1. INTRODUCCIÓN
1.1. Los galardonados
El premio Nobel de Química de 2021 ha sido concedido a los profesores Benjamin List (Universidad de Colonia e Instituto Max Planck de investigación en el carbón, Mülheim) y David MacMillan (Universidad de Princeton) (figura 1) “por el desarrollo de la organocatálisis asimétrica”, según indica el comunicado oficial de la Academia Sueca de Ciencia. En este artículo se explicarán los logros de estos investigadores que dieron lugar al nacimiento de una nueva rama de la síntesis orgánica y se hará también un breve resumen de su evolución posterior, debida al trabajo de estos pioneros y de otros investigadores.

Figura 1. Benjamin List y David MacMillan
1.2. Los conceptos de quiralidad y síntesis asimétrica
El término quiralidad procede del griego kheir (mano) y fue introducido en las ciencias experimentales por el físico William Thomson (Lord Kelvin), profesor de Filosofía Natural en la Universidad de Glasgow (figura 2), en una conferencia que se publicó por primera vez en 1905, aunque hay evidencias de que utilizaba el término ya en 1875 (1). La definición de Kelvin fue la siguiente: “Llamo «quiral» a cualquier figura geométrica, o grupo de puntos, y digo que tiene quiralidad si su imagen en un espejo plano, idealmente realizada, no puede coincidir con ella misma”. En el mismo escrito, utilizó la palabra “quiroide” para describir una molécula dotada de quiralidad. Previamente, Louis Pasteur, tras su famosa separación en 1848 de los enantiómeros del tartrato sódico amónico, había introducido el término “disimetría”.

Figura 2. Louis Pasteur y William Thomson (Lord Kelvin)
En términos de estructura molecular, los elementos que con mayor frecuencia condicionan la existencia de quiralidad son los centros y los ejes estereogénicos (figura 3). Una molécula quiral y su imagen en un espejo se llaman enantiómeros, mientras que moléculas que que presentan combinaciones de centros estereogénicos en las que algunos tienen configuraciones opuestas y otros la misma se llaman diastereómeros.Los enantiómeros son idénticos en sus propiedades físicas y químicas, mientras que los diastereómeros pueden presentar diferencias.
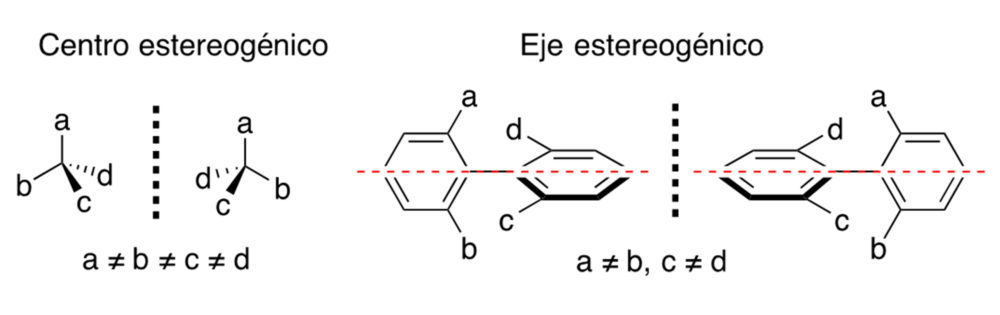
Figura 3. Principales elementos de quiralidad en moleculas orgánicas
Es habitual que dos enantiómeros tengan diferente comportamiento biológico. Un ejemplo muy conocido es el del limoneno, cuyo enantiómero (S ) tiene olor a pino mientras que el (R ) huele a limón (Figura 4A). La explicación es que las proteínas receptoras del olor están constituidas por aminoácidos quirales y por tanto las proteínas también lo son, por lo que las combinaciones enantiómero S-proteína y enantiómero R-proteína son diastereómeras y tienen diferentes propiedades. Análogamente, puesto que las dianas terapéuticas son básicamente proteínas y por tanto especies quirales, en principio pueden diferenciar ligandos enantioméricos. Esto conduce a varias posibles situaciones, incluyendo: (a) Solo uno de los enantiómeros es activo. Por ejemplo: la (R )-isoprenalina es más de 800 veces más activa como broncodilatadora que su enantiómero (S ) (figura 4B). (b) Los dos enantiómeros tienen actividades distintas. Por ejemplo, el dextropropoxifeno es analgésico, mientras que el levopropoxifeno es antitusivo (figura 4C). (c) Un enantiómero posee la acción terapéutica buscada, mientras que el otro es responsable de un efecto tóxico. El ejemplo más conocido es la talidomida, cuyo enantiómero (S ) es teratógeno mientras que el (R ) no lo es (figura 4D). Además de con sus dianas terapéuticas, los enantiómeros presentan diferente interacción con enzimas metabólicas, proteínas de transporte, etc.
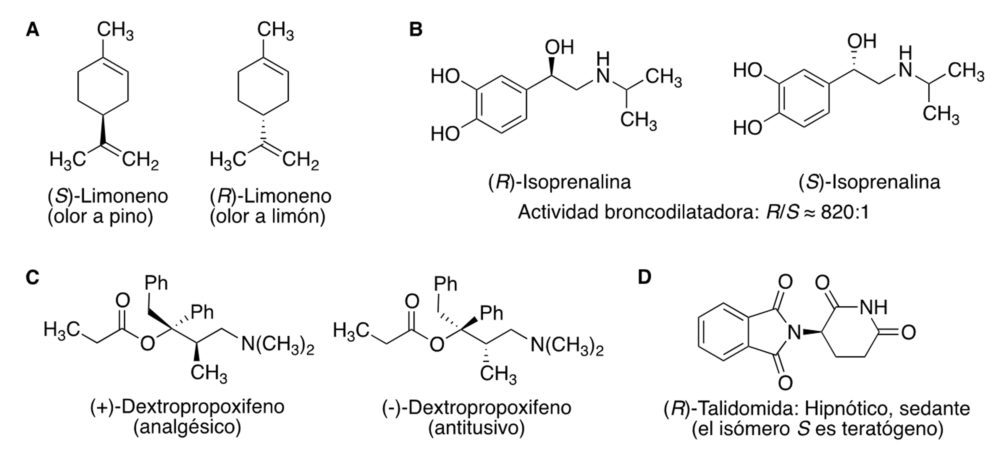
Figura 4. Algunos ejemplos de enantioespecificidad en la interacción de moléculas quirales con proteínas
Debido a las diferencias de comportamiento biológico de los enantiómeros, hace tiempo que existe la tendencia a comercializar los fármacos quirales en forma enantioméricamente pura. Esto require el desarrollo de métodos enantioselectivos de síntesis, tradicionalmente descritos como síntesis asimétrica. Estos métodos requieren que haya una influencia quiral sobre el transcurso de la reacción, y de acuerdo con la naturaleza de la especie quiral, es frecuente clasificar los métodos de síntesis asimétrica en cuatro categorías:
- Métodos de primera generación, controlados por el sustrato. En ellos, la molécula sustrato de la reacción (S*) es quiral, y se transforma de forma estereoselectiva en un producto quiral (P*) (figura 5A).
- Métodos de segunda generación, controlados por un grupo auxiliar quiral. Se basan en la incorporación temporal al sustrato de un fragmento quiral (auxiliar quiral), que es eliminado después de llevar a cabo la transformación deseada. Idealmente, el auxiliar quiral puede recuperarse al final del proceso, directamente o tras una transformación química sencilla (figura 5B).
- Métodos de tercera generación, controlados por un reactivo quiral, en los que la inducción asimétrica procede de uno de los reactivos utilizados (figura 5C).
- Métodos de cuarta generación, controlados por un catalizador quiral. En este caso, la influencia quiral en el curso de la reacción se debe a un catalizador. El diseño de catalizadores de este tipo constituye el objetivo ideal de la síntesis asimétrica, puesto que permite emplear una cantidad pequeña de la especie quiral, que es generalmente la más costosa (figura 5D).

Figura 5. Estrategias en síntesis enantioselectiva
1.3. Concepto de organocatálisis
La organocatálisis puede definirse como la utilización de catalizadores con naturaleza de moléculas orgánicas pequeñas. Durante mucho tiempo, las únicas reacciones asimétricas catalíticas viables fueron las enzimáticas. En los años 80 se empezaron a desarrollar algunos catalizadores enantioselectivos basados en metales de transición y solo a partir de 2000 se empezó a trabajar de forma sistemática en catalizadores orgánicos, como consecuencia del trabajo galardonado con el premio Nobel de Química de 2021. Las principales ventajas de los catalizadores orgánicos sobre los basados en metales de transición son: (a) Estabilidad frente a la humedad y el oxígeno; (b) mucha mayor facilidad de manipulación; (c) baja toxicidad y menor posibilidad de contaminación de los productos finales; (d) residuos más fáciles de tratar; (e) se obtienen a partir de materiales biológicos y son en general más baratos; (f) las reacciones son generalmente más reproducibles.
2. LA CONTRIBUCIÓN DE MCMILLAN: ORGANOCATÁLISIS DE TIPO IMINIO
David William Cross MacMillan nació en la ciudad escocesa de Bellshill en 1968, y estudió Química en la Universidad de Glasgow, graduándose en 1990. Se trasladó a la Universidad de California, Irvine, para realizar su tesis doctoral sobre la síntesis enantioselectiva de heterociclos oxigenados bajo la dirección de Larry Overman, presentándola en 1996. Su primer contacto con la catálisis enantioselectiva tuvo lugar durante sus estudios postdoctorales en el grupo de David Evans, en la Universidad de Harvard, utilizando como catalizadores complejos quirales de estaño. En 1998, consiguió una plaza de Assistant Professor en la Universidad de Berkeley, comenzando su carrera como investigador independiente.
MacMillan se planteó si sería posible emular el efecto catalítico de los ácidos de Lewis que había manejado durante su estancia postdoctoral con otros tipos de catalizadores. En la figura 6A se muestra la activación de la acroleína por un ácido de Lewis, que genera una carga positiva en el oxígeno. Análogamente, si la acroleína reacciona con una amina secundaria se forma un derivado de iminio con una distribución de carga parecida, y MacMillan se planteó emplear este tipo de aminas como catalizadores. Encontró algunos problemas iniciales de falta de enantioselectividad asociados a equilibrios conformacionales en el iminio intermedio, lo que le llevó a diseñar derivados de imidazolidinona del tipo del compuesto 1, que actualmente se conocen como catalizadores de MacMillan. Comprobó computacionalmente que los derivados de iminio derivados de estas aminas secundarias son conformacionalmente rígidos, dejando accesible solo una de las caras de su doble enlace C=C (figura 6B). Utilizando sus imidazolidinonas como catalizadores, MacMillan desarrolló la primera reacción de Diels-Alder enantioselectiva y organocatalítica (figura 6C) (2). Cabe destacar que en la publicación donde documentó este descubrimiento se utilizó por primera vez la palabra “organocatálisis”.
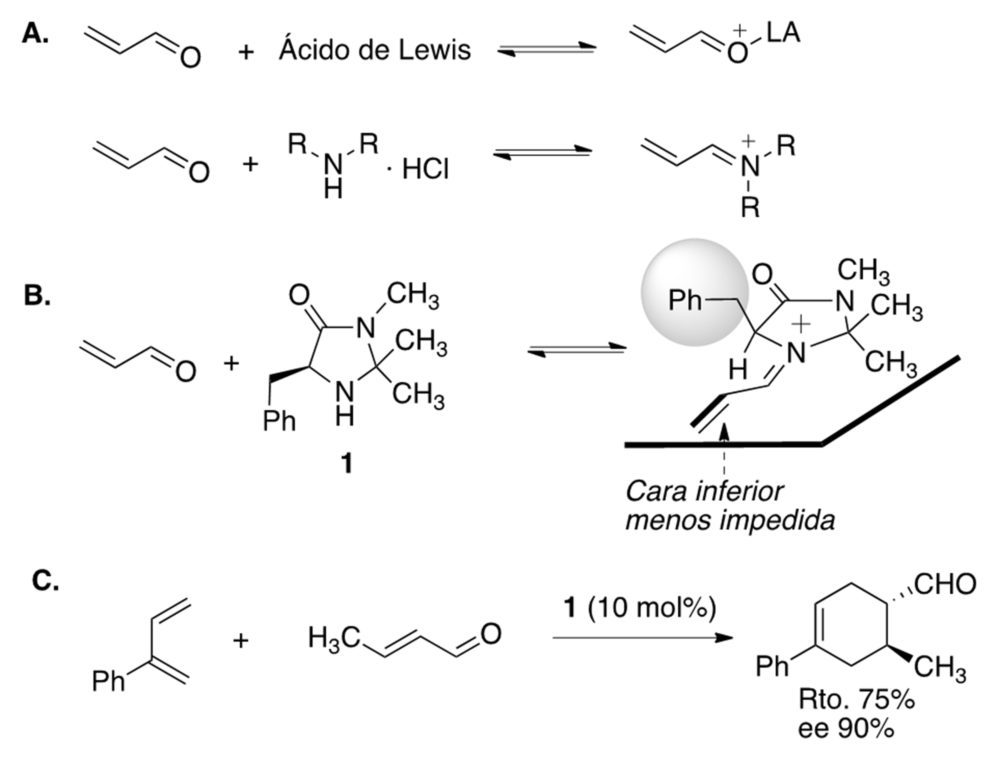
Figura 6. A. Analogía entre la catálisis por ácidos de Lewis y la catálisis de tipo iminio. B. Activación de la acroleína por una de las imidazolidinonas de MacMillan. C. Un ejemplo de una reacción de Diels-Alder enantioselectiva organocatalítica.
3. LA CONTRIBUCIÓN DE LIST: ORGANOCATÁLISIS DE TIPO ENAMINA
Benjamin List nació en Frankfurt en 1968, el mismo año que MacMillan. Estudió en la Universidad Libre de Berlín, de la que salió en 1993 con un grado en Química para dirigir sus pasos a la Universidad Goethe de Frankfurt. En el período 1993-1997 realizó allí su tesis doctoral, dirigida por Johann Mulzer, sobre un proyecto de síntesis total de corrinas. Para sus estudios postdoctorales seleccionó el grupo de Richard Lerner, en el Scripps Research Institute de California, que en aquel momento era líder mundial en el estudio de los anticuerpos como catalizadores. En el grupo de Lerner nuestro protagonista conoció a Carlos Barbas III, que tendría un papel muy importante en los descubrimientos que condujeron al premio Nobel y que es probable que lo hubiera compartido de no ser por su prematuro fallecimiento en 2014. En 1999, List ocupó una plaza de Assistant Professor en el Scripps y, como estaba haciendo MacMillan casi al mismo tiempo, se planteó el uso de aminas como catalizadores, en este caso a través de catálisis de tipo enamina. Existían precedentes bioquímicos de este tipo de reactividad, y así se había propuesto, ya en los años 60 (3), que el ciclo catalítico de las aldolasas transcurría a través de un intermedio de tipo enamina formado a partir del fosfato de dihidroxiacetona, uno de los sustratos de la reacción, y un residuo de lisina, lo que se ha verificado posteriormente por técnicas cristalográficas (4) (figura 7). Unos años antes de la llegada de List al grupo, Lerner y Barbas habían descrito la existencia de anticuerpos con actividad aldolasa, para los que se también propusieron un mecanismo catalítico a través de la formación de una enamina (5). Más aún, Hajos y Parrish, que trabajaban en la industria farmacéutica (Hoffmann-La Roche y Schering), habían publicado en 1974 un primer ejemplo del empleo de un aminoácido, la L-prolina, como catalizador enantioselectivo de una reacción orgánica, concretamente una reacción aldólica intramolecular (6), aunque no llegaron a percibir la importancia de su resultado como primer ejemplo de un nuevo tipo de catálisis.
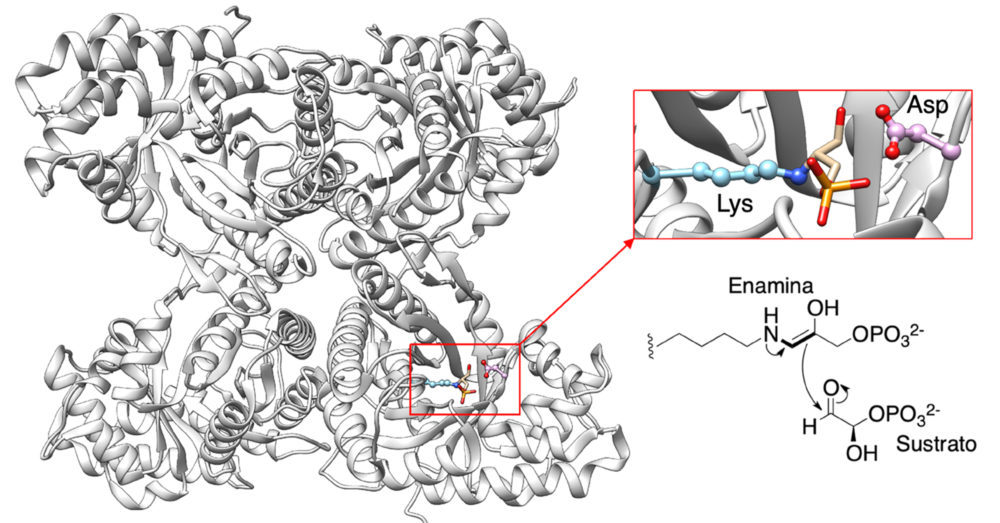
Figura 7. Estructura de la fructosa-1,6-(bis)fosfato aldolasa (PDB 1J4E) y expansión de su sitio activo, que muestra la enamina intermedia de su ciclo catalítico.
Buscando promover la reacción aldólica intermolecular, de mucha mayor versatilidad sintética, List, Lerner y Barbas describieron el uso de la L-prolina para este propósito, considerándose su artículo publicado en 2000 (7), junto con el de MacMillan del mismo año, el evento fundacional de la organocatálisis. Una de las reacciones descritas en esta publicación seminal está representada en la figura 8A, en la que también se recogen algunas reacciones relacionadas descritas en los años inmediatamente posteriores, como la de Mannich descrita por Barbas (figura 8A) (8), la adición aldólica entre cetonas y aldehídos alifáticos desarrollada por List (figura 8B) (9) y la reacción de α-aminación con azodicarboxilatos debida a Jørgensen (figura 8C) (10).

Figura 8. Primeros ejemplos del estudio de la prolina como organocatalizador enantioselectivo. A. Adición aldólica de cetonas a aldehídos aromáticos y reacción de Mannich. B. Adición aldólica de cetonas a aldehídos alifáticos. C. Adición de cetonas a azodicarboxilatos.
La elevada eficacia de la prolina como catalizador ha impulsado la síntesis y estudio de un elevado número de análogos y derivados de este aminoácido. Aunque hay literalmente cientos de ellos (11), destacaremos por su amplia utilización los derivados de diarilprolinol 3 y 4, conocidos, respectivamente, como catalizadores de Hayashi y Jørgensen. También es de interés el compuesto 5, desarrollado por Ley, en el que aplicó el isosterismo entre el grupo carboxílico y el anillo de tetrazol para diseñar un análogo de prolina dotado de mayor lipofilia (figura 9).
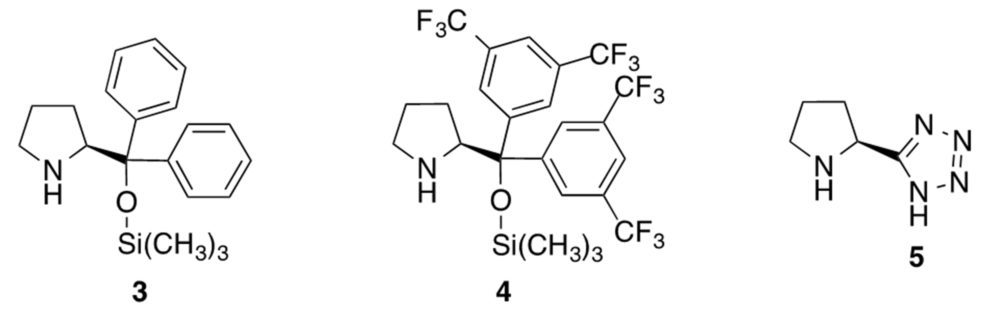
Figura 9. Organocatalizadores de Hayashi, Jørgensen y Ley (3-5, respectivamente)
4. ASPECTOS MECANÍSTICOS DE LA AMIOCATÁLISIS
Las reacciones catalizadas por aminas transcurren a través de intermedios covalentes. En la figura 10A se resume, como ejemplo de catálisis de tipo iminio, el ciclo catalítico de una adición de Michael promovida por el catalizador de MacMillan 1 a través de un mecanismo de tipo iminio, mientras que en la figura 10B se puede encontrar un ciclo catalítico de tipo enamina, correspondiente a la adición aldólica llevada a cabo en presencia de L-prolina 2.
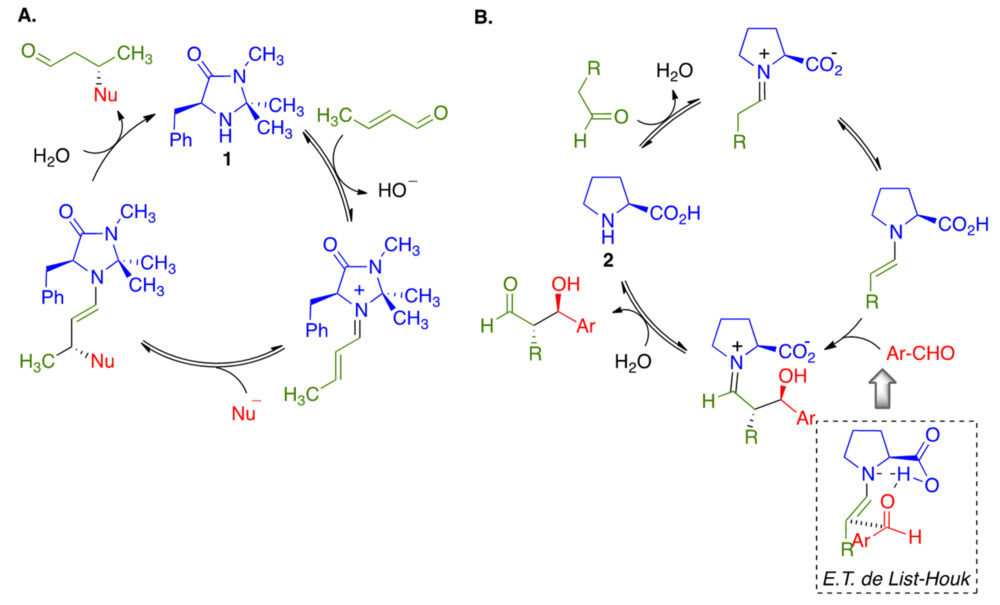
Figura 10. A. Catálisis de tipo iminio: Adición de Michael en presencia del catalizadador de MacMillan 1. B. Adición aldólica catalizada por L-prolina.
Desde el punto de vista de la teoría de orbitales moleculares, la catálisis de tipo iminio se describe como catálisis LUMO y la de tipo enamina como catálisisis HOMO. El HOMO es el orbital ocupado de mayor energía de una molécula, mientras que el LUMO es el orbital desocupado de menor energía. Se admite que las reacciones de tipo nucleófilo-electrófilo entre dos moléculas tienen lugar por transferencia de densidad electronica desde el HOMO de una de ellas, que actúa como donador, al LUMO de la otra, que actúa como aceptor (figura 11A). Cualquier factor que reduzca la diferencia de energía entre estos dos orbitales acelera la reacción. Así, en la catálisis de tipo iminio hay una reducción de la energía del LUMO, mientras que en la de tipo enamina hay un aumento de la energía del HOMO, reduciéndose en ambos casos la diferencia HOMO-LUMO (figura 11B).
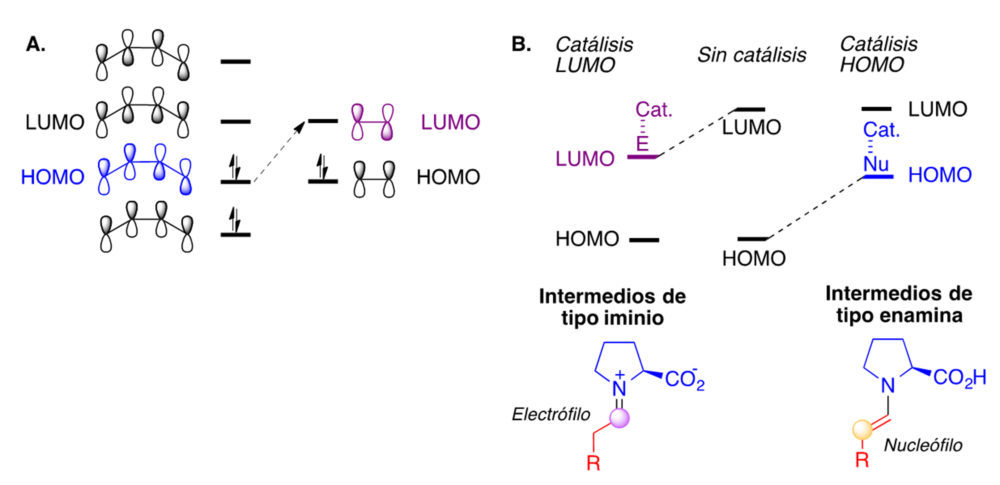
Figura 11. A. Orbitales HOMO y LUMO de un dieno conjugado y una olefina. B. La aminocatálisis desde el punto de vista de la teoría de orbitales moleculares.
5. EVOLUCIÓN POSTERIOR DE LA ORGANOCATÁLISIS
5.1. Otras modalidades de organocatálisis
Aunque la aminocatálisis ha sido muy importante en el desarrollo de la catálisis orgánica y todavía se utiliza muy ampliamente, tiene algunas limitaciones, en particular la de requerir cantidades relativamente elevadas de catalizador. Esto ha impulsado el desarrollo de otras modalidades de la organocatálisis, que se resumen brevemente en la figura 12. Se ha destacado, en primer lugar, el uso de carbenos generados a partir de heterociclos nitrogenados, que tienen un mecanismo de actuación similar al de la tiamina (vitamina B1) y que implica la formación de enlaces covalentes (figura 12A). Por otra parte, hay un número elevado de catalizadores que interaccionan con los sustratos a través de interacciones no covalentes tales como enlaces de hidrógeno, incluyendo, entre otros, numerosos derivados de urea y tiourea, ácidos de Brønsted derivados de estructuras quirales de binaftilo y catalizadores básicos relacionados con alcaloides de Cinchona, incluyendo derivados de una estructura conocida como escuaramida (figura 12B).

Figura 12. Algunas modalidades de organocatálisis desarrolladas con posterioridad a la catálisis por aminas.
5.2. Otros modos de activación
Jørgensen ha extendido la catálisis de tipo enamina a la activación de la posición γ de aldehídos α,β-insaturados y su posterior reacción con electrófilos mediante la formación de una dienamina intermedia (figura 13) Esta química se ha extendido por otros grupos a la obtención de arquitecturas complejas mediante una amplia variedad de estrategias.
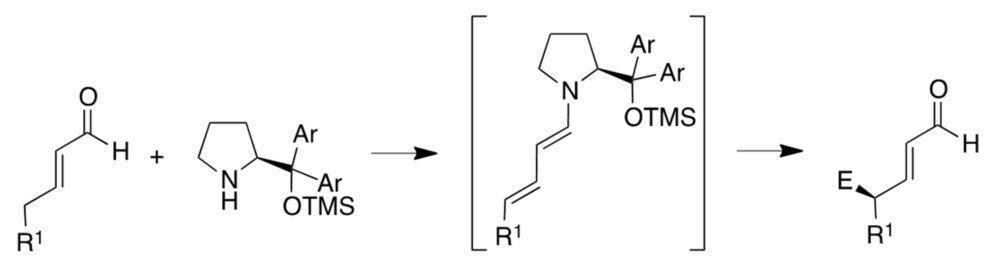
Figura 13. Activación de tipo dienamina
MacMillan fue el primero en proponer una estrategia organocatalítica que describió como activación SOMO, basada en la oxidación in situ de las enaminas generadas a partir de sustratos carbonílicos y aminas quirales para dar lugar a cationes radicales (figura 14) (15). El acrónimo SOMO hace referencia a un orbital molecular ocupado por un solo electrón (singly occupied molecular orbital). Estas especies pueden ser fácilmente captadas por una serie de reactivos más amplia que los que son accesibles a las estrategias de tipo iminio o enamina, ampliando el ámbito de aplicación de los métodos de aminocatálisis (16). En la figura 14 se representa el ciclo catalítico de una reacción de alilación asimétrica de un aldehído en α con aliltrimetilsilano en presencia de nitrato cérico amónico, para llevar a cabo las etapas de oxidación características de la activación SOMO, y la imidazolidinona de MacMillan de segunda generación 6.
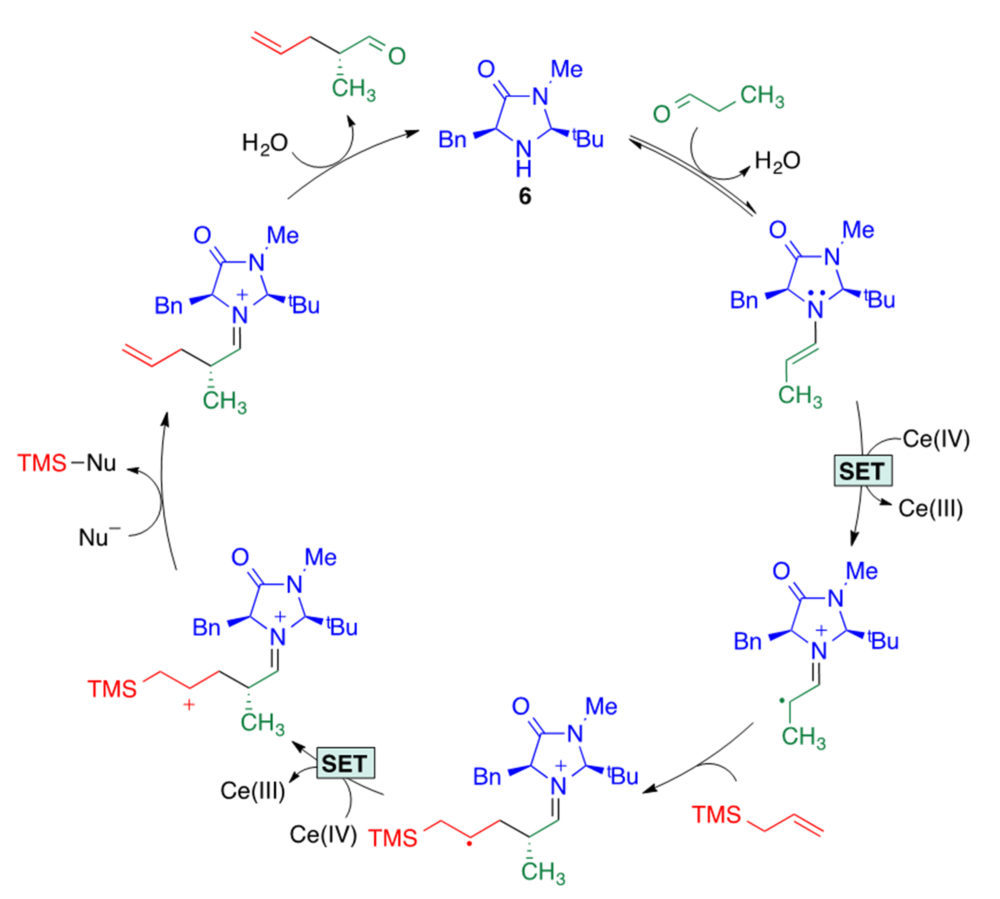
Figura 14. Un ejemplo de un ciclo catalítico de una reacción que transcurre con activación SOMO. SET, single electron transfer.
MacMillan ha sido también pionero en la combinación de la organocatálisis asimétrica con la catálisis foto-redox. La publicación de su grupo que inició el campo de la foto-organocatálisis permitió conseguir la alquilación directa de aldehídos con haluros electrodeficientes, utilizando catálisis de tipo enamina para el ciclo organocatalítico y la activación fotoquímica de un complejo de Ru(II) como etapa inicial del ciclo fotocatalítico acoplado (figura 15) (17). El primer proceso que combinó un ciclo de tipo iminio con activación fotocatalítica procede del grupo de Melchiorre y permitió la síntesis enantioselectiva de enonas cíclicas β,β-disustituidas con un estereocentro cuaternario (18).
Un hito más reciente es el trabajo de Powers en electro-organocatálisis, que combina una oxidación organocatalítica basada en especies de iodo hipervalente con acoplamientos oxidativos C–H/N–H, permitiendo una aminación intramolecular de arenos a partir de o-acetilaminobifenilos para generar carbazoles (figura 16) (19).
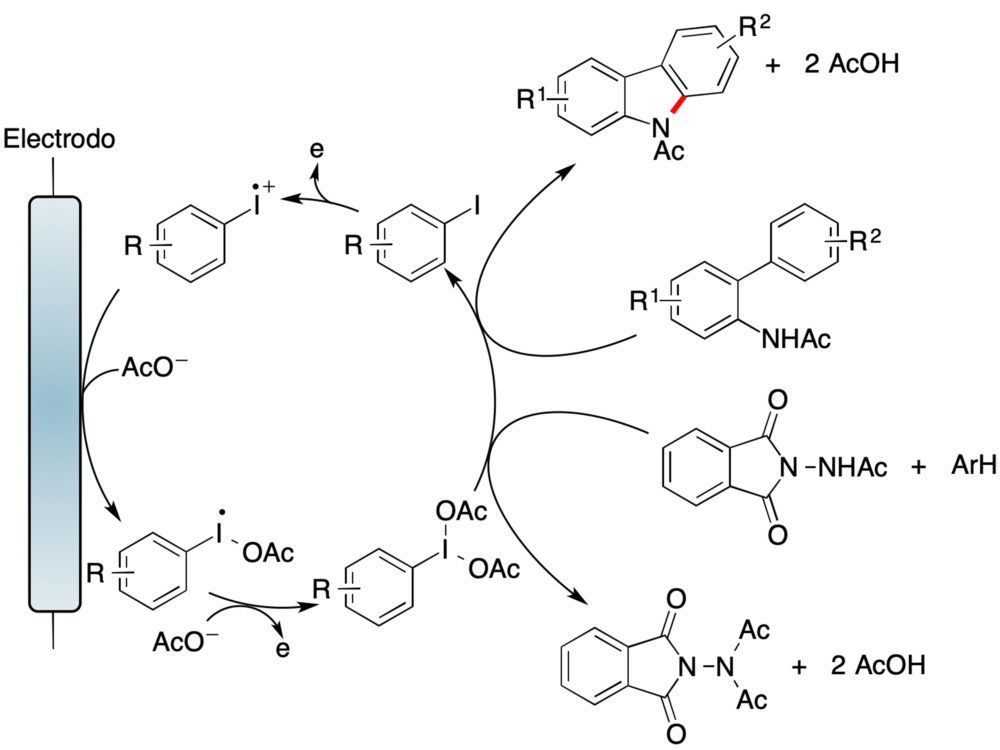
Figura 16. Un ejemplo de un proceso electro-organocatalítico
6. ALGUNAS APLICACIONES SINTÉTICAS DE LA ORGANOCATÁLISIS
Para ilustrar la aplicación de métodos organocatalíticos a la síntesis de moléculas complejas, se comentará en primer lugar su uso en reacciones dominó y multicomponente, capaces de generar varios enlaces en una única operación sintética mediante combinaciones de varios procesos catalíticos. En la figura 17A se muestra el uso por parte de Enders del catalizador de Hayashi para promover la síntesis de ciclohexenocarbaldehídos altamente sustituidos gracias a la formación de tres enlaces C-C y cuatro centros estereogénicos a partir de materiales de partida muy sencillos (20). El segundo ejemplo (figura 17B) ilustra el uso de prolina como catalizador capaz de promover la síntesis de heterociclos aquirales, una aplicación no siempre bien apreciada de la organocatálisis. Así, Perumal y Menéndez han descrito la síntesis en un paso de orto-quinonas tetracíclicas en un proceso que tiene lugar en agua y genera cinco enlaces (21).
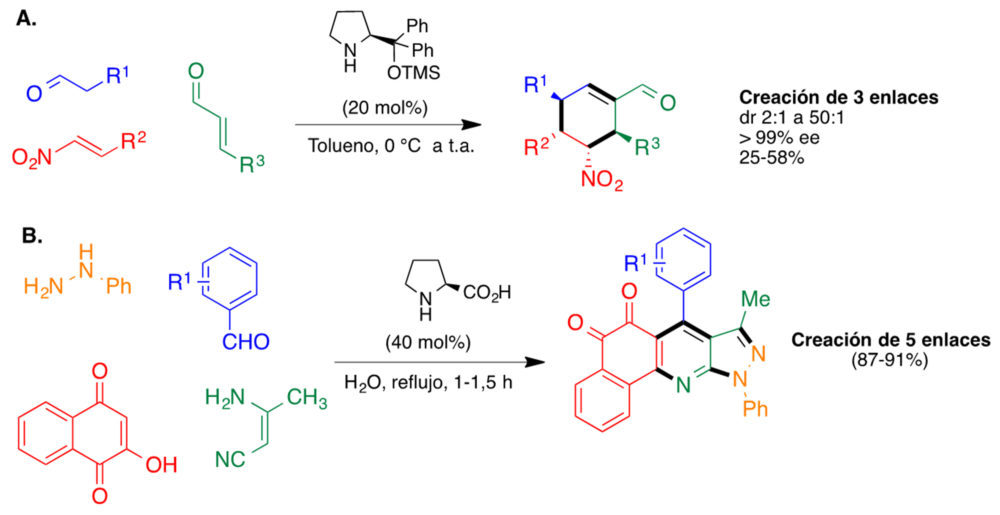
Figura 17. Dos reacciones multicomponente organocatalíticas
La organocatálisis se ha utilizado también como etapa clave de numerosas síntesis totales de productos naturales de interés biológico. Una de las primeras, debida a MacMillan, aprovecha la catálisis de tipo iminio para promover un proceso dominó de tipo adición de Michael-ciclación a partir de un derivado de triptamina. Esta reacción generó un sistema tricíclico derivado del sistema de hexahidropirrolo[2,3-b]indol, que se transformó en la flustramina B, un producto natural de origen marino con propiedades antibacterianas, mediante una sencilla secuencia de cuatro pasos (figura 18) (22).
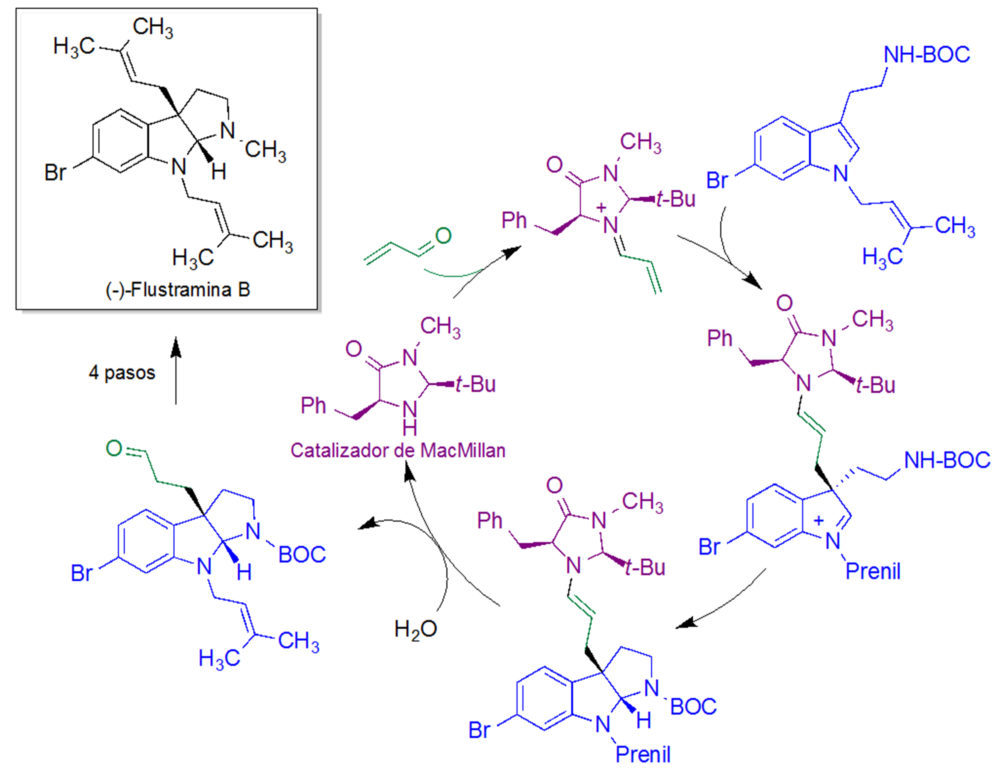
Figura 18. Síntesis de la flustramina B de MacMillan, basada en una reacción organocatalítica
Posteriormente, MacMillan ha publicado una síntesis unificada de seis alcaloides indólicos complejos a partir de un único precursor obtenido por métodos organocatalíticos (23). El proceso consta de dos ciclos catalíticos de tipo iminio, en el primero de los cuales tiene lugar una reacción de Diels-Alder enantioselectiva y en el segundo una reacción aza-Michael intramolecular (figura 19).
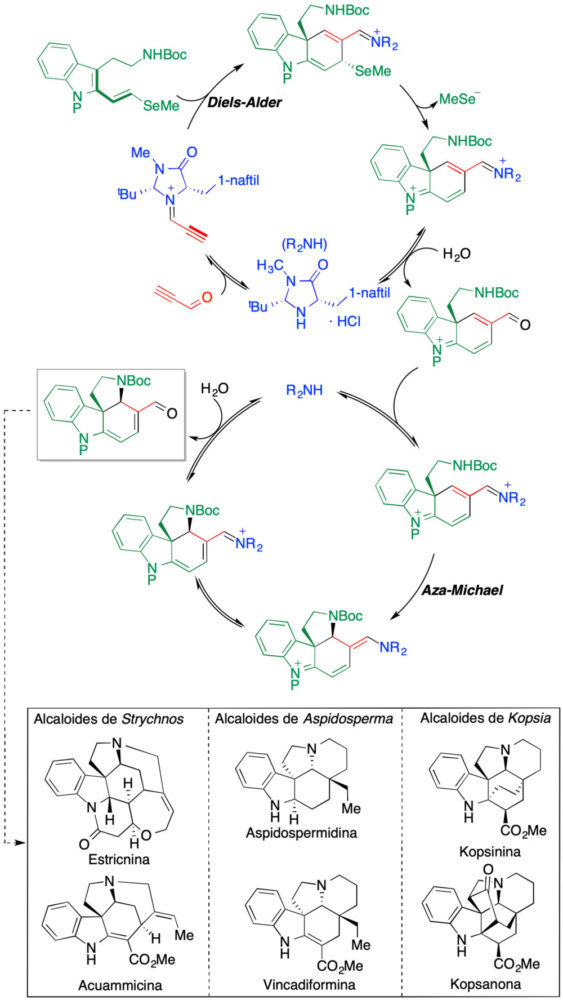
Figura 19. Síntesis colectiva organocatalítica de alcaloides indólicos a partir de un intermedio común
Finalmente, también se han utilizado reacciones organocatalíticas como etapas clave de la síntesis de fármacos (24). En la figura 20 se resume una ruta desarrollada por Hayashi para la síntesis del antigripal oseltamivir, que tiene la peculiaridad de poderse realizar en un único recipiente, sin necesidad de procesos de aislamiento y purificación intermedios. Destaca en ella la primera etapa, una adición de Michael enantioselectiva llevada a cabo en presencia del catalizador de Hayashi y una tiourea aquiral, en medio ácido (figura 20). También es interesante la síntesis de List de la estrona, un estrógeno natural, basada en la ciclación de un precursor aquiral en presencia de un catalizador derivado de binaftilo (figura 21).
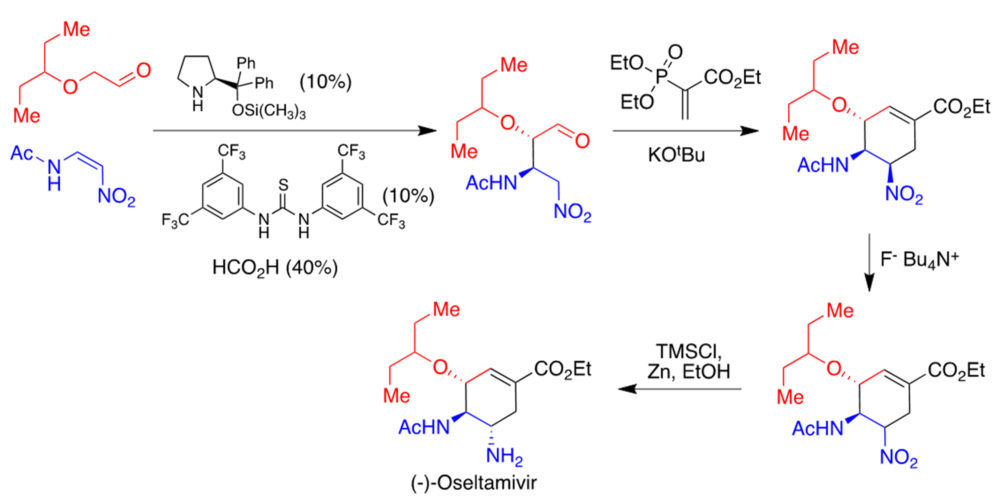
Figura 20. Síntesis del oseltamivir desarrollada por Hayashi
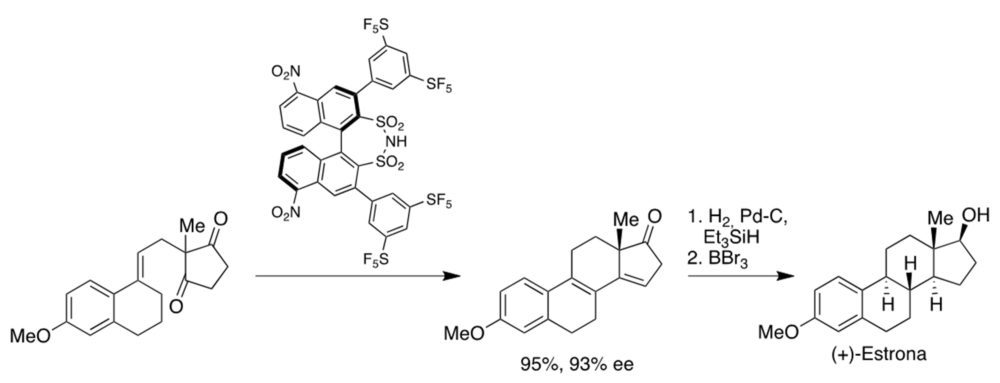
Figura 21. Síntesis de la estrona desarrollada por List
7. CONCLUSIONES
El trabajo de Benjamin List y David MacMillan permitió la identificación de algunas moléculas orgánicas sencillas capaces de actuar como catalizadores enantioselectivos. Lo que es aún más importante, fueron capaces de racionalizar los resultados dentro de un cuerpo de doctrina general, lo que permitió una respuesta inmediata de la comunidad científica y como consecuencia la apertura de un nuevo campo de la síntesis orgánica. Una consecuencia de este trabajo pionero ha sido el desarrollo de una nueva herramienta para la construcción de moléculas quirales, con características mejoradas en cuanto a eficiencia e impacto ambiental respecto a las previamente conocidas.
8. REFERENCIAS
- Barron LD. From cosmic chirality to protein structure: Lord Kelvin’s legacy. Chirality 2012; 24: 879-893.
- Ahrent KA, Borths CJ, MacMillan DWC. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels-Alder reaction. J Am Chem Soc 2000; 122:4243-4244.
- Rutter WJ. Evolution of aldolase. Fed Proc Am Soc Exp Biol 1964; 23: 1248-1257.
- Choi KH, Shi J, Hopkins CE, Tolan DR, Allen KN. Snapshots of catalysis: The structure of fructose-1,6-(bis)phosphate aldolase covalently bound to the substrate dihydroxyacetone phosphate. Biochemistry 2001; 40: 13868-13875.
- Wagner J, Lerner RA, Barbas III C. Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995; 270: 1797-1800.
- Hajos ZJ, Parrish DR. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J Org Chem 1974; 39: 1615-1621.
- List B, Lerner RA, Barbas III CF. Proline-catalyzed direct asymmetric aldol reactions. J Am Chem Soc 2000; 122: 2395-2396.
- Notz W, Sakthivel K, Bui T, Zhong G, Barbas III CF. Amine-catalyzed direct asymmetric Mannich-type reactions. Tetrahedron Lett 2001; 42: 99–201.
- List B, Pojarliev P, Castello C. Proline-catalyzed asymmetric aldol reactions between ketones and -unsubstituted aldehydes. Org Lett 2001; 3: 573-575.
- Kumaragurubaran N, Juhl K, Zhuang W, Bøgevig A, Jørgensen KA. Direct L-proline-catalyzed asymmetric -amination of ketones. J Am Chem Soc 2002; 124: 6254-6255.
- Vachan BS, M. Vinoth KP, Sridharan V, Menéndez JC. Stereoselective organic synthesis in water: Organocatalysis by proline and its derivatives, in: Inamuddin R, Boddula R, Asiri AM, Eds. Green sustainable process for chemical and environmental engineering and science: Organic synthesis in water and supercritical water, chapter 6. Elsevier, 2020.
- Melchiorre P. Cinchona-based primary amine catalysis in the asymmetric functionalization of carbonyl compounds. Angew Chem Int Ed 2012; 39: 9748-9770.
- Bertelsen S, Marigo M, Brandes S, Dinør P, Jørgensen KA. J Am Chem Soc 2006; 128: 12973-12980.
- Anebouselvy K, Ramachary DB, Kumar I. Dienamine catalysis for organic synthesis. Royal Society of Chemistry, 2018.
- Wilson JE, Casarez AD, MacMillan DWC, J Am Chem Soc 2009; 131: 11332–11334.
- Mečiarová M, Tisovský P, Šebesta R. Enantioselective organocatalysis using SOMO activation. New J Chem 2016; 40: 4855-4864.
- Nicewicz DA, MacMillan DWC. Merging photoredox catalysis with organocatalysis: the direct asymmetric alkylation of aldehydes. Science 2008; 322: 77–80.
- Silvi M, Melchiorre P. Enhancing the potential of enantioselective organocatalysis with light. Nature 2018; 554: 41-49.
- Maity A, Frey BL, Hoskinson ND, Powers DC. Electrocatalytic C–N coupling via anodically generated hypervalent iodine intermediates. J Am Chem Soc 2020; 142: 4990–4995.
- Enders D, Huüttl MRM, Grondal C, Raabe G. Control of four stereocentres in a triple cascade organocatalytic reaction. Nature 2006; 441: 861–863.
- Rajesh SM, Bala BD, Perumal S, Menéndez JC. L-Proline-catalysed four-component “on water” protocol for the synthesis of structurally complex heterocyclic ortho-quinones. Green Chemistry 2011; 13: 3248–3254.
- Austin JF, Kim S-G, Sinz CJ, Xiao W-J, MacMillan, DWC. Enantioselective organocatalytic construction of pyrroloindolines by a cascade addition–cyclization strategy: Synthesis of (-)-flustramine B. Proc Natl Acad Sci USA 2004, 101: 5482–5487.
- Jones SB, Simmons B, Mastracchio A, MacMillan DWC. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475: 183-188.
- Hayashi, Y. Domino and one-pot syntheses of biologically active compounds using diphenylprolinol silyl ether. Phys. Sci. Rev. 2020, 20180088.
