1. INTRODUCCIÓN
Benzothiazepines are structures that present a large projection at the pharmacological level. Although there is currently only one benzothiazepine structure marketed as a medicine, there are numerous scientific studies proposing new possible applications of very different benzothiazepine derivatives (1). Recently, five papers reporting the synthesis of 2,4-diaryl-1,5-benzothiazepines have been published (2,3,4,5,6) as well as a review (7). In these papers are reported some structures of representative drugs, in clinical use or undergoing clinical trials (Figure 1). Therefore, it seems appropriate to search for simple, efficient and reproducible synthesis of benzothiazepines.
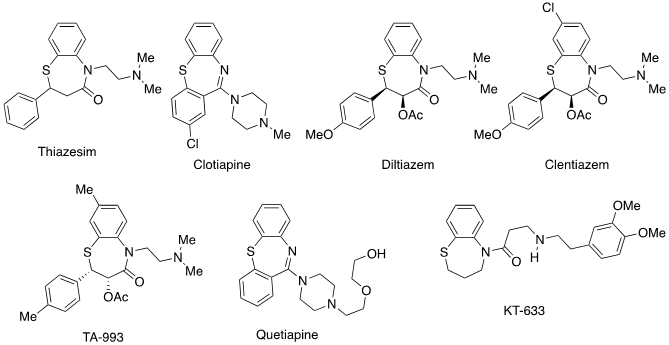
Figure 1. Some structures of representative drugs with a 1,5-benzothiazepine skeleton
On the other hand, curcumin is a widely studied compound, and its accessibility and chemical properties make it an easily manipulated structure, with no health risks and little environmental contamination. Likewise, it has defined and studied pharmacological properties, and obtaining new curcumin derivatives could lead to new structures with the same or other pharmacological properties (8).
The reactivity of curcumin and curcuminoid derivatives with o-aminothiophenol analyzed in this work generates benzothiazepines as final structures. Although reaction of o-aminothiophenol with α,β-ethylenic ketones, in particular chalcones, is much more common than with β-diketones, these last compounds have also been studied (9,10,11,12). The compounds that we are going to study in the present work have both functionalities making them interesting. In addition, the use of different reaction conditions is important when determining which route is best suited for future applications.
The objectives of the present work can be summarized in the following three points:
1. Preparation of the starting curcuminoid derivatives, β-diketones, represented in (Figure 2) as the major existing keto-enol tautomer (13).
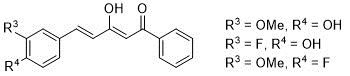
Figure 2. Curcuminoid diketones for use as starting compounds
2. Synthesis of benzothiazepine derivatives by reaction of the β-diketones with o-aminothiophenol. If curcuminoid diketones react as α,β-unsaturated ketones (chalcones) isomers A and B will be formed, but if they do so as 1,3-dicarbonyl compounds pair of isomers C and D could result (Figure 3).
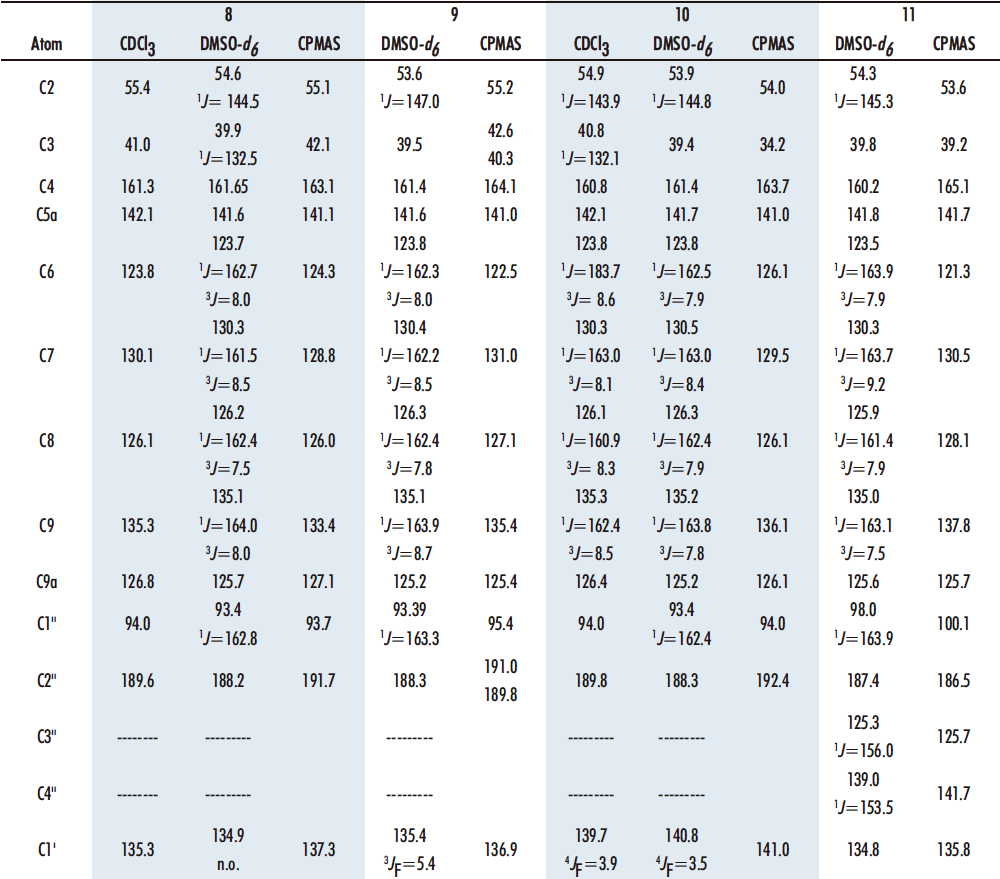
3. Establish the structure of all compounds obtained by 1H, 13C, 15N and 19F NMR in solution, as well as in solid state by 13C and 15N CPMAS.
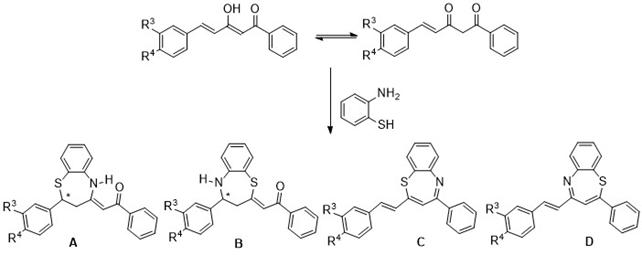
Figure 3. Possible benzothiazepines that can be obtained in the proposed reactions
2. RESULTS AND DISCUSSION
2.1. Synthesis of b-diketones
The synthesis of the starting diketones has been carried out following the Pabon method (14) and the procedure previously used in our research group (13,15,16,17). First, a dicarbonyl compound, in our case 1-phenyl-1,3-butanedione, is complexed with boron oxide. Next, a disubstituted benzaldehyde is added, which reacts with the terminal methyl group of the diketone, and n-butylamine is added dropwise. As a result of the condensation of benzaldehyde and boron complex, water is formed, so n-tributyl borate is used to remove it. Finally, hydrochloric acid is added, thus creating an acid medium in which the boron complex dissociates from the curcuminoid derivatives.
Using 1-phenyl-1,3-butanedione as initial compound and the benzaldehydes selected for this work: 4-hydroxy-3-methoxybenzaldehyde (vanillin) (1), 3-fluoro-4-hydroxybenzaldehyde (2) and 4-fluoro-3-methoxybenzaldehyde (3), curcuminoid diketones (4-6) are obtained with acceptable yields, 50-60% (Scheme 1).
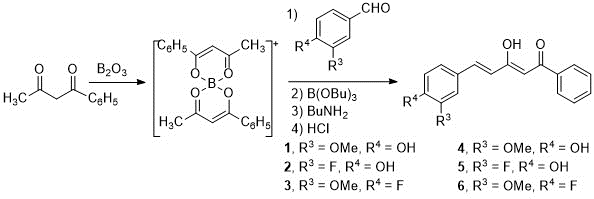
Scheme 1. Synthesis of the starting diketones
2.2. Study of the reactivity of b-diketones with o-aminothiophenol
In previous works carried in out laboratory (18,19), the reaction of curcuminoid diketones with ethylenediamine has shown a double reactivity depending on the reaction medium (Scheme 2), giving rise to different diazepine structures (Scheme 2). In methanol, ethylenediamine attacked the chalcone via a Michael addition forming a diazepine analogue to A (Figure 3) with a C=O group hydrogen bonded to the enamine NH stabilizing the structure. However, when the reaction medium was acetic acid, ethylenediamine attacked both keto carbons, generating another diazepine, related to C (Figure 3) without altering the double bond of the initial diketone.
Therefore, we have analyzed the reactivity of curcumin and curcuminoid diketones with o-aminothiophenol, both in methanol and in acetic acid, to see if similar results will be obtained.
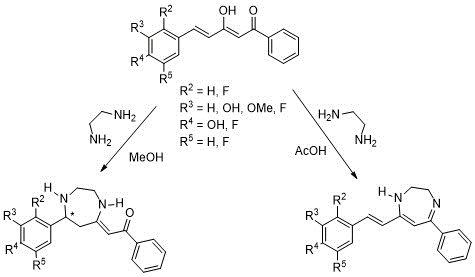
Scheme 2. Reactivity of styrenic diketones with ethylenediamine
2.2.1. Reactivity in methanol
The reaction of diketones 4-6 and curcumin 7 with of o-aminothiophenol, in methanol, leads only to benzothiazepines 8-11 by a Michael-type reaction, in which the SH group attacks to the double bond, accompanied in all cases by the disulfide 12 formed by oxidation of o-aminothiophenol (Scheme 3). Structures 8-10 correspond to type A of Figure 3.
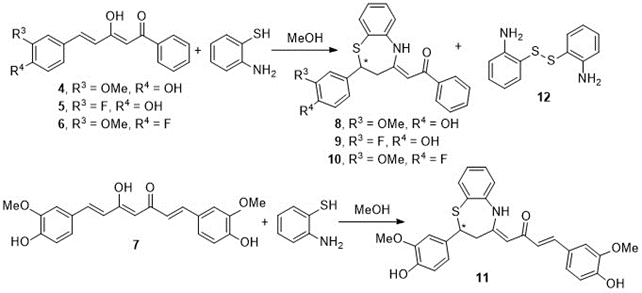
Scheme 3. Benzothiazepines obtained by reaction of curcumin and curcuminoid diketones with o-aminothiophenol in methanol
The evolution of the reactions has been followed by 1H NMR, since in our hands thin layer chromatography has not allowed to discriminate between the diketone and the benzothiazepine, clearly observing the formation of the ABX system of the benzothiazepine ring (∼2.70, ∼2.95 and ∼4.85 ppm), and the presence of a new double bond. As shown in the case of diketone 4 (Figure 4) the enamine double bond appears as a singlet at 5.9-6 ppm, more shielded than that belonging to the enol form of the diketone; at the end of the reaction (six h) the diketone/benzothiazepine ratio is 1/2.5-4.1, which means a yield of 71-80%. This proportion is not modified by leaving the reaction longer or adding more quantity of o-aminothiophenol.
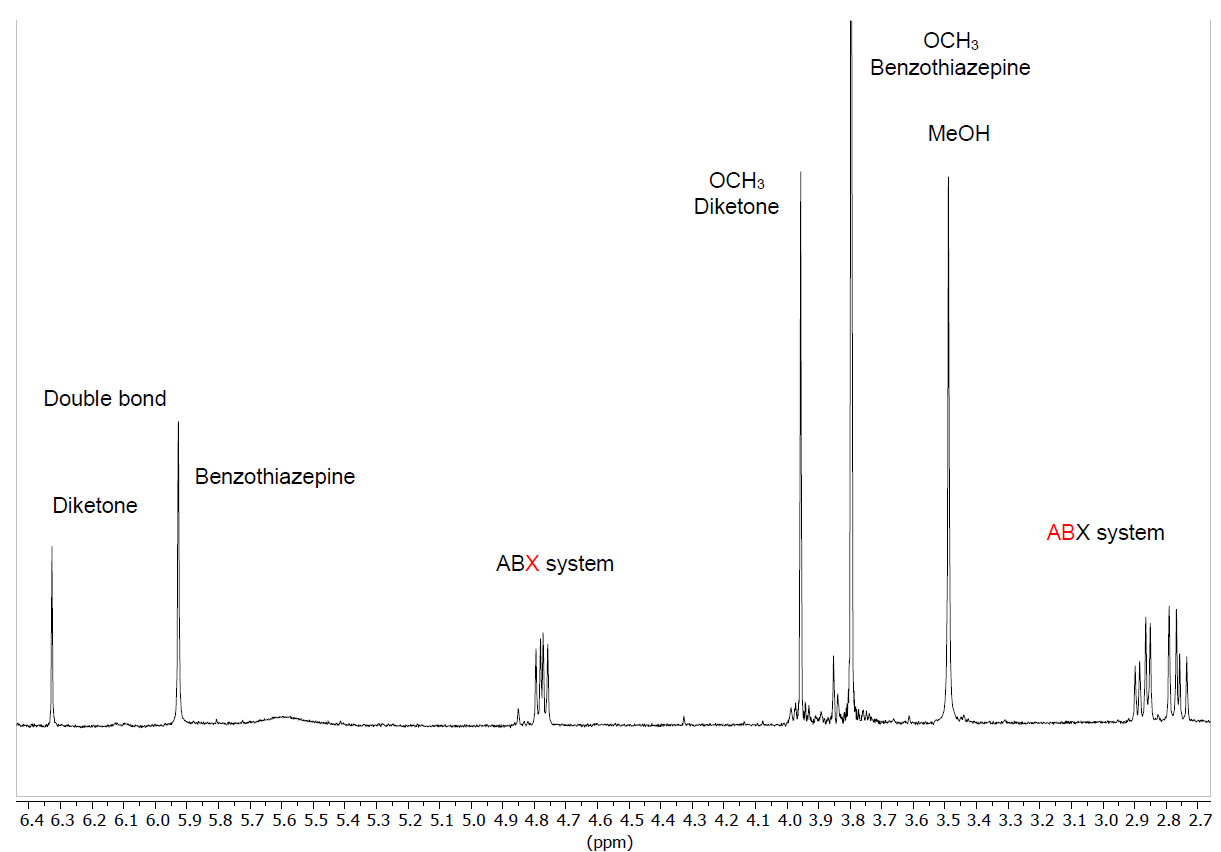
Figure 4. Enlargement of the 1H-NMR spectrum in CDCl3 of the reaction crude of diketone 4 with o-aminothiophenol
The isolation of the products is very difficult because benzothiazepine and diketone have the same Rf in the solvents used: mixtures in different proportions of Cl2CH2/EtOH and hexane/AcOEt.
For this reason, it has been necessary to carry out several chromatographies, in order to isolate the final product pure, with fair yields. The o-aminothiophenol dimer 12 has also been isolated, and its structure confirmed by NMR (see Supporting Information figures S-36-S39).
2.2.2. Reactivity in acetic acid
Curcumin and the aforementioned curcuminoid derivatives were also reacted in glacial acetic acid with o-aminothiophenol. Although the same benzothiazepines 8-11 are formed as those generated in methanol (Scheme 3), dimer 12 is not obtained, and on the other hand the reaction is quantitatively complete in 2 h. In this case, the problem was that when removing the acetic acid in the rotary evaporator, the benzothiazepine decomposes due to the temperature effect forming benzothiazoles (Figure 5). In this way, it has been possible to isolate and characterize a new structure, the 2-(4-fluoro-3-methoxystyryl)benzo[d]thiazole 13, a solid with mp = 127ºC. (Scheme 4).
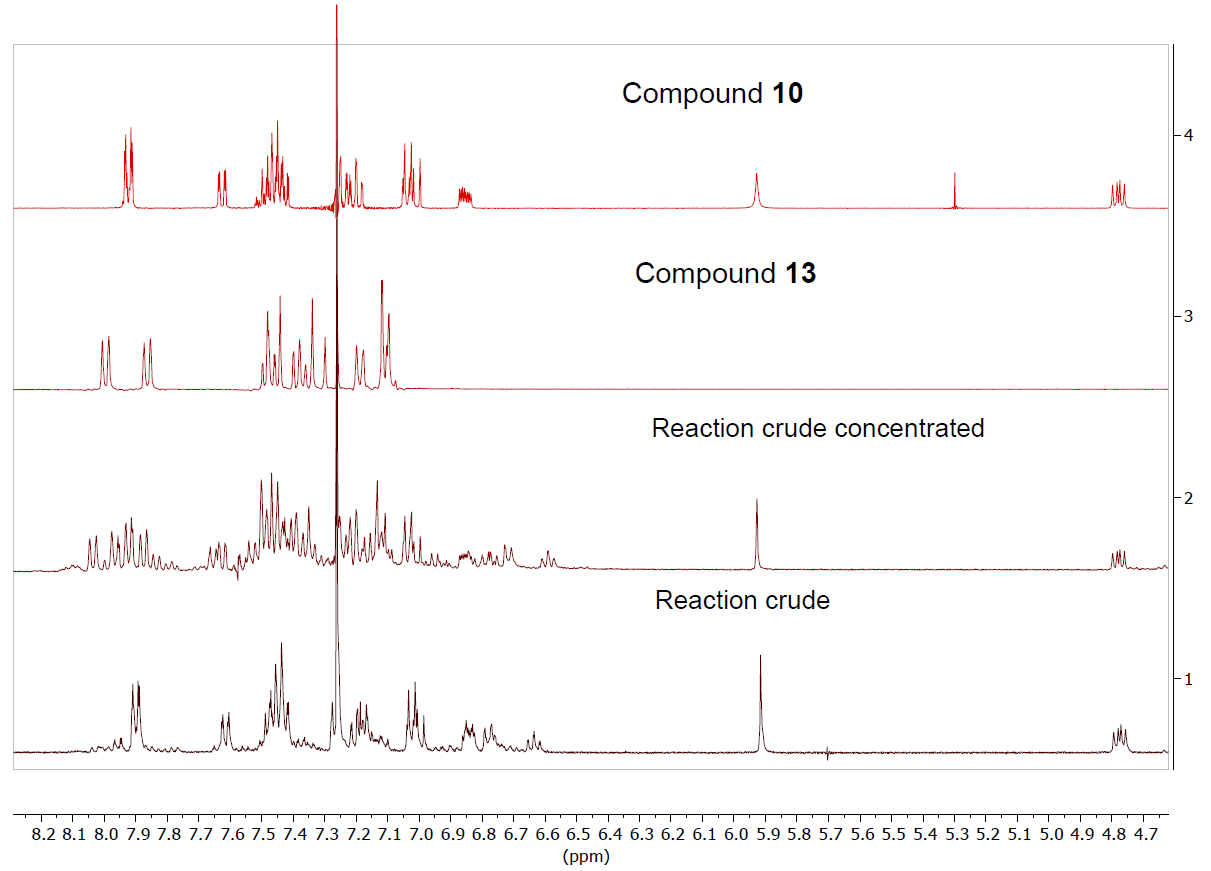
Figure 5. 1H NMR spectrum in CDCl3 of the reaction crude together with the isolated products
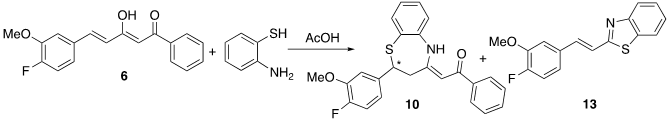
Scheme 4. Reactivity of curcuminoid diketone 6 with o-aminothiophenol in acetic acid
Since the formation of benzothiazole 13 was unexpected, we will discuss in detail the characterization of its structure, clearly different from that of the rest of the isolated compounds, which are described in section 2.3. To number the compound, the benzothiazole ring has been prioritized, then the double bond, and finally the substituted phenyl (Figure 6).
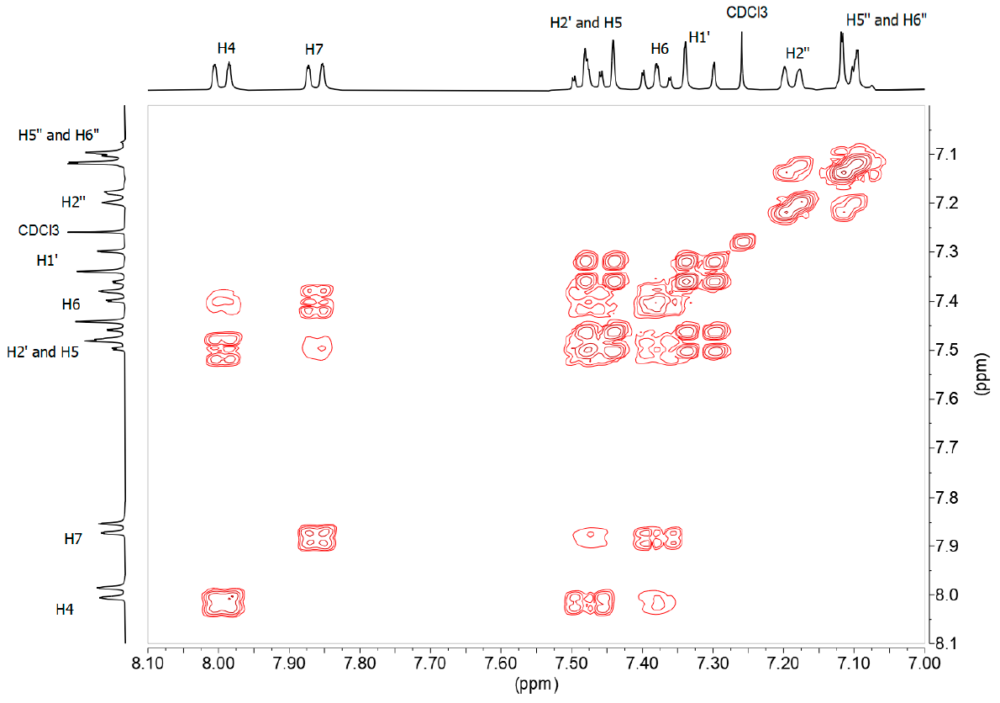
Figure 6. Compound 13 [(E)-2-fluoro-3-methoxystyryl)benzo[d]thiazole
The 1H-NMR (Figure 5) and 13C-NMR spectra of this derivative are simpler than those of benzothiazepines, taking into account that this derivative lacks the signals corresponding to the ABX system. From the 1H-1H-COSY spectrum (Figure 7) we observe that the sequence of the H of the benzo part is: 8.00 (ddd, 3JH5 = 8.2, 4JH6 = 1.2, 5JH7 = 0.7, H4), 7.48 (ddd, 3JH4 = 8.5, 3JH6 = 7.2, 4J7 = 1.3, H5). 7.38 (ddd, 3JH7 = 8.0, 3JH5 = 7.2, 4JH4 = 1.2, H6), 7.86 (ddd, 3JH6 = 8.0, 4JH5= 1.3, 4JH4 = 0.6, H7); the double bond signals, 7.46 (d, 3JH1‘ = 16.3, H2’), 7.32 (d, 3JH2‘ = 16.3 H1’) and those belonging to the fluorinated ring 7.19 (d, 4JF ≈ 9, H2»), 7.11 (m, H5», H6»), 3.95 (s, OMe).
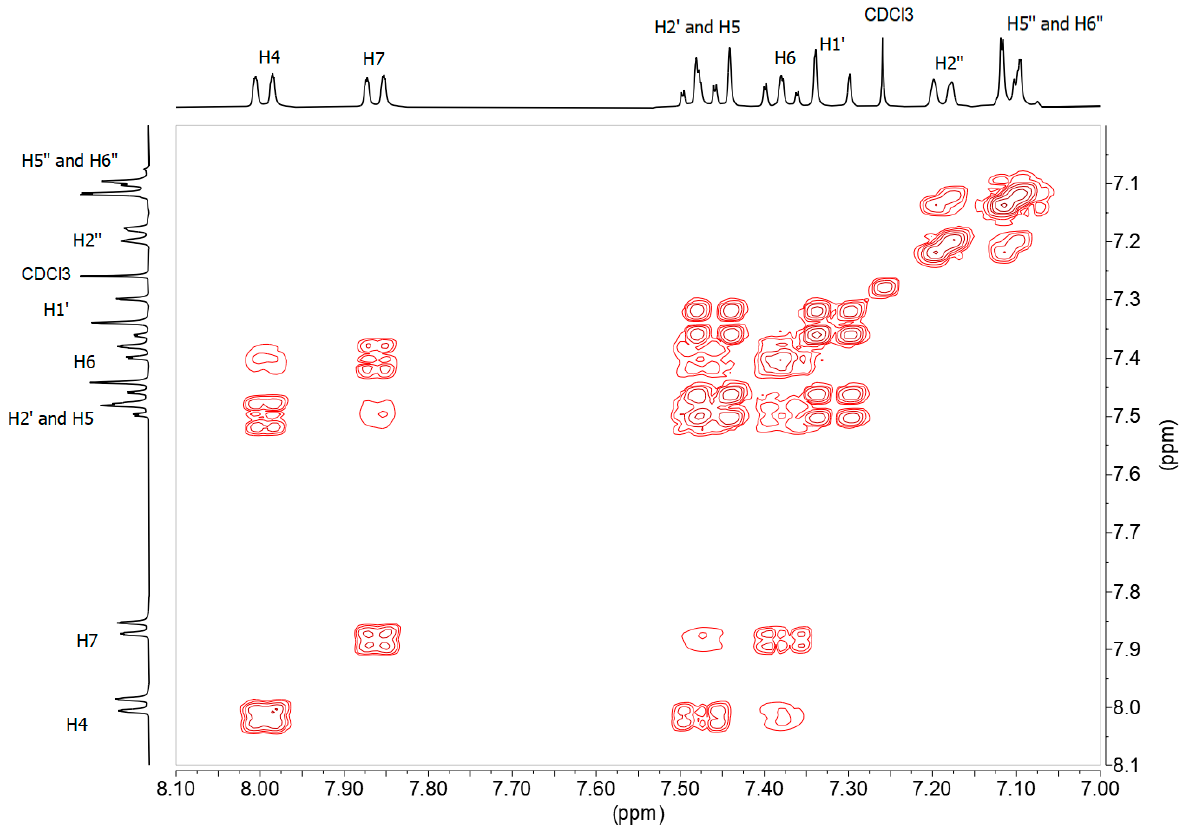
Figure 7. 1H-1H-COSY spectrum in DMSO-d6 of compound 13
To assign the quaternary carbons we have used the correlations observed in the 1H-13C HMBC correlation spectrum (Table 1). The rest of the carbons can be assigned using the correlations in the 1H-13C HMQC spectrum, as well as the C-F couplings in the 4-fluoro-3-methoxyphenyl ring, of carbons C4» (1JF= 250.5), C5» (2JF = 18.8), C3» (2JF = 11.3), C6» (3JF = 7.5), C2» (3JF = 2.5), C1» (4JF = 3.8), C2′ (5JF = 1.9) and C1′ (6JF = 3.1), with coupling constants from one to six bonds away. The values of the chemical shifts and coupling constants C-F are similar to those described for the aldehyde 3 and the starting diketone 6 (16,20).
Table 1. 13C chemical shifts and correlations observed in the HMQC and HMBC spectra of compound 13 in DMSO-d6 as well as 15N and 19F data
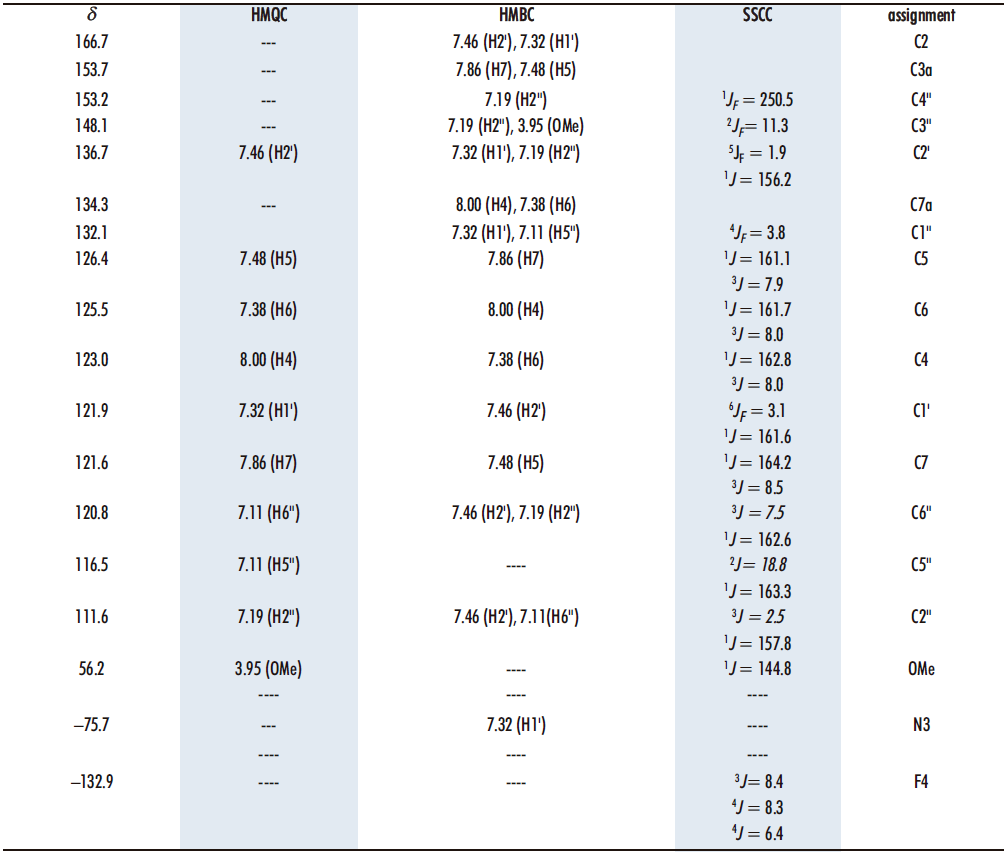
Regarding 15N, the chemical shift is –75.7 ppm, a typical value of a nitrogen atom with sp2 hybridization (19), as it corresponds to the one (N3) present in the benzothiazole structure. In addition, it is coupled to H5‘ (7.32) as observed in the 1H-15N HMBC spectrum in DMSO-d6.
On the other hand, there is in the 19F NMR spectrum in DMSO-d6 (Figure 8), where we observe that the signal obtained, at –132.9, is a ddd, with two coupling constants 4J with H2‘ and H6‘, 4JH2‘ = 8.3 and 4JH6‘ = 6.4, and a 3J with H5‘ 3JH5= 8.4.
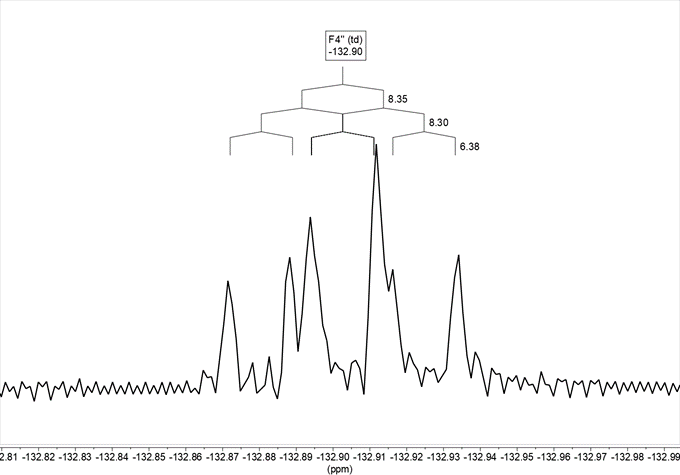
Figure 8. 19F NMR spectrum in DMSO-d6 of compound 13
2.3. NMR structural characterization
In this section, we will show that the benzothiazepine structure formed in the reaction corresponds to type A (Figure 3). The isomeric type B, which also presents an ABX system, has been ruled out on the basis of the 15N chemical shift value, as discussed later in section 2.3.3.
The four benzothiazepines obtained 8-11 have been analyzed by 1H, 13C, 15N and 19F NMR spectroscopies and the values of the chemical shifts are gathered in Tables 2 and 3. The assignments are based on the multiplicity of the signals and the 1H-13C correlations encountered in the HMQC and HMBC correlation spectra. DMSO-d6 has been used as solvent for the NMR analysis in all cases, and also CDCl3 in compounds 8 and 10. For the sake of clarity, when numbering the compounds rather than following IUPAC nomenclature, priority has been given to the benzothiazepine nucleus, then to the chalcone, and finally to the pendant aryl groups (Figure 9).
Figure 9. Atom numbering of compounds 8-11
2.3.1. 1H NMR
Regarding the 1H NMR data (Table 2), note the presence of the ABX system (Figure 10), in the four benzothiazepine isomers 8-11. The protons at position 3 are anisochronous (diastereotopic) because C2 is chiral; note that all our compounds are racemic.
Table 2. 1H NMR parameters (δ in ppm and J in Hz) of benzothiazepines 8-11

In Figure 10, corresponding to the ABX system of the 1H NMR spectrum of compound 8, we observe a dd at 4.86 ppm, which corresponds to nucleus X, which is coupled to two neighboring hydrogens (A and B), with different coupling constants 3J = 10.6 and 3J = 5.5 Hz. On the other hand, the protons of position 3 that form the AB part, resonate at 2.71 and 2.94 ppm coupled with HX, and a geminal constant JAB = 13.1 Hz.
The analysis of the spectrum of compound 9 (Figure 11) shows the following features: the ABX system between 2.6 and 4.9 ppm; a singlet at 6.27 ppm, which corresponds to the proton in position 1’’; H6 and H9 appear as doublets with a 3J ≈ 7 Hz, and in all cases δH9 (~7.6) > δH6 (~7.3). The signals of H7 and H8 are triplets, H8 being more shielded than H7, and H7 coming out together with Hm and Ho.
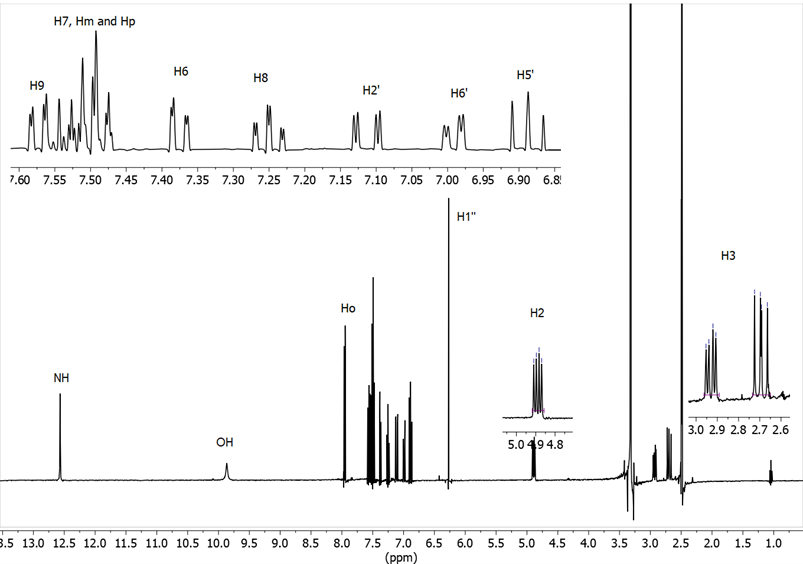
Figure 11. 1H NMR spectrum of benzothiazepine 9
Concerning the aryl group that comes from the aldehyde, in the fluorinated compounds 9 and 10 it has been possible to measure all the H-F couplings (Table 2). The signal of the NH proton appears at 12.57 ppm, due to the fact that it forms a hydrogen bond with CO. Finally, the signals of the protons of the lateral phenyl group are: Ho ~ 7.9 ppm and Hm and Hp ~ 7.5-7.4 ppm. It should be noted that the OH signal in the 4’ position has only been observed in DMSO-d6 at around 9 ppm.
2.3.2. 13C NMR
In the 13C NMR spectra (Table 3), the chemical shifts of the carbons have similar values in the four benzothiazepines 8-11, facilitating their assignment once the pattern of the different signals has been established by 1D and 2D experiments.
Table 3. 13C NMR parameters (δ in ppm and J in Hz) of benzothiazepines 8-11
For the side chain we have the signal corresponding to C1’’ (it can be seen attached to H1’’ in HMQC) at ~94 ppm, very different from C2’’, which bonded to an oxygen nucleus, gives a signal at 188 ppm. For the phenyl group, C6H5, in diazepines 8, 9, and 10 the signals of Co ~127, Cm ~128 and Cp ~131 ppm are observed, with coupling constants 3J = 3J = 7 Hz in Co and Cp and 3J = 7.5 Hz in Cm. Ci appears at ~139 ppm. In all three benzothiazepines, δCi > δCp > δCm > δCo.
At ~54 ppm we find a signal corresponding to C2, and at ~40 ppm there is the signal of C3, both carbons belonging to the nuclei of seven membered ring. The C4 signal appears at ~161 ppm as it is sp2 hybridized and bound to a nitrogen. The rest of the carbons of the benzothiazepine show shifts δC5a (~142) > δC9 (~135) > δC7 (~130) > δC8 (~126) > δC9a (~126) >δC6 (~123), as occurs in related benzothiazepines (21), the assignment of the carbons that appear at ~126 ppm does not present problems since they are a CH and a quaternary carbon.
Next, we find the signals of the 13C spectrum of the substituted phenyl: all of them show the same pattern as the precursor diketones and aldehydes (16,20) both in chemical shifts and in C-F coupling constants. Finally, the 3’ OMe carbon signal for 8, 10 and 11 appears around 56 ppm.
We will discuss in more detail the assignments in [4-(4-hydroxy-3-methoxy-phenyl)-1-(2-(4-hydroxy-3-methoxyphenyl)-2,3-dihydrobenzo[b ][1,4]thiazepin-4(5H)-ylidene)but-3-en-2-one] 11, which presents different 1H and 13C signals for the two 4-hydroxy-3-methoxyphenyl groups.
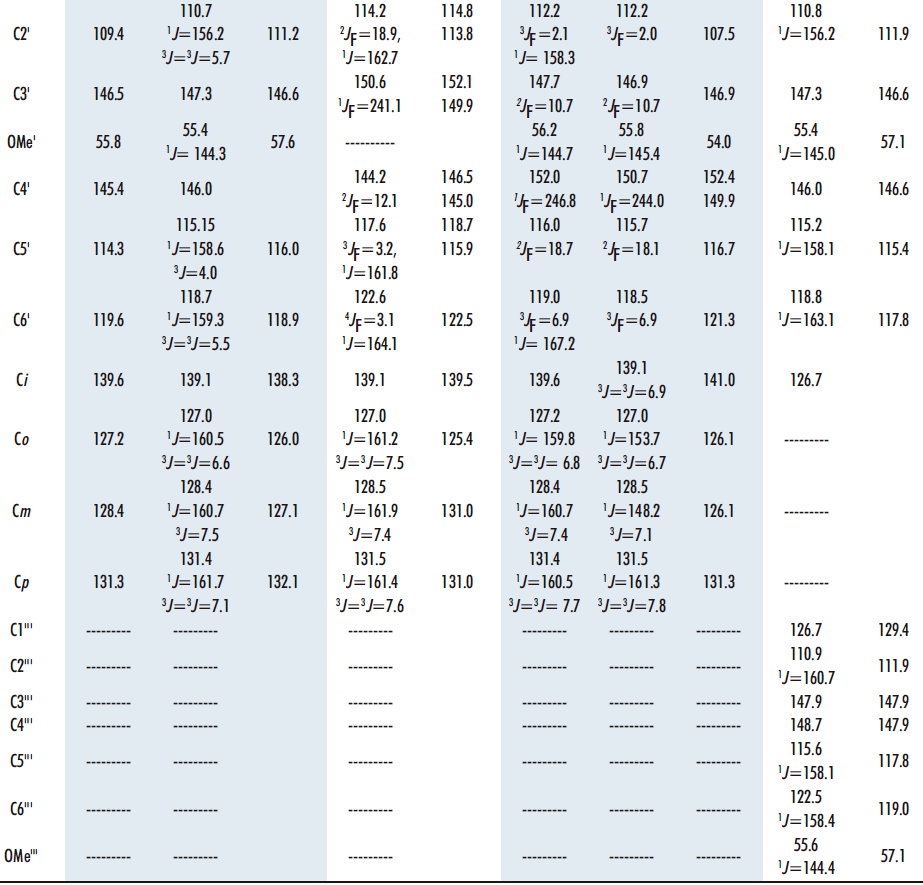
The following connections arise from the correlations observed in the 1H-1H COSY spectrum (Figure 12).
• 6.69 (H5’) ⇔6.74 (H6’) ⇔ 6.92 (H2’) 4-hydroxy-3- methoxyphenyl, thiazepine side
• 7.41 (H4”) 6.78 (H3”) double bond
• 6.79 (H5”‘) ⇔ 7.08 (H6”‘) ⇔ 7.27 (H2”) 4-hydroxy-3-methoxyphenyl, styrene side
• 7.59 (H9) ⇔7.22 (H8) ⇔ 7.47 (H7)⇔ 7.31 (H6) benzo moiety.
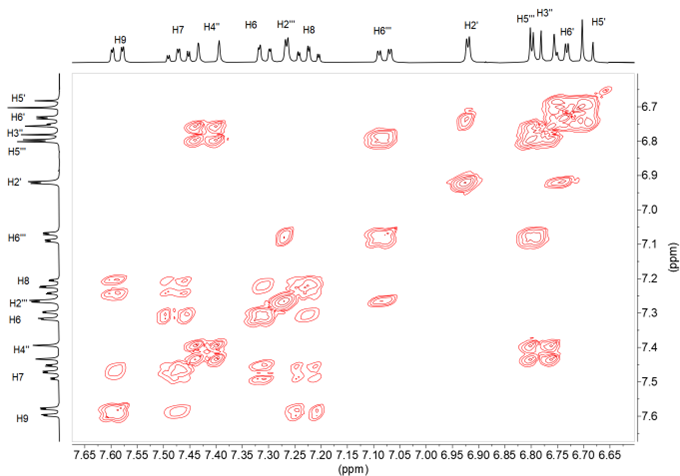
Figure 12. 1H-1H COSY NMR spectrum of compound 11 in DMSO-d6
From the connections observed in the HMQC spectrum all pairs of CH signals are found (see Supplementary material). On the other hand, the 1H-13C HMBC correlation spectrum also provides useful information, especially about the quaternary carbons, we observe that the OH signal that appears at 8.97 ppm is correlated with the carbons at 147.3 (C3’), 146.0 (C4’) and 115.2 (C5’) (Figure 13). The 146.0 signal shows long distance correlation (3J ) with 6.92 (H2’) and 6.74 (H6’) and the 147.3 signal (C3’) shows crossing peaks with 3.70 (OMe’) and 6.69 (H5’), so we know the signals that belong to each 4-hydroxy-3-methoxyphenyl ring (Figure 13); to know which side they are on we rely on the correlation that the signal of 139.0 (C4’’) presents with 7.08 (H6’’’) and 7.27 ppm (H2’’’) (see Supplementary material).
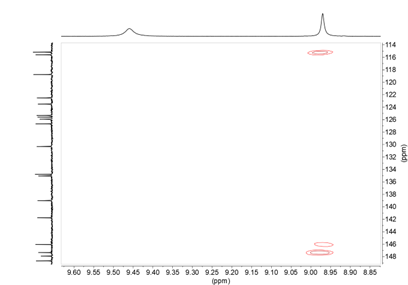
Figure 13. 1H-13C HMBC NMR spectrum enhancement of compound 11 in DMSO-d6
Finally, the 13C solid-state NMR spectra of the four benzothiazepines were also recorded and the assignment of the signals achieved by analogy with the data obtained in solution.
2.3.3. 15N and 19F NMR
The benzothiazepines studied in this work can exist as two tautomers: the keto-enamine and the imino-enol forms (Figure 14).
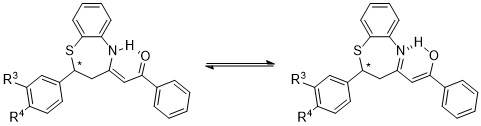
Figure 14. Ketoenamine-iminoenol tautomerism of synthesized benzothiazepines
From the 15N chemical shift value, observed at –255.0 in solution and –251.2 ppm in solid state for 8; –255.1 in solution and –246.2 in solid state for 9 (Figure 15); –255.2 in solution and –253.3 in solid state for 10, and –257.6 in solution and –245.7 in solid state for 11, it can be deduced that the tautomer that predominates is the keto-enamine, since in the imino-enol form the chemical shift should be in the order of –70 to –100 ppm (22).
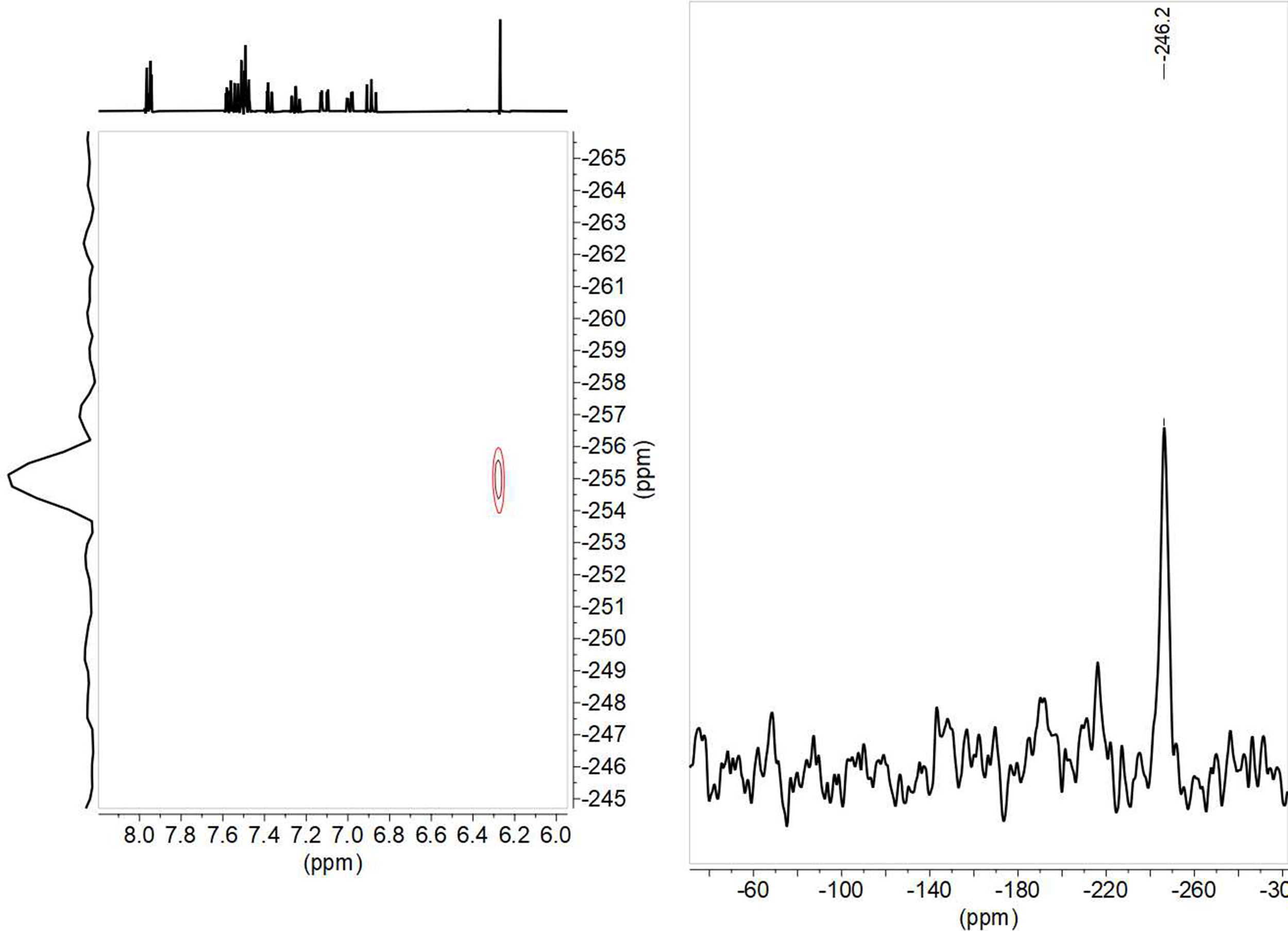
Figure 15. 1H-15N HMBC in DMSO-d6 and 15N-CPMAS NMR spectra of compound 9
The spectra of 19F NMR have also been recorded for benzothiazepines 9 and 10 in DMSO-d6, determining both the chemical shifts and the 1H-19F coupling constants, which agree with the values obtained from the 1H NMR spectra. In compound 9, the chemical shift is δ= –135.8 and the signal corresponds to a doublet of doublets due to the coupling of fluorine with the hydrogens in H2’ and H5’, 3JH2’ = 12.5, 4JH5’ = 9.2 Hz (Figure 16) In the solid-state it appears at –127.7/–130.7. In compound 10, the chemical shift is δ= –136.7 (ddd, 3JH5’ = 12.1, 4JH2 ‘= 8.4, 4JH6’ = 4.2 Hz) (Figure 17). In the solid-state it appears at –136.7.
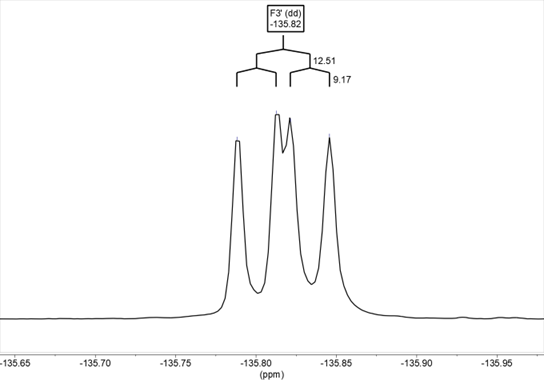
Figure 16. 19F NMR in DMSO-d6 of compound 9. The signal corresponds to a dd due to the coupling of fluorine with the hydrogens in H2′ and H5′ (3JH2‘ = 12.5, 4JH2‘ = 9.2 Hz)
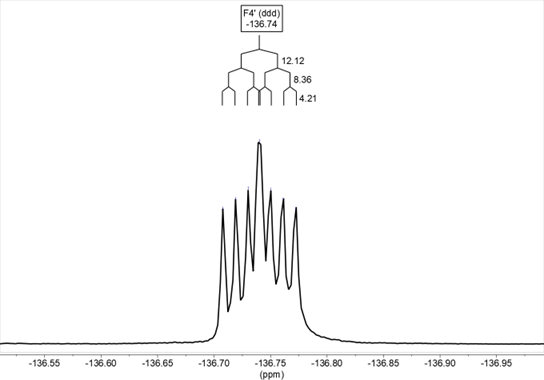
Figure 17. 19F NMR of compound 10 in DMSO-d6: δ = -136.74 (ddd, 3JH5‘ = 12.1, 4JH2 ‘= 8.4, 4JH6‘ = 4.2 Hz)
3. CONCLUSIONS
From the present work the following conclusions can be driven:
i) Benzothiazepines can be synthesized by reaction of o-aminothiophenol with curcumin and curcuminoid β-diketones, in methanol and in acetic acid.
ii) Both in methanol and in acetic acid, the β-diketones react as α,β-unsaturated ketones through a Michael addition, affording the same structural type, a benzothiazepine with two pendant groups, an aryl group adjacent to the sulfur atom and a 1-phenylethanone adjacent to the NH of the seven membered ring.
iii) Reaction times, yields and isolation of the compounds depend on the reaction media. Long reaction times and easy isolation of the final compound occur in methanol; conversely, short reaction times and difficult purification of the benzothiazepines, plus the formation in some cases of benzothiazoles, take place in acetic acid.
iv) 1D and 2D Multinuclear NMR (1H, 13C, 15N, 19F) in solution and 13C NMR in solid state have proved to be essential to elucidate the structures and tautomerism of the benzothiazepines and benzothiazoles.
4. EXPERIMENTAL PART
4.1. General remarks
All reagents cited in the experimental procedure are commercial compounds. For thin-layer chromatography, Merck Kiesegel 60F254 chromatofoils with a fluorescence indicator on aluminum and a layer thickness of 0.2 mm were used. For column chromatography, Merck silica gel 60 (70-230 mesh) was used with the eluent indicated in each case. Melting points have been determined in capillary tube on a Gallekamp apparatus and are uncorrected.
4.2. NMR
Solution NMR spectra were recorded on a 9.4 Tesla Bruker spectrometer (400.13 MHz for 1H, 376.50 MHz for 19F, 100.62 MHz for 13C and 40.54 MHz for 15N) at 300 K with a 5-mm inverse detection H-X probe equipped with a z-gradient coil, or with a QNP 5 mm probe (19F). Chemical shifts (δ in ppm) are given from internal solvents: CDCl3 7.26 for 1H and 77.0 for 13C, DMSO-d6 2.49 for 1H and 39.5 for 13C. External references were used for 15N and 19F, nitromethane and CFCl3. Coupling constants (J in Hz) are accurate to ±0.2 Hz for 1H, ±0.8 Hz for 19F and ±0.6 Hz for 13C.
Typical parameters for 1H NMR spectra were spectral width 3000-6000 Hz and pulse width 9.5 μs at an attenuation level of 0 dB. Typical parameters for 13C NMR spectra were spectral width 21 kHz, pulse width 10.6 ms at an attenuation level of –6 dB and relaxation delay 2 s. WALTZ 16 was used for broadband proton decoupling; the FIDs were multiplied by an exponential weighting (lb = 2 Hz) before Fourier transformation. Typical parameters for 19F-NMR spectra were spectral width 55 kHz, pulse width 13.75 ms at an attenuation level of −6 dB and relaxation delay 1 s. In some cases, for resolution enhancement processing a gaussian multiplication of the FID prior to Fourier transformation was applied.
2D (1H-1H) gs-COSY, (1H-13C) gs-HMQC, (1H-13C) gs-HMBC, (1H-15N) gs-HMQC and (1H-15N) gs-HMBC, were acquired and processed using standard Bruker NMR software and in non-phase-sensitive mode. Gradient selection was achieved through a 5% sine truncated shaped pulse gradient of 1 ms.
Selected parameters for (1H-13C) gs-HMQC and gs-HMBC spectra were: spectral width 3000-6000 Hz for 1H and 20 kHz for 13C, 1024 x 256 data set, number of scans 2 (HMQC) or 4 (HMBC) and relaxation delay 1s. In the gs-HMQC experiments GARP modulation of 13C was used for decoupling. The FIDs were processed using zero filling in the F 1 domain and a sine-bell window function in both dimensions was applied prior to Fourier transformation.
Selected parameters for (1H-15N) gs-HMQC and gs-HMBC spectra were: spectral width 3000-6000 Hz for 1H and 15 kHz for 15N, 1024 x 1024 data set, number of scans 4, relaxation delay 1s. In the gs-HMBC delays of 27-200 ms for the evolution of the 15N-1H long-range coupling were used. The FIDs were processed using zero filling in the F 1 domain and a sine-bell window function in both dimensions was applied prior to Fourier transformation.
Solid-state 13C (100.73 MHz) and 15N (40.60 MHz) CPMAS NMR spectra have been obtained on a 9.4 Tesla Bruker spectrometer at 300 K (100.73 MHz for 13C and 40.60 MHz for 15N) using a 4 mm DVT probehead. Samples were carefully packed in a 4-mm diameter cylindrical zirconia rotor with Kel-F end-caps. Operating conditions involved 2.9 µs 90° 1H pulses and decoupling SPINAL 64 sequence.
13C spectra were originally referenced to a glycine sample and then the chemical shifts were recalculated to the Me4Si (for the carbonyl atom (glycine) δ = 176.1 ppm) and 15N spectra to 15NH4Cl and then converted to nitromethane scale using the relationship: δ15N (nitromethane) = δ15N (ammonium chloride) − 338.1 ppm.
Typical acquisition parameters for 13C-CPMAS were: spectral width, 40 kHz; recycle delay, 5–60 s; acquisition time, 30 ms; contact time, 4 ms; and spin rate, 12 kHz. In order to distinguish protonated and unprotonated carbon atoms, the NQS (Non-Quaternary Suppression) experiment by conventional cross-polarization was recorded; before the acquisition the decoupler is switched off for a very short time of 25 µs (23,24,25). For 15N, CPMAS were: spectral width, 40 kHz; recycle delay, 5–60 s; acquisition time, 35 ms; contact time, 7 ms; and spin rate, 6 kHz.
Solid-state 19F (376.94 MHz) NMR spectra have been obtained on a 9.4 Tesla using a MAS DVT BL2.5 X/F/H double resonance probehead. Samples were carefully packed in 2.5 mm diameter cylindrical zirconia rotors with Kel-F end-caps. Samples were spun at the magic angle at rates of 25 kHz and the experiments were carried out at ambient probe temperature.
The typical acquisition parameters 19F {1H} MAS were: spectral width, 75 kHz; recycle delay, 10 s; acquisition time, 25 ms; pulse width, 2.5 µs and proton decoupling field strength of 100 kHz by SPINAL-64 sequence; 128 scans. The 19F spectra were referenced to ammonium trifluoroacetate sample and then the chemical shifts were recalculated to the CFCl3 (δCF3CO2−NH4+ = −72.0 ppm).
General procedure for the synthesis of curcuminoid β-diketones.
First 20 mmol of 1-phenylbutane-1,3-dione, and then 20 mmol of boric anhydride, were dissolved in 20 ml anhydrous ethyl acetate in a three-neck round-bottom flask equipped with a reflux condenser and magnetic stirring; the mixture being stirred for 90 min at room temperature (25°C).
Thereafter, using an addition funnel, a mixture formed by 20 mmol of the corresponding starting aldehyde [vanillin (1) for the diketone 4, 3-fluoro-4-hydroxybenzaldehyde (2) for the diketone 5, and 4-fluoro-3-methoxybenzaldehyde (3) for the diketone 6] and 40 mmol of tributylborate dissolved in anhydrous ethyl acetate (30 ml) was added and left stirring at room temperature for 60 min. Then, 2 ml of n-butylamine were added dropwise over 5 min and the resulting mixture was stirred at room temperature for 20 h. To hydrolyze the formed boron complex, 30 ml of 0.4 M HCl were added to the reaction mixture previously heated to 80 ºC, and continuous stirring at such temperature was maintained for 60 min. After cooling for 30 min, the organic phase was separated from the aqueous phase. The aqueous phase was extracted with (3 x 15 ml) of ethyl acetate and the organic phases washed with (2 x 10 ml) of water and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure, using a rotary evaporator, and the crude purified by column chromatography and/or crystallization.
5-(4-Hydroxy-3-methoxyphenyl)-1-phenyl-4-penten-1,3-dione (4). The reaction crude was treated with 100 ml of 2M HCl, forming a precipitate that was filtered; the solid obtained was recrystallized using hot methanol with a few drops of dichloromethane. In this way compound 4 with a yield of 45% was obtained. M.p. (MeOH): 195.8ºC [lit. 198 ºC (16)].
5-(3-Fluoro-4-hydroxyphenyl)-1-phenyl-4-penten-1,3-dione (5). The reaction crude was treated with 100 ml of 2M HCl, forming a precipitate that was filtered; the solid obtained was recrystallized using hot methanol with a few drops of chloroform. Compound 5 was obtained as an orange crystalline solid, with a yield of 62.2%. M.p. (MeOH): 192.8ºC [lit. 193.3 ºC (16)].
5-(4-Fluoro-3-methoxyphenyl)-1-phenyl-4-penten-1,3-dione (6). The reaction crude was purified by column chromatography, eluting with CH2Cl2/EtOH (99:1). Further purification was achieved using hot methanol and chloroform to give a light orange crystalline solid, 6, in 32.4% yield. M.p. (MeOH): 126.5ºC [lit. 127.2 ºC (16)].
General procedure for the reaction of curcuminoid diketones with o-aminothiophenol in methanol.
2 Mmol of the corresponding diketone was dissolved in 30 ml of methanol, and subsequently a solution of 4 mmol of o-aminothiophenol in 5 ml of methanol (b.p. 64.7 °C) was slowly added. The reaction was heated under reflux with magnetic stirring for 5.5 h. After that time, heating was stopped and the stirring was continued for another 15 h at room temperature (25 ºC). We concentrated under reduced pressure using a rotary evaporator. The crude product was purified by means of one or more chromatographic columns, and subsequent recrystallization.
2-[2-(4-Hydroxy-3-methoxyphenyl)-2,3-dihydrobenzo[b][1,4]thiazepin-4(5H)-ylidene)-1-phenylethanone (8). The reaction crude is successively chromatographed, eluting with CH2Cl2/EtOH 95:5; to isolate the product which is purified by recrystallization from hot methanol, obtaining a light yellow crystalline solid (yield 43%). M.p. (MeOH): 163.1 °C. Anal. Calc. for C24H21NO3S: C, 71.44; H, 5.25; N, 3.47. Found: C, 71.52; H, 5.50; N, 3.18.
2-[2-(3-Fluoro-4-hydroxyphenyl)-2,3-dihydrobenzo [b][1,4]thiazepin-4(5H)-ylidene)-1-phenylethanone (9). The reaction crude is successively chromatographed, eluting with Hex/AcOEt 9:3; finally we recrystallized in hot methanol and chloroform (yield. 52%). M.p. (MeOH): 196.3 °C. Anal. Calc. for C23H18FNO2S: C, 70.57; H, 4.63; N, 3.58. Found: C, 70.49; H, 4.61; N, 3.70. Found: C, 70.79; H, 4.29; N, 3.58.
2-[2-(4-Fluoro-3-methoxyphenyl)-2,3-dihydrobenzo [b][1,4]thiazepin-4(5H)-ylidene)-1-phenylethanone (10). The reaction crude is successively chromatographed eluting with Hex/AcOEt 9:3 and recrystallized from hot methanol (yield 69.4%). M.p. (MeOH): 163.4 °C. Anal. Calc. for C24H20FNO2S: C, 71.09; H, 4.97; N, 3.45. Found: C, 70.88; H, 5.02; N, 3.44.
4-(4-hydroxy-3-methoxyphenyl)-1-(2-(4-hydroxy-3-methoxyphenyl)-2,3-dihydrobenzo[b][1,4]thiazepin-4(5H)-ylidene)but-3-en-2-one (11). The crude is purified after several chromatographic columns eluted with CH2Cl2/EtOH in different proportions (9:1, 95:5 and 9:3), and the final product is recrystallized from hot methanol and chloroform (yield 53.9%). M.p. (MeOH): 181.5 °C. Anal. Calc. for C27H25NO5S: C, 68.19; H, 5.30; N, 2.95. Found: C, 68.23; H, 5.22; N, 3.01.
General procedure for the reaction of curcuminoid diketones with o-aminothiophenol in acetic acid.
In a three-neck flask equipped with a reflux condenser and magnetic stirring, 1 mmol of the corresponding diketone was dissolved in 12 ml of commercial glacial acetic acid. Then a solution of 2 mmol of o-aminothiophenol in 1 ml of glacial acetic acid was slowly added. The reaction mixture was heated at 80 °C with magnetic stirring for 2 h. After this time, the reaction was stopped, either allowing it to cool to room temperature and concentrating using a rotary evaporator, or pouring the crude oil onto ice and afterwards extracting or filtering. Other attempts with longer reaction times afforded lower yields in all cases.
2-[2-(4-Hydroxy-3-methoxyphenyl)-2,3-dihydrobenzo [b][1,4]thiazepin-4(5H)-ylidene)-1-phenylethanone (8). Starting from diketone 4, the reaction crude poured onto ice, extracted with CH2Cl2, and then column chromatographed with CH2Cl2. Finally, benzothiazepine 8 was recrystallized from methanol (43% yield). M.p. (MeOH): 163.1°C.
2-[2-(3-Fluoro-4-hydroxyphenyl)-2,3-dihydrobenzo [b][1,4]thiazepin-4(5H)-ylidene)-1-phenylethanone (9). Starting from diketone 5, the reaction crude poured onto ice. The solid obtained was filtered and purified by column chromatography using hexane/AcOEt (9:3) as eluent. Finally, benzothiazepine 9 was recrystallized from methanol (40% yield). M.p. (MeOH): 196.3 ºC.
2-[2-(4-Fluoro-3-methoxyphenyl)-2,3-dihydrobenzo [b][1,4]thiazepin-4(5H)-ylidene)-1-phenylethanone (10). Starting diketone 6, the reaction was stopped pouring the crude onto ice, then extracted with CH2Cl2, concentrated on a rotary evaporator and recrystallized from methanol, benzothiazepine 10 (48% yield) was obtained. M.p. (MeOH): 163.4 °C. Anal. Calc for C16H12FNOS C, 67.35; H, 4.24; N, 4.91. Found: C, 67.22; H, 4.31; N, 4.88.
4-(4-hydroxy-3-methoxyphenyl)-1-(2-(4-hydroxy-3-methoxyphenyl)-2,3-dihydrobenzo[b][1,4]thiazepin-4(5H)-ylidene)but-3-en-2-one (11). Starting from diketone 7, the reaction crude was poured onto ice, extracted with CH2Cl2 and concentrated on a rotary evaporator. It was purified by column chromatography using CH2Cl2/EtOH (95:5) as eluent and recrystallized from methanol (21% yield). M.p. (MeOH): 181.5 °C.
Acknowledgements
We are grateful to the Ministerio de Economía y Competitividad (CTQ2014-56833-R), Ministerio de Ciencia, Innovación y Universidades (PID2021-125207NB-C32) and to the Master “Ciencia y Tecnología Química” of the UNED for financial support.
Supporting Information
NMR spectra in solution (Figures S1 to S46) and NMR spectra in solid state (Figures S47 to S56).
ORCID
Dionisia Sanz https://orcid.org/0000-0001-9672-4684
Rosa M. Claramunt https://orcid.org/0000-0001-6634-2677
José Elguero https://orcid.org/0000-0002-9213-6858
5. REFERENCIAS
- Zamani, F, Doustkhah E, Luque R. Structural features of 1,5-benzodiazepines and 1,5-benzothiazepines, Chapter 6 In book: Benzodiazepine-Based Drug Discovery, pages 183-199, 2022.
- Mehmood R, Mughal EU, Elkaeed EB, et al. 2022, Synthesis of novel 2,3-dihydro-1,5-benzothiazepines as α-glucosidase inhibitors: in vitro, in vivo, kinetic, SAR, molecular docking, and QSAR studies, ACS Omega, 7:30215–30232.
- Zhu, GY, Zhou JJ, Liu, LG, et al. 2022, Catalyst-dependent stereospecific [3,3]-sigmatropic rearrangement of sulfoxide-ynamides: divergent synthesis of chiral medium-sized N,S-heterocycles, Angew Chem Int Ed., 61:e202204603.
- Pinate P, Makone S. 2022, Synthesis and study of catalytic perspectives of DABCO based ionic liquid for the synthesis of 2,3-dihydro-1,5-benzothiazepines and 2-phenylbenzo-thiazoles, Catal Lett., in press. https://doi.org/10.1007/s10562-022-04033-z
- Sarhan AE, Sediek AA, Khalifa NM, et al. 2022, Novel pyrazolines and benzothiazepines as tubulin polymerization inhibitors: synthesis, biological evaluation, and molecular docking, Heterocycles, 104: 447–469.
- Haroun M, Chobe SS, Rajasekhar RA, et al. 2022, 1,5-Benzothiazepine derivatives: green synthesis, in silico and in vitro evaluation as anticancer agents, Molecules, 27:3757.
- Shabir, G.; Shafique, I.; Saeed, A. 2022, Ultrasound assisted synthesis of 5–7 membered heterocyclic rings in organic molecules, J Heterocycl Chem., 59:1669-1702
- González-Albadalejo J, Sanz D, Claramunt RM, et al. (2015) Curcumin and curcuminoids: chemistry, structural studies and biological properties. An Real Acad Farm., 81:278–310.
- Nigam S, Joshi YC, 2003, Synthesis of novel derivatives of 1,5-benzothiazepines, Phosphorus Sulfur Silicon, 178:1583–1586.
- Kumari N, Saini RK, Joshi YC, 2007, Synthesis and novel derivative of 1,5-benzothiazepines from 1,3-diketones and their antibacterial activity, Oriental J Chem., 23:2.
- Chhakra S, Mukherjee A, Singh HL, et al. 2019, Synthesis of novel substituted 1,5-benzothiazepines containing 1,4-benzodioxane sulfonyl moiety, Asian J Org Med Chem., 4:70–76.
- Deshmukh R, Shinde V, Kanase DA, 2021, Review on 1,5-benzothiazepines as a versatile pharmacophore, J Emerging Technol Innovative Res., 8:2109333.
- Nieto CI, Cornago MP, Cabildo MP, et al. 2018, Evaluation of the antioxidant and neuroprotectant activities of new asymmetrical 1,3-diketones, Molecules, 23:1837 (27 pages).14
- Pabon HJJ. 1964, A synthesis of curcumin and related compounds, Recl Trav Chim Pays-Bas, 83:379.
- Claramunt RM, Bouissane L, Cabildo MP, et al. 2009, Synthesis and biological evaluation of curcuminoid pyrazoles as new therapeutic agents in inflammatory bowel disease: effect on matrix metalloproteinases. Bioorg Med Chem., 17:1290–1296.
- Cornago P, Cabildo P, Sanz D, et al. 2013, Structures of hemi-curcuminoids in the solid state and in solution, Eur J Org Chem., 6043–6054.
- Nieto CI, Cabildo P, Claramunt RM, et al. 2016, The structure of β-diketones related to curcumin determined by X-ray crystallography, NMR (solution and solid state) and theoretical calculations. Struct Chem., 27:705–730.
- Nieto CI, Andrade A, Sanz D, et al. 2017, Curcumin related 1,4-diazepines: regioselective synthesis, structure analysis, tautomerism, NMR spectroscopy, X-ray crystallography, density functional theory and GIAO calculations, ChemistrySelect, 2:3732–2738.
- Nieto CI, Sanz D, Claramunt RM, et al. 2018, Molecular structure in the solid state by X-ray crystallography and SSNMR and in solution by NMR of two 1,4-diazepines; J Mol Struc., 1155:205-214.
- Sanz D, Nieto C, Claramunt, RM, et al. 2015, A multinuclear magnetic resonance study of fluoro derivatives of hydroxybenzaldehydes, Magn Reson Chem., 53:624–631.
- Prakash O, Kumar A, Sadana A, et al., 2005, Study of the reaction of chalcone analogs of dehydroacetic acid and o-aminothiophenol: synthesis and structure of 1,5-benzothiazepines and 1,4-benzothiazines, Tetrahedron, 61:6642–6651.
- Martin GJ, Martin ML, Gouesnard JP, 15N-NMR Spectroscopy, NMR Basic Principles and Progress (NMR, volume 18), Springer-Verlag, Berlin-Heidelberg, 1981.
- Murphy PD, 1983, Improvement in the cross-polarization NMR experiment for suppression of rigid protonated carbons, J Magn Reson., 52:343–345.
- Murphy PD, 1985, Pulse sequences for the selective observations of nonprotonated and methyl carbon NMR resonances in solids, J Magn Reson., 62;303–308.
- Alemany LB, Grant DM, Alger TD, et al., 1983, Cross polarization and magic angle sample spinning NMR spectra of model organic compounds. 3. Effect of the 13C-lH dipolar interaction on cross polarization and carbon-proton dephasing, J Am Chem Soc., 105:6697–6704.
