1. INTRODUCCIÓN
La investigación clínica aporta conocimiento médico a través del estudio con medicamentos en personas siendo el nexo entre la investigación básica y la asistencia a los pacientes. Los datos de este tipo de investigaciones permiten obtener la autorización de comercialización para nuevos fármacos de las diferentes agencias reguladoras (1,2,3). Este tipo de investigación es clave en el desarrollo de nuevos fármacos, ya que sin ella la aprobación de nuevas terapias y el desarrollo de las guías farmacoterapéuticas quedan limitadas al conocimiento ya disponible.
La investigación clínica ha ayudado a posicionar a España como una de las mayores potencias para el desarrollo de ensayos clínicos gracias al trabajo de colaboración entre la administración, sistema y profesionales sanitarios, pacientes y compañías farmacéuticas (4). Durante el año 2020, se aprobaron hasta un total de 690 ensayos clínicos nuevos de acuerdo al Registro Español de ensayos clínicos (en adelante, REec). Los últimos resultados en la 30ª publicación Medicamentos Innovadores (3) los estudios en fases tempranas (Fase I y II) son un 46,8% de los ensayos realizados y los ensayos fase III un 46.5% del total.
Los servicios de farmacia, además del almacenamiento y dispensación del fármaco de estudio son los encargados en un elevado número de ensayos clínicos de la preparación del fármaco antes de su dispensación y administración al paciente. Es importante resaltar la importancia que tienen las instalaciones y personal encargado de preparar el fármaco, en especial, si es una administración parenteral. Unas buenas instalaciones, personal cualificado y procedimientos normalizados de trabajo para el manejo de las mezclas de administración parenteral reducen el riesgo de que un paciente pueda sufrir eventos adversos por posibles pérdidas de estabilidad fisicoquímica o contaminación microbiológica durante la preparación, sin tener esta relación con la naturaleza del fármaco (6). Desde la preparación hasta la administración de la mezcla de administración parenteral, el farmacéutico y/o el personal bajo su supervisión deben garantizar, no sólo las condiciones idóneas, sino también que se mantengan las condiciones ideales de estabilidad, compatibilidad y esterilidad del producto terminado. Las mezclas deben ser terapéutica y farmacéuticamente apropiadas para el paciente, preparadas con un mínimo de manipulaciones posibles y que presenten condiciones óptimas de eficacia y seguridad, garantizando así que se alcance el objetivo terapéutico deseado (7). En el marco de un ensayo clínico, todo acontecimiento adverso será monitorizado, incluidos aquellos que ocurran durante la administración o después de la administración debido a que pudiesen estar relacionados con el fármaco de estudio.
Todos los motivos expuestos ayudan a entender el motivo por el cual los promotores de ensayos clínicos evalúan todos los procesos, personal implicado e instalaciones de cada uno de los centros en cuanto a las mezclas de administración parenteral. Los promotores basan sus selecciones en las instalaciones, personal y procedimientos en centros de investigación en los que se persigue reducir al máximo cualquier posible evento que pueda poner en tela de juicio la seguridad del fármaco de estudio por un motivo que no esté relacionado directamente con la idioincrasia de la molécula, tal y como se reseña.
2. MATERIAL Y MÉTODOS
Se ha completado una revisión bibliográfica de las normas, guías e informes de diferentes organismos y artículos de investigación publicados relacionados con la preparación de productos estériles en los servicios de farmacia en España con la finalidad de conocer cuál es el proceso a seguir durante la preparación de mezclas de administración parenteral en los centros sanitarios en la actualidad.
Por otro lado, se ha completado un análisis del Registro Español de ensayos clínicos para analizar el número de estudios registrados en él, y de estos, cuáles de ellos contienen fármacos que pudieran ser susceptibles de ser preparados en ambientes estériles para obtener datos acerca del número de ensayos clínicos que contienen fármacos que requieren ser preparados en condiciones de esterilidad.
3. RESULTADOS
Los ensayos clínicos que se llevan a cabo en España promovidos por la industria farmacéutica, además de realizar ensayos clínicos multicéntricos (participación de varios centros de investigación en el mismo país para un mismo ensayo clínico), el 94,1% son de carácter internacional (3). Con este dato, podemos comprender que los laboratorios farmacéuticos necesitan que sus equipos de operaciones clínicas, en los países en los que desarrollan su actividad, estén compuestos por personal suficientemente cualificado, además de en asuntos de carácter científico y médico, también en la normativa local de cada país permitiéndose así tener las máximas garantías en la obtención de datos de calidad en el desarrollo de su fármaco en investigación.
Las mezclas de administración parenteral deben prepararse en ambientes controlados para garantizar su esterilidad y condiciones de seguridad apropiadas a la hora de ser administradas al paciente. A pesar de que este concepto es de carácter general para cualquier mezcla de administración parenteral a nivel mundial, no estará recogido del mismo modo en las normativas, farmacopeas o guías de buenas prácticas en cuanto a cómo han de ser las instalaciones, formación del personal o procedimientos a seguir con este tipo de preparaciones en los diferentes países o regiones.
En enero de 2011, por la resolución CM/ResAP10, se recomendó la necesidad de elaborar directrices prácticas sobre la preparación de medicamentos con la finalidad de evitar diferencias de calidad y seguridad entre las preparaciones de medicamentos en centros sanitarios y aquéllos que fuesen elaborados a escala industrial. En el caso de España, la normativa que se debe conocer relativa a este asunto y las preparaciones estériles de este tipo es el Real Decreto Ley 16/2002, de 20 de abril, de medidas urgentes para garantizar la sostenibilidad del Sistema Nacional de Salud y mejorar la calidad y seguridad de sus prestaciones (8). Esta norma regula en su artículo 7, la “manipulación y adecuación de preparaciones de medicamentos” permitiendo así acreditar a los servicios de farmacia hospitalaria a operaciones de fraccionamiento, personalización de dosis y otras operaciones de reacondicionamiento y transformación de medicamentos garantizando que se cumplen las guías técnicas de buena práctica. Estas guías de buena práctica de manipulación, fraccionamiento y dosificación personalizada son mencionadas en su artículo 7.3 y atribuye la elaboración de dicha guía técnica a la Dirección General de Cartera Básica del Servicio del Sistema Nacional de Salud y Farmacia con la colaboración de la Agencia Española de Medicamentos y
Productos Sanitarios y expertos de reconocido prestigio. Desde la publicación de este RD ley en el BOE el 24 de abril de 2012 hasta junio de 2014 no se publicó la “guía de buenas prácticas de preparación de medicamentos en servicio de farmacia hospitalaria” (9).
De acuerdo al Real decreto legislativo por el que se aprobó el texto refundido de la ley de garantías y uso racional del medicamento (11) se continúa atribuyendo lo ya previsto en el Real Decreto Ley 16/2002, de 20 de abril, con relación a las funciones de modificación, acondicionamiento, ajuste de dosis etc. al farmacéutico especialista en farmacia hospitalaria a través del servicio de farmacia en la publicación de 2006 y sin cambio o actualización en el texto refundido de dicha ley en 2015 (artículo 84.2 y 85.1).
Es importante, dado el objeto de estudio de este trabajo, mencionar que las guías de buena práctica, contemplan tanto la práctica diaria como la investigación clínica. Es bastante habitual que en los centros sanitarios en los que se lleve a cabo investigación clínica, dentro del servicio de farmacia suele existir una unidad de ensayos clínicos de farmacia. La guía de buena práctica de preparación de medicamentos evalúa los procesos a seguir, la vía de administración, el perfil de seguridad, las cantidades a preparar y la vulnerabilidad de la preparación y distribución. Si el fármaco a preparar es de ensayo clínico, es un aspecto a tener en cuenta en el perfil de seguridad, ya que por encontrarse en el contexto de un ensayo clínico es considerado un fármaco de riesgo medio ya que durante el desarrollo clínico debe hacerse un continuo seguimiento a la seguridad del fármaco, buscando en todo momento reducir al mínimo cualquier variable que pueda comprometer la seguridad del fármaco de estudio.
La normativa actual de ensayos clínicos en España (RD1090/2015 (Art.39 ñ) (2), recoge que no siempre es necesaria la participación del servicio de farmacia como servicio colaborador en el ensayo clínico. En aquellos casos en los que el envío de la medicación de investigación se realice directamente al investigador principal sin pasar por un servicio de farmacia, será éste quien asumirá la responsabilidad de correcta preparación, administración, custodia y dispensación. Ante este artículo novedoso en el RD1090/2015, podría interpretarse como una contradicción entre la normativa actual de ensayos clínicos y la guía de buena práctica de preparación en los servicios de farmacia. No obstante, no cabe lugar para dicha contradicción, ya que por normativa local puede seleccionarse un centro en el que el servicio de farmacia pueda no participar como servicio colaborador en un ensayo clínico o participar centros de investigación sin servicio de farmacia teniendo en cuenta que el promotor, debe cerciorarse que es seguro seleccionar un centro con estas características en base a los requerimientos de su fármaco en estudio mediante la firma de la idoneidad del investigador de acuerdo al RD1090/2015 por la persona física designada por el promotor del ensayo clínico. A su vez, además de la conformidad por parte del promotor, la dirección del centro y el Comité de Ética e Investigación con Medicamentos (CEIm) debe emitir sus correspondientes conformidades mediante la idoneidad de las instalaciones y dictamen aprobación respectivamente. Por tanto, la ausencia del servicio de farmacia en un ensayo clínico, es posible, pero debe ser evaluada responsablemente con el conocimiento de toda la normativa vigente revisada en este trabajo por todos los intervinientes antes de comenzar cualquier actividad de reclutamiento e inclusión de paciente en el centro. Algunos estudios (7) recogen que la preparación de medicamentos inyectables estériles realizados por profesionales de salud, es uno de los procedimientos más sujeto a errores tanto por su complejidad como por su elevado riesgo de contaminación microbiológica. Se ha visto que la frecuencia de errores en la preparación de mezclas es ocho veces mayor en la planta que en una unidad de mezclas parenterales por diferentes causas como procedimientos inadecuados de chequeo, roturas de la técnica aséptica y errores de cálculo y preparación. Por este motivo, a nivel internacional, se recomienda que la preparación de los medicamentos parenterales se realice de forma centralizada en los servicios de farmacia y se entreguen listos para su administración a enfermería.
Apoyado en la revisión bibliográfica previa, se ha realizado un análisis de los ensayos clínicos que se encuentran publicados en el Registro Español de ensayos clínicos (REec). En este registro público se recoge la información de todos los ensayos clínicos autorizados en España desde el 1 de enero de 2013.
La revisión del Registro Español de ensayos clínicos (REec) para este trabajo ha consistido en generar un informe de todos los estudios clínicos publicados en la plataforma desde el 1 de enero de 2013 a 31 de agosto de 2021. El total de estudios clínicos incluidos es de 7181 estudios. Basados en este informe general se ha completado un análisis de los datos obteniéndose como primera conclusión que un total de 3012 estudios (figura 1) contiene, al menos una entre las diferentes formas farmacéuticas que se pueden utilizar en ese estudio, sería necesario pre-pararla en condiciones de esterilidad en una unidad de mezclas de administración parenteral.
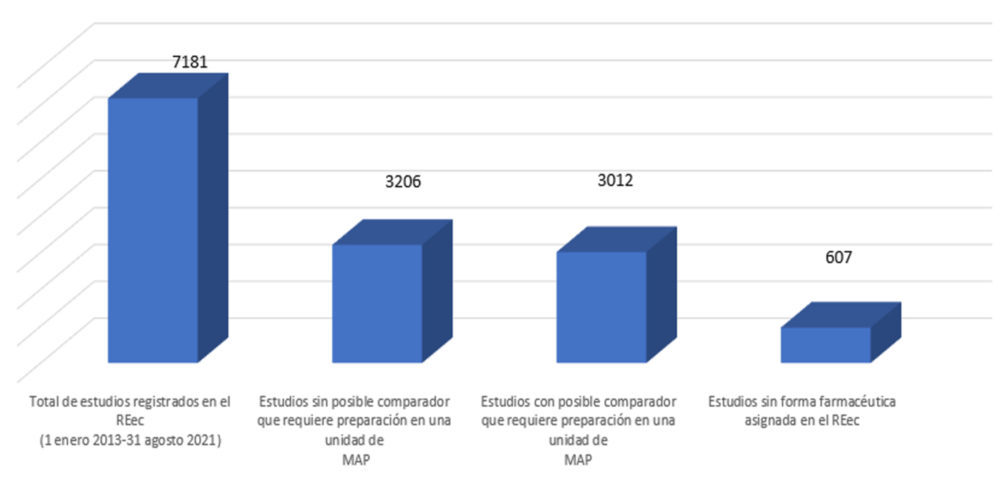
Figura 1. Distribución de los estudios incluidos en el Registro Español de ensayos clínicos (REec) desde 1 de enero de 2013 a 31 de agosto de 2021
Los datos obtenidos de la revisión del Registro Español de ensayos clínicos (REec) indican que cerca del 50% de los estudios de acuerdo a la figura 1 (3012 estudios) contienen fármacos susceptibles de ser preparados en unidades de mezclas de administración parenteral. Además, de todos estos estudios, 2395 estudios son de carácter internacional (figura 2) reafirmándose así la idea mencionada anteriormente acerca de que los laboratorios farmacéuticos necesitan que sus equipos de operaciones clínicas estén compuestos por personal suficientemente cualificado, además de en asuntos de carácter científico y médico, también en la normativa local de cada país.

Figura 2. Distribución de los estudios internacionales frente a nacionales que contienen un posible comparador que requiere preparación en una unidad de mezclas de administración parenteral.
Por último, de los ya 3012 estudios mencionados, 2806 estudios incluyen entre sus objetivos la seguridad del fármaco de estudio (Figura 3). Este objetivo dentro de los estudios, y en especial los de fases tempranas, es decisivo ya que, si un fármaco en investigación muestra durante estas fases un balance beneficio riesgo desproporcionado, su desarrollo clínico puede llegar a ser interrumpido por falta de seguridad de acuerdo a las normas de buena práctica clínica (5).

Figura 3. Distribución de estudios que tienen entre sus objetivos de estudio eficacia y/o seguridad con un posible comparador que requiere preparación en una unidad de mezclas de administración parenteral.
4. DISCUSIÓN
La investigación clínica en España es un sector de elevado interés socio-económico por el desarrollo de nuevas terapias beneficiosas para los pacientes como del gran impacto económico que posee este sector en nuestro país. A pesar de estos intereses económicos, es primordial velar por la seguridad de los pacientes que participen en los ensayos clínicos, así como de obtener datos de calidad que avalen ante las agencias reguladoras la solicitud de autorización de comercialización de los fármacos estudiados.
Tanto por toda la información aportada de la revisión bibliográfica como de la revisión del Registro Español de ensayos clínicos (REec), se pretende remarcar la importancia del especialista en operaciones clínicas encargado de la selección y puesta en marcha de un ensayo clínico. El especialista en operaciones clínicas debe conocer y saber aplicar este entramado legislativo que le permitirá justificar que todo aquel servicio de farmacia hospitalaria que esté acreditado por la comunidad autónoma en la que se encuentre el centro, de acuerdo al artículo 7 del Real Decreto ley 16/2012 de 20 de abril, y por tanto tendrá la suficiente consistencia en sus procesos y medios para poder realizar todas aquellas actividades de fraccionamiento, personalización de dosis y otras operaciones de reacondicionamiento y transformación de medicamentos tan seguros como si se hubiese realizado a escala industrial en el contexto de un ensayo clínico. Si se optase por seleccionar centros en los que no existe un servicio de farmacia y el fármaco pudiera ser preparado en una localización diferente a una unidad de mezclas parenterales, se debe evaluar los potenciales riesgos durante la preparación basado en la matriz de decisión de la guía de buenas prácticas de preparación la preparación de medicamentos inyectables estériles realizados por profesionales de salud es uno de los procedimientos más sujeto a errores tanto por su complejidad, como por su elevado riesgo de contaminación microbiológica y mayor frecuencia de errores durante la preparación en planta, así como otros errores durante la manipulación. Se recomienda que la preparación de los medicamentos parenterales se realice de forma centralizada en los servicios de farmacia y se entreguen listos para su administración a enfermería.
Con relación a los resultados obtenidos de la revisión del Registro Español de ensayos clínicos (REec) aproximadamente el 50% de los estudios que tenían correctamente identificados los comparadores que se utilizaban en los estudios clínicos podrían requerir que estos fármacos fuesen preparados en condiciones de esterilidad por contener una forma farmacéutica de administración parenteral, dado que su presentación final no era apta para administración directa al paciente. De este grupo de estudios que podría contener comparadores que requerirían ser preparados en unidades de mezclas de administración parenteral, el 79% son de carácter internacional reafirmándose de nuevo la importancia de que los promotores de ensayos clínicos con presencia en diferentes localizaciones geográficas requieren especialistas en investigación clínica que conozcan las necesidades normativas de cada región para su puesta en marcha. Finalmente, de todos los estudios que podrían contener un comparador que necesitase ser preparados en condiciones de esterilidad, el 93% de los estudios contenía entre los objetivos de estudio la evaluación de la seguridad del fármaco de estudio, y como se menciona, todo promotor buscará la máxima protección de todos los pacientes participantes en su estudio, y siempre buscará evitar cualquier posible causa que pudiera poner en tela de juicio la seguridad de su fármaco de estudio, ya que de acuerdo a las normas de buena práctica clínica si el balance beneficio riesgo no es proporcionado el desarrollo clínico podría interrumpirse para dicha molécula.
5. CONCLUSIONES
España es en la actualidad es un país de elevado interés para los promotores de ensayos clínicos de carácter internacional. Desde el punto de vista de este trabajo, España tiene un marco normativo robusto en materia de preparación de medicamentos estériles tanto dentro de la práctica habitual como para fármacos en investigación clínica. Este marco normativo nos permite posicionarnos como una nación que vela por la seguridad de los pacientes, siendo esto imprescindible para el desarrollo de ensayos clínicos que permitan obtener unos buenos datos de seguridad y eficacia que serán presentados ante las agencias reguladores para la obtención de la autorización de comercialización.
Por último, es conveniente reseñar que todos los profesionales involucrados en investigación clínica, ya sea bien en los centros sanitarios o como parte de los promotores de ensayos clínicos, deben conocer las normativas revisadas en este trabajo para seguir garantizando la posición de excelencia que tiene España en investigación clínica a nivel internacional.
Agradecimientos
Al Prof. Dr. Carlos del Castillo Rodríguez como director de la tesis doctoral del autor y a sus supervisoras, Dra. Ana Esther García Cadenas y Dra. María Isabel Vázquez Calleja por el soporte y asesoramiento en este trabajo.
6 . REFERENCIAS
- Instituto Nacional de la Salud Infantil y Desarrollo Humano Eunice Kennedy Shriver [página Web]. Oficina de Comunicaciones [16/10/2020; 09/11/2021]. Disponible en: https://espanol.nichd.nih.gov/salud/investigacion-clinica
- Real Decreto 1090/2015, de 4 de diciembre, por el que se regulan los ensayos clínicos con medicamentos, los Comités de Ética de la Investigación con medicamentos y el Registro Español de Estudios Clínicos. Boletín Oficial del Estado, nª 307, 24 de diciembre de 2015.
- BD Metrics. Datos y Análisis de la 30ª Publicación. Proyecto BEST (Investigación Clínica en Medicamentos). Farmaindustria y Plataforma Tecnología Española Medicamentos Innovadores. 15 de noviembre de 2021.
- Farmaindustria [página Web]. Comunicación de Farmaindustria [19/05/2021; 09/11/2021]. Disponible en: https://www.farmaindustria.es/web/otra-noticia/dia-internacional-del-ensayo-clinico-pacientes-espanoles-ya-se-estan-tratando-con-los-farmacos-del-futuro/
- Normas de Buena Práctica Clínica E6(R2). 1 de diciembre de 2016. EMA/CHMP/ICH/135/1995. Comité de medicamentos de uso humano.
- Centers for Disease Control and Prevention. [página Web]. Centers for Disease Control and Prevention, National Center for Emerging and Zoonotic Infectious Diseases (NCEZID), Division of Healthcare Quality Promotion (DHQP) [2/05/2012; 09/11/2021]. Disponible en: https://www.cdc.gov/injectionsafety/index.html
- Gaspar Carreño, M, Torrico Martín, F, Novajarque Sala, L, Batista Cruz, M, Ribeiro Gonçalves, P, Porta Oltra, B, Sánchez Santos, J. C. Medicamentos de Administración Parenteral: Recomendaciones de preparación, administración y estabilidad. Farmacia Hospitalaria [en línea]. 2014, 38(6), 461-467. ISSN: 1130-6343.
- Real Decreto-ley 16/2012, de 20 de abril, de medicas urgentes para garantizar la sostenibilidad del Sistema Nacional de Salud y mejora la calidad y seguridad de sus prestaciones. Boletín Oficial del Estado, nª 98, 24 de abril de 2012.
- Ministerio de Sanidad, Servicios Sociales e Igualdad. Guía de Buenas Prácticas de Preparación de Medicamentos en Servicios de Farmacia Hospitalaria. Junio 2014.
- Consejo de Europa. Resolución CM ResAp 1 sobre las exigencias relativas a la garantía de calidad y de inocuidad de los medicamentos preparados en las farmacias para las necesidades especiales de los pacientes. 1103ª reunión de los ministros delegados del Comité de ministros. 19 de enero de 2011.
- Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios. Boletín Oficial del Estado, nº 177, 25 de julio de 2015.
