1. LA FAMILIA DE MICROARNs LET-7 Y SU PAPEL EN EL CÁNCER
Los micro-ARN (miARNs) son transcritos de genes que no se traducen a proteínas, y que ejercen papeles de regulación sobre la traducción de otros transcritos. El descubrimiento de la presencia de estos genes en el genoma humano, ya entrado en el siglo XXI, ha supuesto un cambio en el paradigma y función que se le otorgaba a los genes no codificantes de proteínas y sus transcritos (los ARN no codificantes).
El primer miembro de estos genes se descubrió al comienzo de la década de los ochenta en el gusano C. elegans (Ambros, 1989; Chalfie et al., 1981). En principio se pensó que era una peculiaridad de este nematodo, sin embargo, años después se empezaron a encontrar secuencias homólogas en otros organismos, incluidos los humanos, hasta el punto de que se ha estimado que corresponden al 1-4% de todos los genes transcritos humanos (Bentwich et al., 2005; Lim et al., 2003), siendo una de las familias de genes reguladores más abundante del genoma. Actualmente, en octubre de 2023 se han censado en la base de datos miRbase (https://mirbase.org/) 1917 secuencias encontradas en el genoma humano que podrían codificar a microARNs, de entre ellas existe evidencia alta de que lo hagan en más de quinientas. El papel de los microARNs sobre la síntesis de proteínas ha abierto un nuevo campo en la patología molecular, ya que disfunciones en la actividad de estos reguladores puede generar diversas patologías cuya causa no se había podido hallar hasta ahora.
El primer microARN descubierto en humanos se trató de let-7a. Su descubrimiento se hizo evidente cuando se compararon los genomas de humanos y del nematodo C. elegans y se halló que la secuencia funcional del gen lethal-7 de C. elegans tenía una secuencia ortóloga en humanos (hsa-let-7a). De hecho, el genoma humano presenta, una, familia de microARNs let-7, compuesta por 12 genes que codifican para 9 microARN maduros, esto es así porque tres de ellos codifican para el mismo microARN maduro. Estos microARNs se agrupan en una familia porque presentan unas secuencias muy similares de 21-22 nucleótidos, que presentan en el extremo 5’ una región conservada llamada semilla de familia (family seed), implicada en su funcionalidad y en el reconocimiento de dianas (Figura 1).

Figura 1: Familia de microARNs let-7: en ratón, al igual que en humanos, existen 12 genes (situados en diferentes posiciones cromosómicas) que codifican para 9 microARNs maduros (representados en la derecha). Las familias de microARNs comparten una secuencia (family seed) que está implicada en el reconocimiento de sus dianas y su funcionalidad, para la familia de let-7 esta secuencia es (GAGGTAG).
En este trabajo se expondrán resultados que intentan profundizar en el conocimiento de la implicación de los microARNs en el desarrollo del cáncer de pulmón con el fin de diseñar terapias experimentales que sirvan como prueba de concepto para su futuro aplicación en la clínica.
El cáncer de pulmón es el tumor más importante en cuanto a mortalidad en el mundo occidental. En España en el 2022 se diagnosticaron alrededor de 31000 casos nuevos de cáncer de pulmón y ha sido responsable de alrededor de 22000 muertes. Esto le hace ser el segundo tipo de cáncer en cuanto a incidencia (tras el cáncer colorrectal) y el primero en cuanto mortalidad. Como enfermedad, el cáncer de pulmón sigue siendo altamente letal, ya que menos del 15% de los pacientes españoles logran sobrevivir cinco años tras el diagnóstico. La letalidad de los cánceres de estómago, hígado, páncreas, esófago y estómago son comparables a la que tiene el cáncer de pulmón, aunque la suma de muertes que estos producen es la mitad de las que este produce el cáncer de pulmón.
Actualmente existen un número de evidencias creciente que vinculan la carcinogénesis pulmonar con aberraciones en los microARNs y en concreto con la familia let-7, algunas de las más significativas son:
- Se ha observado que la ARNasa encargada de la maduración citoplasmática de los trascritos de miARNs (DICER) presenta una pérdida de expresión en cáncer de pulmón relacionada con un peor pronóstico (Karube et al., 2005).
- La expresión de los genes de la familia de microARNs let-7 está reducida significativamente en tumores pulmonares pareados (Takamizawa et al., 2004).
- Varios genes de la familia de microARNs let-7 mapean en regiones genómicas que están delecionadas tumores primarios pulmonares (Calin et al., 2004)
- La de la familia de microARNs let-7 puede inhibir el crecimiento tumoral en algunos modelos celulares experimentales (Johnson et al., 2007)
- La expresión de genes importantes para el desarrollo tumoral como el oncogén KRAS (un oncogén significativamente alterado en cáncer de pulmón) está regulado por los genes de la familia de microARNs let-7 (Johnson et al., 2005).
A continuación, se expondrán evidencias experimentales desarrollas en preclínicos in vivo (Trang P, Medina P.P. et al., 2010) que pueden utilizarse para determinar el papel biológico de let-7 en el desarrollo tumoral y para desarrollar terapias experimentales antitumorales.
2. LA RE-INTRODUCCIÓN DE LET-7 EN TUMORES HUMANOS TRANSPLANTADOS EN RATONES INMUNODEFICIENTES REDUCE EL DESAROLLO TUMORAL
Estos estudios tenían como objetivo, estudiar qué pasa cuando se recupera la actividad de let-7, que se pierde en pacientes de cáncer de pulmón.
Para realizar estos estudios se utilizaron ratones severamente inmunodeprimidos del tipo NOD/SCID. Dichos ratones carecen de linfocitos funcionales, por lo que tienen muy dificultado el rechazo de células tumorales humanas, pudiéndose establecer en ellos trasplantes de tumores humanos (xenografts).
Se utilizó la línea celular H460, derivada de un tumor humano de células no pequeñas de cáncer de pulmón. Esta línea celular está entre las líneas celulares más agresivas y refractarias a terapias antitumorales convencionales (Hohla et al., 2007; Isobe et al., 2005).
Tres millones de células H460 fueron inyectadas subcutáneamente a cada ratón NOD/SCID (n=6). Once días después de la inoculación, los ratones desarrollaron tumores palpables de entre 100 y150 mm3 y fueron inyectados intratumoralmente con suspensiones que contenían el microARN let-7b sintético (oligonucleótidos obtenidos de Ambion, Austin, TX, USA) o un microARN sintético de secuencia aleatoria (miR-NC utilizado como control). Dichos microARN se acomplejaron previamente con un agente de transfección lipídico desarrollado específicamente para ARNs pequeños (siPORT, obtenido de Ambion, Austin, TX, USA)
El tratamiento fue repetido cada tres días (a los días 11, 14,17, 20), y la dinámica de crecimiento tumoral fue analizada durante las siguientes semanas, hasta que los ratones presentaron un tumor que excedía los 600 mm3 momento en el cual los ratones fueron sacrificados. Los tumores tratados solamente con el agente de transfección (siPORT), con suero salino (PBS) o con el microARN control (miR-NC) desarrollaron una dinámica de crecimiento similar. Sin embargo, los tumores que fueron tratados con let-7b, sufrieron una significativa reducción en su tasa de crecimiento (Figura 2).
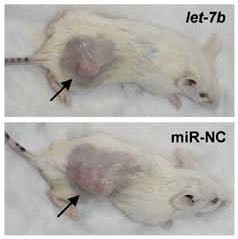
Figura 2: Izquierda Dinámica de crecimiento tumoral de xenografts en grupos de ratones (n=6) tratados con let-7b (cuadrados blancos), microARN control (rombos), el agente de transfección siPORT solamente (cuadrados negros) y suero salino (PBS) (triángulos). En ordenadas se representa el tamaño tumoral en milímetros cúbicos, en abscisas el tiempo en días. Las flechas indican los días en los cuales los ratones fueron tratados (11,14,17,20). Derecha: una pareja de ratones representativos cuyo tumor (flechas) fue tratado con let-7b o con el microARN control.
Trascurridos 21 días los ratones fueron sacrificados y sus tumores fueron estudiados histológicamente. Un análisis de micro-secciones teñidas con hemotoxilina y eosina (H&E) mostraron que los ratones controles (bien los tratados con el microARN control, o bien los tratados únicamente con el agente de transfección, siPORT) mostraron células densamente empaquetadas y sanas (marcadas como v), con pocas regiones necróticas (marcadas como d), en contraposición con los tumores que recibieron let-7b que presentaron amplias regiones con células muertas (Figura 3).
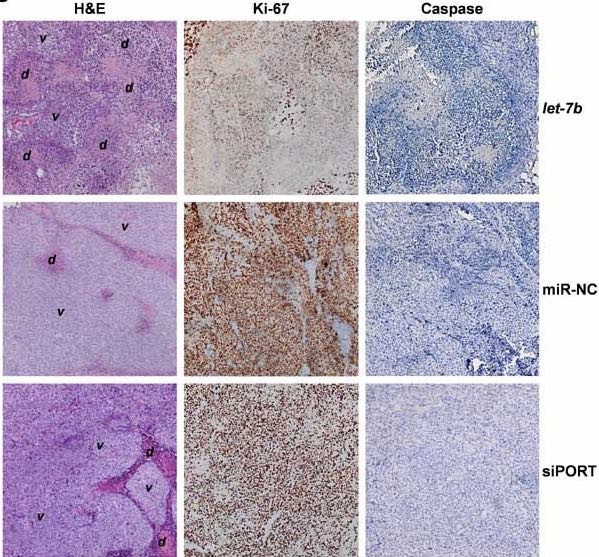
Figura 3: Histología de los tumores xenografts : Microsecciones histológicas teñidas con hematoxilina-eosina (H&E), el marcador de proliferación tumoral (Ki-67) y el marcador de apoptosis (caspase-3). Las células controles (tratadas con el agente de transfección siPORT) o con el microARN control (miR-NC) presentan una morfología viable (marcado como v en H&E) y unos niveles de proliferación (Ki67) sensiblemente superior a las tratadas con let-7b, que presentan abundante necrosis (marcada como d). Sin embargo, los niveles de apoptosis (que se deberían de marcar como marrón en el cuadro caspase) son similares y bajos en todos los tumores.
Las microsecciones tumorales fueron analizadas utilizando el marcador de proliferación celular Ki-67 y el marcador apoptótico (caspase-3), con el fin de evaluar la dinámica de crecimiento tumoral y de apoptosis. Los tumores controles (miR-NC y siPORT) presentaron una marcada presencia del antígeno ki-67 (teñido en marrón oscuro), mientras los tumores tratados con let-7b, presentaron una considerable reducción de este marcador. Sin embargo, los niveles de caspase-3 eran similares y bajos en todos los tumores. Así se puede concluir, que los efectos de la reintroducción de let-7b se deben en mayor medida a un efecto en una disminución en la proliferación tumoral y no en un aumento en la apoptosis de esta.
Con el fin de comprobar que dichos tumores incrementaban los niveles de let-7b tras la inyección del preparado de oligos sintéticos se realizó una PCR cuantitativa capaz de detectar al microARN let-7b maduro (Taqman miRNA, AB). Se observó que los tumores inyectados con los oligos let-7b tenían unos 14 veces más niveles de let-7b en comparación con los tumores provenientes de los controles (miR-NC, siPORT, PBS), confirmándose la adquisición de let7b por parte de las células tumorales inyectadas (Figura 4).
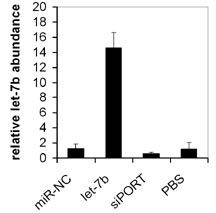
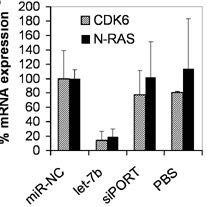
Figura 4: Izquierda: Niveles de let-7b en tumores tratados medido por PCR cuantitativa utilizando cebadores específicos para detectar let-7b. Derecha: niveles de conocidas dianas de let-7 (CDK6, N-RAS) medidos por PCR cuantitativa utilizando cebadores específicos. Los datos muestran medias y errores de tres ratones analizados.
La familia de microARNs let-7 actúa como tumor supresor, inhibiendo la expresión de importantes oncogenes (Johnson et al., 2007), por lo que, con el fin de ver la actividad del microARN transfectado se midió la cantidad de mensajero de dos de sus dianas: CDK6 y N-RAS. Se observó una sensible disminución de esta en los tumores tratados (Figura 4). Concluyéndose que el let-7 inyectado intratumoralmente, no es solo capaz de penetrar en las células tumorales, sino que es capaz de ejercer su función inhibiendo rutas oncogénicas conocidas para impedir el crecimiento tumoral.
3. La expresión de let-7 en tumores desarrollados en ratones que tienen activado el oncogén RAS provoca regresión tumoral
Para observar si let-7 podía reducir la carga tumoral actuando como un supresor tumoral in vivo, se utilizó un modelo de ratón transgénico (Jackson et al., 2001) que desarrolla tumores pulmonares en pocas semanas cuando se activa la forma mutada del oncogén K-RAS (G12D). Dicha activación se produce a través de la administración de la recombinasa cre mediante adenovirus inoculados al pulmón por el pasaje nasofaríngeo (Figura 5). Se utilizó este modelo debido a que previamente se había demostrado que let-7 era capaz de inhibir los niveles de expresión de K-RAS (Johnson et al., 2005), por lo que este modelo puede poner más fácilmente el papel de let-7 en el desarrollo tumoral.
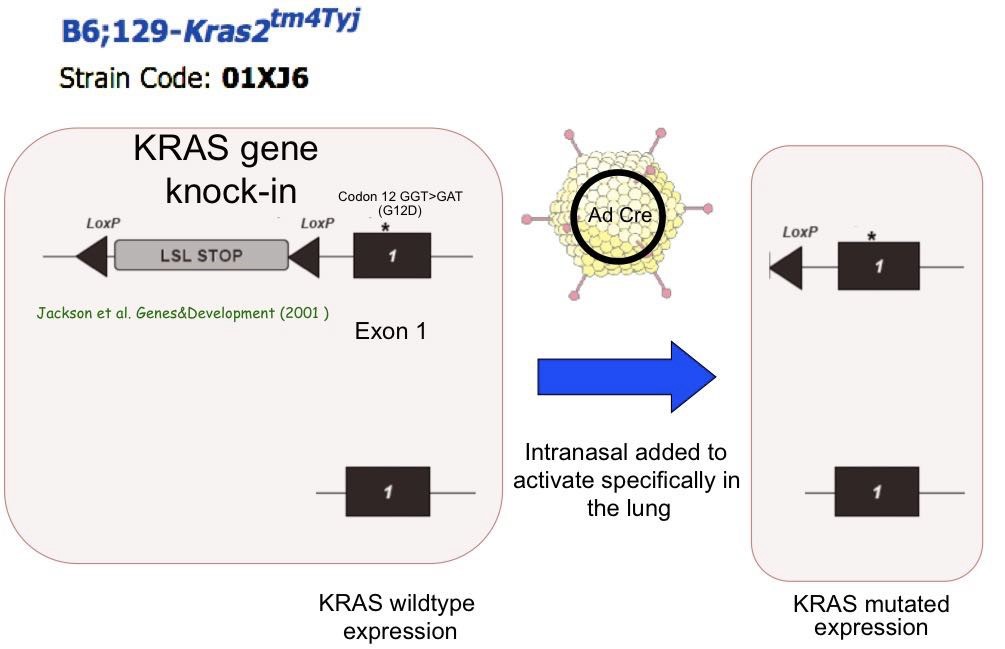
Figura 5: Ratón tansgénico LSL-Kras-G12D (Strain 01X6). La forma mutada de KRAS (G12D) se activa mediante la inoculación de adenovirus intranasalmente que codifican para la recombinasa Cre (Jackson et al., 2001). La actividad recombinasa elimina la secuencia parada (LSL STOP), por lo que la forma mutada de K-RAS (G12D) se transcribe.
Se utilizaron 18 ratones LSL-Kras-G12D de 6 semanas, a los que se administraron 5×108 PFUs unidades de adenovirus intranasalmente. Después de 10 semanas de incubación, se dividieron en tres grupos de seis ratones (0, L, C). Un grupo de ratones (0) se sacrificaron para obtener una idea de la carga tumoral en el momento del tratamiento, los dos restantes grupos (L y C) se inocularon con 1×106 PFUs unidades de lentivirus que codificaban para let-7 y un plásmido vacío (utilizado como control) respectivamente. Los dos grupos de ratones en tratamiento se mantuvieron durante cuatro semanas más y posteriormente fueron sacrificados para analizar su carga tumoral.
La carga tumoral fue medida en los diferentes grupos. El grupo de ratones tratado con let-7 (L) presentó una carga tumoral significativamente menor a los grupos 0 (p=0.04) y C (p=0.01) (Figura 6). De esta forma, el grupo L tenía una carga tumoral un 75% menor que el grupo C y un 64% menor que el grupo 0. Por lo que se puede concluir que el tratamiento de los tumores con let-7 induce una sensible regresión tumoral y que la administración de let-7 puede tener un valor terapéutico en el cáncer de pulmón.
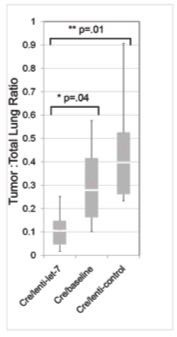
Figura 6: Carga tumoral de los grupos de ratones justo antes del tratamiento (0, Cre/basilene), el grupo de los ratones tratados con let-7 (L, Cre-lenti-let-7) y el grupo de los ratones tratados con el control (C, Cre-lenti-control). Los tumores tratados con let-7 presentan una carga tumoral significativamente menor que los grupos 0, observándose por lo tanto una regresión tumoral tras el tratamiento con let-7. El método de medición de carga tumoral usado se describe en (Esquela-Kerscher et al., 2008).
Los tumores fueron analizados histológicamente con el fin de evaluar los efectos de let-7 en la proliferación (observando la expresión de ki-67). Al igual de lo observado en los xenografts, los tumores tratados con let-7 presentan una reducción en los niveles de proliferación (Figura 7).
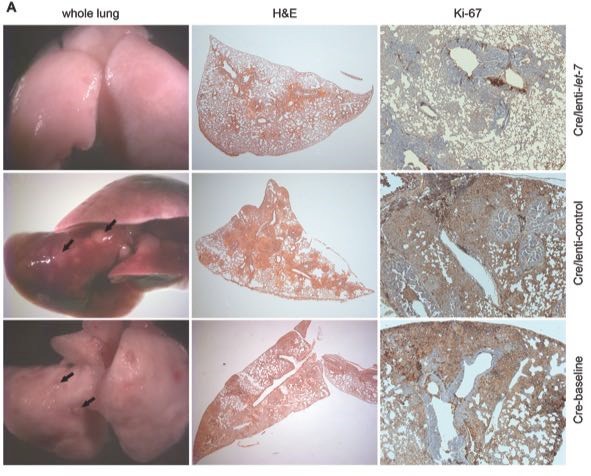
Figura 7: Izquierda: superficie pulmonar de los ratones justo antes del tratamiento (Grupo 0, Cre-baseline), tratados con el control (cre/lenti-control) y tratados con let-7 (cre/lenti-let-7). Las flechas negras indican abscesos tumorales. Centro: sección histológica pulmonar teñida con hematoxilina-eosina (H&E), nótese la hiperplasia tumoral más pronunciada en el grupo 0 y C. Izquierda: sección histológica pulmonar que revela la presencia del marcador de proliferación Ki-67, obsérvese una sensible disminución de proliferación en los tumores tratados con let-7.
4. El bloqueo de la actividad de let-7 promueve el desarrollo tumoral pulmonar
Aunque hay bastantes evidencias de que let-7 funciona como supresor tumoral en cáncer de pulmón (Johnson et al., 2005; Takamizawa et al., 2004) existían pocas evidencias in vivo de que la perdida de función de let-7 promovía el desarrollo pulmonar.
Para realizar este experimento se utilizó unos inhibidores que se habían utilizado con éxito para bloquear funcionalmente a microARNs (Krutzfeldt et al., 2005). Estos inhibidores (anti-miRs), consisten en secuencias inversas complementarias a los microARNs maduros, que presentan modificaciones químicas (enlaces fosfotioato y bases metiladas en la ribosa) que aumentan su estabilidad in vivo (Figura 8).
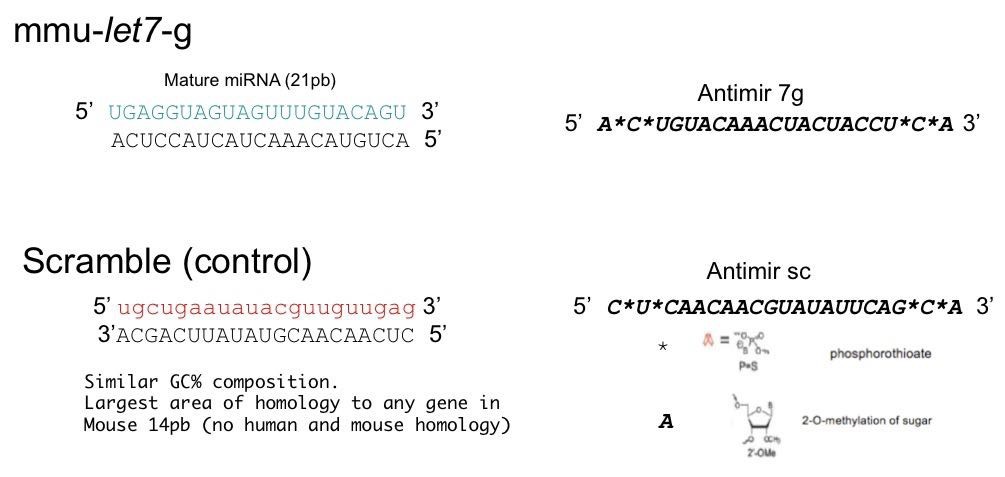
Figura 8: Anti-miRs: moléculas inhibitorias dirigidas contra let-7g se tratan de secuencias reversas complementarios al microARN (mmu-let-7, en azul este caso) modificadas químicamente para aumentar su estabilidad utilizando enlaces fosfotioato (*) y ribosas metiladas (marcadas en cursiva) de igual forma que se describe en (Krutzfeldt et al., 2005). Se utilizó una secuencia aleatoria control (scramble) de similar composición GC al microARN experimental pero sin homología con el genoma.
De entre los 9 miembros de la familia de microARNs let-7, se utilizó el let-7g por el hecho de que se había observado que let-7g se encuentra en la región cromosómica 3p21, implicada en la iniciación del cáncer (Calin et al., 2004) y porque existen evidencias que let-7g tiene efectos especialmente supresores en tumores que albergan mutaciones K-RAS (Kumar et al., 2008). Sin embargo, dada la alta homología presente en la familia de genes let-7, no podemos descartar que este anti-miR pueda tener una reacción cruzada para inhibir a otros miembros de la familia let-7.
El protocolo de administración de de los anti-miRs se basó en un trabajo anterior en el cual se inoculaba intranasalmente de forma eficaz siRNA (Bitko and Barik, 2008; Bitko et al., 2005). Para cerciorarse que los anti-miRs eran capaces de acceder a las células pulmonares, se diseñó unos anti-miRs reporteros, que tenían en su extremo fluoresceína amidato (FAM) de forma que pudiera revelarse su presencia iluminando con luz azul (que la fluoresceína refleja como luz verde). De esta forma se pudo detectar que los inhibidores eran capaces de penetrar en las células pulmonares (figura 9).
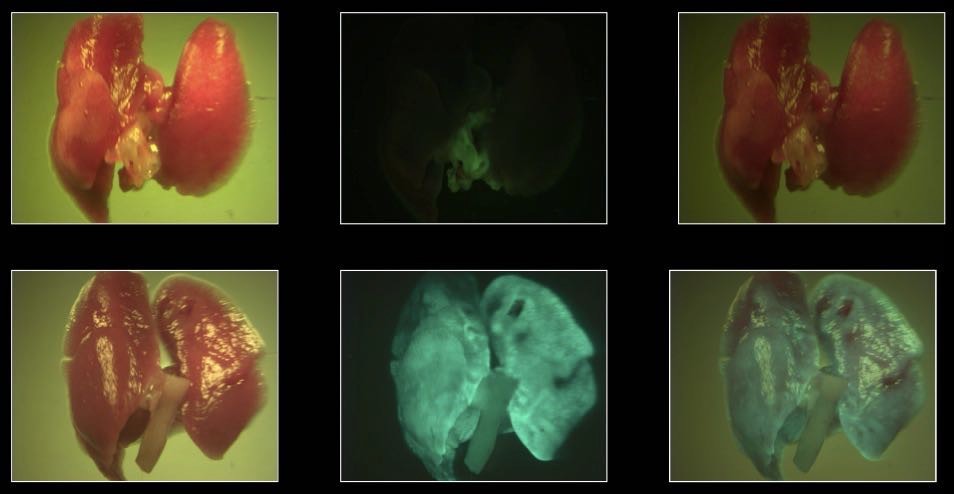
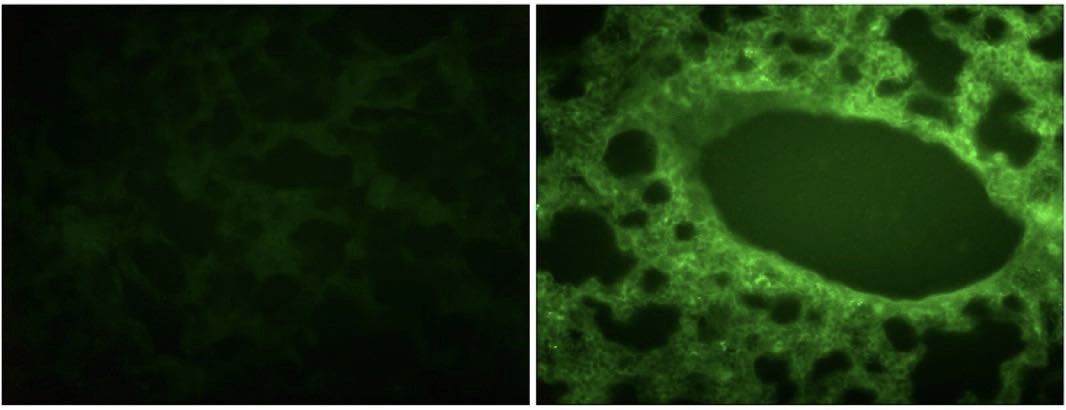
Figura 9: Superior: Pulmones de ratones a los que se había inoculado anti-miR-FAM reporteros (abajo) y pulmones normales (arriba) 16 horas después de la inoculación. Obsérvese que los pulmones inoculados con anti-miR reporteros presentan fluorescencia verde cuando se les ilumina con luz azul (centro). A la derecha puede observarse la superposición de imágenes. Inferior: fluorescencia en el tejido pulmonar (x400) donde puede observarse las células del parénquima bronquial de los pulmones sin tratar (izquierda) y tratados con los anti-miRs reporteros (derecha).
Para ver si estos inhibidores de let-7 eran capaces de modificar el desarrollo tumoral se administraron a ratones LSL-Kras-G12D (ver figura 5), de esta forma se podría validar la hipótesis de si el bloqueo funcional de let-7 promovía el desarrollo tumoral.
Se administró 60µg de anti-let7-g o anti-control (n=5) a los ratones LSL-Kras-G12D de forma simultánea que se les activaba la forma mutante de K-RAS. A las siete semanas los ratones fueron sacrificados para analizar la carga tumoral de sus pulmones.
Los pulmones tratados con los inhibidores de let-7 presentaban una significativa mayor carga tumoral en comparación con los tumores tratados con el control (Figura 10) y una histología tumoral más avanzada (Figura 10).
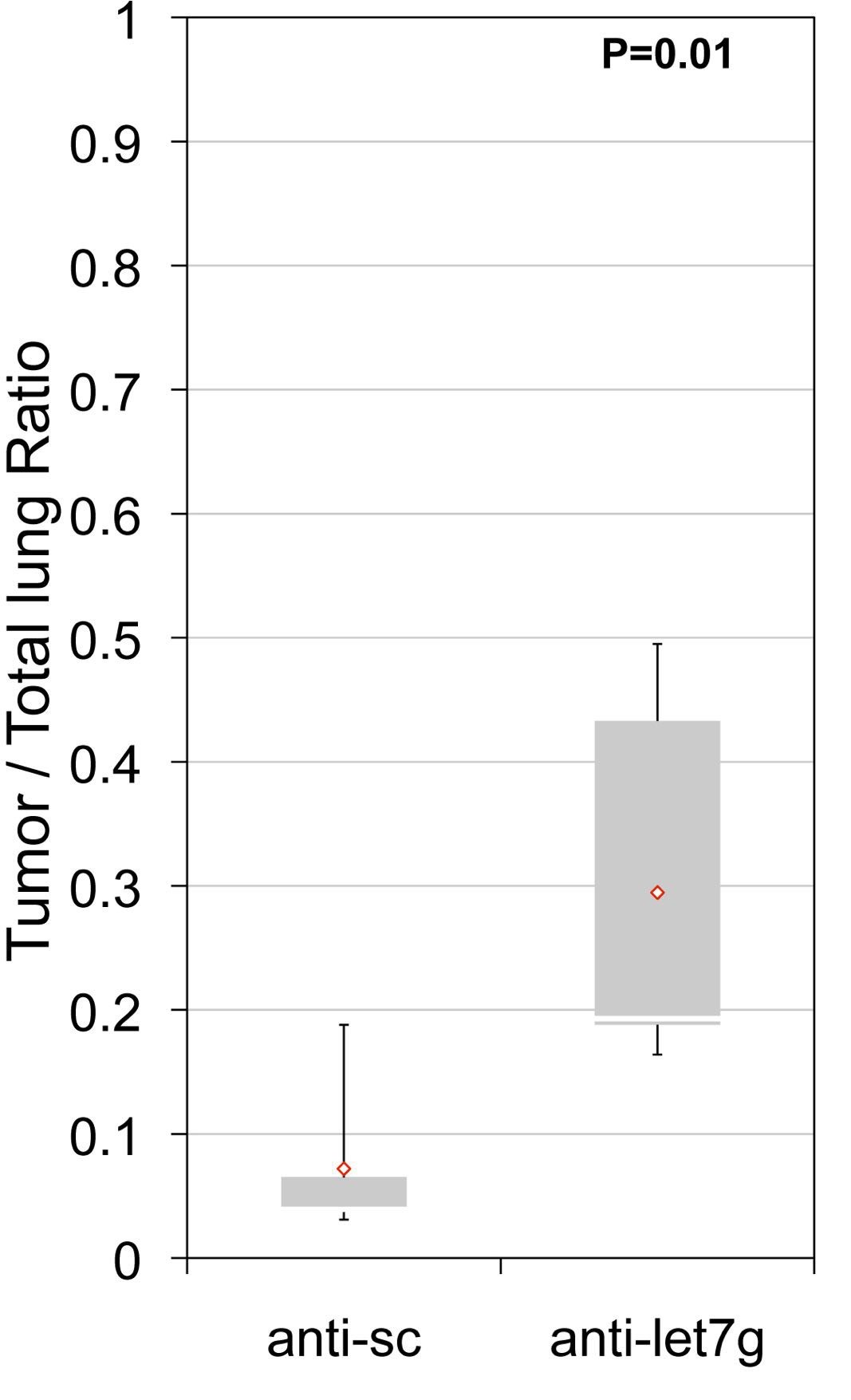
Figura 10: Carga tumoral de los ratones LSL-Kras-G12D tratados con el inhibidor de let-7g (anti-let-7g) y el control anti-sc. Obsérvese una significativa mayor carga tumoral en los tumores en los que se inhibe anti-let-7g.
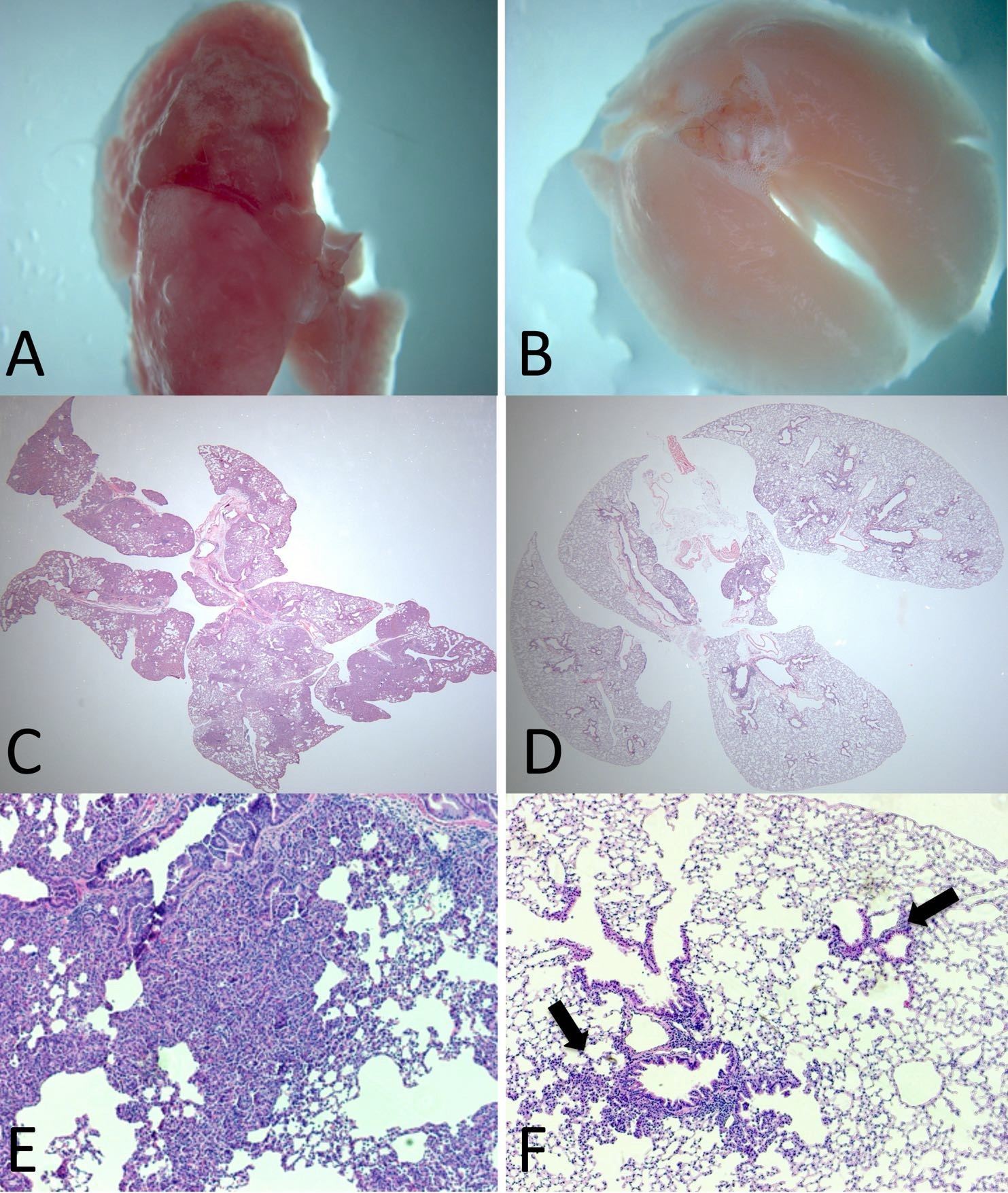
Figura 11: A-B: Pulmones de ratones tratados con inhibidores de let-7 (izquierda) y con el control (derecha). Obsérvese la superficie pulmonar llena de abscesos tumorales en los pulmones tratados con let-7. C-D: Secciones histológicas de los pulmones mostrados arriba (x100) teñidas con hematoxilina-eosina. E-F: Secciones histológicas de los pulmones mostrados arriba (x400): Nótese una hiperplasia-displasia avanzada en los pulmones tratados con inhibidores de let-7. Las flechas negras indican una hiperplasia incipiente en el tejido tratada con el control, una etapa temprana en el desarrollo tumoral.
5. DISCUSIONES Y CONCLUSIONES
Las evidencias presentadas indican que la inhibición de genes de la familia let-7 promueve el cáncer en un modelo preclínico in vivo dirigido por el oncogén K-ras, mientras que el aumento de su expresión suprime el crecimiento tumoral en tumores xenotrasnplantados e incluso provoca su regresión. Esta serie de observaciones cimentan que se pueda considerar a la familia de microRNAs let-7 como genes supresores tumorales relevantes en el desarrollo de cáncer de pulmón.
La relevancia de let-7 en la patología del cáncer se debe a su función como modelador de oncogenes importantes como ras, myc o cdk6 como se había visto anteriormente (Johnson et al., 2007; Johnson et al., 2005). Así se ha observado que la supresión tumoral ejercida por let-7 se debe principalmente a una bajada de la señal proliferativa, más que por un aumento de las rutas apoptóticas.
De forma más transcendente para la clínica, estos estudios sugieren que la re-introducción de let-7 puede tener utilidad terapéutica para tratar el cáncer de pulmón. Estos resultados se unen a otros en las que el valor terapéutico de los microARNs se había puesto de manifiesto en modelos de cáncer de hígado (Kota et al., 2009) y de próstata (Takeshita et al.). Si bien estos estudios son prometedores y sientan las bases para fundamentar estudios traslacionales clínicos, es necesario continuar investigando para utilizar a los microARNs como terapias efectivas en la clínica.
Los nuevos conocimientos alcanzados durante la optimización de las vacunas de ARN, podrían impulsar y mejorar las posibilidades para llevar a la clínica el uso de fármacos basados en la actividad de los microARNs.
6. REFERENCIAS
- Ambros, V. (1989). A hierarchy of regulatory genes controls a larva-to-adult developmental switch in C. elegans. Cell 57, 49-57.
- Bartel, D.P. (2004). MicroARNs: genomics, biogenesis, mechanism, and function. Cell 116, 281-297.
- Bentwich, I., Avniel, A., Karov, Y., Aharonov, R., Gilad, S., Barad, O., Barzilai, A., Einat, P., Einav, U., Meiri, E., et al. (2005). Identification of hundreds of conserved and nonconserved human microARNs. Nat Genet 37, 766-770.
- Bitko, V., and Barik, S. (2008). Nasal delivery of siRNA. Methods Mol Biol 442, 75-82.
- Bitko, V., Musiyenko, A., Shulyayeva, O., and Barik, S. (2005). Inhibition of respiratory viruses by nasally administered siRNA. Nat Med 11, 50-55.
- Calin, G.A., Dumitru, C.D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., Aldler, H., Rattan, S., Keating, M., Rai, K., et al. (2002). Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99, 15524-15529.
- Calin, G.A., Ferracin, M., Cimmino, A., Di Leva, G., Shimizu, M., Wojcik, S.E., Iorio, M.V., Visone, R., Sever, N.I., Fabbri, M., et al. (2005). A MicroARN signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353, 1793-1801.
- Calin, G.A., Sevignani, C., Dumitru, C.D., Hyslop, T., Noch, E., Yendamuri, S., Shimizu, M., Rattan, S., Bullrich, F., Negrini, M., et al. (2004). Human microARN genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101, 2999-3004.
- Chalfie, M., Horvitz, H.R., and Sulston, J.E. (1981). Mutations that lead to reiterations in the cell lineages of C. elegans. Cell 24, 59-69.
- Cimmino, A., Calin, G.A., Fabbri, M., Iorio, M.V., Ferracin, M., Shimizu, M., Wojcik, S.E., Aqeilan, R.I., Zupo, S., Dono, M., et al. (2005). miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102, 13944-13949.
- Esquela-Kerscher, A., and Slack, F.J. (2006). Oncomirs – microARNs with a role in cancer. Nat Rev Cancer 6, 259-269.
- Gregory, R.I., Yan, K.P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., and Shiekhattar, R. (2004). The Microprocessor complex mediates the genesis of microARNs. Nature 432, 235-240.
- He, L., Thomson, J.M., Hemann, M.T., Hernando-Monge, E., Mu, D., Goodson, S., Powers, S., Cordon-Cardo, C., Lowe, S.W., Hannon, G.J., et al. (2005). A microARN polycistron as a potential human oncogene. Nature 435, 828-833.
- Hohla, F., Schally, A.V., Kanashiro, C.A., Buchholz, S., Baker, B., Kannadka, C., Moder, A., Aigner, E., Datz, C., and Halmos, G. (2007). Growth inhibition of non-small-cell lung carcinoma by BN/GRP antagonist is linked with suppression of K-Ras, COX-2, and pAkt. Proc Natl Acad Sci U S A 104, 18671-18676.
- Hutvagner, G., McLachlan, J., Pasquinelli, A.E., Balint, E., Tuschl, T., and Zamore, P.D. (2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834-838.
- Isobe, T., Herbst, R.S., and Onn, A. (2005). Current management of advanced non-small cell lung cancer: targeted therapy. Semin Oncol 32, 315-328.
- Jackson, E.L., Willis, N., Mercer, K., Bronson, R.T., Crowley, D., Montoya, R., Jacks, T., and Tuveson, D.A. (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15, 3243-3248.
- Johnson, C.D., Esquela-Kerscher, A., Stefani, G., Byrom, M., Kelnar, K., Ovcharenko, D., Wilson, M., Wang, X., Shelton, J., Shingara, J., et al. (2007). The let-7 microARN represses cell proliferation pathways in human cells. Cancer Res 67, 7713-7722.
- Johnson, S.M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K.L., Brown, D., and Slack, F.J. (2005). RAS is regulated by the let-7 microARN family. Cell 120, 635-647.
- Karube, Y., Tanaka, H., Osada, H., Tomida, S., Tatematsu, Y., Yanagisawa, K., Yatabe, Y., Takamizawa, J., Miyoshi, S., Mitsudomi, T., et al. (2005). Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 96, 111-115.
- Kota, J., Chivukula, R.R., O’Donnell, K.A., Wentzel, E.A., Montgomery, C.L., Hwang, H.W., Chang, T.C., Vivekanandan, P., Torbenson, M., Clark, K.R., et al. (2009). Therapeutic microARN delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137, 1005-1017.
- Krutzfeldt, J., Rajewsky, N., Braich, R., Rajeev, K.G., Tuschl, T., Manoharan, M., and Stoffel, M. (2005). Silencing of microARNs in vivo with ‘antagomirs’. Nature 438, 685-689.
- Kumar, M.S., Erkeland, S.J., Pester, R.E., Chen, C.Y., Ebert, M.S., Sharp, P.A., and Jacks, T. (2008). Suppression of non-small cell lung tumor development by the let-7 microARN family. Proc Natl Acad Sci U S A 105, 3903-3908.
- Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microARNs from mouse. Curr Biol 12, 735-739.
- Lim, L.P., Glasner, M.E., Yekta, S., Burge, C.B., and Bartel, D.P. (2003). Vertebrate microARN genes. Science 299, 1540.
- Liu, J., Valencia-Sanchez, M.A., Hannon, G.J., and Parker, R. (2005). MicroARN-dependent localization of targeted mRNAs to mammalian P-bodies. Nat Cell Biol 7, 719-723.
- Medina, P.P., and Slack, F.J. (2008). microARNs and cancer: an overview. Cell Cycle 7, 2485-2492.
- O’Donnell, K.A., Wentzel, E.A., Zeller, K.I., Dang, C.V., and Mendell, J.T. (2005). c-Myc-regulated microARNs modulate E2F1 expression. Nature 435, 839-843.
- Richardson, G.E., and Johnson, B.E. (1993). The biology of lung cancer. Semin Oncol 20, 105-127.
- Shiraishi, M., Noguchi, M., Shimosato, Y., and Sekiya, T. (1989). Amplification of protooncogenes in surgical specimens of human lung carcinomas. Cancer Res 49, 6474-6479.
- Slack, F.J., and Weidhaas, J.B. (2006). MicroARNs as a potential magic bullet in cancer. Future Oncol 2, 73-82.
- Takamizawa, J., Konishi, H., Yanagisawa, K., Tomida, S., Osada, H., Endoh, H., Harano, T., Yatabe, Y., Nagino, M., Nimura, Y., et al. (2004). Reduced expression of the let-7 microARNs in human lung cancers in association with shortened postoperative survival. Cancer Res 64, 3753-3756.
- Takeshita, F., Patrawala, L., Osaki, M., Takahashi, R.U., Yamamoto, Y., Kosaka, N., Kawamata, M., Kelnar, K., Bader, A.G., Brown, D., et al. Systemic delivery of synthetic microARN-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther 18, 181-187.
- Trang P, Medina P P, J F Wiggins, L Ruffino, K Kelnar, M Omotola, R Homer, D Brown, A G Bader, J B Weidhaas, F J Slack. (2010). Oncogene 2010 Mar 18;29(11):1580-7.
- Yi, R., Qin, Y., Macara, I.G., and Cullen, B.R. (2003). Exportin-5 mediates the nuclear export of pre-microARNs and short hairpin RNAs. Genes Dev 17, 3011-3016.

{kind=link}