1. INTRODUCCIÓN
Los productos sanitarios son, junto con los medicamentos, la herramienta terapéutica más importante en la asistencia sanitaria y cuidados de salud de la población. Abarcan un amplio rango de productos de características muy diversas tanto por sus especificaciones técnicas como por sus mecanismos de acción o sus fines. A pesar de que llevan a cabo sus funciones de formas muy variadas, comparten una serie de características que nos permiten englobarlos bajo una definición común, diferenciándolos de otros artículos como los medicamentos, los cosméticos, los complementos alimenticios o los biocidas. Se clasifican, a su vez, en función de su complejidad tecnológica, su fin y sus potenciales riesgos, lo que condiciona los procedimientos de evaluación de la conformidad en la Unión Europea.
A principios de los años 90, la Comunidad Económica Europea (CEE) aprobó sus primeras directivas relativas a la comercialización de productos sanitarios. Por un lado, la Directiva 90/385/CEE tenía como objetivo el acercamiento de las diferentes legislaciones de los Estados miembros, con el fin de tener un marco normativo similar en lo referente a los productos sanitarios implantables activos (1). Por otro lado, la Directiva 93/42/CEE, tomaba de ejemplo la anterior y trataba de hacer lo propio con los productos sanitarios (2).
Estas directivas buscaban facilitar la comercialización de productos sanitarios en el mercado interior de la CEE y, para ello, seguían una estructura muy similar a las directivas relativas a los productos industriales. Sin embargo, y debido a las particularidades de estos productos, como su uso sobre las personas o sus fines médicos, la CEE no sólo pretendía lograr una aproximación de las diferentes normativas de los Estados miembros, sino que además establecía una serie de requerimientos técnicos que perseguían garantizar la seguridad del paciente y minimizar los riesgos asociados a los productos sanitarios.
Así, estas normativas tenían rango de directivas (actos legislativos en los que se establecían los objetivos que todos los Estados miembros debían cumplir), y precisaban de su transposición al ordenamiento jurídico nacional. En España, la Directiva 90/385/CEE, relativa a los productos sanitarios implantables activos, se transpuso mediante el Real Decreto 634/1993, derogado posteriormente a través del Real Decreto 1616/2009 (3, 4); y la Directiva 93/42/CEE, relativa a los productos sanitarios, se transpuso mediante el Real Decreto 414/1996, derogado posteriormente a través del Real Decreto 1591/2009 (5, 6), pasando así a contemplar los objetivos comunes europeos dentro de su propio marco legislativo. (Figura 1)
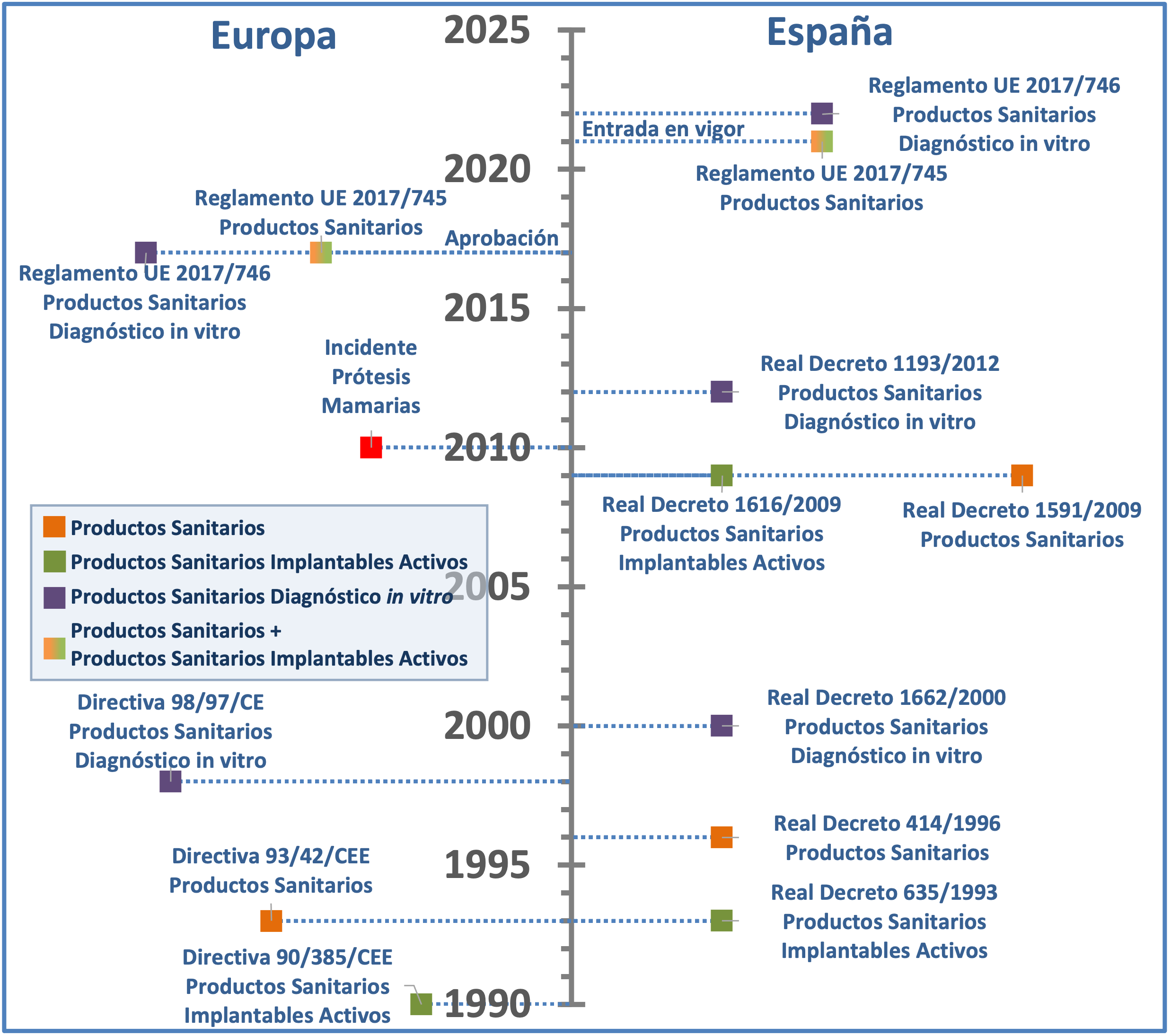
Figura 1. Línea temporal de las normativas europeas sobre productos sanitarios (izquierda) y sus transposiciones al marco jurídico español -cuando procede- (derecha).
En 2010, a través del Sistema de Vigilancia de Productos Sanitarios, las autoridades francesas informaron al resto de Estados miembros de un aumento en las notificaciones de incidentes de ruptura y complicaciones de los implantes mamarios de silicona de un determinado fabricante (7). El incidente se debió a que los implantes mamarios estaban siendo fabricados con un gel de silicona diferente al que había sido aprobado por el organismo notificado correspondiente (8, 9), y se procedió a la suspensión de la puesta en el mercado, distribución, exportación y utilización de las prótesis comercializadas por dicha empresa (10). Este incidente evidenció la necesidad de reforzar significativamente diversos elementos del enfoque normativo entonces vigente, a fin de, por un lado, promover unas normas elevadas de calidad y seguridad que garantizaran un alto nivel de protección de la salud de los pacientes y, por otro lado, garantizar el buen funcionamiento del mercado interior de la Unión Europea.
Dado que estos objetivos no pueden alcanzarse de manera suficiente por los Estados miembros, sino que, debido a su amplitud y sus efectos, pueden lograrse mejor a escala de la Unión, esto supuso el germen para la aprobación del nuevo Reglamento (UE) nº. 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, por el que se modifican la Directiva 2001/83/CE, el Reglamento (CE) nº. 178/2002 y el Reglamento (CE) nº. 1223/2009 y por el que se derogan las directivas 90/385/CEE y 93/42/CEE del Consejo (11), y cuya entrada en vigor estaba planeada para mayo 2020. Sin embargo, la crisis sanitaria ocasionada por la pandemia de SARS-CoV-2 ha supuesto un incremento sin precedentes en la demanda de recursos sanitarios como guantes, mascarillas, geles hidroalcohólicos o respiradores, entre otros (12-14). Por ello, y para prevenir una situación de desabastecimiento y/o una incorrecta implementación del nuevo reglamento en estas circunstancias extraordinarias, el Parlamento Europeo y el Consejo aprobaron una extensión de un año del periodo transitorio, posponiendo la entrada en vigor del reglamento al 26 de mayo de 2021 (15). A diferencia de las directivas hasta ahora vigentes, en tanto que reglamento, la nueva normativa es de aplicación directa sin transposición previa al ordenamiento jurídico de los Estados miembros. Asimismo, este reglamento aúna por primera vez en un único acto legislativo todos los productos sanitarios distintos de los productos para diagnóstico in vitro (cubiertos por el nuevo Reglamento (UE) nº. 2017/746, de aplicación a partir del 26 de mayo de 2022 (15, 16)).
Por otro lado, si bien su definición sigue sin aparecer recogida de forma explícita en el nuevo reglamento, la Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) reconoce la existencia de los denominados ‘productos frontera’, que constituyen “productos complejos para los que existe incertidumbre sobre qué marco legislativo se les aplica” (17). Así, las principales fronteras de productos sanitarios podrán establecerse con: los medicamentos para uso humano (Directiva 2001/83/CE (18)), los complementos alimenticios (Directiva 2002/46/CE (19)), los productos cosméticos (Reglamento (CE) nº. 1223/2009 (20)), los productos de cuidado personal (Real Decreto 1599/1997 (21)), los biocidas (Reglamento (UE) nº. 528/2012 (22)), los equipos de protección individual (Reglamento (UE) nº. 2016/425 (23)) y los productos de consumo (Directiva 2001/95/CE (24)). (Figura 2)

Figura 2. Potenciales fronteras con los productos sanitarios.
Por todo ello, el presente trabajo tiene por objeto analizar la situación reglamentaria de los productos frontera en el actual marco legislativo español. En particular, se analizan las novedades más relevantes del nuevo Reglamento (UE) nº. 2017/745 y se esclarece su ámbito de aplicación con relación a diversos potenciales productos frontera. Además, debido a su relevancia terapéutica, se profundiza en la frontera entre los medicamentos y los productos sanitarios, y se diferencia de aquellas presentaciones en que se usan de manera conjunta, sin constituir una frontera.
2. ANÁLISIS DEL REGLAMENTO (UE) Nº. 2017/745 SOBRE LOS PRODUCTOS SANITARIOS
El Reglamento (UE) nº. 2017/745 sobre los productos sanitarios es una extensa norma que abarca todos los ámbitos relacionados con este tipo de artículos. Se resaltan en este apartado algunos de sus principales epígrafes, concretamente: su definición, su clasificación, sus procedimientos de evaluación de la conformidad, los requerimientos de las investigaciones clínicas que evalúan su seguridad y/o funcionamiento y su base de datos europea.
2.1. Definición de producto sanitario
Según su artículo 2, a efectos del nuevo Reglamento (UE) nº. 2017/745, se entiende por producto sanitario “todo instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo destinado por el fabricante a ser utilizado en personas, por separado o en combinación, con alguno de los siguientes fines médicos específicos:
- diagnóstico, prevención, seguimiento, predicción, pronóstico, tratamiento o alivio de una enfermedad,
- diagnóstico, seguimiento, tratamiento o alivio de una lesión o de una discapacidad,
- investigación, sustitución o modificación de la anatomía o de un proceso o estado fisiológico o patológico,
- obtención de información mediante el examen in vitro de muestras procedentes del cuerpo humano, incluyendo donaciones de órganos, sangre y tejidos.
Y que no ejerce su acción principal prevista en el interior o en la superficie del cuerpo humano por mecanismos farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.
Los siguientes productos también se considerarán productos sanitarios:
- los productos de control o apoyo a la concepción,
- los productos destinados específicamente a la limpieza, desinfección o esterilización de los productos sanitarios, los accesorios de productos sanitarios y los productos sin finalidad médica pero basados en una tecnología similar a productos análogos con finalidad médica (recogidos estos últimos en el anexo XVI del citado reglamento)” (11).
Así, podemos dividir la definición de producto sanitario en tres partes:
La primera parte describe la naturaleza de los productos sanitarios. Sin embargo, en su intención de abarcar lo máximo posible y no dejar ningún tipo de producto fuera de la definición, utiliza un término inespecífico como es el de “materiales”, en el que tienen cabida las sustancias y mezclas químicas, sea cual sea su estado físico. Esto hace que la naturaleza del producto no sea relevante en la delimitación de la frontera, por ejemplo, con la definición legal de medicamento, que incluye “sustancias o combinación de sustancias”. Además, el párrafo incluye en su definición que estos productos deben estar “destinados por el fabricante a ser utilizado en personas” con lo que, esta parte solamente concreta que aquellos que sean exclusivamente para uso veterinario quedarán excluidos de la norma.
La segunda parte incluye una lista de fines médicos específicos que puede tener un producto sanitario.
- El primer guion hace referencia a todas aquellas finalidades relacionadas con la enfermedad. De nuevo, estas finalidades se solapan con aquellas propias de los medicamentos y, por tanto, vuelve a constituir una frontera producto sanitario-medicamento.
- El segundo guion hace referencia a las lesiones o discapacidades. Si bien la definición de medicamento no contempla este objetivo, sí pueden constituir una frontera con otro tipo de artículos denominados ‘ayudas técnicas’. Estas ayudas técnicas, cuyo fin es la integración social de las personas con alguna discapacidad, están constituidas por elementos como las rampas de acceso o los elementos sonoros de los semáforos y, en cierto modo, también tratan de compensar una lesión o discapacidad.
- El tercer guion incluye aquellos productos que pretenden sustituir o modificar la anatomía y, por tanto, pueden no tener una finalidad puramente sanitaria, sino estética. Esto hace que las prótesis mamarias se consideren productos sanitarios y que, en otro tipo de artículos con finalidad estética, el fin que le haya atribuido el fabricante sea clave en la determinación de si se trata de un producto sanitario o no.
- El cuarto guion incluye los productos de examen in vitro. El nuevo reglamento incluye este guion para enfatizar que los productos sanitarios de diagnóstico in vitro son un tipo de productos sanitarios, y deben formar parte de la definición. Sin embargo, siempre ha existido una separación normativa entre los productos sanitarios y los productos sanitarios para diagnóstico in vitro, que se mantiene con el nuevo Reglamento (UE) nº. 2017/746 sobre productos sanitarios para diagnóstico in vitro (16).
- Los siguientes dos guiones hacen referencia a los productos de control o apoyo a la concepción (finalidad que también comparten muchos medicamentos, así como técnicas de reproducción asistida), y los destinados a la limpieza de los productos sanitarios.
La tercera parte especifica que la acción principal de los productos sanitarios no debe llevarse a cabo por mecanismos “farmacológicos, inmunológicos ni metabólicos”. Por tanto, la diferencia entre los medicamentos y los productos sanitarios y, por tanto, la legislación de aplicación a un determinado artículo se establecerá, fundamentalmente, en función de su mecanismo de acción y de cómo logra su fin médico previsto por el fabricante. Cabe destacar que la norma contempla la posibilidad de que a la acción principal llevada a cabo por el producto sanitario puedan contribuir de forma auxiliar mecanismos farmacológicos, inmunológicos o metabólicos.
2.2. Clasificación de productos sanitarios
Los productos sanitarios se clasifican atendiendo a las Reglas de clasificación del capítulo III del anexo VIII del Reglamento (UE) nº. 2017/745. En ellas, se determina el riesgo del producto al valorar el tiempo de contacto (uso pasajero, a corto plazo o prolongado), el grado de invasividad (no invasivo, invasivo por orificio corporal, invasivo quirúrgico o implantable), la fuente de energía de la que depende su funcionamiento (activo o no activo) y la parte del cuerpo con la que se establece el contacto. En función de estos criterios, todos los productos sanitarios pertenecen a una de las cuatro clases: Clase I, Clase IIa, Clase IIb o Clase III, ordenadas de menor a mayor riesgo.
Si bien el nuevo reglamento no ha cambiado el principio de clasificación de los productos sanitarios en función del riesgo, la incorporación de cuatro nuevas reglas a las dieciocho ya existentes en la anterior directiva ha provocado el movimiento de algunos productos a una clase de mayor riesgo (25). Dado que la clasificación de un producto en una clase de mayor riesgo implica una evaluación de la conformidad más exhaustiva, esto puede repercutir de forma positiva en el paciente, ya que recibirá un producto de mayor calidad. Sin embargo, también puede motivar al fabricante a interrumpir la comercialización de un producto en la Unión Europea, debido a los mayores requerimientos administrativos y técnicos que ello supone, repercutiendo negativamente en el paciente.
2.3. Evaluación de la conformidad, Declaración UE de conformidad y Marcado CE de conformidad
Los fabricantes de productos sanitarios deberán realizar el correspondiente procedimiento de evaluación de la conformidad, donde se demuestre si un producto satisface los requisitos del Reglamento (UE) nº. 2017/745. Para productos sanitarios que se consideran de bajo riesgo, como los de clase I no estériles, sin función de medición y/o instrumentos quirúrgicos no reutilizables, debe llevarse a cabo bajo exclusiva responsabilidad de los fabricantes, mientras que para el resto de las clases será necesaria la intervención de un organismo notificado. Los organismos notificados, designados por las distintas autoridades sanitarias de los Estados miembros, son los encargados de llevar a cabo los procedimientos de evaluación de la conformidad y, en función del riesgo del producto, esta evaluación puede basarse en: un análisis del sistema de gestión de la calidad y de la documentación técnica (Anexo IX del Reglamento (UE) nº. 2017/745), un examen de tipo (Anexo X del Reglamento (UE) nº. 2017/745) y/o una verificación de la conformidad del producto (Anexo XI del Reglamento (UE) nº. 2017/745) (por un aseguramiento de la calidad de la producción (Anexo XI, parte A del Reglamento (UE) nº. 2017/745) y/o por una verificación de los productos (Anexo XI, parte B del Reglamento (UE) nº. 2017/745)) (26).
Posteriormente, el fabricante de un producto sanitario asumirá su responsabilidad en el cumplimiento de los requisitos especificados en toda la legislación aplicable al producto, y lo hará constar en una declaración UE de conformidad, que actualizará continuamente.
Finalmente, antes de su introducción al mercado, el fabricante deberá certificar con un marcado CE de conformidad el cumplimiento de los requisitos comunitarios aplicables (27). En aquellos productos que, por su clasificación, requieran de la intervención de un organismo notificado, el marcado CE irá seguido de un código de cuatro dígitos que identifique a este último.
2.4. Evaluación Clínica e Investigaciones Clínicas
Una importante novedad en el Reglamento (UE) nº. 2017/745 es el capítulo VI, relativo a la Evaluación Clínica e Investigaciones Clínicas. En él, se fijan unas normas elevadas de calidad y seguridad, para garantizar, no solo que los datos generados en investigaciones clínicas sean fiables y sólidos, sino que además se proteja la seguridad de los participantes. Mediante esta incorporación a la normativa, los fabricantes están obligados a especificar y justificar el nivel de las pruebas clínicas necesario para demostrar la conformidad con los requisitos de seguridad y funcionamiento del producto sanitario a evaluar.
Por ello, a fin de demostrar su seguridad y funcionamiento, todo fabricante de productos sanitarios implantables o de clase III está ahora bajo la obligación de realizar investigaciones clínicas.
Además, en el caso de los productos sanitarios implantables de clase III se debe llevar a cabo un ‘procedimiento de escrutinio’, mediante el que un panel de expertos independiente del fabricante, del organismo notificado y de las autoridades sanitarias da su opinión sobre las conclusiones del organismo notificado en relación a la evidencia clínica (28).
Sin embargo, cabe destacar que, si el fabricante de un determinado producto demuestra que ya existe un producto equivalente comercializado, aunque no pertenezca al mismo fabricante, y que éste ya ha llevado a cabo la evaluación clínica, el fabricante del segundo producto podrá no realizar la evaluación clínica, valiéndose de los resultados obtenidos por el primero. Por otro lado, en lo relativo a la seguridad de los participantes, se especifican las características del consentimiento informado y se detallan las particularidades de las investigaciones clínicas en grupos vulnerables.
Además, la normativa introduce la creación de un sistema electrónico de investigaciones clínicas con el fin de mantener un registro de los estudios realizados, sus resultados y evitar la duplicación innecesaria de investigaciones clínicas.
2.5. Base de Datos Europea sobre Productos Sanitarios (Eudamed) y Transparencia
El nuevo reglamento contempla, en los artículos 33 y 34, la gestión de una Base de Datos Europea sobre Productos Sanitarios (Eudamed). Esta base de datos tiene, fundamentalmente, dos objetivos: por un lado, permitir la identificación única de los productos, facilitando así su trazabilidad y, por otro, garantizar el acceso a la información relativa a los productos sanitarios.
El primer objetivo se logra mediante la creación de un sistema de Identificación Única de Productos (UDI, por sus siglas en inglés) que será internacional y permitirá la identificación de cualquier producto sanitario comercializado en el mercado interior de la Unión Europea.
El segundo objetivo pretende garantizar uno de los valores clave de la Unión Europea como es la transparencia (29). Su finalidad es facilitar el intercambio de información entre los Estados miembros, reforzando así la colaboración y otorgando las herramientas necesarias para un correcto funcionamiento del mercado interior. Este acceso a la información, no solo de los productos sanitarios, sino de las investigaciones clínicas que los avalan, debe ser posible, no solo por los organismos notificados, los agentes económicos o los promotores, sino por el público en general, y deberá presentarse “en un formato de uso fácil y en el que resulte sencillo realizar búsquedas”.
En este sentido, el preludio al nuevo Reglamento introduce la voluntad de que la transparencia y un acceso adecuado a la información permitan a los pacientes y profesionales de la salud una toma de decisiones más objetiva. Además, estas ideas de transparencia y libertad de información son principios fundamentales de la Unión, y se encuentran expresados en la Carta de los Derechos Fundamentales de la Unión Europea, que en su artículo 42 garantiza el derecho al acceso a documentos en posesión de la Unión (30). Sin embargo, el Reglamento (UE) nº. 2017/745 limita la transparencia al definir la necesidad de confidencialidad, especialmente en los artículos 12, 73, 92 y 109, y en el anexo VII, en las partes 1, 2 y 4.
De hecho, en Estados Unidos, el acceso público a los documentos de la Administración de Alimentos y Medicamentos (FDA, por sus siglas en inglés) reveló en 2009 que solo el 31% de las 78 aprobaciones pre-comercialización de nuevos dispositivos cardiovasculares estaban respaldadas por evidencia científica en más de un ensayo aleatorizado controlado (29, 31, 32). En contraposición, en la Unión Europea todavía no es posible realizar estimaciones similares, dado que la base de datos Eudamed, cuyo desarrollo e implementación son calificados de “alta prioridad” por la Comisión Europea, solo tiene la mitad de los seis módulos que la compondrán operativos (disponibles a junio de 2022: Actors registration, UDI/Devices registration y Notified Bodies and Certificates; pendientes a junio de 2022: Clinical Investigations and performance studies, Vigilance and post-market surveillance y Market Surveillance) (33). En consecuencia, muchos productos sanitarios comercializados en la Unión, con plena autorización y certificación, no tienen sus datos de seguridad, eficacia o ensayos clínicos publicados.
Además, “solo el 13% de los 13327 ensayos clínicos registrados en ClinicalTrials.gov y completados entre 2008 y 2012 (el 79% de ellos sobre medicamentos y el 11% sobre productos sanitarios) comunicaron sus resultados en los 12 meses tras su finalización” y “solo el 49% de 177 estudios sobre nuevos dispositivos cardiovasculares ha sido publicado hasta 7 años tras su finalización” (29, 34). Esto choca con las recomendaciones de la Asociación Médica Mundial que, en su Declaración de Helsinki, publicada por primera vez en 1964 (y revisada por séptima vez en 2013), expresa en sus párrafos 35 y 36 la necesidad de que todo estudio con seres humanos deba ser inscrito en una base de datos disponible al público y de que todos los resultados, incluidos los negativos o no concluyentes, sean publicados (35). La Organización Mundial de la Salud coincide en este sentido y describe el registro de todos los ensayos de intervención como una “responsabilidad científica, ética y moral” (36).
En materia de medicamentos, tres juicios del Tribunal General de la Unión Europea, en relación al acceso a los datos de la autorización de comercialización, confirmaron que la política de la EMA de divulgación en virtud del interés público está respaldada por las normas de transparencia de la Unión, y que esta siempre prevalece sobre cualquier argumento en favor de la confidencialidad (29, 37). Por tanto, aunque la confidencialidad es entendible durante el desarrollo de un producto, esta carece de sentido una vez este ha sido aprobado y fijado con un marcado CE, dado que resultados clínicamente relevantes en la evaluación de un producto sanitario rara vez coinciden con datos comercialmente sensibles (29).
3. PRODUCTOS FRONTERA
Si bien los productos frontera no cuentan con una definición legal recogida en ninguna normativa europea, la EMA los define como “productos complejos para los que existe incertidumbre sobre qué marco legislativo se les aplica” (17). El Working Group on Borderline and Classification, presidido por la Comisión Europea y formado por representantes de todos los Estados miembros y partes interesadas de la industria, es un grupo de trabajo que se reúne regularmente con el fin de intercambiar opiniones y, si es posible, alcanzar un consenso en cuanto a la situación reglamentaria de diversos artículos, con el objetivo último de asegurar un enfoque uniforme (38, 39). Este grupo ha elaborado un manual que, si bien no es vinculante, permite la consulta de diversos ejemplos que han ido surgiendo y cómo han sido abordados, y se actualiza periódicamente (39).
Por otro lado, dadas las particularidades de este tipo de artículos, y para garantizar unas decisiones coherentes al respecto en todos los Estados miembros, la Comisión Europea podrá consultar al Grupo de Coordinación de Productos Sanitarios (MDCG, por sus siglas en inglés), cuya creación se impulsa por el artículo 103 del propio Reglamento (UE) nº. 2017/745, así como a la EMA, a la Agencia Europea de Sustancias y Mezclas Químicas y a la Autoridad Europea de Seguridad Alimentaria, según proceda.
La frontera adquiere especial relevancia a la hora de certificar un determinado producto para su salida al mercado, dado que el lado de la frontera en el que un producto se establezca determinará la regulación de aplicación. A continuación, se analizan las fronteras de los productos sanitarios con los complementos alimenticios, los cosméticos, los productos de cuidado personal, los biocidas, los equipos de protección individual y los productos de consumo. El análisis de la frontera entre productos sanitarios y medicamentos se posterga al siguiente apartado por su especial relevancia. (Ver apartado 4)
3.1. Frontera con Complementos alimenticios
Los complementos alimenticios se definen en la Directiva 2002/46/CE como “los productos alimenticios cuyo fin sea complementar la dieta normal y consistentes en fuentes concentradas de nutrientes o de otras sustancias que tengan un efecto nutricional o fisiológico, en forma simple o combinada, comercializados en forma dosificada, es decir cápsulas, pastillas, tabletas, píldoras y otras formas similares, bolsitas de polvos, ampollas de líquido, botellas con cuentagotas y otras formas similares de líquidos y polvos que deben tomarse en pequeñas cantidades unitarias” (19). Si bien esta directiva ya concreta una lista de vitaminas y minerales que podrán contener este tipo de artículos, la variedad de sustancias que se han ido incorporando, debido a la innovación de los fabricantes a lo largo de los años, ha obligado a publicar nuevas normativas que actualicen dicho listado. Estas normativas son: el Reglamento (CE) nº. 1170/2009, relativo a las listas de vitaminas y minerales y sus formas, y el Reglamento (UE) nº. 1161/2011, relativo a las listas de sustancias minerales (40, 41).
Sin embargo, la Directiva 2002/46/CE no establece la regulación relativa a otras sustancias distintas de las vitaminas y los minerales, y deja a criterio de cada Estado miembro su determinación.
Por tanto, en función de las distintas legislaciones nacionales en materia de alimentación, un mismo componente puede considerarse medicamento, producto sanitario o complemento alimenticio, dependiendo del Estado miembro que tramite su autorización. Esta diferencia viene motivada, en ocasiones, por el uso tradicional de determinados componentes (por ejemplo, extractos de determinadas plantas) en unos territorios de la Unión Europea, en contraposición con un uso inexistente en otros (42).
En la práctica, estas diferencias han llevado a que sea el propio fabricante el que decida en qué territorio iniciar su regulación, con el fin de obtener la autorización de comercialización que resulte de su interés. Posteriormente el fabricante llevará a cabo un proceso de reconocimiento mutuo con el que su artículo, con una determinada sustancia diferente de vitaminas y minerales, será comercializado en el resto de los territorios de la Unión Europea con la misma consideración que en el Estado inicial (42). Sin embargo, si bien el proceso de reconocimiento mutuo es una importante herramienta en la libre circulación de mercancías y el correcto funcionamiento del mercado interior de la Unión Europea, el artículo 36 del Tratado de Funcionamiento de la Unión Europea facilita que un Estado miembro prohíba o restrinja la importación por razones de protección de la salud y vida de las personas, lo que permite un control más estricto de este tipo artículos por parte de aquellos Estados miembros que así lo deseen (43).
Además, en determinados casos se han publicado reglamentos relativos a familias de complementos alimenticios concretas, con el fin de intentar armonizar las decisiones en el territorio europeo con respecto a sustancias con características controvertidas (como el Reglamento (UE) nº. 119/2014, relativo a la levadura enriquecida con cromo y el lactato de cromo (III) trihidrato; el Reglamento (UE) nº. 2015/414, relativo al ácido (6S)-5-metiltetrahidrofólico y la sal de glucosamina; y el Reglamento (UE) nº. 2017/1203, relativo al silicio orgánico (monometilsilanotriol) y a los oligosacáridos fosforilados de calcio (POs-Ca®)), pero se trata de actuaciones puntuales (44-46).
• Se ilustra esta frontera con los productos para la prevención de infecciones del tracto urinario a base de proantocianidinas:
Entre los productos destinados a la prevención de infecciones del tracto urinario, encontramos una subcategoría a base de proantocianidinas del arándano rojo americano. Este tipo de productos inhibe la adhesión de Escherichia coli uropatogénica a las células epiteliales del tracto urinario, principal causante de las infecciones urinarias (47-49).
Podría argumentarse que este tipo de productos lleva a cabo su acción principal por un mecanismo farmacológico, a pesar de que no interaccionan con las células del paciente. Por ello, en 2012 el Tribunal de Justicia Alemán, y en nombre de la Comisión Europea, tuvo que hacer una interpretación de las definiciones de ‘medicamento’ y de ‘acción farmacológica’ de la Directiva 2001/83/CE sobre medicamentos para uso humano. En ella, el Tribunal determinaba que la acción farmacológica de un determinado medicamento puede establecerse con cualquiera de los constituyentes celulares del paciente, independientemente de que se trate de células humanas o, como es el caso, de células de la microbiota presentes en el organismo (50). Esto llevó a que la Comisión Europea emitiera la Decisión de Ejecución (UE) 2017/1445 que materializaba la exclusión como productos sanitarios de aquellos artículos cuya acción principal prevista está basada en las proantocianidinas del arándano rojo (51).
Por tanto, no podrán ser regulados como productos sanitarios según el nuevo Reglamento (UE) nº. 2017/745. Actualmente, varios de estos artículos se encuentran comercializados como complementos alimenticios.
3.2. Frontera con Cosméticos
Desde su entrada en vigor en 2013, los productos cosméticos se regulan bajo el Reglamento (CE) nº. 1223/2009, y constituyen “toda sustancia o mezcla destinada a ser puesta en contacto con las partes superficiales del cuerpo humano (epidermis, sistema piloso y capilar, uñas, labios y órganos genitales externos) o con los dientes y las mucosas bucales, con el fin exclusivo o principal de limpiarlos, perfumarlos, modificar su aspecto, protegerlos, mantenerlos en buen estado o corregir los olores corporales” (20). Por otro lado, si bien el reglamento es de aplicación directa, éste deja que los Estados miembros regulen una serie de aspectos nacionales tales como las autoridades competentes, los procedimientos de comunicación de incidentes y las actividades de control del mercado en materia de productos cosméticos, que en España están cubiertos por el Real Decreto 85/2018 (52).
• Se ilustra esta frontera con los lápices estípticos de alumbre:
Los lápices estípticos de alumbre son unas barras formadas por sales de azufre que, al aplicarse sobre heridas abiertas, producen su cierre, interrumpiendo el sangrado. Si bien podría argumentarse que se trata de productos cosméticos puesto que se aplican sobre la superficie corporal, no gozan de tal consideración dado que la intención del fabricante es el contacto directo con piel herida (53). Por otro lado, no se trata de medicamentos dado que no actúan por medios farmacológicos, inmunológicos ni metabólicos, sino que su mecanismo de acción principal es una simple reacción química que provoca la precipitación de proteínas de la capa superficial de la piel, contrayendo el tejido y, por tanto, interrumpiendo el sangrado (54).
Por ello, se considerarán productos sanitarios de clase IIa según la regla 4 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
3.3. Frontera con Productos de cuidado personal
La Unión Europea establece un marco legislativo común para la mayoría de los artículos comercializados en el mercado interior. Sin embargo, una serie de ellos no se acoge a ninguna de las normativas comunitarias, lo que hace que cada Estado miembro los regule de diferente manera. Este es el caso de los productos de cuidado personal.
En España, el Real Decreto 1559/1997 sobre productos cosméticos, en su disposición adicional segunda, modificada a través de los Reales Decretos 2131/2004 y 209/2005, define los productos de cuidado personal como “sustancias o preparados, que sin tener la consideración legal de cosméticos, biocidas, productos sanitarios o medicamentos, están destinados a ser aplicados sobre la piel o mucosas del cuerpo humano con la finalidad de higiene o de estética, o para neutralizar o eliminar ectoparásitos, tales como dentífricos, productos de estética, pediculicidas, hidratantes vaginales, limpiadores anales en caso de hemorroides, productos para el masaje deportivo, limpiadores nasales o limpiadores oculares, o cualquier otro producto que pueda ser calificado como tal” (21, 55-57).
• Se ilustra esta frontera con los productos de blanqueamiento dental:
Los productos de blanqueamiento dental tienen como objetivo el cambio de coloración de los dientes a un tono más blanco (58, 59). Si bien es cierto que en este proceso pueden eliminar placa u otros residuos presentes en la cavidad bucal, eliminar olores e incluso eliminar agentes causantes de la caries, su objetivo principal es claramente estético. Por tanto, en ocasiones podrán ayudar a la prevención de determinadas afecciones bucodentales, sin ser ese el fin destinado por el fabricante. Además, el cambio de color no se considera el tratamiento de los síntomas de una afección bucodental, sino su enmascaramiento.
Por ello, en España se encuentran comercializados como productos de cuidado personal según el Real Decreto 1559/1997.
3.4. Frontera con Biocidas
En 2009 entró en vigor una nueva normativa, relativa a las sustancias químicas (Reglamento (CE) nº. 1907/2006 (60)), que obligó a redefinir los biocidas. Esto dio lugar a un nuevo reglamento: el Reglamento (UE) nº. 528/2012 relativo a la comercialización y el uso de los biocidas, que los define como “toda sustancia o mezcla, en la forma en que se suministra al usuario, que esté compuesto por, o genere, una o más sustancias activas, con la finalidad de destruir, contrarrestar o neutralizar cualquier organismo nocivo, o de impedir su acción o ejercer sobre él un efecto de control de otro tipo, por cualquier medio que no sea una mera acción física o mecánica” (22).
Por otro lado, en España los productos desinfectantes se han comercializado tradicionalmente bien como biocidas, si su función prevista es como antiséptico para piel sana y/o como desinfectante de ambientes clínicos y quirúrgicos; como medicamentos, si su función prevista es como antiséptico de piel dañada; o bien como productos sanitarios, si su función prevista es como producto de desinfección de productos sanitarios (61). Sin embargo, desde el 1 de junio de 2022, se atribuye la condición de medicamento a cualquier producto desinfectante cuya función prevista sea como antiséptico destinado al campo quirúrgico preoperatorio y/o como desinfectante del punto de inyección (62).
• Se ilustra esta frontera con los geles hidroalcohólicos de desinfección de manos:
En el Reglamento (UE) nº. 2017/745 se recogen como productos sanitarios aquellos destinados a la limpieza, desinfección o esterilización de otros productos sanitarios, pero no aquellos destinados a la desinfección de manos. Además, dado que la acción principal de este tipo de geles hidroalcohólicos no es una simple limpieza de manos con una finalidad estética, no podrán considerarse productos cosméticos (63, 64). Por otro lado, los geles hidroalcohólicos de desinfección de manos tampoco tienen como finalidad la prevención de una enfermedad, sino que se trata de “antisépticos para la piel sana” (65), es decir, biocidas desinfectantes para la higiene humana, conforme al anexo V del Reglamento (UE) nº. 528/2012.
Por ello, y dado que la prevención de enfermedad sería una consecuencia de la destrucción o neutralización del organismo nocivo, y no su objetivo principal, estos productos deberán comercializarse como biocidas (66, 67).
A raíz de la crisis ocasionada por la pandemia de SARS-CoV-2, la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) mantiene una lista, actualizada regularmente, en la que publica aquellos geles hidroalcohólicos de desinfección de manos con eficacia viricida demostrada (autorizados atendiendo a la norma UNE-EN 14476) (68, 69).
3.5. Frontera con Equipos de protección individual (EPI)
El Reglamento (UE) nº. 2016/425 relativo a los equipos de protección individual los define como “el equipo diseñado y fabricado para ser llevado puesto o ser sostenido por una persona para protegerse contra uno o varios riesgos para su salud o seguridad” y se clasifican en diferentes categorías en función del riesgo frente al que protegen (23).
• Se ilustra la frontera con los distintos tipos de mascarillas:
En el ámbito sanitario podemos encontrar infinidad de tipos de mascarillas, cada una de ellas concebida para una situación distinta. Algunas de ellas son, al mismo tiempo, productos sanitarios y equipos de protección individual (70). Este fenómeno es específico de la frontera con los EPIs, siendo así la única categoría de productos que presenta esta dualidad y donde la denominación no es excluyente.
Sin embargo, a raíz de la crisis sanitaria ocasionada por la pandemia de SARS-CoV-2, se han popularizado fundamentalmente dos tipos: las mascarillas quirúrgicas y las medias máscaras filtrantes de protección contra partículas (mascarillas autofiltrantes o FFP2, por sus siglas en inglés de Filtering Face Piece) (71) (Tabla 2).

Tabla 2. Tipos de mascarillas y las normativas europeas y especificaciones que les son de aplicación.
Las mascarillas quirúrgicas están diseñadas para filtrar el aire exhalado y, por tanto, su objetivo es evitar la dispersión vírica (70, 72). Su fin médico específico es la prevención de la enfermedad dado que, al llevarla puesta, se evita la transmisión de agentes infecciosos. Por ello, no pueden clasificarse como equipos de protección individual, y pertenecen a la categoría de productos sanitarios (73). Concretamente, se trata de productos sanitarios de clase I según la regla 1 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
Las mascarillas FFP2 están diseñadas para filtrar el aire inhalado y, por tanto, su objetivo es evitar la entrada de partículas contaminantes en el organismo (70, 72). Están clasificadas como EPIs de categoría III puesto que protegen frente a “agentes biológicos nocivos”, según lo establecido en las Categorías de riesgos con respecto a los EPIs del anexo I del Reglamento (UE) nº. 2016/425 (23, 74).
Por tanto, tal y como establece el Ministerio de Consumo en sus recomendaciones frente a la COVID-19, las mascarillas quirúrgicas están indicadas preferentemente en “personas sintomáticas o asintomáticas positivas”, por esa misión de proteger a los demás de la dispersión vírica en los aerosoles generados al estornudar, toser o hablar; mientras que las mascarillas EPI están indicadas preferentemente en “quienes cuiden o estén en contacto con personas sintomáticas o positivos por COVID-19”, por ofrecer esa protección personal (75).
3.6. Frontera con Productos de consumo
La Directiva 2001/95/CE relativa a la seguridad general de los productos, requiere que los fabricantes se aseguren de que los artículos en venta sean seguros y que se tomen las medidas correctivas cuando no sea el caso (24). Se encuentra transpuesta al marco legislativo español a través del Real Decreto 1801/2003 sobre seguridad general de los productos (76).
• Se ilustra esta frontera con las mascarillas higiénicas:
En el contexto de la pandemia de SARS-CoV-2 se ha popularizado el uso de mascarillas higiénicas por parte de la población en general. Esto es debido, en parte, a que el Ministerio de Consumo las recomienda en “personas sanas” (75).
Si bien existen unas normativas UNE relativas a sus especificaciones técnicas, que establecen si son reutilizables o no, su eficacia de filtración bacteriana y su respirabilidad, estas no son de obligado cumplimiento (75). Así, en el mercado podemos encontrar mascarillas higiénicas de tres tipos: que cumplen las especificaciones UNE (77, 78); que cumplen otras especificaciones (porque no alcanzan los estándares de calidad UNE o han sido fabricadas en base a otras normas); y sin especificaciones (no sometidas a ensayos ni verificaciones) (Tabla 2).
Por tanto, al no suponer una barrera entre un riesgo potencial y el usuario (FFP2, EPI), ni evitar la transmisión de agentes infecciosos (quirúrgicas, producto sanitario), se comercializarán como productos de consumo.
4. FRONTERA PRODUCTOS SANITARIOS – MEDICAMENTOS
En 1965 surge la primera directiva de carácter europeo que pretende armonizar las diferentes legislaciones en materia de medicamentos de los Estados miembros (Directiva 65/65/CEE (79)).
Desde entonces, diferentes normativas se han ido sucediendo hasta la entrada en vigor en 2001 de la actual Directiva 2001/83/CE sobre medicamentos para uso humano, que regula la autorización, importación y producción de éstos en la Unión Europea. Esta directiva define los medicamentos como “toda sustancia o combinación de sustancias que se presente como poseedora de propiedades curativas o preventivas con respecto a las enfermedades humanas” o “todas las sustancias o combinación de sustancias que puedan administrarse al hombre con el fin de establecer un diagnóstico médico o de restablecer, corregir o modificar las funciones fisiológicas del hombre” (18), y establece los procedimientos de autorización para demostrar que se cumplen las normas de alta calidad y seguridad.
Por otro lado, además de la Directiva 2001/83/CE existen otras normativas relativas a otros aspectos relacionados con los medicamentos para uso humano: la Directiva 2003/94/CE, relativa a las prácticas de correcta fabricación de medicamentos (80); las directrices de 2013 y 2015, relativas a la correcta distribución de medicamentos y de principios activos, respectivamente (81, 82); y las directrices de 2015, relativas a la evaluación de riesgos en la fabricación de excipientes para medicamentos para uso humano (83).
Cabe destacar que la seguridad y el principio de protección de la salud de los ciudadanos quedan salvaguardados por la modificación del artículo 2, apartado 2, de la Directiva 2001/83/CE, reflejado en la Directiva 2004/27/CE, que establece que “en caso de duda, cuando, considerando todas las características de un producto, este pueda responder a la definición de medicamento y a la definición de producto contemplada por otras normas comunitarias, se aplicará la presente Directiva” (84).
Se ilustra esta frontera con los siguientes ejemplos (Tabla 3):

Tabla 3. Ejemplos de frontera productos sanitarios – medicamentos, de medicamentos que incluyen un producto sanitario (integrados y no integrados) y de productos sanitarios con sustancia medicinal auxiliar.
• Cremas de óxido de zinc:
Las cremas con óxido de zinc llevan mucho tiempo comercializadas. Sin embargo, recientemente han experimentado una especie de “resurgir” tras una extensa campaña publicitaria por parte de determinados laboratorios que las califican como “cremas botiquín” (85, 86).
Estas cremas contienen sustancias para el tratamiento o la prevención de irritaciones menores de la piel, tales como quemaduras, cortes, erupciones del pañal o eczemas, entre otros. Sin embargo, dependiendo del fin previsto por el fabricante y el mecanismo por el que alcanzan su acción principal prevista, podrán clasificarse como productos sanitarios o como medicamentos.
Ciertamente, el óxido de zinc tiene una acción farmacológica y metabólica, lo que clasificaría a estos productos como medicamentos (87, 88). Sin embargo, la normativa contempla que los productos sanitarios puedan tener componentes con acciones farmacológicas, metabólicas o inmunológicas, siempre y cuando estas sean auxiliares a la función principal.
Cobran importancia, por tanto, las afirmaciones del fabricante respecto al mecanismo por el que el producto alcanza su acción principal prevista. Si el fabricante establece que la acción principal prevista es la de barrera, y el óxido de zinc actúa de forma auxiliar, estos productos pueden comercializarse bajo el Reglamento (UE) nº. 2017/745 como productos sanitarios. Concretamente, al llevar incorporada una sustancia que, utilizada por separado, puede considerarse un medicamento, se trataría de productos sanitarios de clase III según la regla 14 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
• Gotas oftálmicas:
En el mercado existen diversos artículos que pueden clasificarse como gotas oftálmicas. Estos artículos se regularán por distintas normativas en función de su composición y del fin establecido por el fabricante, fundamentalmente. Así, aquellos que lleven un antibiótico, un corticoide o cualquier otro principio activo que ejerza su acción principal mediante un mecanismo farmacológico, serán medicamentos (89, 90); mientras que aquellos destinados a la limpieza o desinfección de lentes de contacto serán productos sanitarios (91, 92), según la regla 16 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745. Sin embargo, hay otros tipos de gotas oftálmicas comercializados cuyo fin suele estar relacionado con el alivio del cansancio, la incomodidad o la irritación causada por factores ambientales (como el polvo, el calor o el humo). Será en este tipo de gotas, en las que la acción principal prevista no se alcance por un mecanismo farmacológico claro, donde surja la frontera entre producto sanitario y medicamento, y adquiera especial relevancia el análisis.
Dentro de estas gotas para el alivio del cansancio, la incomodidad o la irritación podemos encontrar una gran variedad de sustancias. La mayoría ellas llevan a cabo una función hidratante, lubricante o de arrastre, y no estimulan el saco lagrimal, o cualquier otra zona ocular, para que se favorezca el lagrimeo (93). Por tanto, en el caso descrito en que las gotas oftálmicas actúan de forma física, proporcionando un aporte externo de humedad, actuando en la zona superficial del ojo, hidratándolo o dando lugar a un arrastre de las posibles partículas dañinas, se comercializarán como productos sanitarios (94).
Concretamente, se trata de productos sanitarios de clase I según la regla 5 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
• Productos para el control del peso:
Uno de los principales factores que contribuyen al aumento de peso en la población es el consumo excesivo de grasas en la dieta, que son absorbidas por el intestino, y acumuladas en el organismo. Con el tiempo, la acumulación de grasa puede dar lugar a un incremento de peso (95).
Determinados productos para el control del peso están compuestos por fibra alimentaria que, mediante adsorción, forma un complejo con las grasas alimentarias en el estómago, evitando que se absorban (96). Así, esta fibra alimentaria es responsable de la alteración en la absorción de nutrientes, logrando este fin mediante un mecanismo de acción mecánico, físico o químico básico.
Por ello, se encuentran comercializados como productos sanitarios de clase IIb, atendiendo a la nueva regla 21 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
• Productos viscoelásticos intraarticulares de ácido hialurónico:
La osteoartritis es la enfermedad articular más común en el mundo y es debida tanto a factores anatómicos como funcionales (97). Su abordaje terapéutico suele incluir una serie de modificaciones del estilo de vida (principalmente aumento del ejercicio físico y disminución del peso corporal), tratamiento farmacológico y, en última instancia, cirugía (98-100).
Los productos viscoelásticos intraarticulares de ácido hialurónico se utilizan en el tratamiento de la osteoartritis. Sin embargo, a pesar de la cantidad de estudios publicados en relación a las diferencias en el mecanismo de acción en función del peso molecular y estado de isomería del ácido hialurónico, no está claro cuál de ellos aporta mejores resultados clínicos en el tratamiento de la osteoartritis articular (101). No obstante, es de esperar que aquellos con un alto peso molecular ejerzan su acción principal prevista mediante la suplementación del líquido sinovial, aportando viscosidad y elasticidad, por lo que su mecanismo de acción principal sería mecánico; mientras que aquellos con un bajo peso molecular ejerzan su acción principal mediante la absorción del ácido hialurónico, estimulando la producción de condrocitos, por lo que su mecanismo de acción principal sería farmacológico (102). Por tanto, los productos viscoelásticos intraarticulares de ácido hialurónico con un bajo peso molecular se regularán bajo la Directiva 2001/83/CE sobre medicamentos para uso humano, mientras que los productos de elevado peso molecular se considerarán productos sanitarios de clase III, atendiendo a la regla 8 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745 (103).
5. MEDICAMENTOS QUE INCLUYEN UN PRODUCTO SANITARIO
Desde hace años, el desarrollo tecnológico viene marcando la mayoría de los aspectos de la sociedad actual. Se han incorporado a la vida diaria de los ciudadanos una gran variedad de dispositivos de toda clase que han modificado muchos de los hábitos de vida. El sector de los cuidados de la salud no ha sido diferente, y se han ido introduciendo diversos sistemas, tanto a nivel hospitalario como ambulatorio, que han permitido una dosificación más precisa, un incremento en la autonomía del paciente o una mayor comodidad, entre otros, suponiendo un avance en la mejora de los tratamientos y en la calidad de vida de los pacientes. De hecho, en 2019, la EMA declaraba en su informe anual que aproximadamente uno de cada cuatro medicamentos aprobados por procedimiento centralizado incluían un producto sanitario en su composición (104). Este dato no se ha actualizado en su último informe anual (105).
Si bien hoy en día ninguna normativa de carácter europeo recoge la definición legal de medicamento que incluye un producto sanitario, también conocidos como ‘productos combinados’, podrían entenderse como medicamentos que contienen uno o más productos sanitarios como parte integral de su composición, así como medicamentos para los que uno o más productos sanitarios y/o componentes son necesarios para el uso del medicamento (106, 107).
Los medicamentos que incluyen un producto sanitario se pueden clasificar en dos categorías: los productos combinados integrados y los productos combinados no integrados. Cabe destacar que, en contraposición con los productos sanitarios con una sustancia medicinal auxiliar, que serán siempre de clase III en virtud de la regla 14 del nuevo reglamento, todas las otras reglas de clasificación son de aplicación a la parte de producto sanitario de los productos combinados y, por tanto, los productos combinados podrán estar formados por productos sanitarios que no sean necesariamente de clase III.
5.1. Productos Combinados Integrados
Los productos combinados integrados son aquellos en los que el medicamento incorpora uno o más productos sanitarios como parte de su composición. Según los segundos párrafos de las secciones 8 y 9 del artículo 1 del Reglamento (UE) nº. 2017/745, los productos combinados integrados se regularán como medicamentos, cumpliendo las exigencias de la Directiva 2001/83/CE, cuando ejerzan su fin médico previsto mediante la sustancia medicamentosa, que no actuará como accesoria a la acción del producto sino como responsable principal de la acción.
Por otro lado, el artículo 117 del nuevo Reglamento (UE) nº. 745/2017 introduce una modificación de la directiva que regula los medicamentos para uso humano (Directiva 2001/83/CE), que exige que el expediente de autorización de comercialización de los productos combinados integrados incluya los resultados de la evaluación de la conformidad de la parte que constituye un producto sanitario. Por tanto, el medicamento cumplirá las exigencias de la Directiva 2001/83/CE sobre medicamentos para uso humano, y a la parte que constituya un producto sanitario se le aplicarán los requisitos generales en materia de seguridad y funcionamiento del Reglamento (UE) nº. 2017/745 sobre productos sanitarios.
Se ilustran los productos combinados integrados con los siguientes ejemplos:
• Jeringas y plumas precargadas
En las jeringas y plumas precargadas, la parte de producto sanitario (la jeringa) tiene únicamente una función de administración, mientras que la acción principal prevista la lleva a cabo el medicamento inyectado. Pueden administrar un amplio rango de terapias (vacunas, tratamientos contra el cáncer, contra la artritis reumatoide o contra la trombosis venosa profunda), y su uso permite una dosificación más individualizada y promueve un rol más activo del paciente en el control de su medicación (108, 109).
La parte del producto sanitario de las jeringas o plumas precargadas que no llevan incorporada la aguja, no están conectadas a un producto activo y están destinadas a administrar un líquido distinto de un fluido corporal, se considera producto sanitario no invasivo de clase I según la regla 2 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
• Inhaladores de polvo seco precargados no recargables
Los inhaladores de polvo seco precargados no recargables permiten la administración local de medicamentos en las vías respiratorias (110). Por tanto, la parte de producto sanitario (el inhalador) actúa como sistema de administración, mientras que el medicamento inhalado lleva a cabo el fin médico previsto de tratamiento de la afección respiratoria (111).
Tomando en consideración el riesgo de este tipo de productos, el nuevo reglamento ha revisado su clasificación y ha añadido la nueva regla 20 específica para los inhaladores. Esta nueva regla establece que todos los inhaladores invasivos en relación con los orificios corporales son de clase IIa, salvo que su modo de acción tenga un impacto decisivo sobre la eficacia y la seguridad del medicamento administrado o estén destinados al tratamiento de afecciones que suponen un riesgo vital, en cuyo caso se clasifican en la clase IIb.
• Dispositivos intrauterinos para liberación de medicamentos
Los dispositivos intrauterinos para liberación de medicamentos, especialmente aquellos dedicados a la liberación de levonogestrel, se usan tanto como anticonceptivos como en afecciones como la menorragia, dado que han demostrado una reducción del dolor en la dismenorrea, la adenomiosis y la endometriosis (112-114). En este caso, la parte de producto sanitario (el dispositivo intrauterino) lleva a cabo una función de soporte o forma de administración del medicamento, que lleva a cabo la acción principal prevista (115).
Dado que se trata de productos invasivos de uso prolongado cuya acción principal prevista es anticonceptiva, la parte de producto sanitario de los dispositivos intrauterinos para liberación de medicamentos se considera de clase III según la regla 15 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
Así, los productos combinados integrados se consideran medicamentos y, por tanto, están regulados bajo la Directiva 2001/83/CE o el Reglamento (EC) 726/2004. Sin embargo, con la introducción del artículo 117 al nuevo reglamento de productos sanitarios, el expediente de autorización de comercialización deberá incluir los resultados de la evaluación de la conformidad de la parte que constituye un producto sanitario con los requisitos generales pertinentes de seguridad y funcionamiento que figuren en la declaración UE de conformidad, o el certificado expedido por un organismo notificado que permita al fabricante colocar el marcado CE.
Alternativamente, cuando esta documentación no se encuentre disponible, el fabricante de un producto combinado podrá presentar un dictamen sobre la conformidad de la parte que constituye un producto sanitario con los requisitos generales de seguridad y funcionamiento emitido por un organismo notificado.
En cualquier caso, por primera vez, se hace indispensable la intervención de un organismo notificado en la autorización de comercialización de un producto combinado integrado. Esto supone un incremento en la seguridad y el funcionamiento de este tipo de productos, dado que deberán cumplir con los requisitos de ambas normativas de medicamentos y de productos sanitarios.
5.2. Productos Combinados No Integrados
Los productos combinados no integrados son aquellos en los que dos o más componentes separados (medicamento y producto sanitario) no se encuentran físicamente unidos durante la fabricación, pero en los que el medicamento y el producto sanitario específico se combinan para la administración del primero. Según el primer párrafo de la sección 9 del artículo 1 del Reglamento (UE) nº. 2017/745, se considerará producto sanitario en pleno derecho a aquel que esté destinado a administrar un medicamento.
En este tipo de productos, el medicamento y el producto sanitario sí se distinguen como entes separados que, si bien necesitan actuar de forma conjunta, cumplirán sus respectivas normativas en cuanto a su regulación y autorización. Si el producto sanitario se distribuye de forma conjunta al medicamento (en el mismo acondicionamiento secundario), se denominarán ‘co-envasados’. Por el contrario, en aquellos casos en los que se comercialicen como entes separados, la información al paciente del medicamento contará con una referencia al producto sanitario, dado que se necesitará indisolublemente para su administración, y se denominarán ‘referenciados’.
Se ilustra la parte de producto sanitario de productos combinados no integrados con los siguientes ejemplos:
• Productos sanitarios para la administración oral de medicamentos:
Los productos sanitarios para la administración oral de medicamentos son un amplio grupo de productos sanitarios de bajo riesgo, generalmente suministrados en el acondicionamiento secundario de formas líquidas orales, compuestos por cubiletes, cucharas o jeringas, entre otros. Si bien en algunos casos pueden contar con una función de medición, la acción principal prevista es claramente llevada a cabo por el medicamento, mientras que el producto sanitario (el cubilete) lleva a cabo una función de administración.
Al ser productos no invasivos y no ser ninguna otra regla de clasificación de aplicación, se trata de productos sanitarios de clase I según la regla 1 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
• Inhaladores de polvo seco reutilizables
Los inhaladores de polvo seco reutilizables permiten una reducción en la cantidad de residuos generados, con el consiguiente impacto medioambiental, durante el tratamiento o mantenimiento de un paciente con afecciones respiratorias (116, 117). El inhalador y su recambio son los productos sanitarios, dado que actúan como sistemas de administración, mientras que la acción principal prevista, el tratamiento de la afección respiratoria, es alcanzada por el medicamento inhalado.
Tanto si se suministran en diferentes acondicionamientos secundarios, como si se encuentran disponibles en el mismo, la nueva regla 20 de clasificación mencionada previamente también es de aplicación para los inhaladores de polvo seco reutilizables y, por tanto, pueden tener la consideración de productos sanitarios de clase IIa o IIb.
• Bombas de insulina
Las bombas de insulina para el manejo de la diabetes suponen una alternativa terapéutica más fisiológica a las múltiples inyecciones diarias y, si bien no es seguro que su eficacia sea superior, sus beneficios psicosociales pueden haber contribuido a un incremento en su adopción (118, 119). Estos sistemas de infusión de insulina están compuestos por un conjunto de productos sanitarios (la bomba, la cánula, el reservorio, el sistema de adhesión, la zona de inserción, el monitor y el software), cuya función es la de control de la administración del medicamento, mientras que la responsable de la acción principal prevista, el manejo de la diabetes, es la insulina (120, 121).
Este tipo de productos tienen una consideración especial al tratarse de productos activos destinados a administrar y/o retirar medicamentos de una forma potencialmente peligrosa y, por tanto, se clasifican como productos sanitarios de clase IIb según la regla 12 del capítulo III de las Reglas de clasificación del anexo VIII del Reglamento (UE) nº. 2017/745.
Así, en contraposición con los productos combinados integrados, para los que un dictamen de un organismo notificado sobre la conformidad con los requisitos generales de seguridad y funcionamiento es suficiente, los fabricantes de productos combinados no integrados deberán llevar a cabo los procedimientos de evaluación de la conformidad pertinentes para poder elaborar una declaración UE de conformidad.
6. PRODUCTOS SANITARIOS CON SUSTANCIA MEDICINAL AUXUILAR
Según el primer párrafo de la sección 8 del artículo 1 del Reglamento (UE) nº. 2017/745, todo producto que incorpore una sustancia que, utilizada por separado, se consideraría un medicamento y tenga una acción accesoria respecto a la del producto, se considerará producto sanitario. Además, atendiendo a la regla 14 del anexo VIII, serán productos sanitarios de clase III y, por tanto, a estos productos se les exigirán unos mayores controles y requisitos para la evaluación de su conformidad, acorde con su clasificación de mayor riesgo.
Se ilustran los productos sanitarios con sustancia medicinal auxiliar con los siguientes ejemplos:
• Cementos óseos con antibiótico
Los cementos óseos con antibiótico alcanzan su fin médico previsto mediante el cemento, que ofrece una fijación física, mientras que la acción farmacológica de las sustancias antibióticas se incorpora para mejorar el perfil de seguridad de estos productos (122-125).
• Preservativos medicados
La acción principal prevista de los preservativos medicados (en esencia, la contracepción y la prevención de la trasmisión de enfermedades de transmisión sexual) se alcanza mediante la barrera física que supone el producto sanitario, mientras que las sustancias anestésicas incorporadas para paliar los efectos de la eyaculación precoz llevan a cabo una función auxiliar (126-128).
• Stents liberadores de fármacos
Los stents liberadores de fármacos ejercen su acción principal prevista de forma mecánica, mientras que la acción farmacológica de la sustancia medicinal es siempre auxiliar a esa apertura física de la arteria (129-131).
Así, dado que en los productos sanitarios con sustancia medicinal auxiliar la acción principal prevista es llevada a cabo por la parte del producto sanitario, mientras que la sustancia medicinal solo asiste en esa función, estos deberán ser certificados por el nuevo reglamento de productos sanitarios. Sin embargo, la incorporación de una sustancia medicinal en un producto sanitario exige que el organismo notificado encargado de la evaluación obtenga un dictamen científico de una autoridad competente sobre la calidad y la seguridad de dicha sustancia, incluida la relación beneficio/riesgo de su incorporación al producto.
Por tanto, a pesar de que se consideran productos sanitarios y están regulados por el nuevo reglamento sobre productos sanitarios, la intervención de una autoridad competente en medicamentos implica que, si el dictamen científico emitido es desfavorable, el organismo notificado no podrá expedir el certificado UE de evaluación de la documentación técnica, impidiendo así la entrada al mercado del producto.
7. DISCUSIÓN
El mercado interior de la Unión Europea cuenta con una gran variedad de artículos de distinta naturaleza y con fines muy diversos, lo que ha llevado a la Comisión Europea a establecer una serie de normativas para regularlos. En este contexto se encuadra el nuevo Reglamento (UE) nº. 2017/745, que aúna todos los productos sanitarios distintos de los de diagnóstico in vitro bajo un mismo marco legislativo. En tanto que reglamento, esta nueva norma permite una toma de decisiones más uniforme por parte de las distintas autoridades sanitarias de los Estados miembros, y además introduce una serie de novedades y mejoras.
Sin embargo, el nuevo Reglamento se solapa con otras normas, lo que lleva a la aparición de fronteras. Este fenómeno no debe ser motivo de conflicto, sino que es un reflejo de la constante innovación en el sector, y debe impulsar aún más la evolución de toda la legislación de la Unión para ir consiguiendo, paulatinamente, un marco normativo capaz de armonizar las decisiones de todos los Estados miembros. Así, las revisiones que se irán sucediendo irán en beneficio de todos los agentes implicados, incluyendo no solo a los propios legisladores, sino también a los fabricantes o a los pacientes, que con definiciones cada vez más claras y exhaustivas podrán tomar decisiones basadas en criterios objetivos.
En esta línea, la determinación de productos frontera adquiere especial relevancia cuando se trata de la frontera entre productos sanitarios y medicamentos, puesto que los segundos son uno de los artículos más regulados y controlados dentro de la Unión Europea. Cuentan con una serie de controles de calidad (en proceso y en producto terminado), de trazabilidad, de ensayos clínicos, de comercialización o de farmacovigilancia que la mayoría de los artículos no presentan. Por ello, no hay que olvidar que, para un fabricante, el que un artículo se autorice dentro de la categoría de producto sanitario o de medicamento, puede suponer una enorme diferencia en la entrada al mercado y, por tanto, la obtención de beneficios tras la inversión inicial en su desarrollo.
Tanto es así que, en España, el Sistema Nacional de Salud contempla la financiación, con cargo a los fondos de la Seguridad Social o fondos estatales afectos a la sanidad, prescritos y suministrados a pacientes no hospitalizados, de una serie de efectos y accesorios recogidos en los anexos I y II del Real Decreto 9/1996 (132). Si bien esta norma, de 1996, ha sufrido dos modificaciones a lo largo de su historia, ninguna de ellas ha sido sobre los anexos, que únicamente recogen una lista de doce grupos de artículos cada uno de ellos. Esta falta de revisión hace que el
Estado lleve financiando los mismos productos sanitarios desde hace más de dos décadas, sin evaluar aspectos relacionados con su seguridad o eficacia, lo que podría llevar a una mejor distribución de los fondos públicos hacia tratamientos más efectivos. Sin embargo, la falta de información disponible en relación con los productos sanitarios, tanto de los ya recogidos en los anexos como de las nuevas alternativas, hace que la actualización de las listas en base a criterios objetivos no sea posible. Además, en un ambiente hospitalario, la práctica totalidad de los medicamentos utilizados tiene su precio regulado y están financiados por la Seguridad Social. En cambio, gran parte de los productos sanitarios que se utilizan en este ámbito, y que en muchas ocasiones suponen el porcentaje más elevado del gasto en el tratamiento de un paciente, no cuentan con un precio regulado. Así, las declaraciones del fabricante, tanto en relación con el fin médico específico como en relación con el mecanismo por el que un nuevo artículo alcanza la acción principal prevista, pueden condicionar la toma de decisiones hacia su certificación como producto sanitario, de forma que el precio no esté regulado. Por todo ello, a la hora de la salida al mercado de un nuevo artículo, los fabricantes y los organismos notificados se encuentran con diversas opciones, y requerimientos, en función de a qué normativa se acojan. (Tabla 4)
Por un lado, si se comercializan como productos combinados, se considerarán medicamentos, y deberán someterse a los requisitos de la Directiva 2001/83/CE. En el caso de productos combinados integrados, además requerirán de una declaración de la conformidad por parte del fabricante o un certificado UE de conformidad de un organismo notificado o, en su defecto, de un dictamen emitido por un organismo notificado, relativo al cumplimiento de los requisitos generales de seguridad y funcionamiento de la parte correspondiente al producto sanitario. En el caso de productos combinados no integrados, los fabricantes deberán someter la parte producto sanitario al procedimiento íntegro de evaluación de la conformidad pertinente para poder elaborar una declaración UE de conformidad. En cualquier caso, por primera vez, con la introducción del nuevo artículo 117 en el Reglamento (UE) nº. 2017/745 se hace indispensable la intervención de un organismo notificado para la autorización de un medicamento que contenga un producto sanitario en su composición, independientemente de que lo incorpore de forma integrada o no.
Por otro lado, aquellos productos que cuenten con una sustancia medicinal con un mecanismo de acción farmacológico, inmunológico o metabólico que actúe de forma auxiliar a la acción principal del producto se considerarán productos sanitarios de clase III por Regla 14 y, por tanto, la sustancia medicinal será evaluada por una autoridad competente de medicamentos, pero el producto sanitario deberá cumplir los requisitos propios de un producto sanitario de la clase de mayor riesgo.
Sin embargo, cabe destacar que el incremento en los requisitos para la aprobación de nuevos productos sanitarios de alto riesgo puede tener un impacto negativo en la investigación y desarrollo de nuevos productos. Esto es debido a que el nuevo reglamento establece la obligatoriedad de realizar investigaciones clínicas con el fin de demostrar beneficio clínico en los productos sanitarios implantables y/o de clase III, lo que incluye los productos combinados implantables, así como todos los productos sanitarios con sustancia medicinal auxiliar. Este beneficio clínico deberá demostrarse, no solo en la evaluación de la conformidad inicial para la entrada al mercado del producto, sino además anualmente como parte del seguimiento clínico poscomercialización (133).
En conjunto, el incremento en el número de investigaciones clínicas y la introducción de nuevas reglas de clasificación aumentarán los perfiles de seguridad y funcionamiento de los productos en el mercado, pero también supondrán mayores costes y tiempos para la certificación de nuevos productos. Esto puede disuadir a los fabricantes de invertir en nuevos productos e incluso motivarlos a abandonar algunos ya aprobados, lo que puede tener impacto negativo en la innovación y la competitividad en Europa (134).
Además, con la introducción del artículo 117 en el nuevo reglamento de productos sanitarios, muchos nuevos productos requerirán la intervención de un organismo notificado. Sin embargo, el número de organismos notificados designados para determinados tipos de productos de muy alta complejidad es limitado, lo que puede suponer un incremento en la demora para la autorización de nuevos productos.
En resumen, el nuevo marco normativo europeo apuesta por establecer elevados requerimientos que aseguren que los productos sanitarios sean eficaces y seguros para los pacientes, y por permitir que los profesionales sanitarios puedan tomar decisiones más informadas y objetivas gracias a un aumento de la transparencia. Sin embargo, los productos sanitarios son un conjunto de artículos muy heterogéneos, al igual que sus fabricantes, y el establecimiento de requerimientos tan estrictos puede suponer un impacto económico demasiado elevado para las pequeñas y medianas empresas.
Por esta razón, los nuevos requisitos podrían desencadenar una disminución en la disponibilidad o variedad de determinados productos sanitarios, con el consiguiente impacto en los pacientes y sistemas sanitarios (135). El impacto real es todavía difícil de estimar, sin embargo, la crisis sanitaria de la COVID-19 ha demostrado la importancia no solo de contar con dispositivos seguros y de alta calidad, sino también de un sistema robusto que asegure su disponibilidad y evite el desabastecimiento en tiempos de necesidad.
8. CONCLUSIÓN
El nuevo reglamento europeo sobre productos sanitarios supone un avance en la armonización normativa y en la toma de decisiones por parte de los Estados miembros. Sin embargo, esto no exime de realizar revisiones constantes que se adapten a la innovación, no solo de estos productos, sino de todos los comercializados en el mercado interior, con el fin de lograr que las fronteras queden bien definidas y se puedan tomar decisiones basadas en criterios objetivos.
La elaboración de guías prácticas y manuales resulta de gran utilidad para los agentes implicados, pero debe de ir acompañado de la revisión constante de las definiciones de los diversos artículos en sus respectivas normativas con el fin de asegurar la protección de la salud de la población. Así, la frontera entre medicamento y producto sanitario seguirá existiendo, pero con una definición en constante desarrollo se puede lograr un marco legislativo que supere estas dificultades y sea capaz de integrar estos productos al mercado interior de la Unión. En este sentido, la implementación del Reglamento (UE) nº. 2017/745 desde el 26 de mayo de 2021, aporta un avance sustancial en la sistematización de los productos sanitarios que incorporan sustancias medicamentosas con función auxiliar, así como de los productos combinados en aras de maximizar las garantías de seguridad para el paciente y de minimizar los riesgos asociados a su uso.
Lista de abreviaturas
• AEMPS: Agencia Española de Medicamentos y Productos Sanitarios
• CE: Conformité Européenne (Conformidad Europea)
• CEE: Comunidad Económica Europea
• COVID-19: CoronaVirus Disease 2019 (Enfermedad del CoronaVirus 2019)
• EMA: European Medicines Agency (Agencia Europea de Medicamentos)
• EPI: Equipo de Protección Individual
• Eudamed: Base de Datos Europea sobre Productos Sanitarios
• FDA: Food and Drug Administration (Administración de Alimentos y Medicamentos)
• FFP: Filtering Face Piece (Mascarilla Autofiltrante)
• MDCG: Medical Devices Coordination Group (Grupo de Coordinación de Productos Sanitarios)
• SARS-CoV-2: Severe Acture Respiratory Syndrome – CoronaVirus – 2 (Síndrome Respiratorio Agudo Severo – CoronaVirus – 2)
• UDI: Unique Device Identification (Identificación Única de Productos)
• UE: Unión Europea
• UNE: Asociación Española de Normalización
9. Referencias
- [Europa]. 1990. «Directiva 90/385/CEE del Consejo, de 20 de junio de 1990, relativa a la aproximación de las legislaciones de los Estados Miembros sobre los productos sanitarios implantables activos». Diario Oficial de las Comunidades Europeas, [20 de julio de 1990], DO L 189 de 2071990, p 17/36. http://data.europa.eu/eli/dir/1990/385/oj
- [Europa]. 1993. «Directiva 93/42/CEE del Consejo, de 14 de junio de 1993, relativa a los productos sanitarios». Diario Oficial de las Comunidades Europeas, [12 de julio de 1993], DO L 169 de 1271993, p 1/43. http://data.europa.eu/eli/dir/1993/42/oj
- [España]. 1993. «Real Decreto 634/1993, de 3 de mayo, sobre productos sanitarios implantables activos». Boletín Oficial del Estado, [27 de mayo de 1993], BOE-A-1993-13672.
https://www.boe.es/eli/es/rd/1993/05/03/634 - [España]. 2009. «Real Decreto 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos». Boletín Oficial del Estado, [06 de noviembre de 2009], BOE-A-2009-17607. https://www.boe.es/eli/es/rd/2009/10/26/1616
- [España]. 1996. «Real Decreto 414/1996, de 1 de marzo, por el que se regula los productos sanitarios». Boletín Oficial del Estado, [24 de abril de 1996], BOE-A-1996-9089.
https://www.boe.es/eli/es/rd/1996/03/01/414 - [España]. 2009. «Real Decreto 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios». Boletín Oficial del Estado, [06 de noviembre de 2009], BOE-A-2009-17606.
https://www.boe.es/eli/es/rd/2009/10/16/1591 - [Francia]. 2010. «Silicone filled breast implants manufactured by Poly Implant Prosthese (PIP). Press Release of 4 January 2010». Agence Française de Sécurité Sanitaire des Produits de Santé, [01 de abril de 2010]. https://archiveansm.integra.fr/var/ansm_site/storage/original/application/ff8f7014c6ee1b6674c8fb7dd2835840.pdf.
- [Francia]. 2010. «Silicone filled breast implants from Poly Implant Prothèse Company. Tests results». Agence Française de Sécurité Sanitaire des Produits de Santé, [28 de septiembre de 2010]. https://archiveansm.integra.fr/var/ansm_site/storage/original/application/38fbe37bdd1897eb09de4f892a317c14.pdf
- [Europa]. 2014. «Opinion on The safety of Poly Implant Prothèse (PIP) Silicone Breast Implants. Update of the Opinion of February 2012». Comité Científico de los Riesgos Sanitarios Emergentes y Recientemente Identificados (SCENIHR), [12 de mayo de 2014]. https://ec.europa.eu/health/scientific_committees/emerging/docs/scenihr_o_043.pdf
- [España]. 2010. «Nota de seguridad sobre prótesis mamarias POLY IMPLANT (PIP). Ref.: 005/Marzo 2010». Agencia Española de Medicamentos y Productos Sanitarios, [31 de marzo de 2010]. https://www.aemps.gob.es/informa/notasInformativas/productosSanitarios/seguridad/2010/docs/005-2010_protesis-Mamarias.pdf?x44154
- [Europa]. 2017. «Reglamento (UE) nº. 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios, por el que se modifican la Directiva 2001/83/CE, el Reglamento (CE) nº. 178/2002 y el Reglamento (CE) n.o 1223/2009 y por el que se derogan las Directivas 90/385/CEE y 93/42/CEE del Consejo». Diario Oficial de la Unión Europea, [5 de mayo de 2017], DO L 117 de 552017, p 1/175. http://data.europa.eu/eli/reg/2017/745/oj
- Emanuel EJ, Persad G, Upshur R, Thome B, Parker M, Glickman A, et al. Fair Allocation of Scarce Medical Resources in the Time of Covid-19. New England Journal of Medicine. 2020;382(21):2049-55. doi: 10.1056/nejmsb2005114
- Worby CJ, Chang H-H. Face mask use in the general population and optimal resource allocation during the COVID-19 pandemic. Nature Communications. 2020;11(1). doi: 10.1038/s41467-020-17922-x
- McGuire AL, Aulisio MP, Davis FD, Erwin C, Harter TD, Jagsi R, et al. Ethical Challenges Arising in the COVID-19 Pandemic: An Overview from the Association of Bioethics Program Directors (ABPD) Task Force. The American Journal of Bioethics. 2020;20(7):15-27. doi: 10.1080/15265161.2020.1764138
- [Europa]. 2020. «Reglamento (UE) nº. 2020/561 del Parlamento Europeo y del Consejo, de 23 de abril de 2020, por el que se modifica el Reglamento (UE) nº. 2017/745 sobre los productos sanitarios en relación con las fechas de aplicación de algunas de sus disposiciones». Diario Oficial de la Unión Europea, [24 de abril de 2020], DO L 130 de 2442020, p 18/22. http://data.europa.eu/eli/reg/2020/561/oj
- [Europa]. 2017. «Reglamento (UE) nº. 2017/746 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión». Diario Oficial de la Unión Europea, [05 de mayo de 2017], DO L 117 de 552017, p 176/332. http://data.europa.eu/eli/reg/2017/746/oj
- 2021. «Human Regulatory Overview: Medical Devices – Borderline Products». Agencia Europea de Medicamentos, https://www.ema.europa.eu/en/human-regulatory/overview/medical-devices#borderline-products-section
- [Europa]. 2001. «Directiva 2001/83/CE del Parlamento Europeo y del Consejo, de 6 de noviembre de 2001, por la que se establece un código comunitario sobre medicamentos para uso humano». Diario Oficial de las Comunidades Europea, [28 de noviembre de 2001], DO L 311 de 28112001, p 67/128. http://data.europa.eu/eli/dir/2001/83/oj
- [Europa]. 2002. «Directiva 2002/46/CE del Parlamento Europeo y del Consejo, de 10 de junio de 2002, relativa a la aproximación de las legislaciones de los Estados miembros en materia de complementos alimenticios». Diario Oficial de las Comunidades Europeas, [12 de julio de 2002], DO L 183 de 1272002, p 51/57. http://data.europa.eu/eli/dir/2002/46/oj
- [Europa]. 2009. «Reglamento (CE) nº. 1223/2009 del Parlamento Europeo y del Consejo, de 30 de noviembre de 2009, sobre los productos cosméticos». Diario Oficial de las Comunidades Europeas, [22 de diciembre de 2009], DO L 342 de 22122009, p 59/209. http://data.europa.eu/eli/reg/2009/1223/oj
- [España]. 1997. «Real Decreto 1599/1997, de 17 de octubre, sobre productos cosméticos». Boletín Oficial del Estado, [31 de octubre de 1997], BOE-A-1997-23067. https://www.boe.es/eli/es/rd/1997/10/17/1599
- [Europa]. 2012. «Reglamento (UE) nº. 528/2012 del Parlamento Europeo y del Consejo, de 22 de mayo de 2012, relativo a la comercialización y el uso de los biocidas». Diario Oficial de la Unión Europa, [27 de junio de 2012], DO L 167 de 2762012, p 1/123. http://data.europa.eu/eli/reg/2012/528/oj
- [Europa]. 2016. «Reglamento (UE) nº. 2016/425 del Parlamento Europeo y del Consejo, de 9 de marzo de 2016, relativo a los equipos de protección individual y por el que se deroga la Directiva 89/686/CEE del Consejo». Diario Oficial de la Unión Europea, [31 de marzo de 2016], DO L 81 de 3132016, p 51/98. http://data.europa.eu/eli/reg/2016/425/oj
- [Europa]. 2001. «Directiva 2001/95/CE del Parlamento Europeo y del Consejo, de 3 de diciembre de 2001, relativa a la seguridad general de los productos». Diario Oficial de las Comunidades Europeas, [15 de enero de 2002], DO L 11 de 1512002, p 4/17. http://data.europa.eu/eli/dir/2001/95/oj
- Martelli N, Eskenazy D, Déan C, Pineau J, Prognon P, Chatellier G, et al. New European Regulation for Medical Devices: What Is Changing? CardioVascular and Interventional Radiology. 2019;42(9):1272-8. doi: 10.1007/s00270-019-02247-0
- Antich-Isern P, Caro-Barri J, Aparicio-Blanco J. The combination of medical devices and medicinal products revisited from the new European legal framework. International Journal of Pharmaceutics. 2021;607:120992. doi: 10.1016/j.ijpharm.2021.120992
- [Europa]. 2008. «Reglamento (CE) nº. 765/2008 del Parlamento Europeo y del Consejo, de 9 de julio de 2008, por el que se establecen los requisitos de acreditación y vigilancia del mercado relativos a la comercialización de los productos y por el que se deroga el Reglamento (CEE) nº. 339/93». Diario Oficial de la Unión Europea, [13 de agosto de 2008], DO L 218 de 1382008, p 30/47. http://data.europa.eu/eli/reg/2008/765/oj
- Fraser AG, Byrne RA, Kautzner J, Butchart EG, Szymański P, Leggeri I, et al. Implementing the new European Regulations on medical devices—clinical responsibilities for evidence-based practice: a report from the Regulatory Affairs Committee of the European Society of Cardiology. European Heart Journal. 2020;41(27):2589-96. doi: 10.1093/eurheartj/ehaa382
- Fraser AG, Butchart EG, Szymański P, Caiani EG, Crosby S, Kearney P, et al. The need for transparency of clinical evidence for medical devices in Europe. The Lancet. 2018;392(10146):521-30. doi: 10.1016/S0140-6736(18)31270-4
- 30. [Europa]. 2012. «Carta de los Derechos Fundamentales de la Unión Europea». Diario Oficial de la Unión Europea, [26 de octubre de 2012], DO C 326 de 26102012, p 391/407.
http://data.europa.eu/eli/treaty/char_2012/oj - Dhruva SS. Strength of Study Evidence Examined by the FDA in Premarket Approval of Cardiovascular Devices. JAMA. 2009;302(24):2679. doi: 10.1001/jama.2009.1899
- Rome BN, Kramer DB, Kesselheim AS. Approval of High-Risk Medical Devices in the US: Implications for Clinical Cardiology. Current Cardiology Reports. 2014;16(6). doi: 10.1007/s11886-014-0489-0
- [Europa]. 2021. «Salud Pública: Eudamed Overview». Comisión Europea, https://ec.europa.eu/health/md_eudamed/overview_es
- Anderson ML, Chiswell K, Peterson ED, Tasneem A, Topping J, Califf RM. Compliance with Results Reporting at ClinicalTrials.gov. New England Journal of Medicine. 2015;372(11):1031-9. doi: 10.1056/nejmsa1409364
- [Brasil]. 2013. «Declaración de Helsinki de la AMM – Principios Éticos para las Investigaciones Médicas en Seres Humanos». [21 de marzo de 2017]. https://pdf-it.dev.acw.website/please-and-thank-you?url=https://www.wma.net/es/policies-post/declaracion-de-helsinki-de-la-amm-principios-eticos-para-las-investigaciones-medicas-en-seres-humanos/&pdfName=declaracion-de-helsinki-de-la-amm-principios-eticos-para-las-investigaciones-medicas-en-seres-humanos
- [Suiza]. 2015. «WHO Statement on public disclosure of clinical trial results». Organización Mundial de la Salud, [09 de abril de 2015]. https://www.who.int/news/item/09-04-2015-japan-primary-registries-network
- [Europa]. 2018. «General Court confirms EMA approach to transparency. Three rulings clarify the scope of commercial confidentiality with regard to authorised medicines. Press release EMA/73690/2018». Agencia Europea de Medicamentos, [06 de febrero de 2018]. https://www.ema.europa.eu/documents/press-release/general-court-confirms-ema-approach-transparency_en.pdf
- [Europa]. 2015. «Medical Devices: Guidance document – Borderline products, drug-delivery products and medical devices incorporating, as an integral part, an ancillary medicinal substance or an ancillary human blood derivative. MEDDEV 2.1/3 rev 3 (2062695)». Dirección General de Empresa e Industria, Comisión Europea, [21 de marzo de 2010]. http://www.meddev.info/_documents/2_1_3_rev_3-12_2009_en.pdf
- [Europa]. 2019. «Manual on borderline and classification in the community regulatory framework for medical devices. Version 1.22». Working Group on Borderline and Classification, [mayo de 2019]. https://ec.europa.eu/health/sites/health/files/md_topics-interest/docs/md_borderline_manual_05_2019_en.pdf
- [Europa]. 2009. «Reglamento (CE) nº. 1170/2009 de la Comisión, de 30 de noviembre de 2009, por la que se modifican la Directiva 2002/46/CE del Parlamento Europeo y del Consejo y el Reglamento (CE) nº. 1925/2006 del Parlamento Europeo y del Consejo en lo relativo a las listas de vitaminas y minerales y sus formas que pueden añadirse a los alimentos, incluidos los complementos alimenticios». Diario Oficial de la Unión Europea, [01 de diciembre de 2009], DO L 314 de 1122009, p 36/42.
http://data.europa.eu/eli/reg/2009/1170/oj - [Europa]. 2011. «Reglamento (UE) nº. 1161/2011 de la Comisión, de 14 de noviembre de 2011, por la que se modifican la Directiva 2002/46/CE del Parlamento Europeo y del Consejo, el Reglamento (CE) nº. 1925/2006 del Parlamento Europeo y del Consejo y el Reglamento (CE) nº. 953/2009 de la Comisión en lo relativo a las listas de sustancias minerales que pueden añadirse a los alimentos». Diario Oficial de la Unión Europea, [15 de noviembre de 2011], DO L 296 de 15112011, p 29/30. http://data.europa.eu/eli/reg/2011/1161/oj
- [Europa]. 2008. «Informe de la Comisión al Consejo y al Parlamento Europeo sobre la utilización de sustancias distintas de las vitaminas y los minerales en los complementos alimenticios. SEC(2008)2976/7». Comisión de las Comunidades Europeas, [05 de diciembre de 2008]. https://eur-lex.europa.eu/legal-content/ES/TXT/PDF/?uri=CELEX:52008DC0824&from=ES
- [Europa]. 2012. «Tratado de Funcionamiento de la Unión Europea». Diario Oficial de la Unión Europea, [26 de octubre de 2012], DO C 326 de 26102012, p 47/390.
http://data.europa.eu/eli/treaty/tfeu_2012/oj - [Europa]. 2014. «Reglamento (UE) nº. 119/2014 de la Comisión, de 7 de febrero de 2014, por el que se modifica la Directiva 2002/46/CE del Parlamento Europeo y del Consejo y el Reglamento (CE) nº. 1925/2006 del Parlamento Europeo y del Consejo, en lo que se refiere a la levadura enriquecida con cromo utilizada en la fabricación de complementos alimenticios y el lactato de cromo (III) trihidrato añadido a los alimentos». Diario Oficial de la Unión Europea, [08 de febrero de 2014], DO L 39 de 822014, p 44/45. http://data.europa.eu/eli/reg/2014/119(1)/oj
- [Europa]. 2015. «Reglamento (UE) nº. 2015/414 de la Comisión, de 12 de marzo de 2015, por el que se modifica la Directiva 2002/46/CE del Parlamento Europeo y del Consejo en lo referente al ácido (6 S)-5-metiltetrahidrofólico, sal de glucosamina, utilizado en la fabricación de complementos alimenticios». Diario Oficial de la Unión Europea, [13 de marzo de 2015], DO L 68 de 1332015, p 26/27. http://data.europa.eu/eli/reg/2015/414/oj
- [Europa]. 2017. «Reglamento (UE) nº. 2017/1203 de la Comisión, de 5 de julio de 2017, por el que se modifica la Directiva 2002/46/CE del Parlamento Europeo y del Consejo y el Reglamento (CE) nº. 1925/2006 del Parlamento Europeo y del Consejo en lo que respecta al silicio orgánico (monometilsilanotriol) y a los oligosacáridos fosforilados de calcio (POs-Ca®) añadidos a los alimentos y utilizados en la fabricación de complementos alimenticios». Diario Oficial de la Unión Europea, [06 de julio de 2017], DO L 173 de 672017, p 9/11.
http://data.europa.eu/eli/reg/2017/1203/oj - González De Llano D, Moreno-Arribas MV, Bartolomé B. Cranberry Polyphenols and Prevention against Urinary Tract Infections: Relevant Considerations. Molecules. 2020;25(15):3523.
doi: 10.3390/molecules25153523 - González De Llano D, Liu H, Khoo C, Moreno-Arribas MV, Bartolomé B. Some New Findings Regarding the Antiadhesive Activity of Cranberry Phenolic Compounds and Their Microbial-Derived Metabolites against Uropathogenic Bacteria. Journal of Agricultural and Food Chemistry. 2019;67(8):2166-74. doi: 10.1021/acs.jafc.8b05625
- Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nature Reviews Microbiology. 2015;13(5):269-84. doi: 10.1038/nrmicro3432
- [Alemania]. 2012. «Sentencia del Tribunal de Justicia (Sala Quinta), de 6 de septiembre de 2012, «Directiva 2001/83/CE — Medicamentos para uso humano — Artículo 1, punto 2, letra b) — Concepto de medicamento “por su función” — Definición del concepto de “acción farmacológica”». Asunto C-308/11. ECLI:EU:C:2012:548″. Tribunal de Justicia (Sala Quinta), [06 de septiembre de 2012]. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX:62011CJ0308
- [Europa]. 2017. «Decisión de Ejecución (UE) 2017/1445 de la Comisión, de 8 de agosto de 2017, sobre el grupo de productos cuya acción principal prevista, basada en las proantocianidinas (PAC) del arándano rojo (Vaccinium macrocarpon), es prevenir o tratar la cistitis [notificada con el número C(2017) 5341]». Diario Oficial de la Unión Europea, [10 de agosto de 2017], DO L 207 de 1082017, p 28/29. http://data.europa.eu/eli/dec_impl/2017/1445/oj
- [España]. 2018. «Real Decreto 85/2018, de 23 de febrero, por el que se regulan los productos cosméticos». Boletín Oficial del Estado, [27 de febrero de 2018], BOE-A-2018-2693.
https://www.boe.es/eli/es/rd/2018/02/23/85 - 2020. «Manual of the Working Group on Cosmetic Products (Sub-group on Borderline Products) on the Scope of Application of the Cosmetics Regulation (EC) nº. 1223/2009 (Art. 2(1)(a)). Version 5.2». Working Group on Cosmetic Products, Sub-group on Borderline Products, [01 de septiembre de 2020]. https://ec.europa.eu/docsroom/documents/42850/attachments/1/translations/en/renditions/native
- 2015. «Guidance document on the demarcation between the cosmetic products Directive 76/768 and the medicinal products Directive 2001/83 as agreed between the commission services and the competent authorities of Member States». Comissión Europea, [12 de octubre de 2015]. https://ec.europa.eu/docsroom/documents/13032/attachments/1/translations/en/renditions/native
- [España]. 2004. «Real Decreto 2131/2004, de 29 de octubre, por el que se modifica el Real Decreto 1599/1997, de 17 de octubre, sobre productos cosméticos». Boletín Oficial del Estado, [30 de octubre de 2004], BOE-A-2004-18574. https://www.boe.es/eli/es/rd/2004/10/29/2131
- [España]. 2005. «Real Decreto 209/2005, de 25 de febrero, por el que se modifica el Real Decreto 1599/1997, de 17 de octubre, sobre productos cosméticos». Boletín Oficial del Estado, [26 de febrero de 2005], BOE-A-2005-3243. https://www.boe.es/eli/es/rd/2005/02/25/209
- [España]. 2010. «Solicitud de autorización de comercialización para productos de higiene personal». Agencia Española de Medicamentos y Productos Sanitarios, [2010].
https://www.aemps.gob.es/cosmeticos-cuidado-personal/docs/soliciAutori_proHigPersonal.pdf?x58524 - Alkahtani R, Stone S, German M, Waterhouse P. A review on dental whitening. Journal of Dentistry. 2020;100:103423.
doi: 10.1016/j.jdent.2020.103423 - Naidu AS, Bennani V, Brunton JMAP, Brunton P. Over-the-Counter Tooth Whitening Agents: A Review of Literature. Brazilian Dental Journal. 2020;31(3):221-35. doi: 10.1590/0103-6440202003227
- [Europa]. 2006. «Reglamento (CE) nº. 1907/2006 del Parlamento Europeo y del Consejo, de 18 de diciembre de 2006, relativo al registro, la evaluación, la autorización y la restricción de las sustancias y preparados químicos (REACH), por el que se crea la Agencia Europea de Sustancias y Preparados Químicos, se modifica la Directiva 1999/45/CE y se derogan el Reglamento (CEE) nº. 793/93 del Consejo y el Reglamento (CE) nº. 1488/94 de la Comisión así como la Directiva 76/769/CEE del Consejo y las Directivas 91/155/CEE, 93/67/CEE, 93/105/CE y 2000/21/CE de la Comisión». Diario Oficial de la Unión Europea, [30 de diciembre de 2006], DO L 396 de 30122006, p 1/852. http://data.europa.eu/eli/reg/2006/1907/oj
- [España]. 2011. «Nota Informativa sobre Productos Desinfectantes». Agencia Española de Medicamentos y Productos Sanitarios, [29 de marzo de 2011].
https://www.aemps.gob.es/informa/notasInformativas/cosmeticosHigiene/2011/docs/NI_01-2011_prod-Desinfectantes.pdf?x10382 - [España]. 2021. «Resolución de 2 de junio de 2021, de la Agencia Española de Medicamentos y Productos Sanitarios, por la que se atribuye la condición de medicamento de uso humano a los antisépticos destinados al campo quirúrgico preoperatorio y a la desinfección del punto de inyección». Agencia Española de Medicamentos y Productos Sanitarios, [19 de junio de 2021].
https://www.boe.es/eli/es/res/2021/06/02/(5) - 2020. «Orientaciones sobre la legislación aplicable a los limpiadores y desinfectantes que no se aclaran para las manos (en forma de gel, solución, etc.)». Comisión Europea, [30 de marzo de 2020]. https://ec.europa.eu/docsroom/documents/40523/attachments/2/translations/es/renditions/native
- 2020. «Documento técnico acerca del ámbito de aplicación del Reglamento (CE) nº. 1223/2009 sobre los productos cosméticos [artículo 2, apartado 1, letra a)]. Reivindicaciones en geles de manos hidroalcohólicos que no se aclaran en el contexto de la pandemia de COVID-19». Subgrupo de Trabajo sobre Productos «Frontera», Grupo de Trabajo sobre Productos Cosméticos, [12 de noviembre de 2020]. https://ec.europa.eu/docsroom/documents/44913/attachments/1/translations/es/renditions/native
- [España]. 2020. «Nota Informativa: La AEMPS informa sobre las soluciones y geles hidroalcohólicos de desinfección de manos con eficacia viricida demostrada. COS 7/2020». Agencia Española de Medicamentos y Productos Sanitarios, [08 de mayo de 2020].
https://www.aemps.gob.es/informa/notasInformativas/cosmeticos-cuidado-personal/biocidas/2020/NI-COS-7-eficacia-viricida.pdf?x44154 - Leslie RA, Zhou SS, Macinga DR. Inactivation of SARS-CoV-2 by commercially available alcohol-based hand sanitizers. American Journal of Infection Control. 2021;49(3):401-2.
doi: 10.1016/j.ajic.2020.08.020 - Golin AP, Choi D, Ghahary A. Hand sanitizers: A review of ingredients, mechanisms of action, modes of delivery, and efficacy against coronaviruses. American Journal of Infection Control. 2020;48(9):1062-7. doi: 10.1016/j.ajic.2020.06.182
- [España]. 2021. «Antisépticos para piel sana autorizados por la AEMPS que han demostrado eficacia frente a virus atendiendo a la norma UNE-EN14476». Agencia Española de Medicamentos y Productos Sanitarios, [17 de septiembre de 2021].
https://www.aemps.gob.es/cosmeticos-cuidado-personal/docs/listado-biocidas-eficacia-viricida-17-09-21.pdf?x44154 - Jairoun AA, Al-Hemyari SS, Shahwan M. The pandemic of COVID-19 and its implications for the purity and authenticity of alcohol-based hand sanitizers: The health risks associated with falsified sanitizers and recommendations for regulatory and public health bodies. Research in Social and Administrative Pharmacy. 2021;17(1):2050-1.
doi: 10.1016/j.sapharm.2020.04.014 - [España]. 2020. «Tabla resumen de productos: equipos de protección individual (EPI), productos sanitarios (PS) y otros. Versión 2». Ministerio de Industria, Comercio y Turismo, https://www.mincotur.gob.es/es-es/COVID-19/industria/GuiaFabricacionEpis/Generalidades/Tabla_resumen_de_productos_EPI_y_PS_v2.pdf
- Coclite D, Napoletano A, Gianola S, del Monaco A, D’Angelo D, Fauci A, et al. Face Mask Use in the Community for Reducing the Spread of COVID-19: A Systematic Review. Frontiers in Medicine. 2021;7(1060). doi: 10.3389/fmed.2020.594269
- Ju JTJ, Boisvert LN, Zuo YY. Face masks against COVID-19: Standards, efficacy, testing and decontamination methods. Advances in Colloid and Interface Science. 2021;292:102435. doi: 10.1016/j.cis.2021.102435
- [España]. 2019. «Mascarillas quirúrgicas. Requisitos y métodos de ensayo. UNE-EN 14683:2019+AC:2019». Asociación Española de Normalización, [05 de diciembre de 2019]. https://www.une.org/encuentra-tu-norma/busca-tu-norma/norma/?c=N0062987
- [España]. 2021. «Dispositivos de protección respiratoria. Medias máscaras filtrantes de protección contra partículas. Requisitos, ensayos, marcado. UNE-EN 149:2001+A1:2010». Asociación Española de Normalización, [27 de enero de 2010]. https://www.une.org/encuentra-tu-norma/busca-tu-norma/norma/?c=N0044643
- [España]. 2020. «¿Qué debes tener en cuenta al comprar una mascarilla?». Ministerio de Consumo, https://www.mscbs.gob.es/en/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/030520_GUIA_COMPRA_MASCARILLAS.pdf
- [España]. 2003. «Real Decreto 1801/2003, de 26 de diciembre, sobre seguridad general de los productos». Boletín Oficial del Estado, [10 de enero de 2004], BOE-A-2004-511.
https://www.boe.es/eli/es/rd/2003/12/26/1801/con - [España]. 2021. «Mascarillas higiénicas no reutilizables. Requisitos de materiales, diseño, confección, marcado y uso. Parte 1: Para uso en adultos. UNE 0064-1:2021». Asociación Española de Normalización, [08 de septiembre de 2021]. https://www.une.org/encuentra-tu-norma/busca-tu-norma/norma/?c=N0066541
- [España]. 2021. «Mascarillas higiénicas reutilizables para adultos y población infantil. Requisitos de materiales, diseño, confección, marcado y uso. UNE 0065:2021». Asociación Española de Normalización, [08 de septiembre de 2021]. https://www.une.org/encuentra-tu-norma/busca-tu-norma/norma/?c=N0066543
- [Europa]. 1965. «Directiva 65/65/CEE del Consejo, de 26 de enero de 1965, relativa a la aproximación de las disposiciones legales, reglamentarias y administrativas, sobre especialidades farmacéuticas». Diario Oficial de las Comunidades Europeas, [26 de enero de 1965], DO 22 de 921965, p 369/373.
http://data.europa.eu/eli/dir/1965/65/oj - [Europa]. 2003. «Directiva 2003/94/CE de la Comisión, de 8 de octubre de 2003, por la que se establecen los principios y directrices de las prácticas correctas de fabricación de los medicamentos de uso humano y de los medicamentos en investigación de uso humano». Diario Oficial de la Unión Europea, [14 de octubre de 2003], DO L 262 de 14102003, p 22/26.
http://data.europa.eu/eli/dir/2003/94/oj - [Europa]. 2013. «Directrices, de 5 de noviembre de 2013, sobre prácticas correctas de distribución de medicamentos para uso humano». Diario Oficial de la Unión Europea, [23 de noviembre de 2013], DO C 343 de 23112013, p 1/14. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX:52013XC1123(01)
- [Europa]. 2015. «Directrices, de 19 de marzo de 2015, sobre prácticas correctas de distribución de principios activos para medicamentos de uso humano». Diario Oficial de la Unión Europea, [21 de marzo de 2015], DO C 95 de 2132015, p 1/9. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=celex:52015XC0321(01)
- [Europa]. 2015. «Directrices, de 19 de marzo de 2015, sobre la evaluación formal de riesgos a efectos de determinar las prácticas correctas de fabricación apropiadas para los excipientes de medicamentos de uso humano». Diario Oficial de la Unión Europea, [21 de marzo de 2015], DO C 95 de 2132015, p 10/13.
https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX:52015XC0321(02) - [Europa]. 2004. «Directiva 2004/27/CE del Parlamento Europeo y del Consejo, de 31 de marzo de 2004, que modifica la Directiva 2001/83/CE por la que se establece un código comunitario sobre medicamentos de uso humano». Diario Oficial de la Unión Europea, [30 de abril de 2004], DO L 136 de 3042004, p 34/57. http://data.europa.eu/eli/dir/2004/27/oj
- Hebert AA. A new therapeutic horizon in diaper dermatitis: Novel agents with novel action. International Journal of Women’s Dermatology. 2021;7(4):466-70.
doi: 10.1016/j.ijwd.2021.02.003 - Verzì AE, Nasca MR, Dall’Oglio F, Cosentino C, Micali G. A novel treatment of intertrigo in athletes and overweight subjects. Journal of Cosmetic Dermatology. 2021;20(S1):23-7. doi: 10.1111/jocd.14097
- Ågren MS, Phothong N, Burian EA, Mogensen M, Hædersdal M, Jorgensen LN. Topical Zinc Oxide Assessed in Two Human Wound-healing Models. Acta Derm Venereol. 2021;101(5):adv00465.doi: 10.2340/00015555-3829
- Pather P, Hines S, Kynoch K, Coyer F. Effectiveness of topical skin products in the treatment and prevention of incontinence-associated dermatitis: a systematic review. JBI Evidence Synthesis. 2017;15(5):1473-96. doi: 10.11124/jbisrir-2016-003015
- Dubald M, Bourgeois S, Andrieu V, Fessi H. Ophthalmic Drug Delivery Systems for Antibiotherapy—A Review. Pharmaceutics. 2018;10(1):10.
doi: 10.3390/pharmaceutics10010010 - Holland EJ, Fingeret M, Mah FS. Use of Topical Steroids in Conjunctivitis: A Review of the Evidence. Cornea. 2019;38(8):1062-7.
doi: 10.1097/ico.0000000000001982 - Kuc CJ, Lebow KA. Contact Lens Solutions and Contact Lens Discomfort: Examining the Correlations Between Solution Components, Keratitis, and Contact Lens Discomfort. Eye & Contact Lens. 2018;44(6):355-66. doi: 10.1097/icl.0000000000000458
- Zaki M, Pardo J, Carracedo G. A review of international medical device regulations: Contact lenses and lens care solutions. Contact Lens and Anterior Eye. 2019;42(2):136-46.
doi: 10.1016/j.clae.2018.11.001 - Zhang X, M VJ, Qu Y, He X, Ou S, Bu J, et al. Dry Eye Management: Targeting the Ocular Surface Microenvironment. International Journal of Molecular Sciences. 2017;18(7):1398.
doi: 10.3390/ijms18071398 - Barabino S, Benitez-Del-Castillo JM, Fuchsluger T, Labetoulle M, Malachkova N, Meloni M, et al. Dry eye disease treatment: the role of tear substitutes, their future, and an updated classification. Eur Rev Med Pharmacol Sci. 2020;24(17):8642-52. doi: 10.26355/eurrev_202009_22801
- Wang L, Du S, Wang H, Popkin BM. Influence of dietary fat intake on bodyweight and risk of obesity among Chinese adults, 1991–2015: a prospective cohort study. The Lancet. 2018;392:S20. doi: 10.1016/s0140-6736(18)32649-7
- Warrilow A, Mellor D, McKune A, Pumpa K. Dietary fat, fibre, satiation, and satiety—a systematic review of acute studies. European Journal of Clinical Nutrition. 2019;73(3):333-44. doi: 10.1038/s41430-018-0295-7
- Glyn-Jones S, Palmer AJR, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. The Lancet. 2015;386(9991):376-87. doi: 10.1016/s0140-6736(14)60802-3
- Gudbergsen H, Boesen M, Lohmander LS, Christensen R, Henriksen M, Bartels EM, et al. Weight loss is effective for symptomatic relief in obese subjects with knee osteoarthritis independently of joint damage severity assessed by high-field MRI and radiography. Osteoarthritis and Cartilage. 2012;20(6):495-502. doi: 10.1016/j.joca.2012.02.639
- Uthman OA, Van Der Windt DA, Jordan JL, Dziedzic KS, Healey EL, Peat GM, et al. Exercise for lower limb osteoarthritis: systematic review incorporating trial sequential analysis and network meta-analysis. BMJ. 2013;347(sep20 1):f5555-f. doi: 10.1136/bmj.f5555
- Kolasinski SL, Neogi T, Hochberg MC, Oatis C, Guyatt G, Block J, et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Care & Research. 2020;72(2):149-62. doi: 10.1002/acr.24131
- Gupta RC, Lall R, Srivastava A, Sinha A. Hyaluronic Acid: Molecular Mechanisms and Therapeutic Trajectory. Front Vet Sci. 2019;6:192. doi: 10.3389/fvets.2019.00192
- Abad Luna MC. La frontera en productos sanitarios. Divulgación Técnica. 2013;175:30-5. doi:
- Arias Pou P, Delgado Latorre A, Aguinagalde Toya A, Gaspar Carreño M, Silberberg Muiño JM, Sobrido Sampedro C. Presentaciones de ácido hialurónico ¿son realmente diferentes? OFIL ILAPHAR. 2017;28(3):265-73. doi:
- 2020. «Annual Report 2019». Agencia Europea de Medicamentos, [15 de junio de 2020]. https://www.ema.europa.eu/documents/annual-report/2019-annual-report-european-medicines-agency_en.pdf
- [Europa]. 2021. «Annual Report 2020». Agencia Europea de Medicamentos, [14 de junio de 2021]. https://www.ema.europa.eu/documents/annual-report/2020-annual-report-european-medicines-agency_en.pdf
- [Europa]. 2021. «Guideline on quality documentation for medicinal products when used with a medical device. EMA/CHMP/QWP/BWP/259165/2019». Comité de Medicamentos de Uso Humano, Agencia Europea de Medicamentos, [23 de julio de 2021]. https://www.ema.europa.eu/documents/scientific-guideline/guideline-quality-documentation-medicinal-products-when-used-medical-device-first-version_en.pdf
- 2021. «Human Regulatory Overview: Medical Devices – Medicinal Products that Include a Medical Device (‘Combination Products’)». Agencia Europea de Medicamentos, https://www.ema.europa.eu/en/human-regulatory/overview/medical-devices#medicinal-products-that-include-a-medical-device-(‘combination-products’)-section
- I’Ons G. Could pre-filled safety syringes facilitate the adoption of home administration for cancer patients? Journal of Oncology Pharmacy Practice. 2020;26(8):1934-6.
doi: 10.1177/1078155220961463 - Sacha G, Rogers JA, Miller RL. Pre-filled syringes: a review of the history, manufacturing and challenges. Pharmaceutical Development and Technology. 2015;20(1):1-11. doi: 10.3109/10837450.2014.982825
- Chandel A, Goyal AK, Ghosh G, Rath G. Recent advances in aerosolised drug delivery. Biomedicine & Pharmacotherapy. 2019;112:108601. doi: 10.1016/j.biopha.2019.108601
- Popov TA, Passalacqua G, González-Díaz SN, Plavec D, Braido F, García-Abujeta J-L, et al. Medical devices in allergy practice. World Allergy Organization Journal. 2020;13(10):100466. doi: 10.1016/j.waojou.2020.100466
- Bofill Rodriguez M, Lethaby A, Jordan V. Progestogen‐releasing intrauterine systems for heavy menstrual bleeding. Cochrane Database of Systematic Reviews. 2020(6).
doi: 10.1002/14651858.CD002126.pub4 - Abbas AM, Samy A, Atwa K, Ghoneim HM, Lotfy M, Saber Mohammed H, et al. The role of levonorgestrel intra‐uterine system in the management of adenomyosis: A systematic review and meta‐analysis of prospective studies. Acta Obstetricia et Gynecologica Scandinavica. 2020;99(5):571-81. doi: 10.1111/aogs.13798
- Li L, Leng J, Jia S, Lang J. Treatment of symptomatic adenomyosis with the levonorgestrel‐releasing intrauterine system. International Journal of Gynecology & Obstetrics. 2019;146(3):357-63. doi: 10.1002/ijgo.12887
- Quarterman JC, Geary SM, Salem AK. Evolution of drug-eluting biomedical implants for sustained drug delivery. European Journal of Pharmaceutics and Biopharmaceutics. 2021;159:21-35. doi: 10.1016/j.ejpb.2020.12.005
- Ortsäter G, Borgström F, Baldwin M, Miltenburger C. Incorporating the Environmental Impact into a Budget Impact Analysis: The Example of Adopting RESPIMAT® Re-usable Inhaler. Applied Health Economics and Health Policy. 2020;18(3):433-42. doi: 10.1007/s40258-019-00540-0
- Janson C, Henderson R, Löfdahl M, Hedberg M, Sharma R, Wilkinson AJK. Carbon footprint impact of the choice of inhalers for asthma and COPD. Thorax. 2020;75(1):82-4. doi: 10.1136/thoraxjnl-2019-213744
- Beck RW, Bergenstal RM, Laffel LM, Pickup JC. Advances in technology for management of type 1 diabetes. The Lancet. 2019;394(10205):1265-73. doi: 10.1016/s0140-6736(19)31142-0
- Mueller‐Godeffroy E, Vonthein R, Ludwig‐Seibold C, Heidtmann B, Boettcher C, Kramer M, et al. Psychosocial benefits of insulin pump therapy in children with diabetes type 1 and their families: The pumpkin multicenter randomized controlled trial. Pediatric Diabetes. 2018;19(8):1471-80. doi: 10.1111/pedi.12777
- Sharma R, Singh D, Gaur P, Joshi D. Intelligent automated drug administration and therapy: future of healthcare. Drug Delivery and Translational Research. 2021;11(5):1878-902.
doi: 10.1007/s13346-020-00876-4 - Minssen T, Mimler M, Mak V. When Does Stand-Alone Software Qualify as a Medical Device in the European Union?—The Cjeu’s Decision in Snitem and What it Implies for the Next Generation of Medical Devices. Medical Law Review. 2020;28(3):615-24. doi: 10.1093/medlaw/fwaa012
- Sebastian S, Liu Y, Christensen R, Raina DB, Tägil M, Lidgren L. Antibiotic containing bone cement in prevention of hip and knee prosthetic joint infections: A systematic review and meta-analysis. Journal of Orthopaedic Translation. 2020;23:53-60. doi: 10.1016/j.jot.2020.04.005
- Singh R, Bathaei MJ, Istif E, Beker L. A Review of Bioresorbable Implantable Medical Devices: Materials, Fabrication, and Implementation. Advanced Healthcare Materials. 2020;9(18):2000790. doi: 10.1002/adhm.202000790
- Kapadia BH, Berg RA, Daley JA, Fritz J, Bhave A, Mont MA. Periprosthetic joint infection. The Lancet. 2016;387(10016):386-94. doi: 10.1016/s0140-6736(14)61798-0
- Arciola CR, Campoccia D, Montanaro L. Implant infections: adhesion, biofilm formation and immune evasion. Nature Reviews Microbiology. 2018;16(7):397-409. doi: 10.1038/s41579-018-0019-y
- Beksinska M, Wong R, Smit J. Male and female condoms: Their key role in pregnancy and STI/HIV prevention. Best Practice & Research Clinical Obstetrics & Gynaecology. 2020;66:55-67. doi: 10.1016/j.bpobgyn.2019.12.001
- Steiner RJ, Pampati S, Kortsmit KM, Liddon N, Swartzendruber A, Pazol K. Long-Acting Reversible Contraception, Condom Use, and Sexually Transmitted Infections: A Systematic Review and Meta-analysis. American Journal of Preventive Medicine. 2021;61(5):750-60. doi: 10.1016/j.amepre.2021.04.032
- Butcher MJ, Zubert T, Christiansen K, Carranza A, Pawlicki P, Seibel S. Topical Agents for Premature Ejaculation: A Review. Sexual Medicine Reviews. 2020;8(1):92-9. doi: 10.1016/j.sxmr.2019.03.003
- Scafa Udriște A, Niculescu A-G, Grumezescu AM, Bădilă E. Cardiovascular Stents: A Review of Past, Current, and Emerging Devices. Materials. 2021;14(10):2498. doi: 10.3390/ma14102498
- Tomberli B, Mattesini A, Baldereschi GI, Di Mario C. A Brief History of Coronary Artery Stents. Revista Española de Cardiología (English Edition). 2018;71(5):312-9. doi: 10.1016/j.rec.2017.11.022
- Kommineni N, Saka R, Khan W, Domb AJ. Non-polymer drug-eluting coronary stents. Drug Delivery and Translational Research. 2018;8(4):903-17. doi: 10.1007/s13346-017-0414-3
- [España]. 1996. «Real Decreto 9/1996, de 15 de enero, por el que se regula la selección de los efectos y accesorios, su financiación con fondos de la Seguridad Social o fondos estatales afectos a la sanidad y su régimen de suministro y dispensación a pacientes no hospitalizados». Boletín Oficial del Estado, [07 de febrero de 1996], BOE-A-1996-2545. https://www.boe.es/eli/es/rd/1996/01/15/9
- Tarricone R, Ciani O, Torbica A, Brouwer W, Chaloutsos G, Drummond MF, et al. Lifecycle evidence requirements for high-risk implantable medical devices: a European perspective. Expert Review of Medical Devices. 2020;17(10):993-1006. doi: 10.1080/17434440.2020.1825074
- Ben-Menahem SM, Nistor-Gallo R, Macia G, von Krogh G, Goldhahn J. How the new European regulation on medical devices will affect innovation. Nature Biomedical Engineering. 2020;4(6):585-90. doi: 10.1038/s41551-020-0541-x
- Lubbers BR, Schilhabel A, Cobbaert CM, Gonzalez D, Dombrink I, Brüggemann M, et al. The New EU Regulation on in vitro Diagnostic Medical Devices: Implications and Preparatory Actions for Diagnostic Laboratories. HemaSphere. 2021;5(5):e568. doi: 10.1097/hs9.0000000000000568