo a los ensayos clínicos es de tal magnitud que se le conoce como un “valle de la muerte” terapéutico. Ante la evidencia de que el enfoque no es el correcto, quizá por actuar cuando el daño neuronal es ya irreversible o porque las dianas a las que se dirigen los tratamientos actuales no se cuentan entre las causas primarias de la enfermedad, hoy en día la comunidad científica se afana en el desarrollo de terapias que modifiquen el curso de las patologías en cuestión, deteniendo y revirtiendo el curso de la enfermedad. (28) Para ello se están aplicando herramientas terapéuticas muy novedosas, como anticuerpos monoclonales, terapias génicas o moléculas pequeñas dirigidas a nuevas dianas más relacionadas con el origen de la enfermedad. Dentro del campo de las moléculas pequeñas merece la pena destacar el diseño de compuestos multidiana que reúnen varias actividades farmacológicas en una única entidad química, ya que podrían suponer una aproximación especialmente adecuada al tratamiento de enfermedades de etiología multifactorial, como es el caso que nos ocupa.
1.2. Enfermedad de Alzheimer y otras demencias
La demencia se puede definir como un síndrome caracterizado por un deterioro cognitivo que supera a aquel inherente al envejecimiento, en el que se observa una grave afectación de funciones cerebrales como la memoria, el pensamiento, el lenguaje, el razonamiento, la personalidad, etcétera. El 60-70 % de los casos de demencia se asocian a la enfermedad de Alzheimer, que será la que describamos con detalle en lo sucesivo. (29)
1.2.a. Epidemiología y transcurso de la enfermedad
La enfermedad de Alzheimer (EA) fue descrita en 1906 por el neurólogo alemán Alois Alzheimer cuando, examinando el cerebro de una paciente fallecida aquejada de demencia, observó que ciertas zonas del cerebro presentaban un volumen reducido, así como unas lesiones que posteriormente se denominarían placas seniles y ovillos neurofibrilares. Es una patología neurodegenerativa crónica en la que, tras una fase preclínica asintomática que puede durar hasta diez años, se va constatando un deterioro cognitivo paulatino que transita desde un estado funcional asimilable al típico del envejecimiento a un cuadro de demencia severa en el que los pacientes pierden la capacidad de llevar a cabo las tareas cotidianas y de comunicarse, además de presentar otros síntomas como agitación, paranoia e importantes alteraciones del estado de ánimo. (Figura 3) El proceso completo, desde que se alteran los primeros biomarcadores de la enfermedad hasta la muerte del paciente, puede llegar a durar más de 20 años (30).
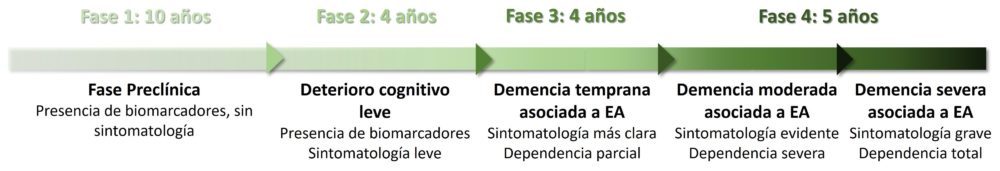
Figura 3: Fases clínicas por las que atraviesa un paciente con EA.
La EA afecta a más de 20 millones de personas hoy en día a nivel mundial, y se espera que esta cifra siga incrementándose drásticamente en las próximas décadas al calor del aumento de la esperanza de vida, ya que el principal factor de riesgo para su aparición es el envejecimiento. Esto se ilustra fácilmente con las siguientes cifras: si entre el 4 y el 6 % de la población mundial mayor de 60 años podría padecer la enfermedad, este porcentaje supera el 15 % en la franja etaria de los 75 a los 85 años, y se eleva hasta un 40 % en los mayores de 90 años. El coste de la enfermedad es inmenso a nivel económico, como ya se explicó en el inicio de esta revisión, pero también supone un terrible problema a nivel emocional y social, dado el elevado grado de dependencia que alcanzan los pacientes y la duración de la enfermedad. Por ello, supone un reto sanitario formidable que será ineludible afrontar en las próximas décadas. (29,31,32)
1.2.b. Etiopatogenia
La verdadera etiología de la EA, al igual que en el caso de las otras enfermedades neurodegenerativas, permanece oculta al saber científico actual. No obstante, se han ido formulando algunas hipótesis según avanzaban los conocimientos sobre la enfermedad. La primera hipótesis sólida sobre el origen de la EA fue la “teoría colinérgica”, formulada tras la observación de que en el cerebro de los enfermos se produce una notable pérdida de la inervación colinérgica en determinadas áreas cerebrales, especialmente en el hipocampo y el neocórtex. La disfunción de la neurotransmisión colinérgica se produce a todos los niveles: hay una pérdida de masa neuronal en esas áreas, además de una disminución en los niveles de acetilcolina y de la enzima responsable de su síntesis, la colinacetiltransferasa. Si bien estos fenómenos están bien documentados y los fármacos que emanan de esta teoría tienen un impacto (muy moderado) sobre la evolución de la enfermedad, parece evidente que esta reducción de la neurotransmisión colinérgica sería más bien la consecuencia de determinados sucesos patológicos previos, y no la causa directa y primigenia de la EA. (33) Más adelante se formuló la “teoría amiloide”, que basaría la enfermedad en la toxicidad provocada por las placas seniles de péptido β-amiloide (Aβ) que ya observara Alois Alzheimer cuando describió esta patología. (34) Más tarde, los ovillos neurofibrilares compuestos por la proteína tau hiperfosforilada se incorporarían a esta hipótesis (Tabla 3) .
Tabla 3: Resumen de la fisiopatología asociada a las enfermedades neurodegenerativas.
Los depósitos de Aβ se originan cuando la proteína precursora amiloide (PPA), por efecto de determinadas mutaciones u otras circunstancias, es procesada erróneamente por las enzimas β-secretasa y γ-secretasa de modo que se generan algunos péptidos, entre los que destaca el Aβ42, que propenden a agregarse formando primero oligómeros muy reactivos, y finalmente grandes agregados que constituyen las placas seniles. La proteína tau, por su parte, es una proteína estabilizadora de los microtúbulos axonales que sufre procesos de hiperfosforilación mediante mecanismos aún no bien elucidados en el contexto de la neurodegeneración. Esta hiperfosforilación induce un cambio conformacional que promueve su agregación en fibras insolubles que constituyen formaciones conocidas como ovillos neurofibrilares. (35) (Figura 4) La correlación entre la gravedad de la enfermedad y la presencia de depósitos proteicos insolubles está mejor establecida para la proteína tau que para el péptido Ab: de hecho, se está cuestionando severamente las bases de la teoría amiloide, bajo acusaciones de fraude en los trabajos originales, y esgrimiendo como argumento adicional la ineficacia de todos los tratamientos dirigidos a la eliminación de las placas seniles. Recordemos que sobre el aducanumab, único tratamiento aprobado que tiene como diana estos depósitos, pesan dudas más que razonables sobre su eficacia real y se teme que hayan sido intereses no estrictamente científicos o médicos los que hayan impulsado su aprobación.
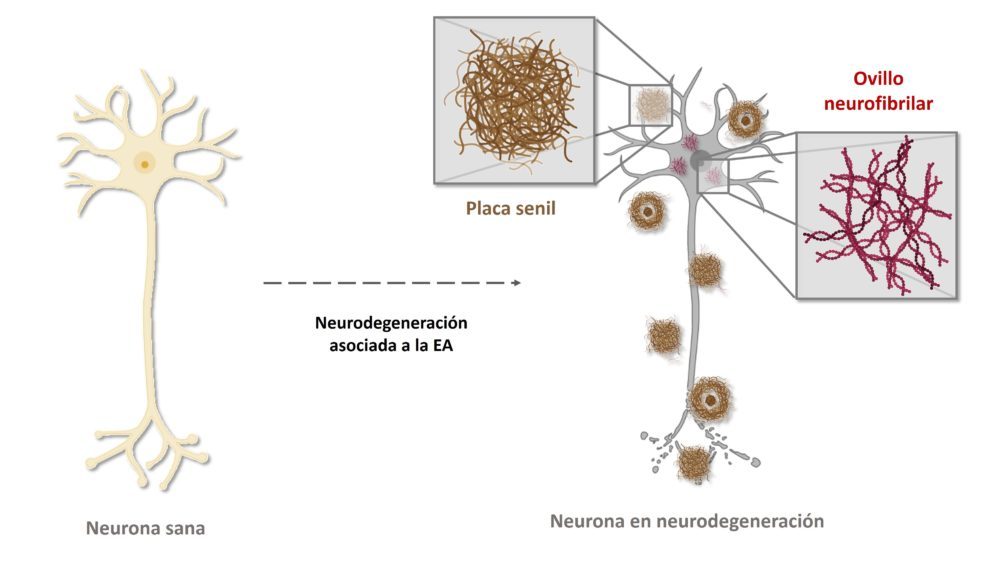
Figura 4: Formación de los macroagregados que actúan como biomarcadores en el proceso neurodegenerativo de la EA, los ovillos fibrilares como agregados de tau, y las placas seniles o neuríticas como agregados de Aβ.
Realmente, lo más probable es que ambas teorías tengan una parte de razón y a la vez ninguna, ni tan siquiera ambas juntas, sean capaces de explicar por sí mismas la enfermedad. Otros actores como la excitotoxicidad, el estrés oxidativo y la neuroinflamación podrían estar también implicados formando una red de fenómenos patológicos que constituya el verdadero origen de la EA.
1.2.c. Tratamiento
El tratamiento actual de la enfermedad de Alzheimer se limita a la atenuación de los síntomas y, si acaso, a una moderada ralentización del deterioro cognitivo. La principal herramienta disponible son los inhibidores de la acetilcolinesterasa, que tratan de reestablecer los niveles de acetilcolina en las zonas cerebrales donde se observa una disminución de este neurotransmisor. Tras la retirada de la tacrina, por problemas de hepatotoxicidad, quedan tres fármacos disponibles con este mecanismo de acción: rivastigmina, galantamina y donepezilo (Figura 5). El otro fármaco aprobado que tiene una experiencia clínica ya contrastada es la memantina, un antagonista del receptor de N-metil-D-aspartato (NMDA) que trata de limitar la excitotoxicidad suscitada por el neurotransmisor glutamato, un fenómeno que muchos estudios han identificado como parte del proceso neurodegenerativo (13). Por último, hay que mencionar al aducanumab, un anticuerpo monoclonal capaz de unirse a los depósitos de péptido β-amiloide y dirigirlos hacia su eliminación. Este fármaco ha sido aprobado recientemente (2021) solo por la agencia reguladora estadounidense FDA (Food and Drug Administration), ya que según muchos expertos y otros organismos reguladores como la Agencia Europea del Medicamento los resultados de los ensayos clínicos en términos de eficacia y seguridad no serían suficientes para permitir su aprobación (36).
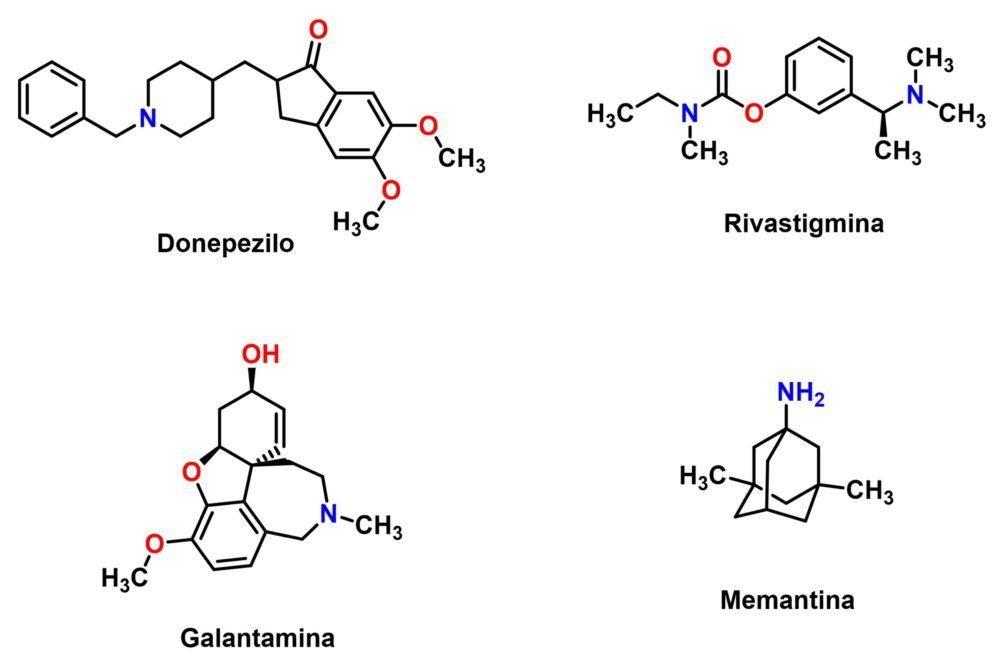
Figura 5: Estructuras de los fármacos empleados en el tratamiento de la EA.
1.3. Enfermedad de Parkinson
1.3.a. Epidemiología y transcurso de la enfermedad
La enfermedad de Parkinson (EP), al igual que la EA, tiende a ser considerada cada vez más como un síndrome que agrupa una serie de manifestaciones clínicas, y no como una entidad clínica fija y bien diferenciada. Su primera descripción corresponde al médico y político inglés James Parkinson, quien describió en 1817 un síndrome al que denominó “parálisis agitante”. Unas décadas más tarde, el estudioso de la enfermedad Jean Martin Charcot nombraría la enfermedad tal cual la conocemos en homenaje a su primer descubridor.
La EP tiene un carácter crónico y progresivo, pudiéndose distinguir varias fases. En primer lugar, se puede reconocer una larga fase preclínica caracterizada por la alteración silenciosa de ciertos biomarcadores y por el comienzo de la neurodegeneración sin que ello suponga manifestación clínica alguna. A esta etapa le sucede la fase prodrómica, en la que comienzan a manifestarse algunas alteraciones motoras leves como la rigidez o los temblores que se pueden controlar satisfactoriamente con terapias de reemplazo dopaminérgico. Por último, cuando la neurodegeneración avanza se instaura la fase clínica de la enfermedad, en la que los síntomas motores ya presentes se agravan y aparecen otros nuevos como la bradiquinesia o la inestabilidad postural, de manera que el paciente va perdiendo la capacidad de realizar sus actividades cotidianas. Además, aparece la sintomatología no motora en forma de alteraciones psiquiátricas, cognitivas y del comportamiento. Es preceptivo aumentar la potencia de la terapia dopaminérgica cuando se alcanza este estadio, pero hay que tener en cuenta las consecuencias: disquinesias, alteraciones del habla, caídas, etc. La degeneración suele llegar a una fase terminal entre 5 y 10 años después del inicio de la fase prodrómica. (37) (Figura 6).

Figura 6: Distintas fases clínicas por las que atraviesa un paciente con EP.
La EP es la segunda enfermedad neurodegenerativa más prevalente, tras la EA. Se estima que más de 7 millones de personas mayores de 60 años la padecen en todo el mundo, lo que supondría una prevalencia de entre el 0,3 y el 1 % en ese grupo etario. Al estar estrechamente relacionada con el envejecimiento, su prevalencia aumenta en los grupos de edad más avanzada, afectando al 4 % de los hombres y al 2 % de las mujeres mayores de 85 años (el sexo es un factor de riesgo en esta patología). Es bastante común, especialmente en los pacientes de mayor edad, que la EP termine generando una demencia secundaria (38).
1.3.b. Etiopatogenia
La principal característica fisiopatológica de la EP es la muerte masiva de neuronas dopaminérgicas en los ganglios basales, concretamente a nivel de la sustancia negra, de modo que queda severamente afectada la vía nigroestriatal que comunica esta región con el cuerpo estriado y que es responsable del control de la actividad motora. A nivel histológico se observa la aparición en la zona afectada de unos cuerpos de inclusión conocidos como cuerpos de Lewy, compuestos por una proteína llamada a-sinucleína que en estado normal desempeña numerosas funciones a nivel sináptico, pero que cuando sufre un procesamiento anormal tiende a formar estos agregados patológicos (Tabla 3). Durante la fase clínica se cree que pueden desaparecer hasta el 80 % de las neuronas dopaminérgicas de la vía nigroestriatal, lo que provoca que los pacientes pierdan el control de sus funciones motoras (38).
La causa de la muerte de las neuronas dopaminérgicas es una incógnita. Se han relacionado con la enfermedad algunas mutaciones que afectan a la estructura de determinadas proteínas como la propia α-sinucleína o proteínas relacionadas con la autofagia (parkina, PINK-1), la defensa antioxidante (DJ-1), la actividad lisosomal (glucocerebrosidasa) o el tráfico intracelular de vesículas (LRRK-2), entre otras. No parece que estas mutaciones sean capaces de explicar por sí solas la enfermedad en la mayoría de los casos, por lo que se les supone un papel adicional al de otros actores comúnmente presentes en el proceso de neurodegeneración: estrés oxidativo, excitotoxicidad, neuroinflamación, etcétera.
1.3.c. Tratamiento
El tratamiento de la EP es eminentemente paliativo, y debe aplicarse bajo un control constante y muy fino debido a las distintas fases por las que pasa la enfermedad y la enorme variabilidad constatable en los cuadros clínicos (39).
La principal estrategia terapéutica pasa por la aplicación de una terapia de reemplazo dopaminérgico, tratando de compensar la pérdida de la neurotransmisión dopaminérgica causada por la muerte de las neuronas nigroestriatales. El fármaco protagonista de esta aproximación es la levodopa, el precursor metabólico de la dopamina, que se puede acompañar de otros fármacos para mejorar su eficacia. Por ejemplo, en el tratamiento de elección para la fase prodrómica de la EP se combina la levodopa con la carbidopa, un inhibidor de la dopa descarboxilasa periférica que disminuye el metabolismo periférico de la levodopa, lo que permite disminuir la dosis de esta última y, por tanto, sus efectos adversos. Este tratamiento de combinación palía significativamente la sintomatología motora asociada a esta fase de la enfermedad.
Se han aprobado análogos sintéticos de la dopamina, entre los que destacan el pramiprexol, el ropinirol o la apomorfina, que se pueden utilizar en monoterapia o en combinación para el tratamiento de la EP. Soslayan los inconvenientes farmacocinéticos de la levodopa pero tienen un mayor potencial de efectos adversos psiquiátricos, eminentemente en forma de alucinaciones o alteraciones del comportamiento. Los inhibidores de la enzima catecol-O-metiltransferasa (COMT), como la entacapona o la tolcapona, se combinan frecuentemente con la levodopa ya que aumentan su semivida, cumpliendo la misma función en el tratamiento que la expuesta anteriormente para la carbidopa. Otra familia de interés son los inhibidores de la enzima monoamino oxidasa B (MAO-B), que inhiben la degradación de la dopamina y la levodopa, además de reducir la recaptación de dopamina en el espacio intersináptico. Gracias a estos efectos la rasagilina está aprobada como monoterapia para la EP, mientras que otros fármacos como la selegilina y la safinamida están incluidas en tratamientos combinados. Por último, hay que mencionar la amantadina, un fármaco comúnmente asociado a la levodopa por su capacidad para limitar la bradiquinesia provocada por el precursor de la dopamina, pero que opera por un mecanismo de acción distinto: es un antagonista de los receptores de NMDA (Figura 7).
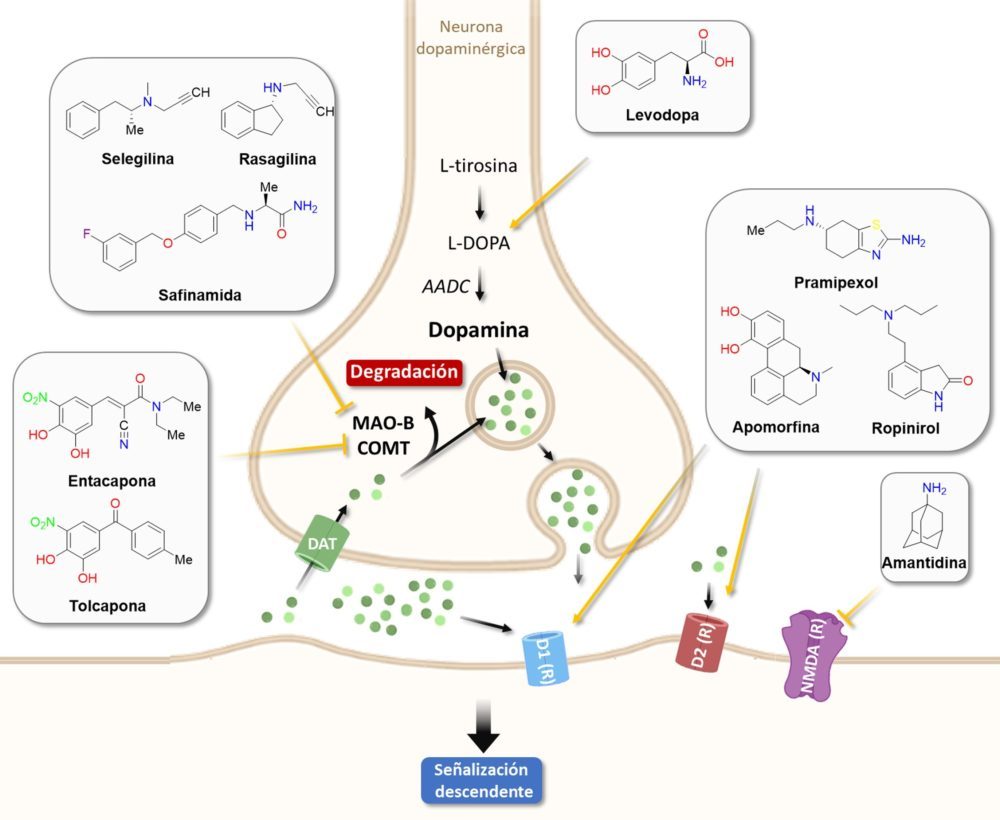
Figura 7: Vía de señalización ascendente de la transmisión dopaminérgica. Las flechas negras señalan la ruta en estado fisiológico. Los efectos de inducción o inhibición de los fármacos se encuentran en color amarillo.
Algunos fármacos anticolinérgicos de acción central, especialmente la benzotropina y el trihexifenidilo, se utilizan ocasionalmente para el tratamiento de los temblores. Su uso se limita a los pacientes más jóvenes, pues se asocian a efectos adversos como estreñimiento, anuria o confusión, que son leves en estos pacientes pero que cobran importancia en los pacientes de edad avanzada.
En EE. UU. y Japón también se ha aprobado un nuevo fármaco, la istradefilina, que se utiliza en combinación con levodopa y carbidopa cuando se producen periodos de ineficacia del tratamiento. Es un antagonista de los receptores de adenosina A2A, si bien no se conoce con exactitud cómo este efecto farmacológico tiene un impacto en la EP (40).
Además de los tratamientos específicos para la sintomatología motora de la EP, los pacientes suelen requerir la administración de otras terapias para el control de los síntomas cognitivos o psiquiátricos, además de intervenciones no farmacológicas para limitar el impacto de la enfermedad en su vida cotidiana. La revisión de Zesiewicz ofrece un buen resumen sobre este tema (37).
1.4. Esclerosis lateral amiotrófica
La esclerosis lateral amiotrófica (ELA) tiene una incidencia, afortunadamente, muy inferior a la de las dos enfermedades analizadas anteriormente, pero tiene una repercusión social muy significativa debido a la relativa juventud de los pacientes a los que puede afectar y al terrible porvenir que les espera, pues se trata de una enfermedad que supone un velocísimo deterioro funcional y físico que culminan con la muerte del paciente, sin que exista tratamiento alguno capaz de frenar significativamente su progresión.
1.4.a. Epidemiología y transcurso de la enfermedad
El cuadro clínico de la ELA viene determinado por la degeneración rápida de las motoneuronas superiores e inferiores, que motiva una parálisis muscular que progresa rápidamente. En la primera fase de la enfermedad el paciente sufre una debilidad muscular general, que puede venir acompañada de otras manifestaciones motoras como espasticidad y fasciculaciones. Según avanza la afectación de las motoneuronas, la debilidad se acentúa y aparecen las dificultades en el habla y la deglución, además de cuadros de insuficiencia respiratoria que son la causa de muerte más común de los pacientes de ELA. Inicialmente considerada una enfermedad neuromuscular, hoy en día se la incluye en el grupo de las patologías neurodegenerativas por la presencia en más de la mitad de los casos de otros síntomas de carácter cognitivo o psiquiátrico. Hasta un 13 % de los pacientes de ELA desarrollan demencia frontotemporal (DFT), una patología con la que parece guardar una relación muy estrecha: de hecho, se propone que ambas enfermedades podrían constituir los extremos fenotípicos de una misma patología, existiendo cuadros clínicos intermedios en los que conviven aspectos característicos de ambas enfermedades (41).
La prevalencia mundial de la ELA oscila entre los 6,1 y los 8,4 casos por cada 100.000 habitantes: en torno a medio millón de enfermos en términos absolutos, con una prevalencia algo mayor en hombres que en mujeres (1,4:1). Se considera que hay dos variantes fundamentales de la ELA: ELA familiar (ELAf), asociada a la herencia genética, y ELA esporádica (ELAe). La ELAf supone un 5-15 % de los casos totales de la enfermedad y tiene una edad de inicio en torno a los 65 años, claramente mayor que los 55 años de edad media de inicio de la ELAe. Ambas variantes destacan en la misma medida por su agresividad, pues el deterioro clínico es muy veloz y la esperanza de vida media tras el diagnóstico oscila entre los dos y los cuatro años, aunque hay algunas variantes muy poco prevalentes en las que la progresión de la enfermedad se alarga incluso décadas (42).
1.4.b. Etiopatogenia
Poco se sabe sobre el origen de la ELA: parece estar relacionado con diversos fenómenos como la alteración de la proteostasis y del transporte axonal, el estrés oxidativo, la excitotoxicidad y la neuroinflamación; pero la evidencia de la que se dispone no es muy firme y está lejos de explicar satisfactoriamente por qué se inicia el proceso de degeneración de las motoneuronas. Se han descubierto mutaciones en más de 30 genes que tienen una relación directa con la ELAf, siendo las más comunes, por este orden, las de los genes c9orf72, SOD1, FUS y TARBP. La expresión de las proteínas mutadas asociadas a estos genes (SOD-1, TDP-43, FUS) (Tabla 3) produce la agregación patológica de las mismas, además de la consecuente toxicidad provocada por la incapacidad de estas proteínas para ejercer su función. El componente genético no es exclusivo de la ELAf, pues hasta el 60 % de los casos de ELAe también estarían asociados en mayor o menor medida con diversas alteraciones genéticas, muchas de las cuales se repiten a lo largo del eje ELA-DFT. Este hallazgo sugiere que la ELA podría ser una enfermedad oligogénica, en la que varias alteraciones de baja penetración génica pueden coexistir para dar también lugar a los casos esporádicos (43,44).
1.4.c. Tratamiento
El panorama terapéutico de la ELA casi merece el apelativo de desolador, pues apenas hay tratamientos disponibles, y la eficacia de los que existen se limita a aumentar en unos meses la esperanza de vida de los pacientes. Detrás de este hecho está el desconocimiento absoluto del origen y desarrollo de la enfermedad, sobre los que pesan aún muchas más incógnitas que en el caso de las enfermedades de Alzheimer y Parkinson.
El primer fármaco aprobado para la ELA, y único en Europa, fue el riluzol, un anticonvulsivante que fue reposicionado en los años 90 tras haber demostrado una relativa utilidad frente a la ELA en un ensayo clínico (Figura 8). No se conoce con exactitud su mecanismo de acción, aunque se le suele atribuir una actividad antiglutamatérgica que podría reducir la excitotoxicidad a nivel de las motoneuronas. Se estima que aumenta la esperanza de vida de los pacientes en torno a seis meses, aunque con poco impacto sobre su sintomatología motora. En 2017 la FDA autorizó en EE. UU. la edaravona, un derivado de pirazolona cuyo mecanismo de acción se suele relacionar con su elevada capacidad antioxidante y captadora de radicales libres, si bien existen muchas dudas al respecto. La edaravona se ha asociado con una cierta ralentización en el progreso de la enfermedad, si bien el impacto sobre la esperanza de vida no parece ser muy significativo (45).
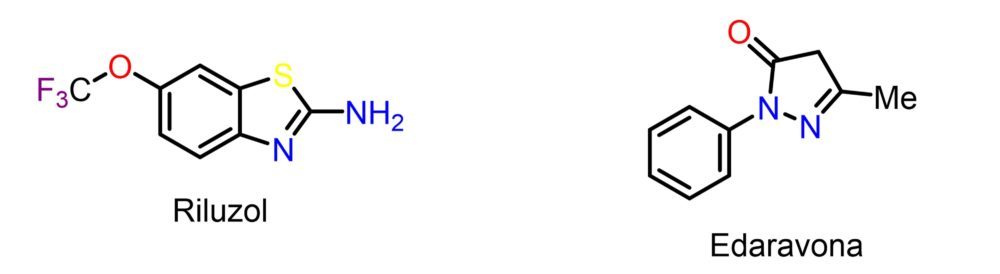
Figura 8: Estructura química de los dos fármacos más representativos del tratamiento farmacológico de la ELA. .
Recientemente se ha aprobado en EE. UU. el uso compasivo del fasudil para el tratamiento de la ELA, aunque una vez más se ha cuestionado ampliamente su eficacia. Actualmente hay varios ensayos clínicos en marcha que tratan de dilucidar si tiene un impacto real en la calidad de vida y la supervivencia de los pacientes. El fasudil es un inhibidor de la Rho-quinasa cuya acción terapéutica se ha asociado a la modulación de la respuesta inflamatoria de la microglía y a la protección de la integridad axonal, si bien no está firmemente establecido su mecanismo de acción. (46)
La posibilidad de que la ELA forme en realidad un continuo patológico con la DFT y tenga un carácter oligogénico abriría las puertas a la medicina personalizada, dirigiendo tratamientos específicos para las particularidades genéticas que presente cada caso. Ya se están ensayando algunas terapias génicas para esta patología, que serán revisadas posteriormente al hablar de la proteostasis como diana terapéutica.
1.5. Enfermedad de Huntington
La enfermedad de Huntington tiene un origen genético claramente establecido, lo que la diferencia de otras enfermedades neurodegenerativas. Desgraciadamente, este mejor conocimiento sobre su origen no se ha traducido en el desarrollo de tratamientos eficaces, de modo que las perspectivas vitales de los afectados continúan siendo muy desfavorables.
1.5.a. Epidemiología y transcurso de la enfermedad
La enfermedad de Huntington (EH) lleva el nombre del médico estadounidense George Huntington, quien la describió por vez primera en el año 1872. Se la conoce también como corea de Huntington, debido a que los pacientes presentan unos movimientos bruscos e involuntarios de las extremidades muy característicos, conocidos como movimientos coreicos. Esta enfermedad, que se suele describir como una tríada de alteraciones motoras, cognitivas y emocionales, suele presentar tres estadios bien diferenciados. En una fase temprana comienzan a observarse alteraciones en la coordinación de los pacientes y algunos pequeños movimientos involuntarios, que se acompañan de un ánimo deprimido o irritable y ciertas dificultades para el pensamiento y la resolución de problemas. La enfermedad progresa hacia una fase donde los movimientos coreicos pasan a ser la manifestación predominante, acompañadas de disartria y disfagia. Las dificultades para controlar los movimientos voluntarios, sumadas al deterioro cognitivo, incapacitan progresivamente a los pacientes para la realización de las tareas cotidianas y laborales. Las manifestaciones motoras en el último estadio de la enfermedad varían desde cuadros de corea muy severos hasta otros en los que predominan la bradiquinesia y la rigidez. Los pacientes suelen desarrollar una demencia severa que los incapacita para comunicarse, aunque a menudo retienen una cierta capacidad de comprensión. Muchos pacientes mueren por las complicaciones derivadas de la disfagia, ya sea por inanición o por asfixia, aunque las caídas e incluso los suicidios representan también un porcentaje significativo de las muertes derivadas de la EH (47).
La EH destaca por su precocidad dentro de las enfermedades neurodegenerativas, pues el inicio de los síntomas suele tener lugar entre los 35 y los 50 años, aunque realmente puede ocurrir en cualquier punto entre la niñez y la vejez. La esperanza de vida media tras el comienzo de la enfermedad ronda los 15 a 20 años, con pacientes que pueden llegar a convivir con ella más de 40 años.
La prevalencia de la EH a nivel mundial presenta una notable variabilidad interregional: ronda los 5 a 10 casos por cada 100.000 habitantes en América del Norte, Europa y Oceanía, mientras que en Asia y África es mucho menor. Esto se puede atribuir al carácter hereditario de la enfermedad, pero también a la menor capacidad diagnóstica que puedan poseer los países en vías de desarrollo, especialmente en el caso de África. Aunque hay una cierta correlación con la edad, la forma juvenil de la EH (inicio de los síntomas antes de los 20 años) supone en torno al 5 % de los casos (48).
1.5.b. Etiopatogenia
La EH es una patología autosómica dominante ligada a una mutación del gen HTT en el cromosoma 4 que codifica para la proteína huntingtina. (Tabla 3) Concretamente, se produce una expansión de las repeticiones de tripletes CAG que se traduce en un fragmento de poliglutamina anormalmente largo. Esta anomalía estructural provoca el plegamiento incorrecto de la proteína, ya que estos fragmentos favorecen el establecimiento de interacciones hidrofóbicas indebidas dentro de la misma proteína o con otras unidades de huntingtina. Las consecuencias a nivel fisiopatológico parecen derivar eminentemente de la generación de agregados patológicos de la proteína anormalmente plegada, que desatan un proceso neurodegenerativo que se inicia en los ganglios basales, concretamente en el núcleo caudado y el putamen, y que se extiende progresivamente hacia otros ganglios basales y zonas adyacentes como el tálamo, el hipotálamo y la corteza cerebral. Este patrón de extensión anatómica explica la evolución del cuadro clínico hacia una tríada de síntomas motores, cognitivos y psiquiátricos. No hay que descartar que la pérdida de las funciones fisiológicas de la huntingtina desempeñe un papel importante en el origen de la enfermedad, aunque al no estar bien caracterizadas estas funciones no se sabe mucho al respecto (49).
1.5.c. Tratamiento
No existe tratamiento para la EH, aunque es común administrar algunos fármacos para el control de los síntomas motores y psiquiátricos. La afectación de varios núcleos a nivel de los ganglios basales y, por ende, de distintos circuitos neuronales interconectados provocan un desajuste muy complejo en los niveles de varios neurotransmisores, imposible de ajustar de forma individual. Como la sintomatología motora parece el resultado de una hiperactividad dopaminérgica imputable a la sustancia negra, la acción farmacológica se orienta a la reducción de dicha neurotransmisión mediante distintos mecanismos. En primer lugar, se pueden utilizar fármacos inhibidores de los transportadores vesiculares de monoaminas, que causan un agotamiento de las reservas neuronales de dopamina. Con este mecanismo de acción actúan la tetrabenazina, uno de los pocos fármacos aprobados específicamente para el tratamiento de la EH; y la reserpina, cuyo uso se está limitando por sus mayores efectos periféricos. También se utilizan antagonistas de los receptores dopaminérgicos postsinápticos (neurolépticos), preferentemente aquellos que exhiben una mayor selectividad hacia los receptores D2: haloperidol, pimozida, tiaprida y perfenazina. Aunque se emplean a dosis menores que cuando se prescriben como antipsicóticos, pueden causar reacciones extrapiramidales y otros efectos adversos que se pueden confundir con los síntomas de la propia enfermedad. Por último, a veces se utilizan también agonistas de receptores dopaminérgicos presinápticos a dosis bajas, entre los que destacan la apomorfina y la bromocriptina. Sea cual sea el tratamiento, es común la administración concomitante de benzodiacepinas como el clonazepam o el diazepam para el control de la sintomatología no motora (26).
El origen genético de la enfermedad fomenta la búsqueda de terapias génicas que permitan evitar la producción errónea de la proteína huntingtina. Esta aproximación, que parece una de las más prometedoras para el abordaje terapéutico de la EH, se analizará con más detalle posteriormente.
2. ESTRÉS OXIDATIVO
El término estrés oxidativo hace referencia a una concentración de especies reactivas de oxígeno y nitrógeno (ROS y RNS, respectivamente) que resulta nociva para las células, llegando a promover la aparición de agregados proteicos tóxicos y de alteraciones genéticas, la disfunción de ciertos orgánulos como la mitocondria y el retículo endoplasmático, e incluso la muerte celular. Estas especies tóxicas se generan de forma fisiológica por ligeros defectos en las reacciones de reducción-oxidación del organismo, que no logran completar la reducción del oxígeno molecular a agua. Normalmente la presencia de estas especies oxidantes en bajas concentraciones no supone un grave problema, ya que el organismo posee múltiples herramientas para neutralizarlas. Sin embargo, bajo ciertas situaciones patológicas como la neurodegeneración, los sistemas que velan por mantener la homeostasis de estas especies se pueden ver saturados, momento en que pueden desatar todo su potencial tóxico (50).
2.1 Las especies oxidantes
A pesar de que es común hablar del estrés oxidativo como ROS, es importante recalcar que el término también incluye a las especies reactivas de nitrógeno (RNS) y a cualquier especie hiperreactiva generada a partir del metabolismo del oxígeno. Estos agentes oxidantes suelen ser de naturaleza radicalaria, pero también incluyen otras especies que, aunque no lo son, generan radicales fácilmente (50).
La formación de ROS comienza con la transferencia de un único electrón al oxígeno, generándose el radical superóxido (O2–), el cual es a su vez precursor de gran parte de las otras especies oxidantes. (Figura 9) Este radical puede evolucionar de dos formas distintas: con la captación de un protón del medio, que dará lugar al radical HO2·, o por mediación de la superóxido dismutasa (SOD), que catalizará su transformación en peróxido de hidrógeno (H2O2). De nuevo, esta molécula puede seguir dos rutas, una enzimática mediada por la catalasa gracias a la cual se convertirá de nuevo en agua y oxígeno; o puede sufrir la reacción de Fenton, catalizada por ciertos cationes metálicos como el catión ferroso, dando lugar a otras especies como el anión hidroxilo (HO–) y el radical hidroxilo (HO·). Esta última especie es una de las más reactivas y, por tanto, de las más nocivas para todos los componentes celulares (51,53).
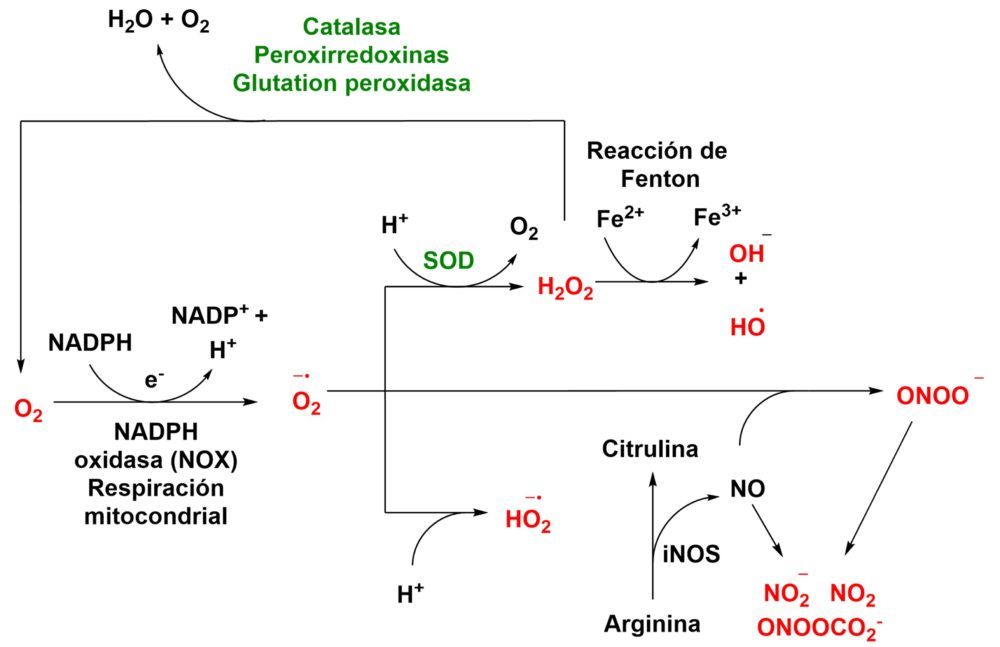
Figura 9: Formación de las principales especies reactivas de oxígeno y nitrógeno. (51,52)
Por otra parte, las especies reactivas de nitrógeno (RNS) se originan por acción de la enzima óxido nítrico sintasa (NOS), que produce óxido nítrico (NO·) durante la transformación de la arginina en citrulina. Este óxido nítrico reacciona de forma espontánea con el radical superóxido, generando el anión peroxinitrito (ONOO–). Además de estas RNS, también es necesario destacar el papel de otras como el dióxido de nitrógeno (NO2), el anión nitrito (NO2–) o el anión nitrosoperoxicarbonato (ONOOCO2–) (52,54).
A pesar de que se ha puesto el foco sobre los efectos tóxicos de estas especies oxidantes, resulta preciso ahondar en el papel fisiológico que también ejercen en el organismo. En primer lugar, cumplen una función imprescindible dentro del sistema inmunitario, ya que muchas de las células de este sistema producen especies oxidantes para atacar a patógenos invasores. Además, se ha comprobado que median en otras muchas funciones esenciales para la supervivencia celular como la regulación de la mitosis y de la señalización del crecimiento celular, e incluso en la producción de ATP. Sin embargo, su función más importante quizás sea la debida al óxido nítrico, el cual además de mediar en la función inflamatoria y el tono vascular, desempeña un papel vital en la plasticidad sináptica actuando prácticamente como un neurotransmisor (55).
Como se ha señalado anteriormente, la formación de estas especies oxidantes tiene su origen en ciertas funciones celulares como el procesamiento postraduccional asociado al retículo endoplasmático, el metabolismo asociado a los peroxisomas, la acción de ciertas enzimas oxidantes como la NADPH oxidasa (NOX) o las monoamino oxidasas A y B (MAO-A, MAO-B), y la actividad de la mitocondria (53). Este orgánulo resulta ser el que más ROS genera, principalmente anión radical superóxido, por la acción de la cadena transportadora de electrones. De hecho, son los complejos I y III de dicha cadena los principales contribuyentes al aumento del estrés oxidativo, especialmente en un órgano que obtiene tanta energía a través de esta ruta como es el cerebro. Cabe destacar que la labor que cumple la mitocondria también se vuelve progresivamente más tóxica, ya que al soportar un ambiente tan oxidante se ven afectados progresivamente a nivel estructural ciertos componentes mitocondriales como las membranas y el ADN mitocondrial, lo cual produce un estado degenerativo que no hace sino maximizar la producción de estas ROS (51,56).
2.2. Respuesta antioxidante endógena
2.2.a. Enzimas y moléculas pequeñas
Dado que estas especies oxidantes se generan de forma fisiológica, el organismo ha logrado desarrollar una serie de mecanismos para defenderse frente a este estímulo nocivo, principalmente mediante moléculas pequeñas y enzimas. En el primer grupo encontramos múltiples ejemplos de sustancias antioxidantes como las vitaminas C y E o el glutatión. Este último ejemplo merece una mención especial, ya que se trata de un sustrato central en la reparación del daño por estrés oxidativo. Se trata de un tripéptido (g-glutamato-cisteína-glicina) con propiedades antioxidantes intrínsecas que además actúa como donador de electrones durante la acción de la glutatión peroxidasa. Por su parte, esta enzima cataliza la reducción de lípidos peroxidados o de peróxido de hidrógeno a agua. Una acción similar presentan las peroxirredoxinas, responsables de la reducción de más del 90 % del peróxido de hidrógeno, además de la de otros peróxidos como el peroxinitrito. Otras enzimas que participan en la neutralización de estas especies oxidantes son la superóxido dismutasa (SOD), la tiorredoxina o las catalasas (50).
2.2.b. Nrf2
Todos estos ejemplos de enzimas con capacidad antioxidante y citoprotectora, además de otras muchas que se han quedado en el tintero, provienen de las secuencias de Elementos de Respuesta Antioxidante (ARE) del ADN, cuya expresión depende de una serie de factores de transcripción, siendo el más relevante el factor de transcripción Nrf2. Nrf2 es una proteína de vida corta sujeta a un control estricto por varios mecanismos, siendo el más estudiado el ejercido por la proteína Keap-1. (57) Usualmente, Nrf2 se encuentra unido a dos monómeros de Keap-1 por sendas regiones presentes en el factor de transcripción (DLG y ETGE) con distinta afinidad por Keap-1. Así, el heterotrímero Nrf2-(Keap-1)2 es reconocido por el complejo ligasa E3 CRL3 de la maquinaria del proteasoma, que dirige la poliubiquitinación y posterior degradación de Nrf2 (58).
Sin embargo, en una situación de elevado estrés oxidativo, las especies oxidantes o electrófilas interactúan con la región sensible de Keap-1, que consiste en una serie de residuos de cisteína presentes en la región intermedia (Cys272 y Cys278) y el dominio BTB (Cys151) de Keap-1. Normalmente los grupos tiol pertenecientes a estos residuos se encuentran estabilizados por iones de zinc, pero en presencia de especies oxidantes o electrófilos producen un cambio conformacional en Keap-1. De este modo se impide que se forme el complejo Nrf2/Keap1/CRL3 y Nrf2 puede translocarse al interior del núcleo, formar un heterodímero con la proteína Maf e iniciar la transcripción de la secuencia ARE (59,60).
Existe una teoría alternativa según la cual Nrf2 no queda completamente libre, sino que solo se rompe la unión a Keap-1 de baja afinidad (mediada por la región DLG de Nrf2), de modo que solo un monómero de Keap-1 queda unido al factor de transcripción. Esto no es suficiente para promover la degradación de Nrf2, pero tampoco para permitirle translocarse al interior del núcleo. Así, Keap-1 quedaría paulatinamente secuestrado, incapaz de unirse al Nrf2 sintetizado de novo, permitiendo por tanto que este se transloque al interior del núcleo (59).
Una vez Nrf2 se encuentra en el interior del núcleo se une a proteínas pequeñas del tipo Maf formando heterodímeros habilitados para la transcripción de los elementos de respuesta antioxidante (ARE), entre los que podemos encontrar enzimas y proteínas con distintas funciones como la neutralización directa de especies oxidantes, el metabolismo de xenobióticos o la regulación de los procesos de supervivencia celular y procesamiento proteico (61).
La función antioxidante es posiblemente la función mejor caracterizada de Nrf2, ya que promueve la expresión de varias enzimas con una potente actividad antioxidante, como son la hemooxigenasa 1 (HO-1), la NAD(P)H deshidrogenasa quinona 1 (NQO1), y otras ya mencionadas como distintas enzimas relacionadas con la homeostasis del glutatión (glutatión peroxidasa, glutation reductasa o subunidades de la glutamato cisteina ligasa), tiorreductasas o peroxirreductasas. (61) La HO-1 es la encargada del catabolismo del grupo hemo, liberando Fe2+, monóxido de carbono, para el cual se han descrito varias actividades neuroprotectoras; (62) y biliverdina, la cual se reduce a bilirrubina, que es otro compuesto antioxidante y neuroprotector. En cuanto a la NQO1, su principal acción es la de reducir moléculas endógenas y exógenas que se encuentran en estado oxidado. Esta enzima también participa en el metabolismo de xenobióticos, al igual que otras proteínas y enzimas cuya expresión está controlada por Nrf2: la aldo-cetorreductasa o la aldehído deshidrogenasa (metabolismo de fase I); la UDP glucuronosiltransferasa o la glutatión-S-transferasa (tipo II); y otras proteínas asociadas con la resistencia y expulsión de fármacos (tipo III) (63).
En cuanto a otras funciones reguladoras, Nrf2 también promueve la expresión de proteínas relacionadas con el sistema autofágico de degradación proteica (p62/SQSTM1) (64); así como de ciertas proteínas antiapoptóticas y reparadoras del ADN, como Bcl-2 (65).
En cuanto a la participación de Nrf2 en el proceso inflamatorio, se debe admitir que existe una cantidad importante de contradicciones entre la expresión de los genes proinflamatorios que estimula este factor de transcripción (CD36, IL-17D), los que inhibe (IL-1β o IL-6), y la actividad de las proteínas antioxidantes cuya expresión también promueve. En cualquier caso, el efecto que se genera finalmente tiene más de efecto antinflamatorio que de proinflamatorio (66).
2.3. Estrés oxidativo y neurodegeneración
La relación entre el estrés oxidativo y la neurodegeneración ha sido estudiada con gran profundidad, ya que es un foco de gran inestabilidad que alimenta otros mecanismos fisiopatológicos de la neurodegeneración, induciendo el plegamiento anómalo de proteínas, magnificando el daño mitocondrial y tisular e incluso promoviendo la apoptosis (Figura 10).
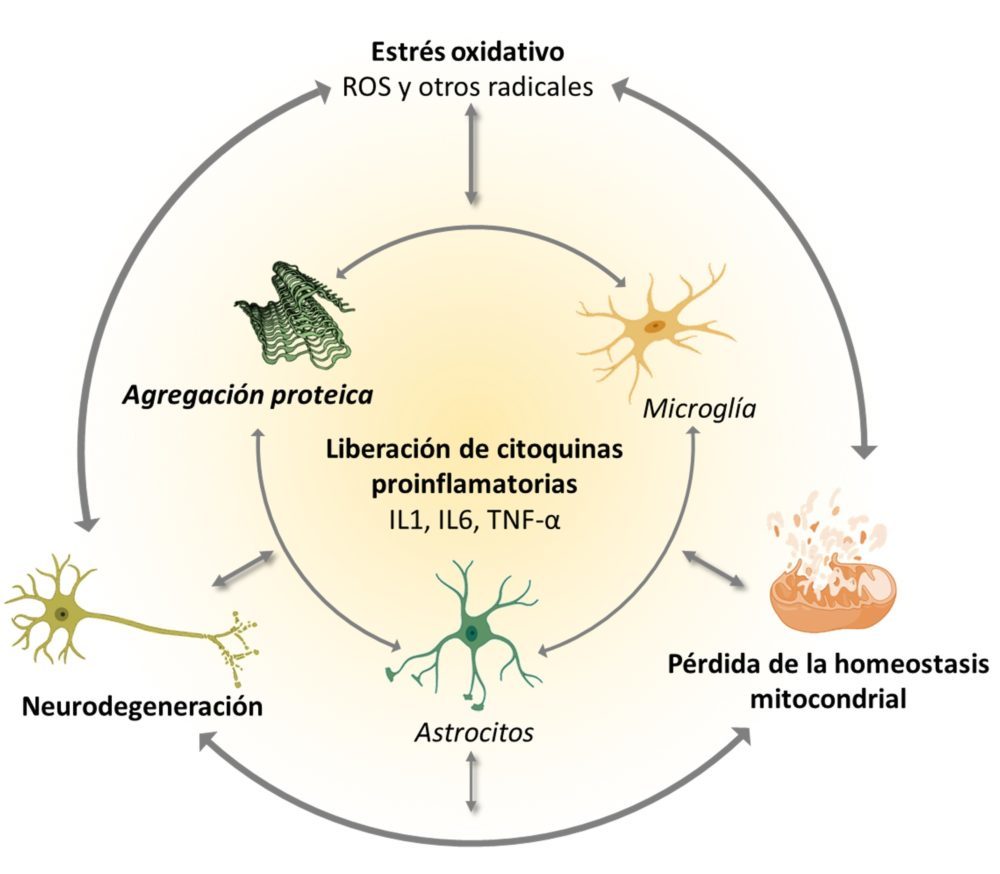
Figura 10: Ciclo de retroalimentación entre los mecanismos fisiopatológicos más generales de la neurodegeneración. (51,56)
La oxidación de ciertos aminoácidos en presencia de una alta concentración de especies oxidantes favorece el plegamiento anómalo de las proteínas, lo que motiva la pérdida de su función fisiológica y su posterior agregación patológica. El estrés oxidativo también puede producir alteraciones en la unidad 26S del proteasoma, disminuyendo considerablemente la capacidad degradadora de proteínas y fomentando aún más la agregación (67). Además, estos agregados proteicos son capaces de quelar iones de cobre o de hierro susceptibles de catalizar la reacción de Fenton, lo que produce un aumento aún mayor de las especies oxidantes.
El estrés oxidativo afecta de forma especialmente potente a la función mitocondrial, un orgánulo de vital importancia para la supervivencia celular. La alteración más básica y profunda sucede sobre el ADN mitocondrial, promoviendo la aparición de proteínas mutadas que no pueden ejercer adecuadamente su función. Este es el caso de las proteínas de la cadena de transporte de electrones que, además, pueden ser directamente oxidadas o nitradas por las ROS/RNS, impidiendo su adecuada función respiratoria (68). Por otra parte, se pueden producir procesos como la peroxidación de lípidos y proteínas de las membranas mitocondriales o un aumento sustancial de la concentración de Ca2+. Estas alteraciones causan cambios drásticos en la permeabilidad de las membranas mitocondriales e incluso la formación de poros de transición de permeabilidad (PTPs) que ponen en riesgo su integridad. La aparición de estos PTPs conlleva la salida al citosol del citocromo C, fenómeno que promueve la apoptosis mediada por la activación de las caspasas. Cabe destacar que todas estas alteraciones de la estructura mitocondrial obstaculizan aún más la respiración celular, aumentando la producción de especies oxidantes y agravando todo el proceso degenerativo (69) .
El estrés oxidativo también promueve una respuesta inflamatoria local, ya que la rotura de barreras biológicas acaba por activar a la microglía y los astrocitos circundantes. Esta inflamación conlleva la producción de citoquinas proinflamatorias y de más ROS y NO, que retroalimentan el proceso de daño tisular (70).
Como se ha podido comprobar, existen varias formas de toxicidad directa e indirecta mediadas por las ROS y RNS que producen un daño celular y tisular enorme y acaban por conducir a la muerte celular, ya sea por alteración de las membranas y la función mitocondrial, por un exceso en la respuesta al estrés del retículo endoplasmático o por la activación directa de vías de señalización apoptóticas mediada por la respuesta inmunitaria (71).
Todas las alteraciones están íntimamente relacionadas entre sí, combinando los mecanismos de toxicidad y agravando el proceso neurodegenerativo en muchas enfermedades del SNC.
En la enfermedad de Parkinson se ha probado la existencia de un círculo vicioso en el cual el ambiente oxidante generado por la disfunción del metabolismo de la dopamina produce la oxidación de la a-sinucleína. La a-sinucleína oxidada es más proclive a la agregación y, además, no es reconocida por el sistema proteasómico, por lo que los niveles de agregados de esta proteína aumentan drásticamente. Esta situación provoca alteraciones en el sistema de almacenaje vesicular que producen la liberación de más dopamina al citosol, lo cual incrementa aún más el estrés oxidativo y conduce a la neurona a una degeneración imparable (72,73). Este ciclo de retroalimentación entre los agregados patológicos de una proteína y el estrés oxidativo se da también en la EA, la EH y la ELA. En la EA se ha comprobado que el estrés oxidativo fomenta la agregación de Ab, y que el acúmulo de sus agregados puede producir daño mitocondrial que conduce a la formación de más especies oxidantes (74). La agregación de la huntingtina (Htt), propia de la enfermedad de Huntington, también es fomentada por las especies oxidantes, y la aparición de estas es promovida por un defecto en el funcionamiento de la cadena transportadora de electrones mitocondrial mediado por la propia Htt. (75) En cuanto a la ELA, se ha comprobado que el acúmulo de especies oxidantes, ya sean de oxígeno o de nitrógeno, conduce a la agregación tanto de SOD-1 como de TDP-43, en función de la mutación que expresen los pacientes (76,77).
2.4. Tratamiento
2.4.a. Productos naturales con propiedades antioxidantes
Existen muchas sustancias de origen natural que poseen cualidades antioxidantes: prácticamente cualquier polifenol, flavonoide e incluso ciertos ácidos grasos poliinsaturados presentes en animales y plantas poseen estas cualidades. A continuación, citaremos aquellos compuestos de mayor potencia o más implementados en la terapia antioxidante (Figura 11).
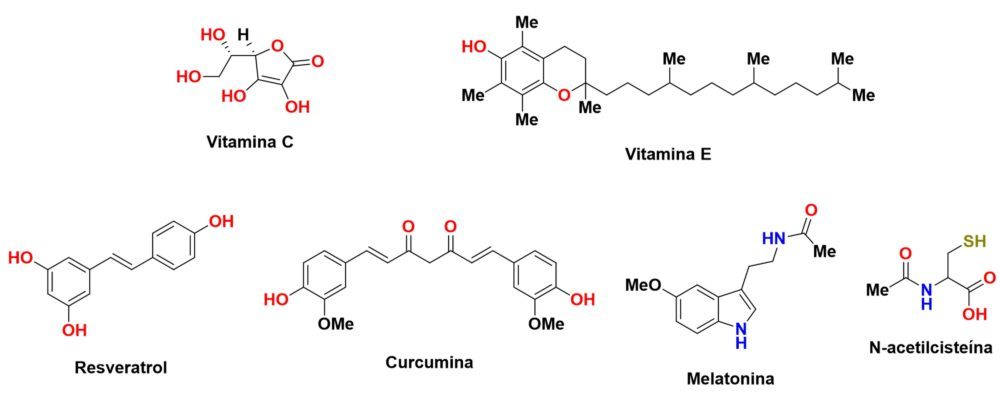
Figura 11: Estructura química de algunos de los antioxidantes más potentes.
Las vitaminas C y E son unos antioxidantes excelentes, con una capacidad de captación de radicales libres que puede incluso detener las reacciones de propagación que tienen lugar en las membranas plasmáticas, además de participar como cofactor de enzimas involucradas en muchos procesos fisiológicos (78). No obstante, sus potentes propiedades antioxidantes no se traducen en un beneficio terapéutico frente a las enfermedades neurodegenerativas, al menos por sí solas (54).
El resveratrol es un flavonoide de origen natural con una más que atestiguada capacidad antioxidante de la que se derivan otras propiedades terapéuticas como antidiabético, hepatoprotector o antiinflamatorio (79,80). Recientemente han aumentado las evidencias que postulan este compuesto como un posible inductor de Nrf2, algo que parece razonable dado el fragmento de carbonilo a,β-insaturado presente en su estructura, así como sus propiedades terapéuticas, muchas de las cuales son atribuibles a este mecanismo (81).
Por su parte, la curcumina es un compuesto fenólico que se puede extraer de la especie Curcuma longa. Sus propiedades antioxidantes y captadoras de radicales libres han sido ratificadas en múltiples ensayos, pero también ha demostrado propiedades terapéuticas antiinflamatorias o incluso antiangiogénicas. Al igual que el resveratrol, existen crecientes indicios de que parte de su acción es debida a la inducción de Nrf2 (82–84).
Otra sustancia de origen natural digna de mención es la melatonina, que normalmente actúa como una hormona reguladora del sueño, pero también participa en la regulación de la respuesta inflamatoria y la apoptosis, así como en el control del estrés oxidativo y la homeostasis mitocondrial. Además, la melatonina es un antioxidante muy potente en sí mismo ya que no solo la propia melatonina sino también varios de los productos de su metabolismo actúan como captadores de radicales libres, llegando a neutralizar hasta tres especies radicalarias por cada molécula de melatonina. Precisamente a este potente efecto antioxidante se le atribuyen las propiedades neuroprotectoras que esta molécula ha demostrado en modelos de EA, EP, ELA y EH (85,86).
2.4.b. Moléculas pequeñas con propiedades antioxidantes
La N-acetilcisteína es un fármaco ampliamente utilizado como mucolítico y como tratamiento de rescate en la intoxicación por paracetamol, precisamente por sus propiedades antioxidantes y protectoras frente a xenobióticos electrófilos (Figura 11). De hecho, la N-acetilcisteína no solo posee capacidad captadora de radicales libres intrínseca, sino que su potencia también radica en su capacidad para promover la regeneración del glutatión. Este fármaco ha demostrado un buen perfil terapéutico para varias enfermedades neurodegenerativas, e incluso ha participado o está participando en varios ensayos clínicos para su uso frente a la EA (NCT04044131, NCT04740580), la EP (NCT02212678, NCT01470027) y la EH (NCT05509153) con distintos resultados, pero en su mayoría positivos (87,88).
2.4.c. Modulación de la respuesta antioxidante endógena
Otra forma de lidiar con una presión oxidativa muy elevada consiste en reforzar las defensas antioxidantes endógenas mediante el uso de miméticos de enzimas antioxidantes como la SOD o la glutation peroxidasa, o mediante la inhibición de enzimas que generan especies oxidantes como pueden ser la NOX, la xantina oxidasa o la iNOS (Figura 12).
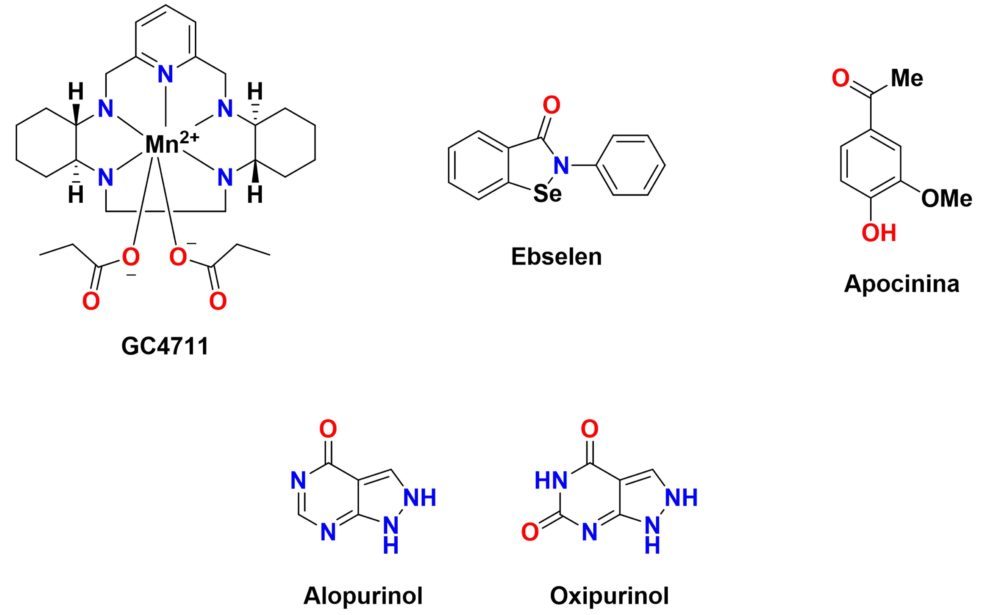
Figura 12: Estructura química de algunos fármacos moduladores de la respuesta antioxidante endógena.
Recientemente ha crecido el interés terapéutico en los miméticos de la enzima SOD para imitar la función de esta enzima y reducir los niveles de radical superóxido. Algunos de ellos (p. ej.: GC4711) han completado exitosamente ensayos clínicos de fase 1 (NCT03194139, NCT03096756) y ya participan en ensayos de fase 2b (NCT04698915) frente a ciertos tipos de cáncer (89).
El ebselén es un compuesto orgánico que incluye un átomo de selenio en su estructura y actúa como mimético de la glutatión peroxidasa, restaurando los niveles normales de glutatión reducido. (90) Esta actividad le confiere un gran potencial antioxidante, también presente en muchos de los análogos de este compuesto que recientemente se están sintetizando. Cabe destacar el interés que suscita el ebselén para el tratamiento de muchas enfermedades asociadas al SNC como el trastorno bipolar (NCT03013400, fase 2), la depresión mayor (NCT05117710, fase 1) o las propias enfermedades neurodegenerativas, para las que, sin embargo, no se ha comenzado ningún ensayo clínico (91).
La apocinina es una acetofenona de origen natural cuya actividad se basa en la inhibición de la NAD(P)H oxidasa (NOX), al impedir que la subunidad p47 de esta proteína migre a la membrana. Esta actividad inhibitoria se traduce en una plétora de efectos terapéuticos en ciertos tipos de cáncer, patologías cardiovasculares o enfermedades neurodegenerativas. (92) De hecho, ya se han realizado una serie de ensayos clínicos de fase uno dirigidos al tratamiento del asma (NCT00992667) y de enfermedades vasculares (NCT03680638), no así de enfermedades neurodegenerativas, a pesar de que existen múltiples indicios que parecen indicar su utilidad frente a estas patologías.
El alopurinol y el oxipurinol son fármacos de elección en el tratamiento de las hiperuricemias y la gota, pero también ejercen una función antioxidante al ser inhibidores de la xantina oxidasa. Por este motivo han sido ensayados en modelos de EA, EP o EH, donde han mostrado una cierta capacidad neuroprotectora (70,93).
Los inhibidores de la iNOS también atraen un creciente interés en el tratamiento de la neurodegeneración, dados los buenos resultados que algunos inhibidores muy potentes están teniendo en estudios preclínicos para el tratamiento de enfermedades neurodegenerativas; y clínicos, para el tratamiento de la migraña (NCT00242866, fase 2) (94,95).
Dada la potencia y el alcance que tiene la respuesta antioxidante controlada por los ARE, la inducción de NRF2 se ha postulado como una opción terapéutica de elección en la búsqueda de nuevos tratamientos antioxidantes. De hecho, ya se han propuestos múltiples estrategias dirigidas a esta diana que podemos clasificar como: interacción directa con Keap-1, inhibición de la interacción entre Nrf2-Keap-1 y modulación independiente de Keap-1 (96).
Como ya se ha mencionado previamente, la interacción de Keap-1 con Nrf2 se rompe en presencia de sustancias oxidantes u electrófilas, de modo que se ha aprovechado esta cualidad para desarrollar moléculas de mayor o menor carácter electrófilo. De entre todas ellas la más característica es el fumarato de dimetilo, el único fármaco inductor de la vía Nrf2/ARE aprobado por la EMA y la FDA para el tratamiento de la esclerosis múltiple. Su mecanismo de acción se basa en el carácter electrófilo de su grupo carbonilo a,β-insaturado, capaz de reaccionar con los tioles de las cisteínas presentes en la región sensible de Keap-1 (97). Existen otros ejemplos de fármacos electrófilos que pertenecen a este grupo, como ciertos triterpenoides (bardaloxona) o polifenoles (resveratrol, curcumina o derivados fenólicos del ácido cinámico), por citar algunos ejemplos. Sin embargo, merece la pena mencionar el potencial tóxico asociado a este tipo de compuestos electrófilos, ya que resultan muy reactivos no solo frente a Keap-1, sino que también pueden formar aductos covalentes con proteínas ricas en cisteína o lisina, o con nitrógenos aromáticos presentes en el ADN (98).
La segunda estrategia más prolífica se basa en inhibir la interacción Nrf2-Keap-1 mediante moléculas capaces de interactuar con las regiones DLG o ETGE de Nrf2, impidiendo su unión a Keap-1. La mayoría de moléculas que poseen esta capacidad son peptidomiméticos y péptidos cíclicos, pero recientemente se han descubierto otras moléculas pequeñas derivadas de urea, tetrahidroquinolinas o triazoles, que también son capaces de inhibir esta interacción (99,100). El principal problema de esta estrategia consiste en lograr la inhibición selectiva de esta interacción, ya que Keap-1 también se une a otras proteínas de gran relevancia para la supervivencia celular como la quinasa del inhibidor del factor nuclear κB (IKK), que regula la señalización del factor de transcripción proinflamatorio NF-κB.
Aprovechando que Keap-1 se une a otras proteínas, una alternativa que también se maneja consiste en el aumento de la expresión de alguna de ellas, de modo que Keap-1 se una en mayor medida a la proteína inducida en detrimento de Nrf2, que queda así libre en mayor proporción. Destaca, en este sentido, la inducción de la proteína autofágica p62/SQSTM-1, cuya afinidad por Keap-1 es superior a la propia de la interacción Nrf2(DLG)-Keap-1, lo que podría traducirse en la expresión de las proteínas de la secuencia ARE, aunque aún hace falta explorar más esta diana para demostrar un beneficio terapéutico (101).
Existen muchas otras alternativas para lograr la inducción de Nrf2, si bien es cierto que algunas de ellas apenas han comenzado a ser exploradas. De entre todas ellas la que mejores resultados ha arrojado hasta el momento es la inhibición de la glucógeno sintasa quinasa 3 b (GSK-3b), una enzima central en la regulación de Nrf2 al mediar su degradación dependiente de fosforilación a través de la proteína b-TrCP, y que además está involucrada en otros procesos fisiológicos y patológicos como la hiperfosforilación de tau (102,103). Otras estrategias menos exploradas son la inhibición de la degradación independiente de Keap-1, la inhibición de la interacción entre Nrf2-b-TrCP, el bloqueo de proteínas que secuestran a las proteínas Maf y el uso de agonistas de receptores nicotínicos α7.
3. PROTEINOPATÍAS
Las enfermedades neurodegenerativas son consideradas a menudo como proteinopatías, pues la mayoría cursan con la aparición de agregados proteicos tóxicos intracelulares y/o extracelulares que participan en la degeneración del tejido nervioso.
3.1. Proteostasis
Para entender cómo se originan estos depósitos, es preceptivo describir brevemente el sistema de síntesis, procesamiento y degradación de proteínas, englobado bajo el concepto de proteostasis. Una vez la cadena peptídica correspondiente a una proteína se sintetiza en el ribosoma mediante la traducción de su ARN mensajero correspondiente, es necesario que esta adopte la conformación tridimensional adecuada al desempeño de su función (104). Este proceso, en el que la proteína tiende a exponer hacia el exterior sus regiones más hidrófilas, se rige por el establecimiento de diversas interacciones de carácter intramolecular (105). Aunque algunas de estas interacciones puedan ser espontáneas, el plegamiento tiene lugar bajo la dirección de otras proteínas llamadas chaperonas, que se unen a la cadena peptídica en procesamiento para controlar el plegamiento correcto de las proteínas y prevenir interacciones indeseadas con otras unidades de la misma proteína u otras biomoléculas.
Hay que destacar que este proceso, aunque fuertemente controlado, no es totalmente fiable, habiéndose estimado que entre un 5 y un 30 % de las proteínas presentan defectos “de fábrica”. Las células disponen de diversos mecanismos para afrontar esta amenaza: primero, se desencadena una respuesta de índole reparadora. Si esta no funciona, existen diversos procesos que permiten la eliminación de la proteína dañada o defectuosa. La respuesta reparadora consiste en el aumento de la expresión de determinadas chaperonas capaces de unirse a las proteínas en cuestión, estabilizándolas de modo que posibiliten la reversión de su anomalía estructural (106). Cuando el daño proteico alcanza un nivel más amplio, el retículo endoplásmico es capaz de desencadenar la conocida como “respuesta al estrés del retículo”, por la que aumenta aún más la expresión de chaperonas a la par que disminuye la tasa de traducción del ARN mensajero, de modo que se reduce la carga de trabajo del retículo endoplásmico y se pueden concentrar los esfuerzos celulares en la reparación de las proteínas implicadas. Hay que decir, no obstante, que esta respuesta de estrés es una solución temporal frente a una situación que amenaza la supervivencia celular, pues si esta se prolonga determina la activación de señales proapoptóticas.
Si la respuesta reparativa no consigue solucionar el problema, la célula dispone de varias alternativas para eliminar los elementos dañados. Entre estas, destaca el sistema ubiquitina-proteasoma, encargado de la degradación de las proteínas anómalas disueltas en el citoplasma. Un sistema de chaperonas se encarga de marcar las proteínas destinadas a su eliminación mediante su conjugación con ubiquitina, tras lo cual son dirigidas al proteasoma, un macrocomplejo proteico donde serán eliminadas y recicladas. Cuando no es posible ubiquitinar la proteína dañada, o bien se trata de hacer frente a grandes agregados proteicos u orgánulos no funcionales, la célula puede marcarlos y englobarlos en una vesícula de doble membrana que se fusiona con un lisosoma, donde son digeridos. Este proceso, conocido como autofagia, provee además a la célula de una fuente extra de ATP y biomoléculas que puede ser muy útil en situaciones extremas o de crecimiento celular (107,108).
Por último, en casos en los que los depósitos proteicos no pueden ser reparados ni eliminados eficazmente, la célula puede confinarlos en cuerpos de inclusión para protegerse de sus efectos citotóxicos. Existen dos tipos principales: los JUNQ, que aíslan sobre todo proteínas ubiquitinadas mal plegadas; y los IPOD, que presentan fundamentalmente fibrillas insolubles no ubiquitinadas. Un ejemplo de estos cuerpos de inclusión serían los cuerpos de Lewy, muy característicos de algunas patologías neurodegenerativas como la demencia con cuerpos de Lewy y la enfermedad de Parkinson.
3.2. Alteración de la proteostasis: Agregación patológica de proteínas
La proteostasis se puede ver comprometida por diversas circunstancias como el estrés celular o determinados factores genéticos, aunque si hay que resaltar una condición fundamental para entender la relación entre la proteostasis y la neurodegeneración, esta es el envejecimiento. En la fase final de la vida la eficacia de los sistemas de síntesis y procesamiento de proteínas disminuye, lo que genera un acúmulo progresivo de proteínas disfuncionales agravado por la menor expresión de chaperonas reparadoras, la caída en el rendimiento de los sistemas de eliminación de proteínas y la mayor presencia de otros factores que incrementan los niveles de proteínas dañadas, como la inflamación o el estrés oxidativo. Todo ello conlleva la acumulación de las proteínas dañadas en depósitos tóxicos que dañan directamente las membranas lipídicas celulares y de los orgánulos, y se unen a diversas biomoléculas con la consecuente afectación funcional, además de la pérdida de las funciones fisiológicas desempeñadas por las proteínas afectadas. Además, se desata una respuesta de estrés reticular prolongada que dirige a la célula hacia la activación de cascadas proapoptóticas y se alteran las modificaciones post-traduccionales de numerosas proteínas, lo cual retroalimenta a su vez todos los procesos descritos anteriormente (109,110).
El proceso de formación de los agregados proteicos tóxicos consta de varias etapas. En primer lugar, la proteína cuya conformación es anómala tiende a presentar una disposición externa de láminas b no pareadas, que actúan como una superficie de nucleación donde otros monómeros proteicos anómalos pueden asociarse. Cuando los niveles de estos monómeros superan la concentración conocida como amiloidogénica comienzan a tener lugar los procesos de nucleación primaria, en los que las zonas hidrófobas anormalmente expuestas hacia la superficie de la proteína permiten la agregación de monómeros o pequeños oligómeros para dar lugar a la formación de oligómeros solubles metaestables. Esta nucleación puede ocurrir de forma espontánea o con la colaboración de otras biomoléculas: cuando son otros agregados de la misma naturaleza los que la propician, se habla de nucleación secundaria. Se considera que este paso del proceso es el que resulta en una mayor toxicidad celular, pues los oligómeros solubles disponen de la mayor relación de superficie hidrófoba respecto a la total, de modo que presentan una elevada reactividad frente a otras proteínas y diversas biomoléculas. No obstante, en su virtud está su pecado: esta gran reactividad motiva que los oligómeros también reaccionen con frecuencia entre sí, formando agregados que se compactan progresivamente con un aumento concomitante de su contenido en láminas b y una disminución de su superficie hidrófoba. Este proceso arroja como resultado la formación de oligómeros con una estructura muy ordenada de láminas b que va mudando desde una morfología esférica hacia otra más lineal, dando lugar finalmente a la formación de unas estructuras conocidas como protofibrillas que aún son solubles pero cuya reactividad es menor. Finalmente, estas protofibrillas terminan confluyendo para formar fibrillas lineales insolubles, que crecen progresivamente por los extremos y que también pueden crecer de volumen a través de procesos de nucleación secundaria (111,112) (Figura 13). Las fibrillas insolubles presentan un grado de toxicidad más bajo debido a una disminuida proporción de superficie hidrofóbica, aunque no por ello deben dejar de considerarse sus efectos deletéreos (113) .
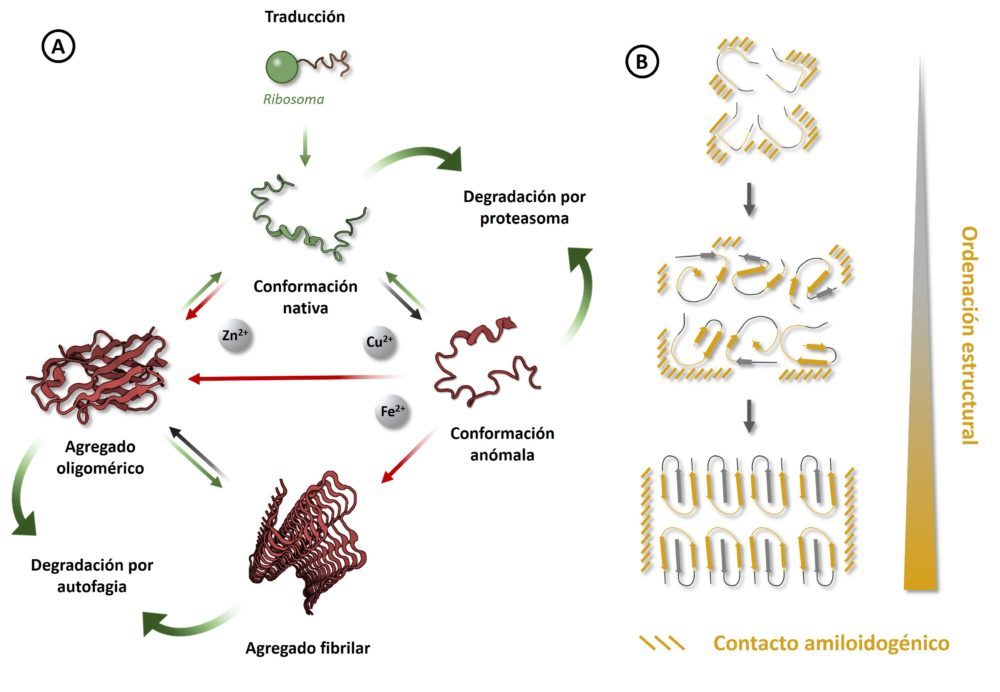
Figura 13: A) Proceso de agregación proteica. Las líneas rojas señalizan los eventos de agregación y las verdes la evolución a estadios menos tóxicos de la agregación, habitualmente mediados por chaperonas. B) Progresión del proceso de agregación de una proteína amiloide. Los agregados van aumentando en ordenación conforme aumenta el número de monómeros y estos van adquiriendo una conformación más rica en lámina β. También se puede observar como las regiones amiloidogénicas (amarillo) se van orientando hacia el interior, permitiendo a las regiones más hidrófilas situarse en la zona externa (gris).
Wells et al. remitieron todos estos procesos y sus consecuencias a la consideración de la ratio entre la eliminación de proteínas dañadas y la formación de fibrillas: si esta ratio es elevada, los sistemas de degradación de proteínas aún son capaces de lidiar con los niveles de proteínas erróneas existentes; si la ratio, por el contrario, es baja, las proteínas mal plegadas se encuentran formando mayoritariamente agregados de toxicidad moderada. Los puntos intermedios, caracterizados por la presencia de altos niveles de oligómeros solubles y por tanto de altas tasas de nucleación primaria y secundaria, serían los que comportarían una mayor toxicidad celular (111).
La composición y la localización tisular de estos depósitos proteicos son específicos para cada enfermedad. De hecho, en cada enfermedad se observa un patrón anatómico característico de estas lesiones, partiendo desde el foco de origen hacia aquellas regiones conectadas anatómicamente. Esta observación motivó la hipótesis de que los fenómenos de agregación proteica podrían extenderse por los tejidos mediante un mecanismo similar al priónico. En esencia, una proteína mal plegada podría actuar como un molde para inducir la misma alteración en otros monómeros vecinos de la misma proteína, e incluso estas proteínas anómalas podrían transferirse a otras células conectadas con la de origen a través de exosomas o nanotubos de membrana, de modo que los territorios afectados por la presencia de depósitos proteicos se irían extendiendo desde el foco inicial hacia otras regiones del cerebro a priori alejadas, pero con las que existe una conexión anatómica. Esta teoría podría explicar los patrones anatómicos característicos de lesión y afectación funcional de cada una de las enfermedades neurodegenerativas (114).
La homeostasis de los metales parece que desempeña un papel significativo en los procesos de agregación proteica (115). Los niveles de algunos cationes metálicos como el Cu2+, el Zn2+ y el Fe2+ aumentan por encima de los niveles habituales durante el envejecimiento, y en pacientes que sufren algún proceso neurodegenerativo se ha observado que este aumento es más marcado en las zonas lesionadas y que la disminución de la concentración de metales puede tener un impacto a nivel clínico, hallazgos todos ellos que sugieren una relación directa de la homeostasis metálica con la etiología de la neurodegeneración (116). En lo que a la proteostasis se refiere, parece que los metales podrían favorecer los procesos de nucleación secundaria implicados en la formación de nuevos oligómeros, aunque también podrían tener cierto papel protector al propiciar la constitución de depósitos fibrilares o de oligómeros de baja solubilidad, como señalan otros estudios (117,118). Los metales también pueden interferir en el procesamiento de las proteínas, como ejemplifica el péptido βamiloide: un exceso de Zn2+ en el medio inhibe la α-secretasa, promoviendo el procesamiento de la proteína precursora amiloide por la vía amiloidogénica, y también inhibe enzimas implicadas en la degradación del péptido Ab (119). Hay que señalar que la elevada concentración de metales también afecta a la proteostasis de forma indirecta mediante otros mecanismos, como el aumento del estrés oxidativo o la disfunción mitocondrial (120).
3.3. Proteostasis y neurodegeneración
A continuación, se expondrán brevemente las alteraciones proteicas más destacadas de las principales enfermedades neurodegenerativas. No hay que olvidar el impacto mayor que pueden tener las proteinopatías a nivel del sistema nervioso central, pues las neuronas son células postmitóticas y por tanto los efectos citotóxicos producen alteraciones funcionales y morfológicas permanentes.
3.3.a. Enfermedad de Alzheimer
La enfermedad de Alzheimer se ha asociado a la presencia de agregados tóxicos de dos proteínas: la proteína β-amiloide y la proteína tau. El péptido β-amiloide se forma a partir de la proteína precursora amiloide (PPA) por acción de las secretasas β y γ, y desempeña funciones importantes relacionadas con la plasticidad sináptica, el aprendizaje y la memoria (121). La longitud del péptido resultante es variable, relacionándose con una especial neurotoxicidad el péptido Aβ42 por su mayor reactividad a la hora de asociarse en oligómeros y un mayor potencial fibrilogénico (122). El aumento de su concentración, como consecuencia de la disfunción de los sistemas de aclaramiento proteico, motiva la formación de macroagregados denominados placas neuríticas o seniles. La relación de estos agregados con la enfermedad de Alzheimer es confusa: su presencia en cerebros sanos sugiere que no son suficientes para constituirse como causa única y directa de la enfermedad, pero hay suficiente evidencia para apoyar la idea de que su proceso de formación sí tiene efectos deletéreos, quizá relacionados con la existencia de formas intermedias oligoméricas altamente reactivas. Los distintos tipos de agregados del péptido Aβparecen participar en otros procesos neurotóxicos como la hiperfosforilación de la proteína tau, la excitotoxicidad o la neuroinflamación (123,124).
La proteína tau, ligada a la estabilización de los microtúbulos celulares, sufre procesos de hiperfosforilación relacionados con la enfermedad de Alzheimer. Además de la pérdida de función que conlleva una grave afectación del transporte axonal, la hiperfosforilación de tau favorece su oligomerización, y finalmente su organización en un tipo de agregados conocidos como ovillos neurofibrilares (125). Aunque parece que estos ovillos exhibirían menos toxicidad que los oligómeros intermedios, su relación con el deterioro cognitivo y anatómico propios de la enfermedad está mejor establecida que en el caso del péptido Aβ, e incluso se postula que se pueden extender desde su foco de origen a través de un mecanismo pseudopriónico (126).
3.3.b. Enfermedad de Parkinson
En el cerebro de pacientes de la enfermedad de Parkinson se han hallado depósitos tóxicos de otra proteína denominada α-sinucleína, cuya función fisiológica entraña la movilización de vesículas sinápticas y la formación de protuberancias sinápticas (127). Esta proteína es capaz de formar oligómeros de manera espontánea, los cuales pueden evolucionar hacia agregados más complejos a través de determinados cambios en el plegamiento proteico. Al contrario que en el caso de la proteína amiloide, la mayor toxicidad se asocia a los estadios protofibrilar y fibrilar, en los que predominarían las regiones dispuestas en láminas β. Estos agregados, que se acumulan por la progresiva disfunción de los sistemas de aclaramiento proteico, destruirían las membranas de las vesículas intracelulares, afectarían a la homeostasis del calcio e inducirían un estatus de disfunción mitocondrial. Cuando son liberados al medio extracelular tras la apoptosis neuronal activan la respuesta inmunitaria local, lo que induce un aumento del estrés oxidativo local y, en definitiva, retroalimenta el proceso neurotóxico. Hay que destacar que los agregados de α-sinucleína pueden ser confinados por las células en unos cuerpos de inclusión muy característicos denominados cuerpos o neuritas de Lewy en función de su localización intracelular o extracelular, donde su toxicidad se vería limitada (73,111).
3.3.c. Esclerosis lateral amiotrófica
La etiopatogenia de la esclerosis lateral amiotrófica sigue siendo un secreto en líneas generales, aunque dentro de las alteraciones que se han asociado con la enfermedad hay algunas proteínas con potencial amiloidogénico.
En primer lugar, podemos citar la proteína TDP-43, una riboproteína implicada en la traducción y el procesamiento del ARN mensajero. Como sucede con la α-sinucleína, TDP-43 tiene una reseñable capacidad para agregarse espontáneamente, aunque hay determinadas modificaciones postraduccionales que incrementan aún más este potencial tóxico. Los oligómeros de TDP-43 son el estadio más reactivo, mientras que su agregación ulterior da lugar a fibrillas con una toxicidad más reducida. No obstante, la elevada concentración de TDP-43 en las motoneuronas asegura la formación constante de oligómeros cuando se instaura el estatus patológico, por lo que la toxicidad asociada a esta proteína no se agota por la formación de los depósitos fibrilares. Aunque la presencia de depósitos de TDP-43 en un 60% de los pacientes de ELA sugiere su asociación con la enfermedad, se desconoce si su potencial tóxico se asocia más con determinadas mutaciones o modificaciones postraduccionales, o bien con la disfunción de los sistemas de degradación de proteínas mal plegadas (128).
La enzima superóxido dismutasa 1 (SOD-1) forma parte del sistema de defensa antioxidante celular. Su relación con la ELA pasa por determinadas mutaciones que motivan su plegamiento incorrecto, dando lugar a un proceso de agregación donde los oligómeros son nuevamente la especie más problemática. La presencia de depósitos tóxicos de SOD-1 ha sido documentada en pacientes tanto de ELA familiar como de ELA esporádico; sin embargo, su relación con la enfermedad sigue siendo un tema irresoluto (111,128).
Otra proteína de unión al ARN relacionada con la ELA es la proteína FUS, que participa en la traducción y reparación del ADN. Su proceso de agregación se aleja del paradigma amiloideo, y sus efectos deletéreos parecen relacionarse más bien con la pérdida de su función fisiológica. La proteína FUS mutada tiende a agregarse secuestrando fragmentos de ARN, además de adoptar una localización citosólica que no se corresponde con su lugar de expresión habitual, el núcleo. Este comportamiento anómalo no solo se relaciona con la ELA, sino también con otras enfermedades neurodegenerativas como la demencia frontotemporal (129).
3.3.d. Enfermedad de Huntington
Es preceptivo finalizar este apartado citando brevemente la enfermedad de Huntington, ya que es una patología autosómica dominante asociada a la mutación del gen que codifica para la proteína huntingtina. Parece que, por un lado, las manifestaciones de la enfermedad estarían relacionadas con la disfuncionalidad de la proteína mutada, pues esta participa en numerosos procesos fisiológicos: transporte de orgánulos y vesículas, coordinación de la división celular, control de la endocitosis y la autofagia, transcripción de numerosos genes…, incluso, desempeña un papel importante en el desarrollo embrionario (130). Por otro lado, la mutación conlleva que la proteína presente numerosos fragmentos de poliglutamina que favorecen la agregación de esta proteína. Los oligómeros de huntingtina exhiben una enorme reactividad, afectando directamente a numerosas funciones celulares esenciales. El proceso de fibrilación, más complejo que en otros casos revisados anteriormente, alumbra agregados menos reactivos que los oligómeros de partida (111).
3.4. Tratamiento
Dado el papel central que parecen tener las proteinopatías en la etiopatogenia de las enfermedades neurodegenerativas, se han propuesto varias estrategias para diseñar terapias modificadoras de las enfermedades basadas en los procesos que ocurren en torno a la generación de los depósitos tóxicos. Así, se han propuesto fármacos capaces de inhibir distintos puntos del proceso de formación de los agregados, inhibidores per se de la agregación de proteínas y, por último, agentes capaces de eliminar los depósitos ya formados.
Los inhibidores de la formación de los depósitos proteicos pueden actuar a distintos niveles del proceso. Por ejemplo, los compuestos quelantes se proponen por su capacidad para capturar los iones metálicos en exceso implicados en la asociación de las proteínas mal plegadas, si bien esta capacidad quelante no debe afectar a los iones metálicos que se encuentran colaborando en diversas funciones fisiológicas como parte de metaloenzimas. Con este mecanismo de acción se pueden destacar dos compuestos: el clioquinol y la deferiprona (Figura 14). El clioquinol es un derivado de 8-hidroxiquinolina con una capacidad quelante media, que cumple el objetivo de capturar los iones metálicos unidos a los agregados proteicos o en exceso sin interferir en las funciones fisiológicas de los mismos. Además, se ha postulado su capacidad para unirse a los agregados de péptido Ab, induciendo su disociación. No obstante, no superó la fase 2 de ensayos clínicos para la enfermedad de Alzheimer por falta de eficacia en lo que al deterioro cognitivo se refiere. (131) Los resultados de fases previas, que sí eran positivos, espolearon el diseño de análogos del clioquinol como los PBT. El compuesto con un perfil preclínico más prometedor, PBT2, tampoco consiguió resultados clínicamente significativos en dos ensayos clínicos de fase 2 para las enfermedades de Alzheimer y de Huntington (132).
Figura 14: Estructuras de fármacos relevantes en la terapia quelante.
Por su parte, la deferiprona es un conocido quelante de hierro usado en clínica para el tratamiento de la talasemia mayor. Dos pequeños ensayos clínicos de fase 2 han mostrado que la deferiprona es capaz de reducir los depósitos de hierro a nivel cerebral y la sintomatología en pacientes con síndromes parkinsonianos y ataxia de Friedreich (133,134). Esta observación, unida al potente efecto inductor de la autofagia y la mitofagia que exhibe este compuesto, le otorgan a la deferiprona un notable potencial para el abordaje de la neurodegeneración a través de la restitución de la proteostasis y la quelación (135).
Otra estrategia ampliamente trabajada, orientada especialmente para la enfermedad de Alzheimer, está representada por los fármacos inhibidores de la BACE-1 (β-site amyloid-precursor-protein-cleaving enzyme 1) (BACE-1), la aspartil-proteasa situada en el sitio activo de la enzima β-secretasa que degrada la PPA por la vía amiloidogénica. Algunas moléculas pequeñas cuya estructura se recoge en la Figura 15, como el verubecestat, JNJ-54861911 o CNP520 han alcanzado las fases 2 y 3 de ensayos clínicos para la enfermedad de Alzheimer, aunque ninguno logró una ralentización significativa del deterioro cognitivo. No obstante, se sigue trabajando en el desarrollo de otros compuestos dirigidos a esta diana, e incluso los grupos farmacóforos inhibidores de BACE-1 se han incluido en algunos compuestos multidiana donde se combinan con otros fragmentos inhibidores de la acetilcolinesterasa (AchE) o la glucógeno sintasa quinasa 3b (GSK-3b), y que han mostrado resultados muy prometedores a nivel preclínico (136).
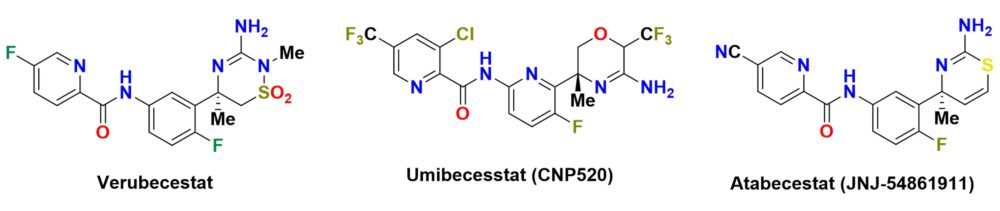
Figura 15: Estructura química de algunos fármacos inhibidores de la enzima BACE-1.
La terapia génica podría ser otra herramienta muy útil para contrarrestar las mutaciones o modular la expresión de las proteínas implicadas en la formación de los depósitos tóxicos. En el caso de la enfermedad de Alzheimer destaca el oligonucleótido antisentido IONIS MAPTRx, dirigido a la inhibición de la expresión de la proteína tau, que ha alcanzado la fase 2 de ensayos clínicos y que está mostrando resultados esperanzadores. Sin abandonar esta patología, también se están desarrollando oligonucleótidos antisentido y métodos de escisión génica por CRISPR-Cas9 para evitar el procesamiento amiloidogénico del PPA. Este abordaje también resultó atractivo para la enfermedad de Huntington, al ser una enfermedad monogénica, de modo que se han desarrollado oligonucleótidos antisentido y escisiones génicas por CRISPR-Cas9 destinadas al silenciamiento completo del gen o al silenciamiento selectivo de los alelos mutados. Algunas de estas aproximaciones han alcanzado las fases más avanzadas de los ensayos clínicos, si bien han aparecido ciertas consideraciones: el silenciamiento completo permitiría el tratamiento universal de todos los pacientes, pero conlleva los efectos adversos vinculados a la pérdida de la función fisiológica de la huntingtina; por el contrario, los silenciamientos selectivos mantienen la funcionalidad, pero al variar las mutaciones implicadas entre pacientes el tratamiento se torna más individualizado. La ELA también se podría beneficiar de terapias génicas, como lo demuestra el hecho de que hay ensayos clínicos en distintas fases para investigar la eficacia y seguridad de oligonucleótidos antisentido para la SOD-1 y la proteína c9orf72, que sobre todo en el primer caso están mostrando resultados muy interesantes (137).
En los últimos años se han caracterizado numerosas moléculas pequeñas con capacidad para unirse a los oligómeros proteicos impidiendo su agregación, o bien a los depósitos proteicos induciendo su desagregación, generalmente gracias a una estructura química tridimensional y/o una funcionalización adecuada para interponerse entre los monómeros u oligómeros. Muchos polifenoles exhiben una destacable capacidad para inhibir o desestabilizar los agregados de distintas proteínas relacionadas con la neurodegeneración, habiendo sido especialmente estudiados para los agregados de péptido Aβ. Merece la pena resaltar entre ellos la curcumina, el resveratrol, la oleuropeína aglicona, la epigalocatequina-3-galato y la fisetina; todos ellos han alcanzado la fase clínica de desarrollo, y a pesar de no haber conseguido la eficacia clínica esperada, han corroborado su actividad antiagregante (138). Dentro de las moléculas pequeñas inhibidoras de la agregación amiloide encontramos también péptidos como el QBP-1 (polyglutamine-binding peptide 1), vitaminas (B12, C, E, K), fármacos ya aprobados para otras patologías como la cicloserina B o pirazinamida, pinzas moleculares como el compuesto CLR01, etcétera (Figura 16). Como ocurre con los inhibidores de BACE, el potencial de estos compuestos antiagregantes ha estimulado su inclusión en nuevos compuestos multidiana con potencial antineurodegenerativo, buscando sinergias con otros fragmentos activos frente a dianas de interés en este ámbito (139).
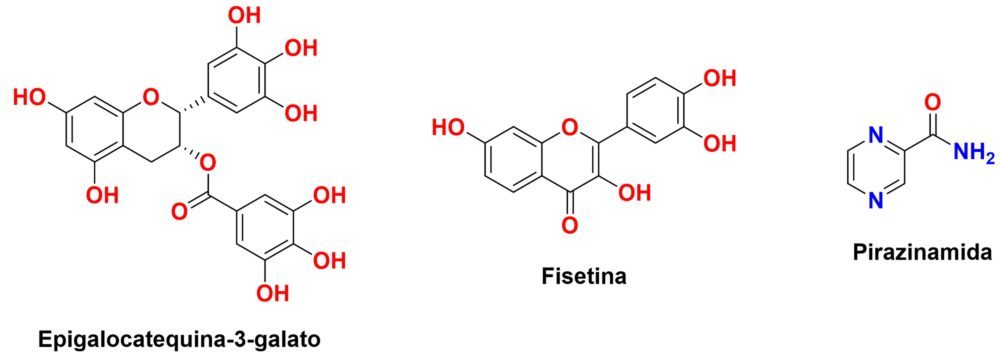
Figura 16: Estructura química de algunas moléculas pequeñas inhibidoras de la agregación patológica de proteínas.
Por último, se ha explorado ampliamente la posibilidad de eliminar los agregados proteicos ya constituidos mediante inmunoterapias activas o pasivas. Dentro de las inmunoterapias pasivas hay que hacer especial mención al aducanumab, un anticuerpo monoclonal contra los oligómeros y las fibrillas de péptido β-amiloide que ha sido recientemente aprobado (2021) por la FDA para el tratamiento de la enfermedad de Alzheimer. Este hecho, que supuso un hito por ser supuestamente la primera terapia modificadora de la enfermedad de Alzheimer, se vio envuelto de una gran polémica, pues aunque los ensayos clínicos avalaron la capacidad de este nuevo anticuerpo para eliminar los depósitos de proteína b-amiloide, esta actividad no parece correlacionarse con un beneficio clínico real, a lo que hay que sumar las dudas acerca de su seguridad. A pesar de todo ello, la FDA aplicó un protocolo de aprobación acelerada estimando que había una “probabilidad razonable” de encontrar un beneficio luego de su aplicación a nivel clínico (36). Los oligómeros y fibrillas de péptido β-amiloide son sin duda la diana más explorada en este campo como demuestra la variedad de anticuerpos monoclonales que han alcanzado la fase de ensayos clínicos avanzados: merece la pena destacar el gantenerumab, que ha llegado a demostrar cierta capacidad para ralentizar el deterioro cognitivo en un ensayo clínico de fase 3. Sin abandonar la enfermedad de Alzheimer, también se han desarrollado anticuerpos monoclonales destinados a reconocer los agregados de proteína tau: hay ensayos clínicos de fase 2 en marcha para evaluar la seguridad y eficacia de fármacos como el gosuranemab, zagotenemab, semorinemab y C2N 8E12 para la enfermedad de Alzheimer u otras patologías neurodegenerativas relacionadas estrechamente con la proteína tau como la parálisis supranuclear progresiva (140).
Existen dos anticuerpos monoclonales dirigidos contra los agregados de α-sinucleína, prasinezumab y BIIB054, que han alcanzado la fase 2 de ensayos clínicos para el tratamiento de la enfermedad de Parkinson. Por el momento, ambos han demostrado seguridad y tolerabilidad, lo que unido a los prometedores resultados en la fase preclínica de desarrollo los convierten en potenciales terapias modificadoras de esta patología neurodegenerativa. Para el resto de las enfermedades de este grupo la estrategia de inmunización pasiva se encuentra más atrasada: se han desarrollado anticuerpos monoclonales contra los agregados de las proteínas SOD-1 (ELA), C9orf72 (ELA y demencia frontotemporal) y huntingtina (corea de Huntington), mas de momento solo se dispone de resultados en modelos murinos de dichas enfermedades.
El desarrollo de vacunas para evitar la formación o fomentar la eliminación de los depósitos proteicos tóxicos trata de superar algunos de los inconvenientes de la inmunización pasiva, como son los efectos adversos, el alto coste y la alta frecuencia de administración que requerirían estos tratamientos. Nuevamente, es en la enfermedad de Alzheimer donde se han conseguido los mayores avances: la Tabla 4 recoge las principales vacunas basadas en el péptido β-amiloide o la proteína tau fosforilada que están siendo actualmente ensayadas, habiendo alcanzado ya algunas la fase 2 de ensayos clínicos. También se ha desarrollado una vacuna prometedora basada en la α-sinucleína humana recombinante que está siendo evaluada en un ensayo clínico de fase 1, mientras que para el resto de proteínas mencionadas en el epígrafe anterior (SOD-1, C9orf72 y huntingtina) existen vacunas experimentales que han demostrado capacidad inmunogénica en modelos murinos pero para las que, por el momento, no se ha pasado a la fase clínica de desarrollo.
Tabla 4: Resumen de las diferentes vacunas basadas en la agregación proteica que se están ensayando frente a la EA
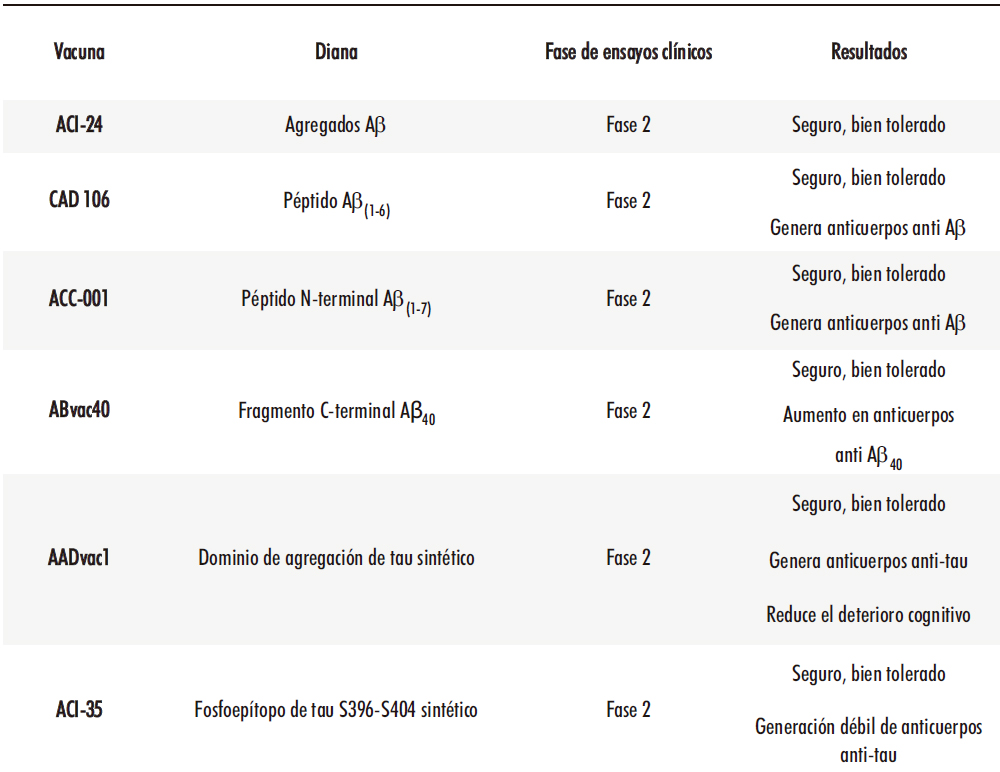
4. NEUROINFLAMACIÓN
La inflamación es un proceso fisiológico por el que las células inmunitarias ejercen una acción agresiva e inespecífica como respuesta a la presencia de estímulos nocivos en el medio y cuya finalidad es la destrucción del origen de ese estímulo. Cuando esta respuesta se da en el entorno del sistema nervioso, ya sea central (SNC) o periférico (SNP), se denomina neuroinflamación. A pesar de ser un proceso fisiológico común en muchas infecciones o enfermedades, su cronificación puede dar lugar a daños celulares y tisulares severos. Esto sucede en situaciones en las que el cerebro expresa cuerpos tóxicos como agregados proteicos, especies oxidantes o ácidos nucleicos libres en el medio extracelular, que transforman esta respuesta que debiera ser protectora en una cascada de daño celular que se retroalimenta a sí misma. A continuación, se irán desgranando los procesos que acontecen en la neurodegeneración que conducen al sistema inmunitario del SNC a este estado patológico.
4.1. Sistema inmunitario del SN
El SNC posee un sistema inmunitario propio, marcado por una serie de particularidades y relativamente aislado del resto del organismo gracias a una red de irrigación sanguínea especial, cuya principal característica es la presencia de la barrera hematoencefálica (BHE) y la ausencia de un sistema linfático propio. La primera supone una barrera física compuesta principalmente de células endoteliales unidas de forma muy íntima unas a otras de modo que se permite un estrecho control de la permeabilidad a sustancias, células o patógenos que puedan acceder al tejido nervioso (141). Este particular aislamiento no permite la presencia de un sistema linfático común al del resto del organismo, sino que requiere de un sistema especializado denominado sistema glinfático, en el cual el líquido cerebroespinal toma las funciones propias de la linfa. (142) Otra cualidad única del sistema inmunitario del SNC es la presencia de células inmunitarias exclusivas de este tejido, como son la microglía y los astrocitos, aunque bajo determinadas circunstancias otras células del sistema inmune como macrófagos o linfocitos T pueden atravesar la BHE y llegar al SNC.
Las células de la microglía son las células inmunes de mayor relevancia para la respuesta inmunitaria del SNC, suponiendo entre el 5 y 12 % del total de células de este tejido. Sus funciones van desde la defensa directa del organismo frente a patógenos externos o tóxicos endógenos asociados a las enfermedades neurodegenerativas, hasta el mantenimiento de la homeostasis en el entorno neuronal en cooperación con los astrocitos. La microglía también ejerce ciertas funciones neuroprotectoras o de mantenimiento del correcto funcionamiento neuronal como el control de la plasticidad neuronal, el mantenimiento de la homeostasis de la mielina o la migración hacia regiones que presentan marcadores de daño o muerte celular.
Entre sus funciones inmunológicas cabe destacar su capacidad de detección de patrones moleculares asociados al daño y muerte celular (DAMP, damage associated molecular patterns), como pueden ser el ADN mitocondrial, las chaperonas o los metabolitos de purinas; u otros patrones asociados a la presencia de patógenos (PAMP, patogen associated molecular patterns), como el lipopolisacárido (LPS). Para ello, estas células poseen numerosos receptores de reconocimiento de estos patrones, como son los receptores tipo Toll (TLR, Toll-like receptors) y los inflamasomas (NLR, Nod-like receptors), entre los que cabe destacar NRLP3 por su papel en la neuroinflamación. La microglía es, además, el principal efector de la respuesta inflamatoria en el SNC, ya que es capaz de secretar citoquinas proinflamatorias o sustancias tóxicas dirigidas a eliminar directamente el estímulo dañino (142,143).
Así, la unión de distintos patrones moleculares a los receptores de la microglía en reposo (M0) desencadena su activación, que puede ser hacia un fenotipo proinflamatorio (M1) o antiinflamatorio (M2) (142) (Figura 17). La existencia de dos fenotipos claramente diferenciados ha sido motivo de discusión en el ámbito científico durante los últimos años, ya que históricamente se ha considerado que las células gliales podían expresar dos fenotipos mutuamente excluyentes. El fenotipo proinflamatorio o M1 se caracteriza por una función más agresiva que incluye la fagocitosis, la presentación de antígenos a otras células inmunes, la secreción de citoquinas proinflamatorias como las interleuquinas IL-1β e IL-18, el factor de necrosis tumoral alfa (TNF-α) o interferones; la activación de la enzima óxido nítrico sintasa inducible (iNOS) y la producción de especies reactivas de oxígeno dirigidas a eliminar el estímulo dañino. Por otra parte, el fenotipo M2 liberaría citoquinas antiinflamatorias como las interleuquinas IL-4 e IL-10, o el factor de crecimiento transformante beta (TFG-β). Además, se sabe que la microglía M2 induce la neurogénesis mediante la activación de la vía mediada por el PPAR-γ (Peroxisome Proliferator- Activated Receptor gamma) y la secreción de factores de crecimiento como el factor de crecimiento insulínico 1 (IGF-1) y el factor neurotrófico derivado de cerebro (BDNF) (145).
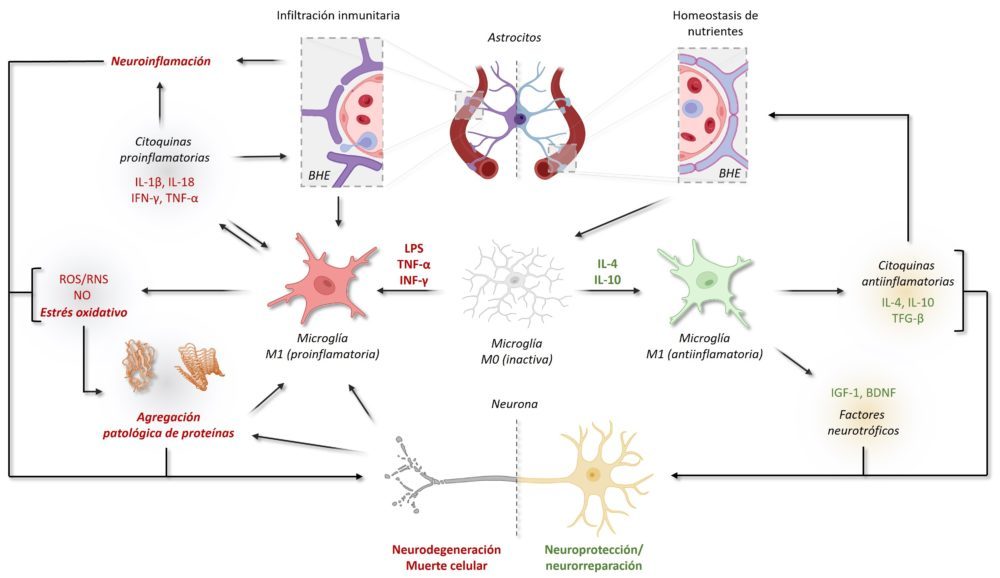
Figura 17:La presencia de citoquinas proinflamatorias como TNF-α, ciertos interferones (IFN-γ, LPS o de agregados proteicos patológicos produce la activación de la microglía hacia un fenotipo proinflamatorio (M1, rojo). La microglía activada producirá a su vez más citoquinas proinflamatorias, incluyendo IL-1β, IL-18, TNF-α e interferones, que activarán más células microgliales y astrocitos alcanzándose un estado proinflamatorio, fagocítico y permisivo con la infiltración de otras células inmunitarias a través de la BHE, favoreciendo la cronificación de la inflamación. Además, la activación de la microglía promueve la secreción de especies oxidantes como ROS y RNS, además de NO por inducción de la iNOS. La presencia de estas especies oxidantes, junto con la toxicidad asociada a la agregación proteica y la neuroinflamación provocan la muerte celular, cuyos residuos fomentan la activación de la microglía (M1). Por otro lado, la presencia de citoquinas antiinflamatorias como IL-4 o IL-10 conduce a un estadio antiinflamatorio de la microglía (M2), que promueve la liberación de citoquinas antiinflamatorias (IL-4, IL-10, TFG-β) y de factores neurotróficos (IGF-1 o BDNF) cuyo efecto es neuroprotector y reparador. En esta situación, los astrocitos mantienen un papel regulador y la permeabilidad de la BHE se recupera (144). Creado con BioRender.com
La lógica subyacente a esta diferenciación parece evidente, y es que en caso de que se produzca un daño tisular, la microglía se activará hacia un fenotipo M1 que instaurará una respuesta inflamatoria. Una vez el origen del daño haya sido eliminado, la microglía evolucionará hacia un fenotipo M2 que revierta la inflamación además de promover la reparación del daño neuronal que haya podido surgir.
Sin embargo, desde 2016 han crecido las voces que afirman que este modelo para explicar la polarización de la microglía no es tan preciso como debería. Y es que, a diferencia de los macrófagos, que sí pueden polarizarse en fenotipos claramente diferenciados, la microglía no presenta los cambios necesarios en la superficie celular como para poder considerar un fenotipo completamente polarizado hacia M1 o M2, por ejemplo, sino que más bien se mueven en un continuum en el que sus funciones son más antiinflamatorias o proinflamatorias. (146) No obstante, tampoco esta hipótesis de un fenotipo variable pero no dicotómico ha podido ser completamente demostrada experimentalmente, de modo que aún es necesario adquirir más conocimientos para poder establecer un modelo claro de comportamiento de la microglía.
Como ya se mencionó anteriormente, existe otro tipo de células que también pertenecen al sistema inmune del SNC, denominadas astrocitos por su morfología. Estas células son las más abundantes del SNC, estimándose que suponen hasta el 70 % del total. Sus funciones principales consisten en el mantenimiento de la homeostasis, el aporte de nutrientes y la asistencia en los procesos sinápticos. Los astrocitos también forman parte de la BHE, de modo que tienen un importante papel en la permeabilidad de esta barrera, siendo piezas clave en procesos como la infiltración de otras células inmunitarias. Los astrocitos también presentan algunas funciones intrínsecamente inmunitarias, como por ejemplo capacidad fagocítica y de presentación de antígenos, aunque muchas de ellas requieren la activación previa por parte de la microglía. Además, los astrocitos poseen receptores de patrones moleculares como los TLR, lo cual les permite desarrollar su propia respuesta inflamatoria, mediada principalmente por el factor de transcripción NF-κB (147).
4.2 Papel patológico de la neuroinflamación
Sin embargo, existen ciertas situaciones en las que la actividad de estas células inmunitarias se puede tornar en una exacerbación del daño tisular, tal como sucede en la neurodegeneración. Como ya se ha explicado anteriormente, en un cerebro en neurodegeneración los DAMPs del medio, tales como agregados proteicos tóxicos o los restos celulares generados por el estrés oxidativo, son reconocidos por las células de la glía a través de los TLR o los NLR, induciéndose su activación hacia un fenotipo proinflamatorio (M1) donde están activadas la fagocitosis y la secreción de citoquinas proinflamatorias. Bajo estas condiciones se produce la activación de los astrocitos e incluso la infiltración de otros tipos de células inmunitarias al interior del SNC. En el caso de que esta respuesta inflamatoria no alcance una eficacia suficiente y comience a cronificarse, tal como sucede en la neurodegeneración, la generación de radicales de oxígeno y nitrógeno así como de citoquinas proinflamatorias acabará por agravar el daño neural, comenzando así una cascada de retroalimentación en la que el estrés oxidativo genera agregados, ambos producen la neuroinflamación y esta produce aún más estrés oxidativo (148,149) (Figura 9). Además de este daño directo, se sabe que la hiperreactividad de las células gliales acaba por activar rutas enzimáticas que conllevan un mayor acúmulo de agregados tóxicos, como es el caso de la Jun N-terminal quinasa (JNK) que promueve la hiperfosforilación de tau, o incluso el aumento de los niveles de estrés oxidativo al liberar depósitos de hierro, muy abundantes en cerebros envejecidos. En los casos más avanzados la inflamación crónica acaba por producir la pérdida de vasos sanguíneos y la consecuente falta de irrigación en regiones adyacentes, que ya no gozan del aporte de nutrientes y oxígeno necesarios para su mantenimiento. De esta manera se acaba por destruir de forma casi completa todo el tejido adyacente, extendiéndose así la neurodegeneración (144).
Esta neurodegeneración asociada a la neuroinflamación se encuentra agravada en la vejez, no solo por el aumento del estrés oxidativo o los defectos de los sistemas de aclaramiento proteico característicos del periodo final de la vida, sino por una adaptación propia de estas células inmunes. Es sabido que el sistema inmunitario cambia su perfil de acción conforme el organismo envejece adecuándose así a la creciente vulnerabilidad asociada al deterioro funcional típico de los sistemas senescentes. Así, el sistema inmunológico adquiere un perfil de activación basal crónica, que si bien suele ser leve y asintomático, puede resultar un trampolín hacia ciertas enfermedades de carácter autoinmune o, como en este caso, hacia la neuroinflamación crónica asociada a la neurodegeneración (inflamm-aging) (150).
4.3 Neuroinflamación en la neurodegeneración
Más allá del componente de la edad como acelerador de la neuroinflamación, existen alteraciones propias de las enfermedades neurodegenerativas que también provocan un refuerzo de este proceso patológico.
En la EA el acúmulo de péptido Aβproduce la activación de los receptores de patrones moleculares de la microglía, promoviendo la producción de citoquinas proinflamatorias y de TNF-α. Además, la presencia de estos agregados induce la activación del inflamasoma NLRP3, que recluta y dirige el ensamblado fibrilar de la proteína ACS (apoptosis-associated speck-like protein containing ACS), la cual a su vez actúa como semilla para la nucleación de nuevos agregados de Aβ, instaurándose así un ciclo de retroalimentación positiva (151).
Complicaciones similares se han observado en modelos murinos de la enfermedad de Parkinson. En este caso, el ciclo de retroalimentación comienza con los agregados de la α-sinucleína, que también son capaces de activar el inflamasoma NLRP3, desencadenando una respuesta inflamatoria que agrava la neurodegeneración y promueve la generación de más DAMPs (142).
En el caso de la ELA, se ha comprobado en varios modelos murinos que existe una correlación entre la neuroinflamación mediada por la liberación de citoquinas dependiente de NLRP3 y la presencia de biomarcadores de la enfermedad, como son los agregados de SOD-1 y TDP-43 (152).
En la enfermedad de Huntington la activación microglial guarda una relación directa con la progresión de la enfermedad, ya que se ha comprobado que la inhibición del inflamasoma NRLP3 en modelos murinos de EH reduce los DAMPs y mejora la sintomatología asociada a esta patología (153,154).
En todas estas enfermedades se repite un patrón por el cual la neuroinflamación desempeña un papel central en la neurodegeneración, pero no como origen de la misma, sino que la etiopatogenia propia de cada enfermedad potencia la respuesta inflamatoria, llevándola a cotas que resultan tóxicas para el tejido nervioso. De esta forma, el tratamiento de la neuroinflamación se debe abordar como una diana inespecífica de interés general para todas estas enfermedades, aunque aparentemente no suponga la causa primaria de ninguna de ellas.
4.4 Tratamiento
El tratamiento de la neuroinflamación se puede abarcar de diversas formas: impedir el reconocimiento de los patrones moleculares por parte de la glía, disminuir la respuesta proinflamatoria o modular vías intermedias que median en la respuesta inflamatoria (155,156).
Para impedir el reconocimiento de los patrones moleculares por parte de la glía se pueden establecer dos estrategias básicas: o bien se reduce la expresión de los receptores de DAMPs, o bien se bloquean directamente. Dentro del primer grupo, encontramos fármacos muy variados como la minociclina, el candesartán o el azeliragon, mientras que el segundo está principalmente representado por anticuerpos monoclonales (Figura 18).
Figura 18: Estructura química de los fármacos más representativos en la inhibición de la neuroinflamación. RRP: receptores de reconocimiento de patrones.
La minociclina es un antibiótico de la familia de las tetraciclinas que ha demostrado actividad antinflamatoria asociada a una reducción en la expresión de ciertos TLRs inducidos por LPS, y por tanto de la activación de la microglía, aunque el mecanismo concreto por el que consigue este efecto no ha sido elucidado todavía (157). A pesar de que el tratamiento con este fármaco en modelos preclínicos de EA, EP, EH y ELA resulta neuroprotector en todos los casos, los ensayos clínicos que se han llevado a cabo demuestran baja eficacia en general y mala tolerabilidad en algunos casos (NCT01463384, NCT00063193, NCT00047723 y NCT00277355). (158)
El candesartán es un antagonista de los receptores tipo 1 de angiotensina II, pero además ha demostrado propiedades antiinflamatorias gracias a que reduce la expresión de TLR-4 e inhibe la liberación de NF-κB, de modo que reduce la producción de citoquinas proinflamatorias en modelos celulares (159). Estos hallazgos motivaron la inclusión de este fármaco en un ensayo clínico frente a la EA (NCT02646982), cuyos resultados aún no han sido publicados.
Otro fármaco de interés es el azeliragon, un inhibidor de un receptor de reconocimiento de patrones denominado RAGE (receptor for advance glication endproducts) capaz de reducir los niveles de Ab del entorno neuronal e incluso de enlentecer la progresión de la EA en modelos de ratón. (160) A pesar de estos prometedores resultados preclínicos, no superó dos ensayos clínicos de fase 3 por falta de eficacia (NCT02080364 y NCT02916056) (155).
Por otra parte, el bloqueo de ciertos receptores de reconocimiento de patrones moleculares con un anticuerpo monoclonal ha demostrado ejercer efectos beneficiosos en modelos murinos de neurodegeneración (161). De hecho, existen anticuerpos monoclonales dirigidos a la reducción de la actividad de TLR-4 que han llegado a ensayos clínicos de fase 2 frente a artritis reumatoide (NCT03241108), (162) si bien aún no se ha logrado un beneficio clínico reseñable.
Otro de los métodos para lidiar con la neuroinflamación es la reducción de las citoquinas proinflamatorias. Una estrategia que ha demostrado ser útil es la inactivación de las citoquinas proinflamatorias interactuando directamente con ellas. Es el caso del etanercept, una proteína recombinante que incluye el dominio de unión a ligando del receptor celular de TNF-α, de modo que puede unirse a él impidiendo su acción (163). Se ha comprobado que la administración de este fármaco es capaz de reducir la neuroinflamación en modelos preclínicos de cerebro envejecido y de EA, llegando incluso a incluirse en algunos ensayos clínicos de fase 1 y 2 frente a esta misma patología (NCT01716637, NCT01068353) (164).
El grupo de moléculas pequeñas derivadas de la talidomida, como la lenalidomida o la pomalidomida, ha demostrado propiedades antiinflamatorias a nivel del SNC mediadas por un mecanismo similar (Figura 18). Este efecto lo logran gracias a su capacidad para truncar parte del ARNm codificante para el TNF-α, acortando su semivida y, por tanto, su acción (165).
Otra estrategia dirigida a lograr un efecto antiinflamatorio es la modulación de las rutas enzimáticas que median entre la activación del receptor de reconocimiento de patrones moleculares y la posterior respuesta proinflamatoria. En este apartado caben muchas moléculas pequeñas con muy distintas dianas, pero a continuación resumiremos las más relevantes en el tratamiento de la neuroinflamación.
El neflamapimod es uno de estos fármacos de interés dirigidos a reducir la producción de citoquinas (Figura 18). Se trata de un inhibidor de la enzima p38 de las MAPK, la cual promueve la producción de citoquinas proinflamatorias como la IL-1β o el TNF-α. Se ha podido comprobar que su aplicación podría resultar beneficiosa en pacientes que padecen enfermedades neurodegenerativas, como demuestran los resultados prometedores en ensayos clínicos frente a la EA y la EP (NCT03402659, NCT04001517) (155,166).
Otro fármaco ha sido recientemente reposicionado en la búsqueda de nuevas dianas para hacer frente a las enfermedades neurodegenerativas: el fasudil (Figura 18). Este fármaco es un inhibidor de la quinasa asociada a Rho (ROCK) cuyo perfil farmacológico en múltiples modelos de enfermedades neurodegenerativas impulsó su inclusión en varios ensayos clínicos (NCT05218668 y NCT03792490, frente a ELA; NCT04734379, frente a taupatías; NCT04793659, frente a demencias). Los resultados están siendo muy prometedores, lo que ha llevado a su aprobación como fármaco de uso compasivo en ciertos casos de ELA, al menos en EE.UU. (46,167,168). Parece ser que la inhibición de ROCK promueve indirectamente la fosforilación de la quinasa Akt y la consecuente respuesta antiinflamatoria, polarizando la microglía hacia un estado protector (M2) (169).
Otra estrategia de acción que va ganando popularidad es la inhibición directa del inflamasoma NLRP3, que permitiría inactivar la ruta de señalización proinflamatoria mediada por este complejo proteico (170). Uno de los compuestos más utilizados para la investigación básica de este componente celular, el MCC950, es además uno de los fármacos más prometedores. Parece ser que actúa inhibiendo el ensamblaje del inflamasoma, aunque aún no se ha logrado identificar el mecanismo de acción concreto. Este compuesto, que ha demostrado muy buenos resultados preclínicos frente a varias enfermedades inflamatorias y a algunos modelos de neurodegeneración, llegó incluso a participar en un ensayo clínico para el tratamiento de la artritis reumatoide que, sin embargo, no llegó a buen término debido a problemas de toxicidad hepática (171). No obstante, esta molécula, así como otras con un mecanismo de acción similar, mantienen la atención de la comunidad científica de cara a nuevos hallazgos. De hecho, existen muchos otros fármacos que se están reposicionando como posibles inhibidores de NLRP3, como sucede con el sulforafano o la estavudina (143) (Figura 18).
Más allá de las rutas propuestas, existen otras muchas moléculas cuya acción reduce la respuesta proinflamatoria, a pesar de que no se haya determinado la manera en que lo logran. Un buen ejemplo lo suponen los antinflamatorios no esteroideos (AINEs), cuya administración produce una reducción de la activación de la microglía y de la producción de citoquinas proinflamatorias, aunque el mecanismo por el que lo hacen no se ha logrado esclarecer todavía (172). Se propone que podría ser a través de su actividad inhibidora de la ciclooxigenasa (COX), considerando el papel central de esta enzima en la producción de prostaglandinas, tromboxanos y otros mediadores de la inflamación. El ibuprofeno ha logrado demostrar beneficio terapéutico en pacientes con EA leve o moderada al ser utilizado en combinación con otro conocido antinflamatorio, el cromoglicato de sodio, según los resultados de dos ensayos clínicos publicados en 2015. (NCT02547818 y NCT04570644) (Figura 18).
Existen muchas más moléculas activas frente a la neuroinflamación, ya sea en desarrollo o como fármacos en reposicionamiento, pero en esta revisión solo buscábamos destacar las moléculas más representativas y poner en evidencia aquellas dianas hacia las que van dirigidas.
5. CONCLUSIONES
El pronóstico asociado a las enfermedades neurodegenerativas continúa siendo muy sombrío hoy en día, con un arsenal terapéutico disponible que aumenta muy lentamente pero que sigue careciendo de fármacos que realmente sean capaces de ejercer un impacto significativo en el curso del proceso degenerativo. Las hipótesis con las que tradicionalmente se había tratado de explicar el origen de estas enfermedades están siendo paulatinamente abandonadas ante la evidencia, cada vez mayor, de que estaban basadas en fenómenos que constituyen más bien consecuencias, y no causas fundamentales, de la degeneración neuronal.
Al tiempo que estas teorías van quedando obsoletas, la ardua investigación científica en este campo está permitiendo señalar nuevos actores cuya relación con el origen de estas enfermedades podría ser mucho más directa. Muchos estudios señalan que el estrés oxidativo, las alteraciones de la proteostasis y la neuroinflamación relacionados con el envejecimiento podrían desempeñar un papel central en el inicio y, sobre todo, la extensión de la degeneración neuronal. Parece, además, que todos estos factores están estrechamente interconectados y son capaces de retroalimentarse constituyendo un círculo vicioso patológico imparable. De la identificación de estos fenómenos como causas primigenias de la neurodegeneración está emergiendo una miríada de nuevas dianas terapéuticas que apenas se han comenzado a explorar, y que ofrecen nuevas oportunidades a antiguos fármacos que buscan ahora reposicionarse como potencial tratamiento de la neurodegeneración. Por otra parte, ante la complejidad que ofrece el abordaje terapéutico de estas patologías multifactoriales, se están incorporando aproximaciones muy novedosas para el diseño de nuevos fármacos. Así, las terapias génicas o inmunológicas ocupan ya un lugar importante en el panorama de los nuevos candidatos a fármaco, mientras que la química médica se adapta al reto mediante el diseño de nuevos compuestos multidiana y la optimización de las herramientas computacionales que, bien utilizadas, otorgan un poder inédito para la concepción de una enorme cantidad de moléculas con potencial actividad frente a muchas de las dianas asociadas a la neurodegeneración.
Aún faltan muchas incógnitas por despejar en lo que se refiere a las causas y el desarrollo del proceso neurodegenerativo a nivel celular, pero los avances conseguidos en los últimos años y la mayor disponibilidad de herramientas para el diseño de nuevos fármacos permiten tener la esperanza de que, paso a paso, nos acercamos al momento en que dispondremos de nuevas terapias realmente capaces de detener el avance de estas patologías e incluso, por qué no, de revertirlas.
LISTA DE ABREVIATURAS
AChE: Acetilcolinesterasa; ACS: apoptosis-associated speck-like protein containing ACS; ADN: Ácido desoxirribonucleico; APP: Amyloid-beta Precursor Protein; ARE: Antioxidant Response Elements; ARN: Ácido ribonucleico; ATP: Adenosín trifosfato; Ab: péptido b-amiloide; BACE-1: Beta-site Amyloid-precursor-protein-Cleaving Enzyme 1; CAG: Citosina-Adenina-Guanina; COMT: Catecol O-metiltransferasa; COX: ciclooxigenasa; CRISPR-Cas9: Clustered Regularly Interspaced Short Palindromic Repeats; DAMP: Damage Associated Molecular Patterns; DCL: Deterioro Cognitivo Leve; DFT: Demencia frontotemporal; EA: Enfermedad de Alzheimer; EH: Enfermedad de Huntington; ELA: Esclerosis Lateral Amiotrófica; ELAe: Esclerosis Lateral Amiotrófica esporádica; ELAf: Esclerosis Lateral Amiotrófica familiar; EP: Enfermedad de Parkinson; FDA: Food and Drug Administration; FUS: Fused in sarcoma; GSK-3b: Glycogen Synthase Kinase 3 beta; HO-1: hemooxigenasa 1; HTT: Huntingtina; IKK: IkappaB kinase; IL: interleuquinas; iNOS: inductible Nitric Oxide Synthase; IPOD: Insoluble protein deposit compartment; JNK: Jun N-terminal Kinase; JUNQ: Juxta nuclear quality control compartment; Keap-1: Kelch-like ECH-associated protein 1; LCR: Líquido cefalorraquídeo; LPS: lipopolisacárido; LRRK-2: Leucine-Rich Repeat Kinase 2; Maf: Musculoaponeurotic fibrosarcoma; MAO-B: Monoamino oxidasa B; NLR: Nod-Like Receptors; NMDA: N-Metil D-Aspartato; NOX: NAD(P)H oxidasa; NQO1: NAD(P)H deshidrogenasa quinona 1; Nrf2: Nuclear factor erythroid 2-Related Factor; PAMP: Patogen Associated Molecular Patterns; PET: Positron Emission Tomography; PINK-1: PTEN-induced kinase 1; PPAR-g: Peroxisome Proliferator- Activated Receptor gamma; PTP: Poros de Transición de Permeabilidad; QBP-1: polyglutamine-binding peptide 1; RAGE: Receptor for Advance Glication Endproducts; RMN: Resonancia Magnética Nuclear; RNS: Reactive Nitrogen Species; ROS: Reactive Oxygen Species ; RRP: Receptores de Reconocimiento de Patrones; SNC: Sistema Nervioso Central; SNP: Sistema Nervioso Periférico; SOD-1: Superóxido dismutasa 1; TARBP: Trans-Activation-responsive RNA-Binding Protein; TDP-43: TAR DNA-binding Protein 43; TGF-b: Transforming Growth Factor beta; TLR: Toll-Like Receptors; b-TrCP: Beta-Transducin repeats-Containing Protein.
7. REFERENCIAS
- Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: What is it and where are we? J Clin Invest. 2003;111(1):3–10.
- Roser M, Ortiz-Ospina E, Ritchie H. Life Expectancy. OurWorldInData.org. 2013. p. Retrieved from: ’https://ourworldindata.org/life-e.
- World Health Organization. Risk Reduction Of Cognitive Decline And Dementia. WHO Guidelines. 2019. Available from: https://www.who.int/mental_health/neurology/dementia/risk_reduction_gdg_meeting/en/
- Rocca WA. The burden of Parkinson’s disease: a worldwide perspective. Lancet Neurol. 2018;17(11):928–9.
- Arthur KC, Calvo A, Price TR, Geiger JT, Chiò A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun. 2016;7(12408):1–6.
- Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019;15:565–81.
- Alzheimer Europe. Dementia in Europe yearbook 2019. Alzheimer Europe. 2019. Available from: https://www.alzheimer-europe.org/sites/default/files/alzheimer_europe_dementia_in_europe_yearbook_2019.pdf
- Hirsch L, Jette N, Frolkis A, Steeves T, Pringsheim T. The Incidence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Neuroepidemiology. 2016;46:292–300.
- Chiò A, Logroscino G, Traynor BJ, Collins J, Simeone JC, Goldstein LA, et al. Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology. 2013;41(2):118–30.
- Logroscino G, Piccininni M. Amyotrophic lateral sclerosis descriptive epidemiology: The origin of geographic difference. Neuroepidemiology. 2019;52(1–2):93–103.
- Dayalu P, Albin RL. Huntington Disease: Pathogenesis and Treatment. Neurol Clin. 2015;33:101–14.
- Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest. 2005;115:1449–57.
- Erkkinen MG, Kim M, Geschwind MD. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb Perspect Biol. 2018;10:1–44.
- Santiago JA, Bottero V, Potashkin JA. Dissecting the molecular mechanisms of neurodegenerative diseases through network biology. Front Aging Neurosci. 2017;9:1–13.
- Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci. 2015;9(124):1–22.
- Lock EA, Zhang J, Checkoway H. Solvents and Parkinson disease: A systematic review of toxicological and epidemiological evidence. Toxicol Appl Pharmacol. 2013;266:345–55.
- Costa LG. Traffic-Related Air Pollution and Neurodegenerative Diseases: Epidemiological and Experimental Evidence, and Potential Underlying Mechanisms. 1st ed. Vol. 1, Advances in Neurotoxicology. Elsevier Inc.; 2017. 1–46 p.
- Kalia L V., Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912.
- Riancho J, Sanchez de la Torre JR, Paz-Fajardo L, Limia C, Santurtun A, Cifra M, et al. The role of magnetic fields in neurodegenerative diseases. Int J Biometeorol. 2021;65:107–17.
- Zubair Alam M, Alam Q, Kamal MA, Jiman-Fatani AA, Azhar EI, Khan M, et al. Infectious Agents and Neurodegenerative Diseases: Exploring the Links. Curr Top Med Chem. 2017;17(12):1390–9.
- Beach TG. A Review of Biomarkers for Neurodegenerative Disease: Will They Swing Us Across the Valley? Neurol Ther. 2017;6(s1):s5–13.
- Jeromin A, Bowser R. Biomarkers in neurodegenerative diseases. In: Advances in Neurobiology. 2017. p. 491–528.
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7(3):280–92.
- Khoury R, Ghossoub E. Diagnostic biomarkers of Alzheimer’s disease: A state-of-the-art review. Biomarkers in Neuropsychiatry. 2019;1:1–6.
- Archetti D, Ingala S, Venkatraghavan V, Wottschel V, Young AL, Bellio M, et al. Multi-study validation of data-driven disease progression models to characterize evolution of biomarkers in Alzheimer’s disease. NeuroImage Clin. 2019;24:1–13.
- Flórez J, Armijo JA, Mediavilla Á. Farmacología humana, 6a ed. 2013.
- Oxford AE, Stewart ES, Rohn TT. Clinical Trials in Alzheimer’s Disease: A Hurdle in the Path of Remedy. Int J Alzheimers Dis. 2020;2020:1–13.
- Cummings J. Disease modification and Neuroprotection in neurodegenerative disorders. Transl Neurodegener. 2017;6(25):1–7.
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020;16(3):391–460.
- Vermunt L, Sikkes SAM, van den Hout A, Handels R, Bos I, van der Flier WM, et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019;15(7):888–98.
- Mayeux R, Stern Y. Epidemiology of Alzheimer Disease. Cold Spring Harb Perspect Med. 2012;2(a006239):1–18.
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census Liesi. Neurology. 2013;80:1178–783.
- Terry Jr A V, Buccafusco JJ. The Cholinergic Hypothesis of Age and Alzheimer ’ s Disease- Related Cognitive Deficits : Recent Challenges and Their Implications for Novel Drug Development. J Pharmacol Exp Ther. 2003;306(3):821–7.
- Hillen H. The Beta Amyloid Dysfunction (BAD) Hypothesis for Alzheimer’s Disease. Front Neurosci. 2019;13(November):1–10.
- Barragán Martínez D, García Soldevilla MA, Parra Santiago A, Tejeiro Martínez J. Alzheimer’s disease. Med. 2019;12(74):4338–46.
- Rabinovici GD. Controversy and Progress in Alzheimer’s Disease: Revisiting FDA Approval of Aducanumab. N Engl J Med. 2021;385(9):771–4.
- Zesiewicz TA. Parkinson Disease. Contin (MINNEAP MINN). 2019;25(4):896–918.
- Simon DK, Tanner CM, Brundin P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin Geriatr Med. 2020;36(1):1–12.
- Armstrong MJ, Okun MS. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA – J Am Med Assoc. 2020;323(6):548–60.
- Pinna A. Adenosine A2A Receptor Antagonists in Parkinson’s Disease: Progress in Clinical Trials from the Newly Approved Istradefylline to Drugs in Early Development and Those Already Discontinued. CNS Drugs. 2014;28:455–74.
- van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084–98.
- Longinetti E, Fang F. Epidemiology of amyotrophic lateral sclerosis: An update of recent literature. Curr Opin Neurol. 2019;32(5):771–6.
- Van Blitterswijk M, Van Es MA, Hennekam EAM, Dooijes D, Van Rheenen W, Medic J, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(17):3776–84.
- Oskarsson B, Gendron TF, Staff NP. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin Proc. 2018;93(11):1617–28.
- Jaiswal MK. Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39(2):733–48.
- Koch JC, Kuttler J, Maass F, Lengenfeld T, Zielke E, Bähr M, et al. Compassionate Use of the ROCK Inhibitor Fasudil in Three Patients With Amyotrophic Lateral Sclerosis. Front Neurol. 2020;11(173):1–8.
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–16.
- Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, Douglas I, Evans SJW, et al. The prevalence of huntington’s disease. Neuroepidemiology. 2016;46:144–53.
- Barker RA, Priller J. Huntington’s Disease. In: Movement Disorders. 2013. p. 213–22.
- Sies H, Berndt C, Jones DP. Oxidative Stress. Annu Rev Biochem. 2017;86:715–48.
- Andersen JK. Oxidative stress in neurodegeneration: Cause or consequence? Nat Rev Neurosci. 2004;10(7):S18.
- Martínez MC, Andriantsitohaina R. Reactive nitrogen species: Molecular mechanisms and potential significance in health and disease. Antioxidants Redox Signal. 2009;11(3):669–702.
- Slimen IB, Najar T, Ghram A, Dabbebi H, Ben Mrad M, Abdrabbah M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int J Hyperth. 2014;30(7):513–23.
- Singh E, Devasahayam G. Neurodegeneration by oxidative stress: a review on prospective use of small molecules for neuroprotection. Mol Biol Rep. 2020;47(4):3133–40.
- Chen X, Guo C, Kong J. Oxidative stress in neurodegenerative diseases. Neural Regen Res. 2012;7(5):376–85.
- Rekatsina M, Paladini A, Piroli A, Zis P, Pergolizzi J V., Varrassi G. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv Ther. 2020;37(1):113–39.
- Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol Rev. 2018;70:348–83.
- Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88(Part B):101–7.
- Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88:93–100.
- Cores Á, Piquero M, Villacampa M, León R, Menéndez JC. NRF2 regulation processes as a source of potential drug targets against neurodegenerative diseases. Biomolecules. 2020;10(6):1–38.
- Dinkova-Kostova AT, Kostov R V., Kazantsev AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018;285(19):3576–90.
- Mahan VL. Neurometabolic effects of carbon monoxide. Med Gas Res. 2012;2(32):1–7.
- Wu KC, Cui JY, Klaassen CD. Effect of Graded Nrf2 Activation on Phase-I and -II Drug Metabolizing Enzymes and Transporters in Mouse Liver. PLoS One. 2012;7(7):1–10.
- Pajares M, Jiménez-Moreno N, García-Yagüe ÁJ, Ceballos ML De, Leuven F Van, Rábano A, et al. Transcription factor NFE2L2 / NRF2 is a regulator of macroautophagy genes. Autophagy. 2016;12(10):1902–16.
- Niture SK, Jaiswal AK. Nrf2 Protein Up-regulates Antiapoptotic Protein Bcl-2 and Prevents Cellular Apoptosis. J Biol Chem. 2012;287(13):9873–86.
- Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, León R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther. 2016;157:84–104.
- Aiken CT, Kaake RM, Wang X, Huang L. Oxidative Stress-Mediated Regulation of Proteasome Complexes. Mol Cell Proteomics. 2011;10:1–11.
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322(1–2):254–62.
- Cenini G, Lloret A, Cascella R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid Med Cell Longev. 2019;2019:1–18.
- Angelova PR. Sources and triggers of oxidative damage in neurodegeneration. Free Radic Biol Med. 2021;173:52–63.
- Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta – Mol Cell Res. 2016;1863(12):2977–92.
- Jemmer P. Oxidative Stress in Parkinson ’ s Disease. Ann Neurol. 2003;53(suppl 3):S23–38.
- Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2018;109:249–57.
- Chen JX, Yan S Du. Amyloid- b -Induced Mitochondrial Dysfunction. J Alzheimer’s Dis. 2007;12:177–84.
- Bossy-Wetzel E, Petrilli A, Knott AB. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008;31(12):609–16.
- Barber SC, Shaw PJ. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010;48(5):629–41.
- Meyerowitz J, Parker SJ, Vella LJ, Ng DC, Price KA, Liddell JR, et al. C-Jun N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol Neurodegener. 2011;6(57):1–22.
- Pehlivan FE. Vitamin C: An Antioxidant Agent. In: Vitamin C. 2017. p. 23–35.
- Gülçin I. Antioxidant properties of resveratrol: A structure-activity insight. Innov Food Sci Emerg Technol. 2010;11(1):210–8.
- Chupradit S, Bokov D, Zamanian MY, Heidari M, Hakimizadeh E. Hepatoprotective and therapeutic effects of resveratrol: A focus on anti-inflammatory and antioxidative activities. Fundam Clin Pharmacol. 2022;36:468–85.
- Farkhondeh T, Folgado SL, Pourbagher-Shahri AM, Ashrafizadeh M, Samarghandian S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed Pharmacother. 2020;127:1–17.
- Hatcher H, Planalp R, Cho J, Torti FM, Torti S V. Curcumin: From ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–52.
- Ak T, Gülçin I. Antioxidant and radical scavenging properties of curcumin. Chem Biol Interact. 2008;174:27–37.
- Shahcheraghi SH, Salemi F, Peirovi N, Ayatollahi J, Alam W, Khan H, et al. Nrf2 regulation by curcumin: Molecular aspects for therapeutic prospects. Molecules. 2022;27(167):1–20.
- Pandi-Perumal SR, BaHammam AS, Brown GM, Spence DW, Bharti VK, Kaur C, et al. Melatonin antioxidative defense: Therapeutical implications for aging and neurodegenerative processes. Neurotox Res. 2013;23:267–300.
- Cardinali DP. Melatonin: Clinical perspectives in neurodegeneration. Front Endocrinol (Lausanne). 2019;10(JULY):1–22.
- Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33(101544):1–8.
- Tenório MCS, Graciliano NG, Moura FA, de Oliveira ACM, Goulart MOF. N-acetylcysteine (Nac): Impacts on human health. Antioxidants. 2021;10(967):1–34.
- Younus H. Therapeutic potentials of superoxide dismutase. Int J Health Sci (Qassim). 2018;12(3):88–93.
- Landgraf AD, Alsegiani AS, Alaqel S, Thanna S, Shah ZA, Sucheck SJ. Neuroprotective and anti-neuroinflammatory properties of ebselen derivatives and their potential to inhibit neurodegeneration. ACS Chem Neurosci. 2020;11:3008–16.
- Li X, Shi Q, Xu H, Xiong Y, Wang C, Le L, et al. Ebselen Interferes with Alzheimer’s Disease by Regulating Mitochondrial Function. Antioxidants. 2022;11(1350):1–14.
- Savla SR, Laddha AP, Kulkarni YA. Pharmacology of apocynin: a natural acetophenone. Drug Metab Rev. 2021;53(4):542–62.
- Kelkar A, Kuo A, Frishman WH. Allopurinol as a cardiovascular drug. Cardiol Rev. 2011;19:265–71.
- Mukherjee P, Cinelli MA, Kang S, Silverman RB. Development of nitric oxide synthase inhibitors for neurodegeneration and neuropathic pain. Chem Soc Rev. 2014;43(19):6814–38.
- Cinelli MA, Do HT, Miley GP, Silverman RB. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med Res Rev. 2020;40(1):158–89.
- Robledinos-Antón N, Fernández-Ginés R, Manda G, Cuadrado A. Activators and Inhibitors of NRF2 : A Review of Their Potential for Clinical Development. Oxid Med Cell Longev. 2019;(1–20).
- Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F, et al. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med Res Rev. 2019;39(5):1923–52.
- LoPachin RM, Geohagen BC, Nordstroem LU. Mechanisms of Soft and Hard Electrophile Toxicities. Toxicology. 2019;418:62–9.
- Kerr F, Sofola-Adesakin O, Ivanov DK, Gatliff J, Gomez Perez-Nievas B, Bertrand HC, et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017;13(3):1–30.
- Mou Y, Wen S, Li YX, Gao XX, Zhang X, Jiang ZY. Recent progress in Keap1-Nrf2 protein-protein interaction inhibitors. Vol. 202, European Journal of Medicinal Chemistry. Elsevier Masson SAS; 2020. p. 1–12.
- Katsuragi Y, Ichimura Y, Komatsu M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Vol. 1, Current Opinion in Toxicology. Elsevier Ltd; 2016. p. 54–61.
- Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, et al. Structural and Functional Characterization of Nrf2 Degradation by the Glycogen Synthase Kinase 3/b-TrCP Axis. Mol Cell Biol. 2012;32(17):3486–99.
- Armagan G, Sevgili E, Gürkan FT, Köse FA, Bilgiç T, Dagci T, et al. Regulation of the Nrf2 pathway by glycogen synthase kinase-3b in MPP+-induced cell damage. Molecules. 2019;24:1–20.
- Davis AA, Leyns CEG, Holtzman DM. Intercellular Sp read of Protein Aggregates in Neurodegenerative Disease. Annu Rev Cell Dev Biol. 2018;34:545–68.
- Nick Pace C, Martin Scholtz J, Grimsley GR. Forces stabilizing proteins. FEBS Lett. 2014;588(14):2177–84.
- Díaz-Villanueva JF, Díaz-Molina R, García-González V. Protein Folding and Mechanisms of Proteostasis. Int J Mol Sci. 2015;16:17193–230.
- Hipp MS, Park S, Hartl FU. Proteostasis impairment in protein- misfolding and -aggregation diseases. Trends Cell Biol. 2014;24(9):506–14.
- Alam P, Siddiqi K, Chturvedi SK, Khan RH. Protein aggregation: From background to inhibition strategies. Int J Biol Macromol. 2017;103:208–19.
- Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. 2019;20(7):421–35.
- Kurtishi A, Rosen B, Patil KS, Alves GW, Møller SG. Cellular Proteostasis in Neurodegeneration. 2019;(February 2018):3676–89.
- Wells C, Brennan S, Keon M, Ooi L. The role of amyloid oligomers in neurodegenerative pathologies. Int J Biol Macromol. 2021;181:582–604.
- Breydo L, Uversky VN. Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 2015;589(19):2640–8.
- Carija A, Navarro S, de Groot NS, Ventura S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 2017;12(February):699–711.
- Frost B, Diamond MI. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 2010;11:155–9.
- Faller P, Hureau C, Berthoumieu O. Role of Metal Ions in the Self-assembly of the Alzheimer ’ s Amyloid b Peptide. Inorg Chem. 2013;52:12193–206.
- Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158:47–52.
- Lee M, Kim JI, Na S, Eom K. Metal ions affect the formation and stability of amyloid baggregates at multiple length scales. Phys Chem Chem Phys. 2018;20(13):8951–61.
- Budimir A. Metal ions , Alzheimer ’ s disease and chelation therapy. Acta Pharm. 2011;61:1–14.
- Kepp KP. Bioinorganic Chemistry of Alzheimer ’ s Disease. Chem Rev. 2012;112:5193–239.
- Cicero CE, Mostile G, Vasta R, Rapisarda V, Santo S, Ferrante M, et al. Metals and neurodegenerative diseases . A systematic review. Environ Res. 2017;159:82–94.
- Morley JE, Farr SA, Nguyen AD, Xu F. What is the Physiological Function of Amyloid-Beta Protein? J Nutr Heal Aging. 2019;23(3):225–6.
- Kuperstein I, Broersen K, Benilova I, Rozenski J, Jonckheere W, Debulpaep M, et al. Neurotoxicity of Alzheimer’s disease Ab peptides is induced by small changes in the Ab42 to Ab40 ratio. EMBO J. 2010;29(19):3408–20.
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, et al. Oligomeric amyloid b associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106(10):4012–7.
- He Y, Cui J, Lee JC m., Ding S, Chalimoniuk M, Simonyi A, et al. Prolonged exposure of cortical neurons to oligomeric amyloid-β impairs NMDA receptor function via NADPH oxidasemediated ROS production: Protective effect of green tea (-)-epigallocatechin-3-gallate. ASN Neuro. 2011;3(1):13–24.
- Saha P, Sen N. Tauopathy: A common mechanism for neurodegeneration and brain aging. Mech Ageing Dev. 2019;178(January):72–9.
- Arriagada P V., Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3):631–9.
- Bellani S, Sousa VL, Ronzitti G, Valtorta F, Meldolesi J, Chieregatti E. The regulation of synaptic function by a-synuclein. Commun Integr Biol. 2010;3(2):106–9.
- Blokhuis AM, Groen EJN, Koppers M, Van Den Berg LH, Pasterkamp RJ. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(6):777–94.
- Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. 2014;10(6):337–48.
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89(5):910–26.
- Jenagaratnam L, McShane R. Clioquinol for the treatment of Alzheimer’s Disease. Cochrane Database Syst Rev. 2006;
- Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, et al. PBT2 rapidly improves cognition in alzheimer’s disease: Additional phase II analyses. J Alzheimer’s Dis. 2010;20(2):509–16.
- Pandolfo M, Arpa J, Delatycki MB, Le Quan Sang KH, Mariotti C, Munnich A, et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann Neurol. 2014;76(4):509–21.
- Martin-Bastida A, Ward RJ, Newbould R, Piccini P, Sharp D, Kabba C, et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci Rep. 2017;7:1–9.
- Villarejo B. Autofagia selectiva en la retina: Fisiología y patología. 2019.
- Prati F, Bottegoni G, Bolognesi ML, Cavalli A. BACE-1 Inhibitors: From Recent Single-Target Molecules to Multitarget Compounds for Alzheimer’s Disease. J Med Chem. 2018;61(3):619–37.
- Sun J, Roy S. Gene-based therapies for Neurodegenerative Diseases. Vol. 24. 2021. 297–311 p.
- Dhouafli Z, Cuanalo K, El C, Hayouni A, Mays CE, Soto C, et al. Inhibition of protein misfolding and aggregation by natural phenolic compounds. Cell Mol Life Sci. 2018;75(19):3521–38.
- Rajan R, Ahmed S, Sharma N, Kumar N, Debas A, Matsumura K. Review of the current state of protein aggregation inhibition from a materials chemistry perspective: Special focus on polymeric materials. Mater Adv. 2021;2:1139–76.
- Mortada I, Farah R, Nabha S, Ojcius DM, Fares Y, Almawi WY, et al. Immunotherapies for Neurodegenerative Diseases. Vol. 12, Frontiers in Neurology. 2021. p. 1–19.
- Daneman R, Prat A. The Blood-Brain Barrier. Cold Spring Harb Perspect Biol. 2008;7:1–23.
- Jessen NA, Munk ASF, Lundgaard I, Nedergaard M. The Glymphatic System: A Beginner’s Guide. Neurochem Res. 2015;40:2583–99.
- Brahadeeswaran S, Sivagurunathan N, Calivarathan L. Inflammasome Signaling in the Aging Brain and Age-Related Neurodegenerative Diseases. Mol Neurobiol. 2022;59:2288–304.
- Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72.
- Tang Y, Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 2016;53(2):1181–94.
- Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–91.
- Morales I, Farías GA, Cortes N, Maccioni RB. Neuroinflammation and neurodegeneration. In: Update on Dementia. 2016. p. 17–47.
- Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. 2019;10(1008):1–17.
- Subramaniam SR, Federoff HJ. Targeting microglial activation states as a Therapeutic Avenue in Parkinson’s disease. Front Aging Neurosci. 2017;9:1–18.
- FRANCESCHI C, BONAFÈ M, VALENSIN S, OLIVIERI F, DE LUCA M, OTTAVIANI E, et al. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann N Y Acad Sci. 2000;908:244–54.
- Lučiūnaitė A, McManus RM, Jankunec M, Rácz I, Dansokho C, Dalgėdienė I, et al. Soluble Ab oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem. 2020;155:650–61.
- Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, Robertson AAB, et al. The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia. 2020;68:407–21.
- Paldino E, D’Angelo V, Sancesario G, Fusco FR. Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: new insight on the inflammasome. Cell Death Discov. 2020;6(69):1–12.
- Paldino E, Fusco FR. Emerging Role of NLRP3 Inflammasome/Pyroptosis in Huntington’s Disease. Int J Mol Sci. 2022;23(8363):1–11.
- Fu WY, Wang X, Ip NY. Targeting neuroinflammation as a therapeutic strategy for Alzheimer’s disease: Mechanisms, drug candidates, and new opportunities. ACS Chem Neurosci. 2019;10(2):872–9.
- Liu CY, Wang X, Liu C, Zhang HL. Pharmacological Targeting of Microglial Activation: New Therapeutic Approach. Front Cell Neurosci. 2019;13:1–19.
- Henry CJ, Huang Y, Wynne A, Hanke M, Himler J, Bailey MT, et al. Minocycline attenuates lipopolysaccharide (LPS)-induced neuroinflammation, sickness behavior, and anhedonia. J Neuroinflammation. 2008;5(15):1–14.
- Cankaya S, Cankaya B, Kilic U, Kilic E, Yulug B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context. 2019;8:1–14.
- Qie S, Ran Y, Lu X, Su W, Li W, Xi J, et al. Candesartan modulates microglia activation and polarization via NF-kB signaling pathway. Int J Immunopathol Pharmacol. 2020;34:1–13.
- Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Slowing of Loss of Cognition in Mild Alzheimer ’ s Disease. J Prev Alzheimer’s Dis. 2018;5(2):149–54.
- Kim C, Spencer B, Rockenstein E, Yamakado H, Mante M, Adame A, et al. Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating a-synuclein transmission and neuroinflammation. Mol Neurodegener. 2018;13(43):1–18.
- Monnet E, Choy EH, McInnes I, Kobakhidze T, De Graaf K, Jacqmin P, et al. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: A phase II study. Ann Rheum Dis. 2019;316–23.
- Gocmez SS, Yazir Y, Gacar G, Demirtaş Şahin T, Arkan S, Karson A, et al. Etanercept improves aging-induced cognitive deficits by reducing inflammation and vascular dysfunction in rats. Physiol Behav. 2020;224:1–9.
- Butchart J, Brook L, Hopkins V, Teeling J, Püntener U, Culliford D, et al. Etanercept in Alzheimer disease. A randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84(21):2161–8.
- Jung YJ, Tweedie D, Scerba MT, Greig NH. Neuroinflammation as a Factor of Neurodegenerative Disease: Thalidomide Analogs as Treatments. Front Cell Dev Biol. 2019;7(December):1–24.
- Alam J, Blackburn K, Patrick D. Neflamapimod: Clinical Phase 2b-Ready Oral Small Molecule Inhibitor of p38a to Reverse Synaptic Dysfunction in Early Alzheimer’s Disease. J Prev Alzheimer’s Dis. 2017;4:273–8.
- Koch JC, Tatenhorst L, Roser AE, Saal KA, Tönges L, Lingor P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol Ther. 2018;189:1–21.
- Lingor P, Weber M, Camu W, Friede T, Hilgers R, Leha A, et al. ROCK-ALS: Protocol for a Randomized, Placebo-Controlled, Double-Blind Phase IIa Trial of Safety, Tolerability and Efficacy of the Rho Kinase (ROCK) Inhibitor Fasudil in Amyotrophic Lateral Sclerosis. Front Neurol. 2019;10(293):1–11.
- Tönges L, Günther R, Suhr M, Jansen J, Balck A, Saal KA, et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia. 2014;62(2):217–32.
- Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17:588–606.
- Li H, Guan Y, Liang B, Ding P, Hou X, Wei W, et al. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur J Pharmacol. 2022;928:1–9.
- Deardorff WJ, Grossberg GT. Targeting neuroinflammation in Alzheimer’s disease: evidence for NSAIDs and novel therapeutics. Expert Rev Neurother. 2017;17(1):17–32.
