1. INTRODUCCIÓN a los microARNs
Los microARNs (miARNs) son una clase de moléculas de ARN pequeñas que regulan la expresión génica a nivel post-transcripcional. Descubiertas inicialmente en el gusano C. elegans (1, 2), se consideraron una peculiaridad de los nematodos hasta que se observó que algunos de ellos estaban filogenéticamente conservados en una amplia variedad de organismos, incluyendo humanos (3-5). Hoy en día se está observando progresivamente el importante valor que desempeñan los microARNs como reguladores de la expresión génica. A nivel celular los microARNs son importantes en el mantenimiento de la identidad celular (6, 7). De hecho, niveles anormales de los microARNs a menudo resultan en una pérdida de la diferenciación celular, un proceso común en el desarrollo tumoral. Por lo tanto, como cabría de esperar, disfunciones en la vía de los miARNs afectan a muchos procesos celulares que están recurrentemente alterados en cáncer como proliferación, diferenciación, apoptosis, metástasis y mantenimiento de los telómeros (8) (9) (10).
1.1. microARNs: Genómica, Biogénesis y modo de acción
Los MicroARNs conforman una extensa y homogénea familia de ARN no codificantes de proteína, con un tamaño comprendido de 19-25 nucleótidos actualmente están creciendo en número, diversidad y función (11, 12). En el 2022 la base de datos de microARNs (13) tenía registradas 48 860 secuencias maduras de microARNs descubiertos en 271 organismos (mirbase.org v22.5). Actualmente (Enero 2022) el genoma humano contiene 2654 secuencias maduras de microARNs documentadas en humanos, pero modelos computacionales predicen más (14).
Alrededor de la mitad de los genes que codifican microARNs están organizados en grupos de transcritos policistrónicos, los cuales son procesados para constituir los microARNs individuales (15, 16). Los restantes microARNs se generan a partir de transcritos individuales. Más de dos terceras partes de los microARNs comparten unidades transcripcionales con genes codificantes para proteína o ARNs largos no codificantes. Estos pueden transcribirse a partir de su propios promotores, promotores de genes cercanos o promotores de sus genes hospedadores (17). Gran parte de los microARNs que surgen de genes codificantes para proteínas están localizados en intrones, mientras que los que surgen de ARN codificantes mas largos pueden estar localizados en intrones o exones (18). Al menos un tercio de las familias de microARNs están altamente conservadas entre especies (19) y el 60% de los loci de microARNs están conservadas de ratones a humanos (13).
El esquema básico de la biogénesis de los microARN se encuentra detallado en la Figura 1. La mayor parte de los microARNs se trascriben a partir de la ARN polimerasa II como transcritos primarios (pri-miARNs) que contienen una estructura 5’ CAP y una cola 3’ poliadenilada (20). Los pri-miARNs que pueden ser de varios miles de nucleótidos, son procesados por la enzima ARNasa III llamada Drosha y la proteína de unión a ARN de doble cadena DGCR8 (21-23) generándose uno o más precursores (pre-miARN).
Los pre-microARNs están formados por alrededor de 65 a 85 nucleótidos y presentan una fuerte complementariedad interna que les hace plegarse sobre si y establecer estructuras de horquilla, que asimilan a las que tienen los ARN de doble cadena. Los pre-miARN son exportados al citoplasma por medio del factor de exportador nuclear Exportin-5 y su cofactor RAN-GTP (24, 25). Una vez en el citoplasma los pre-microARNs son procesados de nuevo por otra enzima ARNasa III llamada Dicer generando ARNs de doble cadena de unos 22 nucleótidos (26-28). La proteína Argonauta 2 es reclutada completándose el complejo silenciador inducido por ARN (RISC) (29, 30). Sólo una de las dos cadenas (cadena guía) quedará en el complejo RISC. El factor que parece determinar cual de estas cadenas permanece en el complejo RISC es la estabilidad del anillamiento formado (31-34).
La represión de la expresión del ARN mensajero (ARNm) puede darse de dos formas dependiendo de la complementariedad que presenten el microARN y su diana. Si el microARN se une con una complementariedad perfecta o casi perfecta al ARNm es fragmentado por el complejo RISC y degradado. Este mecanismo se da principalmente en plantas (35) aunque ha sido también observado en ocasiones en animales (36). Si la unión del microARN y su ARNm es imperfecta se inhibe la traducción proteica seguido por un grado variable de degradación del mensajero. Este último escenario es el que se ha observado más común en células animales y la interacción se da más frecuentemente a nivel de las regiones sin traducir 3’(UTR) (37, 38). Se ha sugerido varios mecanismos mediante los cuales los microARNs producen la inhibición de la traducción que incluyen: a) el bloqueo del inicio de la traducción, el bloque de la elongación, b) el secuestro de los ARNm en los cuerpos P (P-bodies), compartimentos especializados en el citoplasma donde se da la represión transcripcional y c) la degradación de los ARNm (37, 39-41).
Además se ha observado que los microARNs pueden presentar una regulación post-transcripcional en respuesta a un estímulo proliferativo y de diferenciación celular (42). Así, por ejemplo, se ha observado que al menos en el caso de la familia de microARNs let-7, la proteína de unión a ARN llamada Lin28, regula su maduración y es necesaria y suficiente para bloquear el proceso de maduración de let-7 (43).
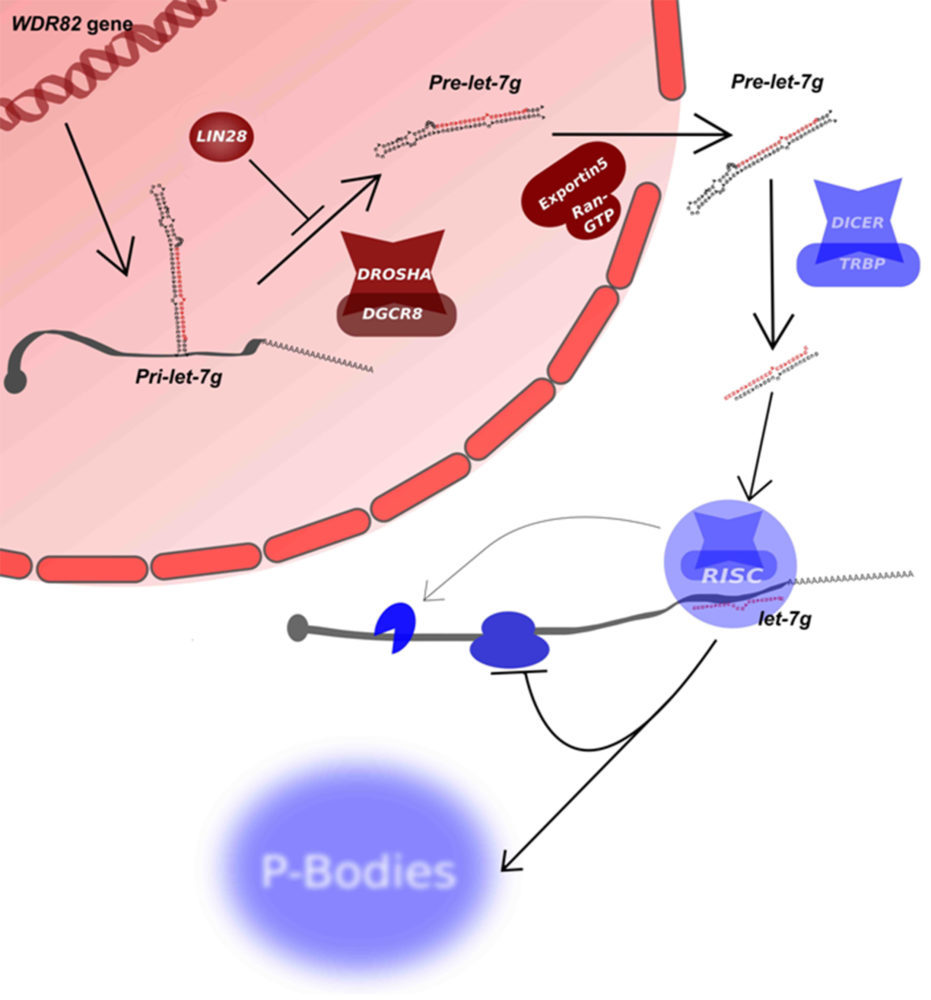
Figura 1. Esquema que muestra la biogénesis básica de los microARNs. Se ha tomado let-7g tomado como ejemplo representativo. let-7g esta codificado en la secuencia intrónica del gen codificante de proteína WDR82. El gen es transcrito por la ARN polimerasa II que añade la modificación 5’ (CAP) y la cola poli-A en la región 3’. Dicho transcrito contiene un ARN primario llamado pri-let-7g (pri-miARN) que es reconocido y cortado por el complejo formado la ARNsa III llamada Drosha y la proteína de unión a ARN DGCR8 resultando en un ARN plegado en horquilla debido a su alta autocomplementariedad de bases de 84 nucleótidos llamado pre-miARN. La Exportina-5 y la Ran-GTP transportan el pre-miARN al citoplasma donde otra ARNsa III, DICER, en asociación con la proteína de unión a ARN TRBP procesan al pre-miARN para formar un ARN de doble cadena con 2 nucleótidos protuberantes que es reclutado por el complejo silenciador inducido por ARN (RISC). A continuación, una de la cadenas, la cadena pasajera, se elimina del complejo y es degradada, y el ARN maduro let-7g (UGAGGUAGUAGUUUGUACAGUU), guía a RISC hacia los ARNm diana para inhibir su traducción (44). La biogénesis de los microARNs puede ser regulada a varios niveles, en este ejemplo LIN28 inhibe la maduración de let-7. La inhibición de la traducción de los ARNm puede realizarse en unas regiones citoplasmáticas especializadas llamados cuerpos P (ver el texto para detalles adicionales). (45).
1.2. Las funciones de los MicroARNs como reguladores: sus dianas.
No todos los genes codificantes de proteínas están regulados por los microARNs. Algunos genes que desempeñan funciones del metabolismo celular fundamental presentan una región 3’UTRs corta que no presenta apenas regiones de unión a los microARNs (46). Sin embargo, se ha estimado que gran parte de los genes codificantes de proteínas presentan regulada su expresión mediante microARNs (47). Por lo tanto, es concebible que los microARNs, regulen gran parte de las rutas biológicas celulares.
La predicción de los mensajeros diana a los que los microARN regulan es difícil debido a que la gran mayoría de las veces la unión microARN-transcrito diana se realiza de forma parcialmente complementaria. Como complejidad añadida, algunos genes presentan 3’UTR alternativas, lo cual puede hacer que estén regulados por grupos de microARNs diferentes. Se han desarrollado varios algoritmos bioinformáticos para predecir dianas de los microARNs (48-50), la mayor parte de los cuales se basan en la presencia de una secuencia llamada semilla de 7 nucleótidos situados entre la posiciones 2 a la 8 del microARN maduro (47). Otro factor utilizado en estos algoritmos es la conservación filogenética de estos sitios de unión a los microARNs de las regiones 3’UTR de las dianas y la ausencia de estructuras secundarias estables (50). Se ha estimado que un único microARN puede regular 200 genes diferentes (47). De esta forma los microARN tienen una cualidad pleiotrópica intrínseca. Así una función aberrante de los microaARN podría desencadenar una ruptura del balance homeostático celular que podría contribuir a una patología sistémica.
2. MICROARNs Y CÁNCER
2.1. Introducción: microARN y cáncer
El cáncer es una enfermedad compleja donde un grupo de células anormales pierden identidad y crecen sin control, siendo capaz de invadir y colonizar otros tejidos. Este comportamiento invasivo puede resultar en una disfunción orgánica que puede desembocar un el fallo orgánico fatal. Existen múltiples líneas de evidencia que indican que la carcinogénesis es un proceso secuencial de múltiples pasos donde las células malignas acumulan alteraciones genéticas y epigenéticas que conducen a una la transformación progresiva en células malignas (51). De esta forma, las poblaciones tumorales seleccionan alteraciones en genes que promueven la progresión tumoral (oncogenes) o genes que la dificultan (genes supresores tumorales). Al final del proceso de transformación, las células malignas pierden la identidad celular, adquieren independencia en el crecimiento, invasión y resistencia a la senescencia y la apoptosis.
Dado a que los microARNs pueden alterar la expresión génica son candidatos al mantenimiento del balance en la proliferación celular (Figura 2). Estudios pioneros han mostrado la existencia de una expresión diferencial de los microARNs entre tejido tumorales y tejidos normales (52-54). Sin embargo, esto no significa que todos los microARNs perturbados estén directamente implicados funcionalmente en el desarrollo tumoral. Muchos de ellos podrían ser simplemente espectadores indirectamente alterados por los cambios genómicos y epigenómicos que surgen durante la carcinogénesis sin ser los agentes causantes de la misma. Los microARNs que están realmente implicados en la carcinogénesis son denominados oncomiRs (55), y su descubrimiento actualmente ha revolucionado la oncología molecular actual, y son el título de este trabajo.
Figura 2:
No sólo microARN específicos se han relacionado con el cáncer (como veremos más adelante), también componentes de la maquinaria de biogénesis de los microARN han sido implicados en el desarrollo tumoral. Así, se han observado que mutaciones de Drosha, DGCR8 o Dicer (proteínas básicas de la biogénesis) colaboran en la transformación tumoral (56). Estos hallazgos concuerdan con resultados anteriores de la pérdida de expresión de Dicer en algunos tumores (57), y una perdida global de expresión de microARNs en tumores cuando se comparan con tejidos normales (53). Por otra parte, la pérdida condicional de Dicer (58, 59) o DGCR8 (60) en células madre embrionarias de ratón (ES) impiden la diferenciación y proliferación celular lo que podría conducir a procesos neoplásicos.
Otros componentes de la maquinaria de biosíntesis de los microARNs han sido implicados en cáncer incluyendo los miembros de la familia de proteínas Argonauta: hAgo1/EIF2C1, hAgo3/EIF2C3, hAgo4 e Hiwi. hAgo1, hAgo3 y hAgo4 se agrupan en la región cromosómica 1p34-35 que es frecuentemente delecionada en algunos tumores humanos que incluyen: tumor de Wilms, neuroblastoma y carcinomas de pecho, hígado y colon (61). Por otra parte, la sobreexpresión de Hiwi (que pertenece al grupo de la familia de proteínas Argonauta implicadas en el mantenimiento de la línea germinal) (62) ha sido relacionada, entre otros, con tumores germinales, (63-65).
Los microARNs pueden clasificarse en oncogenes o supresores tumores desde un punto de vista clásico según promuevan o impidan el desarrollo tumoral (Figura 3). Sin embargo, ya que los efectos de los microARN son intrínsecamente pleotrópicos, esta clasificación debería considerarse flexible.
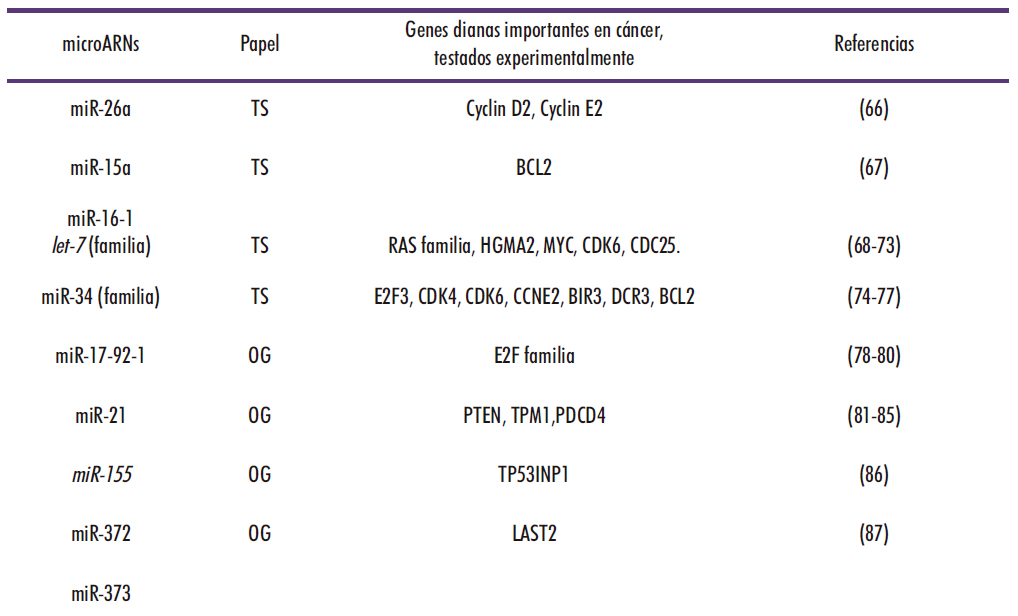
Figura 3: microARNs representativos implicados en cáncer, papel tumoral propuesto (TS: tumor supresor, OG: oncogén) y genes dianas importantes en cáncer que pueden explicar su actividad en cáncer. (45).
2.2. MicroARNs como supresores tumorales
MicroARN-15a y MicroARN-16-1
Estudios sobre la leucemia linfocítica crónica (CLL) revelaron la primera evidencia directa que relacionaba a los microARNs con el cáncer. La mayor parte de las CLL de células B presentaban una deleción recurrente en la región cromosómica 13q14 que sugerían la presencia en dicha región de un gen supresor tumoral. Durante años varios grupos estudiaron esta región cromosómica en busca de genes supresores tumorales codificantes, aunque sin hallar un candidato sólido. Un estudio pormenorizado de dicha región delató una secuencia delecionada mínima común de 30kb. En esta región se hallaba el gen no codificante de proteínas LEU2 (88). Calin y colaboradores se percataron que LEU2 contenían a miR-15a y miR-16-1 en su primer intrón.
Consecuentemente, la pérdida de expresión de estos dos microARNs fue documentada en el 68% de los casos de CLL analizados (89). Por otra parte, se encontraron mutaciones asociadas con la pérdida de expresión de estos microARNs en la línea germinal de pacientes CLL (90).
Un análisis funcional encontró al gen anti-apoptótico BCL2 como una de las dianas reguladas por miR-15a y miR-16-1. Los niveles de estos microARNs se correlacionaban inversamente con los niveles de expresión de BCL2 y los ensayos de fusión de UTR con proteínas reporteras determinaron que ambos microARN son capaces de controlar transcripcionalmente a BCL2. Además la represión de Bcl2 por estos microARNs inducía la apoptosis en líneas celulares de CLL (67).
Ya que la región 13q14 se pierde en otros tipos de cáncer es concebible que la función supresora tumoral de ambos microARNs miR-15a and miR-16-1 se extienda a otros tipos de tumores.
Familia de microARNs let-7
Let-7 fue inicialmente descubierto en el nematodo (C. elegans) donde su expresión está regulada durante en el desarrollo y controla el desarrollo temporal de la diferenciación, actuando como un gen importante que regula a múltiples genes requeridos para la salida del ciclo celular (3). Let-7 fue el primer microARN identificado en humanos y su familia comprende a 12 genes en el genoma humano. Varios estudios han mostrado que la expresión de let-7 se pierde en muchos tumores (52, 68, 91, 92). Además, los niveles de expresión de let-7 se han correlacionado con el pronóstico tumoral (52, 91, 93, 94). El papel biológico de supresor tumoral de let-7 fue provisto por el descubrimiento que importantes oncogenes como RAS (68), HGMA2 (69, 70), myc (71), CDK6 y CDC25 (72).
Familia MicroARN-34
En vertebrados la familia mir-34 está compuesta de tres miembros evolutivamente conservados (miR-34a, miR-34b and miR-34c). En humanos, miR-34a se encuentra en la región cromosómica 1p36 y mir-34b y mir-34c se transcriben de mismo transcrito situado en la región cromosómica 11q23.1. Su relación con el cáncer se encontró inicialmente mediante una pérdida de expresión diferencial en neuroblastomas (75). En este trabajo se reintrodujo miR-34 en una línea celular de neuroblastoma lo que condujo a una pérdida drástica en la proliferación debido a una activación de apoptosis dependiente de caspasas. Poco después de esta observación inicial diferentes laboratorios publicaron casi simultáneamente trabajos que vinculaban mir-34 a la vía de p53 (74, 77, 95-97). Así la expresión de los miembros de la familia de mir-34 está regulada directamente con p53, y consecuentemente la expresión de los miembros de la familia de mir-34 puede reflejar la actividad de p53. Coherentemente, estos microARNs actúan como genes supresores de tumores y su reintroducción en células defectivas promueve el arresto del ciclo celular, la senescencia y la apoptosis dependiendo del fondo genético. Análisis experimentales y predicciones bioinformáticas han implicado a la familia de miR-34 en la regulación de importantes genes implicados en el control del ciclo celular que incluyen E2F3, CDK4, CDK6, CCNE2, BIR3, DCR3 y BCL2 (74-77).
MicroARNs-26a
miR-26a es un microARN que se expresa a niveles altos en diversos tejidos. Inicialmente se observó que este microARN perdía su expresión en hepatocarcinomas (66). La administración sistémica de miR-26a usando adenovirus en un modelo de hepatocarcinoma en ratones resultó en una dramática protección contra el desarrollo tumoral sin efectos tóxicos, inhibiendo la proliferación tumoral y promoviendo una apoptosis específica del tumor (66). miR-26a parece realizar estos efectos uniéndose a las regiones 3’ UTR de las ciclinas D2 y E2 y regulando su expresión. Estos resultados sugieren que la reintroducción de la expresión de determinados microARNs pueden ser de utilidad terapéutica.
2.3. MicroARNs como oncogenes
MicroARNs 17-92-1
Mir-17-92-1 es una agrupación de microARNs (miR-17-5p, miR-17-3p, miR-18, miR-19a, miR-20a, miR-19b-1 y miR-92-1) que nacen del mismo transcrito policistrónico situado en la región cromosómica 13q31.3. Esta región cromosómica se observó frecuentemente amplificada en linfomas de células B y otros tumores, lo que se correlacionaba con una sobreexpresión de los microARNs de la agrupación mir-17-92-1 (98, 99). Se ha observado que los microARNs miR-17-92-1 actúan en sinergia aumentando las propiedades oncogénicas del factor de transcripción MYC acelerando el desarrollo tumoral e incrementado la resistencia a la apoptosis celular (79, 99, 100).
Por otra parte, se observó que MYC se unía directamente al locus de los microARNs mir-17-92-1 activando su expresión. Además se mostró que E2F1 es regulado negativamente por dos microARNs de la agrupación mir-17-92-1: miR-17-5 y miR-20a (79). E2F1 es un miembro de la familia de factores de transcripción E2F implicados en la transición del ciclo celular de las fases G1 a S. Además, E2F1 es una diana transcripcional de MYC. Así MYC regula la expresión de E2F1 a diferentes niveles, por una parte, directamente activando la transcripción de E2F1 pero por otra parte limita su traducción indirectamente a través de la actividades de los microARNs miR-17-92-1. Por otra parte, los factores de transcripción E2F1, E2F2, y E2F3 en sistema de retro-regulación pueden unirse al promotor de los microARNs miR-17-92-1 activando su transcripción (78-80). De esta forma se consigue una férrea regulación del control de la señal proliferativa
Aunque MYC aumenta la expresión de los microARN miR-17-92-1 la actividad predominante de MYC es una represión general de la expresión de los microARNs. Gran parte de esta represión es probablemente debida al resultado de la unión directa de MYC a los promotores de los microARNs. Coherentemente, la re-activación de la expresión de los microARNs reprimidos por MYC, disminuyen su tumorogenicidad en línea celulares (101).
MicroARN-372 & MicroARN-373
Un estudio genético donde se investigaban microARNs que cooperaban con oncogenes en la transformación tumoral identificó propiedades oncogénicas de miR-372 y miR-373 en tumores testiculares de células germinales humanas. (102). Estudios mecanísticos determinaron que estos microARNs podrían interferir en la vía de p53 a través de la inhibición de CDK2, posiblemente mediante de la inhibición directa del supresor tumoral LATS2 (large tumor suppressor homologue 2). De esta forma estos microARNs pueden hacer que las células sean inmunes a los acciones supresoras de p53 y así ser capaces de resistir a la senescencia o apoptosis. Las habilidades de miR-372 y miR-373 para promover el desarrollo tumoral en células que presentan un p53 activo que podría explicar el por qué las mutaciones de p53 no son frecuentes en cánceres testiculares (87).
MicroARN-21
El microARN-21 se encuentra frecuentemente sobrexpresado en los perfiles de expresión de gran variedad de tumores, incluyendo: neuroblastoma, glioblastoma, cáncer de colon, de pulmón, de mama o de páncreas (103-108). Además, diversos estudios han encontrado de utilidad los niveles de expresión de mir-21 para el diagnostico y pronóstico tumoral (108). Estudios in vitro mir-21 han sugerido que miR-21 tiene habilidades anti-apoptóticas ya que su bloqueo mediante tecnología antisentido desencadena en un aumento de apoptosis (103, 107). Estudios funcionales han identificado varias dianas de miR-21, que incluyen supresores tumorales como PTEN (81), TPM1 (82) y PDCD4 (83, 84), lo que podría explicar su papel oncogénico en la carcinogénesis.
MicroARN-155
Estudios pioneros detectaron que el microARN-155 forma parte de una secuencia conservada evolutivamente que formaba parte de un gen no codificante de proteínas llamado BIC (B-cell integration cluster). BIC se había relacionado con la patología tumoral ya que se había identificado como un sitió frecuente de integración de un virus (llamado de la leucosis aviar), que induce linfomas de células B (109-111). Estudios posteriores, relacionaron un aumento de la expresión de miR-155 tanto en neoplasias hematológicas (112, 113) como en tumores sólidos (52, 54). Posteriormente estudios desarrollados en modelos animales, le atribuyéndosele actividades oncogénicas. Así, cuando se aumentaron experimentalmente los niveles de expresión de miR-155 en modelos animales se observó el desarrollo de leucemia de células B. Demostrándose experimentalmente que altos niveles de miR-155 pueden inducir expansión policlonal, facilitando la posterior adquisición de modificaciones genéticas que desemboquen en la transformación tumoral (114). Otros trabajos pusieron de manifiesto que la importancia funcional de miR-155 en el mantenimiento del sistema inmune (115-117). Así, modelos experimentales in vivo, en los que se eliminaba genéticamente a miR-155 se demostraron inmunodeficientes y con anormalidades en la maduración de su linaje linfocítico. Un análisis de perfiles de expresión alterados en estos modelos experimentales puso de manifiesto que un amplio abanico de genes relacionados con el sistema inmune está regulado por miR-155. En otra línea de investigación se ha desvelado que el gen no codificante de proteína miR-K12-11 del virus KSHV (Kaposi’s-sarcoma-associated herpes virus) está relacionado evolutiva y funcionalmente con miR-155. Así, se ha observado que ambos comparten gran parte de las dianas a las que regulan, muchas de las cuales están relacionadas con procesos carcinogénicos (118).
2.4. MicroARNs: utilidad clínica y aplicaciones farmacéuticas
Aunque la era del estudio y descubrimiento de los microARN abarca sólo unas décadas, ha sido muy prometedora para el diagnóstico, el pronóstico y la terapia en biomedicina. Se espera que el rápido desarrollo de potentes técnicas para su análisis y estudio, como la realización de secuenciación profunda del microRNAnome celular, PCR cuantitativa específica de microARN y tecnologías antisentido de utilidad in vivo, tenga una importancia significativa en la oncología clínica en un futuro próximo. Ya que los microARNs juegan un papel importante en la determinación de la identidad celular, uno de los procesos afectados en la carcinogénesis se pensó que podrían tener un valor significativo en el diagnóstico del cáncer. Estudios pioneros realizados comparando perfiles de expresión de microARNs entre muestras sanas y tumorales identificaron patrones únicos que podrían discernir entre células tumorales y no tumorales (52, 54, 119). De hecho, los perfiles de expresión de microARN parecen ser más informativos, y más potentes a la hora de clasificar las muestras tumorales por su origen tisular (algo que puede ser complicado cuando se diagnostica tumores en etapas avanzadas), su tumorigenicidad y su grado de diferenciación que los perfiles de ARN mensajero (ARNm) usados tradicionalmente. Así, por ejemplo, en trabajos pioneros a este respecto se ha observado que el perfil de tan sólo doscientos microARNs es suficiente para clasificar tumores poco diferenciados (una problemática frecuente en la clínica) con mayor precisión que utilizando la información de más de dieciséis mil ARN mensajeros (53). Otro de estos estudios consiguió clasificar con una mayor precisión el tejido de origen de cuatrocientas muestras tumorales proveniente de veintidós tipos de tejido diferentes (120). De igual forma, otro trabajo demostró la efectividad de los perfiles de microARNs para determinar el tejido de origen de cánceres secundarios de origen desconocido, un problema frecuente en la clínica actual (121).
Sin embargo, los perfiles de expresión de los microARNs también proveen una importante información clínica sobre el pronóstico de los pacientes. De esta forma, trabajos pioneros demostraron que los perfiles de microARN se correlacionaban con la supervivencia de diversos tipos tumorales incluyendo aquellos en estados patológicos precoces. De esta forma, niveles bajos de expresión de genes de la familia let-7 y altos de de miR-155 mostraron una correlación con mal pronóstico en tumores de cáncer de pulmón (52). Otro estudio en el mismo tipo tumoral, identificó la importancia pronostica de 5 microARNs: así altos niveles de miR-137, miR-372, and miR-182 se correlacionaron con mal pronóstico mientras que altos niveles de miR-221 y let-7a parecen ser protectores. Además, los niveles de este conjunto de microARNs fueron de utilidad para predecir la recaída tumoral (93). De forma similar, otro estudio pionero demostró que niveles altos de expresión de miR-21 se asociaron a una baja respuesta terapéutica y con mal pronóstico de los pacientes (108).
Pero, además, de forma más relevante para la clínica y la farmacia multitud de trabajos científicos han puesto de manifiesto el potencial terapéutico de los microARNs. Así trabajos pioneros que restituían la función supresora tumoral de microARNs cuya expresión se perdía en la carcinogénesis han mostrado su eficacia terapéutica en modelos animales (66, 122-124). De igual forma, la inhibición de la actividad de microARNs oncogénicos ha demostrado tener un valor clínico. Esta inhibición específica de los microARNs oncogénicos se basan en tecnologías de oligonucleótidos anti-sentido que pueden bloquear su actividad patogénica. Los llamados generalmente anti-miRs son oligonucleótidos complementarios a la secuencia de los microARNs con modificaciones químicas que mejoran su estabilidad y capacidad inhibitoria (Figura 4). Entre las modificaciones más utilizadas que se encuentran: la metilación del segundo oxigeno del anillo de la ribosa (2’-O-Methyl), la sustitución del enlace fosfato por el enlace fosforotioato, o la adicción de un enlace puente adicional entre dos carbonos del anillo de la ribosa (los llamados locked nucleic acid, LNA) (121). Los anti-miRs han sido ampliamente utilizados por la comunidad científica para realizar ensayos funcionales para determinar las repercusiones biológicas de la inhibición de microARN específicos (73). Esta tecnología antisentido ha demostrado una alta eficacia en cultivo celulares, aunque algunos estudios pioneros han mostrado su eficacia en modelos animales, utilizándose diferentes estrategias para mejorar su eficacia. Así, se ha observado que la adición de una molecular de colesterol al extremo 3’ del anti-miRs aumentaba su efectividad en modelo de ratones (125).
De esta forma, una inyección intravenosa de estos anti-miRs (llamados antagomirs) resultaba en una eficaz reducción del correspondiente miARN en todos los tejidos analizados, con la excepción del cerebro. Estudios pioneros que usaron anti-miRs modificados mediante LNA (locked nucleic acid) demostraron su eficacia en primates (126, 127) por lo que se presume que pueden tener utilidad en humanos. No obstante, como cada microARN puede regular la expresión de múltiples ARN mensajeros, la utilización de drogas inhibitorias podría causar efectos secundarios, con lo que la industria farmacéutica tendría que trabajar. Asimismo, diversos trabajos científicos han puesto de manifiesto que la saturación de la maquinaria de ARN de interferencia, que comparten los siARN y los microARNs, puede causar efectos tóxicos (128). A pesar de estas posibles dificultades, intrínsecas al desarrollo de fármacos, las aplicaciones clínicas de los microARNs arrojan una prometedora esperanza. De hecho, en 2018, se aprobó por primera vez por la entidad reguladora de fármacos de Estados Unidos (la FDA) el uso terapéutico de un ARN de interferencia pequeño (siARN) llamado patisiran (129) Este fármaco tiene su utilidad terapéutica para el tratamiento de una enfermedad poco frecuente (una polineuropatía causada por amiloidosis hereditaria mediada por transtiretina, hATTR) y funciona uniéndose y degradando el transcrito de ARN mensajero del gen de la transtiretina (Kristen et al., 2018; Yang, 2019). Esta noticia ha supuesto un impulso relevante para las terapias farmacológicas basadas en el ARN de interferencia, donde se engloban a los microARNs. Así, aunque la aparición de la terapéutica mediada directamente por los microARNs aún no se ha traducido en candidatos aprobados por la FDA para intervención médica, existen fármacos en desarrollo clínico y se prevén que puedan seguir el camino del patisiran. De hecho, actualmente existen compañías de biotecnología centradas exclusivamente en las aplicaciones de fármacos relacionados con miARNs, como Miragen, MiRNA Therapeutics (ahora Synlogic) y Regulus Therapeutics (129). Actualmente laboratorios académicos, empresas de biotecnología y la industria farmacéutica están involucrados en los esfuerzos de investigación clínica relacionada con la actividad biológica de los microARNs. Esperemos que este desarrollo se traduzca en una utilidad farmacológica tangible para su uso en la clínica en un futuro cercano.
3. REFERENCIAS
- B. Wightman, I. Ha, G. Ruvkun, Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 75, 855-862 (1993).
- R. C. Lee, R. L. Feinbaum, V. Ambros, The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843-854 (1993).
- B. J. Reinhart et al., The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901-906 (2000).
- A. E. Pasquinelli et al., Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408, 86-89 (2000).
- M. Kato, F. J. Slack, microRNAs: small molecules with big roles – C. elegans to human cancer. Biol Cell 100, 71-81 (2008).
- B. M. Stadler, H. Ruohola-Baker, Small RNAs: keeping stem cells in line. Cell 132, 563-566 (2008).
- G. Stefani, F. J. Slack, Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 9, 219-230 (2008).
- R. Benetti et al., A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol 15, 268-279 (2008).
- S. F. Tavazoie et al., Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147-152 (2008).
- L. Sinkkonen et al., MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol 15, 259-267 (2008).
- K. C. Pang et al., RNAdb 2.0–an expanded database of mammalian non-coding RNAs. Nucleic Acids Res 35, D178-182 (2007).
- S. He et al., NONCODE v2.0: decoding the non-coding. Nucleic Acids Res 36, D170-172 (2008).
- Ana Kozomara, Maria Birgaoanu, Sam Griffiths-Jones. Nucleic Acids Research, Volume 47, Issue D1, 08 January 2019, Pages D155–D162,
- E. Berezikov et al., Many novel mammalian microRNA candidates identified by extensive cloning and RAKE analysis. Genome Res 16, 1289-1298 (2006).
- M. Lagos-Quintana, R. Rauhut, W. Lendeckel, T. Tuschl, Identification of novel genes coding for small expressed RNAs. Science 294, 853-858 (2001).
- N. C. Lau, L. P. Lim, E. G. Weinstein, D. P. Bartel, An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294, 858-862 (2001).
- Y. Zeng, Principles of micro-RNA production and maturation. Oncogene 25, 6156-6162 (2006).
- A. Rodriguez, S. Griffiths-Jones, J. L. Ashurst, A. Bradley, Identification of mammalian microRNA host genes and transcription units. Genome Res 14, 1902-1910 (2004).
- L. P. Lim et al., The microRNAs of Caenorhabditis elegans. Genes Dev 17, 991-1008 (2003).
- D. P. Bartel, MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281-297 (2004).
- R. I. Gregory, R. Shiekhattar, MicroRNA biogenesis and cancer. Cancer Res 65, 3509-3512 (2005).
- J. Han et al., The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 18, 3016-3027 (2004).
- A. M. Denli, B. B. Tops, R. H. Plasterk, R. F. Ketting, G. J. Hannon, Processing of primary microRNAs by the Microprocessor complex. Nature 432, 231-235 (2004).
- E. Lund, S. Guttinger, A. Calado, J. E. Dahlberg, U. Kutay, Nuclear export of microRNA precursors. Science 303, 95-98 (2004).
- R. Yi, Y. Qin, I. G. Macara, B. R. Cullen, Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17, 3011-3016 (2003).
- G. Hutvagner, P. D. Zamore, A microRNA in a multiple-turnover RNAi enzyme complex. Science 297, 2056-2060 (2002).
- R. F. Ketting et al., Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev 15, 2654-2659 (2001).
- S. M. Hammond, Dicing and slicing: the core machinery of the RNA interference pathway. FEBS Lett 579, 5822-5829 (2005).
- R. I. Gregory, T. P. Chendrimada, N. Cooch, R. Shiekhattar, Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123, 631-640 (2005).
- T. P. Chendrimada et al., TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 436, 740-744 (2005).
- T. A. Rand, S. Petersen, F. Du, X. Wang, Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 123, 621-629 (2005).
- C. Matranga, Y. Tomari, C. Shin, D. P. Bartel, P. D. Zamore, Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 123, 607-620 (2005).
- J. Martinez, A. Patkaniowska, H. Urlaub, R. Luhrmann, T. Tuschl, Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 110, 563-574 (2002).
- D. S. Schwarz et al., Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199-208 (2003).
- G. Tang, B. J. Reinhart, D. P. Bartel, P. D. Zamore, A biochemical framework for RNA silencing in plants. Genes Dev 17, 49-63 (2003).
- S. Yekta, I. H. Shih, D. P. Bartel, MicroRNA-directed cleavage of HOXB8 mRNA. Science 304, 594-596 (2004).
- R. S. Pillai et al., Inhibition of translational initiation by let-7 MicroRNA in human cells. Science 309, 1573-1576 (2005).
- P. H. Olsen, V. Ambros, The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol 216, 671-680 (1999).
- S. R. Viswanathan, G. Q. Daley, R. I. Gregory, Selective blockade of microRNA processing by Lin28. Science 320, 97-100 (2008).
- S. Bagga et al., Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122, 553-563 (2005).
- P. P. Medina, F. J. Slack, microRNAs and cancer: an overview. Cell Cycle 7, 2485-2492 (2008).
- A. Stark, J. Brennecke, N. Bushati, R. B. Russell, S. M. Cohen, Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3’UTR evolution. Cell 123, 1133-146 (2005).
- B. P. Lewis, C. B. Burge, D. P. Bartel, Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15-20 (2005).
- B. John et al., Human MicroRNA targets. PLoS Biol 2, e363 (2004).
- A. Krek et al., Combinatorial microRNA target predictions. Nat Genet 37, 495-500 (2005).
- A. Grimson et al., MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27, 91-105 (2007).
- D. Hanahan, R. A. Weinberg, The hallmarks of cancer. Cell 100, 57-70 (2000).
- N. Yanaihara et al., Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9, 189-198 (2006).
- J. Lu et al., MicroRNA expression profiles classify human cancers. Nature 435, 834-838 (2005).
- S. Volinia et al., A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103, 2257-2261 (2006).
- A. Esquela-Kerscher, F. J. Slack, Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer 6, 259-269 (2006).
- M. S. Kumar, J. Lu, K. L. Mercer, T. R. Golub, T. Jacks, Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 39, 673-677 (2007).
- Y. Karube et al., Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci 96, 111-115 (2005).
- E. P. Murchison, J. F. Partridge, O. H. Tam, S. Cheloufi, G. J. Hannon, Characterization of Dicer-deficient murine embryonic stem cells. Proc Natl Acad Sci U S A 102, 12135-12140 (2005).
- C. Kanellopoulou et al., Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev 19, 489-501 (2005).
- Y. Wang, R. Medvid, C. Melton, R. Jaenisch, R. Blelloch, DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet 39, 380-385 (2007).
- R. Koesters et al., Human eukaryotic initiation factor EIF2C1 gene: cDNA sequence, genomic organization, localization to chromosomal bands 1p34-p35, and expression. Genomics 61, 210-218 (1999).
- A. Aravin et al., A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203-207 (2006).
- H. Taubert et al., Expression of the stem cell self-renewal gene Hiwi and risk of tumour-related death in patients with soft-tissue sarcoma. Oncogene 26, 1098-1100 (2007).
- X. Liu et al., Expression of hiwi gene in human gastric cancer was associated with proliferation of cancer cells. Int J Cancer 118, 1922-1929 (2006).
- D. Qiao, A. M. Zeeman, W. Deng, L. H. Looijenga, H. Lin, Molecular characterization of hiwi, a human member of the piwi gene family whose overexpression is correlated to seminomas. Oncogene 21, 3988-3999 (2002).
- J. Kota et al., Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell 137, 1005-1017 (2009).
- A. Cimmino et al., miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102, 13944-13949 (2005).
- S. M. Johnson et al., RAS is regulated by the let-7 microRNA family. Cell 120, 635-647 (2005).
- C. Mayr, M. T. Hemann, D. P. Bartel, Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315, 1576-1579 (2007).
- Y. S. Lee, A. Dutta, The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21, 1025-1030 (2007).
- V. B. Sampson et al., MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res 67, 9762-9770 (2007).
- C. D. Johnson et al., The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res 67, 7713-7722 (2007).
- P. P. Trang* & Medina* et al., Regression of murine lung tumors by the let-7 microRNA. Oncogene 29, 1580-1587 (2010).
- G. T. Bommer et al., p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 17, 1298-1307 (2007).
- C. Welch, Y. Chen, R. L. Stallings, MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 26, 5017-5022 (2007).
- H. Tazawa, N. Tsuchiya, M. Izumiya, H. Nakagama, Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A 104, 15472-15477 (2007).
- L. He et al., A microRNA component of the p53 tumour suppressor network. Nature 447, 1130-1134 (2007).
- K. Woods, J. M. Thomson, S. M. Hammond, Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. J Biol Chem 282, 2130-2134 (2007).
- K. A. O’Donnell, E. A. Wentzel, K. I. Zeller, C. V. Dang, J. T. Mendell, c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435, 839-843 (2005).
- Y. Sylvestre et al., An E2F/miR-20a autoregulatory feedback loop. J Biol Chem 282, 2135-2143 (2007).
- F. Meng et al., MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133, 647-658 (2007).
- S. Zhu, M. L. Si, H. Wu, Y. Y. Mo, MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 282, 14328-14336 (2007).
- L. B. Frankel et al., Programmed Cell Death 4 (PDCD4) Is an Important Functional Target of the MicroRNA miR-21 in Breast Cancer Cells. J Biol Chem 283, 1026-1033 (2008).
- I. A. Asangani et al., MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene, (2007).
- P. P. Medina, M. Nolde, F. J. Slack, OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 467, 86-90 (2010).
- M. Gironella et al., Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A 104, 16170-16175 (2007).
- H. Q. Peng et al., Mutations of the p53 gene do not occur in testis cancer. Cancer Res 53, 3574-3578 (1993).
- F. Bullrich et al., Characterization of the 13q14 tumor suppressor locus in CLL: identification of ALT1, an alternative splice variant of the LEU2 gene. Cancer Res 61, 6640-6648 (2001).
- G. A. Calin et al., Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99, 15524-15529 (2002).
- G. A. Calin et al., A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353, 1793-1801 (2005).
- J. Takamizawa et al., Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64, 3753-3756 (2004).
- G. A. Calin et al., Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101, 2999-3004 (2004).
- S. L. Yu et al., MicroRNA Signature Predicts Survival and Relapse in Lung Cancer. Cancer Cell 13, 48-57 (2008).
- S. Shell et al., let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci U S A 104, 11400-11405 (2007).
- V. Tarasov et al., Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 6, 1586-1593 (2007).
- D. C. Corney, A. Flesken-Nikitin, A. K. Godwin, W. Wang, A. Y. Nikitin, MicroRNA-34b and MicroRNA-34c Are Targets of p53 and Cooperate in Control of Cell Proliferation and Adhesion-Independent Growth. Cancer Res 67, 8433-8438 (2007).
- T. C. Chang et al., Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26, 745-752 (2007).
- A. Ota et al., Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 64, 3087-3095 (2004).
- L. He et al., A microRNA polycistron as a potential human oncogene. Nature 435, 828-833 (2005).
- M. Dews et al., Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38, 1060-1065 (2006).
- T. C. Chang et al., Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40, 43-50 (2008).
- P. M. Voorhoeve et al., A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Adv Exp Med Biol 604, 17-46 (2007).
- M. L. Si et al., miR-21-mediated tumor growth. Oncogene 26, 2799-2803 (2007).
- M. F. Corsten et al., MicroRNA-21 Knockdown Disrupts Glioma Growth in vivo and Displays Synergistic Cytotoxicity with Neural Precursor Cell Delivered S-TRAIL in Human Gliomas. Cancer Res 67, 8994-9000 (2007).
- C. Roldo et al., MicroRNA expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J Clin Oncol 24, 4677-4684 (2006).
- M. V. Iorio et al., MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65, 7065-7070 (2005).
- J. A. Chan, A. M. Krichevsky, K. S. Kosik, MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65, 6029-6033 (2005).
- A. J. Schetter et al., MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. Jama 299, 425-436 (2008).
- W. Tam, D. Ben-Yehuda, W. S. Hayward, bic, a novel gene activated by proviral insertions in avian leukosis virus-induced lymphomas, is likely to function through its noncoding RNA. Mol Cell Biol 17, 1490-1502 (1997).
- T. Zhang, K. Nie, W. Tam, BIC is processed efficiently to microRNA-155 in Burkitt lymphoma cells. Leukemia, (2008).
- B. E. Clurman, W. S. Hayward, Multiple proto-oncogene activations in avian leukosis virus-induced lymphomas: evidence for stage-specific events. Mol Cell Biol 9, 2657-2664 (1989).
- J. Kluiver et al., BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol 207, 243-249 (2005).
- P. S. Eis et al., Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 102, 3627-3632 (2005).
- S. Costinean et al., Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A 103, 7024-7029 (2006).
- T. H. Thai et al., Regulation of the germinal center response by microRNA-155. Science 316, 604-608 (2007).
- A. Rodriguez et al., Requirement of bic/microRNA-155 for normal immune function. Science 316, 608-611 (2007).
- E. Vigorito et al., microRNA-155 Regulates the Generation of Immunoglobulin Class-Switched Plasma Cells. Immunity 27, 847-859 (2007).
- E. Gottwein et al., A viral microRNA functions as an orthologue of cellular miR-155. Nature 450, 1096-1099 (2007).
- C. G. Liu et al., An oligonucleotide microchip for genome-wide microRNA profiling in human and mouse tissues. Proc Natl Acad Sci U S A 101, 9740-9744 (2004).
- N. Rosenfeld et al., MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 26, 462-469 (2008).
- M. E. Gleave, B. P. Monia, Antisense therapy for cancer. Nat Rev Cancer 5, 468-479 (2005).
- M. S. Kumar et al., Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci U S A 105, 3903-3908 (2008).
- A. Esquela-Kerscher et al., The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 7, (2008).
- F. Yu et al., let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131, 1109-1123 (2007).
- J. Krutzfeldt et al., Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438, 685-689 (2005).
- J. Elmen et al., LNA-mediated microRNA silencing in non-human primates. Nature 452, 896-899 (2008).
- R. E. Lanford et al., Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327, 198-201 (2010).
- D. Grimm et al., Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441, 537-541 (2006).
