1.INTRODUCCIÓN
El receptor mineralocorticoide (MR) es el receptor principal de la hormona aldosterona, la cual regula en el riñón la retención de sodio y agua modulando así la homeostasis iónica y el volumen sanguíneo y, por tanto, la presión arterial. El MR, cuya localización se pensó originariamente que era exclusiva del riñón, se encuentra expresado en multitud de tejidos extra-renales y es funcionalmente activo en los sistemas cardiovascular, inmune y tejido adiposo. La excesiva activación del MR está implicada en varias enfermedades cardiovasculares (1,2) y en los últimos años, se le ha implicado también en enfermedades metabólicas y en la resistencia a insulina. A nivel vascular, el MR se expresa en células endoteliales y células musculares lisas y su activación da lugar a remodelado vascular, fibrosis y disfunción endotelial, lo cual produce daño vascular, rigidez vascular e hipertensión arterial (1-3). El MR también se expresa en células no vasculares pero en contacto directo con la pared vascular, como las procedentes del sistema inmune y los adipocitos mediando diversos procesos inflamatorios y metabólicos (4,5).
El MR es un receptor intracelular que tiene tres dominios: un dominio N-terminal que controla la actividad transcripcional del receptor; el dominio de unión al DNA que influye en la unión de elementos específicos de respuesta a la región promotora de los genes diana del MR y finalmente, el dominio de unión al ligando para aldosterona. Tras la activación, el MR se transloca al núcleo y regula la transcripción génica y la traducción de proteínas, mediante la unión en el DNA a elementos de respuesta estimulantes o represores de la síntesis de hormonas/esteroides (1,2). Además, aldosterona produce efectos no genómicos rápidos mediados por mecanismos dependientes e independientes del MR (6) y estudios recientes han puesto en evidencia que la supresión de la expresión de miRNAs dependiente de MR, ocasiona la sobre-expresión vascular de las dianas de dichos miRNAs (7). Tanto los efectos genómicos como los no genómicos promueven la producción de especies reactivas de oxígeno, principalmente por la enzima NADPH oxidasa, así como inflamación y fibrosis, las cuales intervienen en el remodelado y rigidez vascular así como en la disfunción endotelial. Esta revisión se centra en la relación entre la aldosterona, el receptor mineralocorticoide y el estrés oxidativo así como en los efectos vasculares de dicha relación (Figura 1).
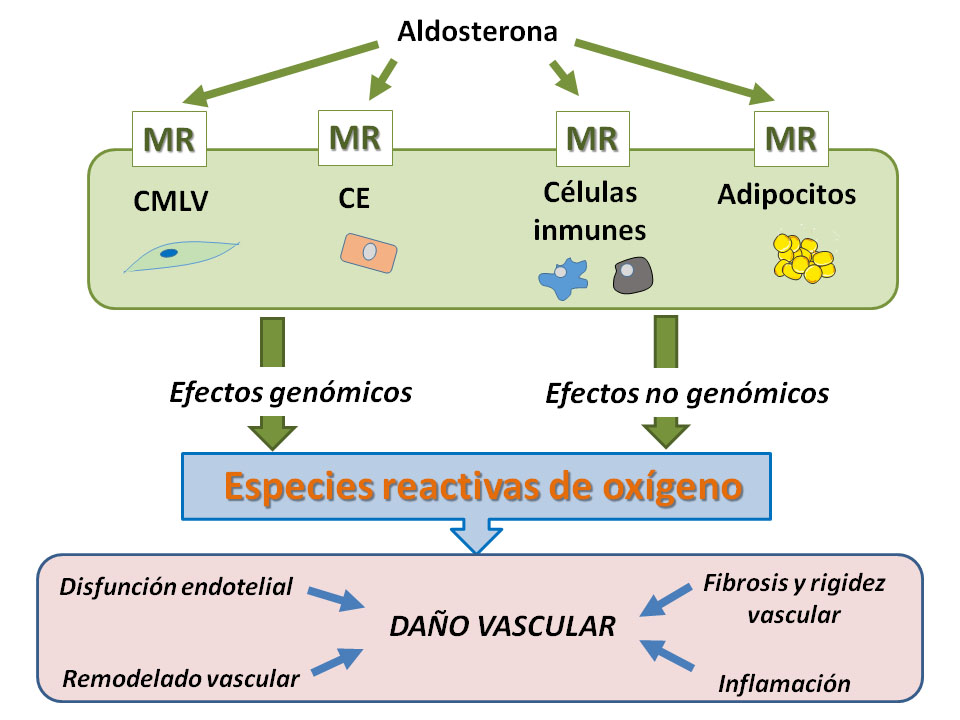
Figura 1. Papel del receptor mineralocorticoide en el daño vascular. A nivel vascular, el receptor mineralocorticoide (MR) se expresa en células endoteliales y vasculares lisas. También se localiza en adipocitos y células inmunes que pueden rodear o infiltrarse en el vaso. La activación del MR a través de mecanismos genómicos y no genómicos da lugar a la producción de especies reactivas de oxígeno, que favorecen la disfunción endotelial, el remodelado, fibrosis y rigidez vascular así como inflamación crónica de bajo grado. Todos estos factores producen daño vascular que favorecen o contribuyen al desarrollo y/o mantenimiento de la hipertensión arterial y otras enfermedades cardiovasculares. CE: Célula endotelial; CMLV: Célula muscular lisa vascular.
2. PAPEL DEL RECEPTOR MINERALOCORTICOIDE EN LA PRODUCCIÓN DE ESPECIES REACTIVAS DE OXÍGENO EN CÉLULAS VASCULARES
Numerosas evidencias demuestran que existe una relación entre el MR y la señalización redox en células vasculares. En modelos animales como el obtenido tras la administración de acetato de deoxicorticosterona (DOCA) y sal o de aldosterona y sal, con o sin nefrectomía, se observa un incremento en el estrés oxidativo vascular. Además, tanto en en modelos experimentales como en humanos, los fármacos bloqueantes del MR reducen los niveles de especies reactivas de oxígeno (ROS, del inglés reactive oxygen species) en patologías cardiovasculares como la hipertensión, obesidad, aterosclerosis o insuficiencia cardiaca, si bien existen ciertas diferencias entre los diversos estudios que bien podrían ser explicadas por una deficiente o no adecuada medida de dichas ROS, lo cual constituye un importante desafío en la actualidad (8,9).
En células musculares lisas vasculares (CMLV) (10-15) y células endoteliales (16-21) en cultivo, la exposición a aldosterona incrementa la producción de ROS. Además, la infusión de aldosterona en roedores incrementa el estrés oxidativo plasmático y vascular y el bloqueo del receptor MR reduce la producción de ROS en el contexto de la hipertensión, la obesidad y otras enfermedades cardiovasculares (21-29). Estudios iniciales identificaron a la enzima NADPH oxidasa como la responsable de la incrementada producción de ROS vasculares, específicamente de anión superóxido (O2.), en aorta de ratas con hipertensión inducida por mineralocorticoides, excluyendo otras fuentes de ROS vasculares (por ejemplo, la eNOS desacoplada, o la xantina oxidasa) como potenciales contribuyentes (30). De hecho, la NAPDH oxidasa se considera la principal fuente productora de ROS en respuesta a aldosterona y activación de MR en vasos.
La familia de la NADPH oxidasa o Nox está compuesta de 7 isoformas que incluyen las Nox1-5 y las Duox1 y 2, varias subunidades regulatorias (p22phox, p47phox, Noxo1, p67phox, Noxa1, p40phox) y el principal factor de unión Rac. La principal función catalítica de las NADPH oxidasas es la generación de ROS y lo hacen reduciendo el oxígeno a O2.- siendo NADPH el donador de electrones. Nox2 es la Nox clásica que fue inicialmente caracterizada en leucocitos. Asimismo, Nox-1, Nox-2, Nox-4 y Nox-5 se expresan en el sistema cardiovascular y, es importante mencionar que Nox5 no se expresa en roedores, lo cual dificulta enormemente su estudio. En relación con la especie oxidante que producen cada una de las isoformas, Nox1, Nox2, Nox3 y Nox5 producen O2.- mientas que Nox4 y las Duox producen H2O2 (31, 32). En las arterias, las células vasculares poseen Noxs funcionales pero además, los macrófagos residentes, los neutrófilos y las plaquetas también expresan NADPH oxidasas, particularmente en situaciones patológicas, que pueden contribuir al daño vascular en diversas patologías (32, 33).
2.1. Mecanismos implicados en la producción de especies reactivas de oxígeno en respuesta a aldosterona y estimulación del MR
En la actualidad, se conoce que el receptor MR puede producir varios de sus efectos a través de efectos genómicos y no genómicos, y la producción de ROS es uno de dichos efectos (Figura 1). Así, diferentes estudios han mostrado modulación de las Nox por aldosterona/MR. Concretamente, a nivel vascular, la combinación de aldosterona y sal produce un aumento en la producción de O2.- en CMLV a través de la sobreexpresión de Nox1 sin afectar al mRNA de Nox4, p22phox o p47phox (11). En células endoteliales de vena umbilical humana, aldosterona aumentó la transcripción de p47phox pero no tuvo efecto en los tránscritos de Nox1, Nox2, Nox4, p22phox, p40phox, o p67phox (16). Sin embargo, otros estudios mostraron que la incubación con aldosterona durante 24 h incrementó de manera dependiente de la dosis la expresión del mRNA de Nox4 en dichas células endoteliales humanas (17). En células endoteliales pulmonares humanas, aldosterona incrementó los niveles de proteína de Nox4 y p22phox así como la producción de H2O2 (20). De manera similar, en células endoteliales de retina bovina, aldosterona incrementó la expresión del gen de Nox4 (19) y otro estudio mostró que la administración de aldosterona moduló exclusivamente la expresión del gen de p22phox en células endoteliales aorticas (21). En estudios in vivo se ha observado que la administración de aldosterona más sal a ratas incrementó la actividad de la NADPH oxidasa y la expresión de p47phox, gp91phox y p22phox (34). Más recientemente, Jia y cols (29) encontraron una incrementada expresión vascular de Nox2 y de nitrotirosina (un marcador de estrés nitrosativo) después de 3 semanas de infusión de aldosterona en ratones. Todos estos resultados claramente sugieren un patrón de sobre-expresión de NADPH oxidasa por aldosterona a nivel vascular, lo cual es además apoyado por el hecho de que antagonistas del MR disminuyen la expresión de distintas subunidades de la NAPDH oxidasa (9). Por ejemplo, el tratamiento con eplerenona disminuyó la expresión de p22phox, p47phox y p40phox en un modelo de dieta alta en grasa (21). Además, la eliminación del MR en distintos tipos de células vasculares específicos disminuye la expresión de las isoformas de la NADPH oxidasa (ampliado más abajo).
En lo que respecta a la activación no genómica rápida del MR por aldosterona, se ha sugerido que MR podría estar localizado cerca de la membrana plasmática pero no directamente insertado en ella. El MR se localizaría en el lado citosólico asociado a proteínas de anclaje que están asociadas a la membrana o insertadas en ella como estriatina o caveolina-1 (6). En esta localización, aldosterona podría interaccionar con receptores con actividad tirosina quinasa tales como el receptor para el factor de crecimiento epidérmico (EGFR), el receptor para el factor de crecimiento derivado de plaquetas (PDGFR) o el receptor para el factor de crecimiento similar a insulina 1 (IGF1R), así como con receptores acoplados a proteínas G (GPCR), tales como el receptor AT1 de angiotensina II o el receptor 1 de estrógenos acoplado a proteínas G (GPER1) (6). La familia Src de proteínas con actividad tirosin quinasa parece estar implicada en la generación no genómica de ROS inducida por aldosterona (9). Concretamente, en CMLV en cultivo, la generación de ROS a través de NADPH oxidasa mediada por los efectos no genómicos de aldosterona está aumentada en células de ratas espontáneamente hipertensas y es dependiente de c-Src (10). También se ha descrito un papel para c-Src y Rac-1 en la activación de NADPH oxidasa en células endoteliales, aunque este efecto podría estar mediado por vías genómicas ya que se analizaron los efectos de la incubación con aldosterona durante tiempos prolongados (18). En CMLV, la transactivación de EGFR y PDGFR pero no de IGFR, inducida por MR y AT1 activa c-Src que a su vez, facilita la activación de la NAPDH oxidasa y la producción de ROS lo que produce migración de CMLV (35).
Además de los receptores acoplados a tirosin quinasas, los GPCR son importantes compañeros implicados en las acciones no genómicas de aldosterona. Así, la interacción entre el MR y el AT1 se ha implicado en la producción de ROS inducida por angiotensina II en vasos ya que el bloqueo del MR puede inhibir dicha producción (22). Más recientemente, se ha mostrado que el subtipo AT1a es necesario para la disfunción endotelial, remodelado vascular, estrés oxidativo e inflamación inducidos por la activación del MR (27), aunque un efecto genómico no puede ser del todo descartado en estos estudios. En miocitos cardiacos, la interacción entre el MR y el AT1 participa en la generación de ROS inducida por aldosterona mediada por Nox4 a través de la quinasa 2 de receptores acoplados a proteínas G (GRK2) probablemente a través de acciones no genómicas (36), aunque si este mecanismo dependiente de GRK2 también ocurre en células vasculares no se conoce. Igualmente, no es conocido el papel del posible nuevo receptor de aldosterona GPER1 (también conocido como GPR30) en la producción de ROS inducida por aldosterona en células vasculares y las evidencias que apoyan esta posibilidad provienen de células cardiacas (37,38).
Finalmente, es importante destacar que la señalización no genómica del MR también puede modular los efectos genómicos (6) perpetuando así la generación de ROS en células vasculares.
2.2. Ruta aldosterona/MR/estrés oxidativo en células endoteliales y musculares lisas
Numerosas evidencias experimentales han demostrado un efecto beneficioso del bloqueo de la ruta aldosterona/MR en el daño vascular, concretamente en la disfunción endotelial, remodelado y rigidez vasculares, así como en el estrés oxidativo (1-3,21,22,39-42). Por tanto, se ha sugerido que muchos de los efectos beneficiosos de los antagonistas de MR podrían deberse, al menos en parte, a su habilidad para disminuir el estrés oxidativo. Esto se apoya también en evidencias directas que proceden de estudios utilizando modelos de infusión de aldosterona o mineralocorticoides junto con tratamientos antioxidantes o por el uso de modelos transgénicos de ratón que sobre-expresan o no expresan el MR.
MR se expresa en células endoteliales de arterias cerebrales donde produce un aumento en la producción de O2.- en respuesta a niveles crónicamente aumentados de aldosterona (26). Además, la expresión del mRNA de p22phox, pero no de gp91phox, se incrementó por aldosterona, siendo este efecto completamente bloqueado en ratones con deleción de MR específicamente en células endoteliales, lo cual se acompañó además de una mejoría en la función endotelial (21). Sin embargo, en ausencia de estímulos, la sobre-expresión condicional de MR en células endoteliales no es suficiente para incrementar los niveles locales o sistémicos de estrés oxidativo (43), lo que sugiere que la sobreexpresión de MR solo no es suficiente para producir un aumento en la generación de ROS. Este hallazgo coincide con la idea de que el MR de las células endoteliales podría ser vasoprotector en estados fisiológicos y que dicha protección se pierde cuando coexisten factores de riesgo cardiovascular tales como hipertensión u obesidad o situaciones asociadas a elevados niveles de aldosterona, como se ha sugerido recientemente (2,44) (Figura 2). En apoyo de esta hipótesis, la deficiencia del MR específicamente en células endoteliales previno el aumento en la expresión de Nox2, Nox4, p22phox y 3-nitrotirosina inducidos por una dieta tipo western y esto fue paralelo a una reducción en la fibrosis aórtica y la rigidez, así como con la normalización de la activación de la eNOS (28). Igualmente, la eliminación de MR en células endoteliales previno en arterias de resistencia, la disfunción endotelial asociada a la hiperlipidemia en hembras, pero no en machos, y esto se asoció a una menor producción de O2.- (45).
En lo que respecta a CMLV, aunque los ratones con deleción específica de MR en CMLV no muestran cambios en los niveles basales de ROS, dichos ratones cuando envejecen producen significativamente menos ROS (7,46). Además, tanto los ratones jóvenes como los envejecidos que no expresan MR en CMLV muestran una atenuada producción de ROS inducida por angiotensina II (46), lo cual podría contribuir a la menor presión arterial que muestran esos animales y a la mejor función vascular (46). Más recientemente, una relación inversa entre el receptor MR de CMLV y el miR-155 se ha descrito en el envejecimiento (7), relación por la cual este miRNA produciría una represión del estrés oxidativo asociado a MR de CML, lo cual tendría un efecto también en la función vascular (7). Sin embargo, la fuente de ROS específica que sería modulada por el miR-155 es desconocida. El MR de CML también se ha implicado en la progresión de la insuficiencia cardiaca post-infarto de miocardio a través de su efecto directo induciendo estrés oxidativo, que favorece la disfunción endotelial y una disminución en la reserva coronaria (47). Así, en este estudio, los antioxidantes apocinina y superoxido dismutasa (SOD), mejoraron la relajación dependiente de endotelio de arterias coronarias de ratones con infarto de miocardio sin afectar a la relajación de las arterias de ratones knockout para MR específicos de CMLV de ratones con infarto ni a las arterias de ratones con infarto tratados con el antagonista del MR finerenona (47), hallazgos que indican un menor efecto del estrés oxidativo cuando el MR está ausente en CMLV o después de un bloqueo general del MR. Todos juntos, estos resultados apuntan a las células endoteliales y las CMLV como fuentes potenciales de ROS en respuesta a aldosterona o condiciones patológicas que impactan sobre la función vascular (Figura 2).

Figura 2. Implicación del receptor mineralocorticoide en el daño vascular en situaciones patológicas. En condiciones patológicas como la diabetes tipo 2 (T2DM), la edad, la hipertensión arterial o la obesidad, la excesiva producción de aldosterona y/o activación del receptor MR en los distintos tipos de celulares vasculares, en los adipocitos o en células inflamatorias infiltradas, produce la activación de la enzima NADPH oxidasa y la generación de especies reactivas de oxígeno (ROS). Estos mediadores afectan a la función de dichos tipos celulares produciendo cambios fenotípicos y la proliferación y migración de células musculares lisas, la excesiva producción de matriz extracelular y de citoquinas inflamatorias. Estos eventos contribuyen al remodelado y rigidez vascular, la disfunción endotelial y al estado inflamatorio característicos de muchas de estas enfermedades.
Respecto al papel del estrés oxidativo en el remodelado y la rigidez vascular, se ha demostrado que la administración de una combinación de aldosterona y sal produjo un aumento en la producción de O2.- e hipertrofia de las CMLV a través de la sobreexpresión de Nox1 (11). Asimismo, diferentes antioxidantes atenuaron la senescencia inducida por aldosterona de CMLV y la expresión del factor proliferativo Ki-ras2A (48). Además, se ha sugerido que aldosterona aumenta los efectos hipertróficos vasculares de la insulina a través de rutas mediadas por MR y estrés oxidativo (49). Igualmente, la hipertrofia y la fibrosis perivasculares inducidas por aldosterona, se disminuyeron significativamente por el tratamiento a largo plazo con espironolactona o antioxidantes (50, 51). Sin embargo, los efectos profibróticos pero no las acciones hipertróficas de aldosterona en arterias de resistencia fueron bloqueados por el antioxidante tempol (23). Los mecanismos responsables de estos efectos posiblemente radican en la capacidad de los ROS producidas por aldosterona para modular diversos genes implicados en el daño vascular, tales como el factor de crecimiento de plaquetas, las metalotioninas 1 y 2 o el factor de crecimiento del tejido conectivo (52). Además, aldosterona incrementó la expresión de los factores profibróticos fibronectina y el inhibidor del activador del plasminógeno (PAI-1) en ratones de fenotipo salvaje pero no en ratones knockout para Nox1 (53,62) (Figura 2). Finalmente, el tratamiento con el antioxidante tempol inhibió la expresión de otros marcadores proinflamatorios y profibróticos tales como osteopontina, la molécula de adhesión intracelular 1, la molécula de adhesión celular vascular 1 o la expresión del gen de PAI-1 inducidos por la infusión de aldosterona en ratas (54).
Entre los mecanismos responsables de la disfunción endotelial, generalmente se asume que la interacción entre NO y O2.- , generalmente procedente de la NADPH oxidasa, da lugar a la formación de ONOO- o al desacoplamiento de eNOS, lo cual facilita una disminución en la disponibilidad de NO, entre otros mecanismos (revisado extensamente en 31-33,55,56). En este escenario, la aldosterona produjo una inhibición de la producción de NO en células endoteliales que fue parcialmente restaurada por la deleción de la subunidad p47phox utilizando un siRNA (16). Otros potenciales mecanismos responsables de la disfunción endotelial inducida por la ruta aldosterona/MR/ROS incluyen: 1) modificaciones postraduccionales oxidativas de la actividad de la guanilato ciclasa, que empeora la capacidad de responder de esta enzima ante el NO (12); 2) disminución en la expresión de la enzima antioxidante Glucosa-6-fosfato deshidrogenasa (57) o 3) modificaciones oxidativas de los grupos tiol de las cisteínas funcionales (Cys405) en el receptor B para endotelina (ETBR), por mecanismos que dependen de la producción e H2O2 de Nox4, que empeoran la activación dependiente de dicho receptor y la consiguiente activación de la eNOS lo que disminuye la producción de NO (20). En este sentido, la activación del MR durante la isquemia renal induce activación de Rac1, que incrementa la producción de ROS en las CMLV, los cuales difunden a las células endoteliales para inducir modificaciones postraduccionales en el ETBR que empeora la activación de la eNOS y disminuye la producción de NO, danto lugar a una sostenida vasoconstricción y reducida perfusión del riñón (58). Además, la sobreexpresión específica en cardiomiocitos del receptor humano MR induce severa disfunción endotelial coronaria con disminuida respuesta relajadora mediada por NO en respuesta a acetilcolina en arterias coronarias, efectos que fueron prevenidos por un mes de tratamiento con un antagonista del MR, con vitamina E/vitamina C o con un inhibidor de la NADPH oxidasa (59). Finalmente, también se ha descrito un papel del canal de sodio epitelial en el efecto de aldosterona incrementando el estrés oxidativo, la rigidez vascular, la disfunción endotelial y la fibrosis (29). Curiosamente, Rac1 no es solo uno de los componentes de la NADPH oxidasa, sino que sirve para aumentar la señalización del MR en el riñón (60). Esta ruta Rac1-MR se activa por ROS en cardiomiocitos (61) y también juega un papel crucial en la producción de ROS y la disfunción cardiaca (62).
3. PAPEL DEL MR COMO PRODUCTOR DE ESTRÉS OXIDATIVO EN CÉLULAS INMUNES
Evidencias cada vez mas abundantes sugieren que aldosterona induce infiltración vascular de monocitos/macrófagos y células T en diferentes situaciones patológicas (24,63,64). El MR se expresa en macrófagos y linfocitos T donde funciona como un importante regulador transcripcional del fenotipo celular y de la función y que puede ser activado incluso con niveles normales o bajos de aldosterona en situaciones patológicas (5,65) (Figuras 1 y 2).
La relación entre las células inmunes, el MR y el estrés oxidativo fue demostrada en ratas uninefrectomizadas y tratadas durante 4 semanas con un 1% de NaCl en la dieta junto con aldosterona. En estas ratas se encontró una incrementada producción de H2O2 por monocitos y linfocitos, una sobreexpresión de la tirosina fosfatasa inducida por estrés oxidativo y de la Mn-SOD en células periféricas mononucleares y la presencia del marcador de estrés oxidativo 3-nitrotirosina en células inflamatorias CD4+ invadiendo arterias coronarias intramurales (66). Asimismo, Guzik y cols (67) mostraron que la hipertensión inducida por DOCA/sal y la producción de O2.- asociada en la aorta se abolieron en ratones deficientes en rag-/- que no poseen linfocitos T y B. Además, una aumentada cantidad de linfocitos T reguladores, que son supresores de las respuestas inmunes innatas y adaptativas, previnieron la disfunción endotelial, el remodelado vascular y el estrés oxidativo inducidos por aldosterona (25). Más recientemente, el papel clave del MR en células inmunes en la generación de estrés oxidativo fue demostrada por Sun y cols (68), que mostraron que arterias de ratones knockout para MR específicamente en células T, mostraban menor producción de O2.- , lo cual además se correlacionó con un menor daño de órgano diana incluyendo mejor función endotelial y menos hipertrofia vascular y fibrosis después de la infusión con angiotensina II.
Esto podría ser debido a una menor producción de células T productoras de IFN-γ en las arterias (68). De hecho, el receptor MR de las células T facilita la activación de dichas células modulando la producción de citoquinas proinflamatorias tales como IFN-γ e IL-6 (69), las cuales es bien conocido que pueden producir producción de ROS a nivel vascular.
Respecto a los macrófagos, se ha demostrado que la estimulación de macrófagos con aldosterona induce un fenotipo proinflamatorio M1 (5). Macrófagos procedentes de ratones que no poseen MR en células mieloides exhiben un perfil transcripcional de activación alternativa de un fenotipo M1 hacia un fenotipo M2 más antiinflamatorio (70). Esto podría modular la función vascular, ya que se ha demostrado que el inflamasoma NLRP3 en macrófagos media la hipercontractilidad inducida por aldosterona, la disfunción endotelial y el remodelado hipertrófico (71), aunque en este estudio la contribución específica de las ROS derivadas de los macrófagos no fue específicamente evaluada. Curiosamente, un desplazamiento en la polarización a un fenotipo M2 en los macrófagos se observó en ratones con deleción específica del MR en células endoteliales cuando fueron expuestos a una dieta de tipo western (28), lo que sugiere un papel del MR endotelial en la función de los macrófagos. Al respecto del estrés oxidativo, aldosterona incrementa la producción de O2.- y la activación de la NADPH oxidasa en macrófagos tanto in vivo como in vitro (72) y también la generación de ROS mitocondriales (71), que podrían contribuir a la activación del inflamasoma en este tipo celular como se ha sugerido previamente (73). Lo que es más importante, la disfunción endotelial y el estrés oxidativo vascular inducidos por aldosterona estaban disminuidos en ratones mcsfOp/+, los cuales poseen un bajo número de monocitos/macrófagos en la pared vascular (24), lo que sugiere que la activación del MR en macrófagos modula el estrés oxidativo vascular. Curiosamente, el estrés oxidativo medido en este caso como expresión génica de las subunidades Nox2 y p22phox se incrementó de manera similar en el corazón de los ratones control y de ratones con deleción específica de MR en macrófagos cuando se expusieron a una combinación del inhibidor de NOS L-NAME y sal, como modelo de hipertensión (74). Asimismo, la deficiencia de MR en macrófagos no influyó en el estado oxidativo en el contexto de la aterosclerosis (75). En conjunto, estos resultados sugieren una contribución diferente de las ROS derivadas de MR en distintas situaciones patológicas. Otros estudios in vivo demostraron que la deficiencia de MR en macrófagos imitó los efectos de los antagonistas del MR y protegió contra el daño vascular causado por L-NAME/angiotensina II (70) y que la eliminación selectiva del MR en células mieloides limitó la acumulación de macrófagos, lo cual dio lugar a menor activación de las CML e inflamación vascular inhibiendo la hiperplasia de la neoíntima y el remodelado vascular (76). Desgraciadamente, en ambos estudios, de nuevo la contribución específica de los ROS derivados de macrófagos no fue evaluada.
En conjunto, todos estos resultados apuntan a una contribución clave del MR presente en células inmunes en la producción de estrés oxidativo en el contexto de daño vascular (Figura 2). Si dicha contribución es debida a ROS producidos localmente por células inmunes infiltradas o a través de la liberación de citoquinas proinflamatorias que afectarían a las CML o células endoteliales adyacentes para inducir estrés oxidativo, o ambos mecanismos, es de momento desconocido.
4. PAPEL DEL MR COMO PRODUCTOR DE ESTRÉS OXIDATIVO EN TEJIDO ADIPOSO
Otro tejido extrarrenal que expresa el MR es el tejido adiposo donde el MR está implicado en procesos esenciales tales como la diferenciación, la autofagia o la secreción de adipoquinas (4,77). La expresión del MR se incrementa en el tejido adiposo de modelos murinos de obesidad y en sujetos humanos obesos y, diferentes estudios utilizando antagonistas del MR o modelos transgénicos con deleción específica del MR en adipocitos, han demostrado un papel clave del MR en la señalización de la insulina y la inflamación (revisado en 4,77,78).
Está bien aceptado que el tejido adiposo, particularmente el tejido adiposo perivascular, modula la salud vascular y la enfermedad a través de la liberación de diversas adipoquinas, que afectan a las propiedades contráctiles y relajadoras del vaso, a la proliferación e hipertrofia de las CML, a la fibrosis y la inflamación (79). Entre las muchas sustancias liberadas, ROS como H2O2 parecen tener un papel principal tanto en condiciones fisiológicas como patológicas. Sin embargo, en algunas situaciones patológicas, tales como obesidad o hipertensión, citoquinas proinflamatorias como IL-1, IL-6 o TNF-α, liberadas de este tejido adiposo perivascular, claramente afectan al tono vascular (79) debido, al menos en parte, a un incremento en la generación de ROS. Así, en arterias mesentéricas de animales sanos, las propiedades anticontráctiles del tejido adiposo perivascular se pierden después de la incubación con aldosterona, propiedades que se restauran con una combinación de los antioxidantes SOD y catalasa y con el antagonista del MR eplerenona, siendo este efecto dependiente de la infiltración de macrófagos en el tejido adiposo perivascular (80). Además, en la línea celular 3T3-L1 de adipocitos, el bloqueo del MR reduce la producción de ROS (81,82).
Estudios iniciales demostraron que las subunidades de la NADPH oxidasa p22 y p47phox estaban significativamente aumentadas en tejido adiposo de ratones obesos ob/ob y db/db comparados con sus controles delgados y que el tratamiento con eplerenona suprimió este incremento (81). El aumento en los niveles de ROS observados en tejido adiposo de estos modelos de obesidad podría ser también debido a una disminución en la expresión génica de las enzimas eliminadoras de ROS catalasa y Cu, Zn-SOD que se encontraban reducidas tanto en los ratones ob/ob como en los db/db y que también fueron restauradas por la administración de eplerenona (81). Otros estudios también demostraron sobreexpresión de enzimas antioxidantes (SOD-1 y catalasa) a nivel vascular tras el bloqueo del MR en obesidad/diabetes (41). Finalmente, en el tejido adiposo de ratas nefrectomizadas, el estrés oxidativo estaba aumentado y esto se revirtió tras el tratamiento con otro antagonista del MR, espironolactona (83).
La sobreexpresión condicional del MR en adipocitos murinos dio lugar a una incrementada producción de H2O2 del tejido adiposo epididimal probablemente debido a una disminución en la expresión del gen de la catalasa y un aumento en la expresión de los niveles de Nox4 sin cambios en las expresiones de Nox1 o Nox2, hallazgos que probablemente explican los cambios en la contractilidad vascular encontrados en este modelo (84). Curiosamente, ratones knockout para MR específicos de adipocitos cuando se alimentaron con una dieta alta en grasa y en sacarosa, mostraron niveles similares del marcador de estrés oxidativo 8-isoprostano, o de los genes de p22phox, SOD-1 o catalasa, y además, no mostraron diferencias en el peso corporal, peso de la grasa, tolerancia a la glucosa, sensibilidad a la insulina o inflamación (85). Resultados similares han sido publicados recientemente por Feraco y cols (86) utilizando un modelo de deleción inducible de MR específica de adipocitos cuanto se les alimentó con una dieta alta en grasa (45%), aunque es importante destacar que en este estudio el estrés oxidativo no fue evaluado. En este contexto, son necesarios estudios futuros para identificar la contribución específica del MR en los diferentes tipos celulares incluidos en el tejido adiposo además de los adipocitos, como preadipocitos o macrófagos y su función como posibles responsables de la inflamación, estrés oxidativo y alteraciones metabólicas dependientes del MR asociadas a la obesidad (Figura 2).
5. RELEVANCIA CLÍNICA DE LA RUTA ALDOSTERINA/MR/ESTRÉS OXIDATIVO.
Como se ha discutido en las secciones anteriores, existe una importante cantidad de evidencia preclínica que demuestra que la ruta aldosterona/MR señaliza a través de estrés oxidativo. Sin embargo, las evidencias en humanos son menos abundantes. Algunos estudios clínicos han sugerido que el hiperaldosteronismo está asociado con una elevada concentración de marcadores circulantes de estrés oxidativo. Así, en pacientes con insuficiencia cardiaca estable y en pacientes con hipertensión, niveles elevados de aldosterona se asociaron con elevados niveles de estrés oxidativo sistémico, inflamación y recambio de matriz extracelular (87). En pacientes con insuficiencia cardiaca, el daño cardiovascular asociado a la aldosterona y la fibrosis renal estaban asociadas a una producción de NO disminuida, incrementado estrés oxidativo y activación de factores de transcripción proinflamatorios como NF-kB (9). A nivel celular, existen también algunas evidencias que demuestran que aldosterona estimula la producción de ROS en humanos. Así, en células endoteliales humanas, espironolactona inhibió el estrés oxidativo dependiente de Nox e incrementó la actividad de la eNOS (88), indicando un papel del MR en la regulación de la producción de ROS . En pacientes con hiperaldosteronismo y adenomas adrenales, diversos estudios han mostrado incrementada expresión de genes y proteínas relacionados con la señalización redox como el factor de transcripción antioxidante Nrf2, p22phox, la enzima antioxidante hemooxigenasa-1 o factores de transcripción proinflamatorios (89-91). Además, en cardiomiocitos humanos, aldosterona empeora la función mitocondrial que es importante en la regulación redox (91).
A pesar de las evidencias que sugieren que el hiperaldosteronismo promueve estrés oxidativo en la enfermedad cardiovascular humana, los estudios existentes utilizando antagonistas del MR, no han mostrado una mejoría significativa en marcadores de estrés oxidativo. Así, Hwang y cols (92) demostraron que la mejoría en la dilatación mediada por flujo (una medida de función endotelial) inducida por eplerenona, no se asociaba con marcadores de estrés oxidativo como los niveles plasmáticos de F2-isoprostanos, o con la expresión proteica de nitrotirosina o p47phox en células vasculares endoteliales. En un grupo pequeño de adultos ancianos con síndrome metabólico, la dilatación dependiente de endotelio mediada por flujo, los niveles de lipoproteínas de baja densidad oxidadas o los F2-isoprostanos, no mejoraron en respuesta al bloqueo del MR, a pesar de observarse una reducción en la presión arterial sistólica de unos 10 mm Hg (93). Además, no hubo efecto del tratamiento durante un mes con eplerenona en el estrés oxidativo (LDL oxidadas) y en la rigidez vascular en adultos ancianos sanos (94). Sin embargo, Chen y cols (95) demostraron recientemente que el aumentado estrés oxidativo dependiente de NAPDH oxidasa, la degradación oxidativa del cofactor de la eNOS tetrahidrobiopterina, el desacoplamiento de la eNOS y la reducida generación de NO, eran los responsables de la empeorada capacidad de reparación endotelial in vivo de células progenitoras endoteliales tempranas de pacientes hipertensos con hiperaldosteronimo primario (95). Las razones de estas discrepancias entre estudios preclínicos y clínicos no son claras, pero podrían incluir la existencia de poblaciones heterogéneas con un prolongado periodo de enfermedad, diferencias en las medidas de los marcadores de estrés oxidativo o la evaluación de grupos pequeños de pacientes. Son necesarios, por tanto, estudios clínicos bien controlados para confirmar el papel de las ROS en el daño cardiovascular mediado por aldosterona y, en este contexto, son imprescindibles ensayos clínicos que determinen los efectos de los antagonistas del MR en los niveles de ROS específicas, en lugar de marcadores de estrés oxidativo como LDL oxidadas o F2-2 isoprostanos. En cualquier caso, no debemos olvidar que los antagonistas del receptor de la aldosterona son fármacos de probada eficacia, que en la actualidad se usan en pacientes seleccionados con hipertensión arterial resistente (96). Además, estos compuestos proporcionan un aumento en la supervivencia en diversas circunstancias como la insuficiencia cardiaca y posiblemente, proporcionan protección renal en pacientes con enfermedad renal
crónica, así como efectos beneficiosos adicionales en otras patologías (97). En base a lo presentado en este artículo, es posible hipotetizar que parte de los efectos beneficiosos de estos compuestos, podrían ser debidos a sus efectos sobre la señalización redox y la mejora del daño cardiovascular.
6. CONCLUSIONES
Las evidencias experimentales muestran claramente que el bloqueo de la ruta aldosterona/MR disminuye el estrés oxidativo vascular y mejora la función, estructura y propiedades mecánicas vasculares alteradas en distintos modelos experimentales. Entre los tipos celulares vasculares implicados en el daño vascular asociado a aldosterona/MR/estrés oxidativo están las células endoteliales y las células musculares lisas. Sin embargo, evidencias cada vez más abundantes sugieren que estos efectos vasculares pueden ser modulados también por el MR expresado en células inmunes infiltradas, como linfocitos y macrófagos, y en el tejido adiposo perivascular que rodea al vaso, células que podrían liberar ROS que impacten en el endotelio y la pared vascular. Alternativamente, estas células pueden generar citoquinas inflamatorias (o adipoquinas) dependientes de MR que actúan de manera paracrina en los vasos adyacentes para inducir estrés oxidativo pudiendo dañar el vaso. Así, el estrés oxidativo asociado a la activación del MR en distintos tipos celulares emerge como una importante ruta que contribuye a la disfunción vascular y al daño asociado a condiciones de elevados niveles de aldosterona o de activación del MR. Por ello, es posible que algunos de los efectos vasoprotectores de los antagonistas del MR, utilizados en la clínica, pueden ser debidos a la inhibición del daño vascular producido por el estrés oxidativo.
7. LISTA DE ABREVIATURAS
AT1: receptor 1 para angiotensina II
CMLV: células musculares lisas vasculares
DOCA: acetato de deoxicorticosterona
ETBR: receptor B para endotelina
GPCR: receptores acoplados a proteínas G
GPER1: receptor 1 de estrógenos acoplado a proteínas G
GRK2: quinasa 2 de receptores acoplados a proteínas G
IGF1R: receptor para el factor de crecimiento similar a insulina 1
MR: receptor mineralocorticoide
Nox: NADPH oxidasa
PDGFR: receptor para el factor decrecimiento derivado de plaquetas
ROS: especies reactivas de oxígeno, del inglés reactive oxygen species
SOD: superoxido dismutasa
T2DM: diabetes mellitus tipo 2
8. AGRADECIMIENTOS
AMB está financiada por el Ministerio de Economía, Industria y Competitividad (SAF2016-80305P), Instituto de Salud Carlos III (PI13/01488; CIBER de Enfermedades Cardiovasculares, CB16/11/00286) y Comunidad de Madrid (B2017/BMD-3676), Fondo Social Europeo (FSE), Fondo Europeo de Desarrollo Regional (FEDER) a way to build Europe. RR-D está financiada por un contrato Juan de la Cierva Reincorporación ((IJCI-2017-31399).
9. REFERENCIAS
- Jaisser F, Farman N. Emerging Roles of the Mineralocorticoid Receptor in Pathology: Toward New Paradigms in Clinical Pharmacology. Pharmacol Rev. 2016; 68(1):49-75.
- DuPont JJ, Jaffe IZ. 30 years of the mineralocorticoid receptor: The role of the mineralocorticoid receptor in the vasculature. J Endocrinol. 2017; 234(1):T67-T82.
- Briet M, Schiffrin EL. Vascular actions of aldosterone. J Vasc Res. 2013; 50(2):89-99.
- Armani A, Marzolla V, Fabbri A, Caprio M. Cellular mechanisms of MR regulation of adipose tissue physiology and pathophysiology. J Mol Endocrinol. 2015; 55(2):R1-10.
- van der Heijden CDCC, Deinum J, Joosten LAB, Netea MG, Riksen NP. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc Res. 2018; 114(7):944-953.
- Ruhs S, Nolze A, Hübschmann R, Grossmann C. 30 years of the mineralocorticoid receptor: Nongenomic effects via the mineralocorticoid receptor. J Endocrinol. 2017; 234(1):T107-T124.
- DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, Christou DD, Hill MA, Jaffe IZ. Vascular mineralocorticoid receptor regulates microRNA-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight. 2016; 1(14):e88942.
- Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A; American Heart Association Council on Basic Cardiovascular Sciences. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System: A Scientific Statement From the American Heart Association. Circ Res. 2016; 119(5):e39-75.
- Queisser N, Schupp N. Aldosterone, oxidative stress, and NF-kB activation in hypertension-related cardiovascular and renal diseases. Free Radic Biol Med. 2012; 53(2):314-27.
- Callera GE, Montezano AC, Yogi A, Tostes RC, He Y, Schiffrin EL, Touyz RM. c-Src-dependent nongenomic signaling responses to aldosterone are increased in vascular myocytes from spontaneously hypertensive rats. Hypertension. 2005; 46(4):1032-8.
- Fan C, Kawai Y, Inaba S, Arakawa K, Katsuyama M, Kajinami K, Yasuda T, Yabe-Nishimura C, Konoshita T, Miyamori I. Synergy of aldosterone and high salt induces vascular smooth muscle hypertrophy through up-regulation of NOX1. J Steroid Biochem Mol Biol. 2008; 111(1-2):29-36.
- Maron BA, Zhang YY, Handy DE, Beuve A, Tang SS, Loscalzo J, Leopold JA. Aldosterone increases oxidant stress to impair guanylyl cyclase activity by cysteinyl thiol oxidation in vascular smooth muscle cells. J Biol Chem. 2009; 284(12):7665-72.
- Fu Y, Shi G, Wu Y, Kawai Y, Tian Q, Yue L, Xia Q, Miyamori I, Fan C. The mitochondria mediate the induction of NOX1 gene expression by aldosterone in an ATF-1-dependent manner. Cell Mol Biol Lett. 2011; 16(2):226-35.
- Muehlfelder M, Arias-Loza PA, Fritzemeier KH, Pelzer T. Both estrogen receptor subtypes, ERα and ERβ, prevent aldosterone-induced oxidative stress in VSMC via increased NADPH bioavailability. Biochem Biophys Res Commun. 2012; 423(4):850-6.
- Zhang X, Liu J, Pang X, Zhao J, Wang S, Wu D. Aldosterone induces C-reactive protein expression via MR-ROS-MAPK-NF-κB signal pathway in rat vascular smooth muscle cells. Mol Cell Endocrinol. 2014; 395(1-2):61-8.
- Nagata D, Takahashi M, Sawai K, Tagami T, Usui T, Shimatsu A, Hirata Y, Naruse M. Molecular mechanism of the inhibitory effect of aldosterone on endothelial NO synthase activity. Hypertension. 2006; 48(1):165-71.
- Hashikabe Y, Suzuki K, Jojima T, Uchida K, Hattori Y. Aldosterone impairs vascular endothelial cell function. J Cardiovasc Pharmacol. 2006; 47(4):609-13.
- Iwashima F, Yoshimoto T, Minami I, Sakurada M, Hirono Y, Hirata Y. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology. 2008; 149(3):1009-14.
- Wilkinson-Berka JL, Tan G, Jaworski K, Miller AG. Identification of a retinal aldosterone system and the protective effects of mineralocorticoid receptor antagonism on retinal vascular pathology. Circ Res. 2009; 104(1):124-33.
- Maron BA, Zhang YY, White K, Chan SY, Handy DE, Mahoney CE, Loscalzo J, Leopold JA. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation. 2012; 126(8):963-74.
- Schäfer N, Lohmann C, Winnik S, van Tits LJ, Miranda MX, Vergopoulos A, Ruschitzka F, Nussberger J, Berger S, Lüscher TF, Verrey F, Matter CM. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity. Eur Heart J. 2013; 34(45):3515-24.
- Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension. 2002; 40(4):504-10.
- Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am J Hypertens. 2004; 17(7):597-603.
- Leibovitz E, Ebrahimian T, Paradis P, Schiffrin EL. Aldosterone induces arterial stiffness in absence of oxidative stress and endothelial dysfunction. J Hypertens. 2009; 27(11):2192-200.
- Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012; 59(2):324-30.
- Dinh QN, Young MJ, Evans MA, Drummond GR, Sobey CG, Chrissobolis S. Aldosterone-induced oxidative stress and inflammation in the brain are mediated by the endothelial cell mineralocorticoid receptor. Brain Res. 2016; 1637:146-153.
- Briet M, Barhoumi T, Mian MOR, Coelho SC, Ouerd S, Rautureau Y, Coffman TM, Paradis P, Schiffrin EL. Aldosterone-Induced Vascular Remodeling and Endothelial Dysfunction Require Functional Angiotensin Type 1a Receptors. Hypertension. 2016; 67(5):897-905.
- Jia G, Habibi J, Aroor AR, Martinez-Lemus LA, DeMarco VG, Ramirez-Pérez FI, Sun Z, Hayden MR, Meininger GA, Mueller KB, Jaffe IZ, Sowers JR. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ Res. 2016; 118(6):935-943.
- Jia G, Habibi J, Aroor AR, Hill MA, Yang Y, Whaley-Connell A, Jaisser F, Sowers JR. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension. 2018; 72(3):731-738.
- Beswick RA, Dorrance AM, Leite R, Webb RC. NADH/NADPH oxidase and enhanced superoxide production in the mineralocorticoid hypertensive rat. Hypertension. 2001; 38(5):1107-11.
- Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012; 110(10):1364-90.
- Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal. 2013; 19(10):1110-20.
- Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011; 34(1):5-14.
- Park YM, Lim BH, Touyz RM, Park JB. Expression of NAD(P)H oxidase subunits and their contribution to cardiovascular damage in aldosterone/salt-induced hypertensive rat. J Korean Med Sci. 2008; 23(6):1039-45.
- Montezano AC, Callera GE, Yogi A, He Y, Tostes RC, He G, Schiffrin EL, Touyz RM. Aldosterone and angiotensin II synergistically stimulate migration in vascular smooth muscle cells through c-Src-regulated redox-sensitive RhoA pathways. Arterioscler Thromb Vasc Biol. 2008; 28(8):1511-8.
- Cannavo A, Liccardo D, Eguchi A, Elliott KJ, Traynham CJ, Ibetti J, Eguchi S, Leosco D, Ferrara N, Rengo G, Koch WJ. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat Commun. 2017; 7:10877.
- De Giusti VC, Orlowski A, Ciancio MC, Espejo MS, Gonano LA, Caldiz CI, Vila Petroff MG, Villa-Abrille MC, Aiello EA. Aldosterone stimulates the cardiac sodium/bicarbonate cotransporter via activation of the g protein-coupled receptor gpr30. J Mol Cell Cardiol. 2015; 89(Pt B):260-7.
- Ashton AW, Le TY, Gómez-Sánchez CE, Morel-Kopp MC, McWhinney B, Hudson A, Mihailidou AS. Role of Nongenomic Signaling Pathways Activated by Aldosterone During Cardiac Reperfusion Injury. Mol Endocrinol. 2015; 29(8):1144-55.
- Sanz-Rosa D1, Oubiña MP, Cediel E, De las Heras N, Aragoncillo P, Balfagón G, Cachofeiro V, Lahera V. Eplerenone reduces oxidative stress and enhances eNOS in SHR: vascular functional and structural consequences. Antioxid Redox Signal. 2005; 7(9-10):1294-301.
- Mulder P, Mellin V, Favre J, Vercauteren M, Remy-Jouet I, Monteil C, Richard V, Renet S, Henry JP, Jeng AY, Webb RL, Thuillez C. Aldosterone synthase inhibition improves cardiovascular function and structure in rats with heart failure: a comparison with spironolactone. Eur Heart J. 2008; 29(17):2171-9.
- Silva MA, Bruder-Nascimento T, Cau SB, Lopes RA, Mestriner FL, Fais RS, Touyz RM, Tostes RC. Spironolactone treatment attenuates vascular dysfunction in type 2 diabetic mice by decreasing oxidative stress and restoring NO/GC signaling. Front Physiol. 2015; 6:269.
- Silva MA, Cau SB, Lopes RA, Manzato CP, Neves KB, Bruder-Nascimento T, Mestriner FL, Montezano AC, Nguyen Dinh Cat A, Touyz RM, Tostes RC. Mineralocorticoid receptor blockade prevents vascular remodelling in a rodent model of type 2 diabetes mellitus. Clin Sci (Lond). 2015; 129(7):533-45.
- Nguyen Dinh Cat A, Griol-Charhbili V, Loufrani L, Labat C, Benjamin L, Farman N, Lacolley P, Henrion D, Jaisser F. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. FASEB J. 2010; 24(7):2454-63.
- Davel AP, Anwar IJ, Jaffe IZ. The endothelial mineralocorticoid receptor: mediator of the switch from vascular health to disease. Curr Opin Nephrol Hypertens. 2017; 26(2):97-104.
- Davel AP, Lu Q, Moss ME, Rao S, Anwar IJ, DuPont JJ, Jaffe IZ. Sex-Specific Mechanisms of Resistance Vessel Endothelial Dysfunction Induced by Cardiometabolic Risk Factors. J Am Heart Assoc. 2018; 7(4). pii: e007675.
- McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, Chambon P, Hill MA, Dorrance AM, Mendelsohn ME, Jaffe IZ. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012; 18(9):1429-33.
- Gueret A, Harouki N, Favre J, Galmiche G, Nicol L, Henry JP, Besnier M, Thuillez C, Richard V, Kolkhof P, Mulder P, Jaisser F, Ouvrard-Pascaud A. Vascular Smooth Muscle Mineralocorticoid Receptor Contributes to Coronary and Left Ventricular Dysfunction After Myocardial Infarction. Hypertension. 2016; 67(4):717-23.
- Min LJ, Mogi M, Iwanami J, Li JM, Sakata A, Fujita T, Tsukuda K, Iwai M, Horiuchi M. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc Res. 2007; 76(3):506-16.
- Sherajee SJ, Fujita Y, Rafiq K, Nakano D, Mori H, Masaki T, Hara T, Kohno M, Nishiyama A, Hitomi H. Aldosterone induces vascular insulin resistance by increasing insulin-like growth factor-1 receptor and hybrid receptor. Arterioscler Thromb Vasc Biol. 2012; 32(2):257-63.
- Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart : role of oxidative stress. Am J Pathol. 2002 161(5):1773-81.
- Nakano S, Kobayashi N, Yoshida K, Ohno T, Matsuoka H. Cardioprotective mechanisms of spironolactone associated with the angiotensin-converting enzyme/epidermal growth factor receptor/extracellular signal-regulated kinases, NAD(P)H oxidase/lectin-like oxidized low-density lipoprotein receptor-1, and Rho-kinase pathways in aldosterone/salt-induced hypertensive rats. Hypertens Res. 2005; 28(11):925-36.
- Newfell BG, Iyer LK, Mohammad NN, McGraw AP, Ehsan A, Rosano G, Huang PL, Mendelsohn ME, Jaffe IZ. Aldosterone regulates vascular gene transcription via oxidative stress-dependent and -independent pathways. Arterioscler Thromb Vasc Biol. 2011; 31(8):1871-80.
- Harvey AP, Montezano AC, Hood KY, Lopes RA, Rios F, Ceravolo G, Graham D, Touyz RM. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: The aldosterone-mineralocorticoid receptor-Nox1 axis. Life Sci. 2017; 179:110-119.
- Hirono Y, Yoshimoto T, Suzuki N, Sugiyama T, Sakurada M, Takai S, Kobayashi N, Shichiri M, Hirata Y. Angiotensin II receptor type 1-mediated vascular oxidative stress and proinflammatory gene expression in aldosterone-induced hypertension: the possible role of local renin-angiotensin system. Endocrinology. 2007; 148(4):1688-96.
- Vanhoutte PM, Zhao Y, Xu A, Leung SW. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res. 2016; 119(2):375-96.
- Förstermann U, Xia N, Li H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ Res. 2017; 120(4):713-735.
- Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. 2007; 13(2):189-97.
- Barrera-Chimal J, Prince S, Fadel F, El Moghrabi S, Warnock DG, Kolkhof P, Jaisser F. Sulfenic Acid Modification of Endothelin B Receptor is Responsible for the Benefit of a Nonsteroidal Mineralocorticoid Receptor Antagonist in Renal Ischemia. J Am Soc Nephrol. 2016; 27(2):398-404.
- Favre J, Gao J, Zhang AD, Remy-Jouet I, Ouvrard-Pascaud A, Dautreaux B, Escoubet B, Thuillez C, Jaisser F, Richard V. Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. Am J Physiol Heart Circ Physiol. 2011; 300(6):H2035-43.
- Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008; 14(12):1370-6.
- Nagase M, Ayuzawa N, Kawarazaki W, Ishizawa K, Ueda K, Yoshida S, Fujita T. Oxidative stress causes mineralocorticoid receptor activation in rat cardiomyocytes: role of small GTPase Rac1. Hypertension. 2012; 59(2):500-6.
- Ayuzawa N, Nagase M, Ueda K, Nishimoto M, Kawarazaki W, Marumo T, Aiba A, Sakurai T, Shindo T, Fujita T. Rac1-Mediated Activation of Mineralocorticoid Receptor in Pressure Overload-Induced Cardiac Injury. Hypertension. 2016; 67(1):99-106.
- McGraw AP, Bagley J, Chen WS, Galayda C, Nickerson H, Armani A, Caprio M, Carmeliet P, Jaffe IZ. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J Am Heart Assoc. 2013; 2(1):e000018.
- Marzolla V, Armani A, Mammi C, Moss ME, Pagliarini V, Pontecorvo L, Antelmi A, Fabbri A, Rosano G, Jaffe IZ, Caprio M. Essential role of ICAM-1 in aldosterone-induced atherosclerosis. Int J Cardiol. 2017; 232:233-242.
- Belden Z, Deiuliis JA, Dobre M, Rajagopalan S. The Role of the Mineralocorticoid Receptor in Inflammation: Focus on Kidney and Vasculature. Am J Nephrol. 2017; 46(4):298-314.
- Ahokas RA, Warrington KJ, Gerling IC, Sun Y, Wodi LA, Herring PA, Lu L, Bhattacharya SK, Postlethwaite AE, Weber KT. Aldosteronism and peripheral blood mononuclear cell activation: a neuroendocrine-immune interface. Circ Res. 2003; 93(10):e124-35.
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007; 204(10):2449-60.
- Sun XN, Li C, Liu Y, Du LJ, Zeng MR, Zheng XJ, Zhang WC, Liu Y, Zhu M, Kong D, Zhou L, Lu L, Shen ZX, Yi Y, Du L, Qin M, Liu X, Hua Z, Sun S, Yin H, Zhou B, Yu Y, Zhang Z, Duan SZ. T-Cell Mineralocorticoid Receptor Controls Blood Pressure by Regulating Interferon-Gamma. Circ Res. 2017; 120(10):1584-1597.
- Li C, Sun XN, Zeng MR, Zheng XJ, Zhang YY, Wan Q, Zhang WC, Shi C, Du LJ, Ai TJ, Liu Y, Liu Y, Du LL, Yi Y, Yu Y, Duan SZ. Mineralocorticoid Receptor Deficiency in T Cells Attenuates Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction Through Modulating T-Cell Activation. Hypertension. 2017; 70(1):137-147
- Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010; 120(9):3350-64.
- Bruder-Nascimento T, Ferreira NS, Zanotto CZ, Ramalho F, Pequeno IO, Olivon VC, Neves KB, Alves-Lopes R, Campos E, Silva CA, Fazan R, Carlos D, Mestriner FL, Prado D, Pereira FV, Braga T, Luiz JP, Cau SB, Elias PC, Moreira AC, Câmara NO, Zamboni DS, Alves-Filho JC, Tostes RC. NLRP3 Inflammasome Mediates Aldosterone-Induced Vascular Damage. Circulation. 2016; 134(23):1866-1880.
- Keidar S, Kaplan M, Pavlotzky E, Coleman R, Hayek T, Hamoud S, Aviram M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. 2004; 109(18):2213-20.
- Kadoya H, Satoh M, Sasaki T, Taniguchi S, Takahashi M, Kashihara N. Excess aldosterone is a critical danger signal for inflammasome activation in the development of renal fibrosis in mice. FASEB J. 2015; 29(9):3899-910.
- Bienvenu LA, Morgan J, Rickard AJ, Tesch GH, Cranston GA, Fletcher EK, Delbridge LM, Young MJ. Macrophage mineralocorticoid receptor signaling plays a key role in aldosterone-independent cardiac fibrosis. Endocrinology. 2012; 153(7):3416-25.
- Shen ZX, Chen XQ, Sun XN, Sun JY, Zhang WC, Zheng XJ, Zhang YY, Shi HJ, Zhang JW, Li C, Wang J, Liu X, Duan SZ. Mineralocorticoid Receptor Deficiency in Macrophages Inhibits Atherosclerosis by Affecting Foam Cell Formation and Efferocytosis. J Biol Chem. 2017; 292(3):925-935.
- Sun JY, Li C, Shen ZX, Zhang WC, Ai TJ, Du LJ, Zhang YY, Yao GF, Liu Y, Sun S, Naray-Fejes-Toth A, Fejes-Toth G, Peng Y, Chen M, Liu X, Tao J, Zhou B, Yu Y, Guo F, Du J, Duan SZ. Mineralocorticoid Receptor Deficiency in Macrophages Inhibits Neointimal Hyperplasia and Suppresses Macrophage Inflammation Through SGK1-AP1/NF-κB Pathways. Arterioscler Thromb Vasc Biol. 2016; 36(5):874-85.
- Jia G, Aroor AR, Sowers JR (2017) The role of mineralocorticoid receptor signaling in the cross-talk between adipose tissue and the vascular wall. Cardiovasc Res 113(9):1055-1063.
- Garg R & Adler GK. Aldosterone and the Mineralocorticoid Receptor: Risk Factors for Cardiometabolic Disorders. Curr Hypertens Rep. 2015; 17(7):52.
- Fernández-Alfonso MS, Somoza B, Tsvetkov D, Kuczmanski A, Dashwood M, Gil-Ortega M. Role of Perivascular Adipose Tissue in Health and Disease. Compr Physiol. 2017; 8(1):23-59.
- Withers SB, Agabiti-Rosei C, Livingstone DM, Little MC, Aslam R, Malik RA, Heagerty AM. Macrophage activation is responsible for loss of anticontractile function in inflamed perivascular fat. Arterioscler Thromb Vasc Biol. 2011; 31(4):908-13.
- Hirata A, Maeda N, Hiuge A, Hibuse T, Fujita K, Okada T, Kihara S, Funahashi T, Shimomura I. Blockade of mineralocorticoid receptor reverses adipocyte dysfunction and insulin resistance in obese mice. Cardiovasc Res. 2009; 84(1):164-72.
- Hirata A, Maeda N, Nakatsuji H, Hiuge-Shimizu A, Okada T, Funahashi T, Shimomura I. Contribution of glucocorticoid-mineralocorticoid receptor pathway on the obesity-related adipocyte dysfunction. Biochem Biophys Res Commun. 2012; 419(2):182-7.
- Hosoya K, Minakuchi H, Wakino S, Fujimura K, Hasegawa K, Komatsu M, Yoshifuji A, Futatsugi K, Shinozuka K, Washida N, Kanda T, Tokuyama H, Hayashi K, Itoh H. Insulin resistance in chronic kidney disease is ameliorated by spironolactone in rats and humans. Kidney Int. 2015; 87(4):749-60.
- Nguyen Dinh Cat A, Antunes TT, Callera GE, Sánchez A, Tsiropoulou S, Dulak-Lis MG, Anagnostopoulou A, He Y, Montezano AC, Jaisser F, Touyz RM. Adipocyte-Specific Mineralocorticoid Receptor Overexpression in Mice Is Associated With Metabolic Syndrome and Vascular Dysfunction: Role of Redox-Sensitive PKG-1 and Rho Kinase. Diabetes. 2016; 65(8):2392-403.
- Hayakawa T, Minemura T, Onodera T, Shin J, Okuno Y, Fukuhara A, Otsuki M, Shimomura I. Impact of MR on mature adipocytes in high-fat/high-sucrose diet-induced obesity. J Endocrinol. 2018; 239(1):63–71.
- Feraco A, Armani A, Urbanet R, Nguyen Dinh Cat A, Marzolla V, Jaisser F, Caprio M. Minor role of mature adipocyte mineralocorticoid receptor in high fat induced obesity. J Endocrinol. 2018; Aug 18. pii: JOE-18-0314. doi: 10.1530/JOE-18-0314. [Epub ahead of print]
- Kotlyar E, Vita JA, Winter MR, Awtry EH, Siwik DA, Keaney JF Jr, Sawyer DB, Cupples LA, Colucci WS, Sam F. The relationship between aldosterone, oxidative stress, and inflammation in chronic, stable human heart failure. J Card Fail. 2006; 12(2):122-7.
- Taye A, Morawietz H. Spironolactone inhibits NADPH oxidase-induced oxidative stress and enhances eNOS in human endothelial cells. Iran J Pharm Res. 2011; 10(2):329-37.
- Calò LA, Pagnin E, Davis PA, Armanini D, Mormino P, Rossi GP, Pessina AC. Oxidative stress-related proteins in a Conn’s adenoma tissue. Relevance for aldosterone’s prooxidative and proinflammatory activity. J Endocrinol Invest. 2010; 33(1):48-53.
- Zhou J, Lam B, Neogi SG, Yeo GS, Azizan EA, Brown MJ. Transcriptome Pathway Analysis of Pathological and Physiological Aldosterone-Producing Human Tissues. Hypertension. 2016; 68(6):1424-1431.
- Ibarrola J, Sadaba R, Martinez-Martinez E, García-Peña A, Arrieta V, Alvarez V, Fernández-Celis A, Gainza A, Cachofeiro V, Santamaria E, Fernández-Irigoyen J, Jaisser F, López-Andres N. Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12. Sci Rep. 2018; 8(1):6801.
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Segal MS, Christou DD. Mineralocorticoid receptors modulate vascular endothelial function in human obesity. Clin Sci (Lond). 2013; 125(11):513-20.
- Hwang MH, Yoo JK, Luttrell M, Meade TH, English M, Christou DD. Effect of Selective Mineralocorticoid Receptor Blockade on Flow-Mediated Dilation and Insulin Resistance in Older Adults with Metabolic Syndrome. Metab Syndr Relat Disord. 2015; 13(8):356-61.
- Hwang MH, Yoo JK, Luttrell M, Kim HK, Meade TH, English M, Nichols WW, Christou DD. Role of mineralocorticoid receptors in arterial stiffness in human aging. Exp Gerontol 2013; 48(8):701-4.
- Chen L, Ding ML, Wu F, He W, Li J, Zhang XY, Xie WL, Duan SZ, Xia WH, Tao J. Impaired Endothelial Repair Capacity of Early Endothelial Progenitor Cells in Hypertensive Patients With Primary Hyperaldosteronemia: Role of 5,6,7,8-Tetrahydrobiopterin Oxidation and Endothelial Nitric Oxide Synthase Uncoupling. Hypertension. 2016; 67(2):430-9.
- Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, Coca A, de Simone G, Dominiczak A, Kahan T, Mahfoud F, Redon J, Ruilope L, Zanchetti A, Kerins M, Kjeldsen SE, Kreutz R, Laurent S, Lip GYH, McManus R, Narkiewicz K, Ruschitzka F, Schmieder RE, Shlyakhto E, Tsioufis C, Aboyans V, Desormais I; ESC Scientific Document Group. Collaborators (182). 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018; 39(33):3021-3104.
- Kolkhof P, Bärfacker L. 30 years of the mineralocorticoid receptor: Mineralocorticoid receptor antagonists: 60 years of research and development. J Endocrinol. 2017; 234(1):T125-T140.
