1. INTRODUCCIÓN
Las Fuerzas Armadas Españolas (FFAA) disponen de un Petitorio que engloba un arsenal terapéutico considerado básico para la atención a la salud de los miembros de las FFAA en los campos logístico-operativos y asistenciales (1). En el Petitorio mencionado está incluido el fármaco antidiarreico loperamida bajo la forma farmacéutica comprimido. En la búsqueda de una mejora de la forma farmacéutica de este fármaco, se realiza un desarrollo galénico que incluye el escalado industrial de un nuevo comprimido bucodispersable de loperamida hidrocloruro (CBD-LH), que permite abordar, con un prisma más operativo dada sus múltiples ventajas (2), los problemas ocasionados por episodios de diarrea en zonas hostiles de despliegue táctico de tropas militares.
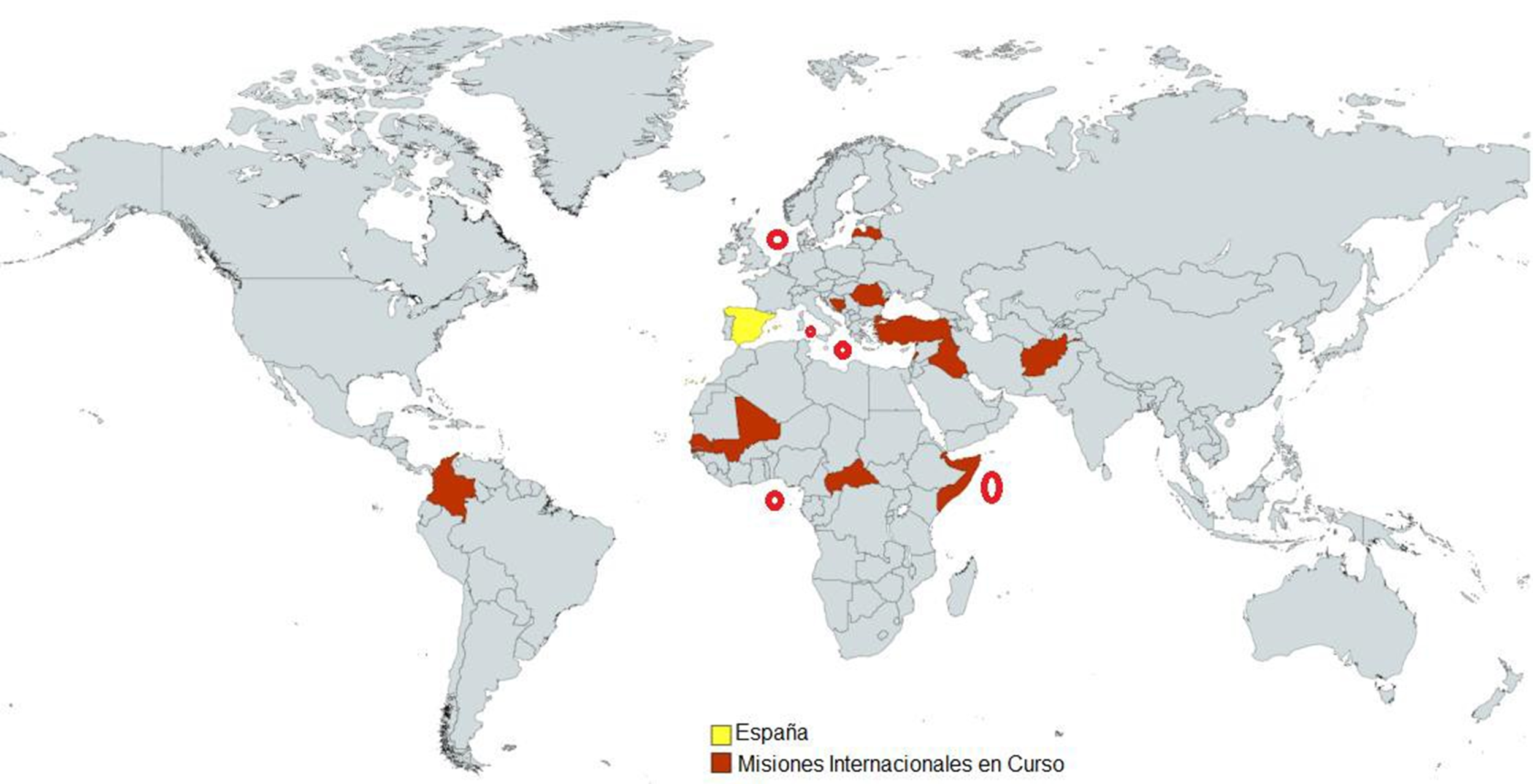
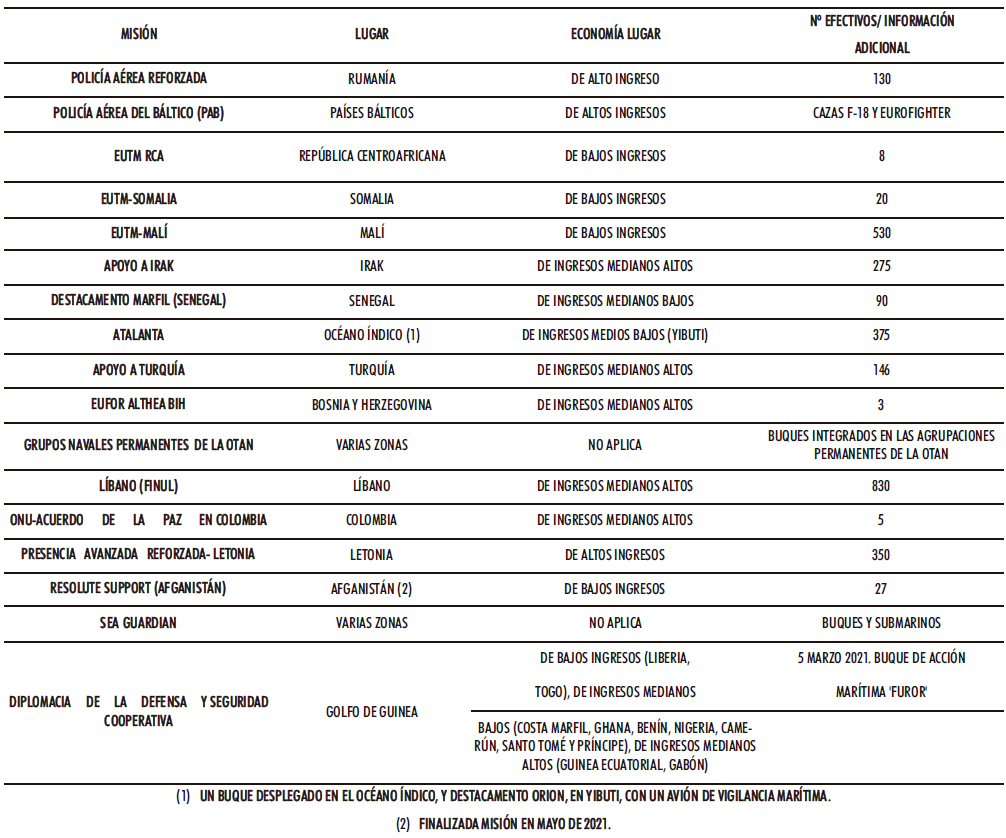
En el año 2021, las FFAA participaron en 17 misiones internacionales desplegadas en cuatro continentes, con la previsión de no superar los 2.900 militares, sin descartar ajustes derivados del ejercicio temporal del mando de las fuerzas internacionales, o bien, debido a la situación sanitaria y de seguridad (3-4). En la Tabla I.1 se detalla la distribución de los efectivos militares españoles desempeñando sus funciones en diversos países, clasificados de acuerdo con el criterio del Banco Mundial (según ingresos nacionales brutos). En base a la clasificación mencionada se distinguen las economías del mundo en cuatro grupos, bajos, medianos bajos, medianos altos y altos (5-6).
Tabla I.1. Misiones internacionales en curso FFAA españolas en el año 2021
Las enfermedades diarreicas constituyeron en el año 2019 la octava causa de muerte en el mundo, siendo la quinta en los países de ingresos bajos y la sexta en los de ingresos medianos- bajos (5), afectando especialmente a la población infantil, que se sitúa como la principal causa de mortalidad y morbilidad a nivel mundial (7-8). Desde una perspectiva militar, el apoyo sanitario es un pilar fundamental para el desarrollo de toda operación (9), y se entiende como el «conjunto de acciones encaminadas a garantizar la protección de la salud de los miembros de la Fuerza, para mantener su capacidad de combate» (10). La novedosa propuesta tratada en el presente estudio, los CBD-LH, puede emplearse sola o combinada con otros fármacos, según el cuadro clínico, como el antibiótico azitromicina, presente también en el actual Petitorio de la Sanidad Militar Española.
La loperamida es un fármaco considerado esencial por parte de la Organización Mundial de la Salud (OMS) (11) y, además, en la actualidad tienen un importante uso clínico en el tratamiento de los problemas asociados a las afecciones intestinales que provoca la Covid-19 (12-14). Por otro lado, los comprimidos bucodispersables (CBD) son formas de dosificación sólidas que se disgregan rápidamente, en menos de un minuto, en la cavidad bucal en presencia de saliva sin ninguna dificultad para tragar.
En un escenario táctico y operativo donde cabe la posibilidad de no disponer de agua potable o de tener que racionar su consumo, los CBD-LH podrían proporcionar una alternativa ideal por no necesitar la ingesta de tal recurso crítico, unido también a un rápido inicio de acción, por su absorción sublingual. Conjuntamente, se evita la deglución de comprimidos que puede resultar de mayor utilidad, no sólo en personal militar, sino en otro abanico de pacientes como niños, ancianos o con patologías, tales como la disfagia, que impide o dificulta la deglución, lo que supone que los CBD tendrían una gran aceptación y una mayor adhesión al tratamiento (2). Además, es preciso mencionar que el aporte hídrico en las fases iniciales de la diarrea aguda inespecífica contribuye a facilitar la evacuación, por lo que, con la toma de CBD-LH sin necesidad de ingesta de agua, supone otra ventaja en una situación de emergencia táctica.
Todo ello puede ser de suma importancia en realidades donde las capacidades sanitarias están limitadas y que, pueden verse mermadas en una situación pandémica como la sufrida actualmente (15). Esto implica otras significativas y variadas ventajas, asociadas a una dosificación precisa, fácil adherencia al tratamiento gracias, en parte, a sus propiedades organolépticas y, finalmente, a la fácil elaboración de la forma farmacéutica propuesta, la compresión directa, en situaciones de fabricación urgentes o dando respuesta a una necesidad imperativa.
Desde un punto de vista clínico la diarrea es una alteración intestinal que se caracteriza por un aumento en la frecuencia, peso y/o contenido de agua en las heces (16). La OMS hace referencia a la frecuencia de deposiciones de baja consistencia (sueltas o líquidas), estableciendo un rango de tres o más veces al día o en las que se observa pus, sangre o moco (16-17). En términos generales, la diarrea suele ser un síntoma de una infección del tracto digestivo que puede estar ocasionada por diversos agentes biológicos, bacterias, virus y parásitos (17), o por la administración de determinados fármacos, o bien, por patologías como la inflamación intestinal (16). En cuanto a su clasificación se puede distinguir entre diarrea aguda y crónica. La diarrea crónica se extiende a un período de más de 3 o 4 semanas de duración y generalmente es una consecuencia derivada de otras patologías de tipo congénito (véase intolerancia a la lactosa, enfermedad celíaca, malabsorción primaria de ácidos biliares, etc.) o adquiridas (enfermedad inflamatoria intestinal, carcinomas, insuficiencia pancreática o hepática, etc.) (16). En cambio, la diarrea aguda es un proceso, generalmente autolimitado, en el que se produce un aumento en el volumen y número de las deposiciones y una disminución de su consistencia habitual con una duración inferior a dos semanas (16, 18), puede ser causada por una infección o por fármacos. Este tipo se produce por una disminución del proceso de absorción, por una alteración en tal flujo bidireccional presentando un gran volumen de agua en el intestino grueso que supera toda capacidad de absorción (16) y suele ir acompañada de una disminución de peso y una pérdida anormal de agua y electrólitos. Cuando el cuadro diarreico dura más de 14 días la OMS la define como diarrea persistente, y diversos autores la consideran crónica cuando se prolonga o cuando ocurren tres o más episodios de diarrea de corta duración en dos meses (19).
Entre los fármacos que producen diarrea aguda, la que se delimita a la presencia de 3 o más deposiciones de consistencia blanda-liquida en 24 h, destacan los antibióticos de amplio espectro, que modifican la microbiota bacteriana comensal en el intestino, lo cual favorece la proliferación de determinados grupos bacterianos que producen diarrea. En el caso, de la azitromicina, entre sus efectos adversos más frecuentes destacan diarrea (3,6%), náuseas (2,9%) y dolor abdominal (2,5%), cefalea y vértigo (1,3%) y aumento de transaminasas (1,5%) (20, 21). Por su parte, determinados fármacos laxantes (lactulosa, sorbitol, etc.) y antiácidos que contienen sales de magnesio pueden producir cuadros diarreicos, al producir fenómenos osmóticos en el lumen intestinal. Finalmente, los fármacos que proveen la secreción hidroeléctrica o la motilidad gastrointestinal, incluyendo laxantes estimulantes, fármacos procinéticos, fármacos con propiedades parasimpaticomiméticas y fármacos citostáticos (16).
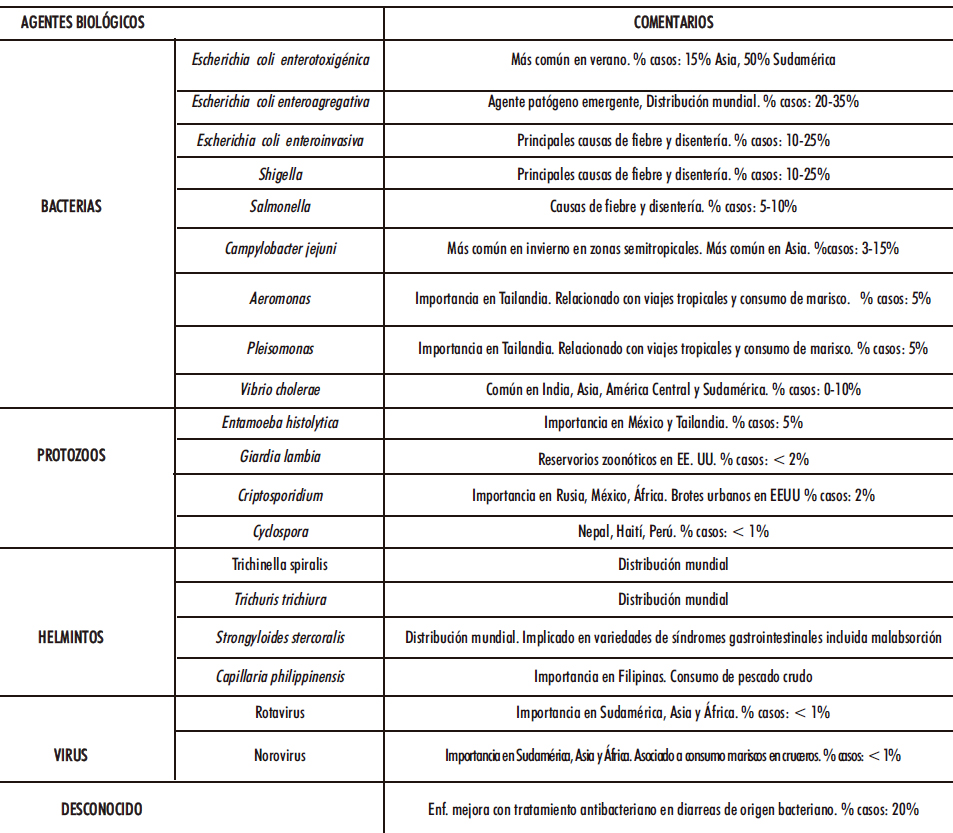
Una vez aclarada la definición, clasificación y etiopatogenia de la diarrea es conveniente abordar la gastroenteritis aguda, también conocida con el nombre diarrea del viajero. Se trata de una infección del tracto gastrointestinal, habitualmente autolimitada, y cuya manifestación clínica principal es la diarrea. Se adquiere principalmente a través de alimentos y bebidas contaminados con patógenos, tales como bacterias, protozoos, helmintos y virus (22-24). En la Tabla I.2 se recogen y comentan los principales responsables (25-26).
Tabla I.2. Agentes biológicos causantes de diarrea del viajero (25-26)
El personal militar español desplegado en misiones internacionales (Tabla I.1) se encuentra, fundamentalmente, en las áreas donde existe una mayor probabilidad de contraer la diarrea del viajero, ver Figura I.1.
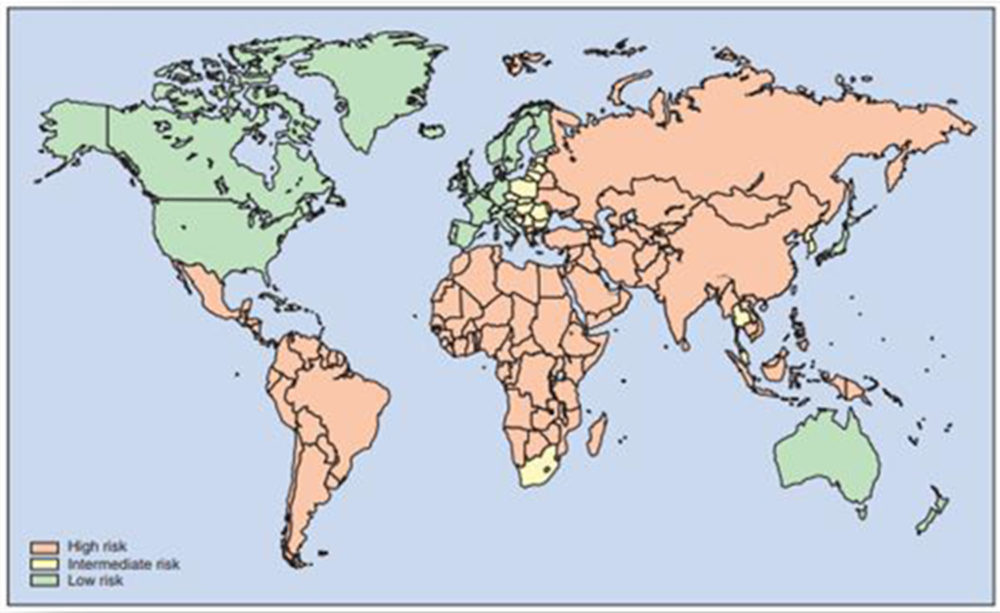
Figura I.1. Misiones internacionales en curso FFAA españolas en el año 2021
Desde una perspectiva sanitaria militar, un síndrome como el expuesto, que a priori cursa sin complicaciones, puede entrañar una disminución de las capacidades del contingente, poniendo en riesgo el correcto desempeño de la misión, lo que se corrobora en diversos estudios de campo en escenarios austeros, tácticos y de combate, donde no siempre se disponen de todos los medios logísticos, de infraestructuras ni de recursos sanitarios (29-31), llegando incluso a situaciones en que se somete al combatiente a situaciones extremas que desencadenan respuestas psicológicas y físicas que incluyen la diarrea (32). Si a lo anterior se le añade el calentamiento climático (33) y los períodos prolongados de tiempo en los que el contingente permanece operativo, se hace evidente el apoyo sanitario requerido para controlar el impacto de la gastroenteritis aguda, ya que puede afectar negativamente, no sólo al transcurso y mantenimiento de la misión, sino a las consecuencias crónicas que se puedan derivar en el paciente o a los costos médicos asociados o a las posibles derivaciones a territorio nacional (27).
Los agentes biológicos se clasifican según la facilidad de diseminación en el medio del agente biológico, su transmisibilidad, impacto sobre la colectividad y sus requerimientos en medios diagnósticos (34), como se observa en la Tabla I.3. Algunos de los agentes biológicos relacionados con la diarrea del viajero pueden ser métodos estratégicos de diseminación por sabotaje en la contaminación de agua de consumo y alimentos, en cuyo caso la principal defensa contra esta forma de diseminación es el control de ambos recursos (35-37).
Tabla I.3. Clasificación de agentes biológicos (34)

En casos de intoxicación alimentaria la ingesta de toxinas con los alimentos, cursan con vómitos y la diarrea que se resuelven espontáneamente en 12 h (38). Las bacterias, como la Escherichia coli enterotoxigénica, son la causa más de la diarrea del viajero y dentro de los fármacos empleados para su tratamiento se encuentra la loperamida (ver Figuras I.2, I.3 y I.4). En la Figura I.2 se visualizan los fármacos empleados en autoterapia, según el grado de gravedad. (38-42).
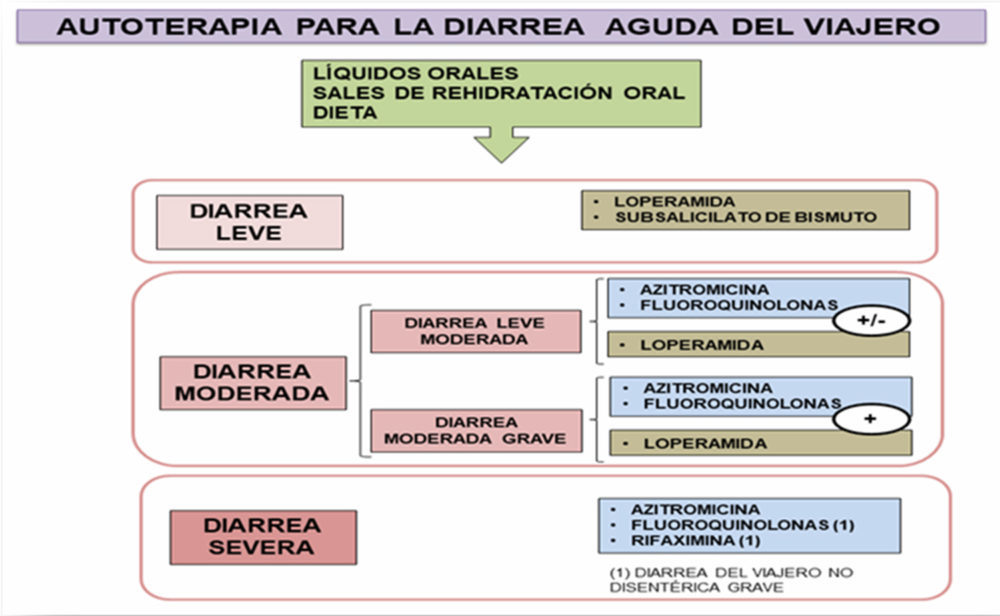
Figura I.2. Autoterapia para la diarrea del viajero (38-39)
El subsalicilato de bismuto presenta propiedades antisecretoras y antimicrobianas, pero no debe combinarse con fluoroquinolonas por sus propiedades quelantes. Además, es un fármaco que presenta una comercialización limitada en el mercado internacional. La loperamida, en cambio, cuenta con una mayor expansión en el mercado internacional (39), no obstante, está contraindicada como único tratamiento cuando se sospecha que la diarrea es exudativa. Como única terapia, estos agentes solo deben usarse en casos de diarrea aguda no febril y no disentérica, al igual que en casos de colitis ulcerosa aguda, colitis pseudomembranosa asociada al tratamiento con antibióticos de amplio espectro, y enterocolitis bacteriana causada por organismos invasivos incluyendo Salmonella, Shigella y Campylobacter (39, 43). En la Figura I.3 se representa un algoritmo de actuación en caso de fallo en la terapia inicial, que consiste en persistencia del cuadro diarreico por más de una semana (38-39). Los antibióticos son la piedra angular del tratamiento de la diarrea del viajero, ya que reducen eficazmente la duración de la enfermedad (40). La azitromicina puede emplearse en las diarreas moderadas siendo el antibiótico de elección en las diarreas severas (38), tal acción debe ser considerada para personas con diarrea clásica del viajero que necesitan una pronta resolución de los síntomas (44).

Figura I.3. Algoritmo tras fallo auto tratamiento inicial diarrea del viajero (39)
Las resistencias a antibióticos son un elemento a tener en cuenta en el planeamiento de la terapéutica, ver ejemplo en la Figura I.4 (40).
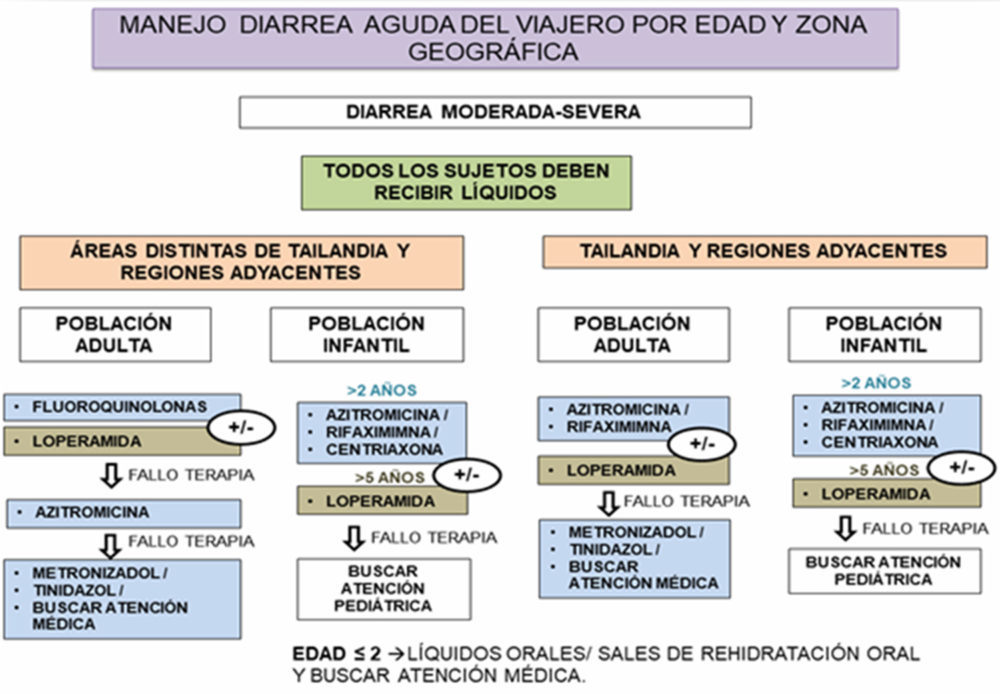
Figura I.4. Algoritmo para manejo de diarrea del viajero según edad y destino (40)
2. OBJETIVOS Y PLAN DE TRABAJO OBJETIVOS
El objetivo general de este trabajo es el desarrollo galénico y escalado industrial de un comprimido bucodispersable de loperamida, CBD-LH, mediante tecnología de compresión directa, para abordar los problemas ocasionados por la diarrea en zonas de operaciones de tropas militares.
Para abordar este objetivo general se presentan los siguientes objetivos específicos:
- Revisión bibliográfica de CBD-LHs comercializados en el mercado farmacéutico español, y de los materiales para el desarrollo galénico de una nueva formulación de CBD-LH.
- Diseño de una nueva formulación de un CBD-LH.
- Diseño del proceso de fabricación de un CBD-LH.
- Análisis de riesgos del proceso de fabricación industrial de un CBD-LH.
- Realización de controles de calidad en procesos y en producto terminado (control físico, químico y microbiológico).
- Estudios de compatibilidad de los materiales seleccionados.
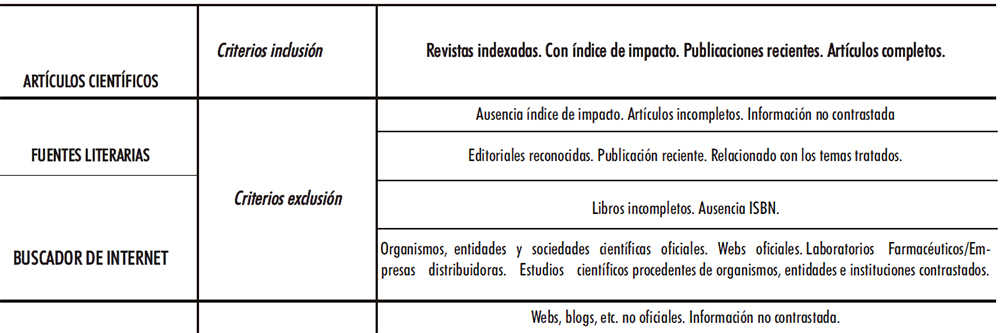
Para dar cumplimiento a los objetivos planteados se consultaron tres fuentes de información principales, cuyos criterios de inclusión/exclusión se ven en la Tabla II.1.
Tabla II.1. Criterios de inclusión/exclusión principales fuentes bibliográficas
2.1. Plan de trabajo
Para obtener una visión global del estudio se desarrolló un mapa de procesos (Tabla II.2) para el desarrollo galénico y el posterior escalado industrial de un CBD-LH, y se definieron las acciones operativas, estratégicas y de soporte con la intención de trabajar siguiendo un flujo eficiente en la consecución de los objetivos expuestos. Para garantizar su cumplimiento se definieron acciones soporte para cada etapa, como la definición de la necesidad terapéutica en el ámbito militar, la propuesta de una nueva forma farmacéutica en CBD-LH y el desarrollo y validación del método analítico por HPLC, que se demostró su utilidad para la determinación de la cantidad de principio activo en los únicos CBD-LH aprobados y comercializados en España al momento del desarrollo del presente trabajo científico, Salvacolina Flas 2 mg comprimidos bucodispersables (43) de Laboratorios Salvat S.A.
Tabla II.2. Mapa de valor objetivos del estudio científico

En cuanto al proceso de elaboración se definieron cada una de las etapas de la fabricación industrial previas a su acondicionamiento primario y secundario (pesada, tamizado, mezclado, compresión), se establecieron los atributos críticos de las materias primas, los parámetros críticos del proceso y los atributos de calidad del producto terminado como antesala a la siguiente etapa, la formulación. La selección de excipientes y sus proporciones se llevó a cabo de acuerdo a su función y a los aspectos tecnológicos propios de la tecnología de compresión directa por lo que se incluyeron aglutinantes, diluyentes, lubrificantes, antiadherentes, aditivos y superdisgregantes, a los que se aplicaron estudios de compatibilidad principio activo- excipiente de cara a garantizar buenos resultados en los ensayos de estabilidad. Para ello se emplearon estudios de espectroscopia infrarroja por transformada de Fourier (FTIR), calorimetría diferencial de barrido (DSC) y microscopía electrónica de barrido (SEM).
También se estudió la densidad aparente, densidad golpeada, fluidez, ángulo de reposo, y disgregación en saliva artificial (45) para determinar el comportamiento de cada materia prima y comprender su influencia en el proceso de mezcla. La selección de una formula u otra (ambas con 2 mg/ CBD) dependió de los tamaños de punzones disponibles, para que la relación del diámetro-altura del comprimido fuese la adecuada, manteniendo los porcentajes de los excipientes.
Una vez garantizada la homogeneidad del principio activo y compatibilidad de la materia prima en ambas formulaciones se efectuó una prueba piloto previa al escalado industrial con 3 kg de mezcla, y con un muestreo representativo en cuatro puntos distintos de ésta donde se evidenció la homogeneidad del fármaco en la mezcla, y su cantidad en los CBD resultantes tras la fase de compresión. Finalmente, se llevó a cabo un escalado industrial con 20 kg de mezcla para demostrar el cumplimiento con los criterios de calidad de las fórmulas seleccionadas y, se realizó un análisis de riesgos del proceso propuesto.
3. MATERIALES Y MÉTODOS
3.1. Reactivos, soluciones, materias primas y equipos
Las materias primas y los reactivos empleados para el estudio fueron adquiridos en proveedores certificados, según la calidad requerida en cada caso. En cuanto a los equipos empleados su selección se hizo conforme a los requerimientos de cada etapa de desarrollo:
- Balanzas de precisión Mettler Toledo AG245, PJ 3600 DeltaRange®, PM 480 DeltaRange®, AE 260 DeltaRange®, Sartorius Cubis MSA323S-100-DI. Tamizadora CISA con tamaño tamiz 0,8 mm, 1,00 mm y 1,5 mm. Mezclador en V P Prat tipo B n◦ 41412 y bicónico Glatt Labortecnic. Máquina de comprimir excéntrica J. Bonals Modelo B nº 508 (punzones 7 mm), rotatoria Kilian RTS 21 (punzones 8 mm).
- Se emplearon adicionalmente los siguientes equipos: Calibrador Vernier y equipo Pharmatest PTB 311E, Tambor Pharma Test PTF E®, Mordaza portátil Bonals, durómetro Pharma Test modelo PTB-411E, aparato de disgregación Turu-Grau, Balanza Moisture Analyzer Mettler Toledo LJ16, Hanson Research SR8 SRII 8-Flask dissolution test station. Equipo Mettler TA 4000 DSC Star System, espectrofotómetro infrarrojo por transformada de Fourier Spectrum 2000 spectrometer Perkin Elmer®, y microscopio electrónico Zeiss DSM 950.
3.2. Métodos de caracterización y cuantificación
3.2.1. Métodos de caracterización materias primas
Calorimetría diferencial de barrido (DSC): El análisis térmico diferencial permite investigar posibles incompatibilidades entre el fármaco y los excipientes seleccionados. El equipo empleado es Mettler TA 4000 DSC Star System, calibrado con indio. Las muestras se someten a un calentamiento programado, bajo purga dinámica de gas nitrógeno conforme a las condiciones consignadas en la Tabla III.1.
Tabla III.1. Condiciones experimentales DSC

Espectrofotometría infrarroja por transformada de Fourier (FT-IR): Los espectros para el fármaco y cada excipiente de las fórmulas seleccionadas, de las mezclas físicas API- excipientes y de los comprimidos finales, se determinaron en el rango de barrido de 500 a 4000 cm−1 en el Espectrómetro Fourier Spectrum 2000 Perkin Elmer® System 20.000 FT-IR con resolución de 1 cm−1.
Las muestras en mezcla 1:100 Muestra:KBr , se trituran en mortero de ágata y el polvo se prensa en discos de unos 13 mm de diámetro.
Microscopía electrónica de barrido (SEM): Con el microscopio electrónico de barrido Zeiss DSM 950 que aplica una señal de electrones secundarios y una señal de retrodispersión a una resolución de 3 nm, se evaluaron las muestras recubiertas con oro en su proceso de metalización.
3.2.2. Métodos de elaboración y ensayos de CBD-LH frente a CBD-LH comercial
Pesada: En esta etapa tiene en cuenta la tolerancia de las balanzas analíticas empleadas y de la correcta calibración de las mismas, con tolerancia de ± 1% para los excipientes y de un ± 0,5% de API.
Tamizado: El propósito de esta operación es una estandarización del tamaño de partícula a fin de asegurar un mezclado controlado y homogéneo del sólido pulverulento caracterizado con estudios de densidad aparente y densidad después del apelmazamiento de los polvos (RFE, 2.9.34), ángulo de reposo y fluidez (RFE, 2.9.36) (46).
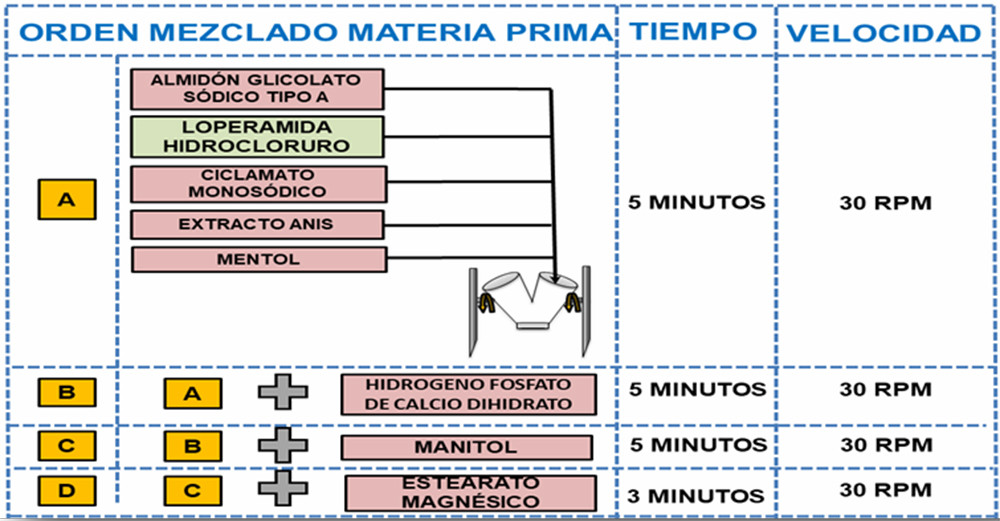
Mezclado Se definieron como parámetros críticos del proceso, el orden de incorporación al mezclador, la velocidad y el tiempo de mezclado, para el tamaño del lote piloto de 3 kg en mezclador VP Prat tipo B n◦ 41412 en el que se muestreó según Figura III.1. Para garantizar una mezcla homogénea y asegurar un enmascaramiento desde un punto de vista organoléptico del principio activo se inicia con el mezclado de los componentes minoritarios (loperamida hidrocloruro, ciclamato sódico, extracto de anís, mentol y almidón glicolato sódico tipo A) 30 rpm,5 min, seguido por hidrogenofosfato de calcio dihidrato 30 rpm, 5 min, manitol 30 rpm, 5 min, y estearato magnésico 30 rpm, 3 min.
En el escalado industrial con 20 kg de mezcla se empleó un mezclador bicónico Glatt Labortecnic. A pesar de ser un equipo distinto al mezclador en uve, presenta una misma rotación en su eje, por lo que los atributos críticos de la mezcla (fluidez, ángulo de reposo, distribución y tamaño de partícula), sus propiedades físicas, biológicas y microbiológicas no se vieron afectadas.
Las condiciones de incorporación fueron: componentes minoritarios 20 rpm, 7 min, hidrogenofosfato de calcio dihidrato 20 rpm, 7 min, manitol 20 rpm, 10 min, y el estearato magnésico 20 rpm, 3 min. Se tomaron las muestras de la parte superior, mitad e inferior de la mezcla en el propio bidón de descarga, ver Figura III.1.
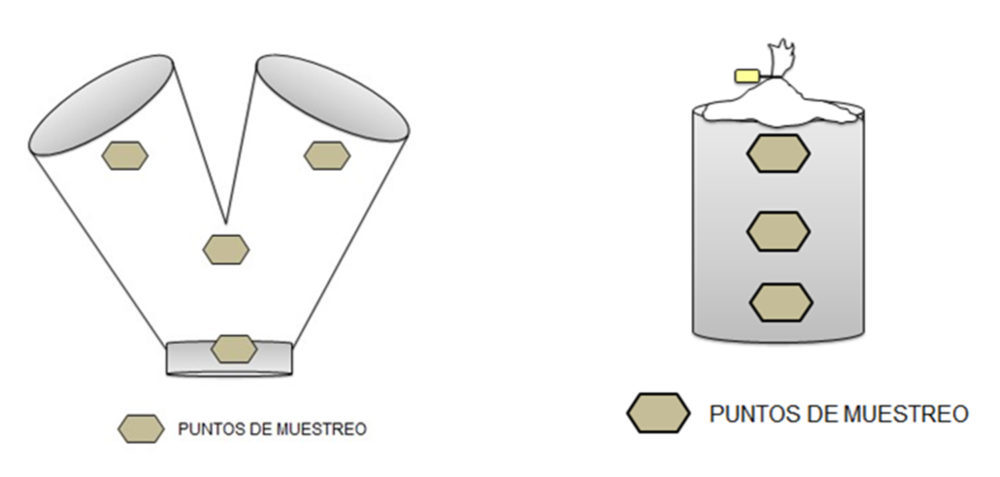
Figura III.1. Puntos muestreo de mezcla en escalado piloto (izda.) y en escalado industrial (dcha.)
Compresión: El factor crítico en esta etapa es la selección de los ocho excipientes atendiendo a las diferentes propiedades organolépticas de gran relevancia en la palatabilidad de la formulación y farmacotécnicas como el flujo, compresión, lubrificación, solubilidad, capacidad de disgregación y de aglutinación (47-61).
La selección del almidón glicolato sódico tipo A y de la hipromelosa fue trascendental, el primero dado su carácter superdisgregante y la segunda por sus propiedades aglutinantes, con miras a evitar posibles problemas tecnológicos, como el «capping» resultante del tipo de proceso de fabricación seleccionado. Para esta etapa se usa la máquina de comprimir excéntrica J. Bonals Modelo B nº 508 (punzones ranurados 7 mm), teniendo en cuenta que para el escalado industrial los punzones serán ranurados de 8 mm, es decir, en las fases previas a la fase industrial se trabajaron comprimidos de menor diámetro y mayor altura.
Caracteres organolépticos
Caracteres geométricos: Se hizo el estudio para los CBD-LH desarrollado localmente frente al comercializado de importación. Para el diámetro y la altura se emplean 10 unidades. El valor de la altura se relaciona con el peso y dureza del CBD-LH.
Palatabilidad: Al tratarse de un CBD la adecuada palatabilidad ayuda a la adherencia del paciente en el tratamiento farmacológico. Los excipientes seleccionados deben enmascarar el sabor amargo propio de la loperamida hidrocloruro. El primer contacto del paciente con el CBD-LH será en la apertura de su envase primario, luego durante la disgregación (desde su ingreso a la cavidad oral hasta la desintegración de su núcleo) y sensación remanente luego de ingerido. Durante la fase previa a la toma se definieron como índices de calidad el aspecto y el olor, y en la disgregación, tres índices de calidad: tiempo, textura y sabor, en la fase posterior un índice de calidad: el gusto latente en la cavidad bucal.
Características farmacotécnicas
Se hizo un estudio de las características farmacotécnicas tanto de los CBD-LH de las formulaciones seleccionadas como de CBD-LH comercial, con el objetivo de comparar los comprimidos propios con el referente en el mercado nacional español, según los métodos de la RFE: Uniformidad de contenido: 2.9.6 (62), Uniformidad de masa: 2.9.5 (63), Friabilidad: 2.9.7 (64), Dureza: 2.9.8 (65), Ensayo de disgregación: 2.9.1 (66), Ensayo de disolución: 2.9.3 (67), Estudio de divisibilidad: según la «divisibilidad de los comprimidos» de la RFE (68). Disgregación en saliva artificial: En seis unidades independientes (45) empleando una placa Petri de 10 cm de diámetro conteniendo 20 mL en unas condiciones de temperatura de 37 ± 0,5ºC de agua purificada.
Contenido en humedad: Por triplicado en balanza Mettler Toledo LJ16 Moisture Analyzer, empleando 1,5 g. de comprimidos pulverizados.
Identificación
Cromatografía líquida de alta resolución (HPLC)
Aplicando la guía ICH Q2 (R1) (CPMP / ICH / 381/95) (74) para la determinación de los CBD- LH, se empleó una columna ACE Excel 5 C18 150 × 4,6; 5 mm, fase móvil fue ACN: ácido acético 1% a un caudal de 1,2 mL/min. La temperatura de la columna en 25 ± 5ºC, la presión en 200 bar, el volumen de inyección de 15 µL, a λ= 224 nm. Equipo Hewlett-Packard GMBH Series 1050. El método tuvo tR de 1,9 min y demostró ser selectivo con una curva de calibración (y = 15,825 × + 45,91), r2 = 0,995 (n = 9), para el rango de concentración de 2,0 a 60,0 µg/mL, con un límite de detección de 0,3 ng/mL y un límite de cuantificación de 1,0 ng/mL. La precisión método entre 97,56 – 102,01%, con RSD < 2,8% (69-73).
4. RESULTADOS Y DISCUSIÓN
4.1. CBD-LH: Desde etapa de preformulación hasta el escalado industrial
Preformulación
Del análisis del mercado de CBD-LH, ver Figura IV.1 y Tabla IV.1, se encontró la loperamida en forma farmacéutica CBD se comercializa en trece países, (75-76).
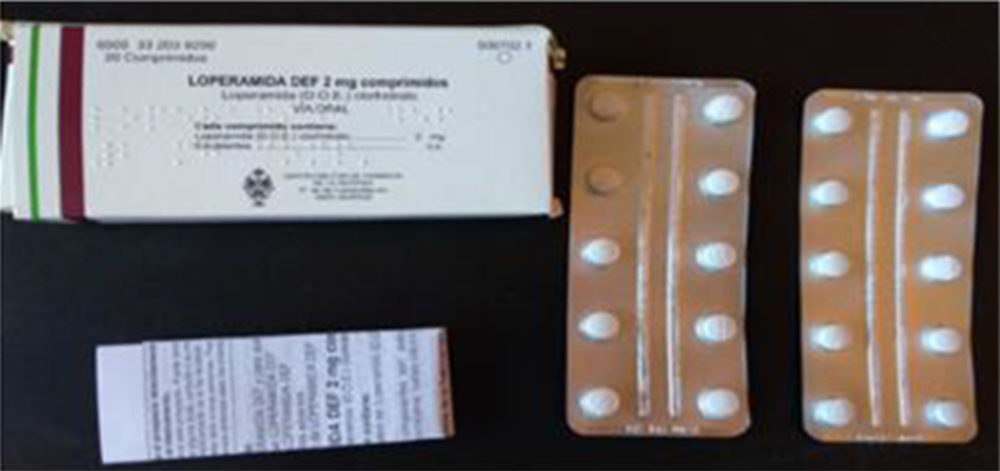
Figura IV.1. Loperamida DEF 2 mg comprimidos: estuche, blíster y prospecto
Tabla IV.1. Comprimidos bucodispersables de loperamida hidrocloruro en el mercado internacional (75-76)

De acuerdo a lo encontrado, el 84,62% de los productos son de la empresa multinacional estadounidense Johnson & Johnson gracias a sus filiales (77), el resto de entidades: Laboratorios Salvat, Eurim-Pharm Arzneimittel GMBH, the Boots Company PLC y Kwng Dong Pharm, abarcan el 15,4% de la totalidad de zona geográfica (75-77), como queda recogido en la Figura IV.2.
Dada la representatividad en el mercado de la loperamida de la entidad líder (78-83), se decidió estudiar detenidamente los excipientes declarados en sus envases (Tabla IV.2).
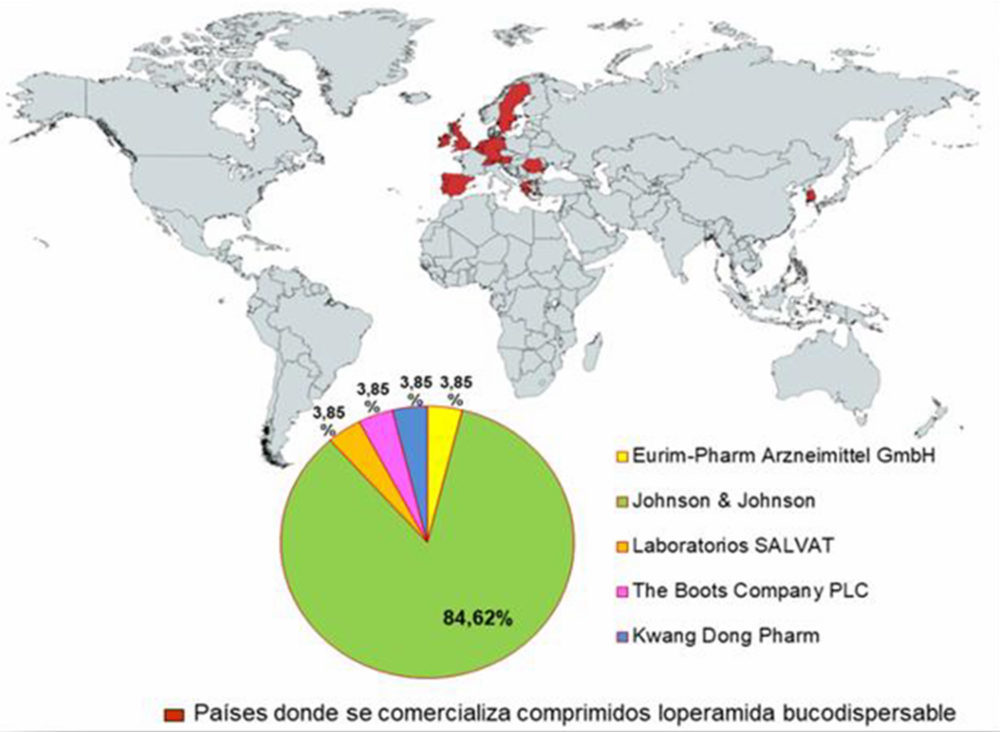
Figura IV.2. Localización geográfica del mercado internacional de comprimidos bucodispersables de Loperamida y sus principales entidades
Tabla IV.2. Excipientes medicamentos de la entidad líder (78-83)
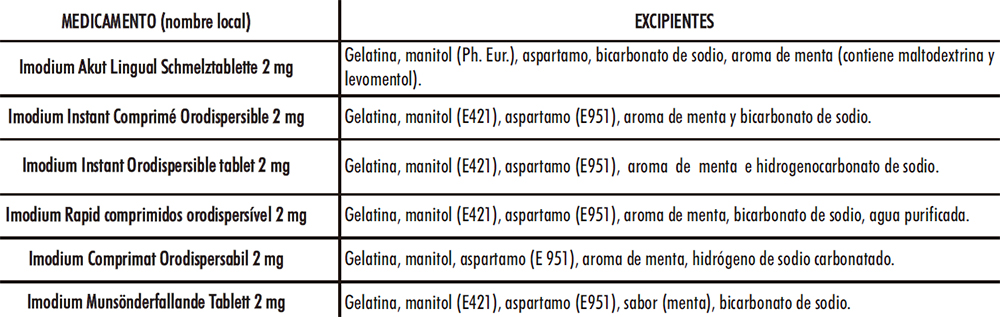
La gelatina puede emplearse como aglutinante en comprimidos (84-85) y en el caso de CBD junto con el manitol son responsables de generar una matriz altamente porosa (86), al igual que el bicarbonato de sodio (87), que facilita la disgregación de este en la cavidad bucal. El manitol podría proporcionar, además, una cristalinidad, aspecto, dureza y adecuadas características organolépticas junto con el aspartamo y el aroma de menta. El bicarbonato de sodio, es un compuesto sólido de color blanquecino y cristalino soluble en agua que actúa a modo de disgregante al generar dióxido de carbono en contacto con ésta provocando efervescencia. El empleo de los excipientes analizados se tuvo en cuenta a la hora de la selección en el desarrollo de CBD-LH. En el caso del mercado español, únicamente hay un medicamento: «Salvacolina Flas 2 mg comprimidos bucodispersables», autorizado desde el 11 de septiembre de 2017, alcanza su comercialización durante el desarrollo del presente trabajo científico (76). Las muestras empleadas como referencia se adquirieron en Oficina de Farmacia, y se compararon sus excipientes con los del comprimido de loperamida hidrocloruro del Petitorio de Farmacia Militar (comprimido loperamida DEF) (Tabla IV.3).
Tabla IV.3. Comparativa excipientes comprimidos bucodispersables loperamida hidrocloruro y comprimido loperamida DEF
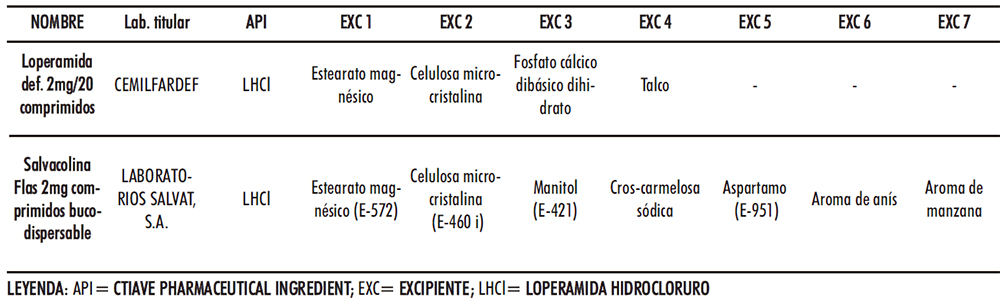
En una primera apreciación la diferencia fundamental en ingredientes se encuentra los modificadores de las propiedades organolépticas. Dentro de estos ingredientes, el manitol se comporta adicionalmente como un agente diluyente ideal para compresión directa.
Entre los ingredientes comunes están la celulosa microcristalina y el estearato magnésico. La primera ejerce una acción aglutinante y diluyente con propiedades lubricantes y disgregantes, por lo que es excipiente ideal, en formulaciones por compresión directa, aunque se debe tener en cuenta su higroscopicidad en la adecuada selección del resto de excipientes. Cabe señalar que tal excipiente puede generar un aspecto arenoso a nivel de cavidad bucal debido a la dificultad de que solubilice a 35ºC, no obstante, desde un punto de vista militar, priman los costes y rapidez en fabricación y no un aspecto publicitario como pueda ser el de una distinción de la competencia. En cuanto al estearato magnésico, es un agente lubricante ideal para un proceso de compresión directa. Para la formulación en desarrollo su incorporación contrarresta el carácter abrasivo del fosfato cálcico dibásico dihidrato, también ingrediente común que presenta buenas propiedades de compactación y de flujo, aparte de una fractura fácil, que le permite una adaptación a la escala de producción industrial, el objetivo deseado.
El talco, dadas sus propiedades antiaglomerantes y deslizantes constituye un gran candidato para este tipo de proceso productivo. Por su parte, la croscarmelosa presente en el comprimido comercial resulta un agente de utilidad en procesos por compresión directa siendo compatible con excipientes higroscópicos.
Formulación
Se desarrollaron, analizaron y estudiaron quince formulaciones de CBD-LH, para la consecución final de las fórmulas seleccionadas. En base a los resultados se decidió que el peso final del CBD-LH fuera de 150 mg. A partir de los resultados de la evaluación integral de la Fórmula 1, se preparó una mezcla para 100 comprimidos con el fin de determinar los problemas tecnológicos que pudiesen surgir, y ameritar modificaciones de ingredientes o de proceso productivo. El fenómeno relevante detectado fue el de capping y chipping, el cual puede ser evitado con la incorporación de hidroxipropilcelulosa, por lo cual se elaboró una mezcla añadiendo ésta al 10%, sobre la fórmula 1 y usando punzones ranurados. En este caso se volvió a presentar el fenómeno de capping y adicionalmente una separación laminar superficial facilitada por la ranura del punzón. El resumen de cada una de las modificaciones realizadas y los resultados obtenidos se encuentran resumidos en la Tabla IV.4, en la que se puede observar que desde las formulaciones 4 en adelante se probaron diferentes proporciones de modificadores organolépticos, y se realizaron ensayos de palatabilidad, las cuales no se incluyeron desde el principio hasta no definir las proporciones de excipientes que daban los mejores resultados farmacotécnicos e incorporando modificaciones en procedo productive según los resultados. Como ejemplo del orden y condiciones de mezclado en la Tabla IV.5. se presenta el usado para la «formula propuesta n° 5.
Tabla IV.5. Orden y tiempos mezclado «fórmula propuesta nº 5»

Escala piloto
Con los resultados prometedores cosechados desde el punto de vista farmacotécnico con la «fórmula propuesta nº 12», y en virtud de la interferencia en lectura por HPLC, se definió la «fórmula propuesta nº 13», sustituyendo la sacarina por el ciclamato mono sódico, usando esta modificación directamente para el primer escalado piloto. El proceso productivo para esta formulación es por compresión directa según el esquema presentado en la Figura IV.3.
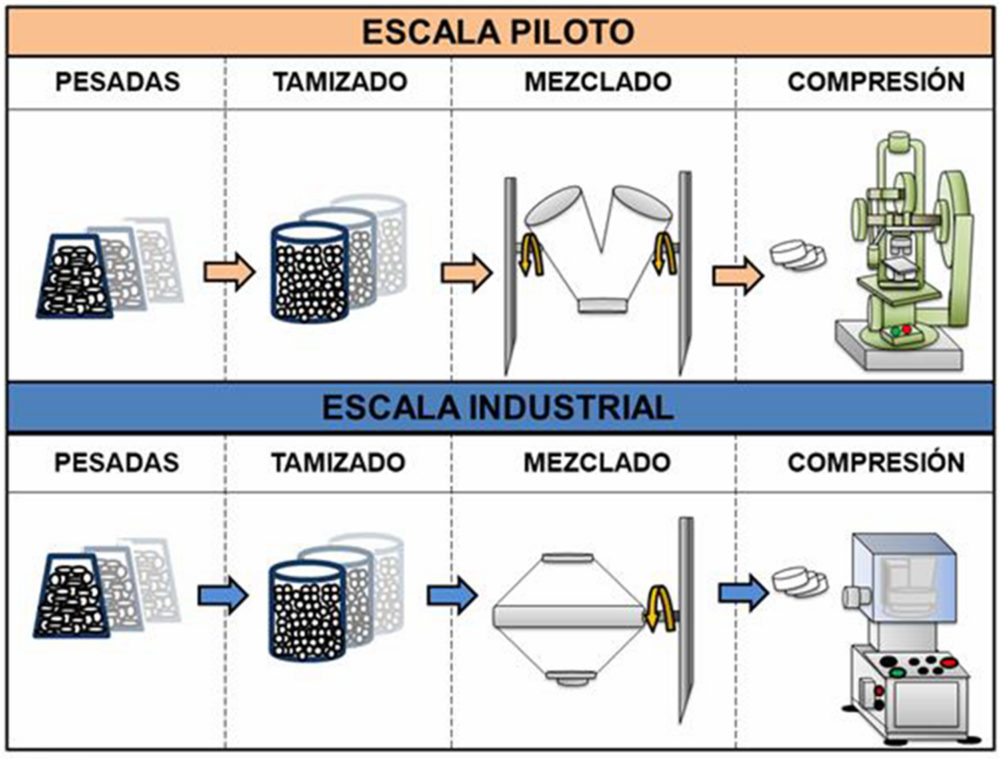
Figura IV.3. Proceso teórico y su comparativa de las etapas de compresión directa entre escala piloto e industrial
La densidad se entiende como la relación existente entre la masa y el volumen que ocupa tal masa. No obstante, esta propiedad en los sólidos pulverulentos puede llegar a ser un parámetro variable puesto que se deben tener en cuenta factores como el tamaño de partícula, forma o capacidad de empaquetamiento. Una densidad constante puede indicar un comportamiento de flujo independiente al manejo (presencia en tolva de alimentación, almacenamiento en bulk, etc.) de la materia prima previo o durante un proceso productivo y este comportamiento puede caracterizarse tanto por la DA como por la densidad apelmazada (DC) (88) y resulta de interés su conocimiento para controlar tales parámetros y disminuir con ello la variabilidad de un futuro proceso productivo de CBD-LH. Paralelamente, se estudió el flujo de la materia prima, se calcularon los índices estrechamente relacionados con las densidades aparente y apelmazada, como son el Índice de Carr y el de Hausner (89). Ver Tabla IV.6.
Tabla IV.6. Caracterización reológica «fórmula propuesta nº 13» (46-47, 49, 51, 89)
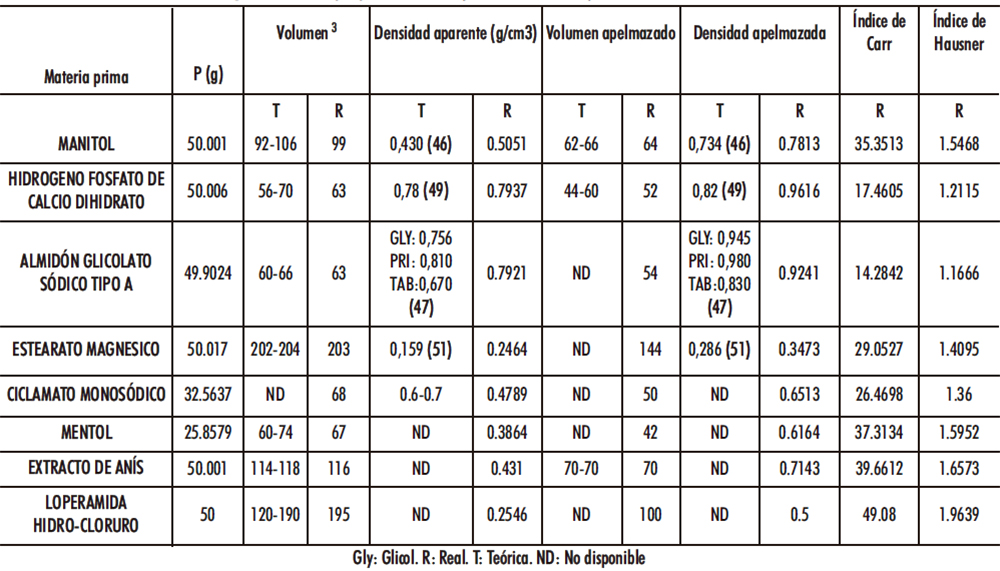
En la loperamida hidrocloruro, mentol y estearato magnésico presentaron una ausencia de fluidez siendo el resto, en orden decreciente ciclamato monosódico, manitol, hidrógeno fosfato de calcio dihidrato y almidón glicolato sódico tipo A.
Se reservó el extracto de anís para el escalado Industrial. Se obtuvo un mayor ángulo de reposo con manitol, seguido de hidrógeno fosfato de calcio dihidrato, ciclamato monosódico y almidón glicolato sódico tipo A. Tabla IV.7.
Tabla IV.7. Estudio ángulo de reposo y fluidez materia prima «fórmula propuesta nº 13»

Para la etapa de mezclado se empleó la teoría de adiciones sucesivas usando de referencia el principio activo loperamida hidrocloruro. Tabla IV.8.
Tabla IV.8. Orden, tiempo y velocidad de mezclado «fórmula propuesta nº 13»
En la fase de mezclado se empleó un mezclador en uve P Prat tipo B n◦ 41412 (Barcelona, España), es decir, un mezclador distinto al que se iba a emplear en la fase industrial (mezclador bicónico), no obstante al presentar una misma rotación en su eje (Figura IV.4) los atributos críticos de la mezcla (fluidez, ángulo de reposo, distribución y tamaño de partícula), sus propiedades físicas, biológicas y microbiológicas no deberían verse afectadas, tal como quedó evidenciado en la etapa del propio escalado industrial.

Figura IV.4. Puntos de muestreo mezcla «fórmula propuesta nº 13» (A), mezcla final en equipo mezclador (B) y detalle (C)
Con el fin de justificar la homogeneidad de la mezcla se tomaron muestras en una serie de zonas representativas en el propio mezclador en uve (Figura IV.5, A), para su análisis de acuerdo con el método HPLC establecido. La mezcla presentó un aspecto homogéneo tanto en el tamaño de partícula como en su color blanquecino. Por otro lado, no se adhirió a las paredes del equipo una vez finalizada la etapa de mezclado (Figura IV.5, B) siendo un indicativo a priori de buena fluidez. En esta línea se estudiaron su DA, DC, IC e IH. Para ello se pesaron 50,0241 g de la mezcla y se obtuvo un volumen en la probeta de 70 mL, DA 0,71469 g/cm3, DC tras 1250 caídas (volumen 56 mL) 0,89329 g/cm3. El IC calculado 19,99 mientras que el IH 1,25.

Figura IV.5. Detalle tolva de alimentación mezcla (A) y CBD-LH «fórmula propuesta nº 13» sin defectos tecnológicos (B
Durante la etapa de compresión se produjo el fenómeno de «capping» en algunos de los comprimidos. En base a los resultados se decidió la adición de un 5% de hidroxipropilmetil celulosa, un excipiente que se empleó en las «fórmulas propuestas» nº 4 a la nº 9 en unas cantidades muy superiores a la planteada para la nueva fórmula propuesta. Para ello se llevó a cabo una nueva mezcla partiendo de un 95,00% de la mezcla de la «fórmula propuesta nº 13» + 5,00% de hidroxipropilmetil celulosa + 10,00% g loperamida hidrocloruro siendo la cantidad de API a pesar de 0,13 g.a mezcla generada («mezcla 1») se llevó a cabo con 95,000 g de mezcla «fórmula propuesta nº 13», con 5,0020 g de hidroxipropilmetil celulosa y con 1,3250 g de loperamida hidrocloruro. Así mismo, se determinó su DA, DC, IC e IH. Para ello se pesaron 50,0623 g de la «mezcla 1» y se obtuvo un volumen en la probeta de 70 mL, DA 0,71517 g/cm3, DC tras 1250 caídas (volumen 56 mL) 0,89397 g/cm3. El IC calculado fue de 20,00 mientras que el IH de 1,25. Valores todos ellos bastante similares a la mezcla de la «fórmula propuesta nº 13». Sin embargo, el proceso de compresión con la mezcla generada, «mezcla 1», aconteció sin ningún defecto tecnológico en los CBD-LH, tal como queda evidenciado en la Figura IV.6.

Figura IV.6. Comprimidos bucodispersables loperamida hidrocloruro de «mezcla 1»
Con los nuevos datos cosechados se abrió un camino prometedor a la futura «fórmula propuesta nº 14» que se elaboró en el escalado industrial, con la incorporación del aglutinante hidroxipropilmetil celulosa al 5,00% y restando 2,50% a cada excipiente mayoritario de la fórmula propuesta anterior (manitol e hidrogenofosfato de calcio dihidrato), y además, teniendo en cuenta la disposición de un punzón de 8,00 mm en el escalado industrial, que iban a generar un CBD-LH de mayor diámetro y menor altura con el empleo de una máquina de comprimir rotatoria y, probablemente, se obtuvieran mejores resultados desde un punto de vista farmacotécnico.
Escala Industrial
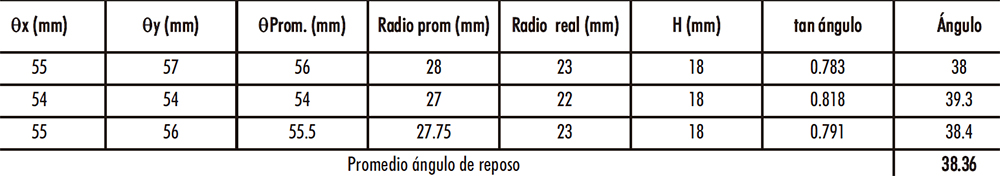
En la Tabla IV.9 se presentan los ingredientes constitutivos de la «fórmula propuesta nº 14» para una mezcla de 20 kg y los resultados del cálculo del ángulo de reposo de la mezcla obtenida se muestran en la Tabla IV.10.
Tabla IV.9. Pesadas materia prima CBD-LH «fórmula propuesta nº 14»

Tabla IV.10. Ángulo reposo mezcla escala industrial
Posteriormente, se tamizó una muestra de mezcla para estudiar la distribución de tamaño de partícula y argumentar con ello su grado de impacto, en los atributos de la calidad de los CBD- LH de la siguiente fase productiva, la compresión (Tabla IV.11).
Tabla IV.11. Distribución tamaño partícula mezcla escala industrial

La etapa de mezclado se realizó con el mismo planteamiento que en la escala piloto introduciendo los ingredientes manualmente, obteniendo los resultados esperados en un mezclador bicónico Glatt Labortecnic y la fase de compresión se efectuó en la máquina de comprimir rotatoria Kilian RTS 21 con punzones ranurados de 8 mm en las siguientes condiciones: se aplicó una profundidad de 3,5 mm y una presión de 1,8 kN, con una velocidad de 35.000 unidades/ hora.
Una vez obtenidos CBD-LH de 150 mg teóricos (1,33% teórico de principio activo) por unidad tras ajuste inicial del equipo se modificaron las condiciones de compresión con el objetivo de generar CBD-LH de 200 mg. En las nuevas condiciones las únicas modificaciones fueron la aplicación de una profundidad de 5,5 mm y una presión de 2,2 kN siendo el resto de los parámetros idénticos a las condiciones de partida.
Estos nuevos cambios se tradujeron en la fabricación de CBD-LH basados en una nueva formulación, la «fórmula propuesta nº 15», prácticamente igual a la anterior salvo el incremento de los 50 mg teóricos del peso final del CBD-LH, lo que se traduce en que el principio activo supondría un 1,00% de la fórmula y los 0,33% restantes se añadirían en los excipientes mayoritarios, es decir, el manitol y el hidrogenofosfato de calcio dihidrato (ambos tendrían un incremento del 0,165% respectivamente con respecto a la anterior formulación).
Fueron necesarios no sólo la caracterización farmacotécnica del producto terminado de las «fórmulas propuestas nº 14 y nº 15», y del medicamento mencionado, sino de una serie de estudios de caracterización y cuantificación, además de la necesidad de desarrollar y validar métodos analíticos por HPLC para los medicamentos estudiados y paralelamente para comprimidos de azitromicina DEF. Ver Figura IV.7.
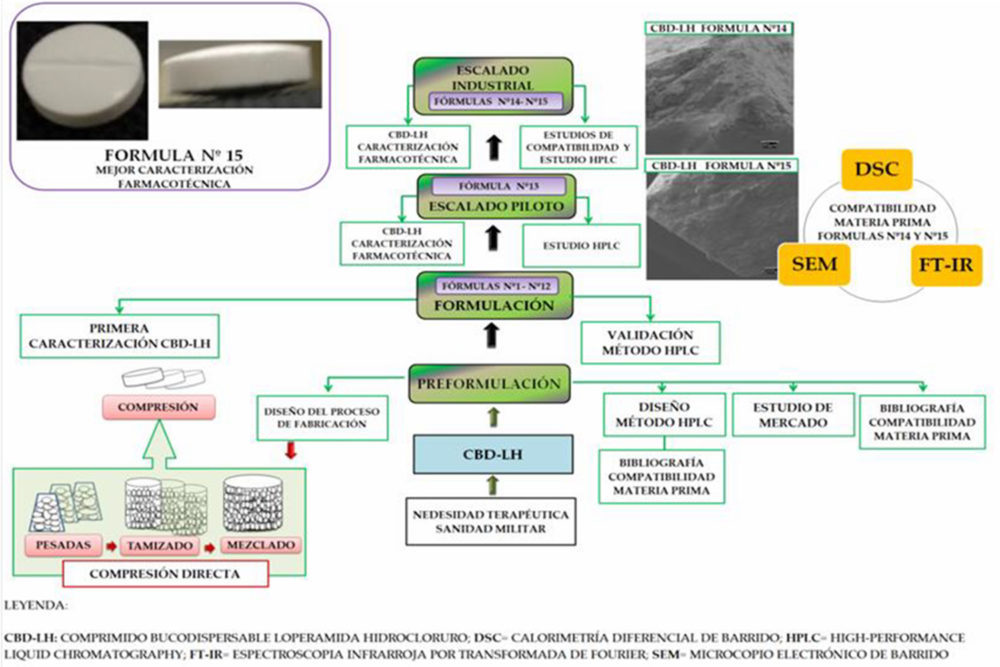
Figura IV.7. Resumen gráfico resultados y discusión
Todo ello, permitió proporcionar una base científica basada en la filosofía «Quality by design» (QbD) que incluye la identificación del perfil de calidad del producto objetivo (QTPP por sus siglas en inglés «quality target product profile»), que consta de atributos críticos de calidad (CQA, por sus siglas en inglés «critical quality attributes»), atributos de críticos de la materia prima (CMA, por sus siglas en inglés «critical material attributes») y parámetros críticos del proceso (CPP, por sus siglas en inglés «critical process parameters») mediante evaluación de riesgos para la calidad de acuerdo a la Figura IV.8.
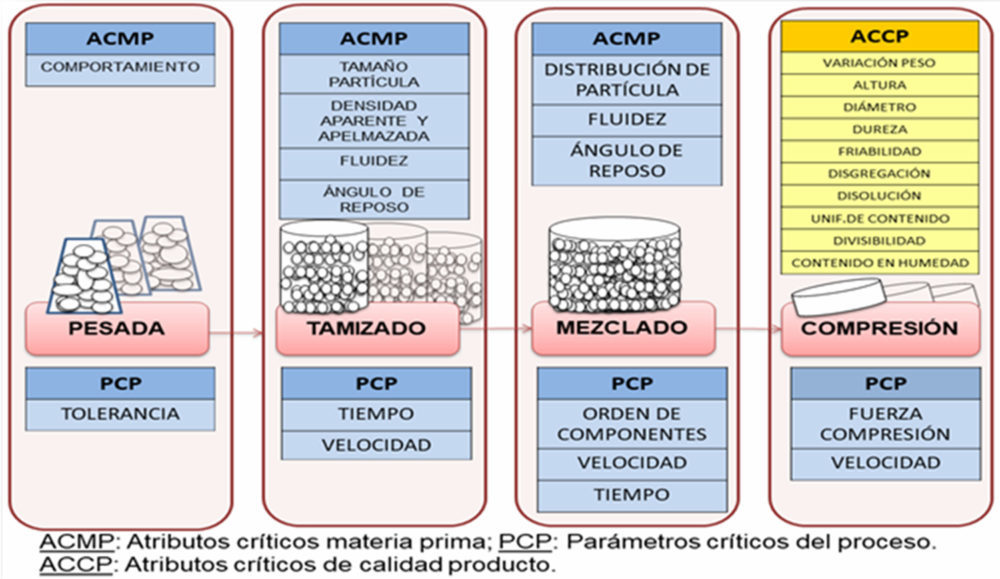
Figura IV.8. Aspectos del proceso productivo CBD-LH por compresión directa
4.2. MÉTODOS DE CARACTERIZACIÓN Y CUANTIFICACIÓN
4.2.1. Métodos de caracterización física de materias primas y producto terminado en comprimidos bucodispersables de loperamida hidrocloruro
Calorimetría diferencial de barrido (DSC)
En la Figura IV.9 se presentan las curvas en DSC de loperamida hidrocloruro polvo original y de cada uno de los excipientes que formaban parte en la composición del CBD-LH de las «fórmulas propuestas nº 14 y nº 15».
El principio activo puede cristalizar bajo tres formas cristalinas diferentes, una forma polimórfica anhidra I, que representa al polimorfo estable de cristales isométricos y a la forma metaestable (punto de fusión alrededor de 224°C), una forma polimórfica anhidra II (punto de fusión de aproximadamente 218°C) (90-92) y una forma tetrahidratada, de punto de fusión alrededor de 190°C (93-94). El DSC-termograma de la loperamida hidrocloruro polvo original seleccionada exhibió un único pico endotérmico localizado a Tonset = 229,48°C (ΛHF = 1480,48 J/g), que indica fusión siendo un evento típico de compuestos anhidros cristalinos, en este caso correspondiente a la forma polimórfica I, seguido de un proceso de descomposición endotérmico a temperaturas superiores a la fusión (Tonset = 259,65°C). En todo el presente estudio se empleó el dato del «onset» en lugar de la temperatura de fusión o descomposición porque en ese caso la masa no influye.

Figura IV.9. DSC de loperamida hidrocloruro y los excipientes manitol, hidroxipropilmetilcelulosa, extracto anís, almidón glicolato sódico tipo A, estearato magnésico, hidrógeno fosfato de calcio dihidrato, mentol y ciclamato monosódico
El manitol presentó un amplio pico endotérmico correspondiente a la fusión a Tonset = 165,81°C y el área bajo pico revela una entalpía de fusión a 286,07 J/g. Es conocido que el manitol coexiste bajo tres modificaciones polimórficas diferentes: forma cristalina I que es termodinámicamente estable y que está monotrópicamente relacionada con la forma II, la forma III es la forma metaestable que revierte a los polimorfos I ó II durante el calentamiento (puntos de fusión 166,5°C, 166°C y 155°C, respectivamente). Se empleó la forma II en esta investigación (Tonset=165,81ºC), asimismo la compactabilidad de este polimorfo es más alta en comparación con las otras dos formas cristalinas. Para caracterizar el comportamiento térmico y determinar la transición vítrea de la hidroxipropilmetilcelulosa, se utilizaron nuevas condiciones experimentales, se diseñó un ciclo de 0 a 200°C a una velocidad de calentamiento de 200°C/min, lo que permitió observar que la Tg de la hidroxipropilmetilcelulosa ocurrió a 178ºC.
El DSC-extracto de anís y DSC-almidón glicolato sódico tipo A exhibieron un único evento endotérmico localizado a 166,29ºC y a 165,83°C, respectivamente. Por otro lado, el estearato magnésico presentó varios picos a 62,57ºC y 92,73ºC debido a la pérdida de agua superficial, y próximo a 112ºC debido a la fusión del palmitato magnésico, ya que en su composición aparece ácido esteárico y ácido palmítico (frecuentemente se presenta esta impureza en los lotes comerciales de estearato magnésico), seguido de una degradación a 178ºC. El hidrógeno fosfato de calcio dihidrato mostró dos eventos endotérmicos, uno alrededor de 110°C correspondiente al «onset» de la evaporación del agua de hidratación y otro alrededor de 135°C que se puede asociar con una transición de fase del cristal. Por su parte, en los estudios de Landin et al. (95) se afirma que la deshidratación tiene lugar en dos pasos y que se produce de acuerdo con el tamaño de partícula.
El mentol se presenta en dos formas L-mentol y dL-mentol, con diferente polimorfismo α, β, γ y δ para L-mentol y los polimorfos α, β, γ para dL-mentol. Las temperaturas de fusión son para el L-mentol, 42,45°C, 36,85°C, 35,55°C y 35,15°C y para el dL-mentol, 32°C, 27,55°C y 22,75°C. Con respecto al DSC-mentol del presente estudio, dada la endoterma tan amplia que se obtuvo, fue necesario diseñar un nuevo ciclo de calor-frío, a elevadas velocidades de calentamiento y de enfriamiento (100ºC/min). Tras el primer ciclo de calentamiento, la muestra se enfrió a una gran velocidad lo que hizo que no se pudiera cristalizar por completo hasta una temperatura de –60°C. En el segundo ciclo de calentamiento se observó una transición vítrea a aproximadamente –27°C seguida de una fusión que comienza alrededor de los 30°C.
En último lugar, con respecto al ciclamato monosódico, en la bibliografía consultada (96), se describen dos formas pseudopolimórficas, como ciclamato de sodio dihidratado y ciclamato de sodio anhidro. En el termograma apareció una señal endotérmica a 154,8°C, con un hombro a 55°C, referente al proceso de deshidratación, por tanto, en este estudio se empleó la forma hidratada. La deshidratación del ciclamato sódico es un proceso que transcurre en múltiples pasos de forma espontánea a temperatura ambiente seguido de un proceso de descomposición alrededor de 190-200°C debido a un proceso de dimerización que conduce a la formación de N,N’-diciclohexilsulfamida y de sulfato de sodio. Las interacciones entre las mezclas en estos estudios calorimétricos se deducen por la aparición o desaparición de picos, saltos de picos especialmente en el asociado a la fusión y/o variaciones en los valores de entalpía (éstas pueden ser mayores o menores), indistintamente, pueden producirse cambios en la forma del pico (97), aunque hay que tener presente que algunas ampliaciones de pico se deben a un descenso de la pureza o de la cristalinidad de cada componente en la mezcla.
En la Figura IV.10, se describen los resultados de las mezclas binarias (1:1) del principio activo con cada uno de los excipientes utilizados. Las curvas exhibieron un comportamiento característico para cada compuesto.

Figura IV.10. DSC de API, manitol y mezcla física (A); DSC de API, estearato magnésico y mezcla física (B); DSC de API, hidroxipropil metil celulosa y mezcla física (C); DSC de API, anís y mezcla física (D); DSC de API, Almidón glicolato sódico tipo A y mezcla física (E); DSC de API, ciclamato sódico y mezcla física (F); DSC de API, hidrogeno fosfato de calcio dihidrato y mezcla física (G); DSC de API, mentol y mezcla física (H)
En el caso de la mezcla física de loperamida HCl con manitol (Figura IV.10, A) y la mezcla física con estearato magnésico (Figura IV.10, B), se observó un salto a temperaturas inferiores del evento endotérmico correspondiente a la fusión del principio activo, desde 229,48°C a 185,73°C y 198,75°C, respectivamente; estos saltos han sido descritos para otros fármacos como la nateglinida o el ibuprofeno con polivinil pirrolidona (PVP) y se pueden atribuir a alguna interacción sólido–sólido o a una reducción de la pureza individual pero no significa necesariamente una incompatibilidad.
Por otro lado, en las Figuras IV.10, F y G se presentan los resultados correspondientes a la mezcla física con ciclamato sódico y el hidrogeno fosfato de calcio dihidrato y se observó un cambio en el evento de fusión correspondiente al principio activo, mucho más notable en el caso del ciclamato sódico, para el hidrogeno fosfato de calcio dihidrato se produjo un salto a temperaturas ligeramente inferiores del evento endotérmico, pero a diferencia de los excipientes manitol y estearato magnésico, el pico presentó una menor área de fusión, lo que claramente indica que el agua de cristalización del hidrógeno fosfato de calcio disuelve parcialmente al fármaco y el entorno básico resultante podría contribuir a ello.
Esto se verificó mediante un nuevo estudio con un segundo calentamiento y éste mostró un amplio pico de fusión correspondiente al fármaco cambiando a una temperatura más baja con un ligero cambio de entalpía asociado. Son muchos los principios activos que son incompatibles con este excipiente como, por ejemplo, la famotidina (100), quinapril (101) o metronidazol (102).
Finalmente, en la Figura IV.10, H se presentan los resultados correspondientes a la mezcla física con mentol, donde la ligera reducción de la temperatura de fusión del fármaco puede representar una interacción física entre ambos elementos sin indicar una incompatibilidad, porque el valor medio de entalpía para la mezcla es estadísticamente igual al encontrado para la loperamida HCl sola. Cambios más significativos en los valores de entalpía indicarían una posible incompatibilidad química entre ellos, lo que podría conducir a la pérdida parcial o total de la actividad farmacológica del futuro medicamento.
Figura IV.10. DSC de API, manitol y mezcla física (A); DSC de API, estearato magnésico y mezcla física (B); DSC de API, hidroxipropil metil celulosa y mezcla física (C); DSC de API, anís y mezcla física (D); DSC de API, Almidón glicolato sódico tipo A y mezcla física (E); DSC de API, ciclamato sódico y mezcla física (F); DSC de API, hidrogeno fosfato de calcio dihidrato y mezcla física (G); DSC de API, mentol y mezcla física (H).
Espectrofotometría infrarroja por transformada de Fourier (FT-IR)
La utilización de los métodos espectroscópicos como FT-IR en los estudios de preformulación ha contribuido significativamente a una caracterización exacta de posibles interacciones físicas o químicas entre fármaco-excipiente y a ayudar en la selección racionalizada de los excipientes más apropiados en el diseño de las formas de dosificación (103, 104). Se examinaron en el rango de barrido de 500 a 4000 cm−1 obtenidos con un Espectrómetro Fourier Spectrum 2000 Perkin Elmer® System 20.000 FT-IR y una resolución de 1 cm−1.
En la Figura IV.11 se presentan los infrarrojos obtenidos para la materia prima correspondiente a las «fórmulas propuestas» nº 14 y nº 15. El espectro IR de la loperamida HCl (Figura IV.11, A) reveló picos de absorción característicos similares a los publicados con anterioridad (105), asegurando la presencia de determinados grupos funcionales.
Se obtuvo un pico muy amplio alrededor de 3200 cm-1, lo que indica la presencia de un estiramiento de protones intercambiables (-OH). Alrededor de 2900 cm-1 aparecieron nuevos picos que indican la presencia de carbonos saturados confirmando la presencia del grupo –CH. Por debajo de 2000 cm-1, que es la región de la huella digital, se observaron muchos picos característicos de distintos grupos funcionales de la molécula, como son los grupos -CO (1475 cm-1), -CH3 (1386 cm-1), -R-Cl (1037 cm-1), y un área característica situada entre 770-735 cm-1 para los hidrocarburos aromáticos.
En las Figuras IV.11, B-I se detallan los infrarrojos de los excipientes seleccionados en las formulaciones mencionadas de los CBD-LH. Tras realizar una comparativa precisa de los infrarrojos de las mezclas físicas principio activo + excipientes y los obtenidos para las materias primas individuales se evidenció que los infrarrojos de manitol, ciclamato sódico e hidrogeno fosfato de calcio mostraron las mayores diferencias con una clara ampliación de la región más alta, posiblemente debida a una superposición fármaco-excipiente (106-107).
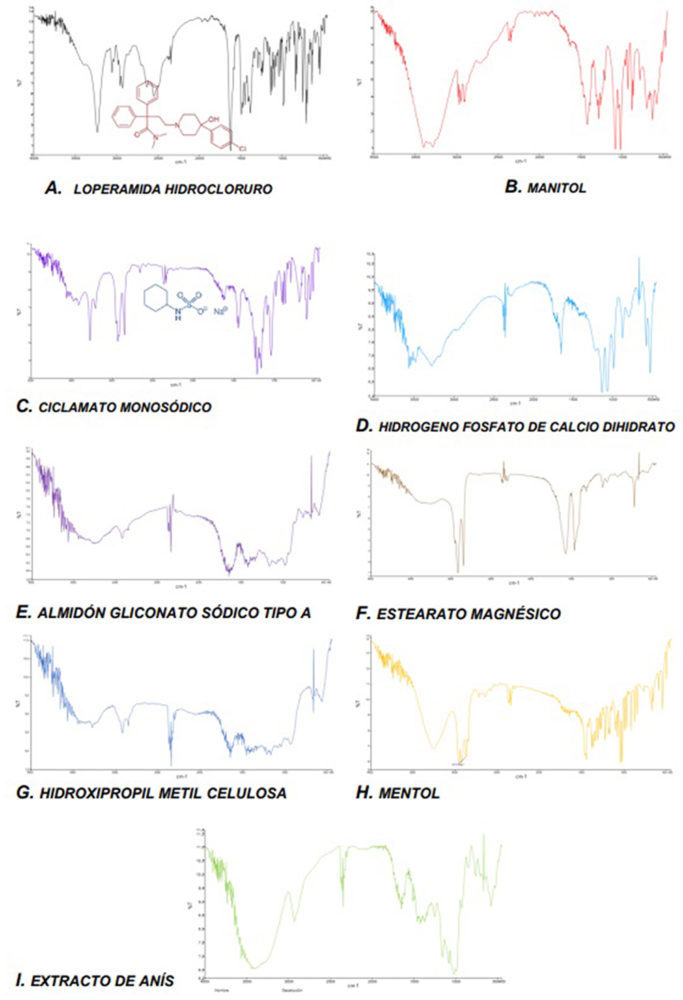
Figura IV.11. IR de loperamida hidrocloruro y los excipientes seleccionados en KBr en la composición del comprimido bucodispersable diseñado «fórmulas propuestas nº 14 y nº 15»
Microscopía electrónica de barrido (SEM)
La combinación de estudios SEM con otras técnicas térmicas y espectroscópicas, como, por ejemplo, las ya expuestas en torno a DSC o FT-IR proporciona interesantes oportunidades para la caracterización de incompatibilidades entre materias primas (103). Por ello, con el fin de asegurar que las partículas mantuvieran las características físicas deseadas durante la fase de compresión directa de acuerdo al diseño de fabricación, se realizó una prueba SEM. Esta técnica también facilitó una evaluación cualitativa de tamaño, forma, morfología, porosidad, distribución de tamaño, forma cristalina y consistencia (108) de tal manera que la información proporcionada por SEM pudiera proporcionar una orientación para asegurar el la QTPP definida para el CBD-LH y con ello afianzar los CQA, CMA y CPP. En la Figura IV.12 se muestran las microfotografías obtenidas de loperamida hidrocloruro y los excipientes seleccionados de las «fórmulas propuestas nº 14 y nº 15».
La técnica esencialmente consiste en hacer incidir en la muestra un haz de electrones. Este bombardeo de electrones provoca la aparición de diferentes señales que, captadas con detectores adecuados, nos proporcionan información acerca de la naturaleza de la muestra.
En este análisis se ha empleado una señal de electrones secundarios (SE) que proporcionó una imagen de la morfología superficial de la muestra y una señal de retrodispersados (BSE) que dio una imagen cualitativa de zonas con distinto número atómico medio. En la Figura IV.11, A se observaron los cristales irregulares del fármaco con superficies regulares planas y los bordes agudos (109). El manitol (Figura IV.11, B) apareció como agujas ortorrómbicas características cuando se cristaliza a partir de alcohol y el excipiente hidrógeno fosfato de calcio dihidrato apareció como un polvo blanco en forma de cristales triclínicos (Figura IV.11, C).
El almidón glicolato sódico tipo A en la Figura IV.11D se observa como un polvo en forma de gránulos irregulares, de tamaño 30–100 mm. El estearato magnésico y ciclamato sódico son polvos muy finos, blancos y con bordes muy irregulares (Figuras IV.11, E y F). El mentol es cristalino acicular o hexagonales, en el que resultó de difícil observación debido a una posible sublimación durante el período de observación en el microscopio (Figura IV.11, G).
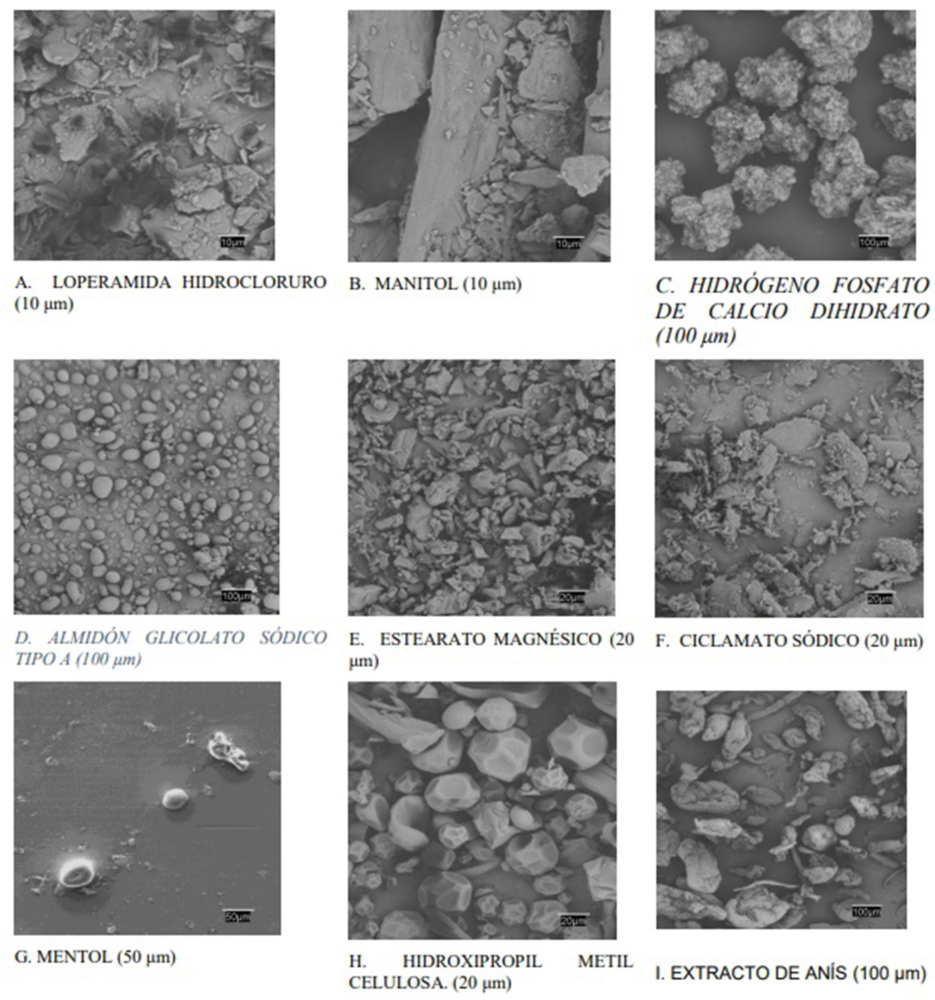
Figura IV.12. SEM de loperamida hidrocloruro y los excipientes seleccionados en la composición del CBD-LH para las «fórmulas propuestas nº 14 y nº 15»
Sin embargo, en la Figura IV.11, H mostró la forma redonda y la superficie lisa y homogénea de la hidroxipropilmetil celulosa, sin duda esto podría permitir una excelente dispersión e influiría como modificador de la liberación del fármaco. Finalmente, el extracto de anís (Figura IV.11, I) se reveló como un polvo muy heterogéneo de formas suaves y con tamaños muy diferentes. También se realizaron estudios microscópicos con las mezclas físicas fármaco- excipiente, pero no proporcionaron ningún dato revelador.
En las Figura IV.12 y IV.13, se exponen los resultados de las microfotografías obtenidas con el corte transversal del CBD-LH de 150 mg, y del CBD-LH de 200 mg, empleando dos aumentos 50 μm y 200 μm que proporcionaron datos complementarios reveladores.
En ambos casos, se observó una mezcla bien compactada en cuya superficie se apreciaron grandes partículas esféricas correspondientes al almidón glicolato sódico tipo A perfectamente disperso en el interior pese a que, en el caso del CBD-LH de la «fórmula propuesta nº 15» no se pudo llevar a cabo un proceso de tamización (99).

Figura IV.13. Corte transversal de CBD-LH «fórmula propuesta nº14» a 50 μm (A) y 200 μm (B) de resolución respectivamente

Figura IV.13. Corte transversal de CBD-LH «fórmula propuesta nº15» a 50 μm (A) y 200 μm (B) de resolución respectivamente
4.2.2. Desarrollo y validación. Método analítico por HPLC para CBD-LH
El desarrollo y validación del método analítico por HPLC para la determinación de loperamida API en CBD-LH fue necesario como acción soporte en las etapas de formulación, escalado piloto y escalado industrial, asentando también en esta última los controles en proceso de la etapa de mezclado y producto terminado. Posteriormente se llevó a cabo un análisis de las fórmulas seleccionadas tras 20,5 meses desde su fabricación, aplicando las condiciones de la Tabla IV.12. El tratamiento de las muestras se explica en la Figura IV.14 y posteriormente el desarrollo, validación del método, y los resultados obtenidos.
Tabla IV.12. Condiciones método HPLC loperamida API en CBD-LH

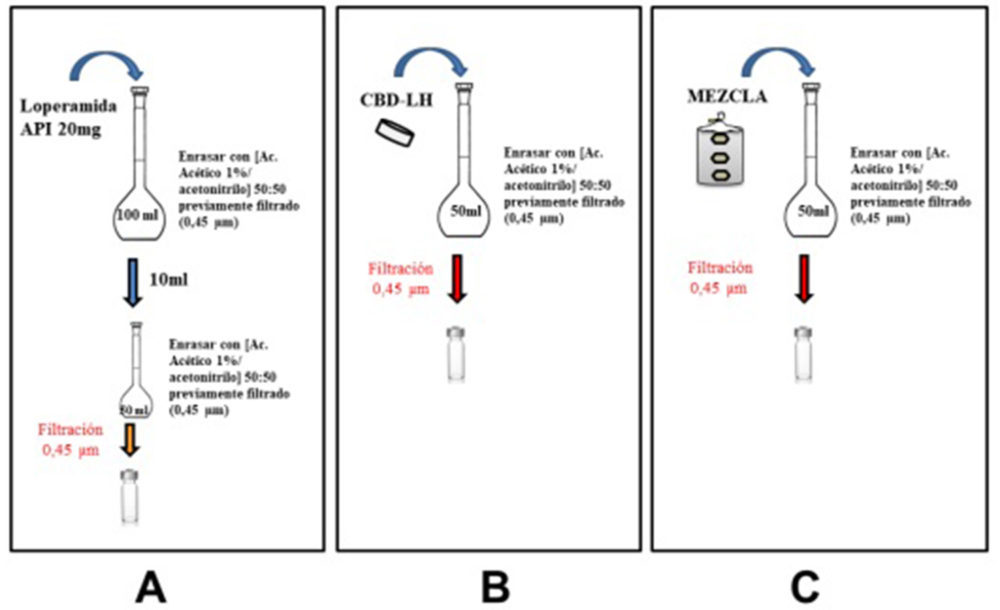
Figura IV.14. Preparación muestra a partir de loperamida API (A), a partir de CBD-LH (B) y a partir de mezcla de la «fórmula propuesta nº 14» y «fórmula propuesta nº 15» (C) empleadas en el método por HPLC
Preparación muestra API loperamida
Se tomaron 20 mg de API y se disolvieron en 100 mL de solución fase móvil previamente filtrada con filtro 0,45 µm (Figura IV.14, A). De la disolución generada se tomaron 10 mL y se enrasó a 50 mL con fase móvil previamente filtrada (0,45 µm). Los viales se prepararon a partir de ésta con filtro 0,45 µm.
Preparación muestras a partir de producto terminado CBD-LH
Sirviendo como guía la Figura IV.14, B, se tomó un CBD-LH, se trituró y se disolvió en 50 mL de fase móvil previamente filtrada (0,45 µm). De la disolución generada se prepararon los viales de muestra filtrando previamente el producto intermedio.
Preparación muestras a partir de producto intermedio mezcla
Según lo especificado en la Figura IV.14, C, se tomaron 150 mg de mezcla (el equivalente en peso a un CBD-LH de la «fórmula propuesta nº 14») y se disolvió en 50 mL de fase móvil previamente filtrada con filtro 0,45 µm. De la disolución generada se prepararon los viales de muestra siendo el producto intermedio previamente filtrado con 0,45 µm.
Validación método analítico
La validación de un método analítico es el proceso por el cual mediante estudios de laboratorio se conocen las características de desempeño de un método (110), es decir, se trata de demostrar mediante pruebas documentadas que la aplicación de un método produce resultados aceptables dentro de unas especificaciones de calidad establecidas (111). Por ello, para la determinación de API loperamida en los CBD-LH se validó el método analítico de HPLC de acuerdo con la ICHQ2 (R1) (74, 112) y para considerar su validez se estudió (113-115):
- Especificidad/selectividad
- Linealidad
- Exactitud
- Precisión: (Repetibilidad, Reproducibilidad, Precisión intermedia)
- Límite de detección
- Límite de cuantificación
- Robustez
Método HPLC aplicado en etapa de formulación
Durante la etapa de formulación en la lectura de los excipientes presentes en la «fórmula propuesta nº11» y «fórmula propuesta nº12» se evidenció una notable distinción en tiempo de la lectura de sacarina sódica con respecto al resto de excipientes.
En las Figuras IV.15 y IV.16 se observa que se podría producir interferencias con la lectura del API loperamida. Este evento resultó significativo puesto que supuso un motivo de cambio que dio lugar a la siguiente fórmula y los problemas tecnológicos en la misma derivaron a las formulaciones finales nº14 y nº15 con la adición de hidroxipropilmetil celulosa.

Figura IV.15. Gráficas obtenidas de los excipientes de la «fórmula propuesta nº11» y «fórmula propuesta nº12»

Figura IV.16. Comparación gráficas sacarina sódica / API loperamida
Finalmente, durante la fase de formulación quedó establecida la validación del método HPLC para la determinación de API loperamida en los CBD-LH «fórmula propuesta nº13», «fórmula propuesta nº14» y «fórmula propuesta nº15», así como en las mezclas para evidenciar su homogeneidad como control en proceso.
Método HPLC aplicado en etapa de escalado piloto y escalado industrial
En el escalado a modo de control en proceso, se determinó la cantidad de API de distintas alturas de la mezcla (superior, medio e inferior). En el escalado industrial de los CBD-LH de las «fórmulas propuestas» nº14 y nº15 se aplicó la correspondiente recta de calibrado calculada tal como se aprecia en la Tabla IV.13.
Tabla IV.13. Determinación cantidad API en mezcla y CBD-LH «fórmulas propuestas» nº14 y nº15

Posteriormente, se tomaron dos CBD-LH de la «fórmula propuesta nº15» siendo la primera muestra un CBD-LH de 201,9 mg la cual representaba (de acuerdo al mismo criterio que el establecido con la mezcla) 2,69 mg teóricos de API loperamida. Se obtuvieron 2,67 mg de API (1,32%). Por su parte, la segunda muestra del CBD-LH de la «fórmula propuesta nº15» partió de un peso de 200,8 mg la cual representaba 2,67 mg teóricos de API loperamida. Se obtuvieron 2,75 mg de API (1,37%). Además, de manera paralela, en el método validado de HPLC se analizaron CBD-LH comerciales de Salvacolina Flas 2 mg comprimidos bucodispersables aplicando de nuevo la recta de calibrado obtenida, con los resultados expuestos en la Tabla IV.14.
Tabla IV.14. Determinación cantidad API en CBD-LH comerciales y detalle gráfica lectura CBD-LH comercial muestra 2

En las tres muestras se obtuvieron un rango de 1,94-2,01 mg de API loperamida, de acuerdo con las especificaciones del medicamento.
Método HPLC aplicado en estudio de estabilidad
Se realizó un análisis de los CBD-LH de las «fórmulas propuestas nº14 y nº15», para determinar la cantidad de API, así como la posibilidad de degradación de acuerdo a las condiciones de la zona climática II (subtropical y climas mediterráneos, 25ºC/60% humedad relativa).
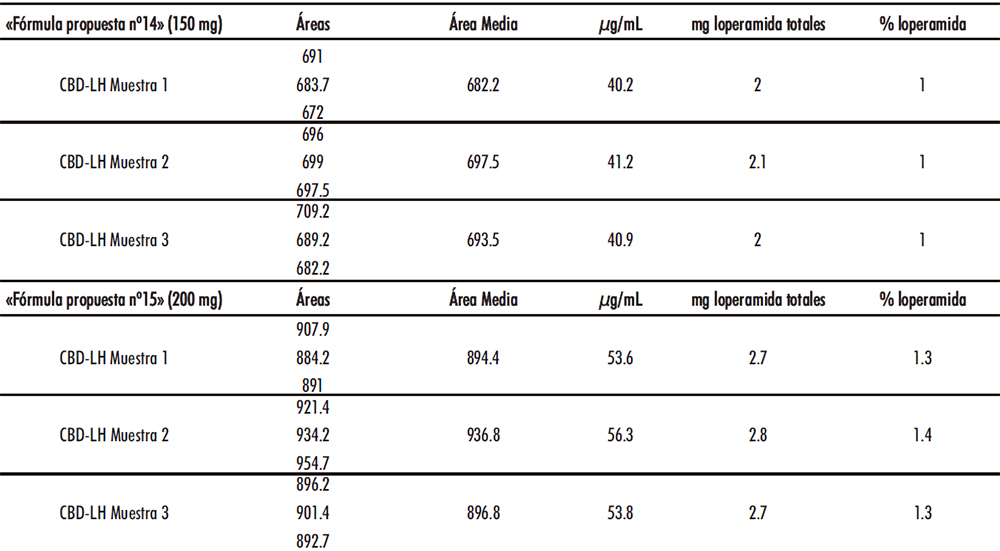
Se tomaron CBD-LH fabricados en el escalado industrial y que fueron debidamente almacenados durante 624 días (Tabla IV.15). Al comparar los resultados anteriores, se observa que la cantidad de API es constante en las formulaciones de CBD-LH no apreciándose anomalías que evidencien una posible degradación.
Tabla IV.15. Estudio estabilidad «fórmulas propuestas nº14 y nº15»
En la Figura IV.17 se compara la gráfica obtenida en el tiempo 0 y la obtenida tras 20,5 meses de la «fórmula propuesta nº14».
Figura IV.17. Comparativa gráfica tiempo «fórmula propuesta nº14
4.2.3. Métodos de elaboración y ensayos de CBD-LH y su comparación con CBD-LH comercial
Una vez obtenidos tanto los CBD-LH de la «fórmula propuesta nº 14» como de la «fórmula propuesta nº 15» en la etapa de escalado industrial, se efectuaron una serie de ensayos para comparar los atributos de calidad de los productos terminados, así como su comparativa con un modelo de referencia, el comprimido bucodispersable Salvacolina flas comp. Bucodispersable 2 mg.
Para esbozar los perfiles de calidad de cada uno de los tres sujetos objeto de estudio se ejecutaron los pertinentes análisis de caracteres geométricos (altura, diámetro), caracteres organolépticos (palatabilidad) y características farmacotécnicas: uniformidad de contenido (RFE 2.9.6), uniformidad de masa (RFE 2.9.5), friabilidad (RFE 2.9.7), dureza (RFE 2.9.8), ensayo de disolución (RFE 2.9.3), ensayo de disolución en saliva artificial, contenido en humedad, disgregación y estudio de divisibilidad (Figura IV.18).
Figura IV.18. Esquema ensayos comprimidos bucodisperables «fórmula propuesta nº 14» (A), «fórmula propuesta nº 15» (B) y comprimido budosipersable comercial (C)
Estudio caracteres geométricos
Los estudios geométricos de los tres tipos de CBD-LH giraron en torno a la medición de la altura y diámetro de diez unidades de estos, la recopilación de datos aparece en la Tabla IV.16.
Tabla IV.16. Resultados estudios caracteres geométricos

La evidencia de la altura es notoria a nivel visual (Figura IV.19) y se trata de un parámetro que se relaciona con el peso y dureza del CBD-LH. Como se evidenciará posteriormente los CBD- LH de mayor altura, es decir, los de la «fórmula propuesta nº 15» y CBD-LH comercial, presentaron un mayor peso y dureza que los de la «fórmula propuesta nº 14». Los datos obtenidos se representan gráficamente y cabe destacar la similitud en cuanto al diámetro de los tres CBD-LH y una ligera variación en cuanto a la altura.
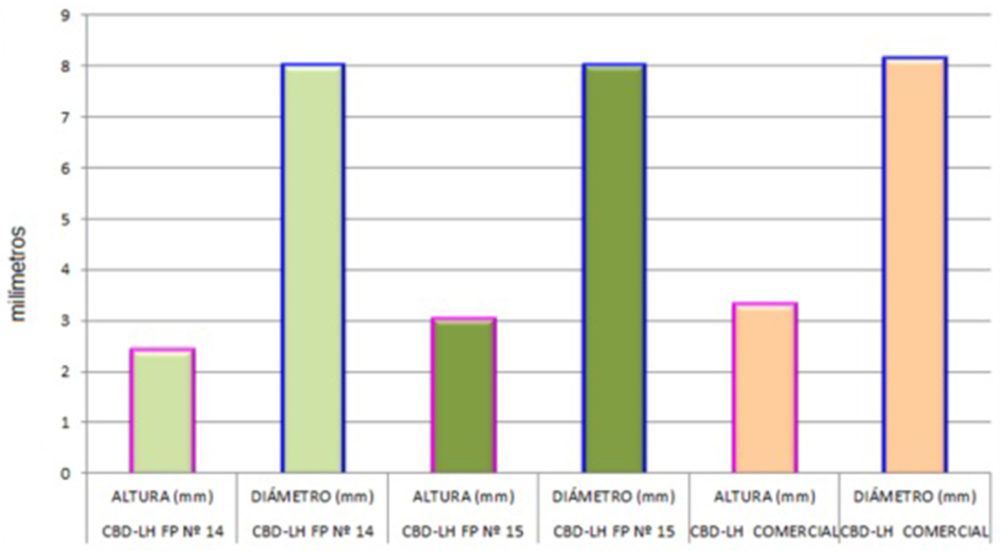
Figura IV.19. Estudio caracteres geométricos CBD-LH «fórmula propuesta nº 14», CBD-LH «fórmula propuesta nº 15» y CBD-LH comercial
Estudio caracteres organolépticos
Los tres tipos de CBD-LH presentaron un aspecto blanquecino y homogéneo como se puede observar en la Figura IV.20.

Figura IV.20. Aspecto CBD-LH de «fórmula propuesta nº 14» (A), «fórmula propuesta nº 15» (B) y comercial (C)
La palatabilidad de los CBD-LH de la «fórmula propuesta nº 14» como de la «fórmula propuesta nº 15» presentó un sabor agradable con la armonización cosechada durante la fase de formulación con los porcentajes de extracto de anís, mentol y sacarina sódica, complementado por la incorporación en gran proporción de manitol. No se detectó una desvirtualización de su carácter farmacoterapéutico en el sentido que enmascaró el amargor del principio activo sin la necesidad de emplear un exceso de anís y mentol, saborizantes que destacan sobre el resto. Desde el punto de vista olfativo, tanto en la «fórmula propuesta nº 14» como en la «fórmula propuesta nº 15» predomina un agradable olor a mentol pese a su baja concentración.
En cuanto al sabor durante la disgregación en la cavidad bucal destaca el mentol sobre el aroma de anís produciéndose una rápida disgregación en la cual se aprecia una ligera sensación arenosa.
En el recuerdo que se percibe minutos posteriores a la pérdida del núcleo cabe destacar la combinación mentol-anís predominando el mentol. Por otro lado, el CBD-LH comercial destacó la presencia olfativa del aroma de manzana. Los excipientes como el aspartamo, aroma de anís, aroma de manzana y manitol lograron enmascarar, al igual que los CBD-LH expuestos, el amargor de la loperamida hidrocloruro. La combinación de estos excipientes resultó agradable durante la administración, subrayando el sabor al aroma de manzana y la sensación arenosa, en menor medida, que los CBD-LH diseñados.
Ensayos farmacotécnicos
Los resultados de uniformidad de masa según la RFE 2.9.5, tomando 20 unidades de cada uno de los tres tipos de CBD-LH, se explican en la Tabla IV.17.
Tabla IV.17. Resultados ensayo uniformidad de masa
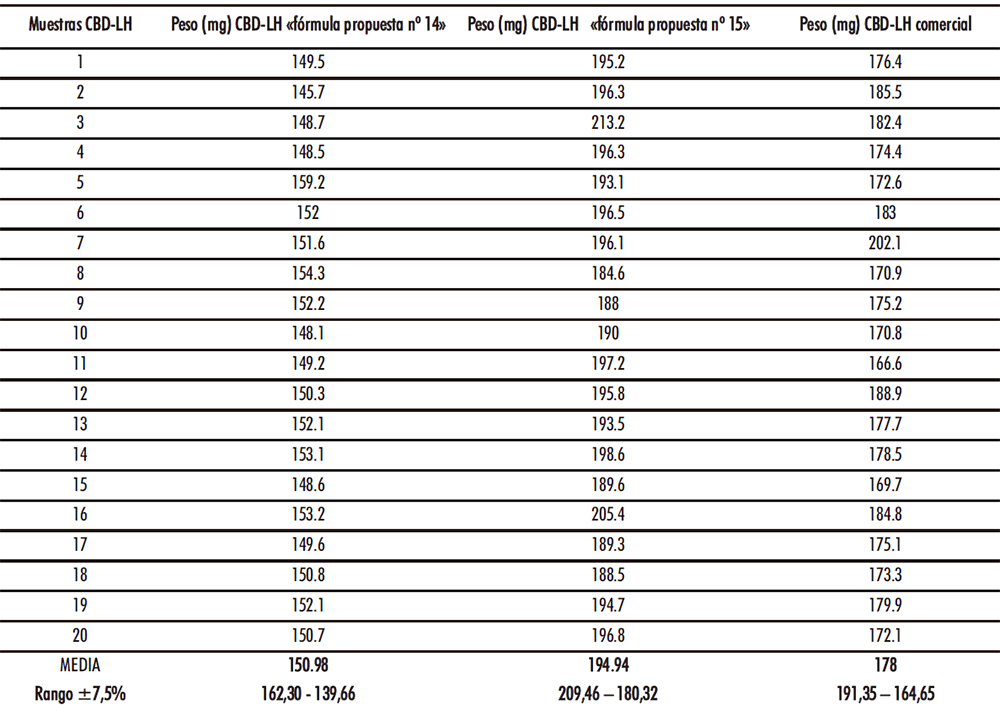
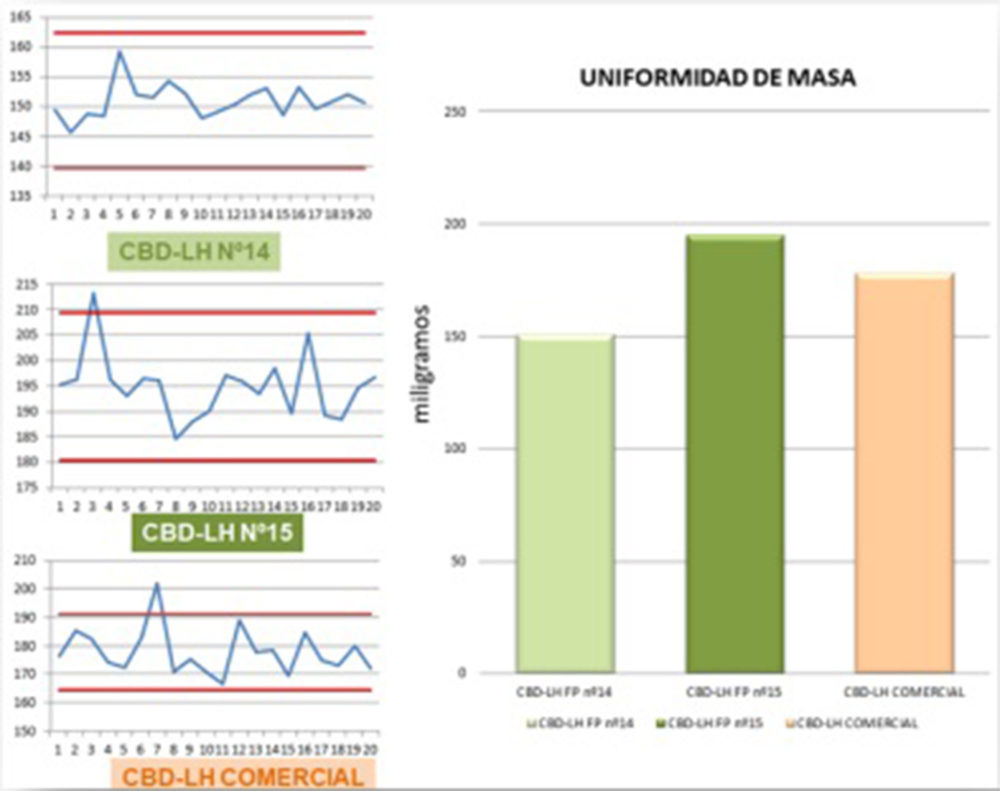
Figura IV.21. Gráficas ensayo uniformidad de masa
El ensayo de cada uno de los tipos de CBD-LH analizados es satisfactorio si no más de dos de las veinte masas individuales se desvían de la masa media en un porcentaje más elevado que el porcentaje de desviación de 7,5% y ninguna en más del doble de tal porcentaje. En el caso de los CBD-LH de la «fórmula propuesta nº 14» todas las unidades analizadas se encontraron dentro del rango ±7,5%. Por otro lado, en las unidades analizadas del CBD-LH «fórmula propuesta nº 15» se observó en una de las lecturas (213,20 mg) una desviación de 7,63% mientras que el resto de las lecturas se situaron dentro del rango ±7,5%. Finalmente, en las unidades analizadas del CBD-LH comercial se evidenció que una de las unidades se desvió un 7,92% (202,10 mg) acotándose el resto de las unidades en el rango ± 7,5% (Figura IV.21). Por tanto, los tres tipos de CBD-LH cumplieron satisfactoriamente con el ensayo de uniformidad de masa ya que las desviaciones fuera del rango ±7,5% que se presentaron en el CBD-LH «fórmula propuesta nº 15» y en el CBD-LH comercial en ningún caso fueron más de una de las veinte masas individuales y ninguna en más del doble de tal porcentaje. Cabe señalar que tanto el CBD-LH de la «fórmula propuesta nº 14» como de la «fórmula propuesta nº 15» se aproximan con una desviación de +0,95% y -2,53% respectivamente al peso ideal.
Uniformidad de contenido
El ensayo de uniformidad de contenido de las preparaciones unidosis de la RFE 2.9.6 y se seleccionaron al azar diez CBD-LH de cada uno de los tres tipos de CBD-LH a analizar. Los resultados quedan expresados en la Tabla IV.18 tras su análisis en el método HPLC.
Tabla IV.18. Resultados ensayo uniformidad de contenido
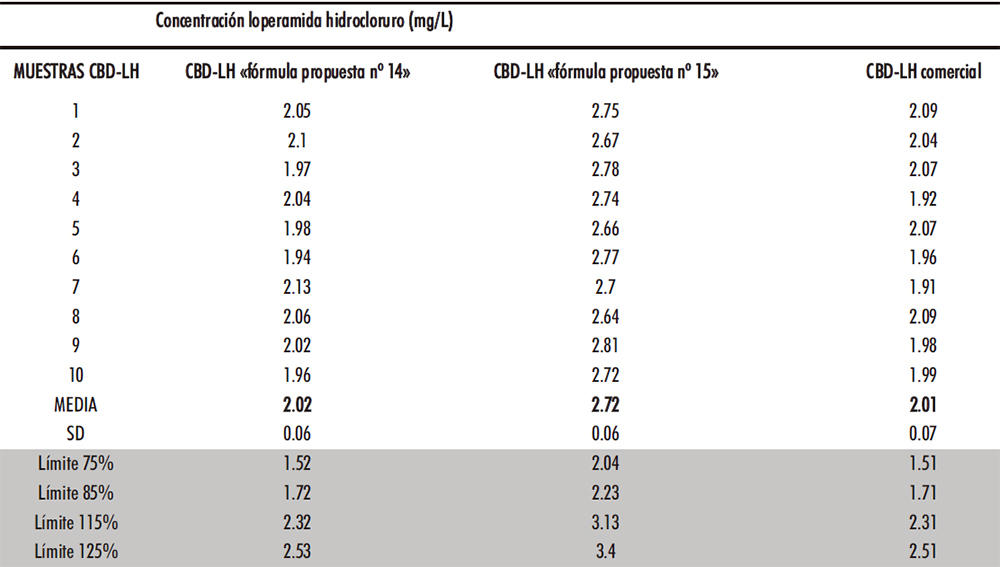
Como se puede observar los tiempos de retención aumentan conforme se obtienen las lecturas de las distintas muestras. Como un método de control en proceso de la fabricación de CBD-LH habría que verificar que es el pico del API añadiendo previamente un patrón. El ensayo cumplió con el ensayo de uniformidad de contenido ya que ningún contenido individual está fuera de los límites del 85% al 115% del contenido medio de acuerdo con lo expuesto para cada una de las formulaciones. Además, ninguno está fuera de los límites del 75% al 125% del contenido medio mencionado.
Friabilidad
El ensayo de friabilidad (Tabla IV.19) se llevó a cabo según la especificación de la RFE 2.9.7, tomando 6,5 g teóricos como peso inicial con la ecuación:
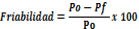
Donde «Po» es el Peso inicial y «Pf» es el peso final
Todos ellos cumplieron satisfactoriamente el ensayo de friabilidad, no obstante, es notoria la mejora de la «fórmula propuesta nº 15» frente a la formulación anterior. Por ello, de las dos formulaciones, el CBD-LH «fórmula propuesta nº 15» se presentó como mejor candidato frente a las posibles pérdidas de masa por abrasión en un mismo proceso de fabricación industrial.
Tabla IV.19. Resultados ensayo de friabilidad

Dureza
El ensayo de dureza se efectuó atendiendo a la especificación de la RFE 2.9.8. Como se puede valorar en la Tabla IV.50, los CBD-LH de la «fórmula propuesta nº 15» presentaron una mayor dureza, lo cual concuerda con el resultado obtenido en el ensayo de friabilidad donde esta formulación obtuvo una pérdida del 0,4975% frente al 0,7879% de la «fórmula propuesta nº 14».
Por su parte, el CBD-LH comercial presentó una dureza notablemente superior a lasformulaciones anteriores, con una dureza media de 34,57 Nw frente a 24,74 Nw de la «fórmula propuesta nº 15» y 23,68 Nw de la «fórmula propuesta nº 14».
Disolución en saliva artificial
En el estudio de disgregación en saliva artificial se empleó una placa Petri de 10 cm de diámetro, en unas condiciones de temperatura de 37 ± 0,5ºC de agua purificada. Se decidió emplear 20 mL en lugar de 30 mL para simular las condiciones de sequedad con un movimiento armónico (leve movimiento:parada, 1:1) simulando la cavidad bucal. Se estudió el tiempo de disgregación tras la dosis inicial (DI) según la posología del paciente adulto (es decir, dos CBD-LH en la primera toma) y tras la dosis continuada (DC) de adulto (1 CBD-LH por toma) de acuerdo a la posología del paciente adulto, coincidiendo además esta última con la DI y DC en el paciente pediátrico (es decir, 1 CBD-LH).
Tabla IV.20. Resultados ensayo disgregación en saliva artificial

Los resultados tras seis lecturas de DI y DC aparecen en la Tabla IV.20, considerando el tiempo de finalización de disgregación el del último CBD-LH en perder la forma farmacéutica.
Disgregación
El ensayo de disgregación se elaboró conforme a la especificación de la RFE 2.9.1. Disgregación de comprimidos y cápsulas, empleando unas condiciones de 37 ± 0,5ºC de agua purificada para 6 comprimidos bucodispersables. Se estudió el tiempo medio de disgregación, siendo satisfactorio el ensayo si la disgregación se produce en menos de tres min.
En el caso de los CBD-LH de la «fórmula propuesta nº 14», se produjo una pérdida de los seis núcleos entre los 9,00-9,57 s con una temperatura de 37ºC aproximada, con una lectura, en el caso del CBD-LH «fórmula propuesta nº 15» entre los 9,30-9,63 s. Por otro lado, el CBD-LH comercial, sujeto a las mismas condiciones, se produjo una pérdida de los núcleos entre los 16,40-17,00 s.
En todos ellos, el ensayo de disgregación fue satisfactorio siendo muy similares los resultados obtenidos en los CBD-LH de la «fórmula propuesta nº 14» y «nº15».
Contenido en humedad
Se pulverizó aproximadamente 1,5 g de CBD-LH con la ayuda de un mortero de cada una de las formulaciones para la realización del ensayo. Los resultados fueron todos <2% (CBD-LH «Fórmula propuesta nº 14» 1,41%, CBD-LH «Fórmula propuesta nº 15» 1,28% y CBD-LH Comercial 0,73%).
Divisibilidad
Los resultados del estudio de divisibilidad de acuerdo con la RFE. En el caso de los CBD-LH de la «fórmula propuesta nº 14» se obtuvo un promedio de 79,94 mg donde una de las masas individuales obtenida fue de 94,2 mg la cual está situada fuera del rango 85-115% (67,95-91,93 mg) pero se encuentra dentro del rango 85-125% (67,95-99,93%) lo que permitió cumplir satisfactoriamente el ensayo de divisibilidad. Por otro lado, los CBD-LH de la «fórmula propuesta nº 15» y los del CBD-LH comercial cumplieron satisfactoriamente el ensayo de divisibilidad, rango fracciones 85-115%. Como se puede observar en la Figura IV.22 (A), la media de las fracciones de cada uno de los CBD-LH se aproxima a la mitad del peso medio obtenido en el ensayo de uniformidad de masa de los mismos. en la Figura IV.22 (B) la desviación sería de un 6,59% para el peso medio de la fracción (79,94 mg) y un 3,43% (103,43 mg) respectivamente.

Figura IV.22. Detalle estudio divisibilidad / peso medio del ensayo de uniformidad de masa (A) y / peso ideal (B)
4.3. Análisis de riesgos. Proceso de Fabricación
La gestión de riesgos para la calidad resulta esencial para identificar los riesgos asociados a cada uno de los procesos que componen la línea de fabricación del nuevo CBD-LH y que pueden repercutir en su calidad, seguridad y eficacia. Cada uno de los procesos está compuesto por una serie de actividades, instalaciones, equipos y sistemas. De acuerdo con los principios básicos de la gestión mencionada la evaluación de los riesgos debe basarse en conocimientos científicos y ha de estar ligada en última instancia a la protección de los pacientes. Así mismo, el nivel de esfuerzo debe estar en relación con el nivel del riesgo. Estos dos principios son la base para la valoración del riesgo, el control de estos hasta niveles aceptados y para futuras revisiones (117).
En el presente trabajo científico, la valoración del riesgo consistió en identificar los peligros, analizar y, finalmente, evaluar los riesgos asociados a la exposición a dichos peligros para cada una de las fases productivas. Posteriormente, se propusieron acciones para controlar aquellos riesgos considerados como no aceptables hasta niveles aceptables. Para ello se empleó la herramienta de calidad «Análisis modal de fallos, efectos y su criticidad» (AMFEC). Esta herramienta permite identificar los fallos y defectos en un proceso antes de que ocurran (118) donde se define «fallo» a lo que podría salir mal. El «efecto» en el presente estudio se centra en el daño que provoca en la calidad, seguridad y eficacia del CBD-LH.
Con el objetivo de cuantificar los riesgos se evaluó el índice de calidad denominado «Índice Prioritario de Riesgos» (IPR) el cual se obtiene de la multiplicación de la «severidad» (S) por la «probabilidad» (P) y por la «detección» (D) (118).
La escala de cuantificación para el parámetro «severidad» se llevó a cabo de acuerdo con la categorización establecida por la Agencia Europea del Medicamento (119) considerando a la «severidad» como la magnitud del daño («efecto») (118) en el paciente (Tabla IV.21).
Tabla IV.21. Escala cuantificación «severidad»

La escala de cuantificación para el parámetro «probabilidad» se configuró de acuerdo con la propuesta de la Asociación de Farmacéuticos de Industria (119) considerando la «probabilidad» como la frecuencia con la que ocurren las causas potenciales (118) (Tabla IV.22).
Tabla IV.22. Escala cuantificación «probabilidad»

Finalmente, la escala de cuantificación del parámetro «detección» se consideró la efectividad de los controles, así como su capacidad para detectar y prevenir el fallo (Tabla IV.23).
Tabla IV.23. Escala cuantificación «detección»

Con el IPR se generó una matriz de valoración del riesgo la cual se aplicó al proceso propuesto por compresión directa del CBD-LH, como se aprecia en la Tabla IV.24. En ésta quedan expuestos los métodos analíticos y farmacotécnicos llevados a cabo.
Tabla IV. 24. Valoración del riesgo

A continuación, se expone el análisis AMFE del proceso de fabricación del medicamento propuesto, CBD-LH (Tabla IV.25).
Tabla IV.25. Estudio gestión de riesgos fabricación industrial CBD-LH

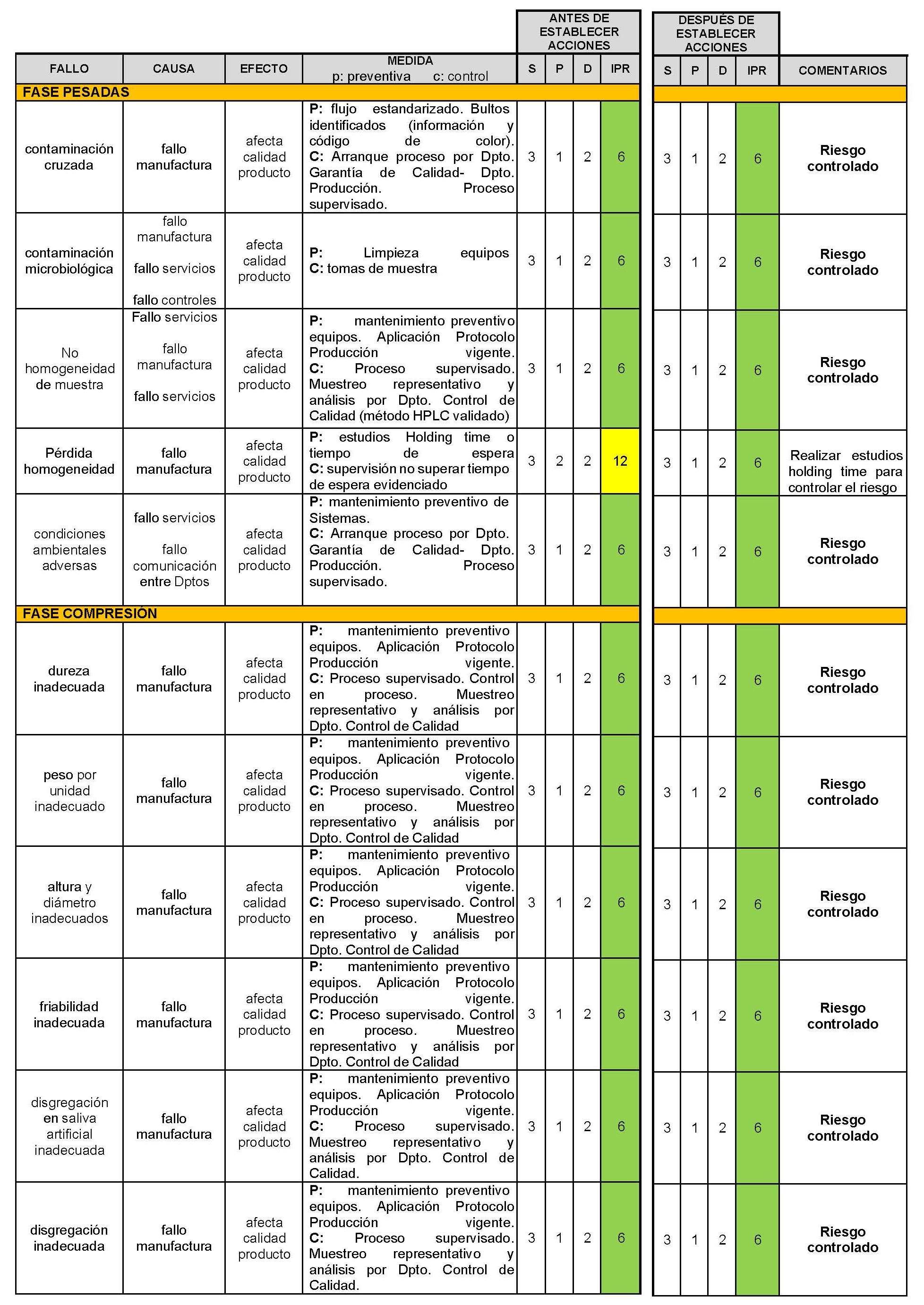

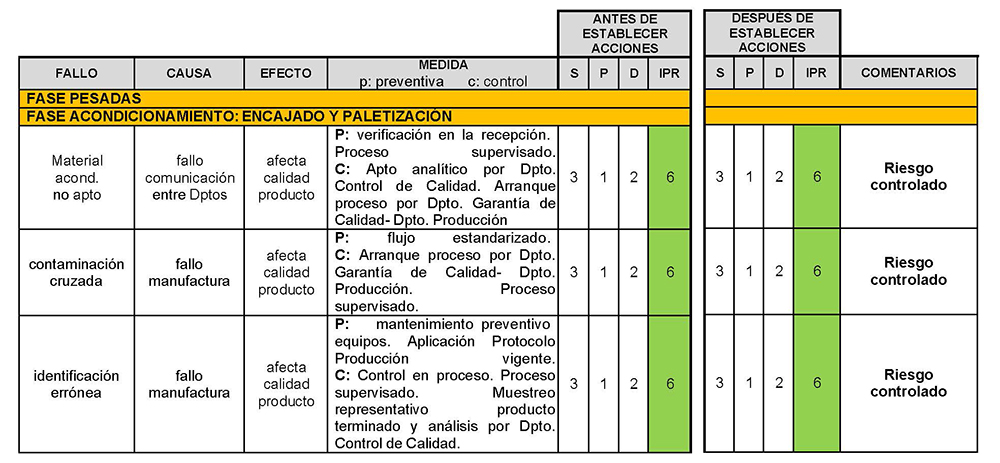
5. CONCLUSIONES
Conclusión general
Las «fórmulas propuestas nº 14 y nº 15» de los comprimidos bucodispersables de loperamida hidrocloruro obtenidos, cumplen tanto con los criterios de calidad requeridos por la farmacopea, como con los criterios de calidad internos, teniendo el aval de un estudio de estabilidad de más de veinte meses. La «fórmula propuesta nº15» resultó la opción más idónea al obtener mejores resultados en su caracterización fisicoquímica, por lo que representa un medicamento cuya forma farmacéutica contribuye a resolver numerosas dificultades encontradas en el tratamiento sintomático de la diarrea aguda inespecífica del combatiente.
Conclusiones específicas
El estudio de mercado de comprimidos bucodispersables de loperamida hidrocloruro ha permitido evidenciar su relevancia terapéutica y ha facilitado la elección de la marca comercial como referente en los estudios realizados.
Las acciones estratégicas y de soporte en torno a las etapas de desarrollo galénico, formulación, escalado piloto y escalado industrial han definido el diseño de un comprimido bucodispersable de loperamida hidrocloruro enfocado a la facilidad de administración en un contexto militar.
El carácter logístico operativo que representa el comprimido bucodispersable de loperamida hidrocloruro se traduce en un proceso de fabricación ágil y robusto de procesos controlados por compresión directa.
El análisis de riesgos aplicado al proceso de fabricación propuesto permitió detectar, analizar, evaluar y controlar los riesgos asociados al mismo para con ello contribuir a la obtención de comprimidos bucodispersables de loperamida hidrocloruro de calidad, seguridad y eficacia.
El método de HPLC para los comprimidos bucodispersables de loperamida hidrocloruro ha demostrado ser un método adecuado como control en producto terminado. Además, ha representado una herramienta para el aseguramiento de la calidad y seguridad del mismo, en las distintas etapas de su desarrollo y en producto intermedio, incluido el estudio de estabilidad del producto terminado.
Las técnicas de espectroscopia infrarroja por transformada de Fourier, calorimetría diferencial de barrido y microscopía electrónica de barrido permitieron conocer la compatibilidad de principio activo y los excipientes finalmente seleccionados en los comprimidos bucodispersables de loperamida hidrocloruro.
6. REFERENCIAS
- Boletín Oficial de Defensa. Orden Ministerial 8/2014, de 30 de enero, por la que se establece el Petitorio de Farmacia del Ministerio de Defensa. Ministerio de Defensa. Gobierno de España. 2014.
- Fernandes Tavares DF., Espada García JI., Martín Sabroso C., Gil Alegre ME., Torres Suárez AI. Comprimidos bucodispersables: ventajas terapéuticas y tecnológicas de elaboración. Ars Pharm, 2009, Vol.50 nº 3; 153-167.
- Misiones en el exterior. Ministerio de Defensa. Gobierno de España. 2021. Disponible en URL: https://www.defensa.gob.es/misiones/en_exterior/
- Infografía de misiones internacionales. Ministerio de Defensa. Gobierno de España. 2021. Disponible en URL: https://www.defensa.gob.es/Galerias/gabinete/red/2021/01/infografia- misiones-379.pdf
- Las 10 principales causas de defunción. Organización Mundial de la salud. 2020. Disponible en URL: https://www.who.int/es/news-room/fact-sheets/detail/the-top-10- causes-of-death
- World Bank Country and Lending Groups. The World Bank. 2021. Disponible en URL: https://datahelpdesk.worldbank.org/knowledgebase/articles/906519
- Enfermedades diarreicas. Organización Mundial de la Salud. 2017. Disponible en URL: https://www.who.int/es/news-room/fact-sheets/detail/diarrhoeal-disease
- Estadísticas sanitarias mundiales. Monitoreando la salud para los objetivos de desarrollo sostenibles. Organización Mundial de la Salud. 2020. Disponible en URL: https://apps.who.int/iris/bitstream/handle/10665/338072/9789240011953- spa.pdf?sequence=1&isAllowed=y%22
- Chereguini E., Blasco F. Sanidad Militar Operacional, en el contexto multinacional. Informe. Sanid. mil. 2015, 71, 2, 109-116.
- Debán J., García A., Castillo JM. Optimización de despliegues militares sanitarios en operaciones mediante técnicas de modelado y simulación. Sanid. mil. 2016; 72, 1, 8-14.
- Model List of essential medicines. World Health Organization. 2021. Disponible ne URL: https://list.essentialmeds.org/
- Siang C., Shahza S. The use of antimotility drugs in COVID-19 associated diarrea. Letter to the editor. J Infect. 2021, 82, 2, e19.
- Teimury A., Mahmoodi E. Current Options in the Treatment of COVID-19: A Review. Risk Manag Healthc Policy. 2020, 13, 1999–2010.
- Manejo clínico del COVID-19: atención hospitalaria. Documento Técnico. España. Ministerio de Sanidad, Servicios Sociales e Igualdad. 2021. Disponible en URL: https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/docume ntos/Protocolo_manejo_clinico_ah_COVID-19.pdf
- Pulse survey on continuity of essential health services during the COVID-19 pandemic. Interim report. World Health Organization. 2020. Disponible en URL: https://www.who.int/publications/i/item/WHO-2019-nCoV-EHS_continuity-survey-2020.1
- Zarzuelo A. Terapéutica del vómito, la diarrea y el estreñimiento. Síndrome del intestino irritable. Trastornos biliares. Programa de actualización en farmacología y farmacoterapia. Terapéutica farmacológica de los trastornos del aparato digestivo, metabolismo y sistema endocrino. Consejo General de Colegios Oficiales de Farmacéuticos. 2013, 203-226.
- Diarrea. Organización Mundial de la Salud. 2020. Disponible en URL: https://www.who.int/topics/diarrhoea/es/
- Calvo C., Marugán J., Bartolomé J. Diarrea aguda. En: Tratado de gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. Sociedad Española de Gastroenterología y hepatología y nutrición pediátrica. 2010. Disponible en URL: https://www.seghnp.org/sites/default/files/2017-06/Trat%20SEGHNP.pdf
- García J., de Manueles J., Ramos J. Diarrea crónica. En: Tratado de gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. Sociedad Española de Gastroenterología y hepatología y nutrición pediátrica. 2010. Disponible en URL: https://www.seghnp.org/sites/default/files/2017-06/Trat%20SEGHNP.pdf
- Medavilla A. Antibióticos macrólidos. En: Farmacología humana. Florez J. 3º Ed. MASSON S.A. 1997.
- FICHA TÉCNICA Azitromicina Ratio 500 mg comprimidos recubiertos con película EFG. España. Ministerio de Sanidad, Servicios Sociales e Igualdad. 2019.
- Vila J., Oliveira I., Zboromyrska Y., Gascon J. Diarrea del viajero. Enfermedades infecciosas y microbiología clínica. 2016, 34, 9, 579-584.
- Melissa Beraun-Villa M., Valdez L. Diarrea del viajero. Rev Med Hered. 2013, 24, 54-61.
- Gavilán C., García B., González R. Gastroenteritis aguda. Protocolos diagnóstico- terapéuticos de la AEP: Infectología pediátrica. 113-124.
- Butterton J., Calderwood S. Acute infectious diarrheal diseases and bacterial food poisoning. En: Harrison´s Infectious disease. Kasper, D., Fauci, A. Ed. Mc Graw Hill Medical. 2010.
- Hashmey R., Genta R., Parasites and Clinton, A. Diarrhea. II: Helmints and Diarrhea. J Travel Med 1997, 4, 72-75.
- Sanders JW., Riddle MS., Taylor DN., DuPont, H. L. Epidemiology of Travelers’ Diarrhea. ,En: Travel Medicine 4ª Ed. Elsevier. 2019, 187–198.
- Connor P., Porter C., Swierczewskic B. Riddleb C. Diarrhoea during military deployment: current concepts and future directions. Curr Opin Infect Dis. 2012, 25, 5, 546-54.
- Walters WA., Reyes F., Soto GM., Reynolds ND., Fraser JA., Aviles R., et al. Epidemiology and associated microbiota changes in deployed military personnel at high risk of traveler’s diarrhea. PLoS ONE. 2020, 15, 8.
- Rodríguez I., Planas E., Grañas A., Herrera P., Sánchez G., Rambla M. ¿De qué enfermamos en Afganistán? Sanidad mil. 2016, 72, 4, 285-287.
- Planas E. E., et al. Yala-Yala: ¿cómo fue la gastroenteritis en Badghis (Afganistán)? Sanid. Mil. 2016, 72, 2,125-128.
- Li X., Kan EM., Lu J., Cao Y., Wong RK., Keshavarzian A., Wilder-Smith CH. Combat- training increases intestinal permeability, immune activation and gastrointestinal symptoms in soldiers. Aliment Pharmacol Ther. 2013, 37, 799–809.
- Cuadernos de estrategia 203. Emergencias pandémicas en un mundo globalizado: amenazas a la seguridad. Instituto Español de Estudios Estratégicos. Ministerio de Defensa. 2019. Disponible en URL: http://www.ieee.es/Galerias/fichero/cuadernos/CE_203_2p.pdf
- Bioterrorism Agents/Diseases. Centers for Disease Control and Prevention. 2018. Disponible en URL: https://emergency.cdc.gov/agent/agentlist-category.asp
- U.S. Army Center for Health Promotion and Preventive Medicine. The Medical NBC Battlebook. 2000. Disponible en URL: https://apps.dtic.mil/dtic/tr/fulltext/u2/a429006.pdf
- Khan AS., Swerdlow DL., Juranek DD. Precautions against biological and chemical terrorism directed at food and water supplies. Public Health Rep. 2001, 116, 1, 3-14.
- Public health response to biological and chemical weapons—WHO guidance. Precautions against the sabotage of drinking-water, food, and other products. World Health Organization. Disponible en URL: https://www.who.int/csr/delibepidemics/annex5.pdf
- Connor BA. Self-treatable conditions. Travelers´ diarrhea. In Centers for Disease Control and Prevention. CDC Yellow Book: Health Information for International Travel. Ed. Oxford University Press. 2018.
- Bouckenooghe A., Kass B. general principles in self – treating travelers’ diarrhea abroad. En: Travelers’ diarrhea. Ed. BC Decker Inc. 2003.
- DuPont H., Mattila L. Antimicrobial treatment: an algorithmic approach. En: Travelers’diarrhea. Ed. BC Decker Inc. 2003.
- Sales para rehidratación oral. Asociación Española de Pediatría. 2018. Disponible en URL:https://www.aeped.es/comite-medicamentos/pediamecum/sales-rehidratacion-oral
- Manrique I., Mora A., Álvarez G. Nuevas pautas de rehidratación en el manejo de la gastroenteritis aguda en urgencias. An Pediatr Contin. 2011, 9, 2, 106-15.
- FICHA TÉCNICA Salvacolina Flas 2 mg comprimidos bucodispersables. España. Ministerio de Sanidad, Servicios Sociales e Igualdad. 2019.
- Hill D., Ryan E. Management of travelers’ diarrhoea. BMJ. 2008, 337.
- Torrado G., Aberturas MR., Molpeceres J., Peña MA., Villarrubia A. Estudio comparativo de disgregación de diferentes formulaciones de olanzapina. An. R. Acad. Farm. 2011, 77,58–75.
- 2.9.34. Densidad aparente y densidad después de asentamiento de los polvos. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 21 de octubre de 2020].
- Moreton R. Calcium Phosphate, Dibasic Dihydrate. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Allen L. y Luner P. Magnesium stearate. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Armstrong NA. Mannitol. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Rogers T. Hypromellose. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Young PM. Sodium starch glycolate. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Langdon BA, Mullarney MP. Menthol. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Chauhan R. Taste masking: A unique approach for bitter drugs. J. Stem.Cell. Bio. Transplant. 2017, 1, 2–12.
- Thoke S., et al. Review On: Taste masking approaches and Evaluation of Taste Masking. Int J Ph Sci. 2012, 3, 1895-1907.
- Sohi H., Sultana Y., Khar RK. Taste Masking Technologies in Oral Pharmaceuticals: Recent Developments and Approaches. Drug Dev Ind Pharm. 2004, 30, 5, 429-448.
- Inoue Y., Funato S., Suzuki R., Morita Y., Murata I., Kanamoto I. Human sensory testing of loperamide hydrochloride preparations for children to improve their palatability. World J. Pharm. Sci. 2015, 3, 570–579.
- Deepak S., Dinesh K. et al. Taste Masking Technologies: A Novel Approach for the improvement of Organoleptic Property of Pharmaceutically Active Substance. Inter Res J Pharm. 2012, 3, 4, 108-116.
- Sayalero ML., Maderuelo, C. Formas sólidas orales de liberación inmediata. En: Tratado de tecnología Farmacéutica. Volumen III: formas de dosificación. Ed. Síntesis. 2017.
- García N., del Barrio P. Correción de propiedades organolépticas. En: En: Tratado de tecnología Farmacéutica. Volumen III: formas de dosificación. Ed. Síntesis. 2017.
- Ficha seguridad 261003 – ANISE 1003 FLAVOUR. Flavours & Technology. 2017.
- Goggin PL. Sodium cyclamate. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- 2.9.6. Uniformidad de contenido de las preparaciones unidosis. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 20 de octubre de 2020].
- 2.9.5. Uniformidad de masa de las preparaciones unidosis. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 20 de octubre de 2020].
- 2.9.7. Friabilidad de los comprimidos no recubiertos. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 20 de octubre de 2020].
- 2.9.8. Resistencia de los comprimidos a la rotura. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 20 de octubre de 2020].
- 2.9.1. Disgregación de comprimidos y cápsulas. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 20 de octubre de 2020].
- 2.9.3. Ensayo de disolución de las formas farmacéuticas sólidas. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 20 de octubre de 2020].
- Divisibilidad de los comprimidos. Comprimidos. Real Farmacopea Española, acceso restringido con clave [Internet]. 2020. [Consultado 21 de octubre de 2020].
- Halder A., Behera A., Si S., Biswal I., Dinda A. Preparation of loperamide hydrochloride chewable tablet: Method validation by HPLC. Int. J. Pharm. Pharmaceut. Sci. 2012, 4, 372–381.
- Sujatha T., Balmuralikrishna K., Raju R. A validated RP-HPLC method for the estimation of loperamide hydrochloride in tablet dosage forms. Int. J. Chem Tech Res. 2014, 6, 1097–1102.
- Sonawane AM., Dudhe PB., Chalke NH., Bhagat KB. Development and validation RP- HPLC method for the simultaneous determination of loperamide hydrochloride and norfloxacin in pharmaceutical formulation. Int. J. Pharm. Sci. Res. 2016, 7, 3441–3445.
- Kabir H., Paul RK., Rahaman MS., Ahmad MF., Bhattacharjya DK., Rahaman MS. Method validation for assay of loperamide hydrochloride by HPLC in loperamide hydrochloride tablets. Int. J. Adv. Res. Chem. Sci. 2017, 4, 11–27.
- Suneetha A., Sharmila N., Purnima M. Stability indicating RP-HPLC method for the determination &validation of loperamide hydrochloride & simethicone in pharmaceutical dosage form. World J. Pharm. Pharm. Sci. 2017, 6, 955–971.
- ICH Q2 (R1) Validation of analytical procedures: text and methodology. (CPMP/ICH/381/95). European Medicines Agency. Disponible en: https://www.ema.europa.eu/en/ich-q2-r1-validation-analytical-procedures-text- methodology#current-effective-version-section
- Centro de Información Online de Medicamentos de la Agencia Española de Medicamentos y Productos Sanitarios. Agencia Española de Medicamentos y Productos Sanitarios. 2021. Disponible en URL: https://cima.aemps.es/cima/publico/lista.html
- Comprimidos bucodispersables de loperamida. Vademécum. 2021. Disponible en URL: https://www.vademecum.es/medicamento- salvacolina+flas+comp.+bucodispersable+2+mg_equivalencias_46398?tri=nameforme&o derSeq=ASC
- Annual report pursuant to section 13 of the securities exchange act for the fiscal year ended December 29, 2019. JOHNSON & JOHNSON. 2019. Disponible en URL: https://johnsonandjohnson.gcs- web.com/node/46106/html#sBA61C6974B02514088A51D14F9CE0D03
- Imodium Akut Lingual Schmelztablette 2 mg. Información especializada. Resumen de las características del medicamento. 2015. Disponible en URL: https://s3.eu-central-1.amazonaws.com/prod-cerebro-ifap/media_all/69600.pdf
- Imodium Instant Comprimé Orodispersible 2 mg. Prospecto: información para el usuario. 2020. Disponible en URL: https://www.pixelpharma.be/files/products/1303338/1303338_fr.pdf
- Imodium Instant Orodispersible Tablet 2 mg. Resumen características medicamento. 2020. Disponible en URL: http://www.hpra.ie/img/uploaded/swedocuments/164215c3-5a8d-428c-845d-bc6e3a1fca38.pdf
- Imodium Rapid Comprimidos Orodispersível 2 mg. Resumen características del medicamento. 2012. Disponible enURL: https://farmacia24.eu/content/MNSRM/2438489.pdf
- Imodium Comprimat Orodispersabil 2 mg. Resumen características del medicamento. 2019 Disponible en URL: https://www.jnjconsumer.ro/sites/brandhub_soe/files/ficha- tecnica-imodium-instant.pdf
- Imodium Munsönderfallande Tablett 2 mg. Resumen características del medicamento. 2020. Disponible en URL: https://docetp.mpa.se/LMF/Imodium%20orodispersible%20tablet%20SmPC_09001bee807a4372.pdf
- Sayalero ML., Maderuelo C. Formas sólidas orales de liberación inmediata. Tratado de Tecnología Farmacéutica. En: Volumen III Formas de dosificación. 1ª Ed. Síntesis. 2017.
- Podczeck F. Gelatin. En: Handbook of pharmaceutical excipients. Rowe R., Sheskey P. y Quinn M. 6ª Ed. Pharmaceutical Press and American Pharmacists Association. 2009.
- Jones RJ., Rajabi-Siahboomi A., Levina M., Perrie Y., Mohammed AR. The influence of formulation and manufacturing process parameters on the characteristics of lyophilized orally disintegrating tablets. Pharmaceutics. 2011, 20, 3(3), 440-57.
- Patil PB., More NS. Tour. Recent trends in orodispersible tablets – an overview of formulation technology and future prospects. IJPSR. 2015, 6, 7, 1056-1066.
- Civera C., Córdoba M. Reología de materias primas de uso farmacéutico. En: Manual de Tecnología Farmacéutica. 1ª Ed. Elsevier. 2012.
- Merck. 2021. Disponible en URL: https://www.merckmillipore.com/ES/es
- Caira, MR., Gerber JJ., Lotter AP. Hydration of drug molecules: Cavity inclusion of water in crystals of loperamide hydrochloride tetrahydrate. Supramol. Chem. 1995, 5, 225–230.
- Newman AW., Andres MC., Stahly GP., Ohannessian L., Adair A. Unsolvated enantiotropic polymorphs of loperamide hydrochloride. AAPS PharmSci. 2002, 4, 1816.
- Woertz C., Kleinebudde P. Development of orodispersible polymer films with focus on the solid-state characterization of crystalline loperamide. Eur. J. Pharm. Biopharm. 2015, 94,52–63.
- Rompay J., Carter JE. Clorhidrato de loperamida. En: perfiles analíticos de sustancias y excipientes de drogas. Florey K. Ed. Academic Press. 1990.
- Carlton RA. Experimental methods in the study of thermodynamic polymorph stability relationships. Am. Pharm. Rev. 2006, 9, 88–93.
- Landin M.; Rowe RC., York P. Structural changes during the dehydration of dicalcium phosphate dehydrate. Eur. J. Pharm. Sci. 1994, 2, 245–252.
- Medina D., Ferreira A., Cavalheiro E., Vargas, D. Polymorphism and thermal behavior of sodium cyclamate. J. Therm. Anal. Calorim. 2019, 137, 1307–1313.
- Ali F., Kumar R., Sahu, P.L., Singh GN. Physicochemical characterization and compatibility study of roflumilast with various pharmaceutical excipients. J. Therm. Anal. Calorim. 2017, 130, 1627–1641.
- Chavan RB., Shastri NR. Polymorphic transformation as a result of atovaquone incompatibility with selected excipients. J. Therm. Anal. Calorim. 2018, 131, 2129–2139.
- Georgy KR., Farid RM., Latif, R., Benda ER. A new design for a chronological release profile of etodolac from coated bilayer tablets: In-vitro and in-vivo assessment. J. Adv. Res.2018, 15, 37–47.
- Indrayanto G., Mugihardjo HR. Compatibility study between famotidine and some excipients using differential scanning calorimetry. Drug Dev. Ind. Pharm. 1994, 20, 911–920.
- Thomas VH., Naath M. Design and utilization of the drug–excipient chemical compatibility automated system. Int. J. Pharm. 2008, 359, 150–157.
- Kiss D., Zelkó R., Novák C., Éhen Z. Application of DSC and NIRS to study the compatibility of metronidazole with different pharmaceutical excipients. J. Therm. Anal. Cal. 2006, 84, 447–451.
- Chadha R., Swati, BS. Drug–Excipient compatibility screening. Role of thermoanalytical and spectroscopic techniques. J. Pharm. Biomed. Anal. 2014, 87, 82–97.
- Sachin TV., Deodhar, M.N.; Prakya, V. Advances in analytical techniques used in predicting drug-excipient interactions. Int. J. Pharm. Tech. 2014, 6, 6388–6417.
- Venkateswarlu K., Preethi JK., Chandrasekhar KB. Enhancement of loperamide dissolution rate by liquid-solid compact technique. Adv. Pharm. Bull. 2016, 6, 385–390.
- Aigner Z., Heinrich R., Sipos E., Farkas G., Ciurba A., Berkesi, O., Szabó-Révész P. Compatibility studies of aceclofenac with retard tablet excipients by means of thermal and FT-IR spectroscopic methods. J. Therm. Anal. Calorim. 2011, 104, 265–271.
- Stulzer HK., Tagliari MP., Cruz AP., Silva MA., Laranjeira M. Compatibility studies between piroxicam and pharmaceutical excipients used in solid dosage forms. Pharm. Chem. J. 2008, 42, 215–219.
- Ríos M. Developments in scanning electron microscopy for tablet and granule characterization. Pharm. Tech. 2008, 2, 36–38.
- Dadhich T., Kumar M., Pathak K. Capsulated surface solid dispersion of loperamide for targeted delivery. Pharm. Chem. 2016, 3, 78–90.
- USP 31 –NF 26. Capítulo General 1225. Validación de métodos farmacopéicos.
- UNE-EN ISO/IEC 17025:2005. Evaluación de la conformidad. Requisitos generales para la competencia de los laboratorios de ensayo y de calibración. 2005.
- Borman P., ElderD. Q2 (R1) Validation of analytical procedures: an implementation guide. Global of Journal for Pharma and Allied Science. 2017. 1127-166.
- Magnusson TB., Örnemark U. Eurachem Guide: the fitness for purpose of analytical methods – a laboratory guide to method validation and related topics. 2a Ed. 2014. Disponible en URL: www.eurachem.org
- Barwick V. Eurachem/CITAC Guide: Guide to Quality in Analytical Chemistry: An Aid to Accreditation. 3a Ed. 2016. Disponible en URL: www.eurachem.org
- U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER) and Analytical PROCEDURES AND METHODS VALIDATION FOR DRUGS AND BIOLOGICS GUIDANCE FOR INDUSTRY. 2015. Disponible en URL: https://www.fda.gov/downloads/drugs/guidances/ucm386366.pdf
- Vessman J., et al. Selectivity in analytical chemistry. Pure Appl. Chem., 2001, 73(8),1381.
- Anexo 20: Gestión de riesgos para la calidad. Guía de Normas de Correcta Fabricación de Medicamentos de Uso Humano y Veterinario. Agencia española de medicamentos y productos sanitarios. MINISTERIO DE SANIDAD, POLÍTICA SOCIAL E IGUALDAD. 2008. Disponible en URL: https://www.aemps.gob.es/industria/inspeccionNCF/guiaNCF/docs/anexos/33_anexo-20.pdf
- Gomez A. Herramientas de gestión de calidad. Con ejemplos prácticos en base a requisitos de la norma ISO 9001:2015. 1ª Ed. 2018.
- Juberías A., Zamanillo A., Cabrera JI., Verón M., Urquía ML., Salado G. Gestión del riesgo en la transferencia de procesos productivos: Aplicación a la fabricación de comprimidos de oseltamivir en la pandemia de gripe A. San. Mil. 2011, 67(4), 334-344.
