1. INTRODUCCIÓN
La conservación de la integridad de los organismos superiores –que alcanza su expresión más elevada en lo que denominamos salud– precisa mantener la estabilidad de su medio interno a pesar de las variaciones del entorno. A la obtención de la estabilidad a través del cambio se la conoce como homeostasia. Y a todo aquello que fuerza un cambio homeostático se lo considera estrés. Los seres vivos se encuentran inevitablemente expuestos a toda clase de estresores frente a los que deben poner en marcha respuestas eficaces. De muchas de estas respuestas no somos conscientes (p. ej., del aumento de la producción hepática de glucosa en los periodos de ayuno, de la dilatación de las pupilas al disminuir la iluminación), y de otras apenas nos damos cuenta (p. ej., de los cambios de la frecuencia y profundidad de la respiración). Suele tratarse de reflejos mediados por el sistema nervioso autónomo (simpático, parasimpático y entérico) o de reacciones implementadas por vía humoral en respuesta a estímulos procedentes tanto del interior como del exterior del organismo. Inicialmente, las respuestas al estrés cumplen una función adaptativa, aunque en ocasiones pueden resultar contraproducentes (respuestas incrementadas, espontáneas, carentes de acomodación, etc.) llegando a generar alteraciones patológicas (1).
Un ejemplo clásico de respuesta global del organismo ante lo que se percibe como una amenaza es la llamada de “lucha o huida”. Se manifiesta por un aumento de la frecuencia y fuerza de contracción cardiacas, broncodilatación con incremento de la frecuencia y volumen respiratorios, mayor aporte de sangre al músculo esquelético, dilatación pupilar, incremento de la sudoración y de la glucogenolisis hepática y muscular, e, incluso, modificaciones en el sistema inmune y en la coagulación sanguínea. La mayor parte de estos cambios se deben al aumento de la liberación de noradrenalina (NA) por las terminaciones nerviosas de las neuronas simpáticas posganglionares que inervan el músculo liso y cardiaco y al incremento de la adrenalina (A) circulante proveniente de la médula adrenal (2). Existen, no obstante, diferencias en la contribución del sistema simpático neural (NA) y adrenal (A) en la respuesta al estrés. Estas diferencias se deben a un diferente control a nivel central de ambos sistemas, al hecho de que la A puede actuar sobre tejidos no inervados por el sistema nervioso simpático y a una distinta actuación de las dos catecolaminas sobre los receptores adrenérgicos, de forma que la A es particularmente potente para activar los receptores adrenérgicos β2. Esto último explicaría las acciones metabólicas (movilización del glucógeno hepático y muscular), el incremento del flujo sanguíneo muscular y la broncodilatación que produce la A. Es de señalar también que en ratones KO para la feniletanolamina N-metil transferasa (PNMT), la enzima encargada de la síntesis de A, no se modifica la presión arterial y la función cardiaca en condiciones de reposo, pero se observa un incremento de la presión arterial en respuesta al ejercicio físico y una disminución del gasto cardiaco en animales sujetos a restricción del movimiento (3). Así mismo, el ejercicio físico prolongado conduciría en estos animales a la hipertrofia ventricular izquierda como consecuencia de un aumento sostenido de la presión arterial (4). La contribución del sistema simpático neural predomina en las respuestas al descenso de la presión arterial (ortostatismo, hemorragia), al frío, la locomoción o el ejercicio físico moderado; por su parte, el sistema simpático adrenal se activaría en situaciones de serio compromiso vital (ejercicio físico extenuante, asfixia, shock circulatorio, hipoglucemia, etc.) (5, 6).
El estrés induce la puesta en marcha de cambios adaptativos del organismo que son precedidos (estrés agudo) y acompañados (estrés crónico) de otros cambios en el sistema nervioso simpático, sin los cuales los primeros no se producirían o lo harían de forma diferente. Podríamos decir por tanto que el sistema nervioso también se adapta.
En este artículo se revisan los cambios de la médula adrenal en respuesta a estímulos estresantes y su contribución a la génesis de algunas patologías. Ciertamente, que en las respuestas al estrés crónico el eje hipotálamo-hipófisis-corteza adrenal (HHA) y, consiguientemente, los esteroides córticoadrenales desempeñan un papel crucial. Además, frente a la mayoría de los estímulos estresantes la secreción de corticosteroides se correlaciona mejor con la de A que con la de NA (7). Ello implica una respuesta integral de la glándula adrenal aunque la regulación de la médula y la corteza obedezca a mecanismos diferentes. Por ello, en este artículo se revisan exclusivamente los mecanismos implicados en la respuesta de la médula adrenal.
2. ACOPLAMIENTO EXCITACIÓN-SECRECIÓN EN LAS CÉLULAS CROMAFINES DE LA MÉDULA ADRENAL
La médula adrenal ocupa la región central de la glándula adrenal, cuya parte externa, la corteza, sintetiza y libera las hormonas corticosteroideas (glucocorticoides o esteroides del estrés, mineralcorticoides y esteroides sexuales). Las células cromafines constituyen el parénquima de la médula adrenal y almacenan catecolaminas (A y, en menor proporción, NA y dopamina) en gránulos de secreción (gránulos cromafines). La médula adrenal está inervada por fibras colinérgicas del nervio esplácnico mayor que forman sinapsis con las células cromafines. En la hendidura sináptica la acetilcolina se combina con receptores colinérgicos nicotínicos y muscarínicos de la membrana produciendo un cambio en el potencial de membrana (potencial sináptico) que desencadenará uno o varios potenciales de acción (8, 9). La liberación de las catecolaminas se produce mediante el mecanismo de la exocitosis, que conlleva la fusión de la membrana del gránulo de secreción con la membrana plasmática, y es desencadenado fundamentalmente por el Ca2+ que entra en la célula con cada potencial de acción a través de canales de Ca2+ dependientes de voltaje (Cav) y alto umbral de activación (de los tipos L, N, P/Q y R; high threshold exocytosis) (10, 11). La respuesta secretora de catecolaminas y, en última instancia, la respuesta al estrés se encuentra regulada también a nivel tisular ya que tanto el patrón de inervación de las células cromafines, que muestra una divergencia elevada (una fibra del nervio esplácnico inerva a 80-100 células cromafines), como la existencia de uniones en hendidura (gap junctions) entre las células cromafines coordinan y aseguran la respuesta secretora del conjunto del tejido adrenomedular.
En todos los aspectos que acabamos de mencionar de la función de la médula adrenal (transmisión sináptica, disparo de potenciales de acción por la célula cromafín e integración tisular) intervienen canales iónicos. Además, muchos de estos canales modifican su actividad y sus niveles de expresión durante las respuestas al estrés, haciendo posible la adaptación del organismo tanto frente a estímulos agudos como crónicos (12, 13) (Figura 1).
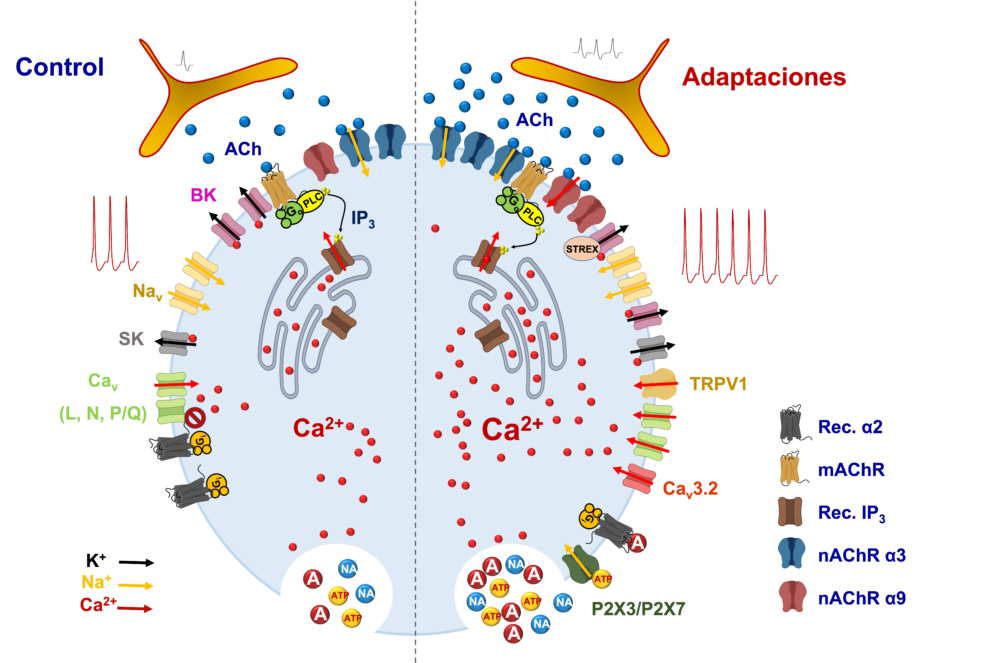
Figura 1. Principales adaptaciones de la médula adrenal al estrés agudo y crónico. Se ilustran los distintos tipos de canales iónicos y mecanismos implicados en el acoplamiento excitación-secreción de adrenalina (A), noradrenalina (NA) y ATP por las células cromafines, tanto en situación control (Control) como de estrés (Adaptaciones). En particular, se destacan las siguientes adaptaciones: a) aumento de la frecuencia de descarga de potenciales de acción en las terminaciones del nervio esplácnico con el consiguiente aumento de la liberación de acetilcolina (ACh); b) aumento de la expresión de receptores nicotínicos (nAChR) α9; c) expresión de canales de Ca2+ dependientes de voltaje (Cav) de tipo T (Cav 3.2); d) expresión de la isoforma STREX de los canales de K+ dependientes de Ca2+ de alta conductancia iónica (BK); e) expresión del canal TRPV1 y de los receptores purinérgicos ionotrópicos P2X3 y P2X7; f) aumento del almacenamiento y liberación de Ca2+ desde el retículo endoplásmico; g) internalización del receptor adrenérgico α2 (Rec. α2) y pérdida de la inhibición de la liberación de A; h) todo ello conduce a un aumento de la frecuencia de descarga de potenciales de acción por las células cromafines y el incremento de la concentración citosólica de Ca2+ con una mayor liberación exocitósica de A, NA y ATP. mAChR: receptor muscarínico; PLC: fosfolipasa C; IP3: inositol 1,4,5-trisfosfato; Rec. IP3; receptor de IP3; Gq: proteína Gq heterotrimérica; Gi: proteína Gi heterotrimérica; Nav: canal de Na+ dependiente de voltaje; Kv: canal de K+ dependiente de voltaje; SK: canal de K+ dependiente de Ca2+ de pequeña conductancia iónica.
3. TRANSMISIÓN COLINÉRGICA EN LA SINAPSIS ESPLACNOCROMAFÍN
En situación de reposo y durante la respuesta inicial al estrés, la señalización en la sinapsis esplacnocromafín depende de la activación de receptores nicotínicos. Se trata de canales iónicos activados por la acetilcolina con arquitectura pentamérica que dejan pasar cationes (Na+, K+ y Ca2+) a favor de su gradiente electroquímico con la consiguiente despolarización de la membrana celular (14). Se trata de un cambio rápido y transitorio denominado potencial sináptico excitador rápido (fEPSP) que, si alcanza el nivel umbral necesario, puede desencadenar un potencial de acción en la célula cromafín. Los receptores nicotínicos de las células cromafines están compuestos por diferentes subunidades. En todas las especies animales estudiadas (hombre, vaca, rata y ratón) se ha descrito la presencia de las subunidades α3 y α7, capaces de formar receptores homoméricos (α7) y heteroméricos (incorporan otras subunidades como la α2, α3,α4,α5, α6, β2 y β4) (15, 16). En las células cromafines humanas se ha descrito que la interacción física entre los receptores α7 y los α3β4 permite prevenir su desensibilización cuando los dos receptores son activados simultáneamente (17). El hecho de que la expresión del receptor α7 se encuentre regulada por los corticosteroides ha llevado a proponer que su participación en la transmisión en la unión esplacnocromafín podría verse incrementada en situaciones de estrés crónico (8). Es posible además que esta mayor contribución pueda en parte deberse a un aumento de la colocalización con los receptores α3β4.
En las células cromafines de roedores (rata y ratón) se ha identificado también la subunidad α9, capaz de constituir receptores tanto homoméricos como heteroméricos (en asociación, entre otras, con la subunidad α10) (18). Curiosamente, los receptores constituidos por las subunidades α9 modificarían el potencial de membrana en sentido hiperpolarizante. Ello se debería a su elevada permeabilidad al Ca2+ y a su colocalización con canales de K+ dependientes de Ca2+ de pequeña conductancia iónica (SK). Por ello, la estimulación nicotínica es capaz activar los canales SK, siendo este efecto bloqueado por antagonistas selectivos de los receptores nicotínicos α9 como la α-conotoxina RgIA. Además, tanto esta toxina como la apamina, un bloqueante de los canales SK, aumentan la duración de los fEPSP inducidos por la estimulación eléctrica de las terminaciones del nervio esplácnico (15), lo que sugiere que, a través de los canales SK, los receptores α9 podrían prevenir el bloqueo por despolarización de la neurotransmisión que aparece en situaciones de estrés. Sin embargo, el papel del receptor nicotínico α9 no se limitaría a las situaciones de estrés agudo. Se ha comprobado que la expresión del receptor α9 aumenta de forma selectiva (en relación a los receptores α3 y α7) en ratas sometidas a estrés crónico (5 días) por frío (4ºC) (18, 19) (Figura 1). Se trata, por tanto, de un ejemplo de plasticidad sináptica, que junto a otros cambios en la dotación y actividad de canales iónicos que se describen a continuación, posibilita la modificación de la actividad eléctrica de las células cromafines para lograr un aumento de la secreción de catecolaminas en condiciones de estrés.
4. EXPRESIÓN Y MODULACIÓN DE CANALES IÓNICOS IMPLICADOS EN EL DISPARO DE LOS POTENCIALES DE ACCIÓN
Como ya se ha mencionado, la respuesta secretora de las células cromafines se desencadena por la entrada de Ca2+ a través de canales Cav activados fundamentalmente durante el potencial de acción. La descarga de un único potencial de acción resulta habitualmente insuficiente para elevar la concentración citosólica de Ca2+ hasta el nivel necesario (rango µM) para inducir la exocitosis de las catecolaminas (20), por lo que se requiere el disparo repetido de potenciales de acción, tanto espontáneos (resultantes de pequeñas despolarizaciones como las producidas por el bloqueo de la corriente de K+ de tipo M o el incremento de la actividad del intercambiador Na+/Ca2+) como inducidos por la actividad sináptica (sucesión de fEPSPs con potenciales de acción asociados) para activar eficazmente la respuesta secretora de las células cromafines.
Un canal iónico que desempeña un papel fundamental en la adaptación de las células cromafines durante el estrés es un canal de Ca2+ de tipo T (en concreto, el Cav3.2). Este canal presenta un umbral de activación bajo (V50 ≈ -55 mV) y una inactivación rápida (decenas de ms) a potenciales positivos. Además, el hecho de que la activación y la inactivación en el estado estacionario se solapen da lugar a una corriente continua (window current) a potenciales próximos al de reposo (≈ -50 mV). El nivel de expresión de estos canales en las células cromafines de animales adultos (vaca, rata y ratón) es muy bajo. Sin embargo, su número y actividad aumentan en situaciones de estrés (stress-induced channels) (21) (Figura 1). Los estímulos capaces de inducir esta respuesta adaptativa en las células cromafines son muy diferentes. Algunos actúan rápidamente (minutos), como el PACAP (polipéptido activador de la adenilato ciclasa hipofisaria) o la estimulación simpática de alta frecuencia, mientras que otros, como la hipoxia o la estimulación de los receptores adrenérgicos β1, lo hacen más lentamente (horas o días) (22). El aumento de los canales Cav de tipo T posibilita la disminución de la corriente necesaria para alcanzar el umbral de disparo de los potenciales de acción (reobase) gracias a la capacidad de estos canales para activarse con pequeñas despolarizaciones y, a su vez, contribuir a despolarizar la membrana celular. Los canales Cav de tipo T serían también capaces contribuir de forma directa a la exocitosis (low-threshold exocytosis) (23), particularmente cuando se producen despolarizaciones pequeñas y sostenidas del potencial de membrana como consecuencia de la activación del intercambiador Na+/Ca2+ o durante la descarga de potenciales de acción en ráfagas de alta frecuencia (24).
Aunque se ha propuesto la participación de diversos factores de transcripción y cascadas de señalización en el incremento de la expresión del canal Cav3.2 (gen CACNA1H), un punto de convergencia de varios de ellos sería la proteína quinasa C (PKC). La actividad de esta enzima aumentaría secundariamente a la activación de la fosfolipasa C, con el consiguiente reclutamiento de Epac (exhange protein activated by cAMP), y por el incremento de especies reactivas de oxígeno. Por su parte, la PKC podría fosforilar a la ERK (extracellular signal-regulated kinase) que, a su vez, podría activar factores de transcripción como CREB (cAMP responsive element binding protein) o actuar sobre factores inducibles por la hipoxia como HIF-1α e HIF-2α para estimular la síntesis del canal Cav3.2 (19).
Los canales Cav (L, N, P/Q y R) de alto umbral de activación (V50 ≈ -10 mV) son los principales mediadores de la elevación de la concentración citosólica de Ca2+ que regula la exocitosis de catecolaminas en las células cromafines. La expresión de los distintos tipos de estos canales varía en función de la especie animal, predominando los canales de tipo L en el hombre, el gato, la rata y el ratón, y los de tipo N y P/Q en la vaca. Todos ellos son modulados de forma autocrina por sustancias coalmacenadas y liberadas por las vesículas de secreción (péptidos opioides, ATP y las propias catecolaminas). Esta modulación suele ser de carácter inhibidor y se ejerce directamente sobre el canal por las proteínas G de los receptores correspondientes (opioides µ, purinérgicos P2Y y adrenérgicos a2). Por otra parte, la activación de receptores de PACAP y adrenérgicos β1 aumenta la corriente de Ca2+ a través del canal de tipo L mediante la elevación del AMPc y la fosforilación del canal dependiente de la proteína quinasa A (PKA) (25).
Los canales de K+ dependientes de Ca2+ de elevada conductancia iónica (canales BK) también influyen en la capacidad de las células cromafines para sostener la descarga de potenciales de acción.
Los canales BK dominan la corriente de salida de K+ de las células cromafines a potenciales en el rango de activación de los canales Cav de alto umbral. En consecuencia, desempeñan un papel esencial en la repolarización de los potenciales de acción que dependen de la entrada de Na+ y de Ca2+. La subunidad α, formadora del poro de los canales BK de las células cromafines, presenta ayuste (splicing) alternativo del exón STREX (stress axis-regulated exon) que codifica una región localizada en el extremo carboxilo del canal. La incorporación de este exón al canal BK conduce a un desplazamiento hacia la izquierda (20-36 mV en sentido hiperpolarizante) de la curva de activación del canal, lo que se asocia a una activación más rápida y una desactivación más lenta del mismo reduciendo la inactivación de los canales de Na+ dependientes de voltaje y posibilitando así la descarga sostenida de potenciales de acción. La dependencia hormonal de este proceso de ayuste se manifiesta por la práctica desaparición de la variante STREX del canal BK en ratas hipofisectomizadas, mientras que la terapia de sustitución con corticotropina previene el efecto de la hipofisectomía (26). Más relevante de cara a su implicación en las respuestas adaptativas al estrés crónico, es el aumento del ARNm correspondiente a la variante STREX del canal BK en la médula adrenal de ratones que desarrollan un comportamiento dominante cuando son sometidos a un modelo de estrés social consistente en el cambio diario de compañero de jaula durante 19 días (27) (Figura 1). Este hallazgo pondría de manifiesto una nueva forma de interacción entre el eje HHA y el sistema simpático adrenal en la coordinación de las respuestas a los estímulos estresantes.
5. ACOPLAMIENTO EXCITACIÓN-SÍNTESIS DE CATECOLAMINAS
La médula adrenal es la gran reserva de A de la que dispone el organismo en situaciones de estrés. El mantenimiento de la respuesta secretora de A requiere de la síntesis continua de catecolaminas y de su rápida incorporación a los gránulos de secreción, y que dicha síntesis se adapte al nivel de estimulación (acoplamiento excitación-síntesis) al objeto de satisfacer adecuadamente las demandas del organismo. De hecho, son los gránulos de secreción mas recientemente formados los que tienen una mayor probabilidad de liberar su contenido en catecolaminas (28, 29). El acoplamiento excitación-síntesis se logra mediante la regulación de las enzimas de la ruta biosintética (tiroxina hidroxilasa –TH–, L-aminoácido aromático descarboxilasa o DOPA descarboxilasa, dopamina β-hidroxilasa y PNMT) de las catecolaminas. Los puntos de regulación fundamentales se sitúan en la primera (TH) y la última (PNMT) enzimas de la vía. La TH (EC 1.14.16.2) cataliza la conversión de la L-tirosina en l-3,4-dihidroxifenilalanina (L-DOPA). Su presencia en un tejido permite catalogarlo como catecolaminérgico, con independencia de que la catecolamina que se encuentre predominantemente en el mismo sea la dopamina (sintetizada por la DOPA descarboxilasa), la NA (sintetizada por la dopamina β-hidroxilasa) o la A (sintetizada por la PNMT). La actividad de la TH está sujeta a regulación neural por las terminaciones del nervio esplácnico (30). Se trata de un mecanismo de regulación rápida a nivel postranscripcional (en 30 min se fosforila por la PKA y/o la ERK), que incrementa la actividad de la enzima, y transcripcional (en 6-8 h se incrementa el ARNm de la proteína) que aumenta la síntesis de la enzima pero que se traslada más lentamente a la actividad enzimática, por lo que no impide una caída transitoria del contenido de catecolaminas en la médula adrenal durante la estimulación de alta intensidad (31, 32, 33). Sin embargo, la repetición de los estímulos estresantes (p. ej., hipoxia, inmovilización) conduce a una elevación persistente del ARNm y al aumento sostenido de la actividad enzimática (34, 35, 36). En tanto que enzima limitante de la ruta biosintética, la TH es inhibida por los productos finales de la vía (las tres catecolaminas ya mencionadas) contribuyendo a la autorregulación del acoplamiento excitación-síntesis de catecolaminas (37, 38).
La PNMT (EC 2.1.1.28) es la enzima responsable de la síntesis de A partir de la NA en una reacción de N-metilación que usa S-adenosil L-metionina (SAM) como cosustrato y se expresa de forma mayoritaria en la médula adrenal (39). Se trata de una enzima regulada predominantemente por los glucocorticoides y consiguientemente por la actividad del eje HHA (40). Los mecanismos implicados en esta regulación son diversos. Uno de ellos sería indirecto, favoreciendo la síntesis de SAM que al unirse a la PNMT la protegería de la degradación. Otro mecanismo consiste en el incremento de la expresión de ARNm codificante de la PNMT dado que el promotor del gen de la PNMT contiene elementos de respuesta a glucocorticoides. Por otra parte, los efectos de los corticoides sobre el gen de la PNMT son sinérgicos con los de factores de transcripción como Egr-1 y AP-2 y pueden ser modulados por la PKA (efecto estimulador) y la PKC (efecto inhibidor), lo que añadiría complejidad a la regulación hormonal de la PNMT al vincularla con la actividad de las células cromafines (41). La PNMT está también sujeta a regulación neural. En particular, por la acetilcolina liberada por la estimulación de las terminaciones del nervio esplácnico que activa la transcripción del gen y la síntesis de PNMT tras combinarse con receptores nicotínicos y muscarínicos. Al igual que en el caso de los glucocorticoides, los efectos de la acetilcolina estarían regulados por Egr-1, PKA y PKC (40). Así mismo, el PACAP, un cotransmisor de la acetilcolina en la sinapsis esplacnocromafín, estimula la transcripción del gen de la PNMT de forma dependiente del incremento de AMPc, con la subsiguiente activación de la PKA y de la entrada de Ca2+ a través de canales Cav de tipo L. Cabe señalar que el efecto de este transmisor sobre la síntesis de A y su liberación exocitósica no manifiesta tolerancia, a diferencia de lo que ocurre con el de la acetilcolina (42). Como acaba de explicarse, en la regulación de esta enzima se integran los dos sistemas efectores fundamentales de la respuesta al estrés en la periferia: el eje HHA y el sistema nervioso simpático. En el modelo experimental de estrés consistente en la inmovilización se produce una marcada activación de ambos sistemas y el aumento de los niveles circulantes de corticosterona y A (43, 44), lo que ha llevado a su utilización para el estudio de los efectos del estrés sobre la ruta biosintética de las catecolaminas en la médula adrenal (32, 45). En este modelo, la inmovilización aguda y crónica (incluyendo la repetición de episodios agudos) se asocia al aumento de la expresión y actividad de todas las enzimas de la vía biosintética. No obstante, la respuesta enzimática parece modificarse también con el tipo de estresor. Así, el aislamiento social crónico se asocia a una menor transcripción del ARNm de la TH, si bien estos animales crónicamente estresados también experimentarían un aumento de la expresión del gen de la TH y de los niveles de A circulante cuando son expuestos a un estrés agudo (46, 47). Un patrón de respuesta similar se observa en las ratas espontáneamente hipertensas (SHR) (véase mas adelante).
6. CONTRIBUCIÓN DE LA MÉDULA ADRENAL A LAS ENFERMEDADES RELACIONADAS CON EL ESTRÉS
Incrementos en la duración de los estímulos estresantes y particularmente de su frecuencia son capaces de convertir una respuesta adaptativa (p. ej., la vasoconstricción y aumento transitorio de la presión arterial) frente estímulos físicos (frío), metabólicos (ejercicio) o psíquicos (miedo, angustia, etc.) en maladaptativa, al favorecer la aparición (influencia en la patogenia) o el desarrollo (influencia en la fisiopatología) de diversas patologías. Como se describe a continuación, se trataría de enfermedades relacionadas con el estrés (ya que en la mayoría de ellas no existe una relación causal entre el estrés y la enfermedad), que pueden producirse tanto por acumulación de los efectos de las respuestas al estrés como por una pérdida de las mismas (hipertensión arterial, insuficiencia cardiaca, dolor, ansiedad, depresión). Por otra parte, es importante diferenciar estas enfermedades de las debidas (parálisis supranuclear progresiva, algunas formas de la enfermedad de Parkinson, hipertrofia ventricular, hipotensión postural, etc.) a alteraciones genéticas (incluyendo cambios en la expresión de isoformas y polimorfismos de un solo nucleótido) de las enzimas de la ruta biosintética de las catecolaminas (4, 48, 49), cuya génesis no reside en una respuesta maladaptativa a los estresores sino en un daño primario del sistema nervioso catecolaminérgico.
6.1. Insuficiencia cardiaca
La insuficiencia cardiaca (IC) es un síndrome con elevada prevalencia y mortalidad para la que en actualidad no se dispone de un tratamiento farmacológico satisfactorio. Presenta una etiología y patogenia variadas pero una fisiopatología dominada por un conjunto de respuestas adaptivas, mediadas por el sistema nervioso simpático y el eje renina-angiotensina-aldosterona (RAA), cuyo objetivo principal es el mantenimiento del gasto cardiaco y la perfusión tisular. La activación simpática es consecuencia de la disminución de la presión arterial y persigue su recuperación en virtud de sus efectos a nivel cardiaco (efecto inotrópico y cronotrópico positivo) y vascular (vasoconstricción). Estos efectos están mediados fundamentalmente por adrenoceptores a (vasculares) y b (cardiacos) activados por la NA, liberada por las terminaciones nerviosas de las fibras simpáticas posganglionares, y por la NA y A provenientes de la médula adrenal. Aunque los efectos de la activación simpática (y también del eje RAA) son beneficiosos a corto plazo sobre la situación hemodinámica, no actúan sobre la causa de la insuficiencia y además pueden agravar su evolución.
Ello se debe a su capacidad para incrementar el consumo de oxígeno miocárdico, aumentar las resistencias vasculares y promover la remodelación mecánica (muerte de los cardiomiocitos y fibrosis compensatoria) y eléctrica (cambios en la expresión de canales iónicos) del tejido cardiaco, con el consiguiente riesgo de aparición de arritmias y disminución de la contractilidad cardiaca. Ello conduce a un círculo vicioso fisiopatológico que afecta negativamente (descompensaciones agudas, empeoramiento del estado funcional y muerte) al curso temporal de la enfermedad. Todo ello justifica el uso de fármacos simpaticolíticos (bloqueantes β adrenérgicos) y antagonistas del eje RAA (inhibidores de la enzima convertidora de la angiotensina y antagonistas del receptor AT1 de la angiotensina) en el manejo de la IC crónica. Pero también el sistema nervioso simpático y en particular la médula adrenal experimenta cambios durante la IC crónica, que afectan a los mecanismos de autorregulación de su actividad y contribuyen al agravamiento de la enfermedad. Un mecanismo de autorregulación fundamental de las células cromafines se basa en la existencia de los receptores adrenérgicos α2, identificados en los años 70 del pasado siglo como receptores presinápticos inhibidores de la liberación de NA por las terminaciones nerviosas de las neuronas simpáticas (autorreceptores) (50, 51). Estos receptores se localizan también en la membrana de las células cromafines donde actuarían como reguladores de la respuesta secretora al ser activados por las catecolaminas (A y/o NA) liberadas por la misma célula (efecto autocrino) o por células vecinas (efecto paracrino) (51). Se trata de receptores acoplados a proteínas G heterotriméricas que presentan diversas vías de señalización intracelular. La mejor conocida consiste en la inhibición directa de los canales Cav de tipo L por las subunidades βγ de la proteína Gi/o (52). La inhibición alcanza un máximo del 50% de las corrientes a través de estos canales, no afecta a la cinética de las mismas, es rápida (<1s) y no se revierte por la actividad eléctrica de la célula, posibilitando por ello la reducción de la respuesta secretora en situaciones de estrés en las que aumenta la frecuencia de descarga de potenciales de acción (53). Cabe señalar también que la activación de los receptores adrenérgicos α2 disminuye la expresión y la actividad de la TH al reducir la concentración intracelular de AMPc y Ca2+, evidenciando que la regulación del acoplamiento excitación-síntesis de catecolaminas es bidireccional (43).
Lo que otorga a los receptores adrenérgicos a2 de la médula adrenal un interés particular es que su función es regulada por la actividad celular y que esta regulación tiene implicaciones fisiopatológicas. Como la mayoría de los receptores acoplados a proteínas G, los receptores adrenérgicos a2 pueden desensibilizare e internalizarse cuando se unen a un agonista. Ello se debe a la fosforilación de la forma activa del receptor por la proteína quinasa 2 de los receptores acoplados a proteínas G (GRK2) (54). En experimentos in vivo se ha observado que la expresión de esta enzima está regulada por actividad de la sinapsis esplacnocromafín, de forma que la esplacnectomía o la administración de antagonistas nicotínicos con efecto bloqueante ganglionar (hexametonio) reduce la expresión de la GRK2 (55). En células cromafines aisladas, son las catecolaminas (A y NA), a concentraciones en el rango nM y actuando a través de los receptores adrenérgicos α2 y β, las que pueden incrementar la expresión de la GRK2, en un proceso que implica a quinasas ERK1/2 y Src (stored response chain) (56, 57). La desensibilización de los receptores α2 adrenérgicos por la GRK2 y la pérdida del control inhibidor de la liberación de catecolaminas adquiere relevancia en situaciones en las que la estimulación crónica de la glándula adrenal y el subsiguiente incremento de la liberación de catecolaminas adquiere un carácter maladaptativo, como es el caso de la IC (58, 59) (Figura 1). En distintos modelos experimentales de IC (infarto de miocardio, constricción aórtica, hipertensión arterial crónica) se observa una correlación entre la hipertrofia cardiaca con disminución del volumen minuto (fracción de eyección reducida) y el aumento del tamaño de la glándula adrenal, de la actividad de la TH, de la GRK2 y de los niveles circulantes de A (55). Es de destacar que la inhibición de la actividad de la GRK2 de la médula adrenal mediante el péptido b ARKct o la galleína reduce la secreción de catecolaminas al tiempo que atenúa la sintomatología de la IC (57, 58, 60, 61, 62); así mismo, el tratamiento con bloqueantes β adrenérgicos (misoprolol, metoprolol) restaura la actividad de la GRK2 y la función de los receptores adrenérgicos α2 en las células cromafines (63), lo que presumiblemente contribuiría a los efectos beneficiosos de estos fármacos en los pacientes con IC. Dado que las vías de señalización implicadas en la inhibición de la secreción de catecolaminas (proteínas Gi/o) y la desensibilización del receptor α2 adrenérgico (GRK2) son diferentes, es de esperar que estos hallazgos estimulen el desarrollo de agonistas sesgados (biased) de los receptores α2 adrenérgicos de acción periférica capaces de inhibir de forma sostenida la secreción de catecolaminas en condiciones de estimulación simpática crónicamente elevada (64).
6.2. Hipertensión arterial
La hipertensión arterial (HTA) esencial es la alteración cardiovascular mas prevalente, afectando al 12% de la población mundial, y relevante al actuar como factor de riesgo de la enfermedad coronaria, la insuficiencia cardiaca, el aneurisma disecante de la aorta, la insuficiencia renal o el accidente cerebrovascular. Si bien en su patogenia intervienen multitud de factores metabólicos (hiperlipemia, hiperglucemia, obesidad, sobrecarga de sal) y mecanismos celulares (estrés oxidativo, inflamación de bajo grado, tensión mecánica de la pared vascular, etc.), una desregulación de los sistemas homeostásicos implicados en el control de las resistencias vasculares periféricas (SNS y eje RAA) ha sido comúnmente implicada en el origen de esta enfermedad (65). Así, en algunos sujetos hipertensos se ha constatado el aumento de la renina y/o catecolaminas circulantes, lo que provee justificación para al uso de fármacos inhibidores del eje RAA (fundamentalmente los inhibidores de la enzima convertidora de la angiotensina I y los antagonistas del receptor AT1 de la angiotensina II) y simpaticolíticos (antagonistas a y b adrenérgicos) en el tratamiento de la HTA (66). En concordancia con las anteriores observaciones, se ha registrado una mayor actividad en los nervios simpáticos en sujetos hipertensos (67).
La rata SHR es el modelo experimental más utilizado para el estudio de la HTA (68). Estos animales, a diferencia de los de la cepa control (Wistar Kyoto; WKY), desarrollan HTA (160-180 mm Hg) a las 5-12 semanas de vida, así como algunas de las alteraciones fisiopatológicas (hipertrofia ventricular izquierda, disfunción endotelial, remodelado vascular, daño renal, etc.) características de la enfermedad en humanos (69). En la patogenia de la HTA en este modelo participaría el SNS por cuanto que la actividad simpática tanto a nivel central como periférico se eleva en paralelo con la presión arterial (70). Además, es posible limitar notablemente el desarrollo de la HTA mediante la simpactectomía química (administración de guanetidina) en ratas recién nacidas (71, 72), si bien esta intervención resulta mucho menos eficaz en rata adultas (73). Ello sugiere que existe un periodo crítico en el desarrollo de la HTA en el que intervendría del sistema simpático neural que, sin embargo, no sería el único factor implicado en la patogenia de la enfermedad. A este respecto, la médula adrenal podría también contribuir al desarrollo de la HTA. Algunos autores han descrito una mayor activación de las neuronas de las que parten las fibras del nervio esplácnico en respuesta a diversos estímulos estresantes (hipoglucemia, estrés mental, etc.), lo que se traduciría en mayores niveles de A en sangre (74). Además, la adrenomedulectomía bilateral en ratas SHR jóvenes (menos de 6 se semanas) pero no en animales adultos atenúa el desarrollo de HTA, mientras que la colocación de implantes subcutáneos de A previene el efecto de la adrenomedulectomía. Cabe señalar también que esa intervención disminuye la respuesta contráctil a la estimulación simpática en las arterias mesentéricas, lo que apunta un efecto facilitador de la A sobre la liberación de NA por las terminaciones nerviosas simpáticas de los vasos (75). Así mismo, la simpatectomía y la adrenomedulectomía parecen ejercer efectos sinérgicos, ya que su combinación normaliza las cifras de presión arterial y revierte la remodelación vascular y cardiaca que se observa en estos animales (76).
En la médula adrenal de animales SHR se producen múltiples adaptaciones funcionales y estructurales (77). La estimulación colinérgica (nicotínica y muscarínica) y la despolarización directa de la membrana celular con una solución extracelular de alto K+ inducen una mayor respuesta secretora de catecolaminas en las ratas SHR que en las WKY (78; ver, no obstante, 79). El incremento de la respuesta secretora sería debido a la exocitosis de un número mayor de gránulos cromafines con, además, un mayor contenido de catecolaminas (80, 81), lo que a nivel estructural se ha relacionado con una mayor densidad de gránulos cromafines en la vecindad de la membrana plasmática (82, 83, 84). Los resultados obtenidos sobre las enzimas de la ruta biosintética de las catecolaminas son contradictorios, habiéndose descrito tanto el aumento como la disminución del ARNm y de la proteína de la TH y la PNMT (85, 86, 87, 88, 89). Análoga situación se produce en relación con el contenido adrenal de catecolaminas (A, NA y dopamina) (72, 73, 76, 87, 89). Estos resultados contradictorios se han tratado de explicar por una elevada vulnerabilidad de las ratas SHR a los estresores frecuentemente implicados en los experimentos (administración de fármacos, incluidos los anestésicos, medida de la presión arterial, método de sacrificio, etc.) y por la capacidad de la HTA para activar a través de los barorreceptores del cuerpo carotídeo respuestas compensadoras de disminución del tono simpático. Estos factores estarían también implicados en la disminución de la actividad de la TH que se observa en ratas SHR no estresadas y el aumento drástico de la misma y de la A en sangre cuando son expuestas de forma aguda o crónica a estímulos estresantes (88).
Por otra parte, se han documentado muy diversas modificaciones en los elementos intervinientes en el acoplamiento excitación-secreción que contribuirían a la mayor liberación de catecolaminas en estos animales. Merecen destacarse la disminución de las corrientes a través de los canales Cav y Kv, lo que unido a una mayor acumulación y liberación de Ca2+ inducida por Ca2+ desde el retículo endoplásmico favorecería elevaciones de la concentración intracelular de Ca2+ de mayor amplitud y duración, con la consiguiente potenciación de la respuesta secretora (81, 90, 91, 92) (Figura 1). Es de destacar que estas alteraciones se producirían también en otros modelos experimentales de HTA como los que consisten en la administración de alcohol, de deoxicorticosterona y sal o de estreptozotocina (induce también diabetes), lo que reforzaría el vínculo entre la médula adrenal y la HTA (83, 92, 93). Además, el hecho de que en ratas SHR prehipertensas se observen también algunas de estas alteraciones, apoyaría una relación causal con la HTA (84). Cabe señalar también que a diferencia de lo que ocurre en la IC, la expresión y función de los receptores adrenérgicos a2 no se ve modificada en la médula adrenal de las ratas SHR (79).
6.3. Hiperalgesia y dolor neuropático
La asociación Internacional para el Estudio del Dolor define el dolor como una experiencia sensorial y emocional desagradable asociada o similar a la asociada con daño tisular real o potencial. La finalidad del dolor es la de alertar al individuo de una lesión orgánica al objeto de poner en marcha estrategias de evitación o protección frente a los posibles agentes capes de agravarla (fuerza mecánica, temperatura, sustancias químicas, etc.). Comúnmente, el dolor cesa cuando la lesión que lo origina desparece (dolor agudo). Sin embargo, en ocasiones la lesión puede perdurar en el tiempo, lo que conduce a la cronificación del dolor. En esta situación, el sistema nervioso nociceptivo modifica su funcionamiento no solo para aumentar su actividad en respuesta a los estímulos dolorosos (hiperalgesia), como ocurre en el dolor agudo, sino también para activarse ante estímulos no dolorosos (alodinia) o incluso en ausencia de cualquier estímulo (dolor espontáneo). Se trata de modificaciones que acompañan al dolor crónico secundario a una lesión tisular (dolor nociceptivo) pero también a aquellas formas de dolor en las que el daño primario acontece sobre el propio sistema nervioso nociceptivo (dolor neuropático) o se produce en ausencia de una lesión claramente identificable (dolor nociplástico). En consecuencia, el dolor crónico pierde cualquier carácter adaptivo y se convierte en una patología en sí misma. De hecho, constituye una importante necesidad médica no cubierta en la actualidad (94).
La relación entre el estrés y el dolor es bidireccional y compleja. Ciertamente que el dolor es una fuente de estrés (de alguna manera, “no hay dolor sin estrés”) pero el estrés también influye sobre el dolor (95, 96). Y lo hace tanto sobre el componente emocional como el sensorial del mismo. Además, esa influencia varía en función de la naturaleza del estrés. Así, el estrés agudo se ha asociado con la analgesia. Es el caso de la disminución de la percepción del dolor en situaciones en las que se produce una reacción de lucha o huida, como las que tienen lugar en determinadas prácticas deportivas y formas de ocio, o se originan en el contexto de accidentes de tráfico, conflictos bélicos, etc. En estas situaciones se suspende temporalmente la función de alerta del dolor y la analgesia cumple una función adaptativa al posibilitar al sujeto concentrarse en una tarea fundamental para su supervivencia o la consecución de algún objetivo del máximo interés. Por el contrario, el estrés crónico conlleva frecuentemente un aumento de la percepción de los estímulos dolorosos. En estas circunstancias, el estrés puede actuar como inductor de hiperalgesia en ausencia de daño tisular o como potenciador de la hiperalgesia en cuadros de dolor nociceptivo (artritis reumatoide), neuropático (lesión de un nervio periférico) y nociplástico (fibromialgia, síndrome del intestino irritable).
El estrés modula la sensación dolorosa actuando a nivel central y periférico. La analgesia asociada al estrés agudo muy probablemente se debe a la interrupción de la señalización dolorosa proveniente de la médula espinal antes de que acceda a las regiones corticales donde se hace consciente. Ello implica a estructuras troncoencefálicas (sustancia gris periacueductal, médula rostral ventral, núcleo parabraquial, locus coeruleus, entre otras) de las que parten las vías descendentes inhibidoras del dolor, de naturaleza opioide y noradrenérgica, que actúan en el asta dorsal de la médula espinal (96, 97). Por otra parte, el hipocampo, la amígdala y el tálamo han sido implicados junto con diversos núcleos del tronco del encéfalo (véase mas arriba) en la hiperalgesia térmica, mecánica, etc. inducidas en animales de experimentación por estresores como el frío, la inmovilización, la hipoglucemia, la natación forzada o la separación materna (98). Tanto el eje HHA como el sistema nervioso simpático participan en la modulación del dolor por el estrés. A veces de forma conjunta, a veces de manera independiente. En algunas formas de dolor, el sistema nervioso simpático neural parece desempañar un papel crucial (99). Corresponden a lo que antiguamente se denominaba dolor mantenido por el sistema nervioso simpático (sympathetically-maintained pain) o distrofia simpática refleja y, actualmente, síndrome doloroso regional complejo, caracterizado por la existencia de dolor espontáneo, alodinia mecánica y al frío en la periferia del organismo, ortostatismo, sudoración, etc. (100). La implicación del sistema simpático neural en la patogenia de este cuadro se basa en la mejoría del dolor que se observa tanto en clínica humana como en medicina experimental tras la práctica de una simpatectomía química (administración de guanetidina) o quirúrgica, y la recurrencia de la sintomatología al ser administrada NA por vía subcutánea (101).
Pero la médula adrenal también modula la sensación dolorosa y, además, lo hace de forma independiente del sistema simpático neural. La médula adrenal parece modular la sensación táctil en la piel en condiciones normales (ausencia de estrés) a tenor del aumento de la percepción de estímulos mecánicos en ratas a las que se ha practicado una vaguectomía subdiafragmática, ya que ese efecto desaparece tras la realización de una adrenomedulectomía bilateral o la sección de los nervios esplácnicos (102). La contribución de la médula adrenal parece depender de la liberación de A, dado que la administración de esta hormona a animales adrenomedulectomizados consigue recuperar la sensibilidad mecánica aumentada (103, 104). La modulación de la sensibilidad dolorosa por la médula adrenal se ha evidenciado también en ratas sometidas a estrés crónico mediante la aplicación de estímulos sonoros de frecuencia variable (11-19 kHz) durante 30 min a lo largo de 4 días. Este procedimiento potencia la hiperalgesia mecánica cutánea y músculoesquelética inducida por la inyección local de bradicinina, prostaglandina E2 (PGE2) o A, sustancias todas ellas con efecto proinflamatorio y proalgésico (105, 106, 107). Este fenómeno se acompaña de un incremento sostenido de los niveles sanguíneos de A y corticosterona, lo que supone la activación conjunta del eje HHA y del sistema simpático adrenal. Ambas hormonas son necesarias para la potenciación de la hiperalgesia puesto que tanto la administración de antagonistas del receptor de glucocorticoides (mifepristona) como la adrenomedulectomía revierten dicha potenciación. Cabe señalar que los efectos de la A estarían mediados por receptores adrenérgicos b2, probablemente localizados en las terminaciones periféricas de las neuronas nociceptivas primarias (nociceptores), ya que pueden bloquearse por la administración de antagonistas b adrenérgicos (propranolol) y reproducirse con fármacos agonistas de los mismos (isoproterenol, salbutamol) (108). Es de destacar que los glucocorticoides aumentan la expresión de estos receptores, lo que explicaría el carácter permisivo de estas hormonas en la potenciación de la hiperalgesia (109). Sin embargo, la potenciación conllevaría no solo un aumento de la expresión de los receptores adrenérgicos β2 sino también un cambio en su mecanismo de transducción, consistente en un desplazamiento del acoplamiento a la proteína Gs en favor de la Gi/o y la activación de la proteína quinasa Ce (110, 106). Curiosamente, los efectos de la A sobre la sensación dolorosa parecen tener dimorfismo sexual en las ratas, ya que son mas acusados en los machos que en las hembras. Ello parece deberse a la acción de los estrógenos sobre la médula adrenal en las hembras, lo que se traduciría en un aumento de la liberación de A y la desensibilización de los receptores adrenérgicos β2 de los nociceptores (111, 112). Los resultados de estos experimentos en animales de experimentación tendrían valor traslacional en humanos, ya que se han relacionado con la capacidad del estrés para agravar cuadros de dolor generalizado como la artritis reumatoide o el síndrome del intestino irritable, en los que se ha descrito un aumento de la producción de diversas interleucinas y PGE2 (113, 114), cuyos efectos algésicos podrían potenciarse por la A y corticosteroides circulantes. La eficacia analgésica de los bloqueantes β adrenérgicos y la capacidad de los corticosteriodes para agravar la sintomatología dolorosa en algunos de estos pacientes otorgarían plausibilidad a esta hipótesis (115, 116, 117).
La médula adrenal contribuiría también de forma importante a modular la sensación dolorosa en un modelo experimental de dolor neuropático, como es el producido por la constricción crónica del nervio ciático de la rata (118). Dicho modelo comporta la aparición de alodinia térmica y mecánica a la estimulación de la superficie plantar de la extremidad afecta. En la médula adrenal de estos animales se producen muchas de las adaptaciones previamente identificadas en un modelo de estrés crónico por frío (5 días a 4 ºC): aumento de la inervación colinérgica con incremento de la frecuencia de las corrientes sinápticas espontáneas de tipo nicotínico, aumento de la expresión de receptores nicotínicos a9 y aumento de la exocitosis de catecolaminas en respuesta a la entrada de Ca2+ a través de canales Cav (119) (Figura 1). Ello se traduce en un aumento de la A circulante y, en particular, del cociente entre la A y la NA, probablemente como consecuencia del incremento de la expresión y actividad de la PNMT en las células cromafines (120). La implicación de la médula adrenal en la fenomenología dolorosa se evidencia por la atenuación de la alodinia mecánica tras la esplacnectomía y la reversión de la misma por un inhibidor de la PNMT, el SKF29661. El efecto de SKF29661 (300 mg/kg, IP) sobre la alodinia mecánica es de rápida instauración (30 min) y no manifiesta tolerancia con la administración repetida. Dado que este fármaco carece de efecto sobre las conductancias iónicas implicadas en el acoplamiento excitación-secreción, su acción antialodínica sería probablemente debida a la inhibición de la síntesis de A y la disminución del cociente entre la A y la NA circulantes (120). Cabe señalar que en este modelo experimental se desarrolla un tono algésico catecolaminérgico dependiente de la activación de receptores adrenérgicos α y β, probablemente localizados en las terminaciones periféricas de las neuronas nociceptivas primarias y que se vería disminuido tras el tratamiento con SKF29661.
Debe destacarse que en las células cromafines de los animales con dolor neuropático se producen modificaciones en la regulación autocrina y paracrina de la liberación de catecolaminas. Al igual que en los animales SHR, en este modelo experimental de dolor neuropático no se observa una disminución la capacidad de los agonistas a2 adrenérgicos para inhibir la entrada de Ca2+ a través de los canales Cav y, consiguientemente, la exocitosis de las vesículas de secreción, lo que descartaría un proceso de desensibilización de los receptores adrenérgicos α2. Sin embargo, se ha constatado la expresión de receptores purinérgicos ionotrópicos P2X3, P2X7 y de canales TRPV1 (transient receptor potential Vanilloid1 channel) (119) (Figura 1). Los dos primeros son canales activados por ligando, que mediarían una despolarización de la membrana celular al combinarse con ATP y diversos dinucleótidos polifosfato almacenados y liberados en las vesículas de secreción juntamente con las catecolaminas (121, 122, 123, 124). El incremento de estos receptores contrarrestaría el efecto inhibidor de los receptores purinérgicos P2Y sobre los canales Cav y la exocitosis, poniendo en marcha un proceso de realimentación positiva de la liberación del contenido de los gránulos cromafines que probablemente contribuye al tono algésico catecolaminérgico observado en estos animales (125). Los canales TRPV1 son receptores polimodales activados por la capsaicina, el pH ácido, el calor nocivo y la estimulación mecánica (126). La expresión de receptores P2X3 y del receptor polimodal TRPV1 en las células cromafines se produce también en las neuronas nociceptivas primarias del nervio ciático lesionado en los animales con dolor neuropático (127, 128).
Ello refleja su común origen embrionario y probablemente la capacidad de las células cromafines para la transducción sensorial. A este respecto, debe mencionarse la liberación de catecolaminas por las células cromafines que tiene lugar durante el momento del parto en respuesta a la hipoxia, y que resulta fundamental para la adaptación cardiovascular de los recién nacidos (129, 23). Desconocemos, no obstante, la función de los receptores TRPV1 de las células cromafines en los animales con dolor neuropático.
7. CONCLUSIONES
En las anteriores páginas hemos pasado revista a las adaptaciones de la médula adrenal en situación de estrés agudo y crónico, y a la participación de esta glándula doblemente interna, tanto por su ubicación como por su secreción, en algunas condiciones patológicas. Los resultados presentados abundan en la idea de que la médula adrenal desempeña un papel propio en el funcionamiento del sistema nervioso simpático, bien diferenciado del del sistema simpático neural. Durante la ontogénesis las células cromafines terminan alejándose de sus parientes cercanos, las neuronas simpáticas posganglionares, convirtiéndose bajo la influencia de los glucocorticoides en células neuroendocrinas, que vierten al torrente sanguíneo sus productos de secreción. La cercanía física con las células adrenocorticales facilita también la integración funcional del eje HHA y del sistema simpático adrenal y un efecto sinérgico particularmente en situaciones de estrés crónico. En la actualidad contamos con diversas opciones farmacológicas para actuar sobre la corteza adrenal y el sistema simpático neural. Las posibilidades de intervenir sobre la médula adrenal son mucho mas limitadas y prácticamente se reducen a los inhibidores de la PNMT. A la vista de las evidencias revisadas en este trabajo, sería conveniente prestar atención a otras particularidades farmacológicas de las células cromafines al objeto de desarrollar compuestos selectivos para el sistema simpático adrenal (antagonistas no peptídicos de los receptores nicotínicos α9, moduladores de la interacción entre los receptores nicotínicos α7 y α3β4, agonistas sesgados de los receptores adrenérgicos α2, etc.) potencialmente útiles en el número creciente de patologías en las que parece estar implicado.
8. ABREVIATURAS
A: adrenalina
BK: canales de K+ dependientes de Ca2+ de elevada conductancia iónica
Cav: canales de Ca2+ dependientes de voltaje
Egr-1: early growth response-1
ERK: extracellular signal-regulated kinase
fEPSP: potencial sináptico excitador rápido
GRK2: proteína quinasa 2 de los receptores acoplados a proteínas G
HHA: hipotálamo-hipófisis-corteza adrenal
HTA: hipertensión arterial
IC: insuficiencia cardiaca
Kv: canales de K+ dependientes de voltaje
NA: noradrenalina
PACAP: polipéptido activador de la adenilato ciclasa hipofisaria
PGE2: prostaglandina E2
PKA: proteína quinasa A
PKC: proteína quinasa C
PNMT: feniletanolamina N-metil transferasa
RAA: renina-angiotensina-aldosterona
SHR: ratas espontáneamente hipertensas
SK: canales de K+ dependientes de Ca2+ de pequeña conductancia iónica (SK).
TH: tiroxina hidroxilasa
TRPV1: transient receptor potential vanilloid1 channel
WKY: ratas Wistar Kyoto
Agradecimientos
CLS está financiada por un contrato predoctoral de la Universidad Complutense de Madrid (UCM). La investigación del grupo al que pertenecen los autores está financiada por el Ministerio de Ciencia e Innovación (PID2019-109155RB-I00 y la UCM (FEI20/35).
9. REFERENCIAS
- McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med 1993; 153: 2093-101.
- Cannon WG, De LaPaz D. Emotional stimulation of adrenal secretion. Am J Physiol 1911; 28: 64-70.
- Bao X, Lu CM, Liu F, et al. Epinephrine is required for normal cardiovascular responses to stress in the phenylethanolamine N-methyltransferase knockout mouse. Circulation 2007; 116: 1024-31.
- Mendes P, Martinho R, Leite S, et al. Chronic exercise induces pathological left ventricular hypertrophy in adrenaline-deficient mice. Int J Cardiol 2018; 253: 113-119.
- Pacak K, Palkovits M, Yadid G, et al. Heterogeneous neurochemical responses to different stressors: a test of Selye’s doctrine of nonspecificity. Am J Physiol 1998; 275: R1247-55.
- Carter JR, Goldstein DS. Sympathoneural and adrenomedullary responses to mental stress. Compr Physiol 2015; 5: 119-46.
- Goldstein DS. Adrenal responses to stress. Cell Mol Neurobiol 2010; 30: 1433-40.
- Sala F, Nistri A, Criado M. Nicotinic acetylcholine receptors of adrenal chromaffin cells. Acta Physiol (Oxf) 2008; 192: 203-12.
- Olivos L, Artalejo AR. Muscarinic excitation–secretion coupling in chromaffin cells. Acta Physiol (Oxf). 2008; 192: 213-20.
- Duan K, Yu X, Zhang C, Zhou Z. Control of secretion by temporal patterns of action potentials in adrenal chromaffin cells. J Neurosci 2003; 23: 11235-43.
- Garcia AG, Garcia-De-Diego AM, Gandia L, et al. Calcium signaling and exocytosis in adrenal chromaffin cells. Physiol Rev 2006; 86: 1093-1131.
- Artalejo AR. Canales iónicos y estrés: el caso de la médula adrenal. En: Simposio Homenaje al Profesor Antonio G. García. Fundación Teófilo Hernando 2016; pp. 126-137.
- Carbone E, Borges R, Eiden LE, et al. Chromaffin Cells of the Adrenal Medulla: Physiology, Pharmacology, and Disease. Compr Physiol. 2019; 9: 1443-1502.
- Artalejo, AR. Electrical properties of adrenal chromaffin cells. En: The Electrophysiology of Neuroendocrine Cells. Hescheler, J. and Scherübl, H. (eds.). CRC PRESS, Boca Raton, Florida, 1995; pp. 259-299.
- Bustillo, D. Caracterización farmacológica y funcional de los receptores nicotínicos de las células cromafines de la médula adrenal de la rata: plasticidad inducida por un modelo de estrés crónico. Universidad Complutense de Madrid 2015.
- Hone AJ, Rueda-Ruzafa L, Gordon TJ, et al. Expression of α3β2β4 nicotinic acetylcholine receptors by rat adrenal chromaffin cells determined using novel conopeptide antagonists. J Neurochem 2020; 154: 158-176.
- Jiménez-Pompa A, Sanz-Lázaro S, Omodolor RE, et al. Cross Talk between a7 and a3b4 Nicotinic Receptors Prevents Their Desensitization in Human Chromaffin Cells. J Neurosci 2022; 42: 1173-1183.
- Colomer C, Olivos-Ore LA, Vincent A, et al. Functional characterization of alpha9-containing cholinergic nicotinic receptors in the rat adrenal medulla: implication in stress-induced functional plasticity. J Neurosci 2010; 30: 6732-42.
- Guérineau NC, Desarménien MG, Carabelli V, et al. Functional chromaffin cell plasticity in response to stress: focus on nicotinic, gap junction, and voltage-gated Ca2+ channels. J Mol Neurosci 2012; 48: 368-86.
- Cárdenas AM, Marengo FD. How the stimulus defines the dynamics of vesicle pool recruitment, fusion mode, and vesicle recycling in neuroendocrine cells. J Neurochem 2016; 137:867-79.
- Mahapatra S, Calorio C, Vandael DH, et al. Calcium channel types contributing to chromaffin cell excitability, exocytosis and endocytosis. Cell Calcium 2012; 51: 321-30.
- Hill J, Chan SA, Kuri B, et al. Pituitary adenylate cyclase-activating peptide (PACAP) recruits low voltage-activated T-type calcium influx under acute sympathetic stimulation in mouse
- Carabelli V, Marcantoni A, Comunanza V, et al. Chronic hypoxia up-regulates alpha1H T-type channels and low threshold catecholamine secretion in rat chromaffin cells. J Physiol 2007; 584:149-165.
- Smith CB, Eiden LE. Is PACAP the major neurotransmitter for stress transduction at the adrenomedullary synapse? J Mol Neurosci 2012; 48: 403-12.
- Carabelli V, Giancippoli A, Baldelli P, et al. Distinct potentiation of L-type currents and secretion by cAMP in rat chromaffin cells. Biophys J 2003; 85: 1326–37.
- Lovell PV, McCobb DP. Pituitary control of BK potassium channel function and intrinsic firing properties of adrenal chromaffin cells. J Neurosci 2001; 21: 3429-42.
- Chatterjee O1, Taylor LA, Ahmed S, et al. Social stress alters expression of large conductance calcium-activated potassium channel subunits in mouse adrenal medulla and pituitary glands. J Neuroendocrinol 2009; 2: 167-76.
- Duncan RR, Greaves J, Wiegand, UK, et al. Functional and spatial segregation of secretory vesicle pools according to vesicle age. Nature 2003; 422: 176-80.
- Estévez-Herrera J, Domínguez N, Pardo MR, et al. ATP: The crucial component of secretory vesicles. Proc Natl Acad Sci U S A 2016; 113: E4098-106.
- Stachowiak MK, Goc A, Hong JS, et al. Neural and hormonal regulation of the tyrosine hydroxylase gene in adrenal medullary cells: Participation of c-fos and AP1 factors. Mol Cell Neurosci 1990; 1: 202-13.
- Zigmond RE, Schwarzschild MA, Rittenhouse AR. Acute regulation of tyrosine hydroxylase by nerve activity and by neurotransmitters via phosphorylation. Annu Rev Neurosci 1989;12: 415-61
- Sabban EL, Kvetnanský R. Stress-triggered activation of gene expression in catecholaminergic systems: dynamics of transcriptional events. Trends Neurosci 2001; 24: 91-8.
- Dunkley PR, Dickson PW. Tyrosine hydroxylase phosphorylation in vivo. J Neurochem 2019; 149: 706-728.
- Kvetnansky R, Weise VK, Kopin IJ. Elevation of adrenal tyrosine hydroxylase and phenylethanolamine-N-methyl transferase by repeated immobilization of rats. Endocrinology 1970; 87: 744-9.
- Mishra RR, Adhikary G, Simonson MS, et al. Role of c-fos in hypoxia-induced AP-1 cis-element activity and tyrosine hydroxylase gene expression. Brain Res Mol Brain Res 1998; 59: 74-83.
- Rusnák M, Jeloková J, Vietor I, et al. Different effects of insulin and 2-deoxy-D-glucose administration on tyrosine hydroxylase gene expression in the locus coeruleus and the adrenal medulla in rats. Brain Res Bull 1998; 46: 447-52.
- Udenfriend S, Zalzman-Nirenberg P, et al. Inhibitors of purified beef adrenal tyrosine hydroxylase. Biochem Pharmacol 1965; 14: 837-845.
- Quinsey NS, Lenaghan CM, Dickson PW. Identification of Gln313 and Pro327 as residues critical for substrate inhibition in tyrosine hydroxylase. J Neurochem 1996; 66: 908-14.
- Kvetnansky R, Micutkova L, Kubovcakova L, et al. Localization and regulation of phenylethanolamine N-methyltransferase gene expression in the heart of rats and mice during stress. Ann N Y Acad Sci 2004; 1018:405-17.
- Wong DL. Epinephrine biosynthesis: hormonal and neural control during stress. Cell Mol Neurobiol 2006; 26: 891-900.
- Tai TC, Wong DL. Protein kinase A and protein kinase C signaling pathway interaction in phenylethanolamine N-methyltransferase gene regulation. J Neurochem 2003; 85: 816-29.
- Wong DL, Tai TC. Neural mechanisms regulating phenylethanolamine N-methyltransferase gene expression. En: Nagatsu T, Nabeshima T, McCarty R, Goldstein DS, Eds. Catecholamine Research: From Molecular Insights to Clinical Medicine. Kluwer Academic, New York, 2002; pp. 135-138.
- Tai TC, Claycomb R, Siddall BJ, et al. Stress-induced changes in epinephrine expression in the adrenal medulla in vivo. J Neurochem 2007; 101: 1108-18.
- Goldstein DS, Kopin IJ. Adrenomedullary, adrenocortical, and sympathoneural responses to stressors: a meta-analysis. Endocr Regul 2008; 42: 111-9.
- Kvetnansky R, Micutkova L, Rychkova N, et al. Quantitative evaluation of catecholamine enzymes gene expression in adrenal medulla and sympathetic ganglia of stressed rats. Ann N Y Acad Sci 2004a; 1018: 356-69.
- McCarty R, Horwatt K, Konarska M. Chronic stress and sympathetic-adrenal medullary responsiveness. Soc Sci Med 1988; 26: 333-341.
- Gavrilovic L, Spasojevic N, Tanic N, et al. Chronic isolation of adult rats decreases gene expression of catecholamine biosynthetic enzymes in adrenal medulla. Neuro Endocrinol Lett 2008; 29: 1015-1020.
- Haavik J, Blau N, Thöny B. Mutations in human monoamine-related neurotransmitter pathway genes. Hum Mutat 2008; 29: 891-902.
- Nagatsu T, Nagatsu I. Tyrosine hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other catecholamine-related enzymes, and their human genes in relation to the drug and gene therapies of Parkinson’s disease (PD): historical overview and future prospects. J Neural Transm (Vienna) 2016; 123: 1255-1278.
- Langer SZ. Presynaptic regulation of the release of catecholamines. Pharmacol Rev 1980; 32(4): 337-62.
- Artalejo AR, Olivos-Oré LA. Alpha2-adrenoceptors in adrenomedullary chromaffin cells: functional role and pathophysiological implications. Pflugers Arch 2018; 470: 61-66.
Starke K. Presynaptic autoreceptors in the third decade: focus on alpha2-adrenoceptors. J Neurochem 2001; 78: 685-93. - Kleppisch T, Ahnert-Hilger G, Gollasch M, et al. Inhibition of voltage-dependent Ca2+ channels via alpha 2-adrenergic and opioid receptors in cultured bovine adrenal chromaffin cells. Pflugers Arch 1992; 421:131-137.
- Hernández-Guijo JM, Carabelli V, Gandía L, et al. Voltage-independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. Eur J Neurosci 1999; 11:3574-584.
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab 2006; 17: 159-165
- Schneider J, Lother A, Hein L, et al. Chronic cardiac pressure overload induces adrenal medulla hypertrophy and increased catecholamine synthesis. Basic Res Cardiol 2011; 106:591-602.
Schneider J, Lother A, Hein L, Gilsbach R. Chronic cardiac pressure overload induces adrenal medulla hypertrophy and increased catecholamine synthesis. Basic Res Cardiol 2011; 106: 591-602. - Jafferjee M, Reyes Valero T, et al. GRK2 up-regulation creates a positive feedback loop for catecholamine production in chromaffin cells. Mol Endocrinol 2016; 30: 372-381
- Cannavo A, Liccardo D, Lymperopoulos A, et al. GRK2 Regulates a2-Adrenergic Receptor-Dependent Catecholamine Release in Human Adrenal Chromaffin Cells. J Am Coll Cardiol 2017; 69: 1515-1517.
- Lymperopoulos A, Rengo G, Funakoshi H, et al. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med 2007a; 13: 315-323.
- Borges JI, Ferraino KE, Cora N, et al. Adrenal G Protein-Coupled Receptors and the Failing Heart: A Long-distance, Yet Intimate Affair. J Cardiovasc Pharmacol 2022; 80:386-392.
- Lymperopoulos A, Rengo G, Koch WJ. Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol Med 2007b; 13: 503-511.
- Lymperopoulos A, Rengo G, Zincarelli C, et al. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther 2008; 16: 302-307.
- Lymperopoulos A, Rengo G, Gao E, et al. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J Biol Chem 2010; 285: 16378-16386.
- Rengo G, Lymperopoulos A, Zincarelli C, et al. Blockade of beta-adrenoceptors restores the GRK2-mediated adrenal alpha(2)- adrenoceptor-catecholamine production axis in heart failure. Br J Pharmacol 2012; 166: 2430-2440.
- Fink EA, Xu J, Hübner H, et al. Structure-based discovery of nonopioid analgesics acting through the 2A-adrenergic receptor. Science 2022; 377: 1509.
- Grassi G, Mark A, Esler M. The sympathetic nervous system alterations in human hypertension. Circ Res 2015;116: 976-90.
- Floras JS. Epinephrine and the genesis of hypertension. Hypertension 1992; 19: 1-18.
- Anderson EA, Sinkey CA, Lawton WJ, et al. Elevated sympathetic nerve activity in borderline hypertensive humans. Evidence from direct intraneural recordings. Hypertension 1989; 14: 177-83.
- Yagil Y, Yagil C. Genetic models of hypertension in experimental animals. Exp Nephrol 2001; 9: 1-9.
- Pintérová M, Kuneš J, Zicha J. Altered neural and vascular mechanisms in hypertension. Physiol Res 2011; 60: 381-402.
- Judy WV, Farrell SK. Arterial baroreceptor reflex control of sympathetic nerve activity in the spontaneously hypertensive rat. Hypertension 1979; 1: 605-614.
- Lee RM, Triggle CR, Cheung DW, et al. Structural and functional consequence of neonatal sympathectomy on the blood vessels of spontaneously hypertensive rats. Hypertension 1987; 10: 328-338.
- Korner P, Bobik A, Oddie C, et al. Sympathoadrenal system is critical for structural changes in genetic hypertension. Hypertension 1993; 22: 243-252.
- Vavřínová A, Behuliak M, Bencze M, et al. Sympathectomyinduced blood pressure reduction in adult normotensive and hypertensive rats is counteracted by enhanced cardiovascular sensitivity to vasoconstrictors. Hypertens Res 2019; 42: 1872-1882.
- Vlachakis ND, Alexander N, Maronde RF. Increased plasma normetanephrine in spontaneously hypertensive rats. Clin Exp Hypertens 1980; 2: 309-319.
- Borkowski KR. Effect of adrenal demedullation and adrenaline on hypertension development and vascular reactivity in young spontaneously hypertensive rats. J Auton Pharmacol 1991; 11: 1-14.
- Lee RM, Borkowski KR, Leenen FH, et al. Combined effect of neonatal sympathectomy and adrenal demedullation on blood pressure and vascular changes in spontaneously hypertensive rats. Circ Res 1991; 69: 714-721.
- Vavřínová A, Behuliak M, Vaněčková I, et al. The abnormalities of adrenomedullary hormonal system in genetic hypertension: Their contribution to altered regulation of blood pressure. Physiol Res. 2021; 70: 307-326.
- Lim DY, Jang SJ, Park DG. Comparison of catecholamine release in the isolated adrenal glands of SHR and WKY rats. Auton Autacoid Pharmacol 2002; 22: 225-232.
- Moura E, Pinto CE, Caló A, et al. α₂-Adrenoceptor-mediated inhibition of catecholamine release from the adrenal medulla of spontaneously hypertensive rats is preserved in the early stages of hypertension. Basic Clin Pharmacol Toxicol 2011; 109: 253-60.
- Miranda-Ferreira R, De Pascual R, De Diego AM, et al. Single-vesicle catecholamine release has greater quantal content and faster kinetics in chromaffin cells from hypertensive, as compared with normotensive, rats. J Pharmacol Exp Ther 2008; 324: 685-693.
- Segura-Chama P, López-Bistrain P, Pérez-Armendáriz EM, et al. Enhanced Ca(2+)-induced Ca(2+) release from intracellular stores contributes to catecholamine hypersecretion in adrenal chromaffin cells from spontaneously hypertensive rats. Pflugers Arch 2015; 467: 2307-23.
- Tabei R, Fujiwara T, Kondo M, et al. Morphological studies on the paraneuron in spontaneously hypertensive rats. Clin Exp Hypertens A 1988;10 Suppl 1: 235-47.
- Musial DC, Bomfim GH, Arranz-Tagarro JA, et al. Altered mitochondrial function, calcium signaling, and catecholamine release in chromaffin cells of diabetic and SHR rats. Eur J Pharmacol 2017; 815: 416-426.
- Peña Del Castillo JG, Segura-Chama P, Rincón-Heredia R, et al. Development of the hypersecretory phenotype in the population of adrenal chromaffin cells from prehypertensive SHRs. Pflugers Arch 2021; 473: 1775-1793.
- Kumai T, Tanaka M, Watanabe M, et al. Elevated tyrosine hydroxylase mRNA levels in the adrenal medulla of spontaneously hypertensive rats. Jpn J Pharmacol 194; 65: 367-369.
- Reja V, Goodchild AK, Phillips JK, et al. Tyrosine hydroxylase gene expression in ventrolateral medulla oblongata of WKY and SHR: a quantitative real-time polymerase chain reaction study. Auton Neurosci 2002; 98: 79-84.
- Moura E, Pinho Costa PM, et al. Decreased tyrosine hydroxylase activity in the adrenals of spontaneously hypertensive rats. Life Sci 2005; 76: 2953-2964.
- Grundt A, Grundt C, Gorbey S, et al. Strain-dependent differences of restraint stress induced hypertension in WKY and SHR. Physiol Behav 2009; 97: 341-346.
- Vavřínová A, Behuliak M, Bencze M, et al. Which sympathoadrenal abnormalities of adult spontaneously hypertensive rats can be traced to a prehypertensive stage? Hypertens Res 2019 42: 949-959.
- Miranda-Ferreira R, de Pascual R, Caricati-Neto A, et al. Role of the endoplasmic reticulum and mitochondria on quantal catecholamine release from chromaffin cells of control and hypertensive rats. J Pharmacol Exp Ther. 2009; 329: 231-40.
- Miranda-Ferreira R, de Pascual R, Smaili SS, et al. Greater cytosolic and mitochondrial calcium transients in adrenal medullary slices of hypertensive, compared with normotensive rats. Eur J Pharmacol 2010; 636: 126-36.
- Bomfim GHS, Méndez-López I, Fernández-Morales JC, et al. Electrophysiological properties and augmented catecholamine release from chromaffin cells of WKY and SHR rats contributing to the hypertension development elicited by chronic EtOH consumption. Eur J Pharmacol 2017; 803: 65-77.
- Fhaner MJ, Galligan, JJ, Swain GM. Increased catecholamine secretion from single adrenal chromaffin cells in DOCA-salt hypertension is associated with potassium channel dysfunction. ACS Chem Neurosci 2013; 4: 1404-13.
- Cohen SP, Vase L, Hooten, WM. Chronic pain: an update on burden, best practices, and new advances. Lance. 2021; 397: 2082-2097.
- Chen J, Abbod M, Shieh, JS. Pain and Stress Detection Using Wearable Sensors and Devices-A Review. Sensors (Basel); 2021; 21:1030.
- Ferdousi M, Finn DP. Stress-induced modulation of pain: Role of the endogenous opioid system. Prog Brain Res 2018; 239: 121-177.
- Caraci F, Merlo S, Drago F, et al. Rescue of Noradrenergic System as a Novel Pharmacological Strategy in the Treatment of Chronic Pain: Focus on Microglia Activation. Front Pharmacol 2019;10: 1024.
- Imbe H, Iwai-Liao Y, Senba E. Stress-induced hyperalgesia: animal models and putative mechanisms. Front Biosci 2006; 11: 2179-92.
- Jänig. Pain and the Sympathetic Nervous System: pathophysiological mechanisms. En: Autonomic Failure: A Textbook of Clinical Disorders of the Autonomic Nervous System (5 edn). Christopher J. Mathias (ed.), Sir Roger Bannister (ed.). Oxford University Press 2013; pg. 236- 246.
- Taylor SS, Noor N, Urits I, et al. Complex Regional Pain Syndrome: A Comprehensive Review. Pain Ther 2021; 10: 875-892.
- Jänig W, Levine JD, Michaelis M. Interactions of sympathetic and primary afferent neurons following nerve injury and tissue trauma. Prog Brain Res 1996; 113: 161-84.
- Khasar SG, Miao JP, Jänig W, et al. Modulation of bradykinin-induced mechanical hyperalgesia in the rat by activity in abdominal vagal afferents. Eur J Neurosci 1998; 10: 435-44.
- Khasar SG, Miao FJ, Jänig W, et al. Vagotomy-induced enhancement of mechanical hyperalgesia in the rat is sympathoadrenal-mediated. J Neurosci 1998;18: 3043-9.
- Jänig W, Khasar SG, Levine JD, et al. The role of vagal visceral afferents in the control of nociception. Prog Brain Res 2000 ;122: 273-87.
- Khasar SG, Green PG, Levine JD. Repeated sound stress enhances inflammatory pain in the rat. Pain 2005; 116: 79-86.
- Khasar SG, Burkham J, Dina OA, et al. Stress induces a switch of intracellular signaling in sensory neurons in a model of generalized pain. J Neurosci 2008; 28: 5721-30.
- Khasar SG, Dina OA, Green PG, et al. Sound stress-induced long-term enhancement of mechanical hyperalgesia in rats is maintained by sympathoadrenal catecholamines. J Pain. 2009; 10: 1073-7.
- Khasar SG, Green PG, Miao FJ, et al. Vagal modulation of nociception is mediated by adrenomedullary epinephrine in the rat. Eur J Neurosci 2003; 17: 909-15.
- Fraser CM, Venter JC. The synthesis of beta-adrenergic receptors in cultured human lung cells: induction by glucocorticoids. Biochem Biophys Res Commun 2006; 94: 390 -397.
- Pavoine C, Behforouz N, Gauthier C, et al. beta2-Adrenergic signaling in human heart: shift from the cyclic AMP to the arachidonic acid pathway. Mol Pharmacol 2003; 64: 1117–1125.
- Dina OA, Aley KO, Isenberg W, et al. Sex hormones regulate the contribution of PKCepsilon and PKA signalling in inflammatory pain in the rat. Eur J Neurosci 2001; 13: 2227-33.
- Khasar SG, Dina OA, Green PG, et al. Estrogen regulates adrenal medullary function producing sexual dimorphism in nociceptive threshold and beta-adrenergic receptor-mediated hyperalgesia in the rat. Eur J Neurosci 2005; 21: 3379-86.
- Raddatz D, Bockemuhl M, Ramadori G. Quantitative measurement of cytokine mRNA in inflammatory bowel disease: relation to clinical and endoscopic activity and outcome. Eur J Gastroenterol Hepatol 2005; 17: 547–557.
- Petrovic-Rackov L, Pejnovic N. Clinical significance of IL-18, IL-15, IL-12 and TNF-alpha measurement in rheumatoid arthritis. Clin Rheumatol 2006; 25: 448–452
- Wollstein R, Chaimsky G, Carlson L, et al. Evaluating short-term pain after steroid injection. Am J Orthop 2007; 36:128 -131.
- Tchivileva IE, Hadgraft H, Lim PF, et al. Efficacy and safety of propranolol for treatment of temporomandibular disorder pain: a randomized, placebo-controlled clinical trial. Pain 2020; 161: 1755-1767.
- Nakafero G, Grainge MJ, Valdes AM, et al. b-blocker prescription is associated with lower cumulative risk of knee osteoarthritis and knee pain consultations in primary care: a propensity score-matched cohort study. Rheumatology (Oxford). 2021; 60: 5686-5696.
- Bennett GJ, Xie Y-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87-107.
- Arribas-Blázquez M, Olivos-Oré LA, Barahona MV, et al. Overexpression of P2X3 and P2X7 Receptors and TRPV1 Channels in Adrenomedullary Chromaffin Cells in a Rat Model of Neuropathic Pain. Int J Mol Sci 2019; 20: 155.
- Arribas-Blázquez M, Olivos-Oré LA, Barahona MV, et al. The Adrenal Medulla Modulates Mechanical Allodynia in a Rat Model of Neuropathic Pain. Int J Mol Sci 2020; 21: 8325.
- Rodriguez del Castillo A, Torres M, Delicado EG, et al. Subcellular distribution studies of diadenosine polyphosphates–Ap4A and Ap5A–in bovine adrenal medulla: presence in chromaffin granules. J Neurochem 1988; 51: 1696-703.
- Castillo CJ, Moro MA, Del Valle M, et al. Diadenosine tetraphosphate is co-released with ATP and catecholamines from bovine adrenal medulla. J Neurochem 1992; 59: 723-32.
- North RA. Molecular physiology of P2X receptors. Physiol Rev 2002; 82:1013-67.
- Salas E, Carrasquero LM, Olivos-Oré LA, et al. Purinergic P2X7 receptors mediate cell death in mouse cerebellar astrocytes in culture. J Pharmacol Exp Ther 2013; 347: 802-15.
- Ulate G, Scott SR, González J, et al. Extracellular ATP regulates exocytosis in inhibiting multiple Ca(2+) channel types in bovine chromaffin cells. Pflugers Arch 2000; 439: 304-14.
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci 2001; 24: 487-517.
- Wilson-Gerwing TD, Dmyterko MV, Zochodne DW, et al. Neurotrophin-3 suppresses thermal hyperalgesia associated with neuropathic pain and attenuates transient receptor potential vanilloid receptor-1 expression in adult sensory neurons. J Neurosci 2005; 25: 758-67.
- Xiang Z, Xiong Y, Yan N, et al. Functional up-regulation of P2X 3 receptors in the chronically compressed dorsal root ganglion. Pain. 2008; 140: 23-34.
- Bao G, Metreveli N, Li R, Taylor A, et al. Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol 1997; 83: 95-101.
