EUROPEAN MEDICINES AGENCY (EMA) – FDA (U.S. FOOD & DRUG ADMINISTRATION)
(A) TRACTO ALIMENTARIO Y METABOLISMO
Donislecel (Lantidra®) CellTrans (FDA; Food & Drug Administration, USA)
Indicación: Tratamiento de adultos con diabetes tipo 1 que no pueden acercarse a la meta de hemoglobina glucosilada debido a episodios repetidos actuales de hipoglucemia grave a pesar de un control y educación intensivos sobre la diabetes.
Tipo: Medicamento de terapia avanzada (celular somática alogénica), constituido por no más de un millón [10(6)] de islotes alogénicos de Langerhans procedentes de páncreas de donantes fallecidos. Los islotes contienen varios tipos de células endocrinas: b, a, PP (péptido pancreático), d y e. Autorizado el 28-6-2023; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Secreción de insulina por las células beta de los islotes alogénicos infundidos. En algunos pacientes con diabetes tipo 1, estas células infundidas pueden producir suficiente insulina, por lo que el paciente ya no necesita administrarse insulina para controlar sus niveles de glucosa en la sangre. El medicamento se administra como una infusión única en la vena porta hepática. Se puede realizar una infusión adicional dependiendo de la respuesta del paciente a la dosis inicial.
Eficacia clínica: Dos estudios de un solo brazo no aleatorizados en los que un total de 30 participantes con diabetes tipo 1 e hipoglucemia recibieron al menos una infusión y un máximo de tres infusiones. En general, 21 participantes (70%) no necesitaron la administración exógena de insulina durante un año o más (11 durante uno a cinco años y 10 durante más de cinco años). Cinco participantes (17%) no lograron ningún día de independencia de la insulina.
Eventos adversos: Los más comunes son náuseas (83%), fatiga (83%), anemia (80%), diarrea (80%), astenia (67%), cefalea (67%) y dolor abdominal (67%). La mayoría de los participantes (90%) experimentó al menos una reacción adversa grave relacionada con el procedimiento de infusión del medicamento en la vena porta hepática (laceración hepática/hematoma, hemorragia y sangrado intraabdominal, 13 %; o elevación de la presión portal, 7 %) y el uso de medicamentos inmunosupresores necesarios para mantener la viabilidad de las células de los islotes (infección, 87%; o neoplasia, 37%). Algunas reacciones adversas graves requirieron la interrupción de los medicamentos inmunosupresores, lo que provocó la pérdida de la función de las células de los islotes y la independencia de la insulina.
Pozelimab (Veopoz®) Regeneron (FDA; Food & Drug Administration, USA)
Indicación: Tratamiento de pacientes adultos y pediátricos de 1 año de edad o mayores con enteropatía perdedora de proteínas (PLE) por deficiencia de CD55, también conocida como enfermedad de CHAPLE.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal recombinante (isotipo IgG4), producido mediante tecnología de ADN recombinante. Autorizado en Estados Unidos (FDA) el 18-8-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track) y con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo dirigido contra la proteína del complemento terminal C5 que inhibe su activación al bloquear la escisión de C5 en C5a (anafilatoxina) y C5b, bloqueando así la formación del complejo de ataque a la membrana (C5b-C9, una estructura que media la lisis celular).
Eficacia clínica: Un estudio de un solo grupo en el que se compararon los resultados con los datos previos al tratamiento en diez pacientes con enteropatía perdedora de proteínas (PLE) con deficiencia de CD55 activa que tenían hipoalbuminemia, con edades comprendidas entre 3 y 19 años. La mediana de tiempo para que la albúmina sérica alcanzara al menos 3,5 g/dl fue de 15,5 días; los 10 pacientes lograron la normalización en la semana 12 y mantuvieron las concentraciones de albúmina sérica dentro del rango normal durante al menos 72 semanas de tratamiento.
Eventos adversos: Los más comunes son infección del tracto respiratorio superior (30%), fractura (30%), urticaria (20%) y alopecia (20%).
(D) DERMATOLOGÍA
Lotilaner (Xdemvy®) Tarsus (FDA; Food & Drug Administration, USA)

Indicación: Tratamiento de blefaritis por Demodex.
Tipo: Medicamento sintético estándar derivado del núcleo de isoxazoliltiofenocarboxamida. Autorizado en Estados Unidos (FDA) el 25-7-2023; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Una de las causas más comunes de la blefaritis es el Demodex folliculorum: un ácaro microscópico alargado, considerado como el ectoparásito permanente más común de los seres humanos, presente en los folículos pilosos de las pestañas de hasta el 85% de los adultos mayores de 60 años. El lotilaner es un inhibidor de los canales de cloruro activado por ácido gamma-aminobutírico (GABA) selectivo para estos ácaros. La inhibición de estos canales de cloruro GABA provoca una acción paralizante en el organismo diana que conduce a su muerte.
Eficacia clínica: Evaluado en un total de 833 pacientes (415 de los cuales recibieron lotilaner) en dos estudios de 6 semanas, aleatorizados, multicéntricos, doble ciego, controlados por vehículo. La eficacia se demostró mediante la mejora en los párpados (reducción de los collares alrededor de los folículos a no más de 2 por párpado superior) para el día 43, con una tasa de éxito del 44 vs. 7%.
Eventos adversos: Los más comunes son escozor y ardor en el lugar de la instilación (10%); chalazión/orzuelo y queratitis puntiforme (<2%). (J) ANTIINFECCIOSOS SISTÉMICOS Vacuna Ántrax (Cyfendus®) Gaithersburg (FDA; Food & Drug Administration, USA) Indicación: Para la profilaxis posterior a la exposición de la enfermedad, tras una exposición sospechada o confirmada a Bacillus anthracis en personas de 18 a 65 años de edad cuando se administra junto con los medicamentos antibacterianos recomendados. Tipo: Medicamento biológico producido a partir de filtrados libres de células de cultivos microaerófilos de una cepa avirulenta y no encapsulada de Bacillus anthracis. Autorizado en Estados Unidos (FDA) el 20-7-2023. Mecanismo: Induce anticuerpos contra la proteína antigénica protectora que puede contribuir a la protección al neutralizar las actividades de la toxina letal citotóxica y la toxina del edema de B. anthracis. La vacuna está adyuvada con CPG 7909, una molécula de ADN sintético de 24 nucleótidos de longitud con un esqueleto de fosforotioato resistente a las nucleasas. Eficacia clínica: Estudio clínico multicéntrico, aleatorizado, con control activo, doble ciego, de grupos paralelos. 3151 participantes recibieron al menos una dosis de Cyfendus, y 533 participantes recibieron al menos una dosis de BioThrax. Se realizó un seguimiento de la inmunogenicidad de los participantes hasta el día 64 (7 semanas después de la última vacunación con Cyfendus, 5 semanas después de la última vacunación con BioThrax). Los objetivos primarios de inmunogenicidad fueron evaluar la respuesta del factor de neutralización del 50 % (NF50) de TNA inducida por la vacuna Cyfendus en el día 64 y la no inferioridad frente a BioThrax después de dos dosis de Cyfendus administradas por vía intramuscular con dos semanas de diferencia (semanas 0 y 2) y tres dosis de BioThrax administrados por vía subcutánea con dos semanas de diferencia (semanas 0, 2 y 4). Los dos criterios de valoración de inmunogenicidad coprimarios y los criterios de evaluación estadística fueron el porcentaje de receptores de Cyfendus que alcanzaron un valor umbral de TNA NF50 ≥0,56 en el día 64 y la diferencia porcentual entre los participantes que alcanzaron un valor umbral de TNA NF50 ≥0,29 en el día 64 en los que recibieron Cyfendus y en los que recibieron BioThrax: 86,6 vs. 61,4%, lo que corresponde a una diferencia del 25,2 %. Eventos adversos: Los más comunes (>10%) en el lugar de la inyección son sensibilidad (88%), dolor (86%), limitación del movimiento del brazo (64%), calor (51%), induración (38%), picazón (22%), hinchazón (20%) y eritema/enrojecimiento (18%). Los reacciones adversas sistémicas más frecuentes (>10%) son dolores musculares (75%), cansancio (67%) y dolor de cabeza (58%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
Tislelizumab (Tevimbra®) Novartis (EMA; European Medicines Agency)
Indicación: Tratamiento de pacientes adultos con carcinoma de células escamosas de esófago irresecable, localmente avanzado o metastásico, tras quimioterapia previa basada en platino.
Tipo: Medicamento biológico; es un anticuerpo monoclonal humanizado variante de la inmunoglobulina G4 (IgG4) Fc. Autorizado por la Unión Europea (EMA) 15-9-2023 como medicamento huérfano (Orphan drug); no autorizado previamente en Estados Unidos.
Mecanismo: Anticuerpo monoclonal humanizado frente a PD-1, que se une al dominio extracelular de PD-1 humano. Bloquea de forma competitiva la unión de PD-L1 y PD-L2, inhibiendo la señalización negativa mediada por PD-1 y aumentando la actividad funcional en células T.
Eficacia clínica: Estudio fase 3 abierto, controlado con quimioterapia escogida por el investigador (paclitaxel, xocetaxel o irinotecán) y aleatorizado sobre un total de 512 pacientes. La variable de eficacia primaria fue la supervivencia global en la población por intención de tratar (ITT): 23% (tislelizumab) vs. 16,8% (control activo), con una mediana de supervivencia de 8,6 vs. 6,3 meses.
Eventos adversos: La reacción adversa más frecuente fue anemia (29,2%), siendo muy frecuentes (>10%) hipotiroidismo, tos, erupción cutánea, prurito, fatiga, disminución del apetito, aumento de valores de enzimas hepáticas y de la bilirrubina. Las reacciones adversas de grado 3/4 más frecuentes fueron anemia (5,0%) y neumonía (4,2%). El 1,17% de los pacientes presentaron reacciones adversas que causaron la muerte: neumonía (0,78%), hepatitis (0,13%), neumonitis (0,07%), disnea (0,07%), disminución del apetito (0,07%) y trombocitopenia (0,07%).
Quizartinib (Vanflyta®) Daiichi Sankyo (FDA; Food & Drug Administration, USA)
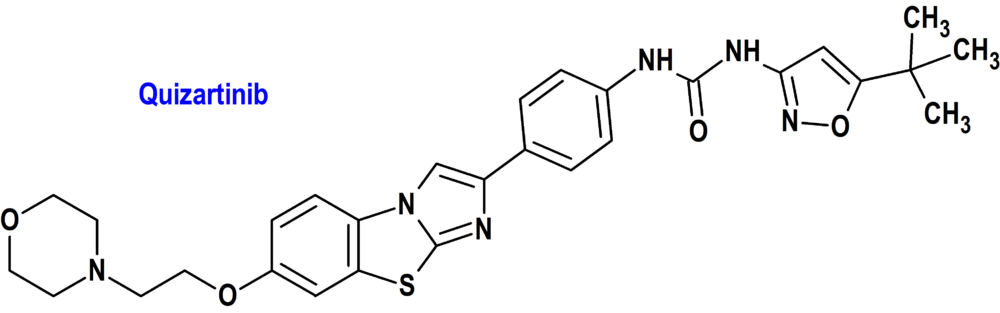
Indicación: En combinación con citarabina estándar y inducción con antraciclina y consolidación con citarabina, y como monoterapia de mantenimiento después de la quimioterapia de consolidación, para el tratamiento de pacientes adultos con leucemia mieloide aguda (LMA) recién diagnosticada que es positiva para duplicación interna en tándem (ITD) de FLT3 detectada por una prueba aprobada por la FDA
Tipo: Medicamento sintético estándar derivado de un núcleo de imidazo[2,1b][1,3]benzotiazol. Autorizado en Estados Unidos (FDA) el 20-7-2023 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor del receptor tirosina cinasa FLT3, codificado por uno de los genes que se encuentra frecuentemente alterado en pacientes con leucemia mieloide aguda. Este gen codifica una cinasa de tirosina, cuya función se relaciona con el proceso ontogénico del desarrollo mieloide. Mutaciones en este gen están relacionados con pobre pronóstico. Quizartinib y su principal metabolito activo AC886 se unen al dominio de unión de trifosfato de adenosina (ATP) de FLT3 con una afinidad comparable, y ambos tenían una afinidad 10 veces menor hacia la mutación FLT3-ITD en comparación con FLT3. Quizartinib y AC886 inhibieron la actividad de la quinasa FLT3, previniendo la autofosforilación del receptor, inhibiendo así la señalización del receptor FLT3 y bloqueando la proliferación celular dependiente de FLT3-ITD.
Eficacia clínica: Un estudio aleatorizado, doble ciego, controlado con placebo de 539 pacientes con LMA positiva para FLT3-ITD recién diagnosticada. La eficacia se estableció sobre la base de la supervivencia general (SG), medida desde la fecha de aleatorización hasta la muerte por cualquier causa. El análisis primario se realizó tras un seguimiento mínimo de 24 meses tras la aleatorización del último paciente. El estudio demostró una mejora estadísticamente significativa en la SG para el brazo de quizartinib vs. placebo [cociente de riesgos instantáneos (HR) 0,78.
Eventos adversos: Los más comunes son (≥10 % con una diferencia entre brazos de ≥2 % en comparación con el placebo), son linfopenia, hipopotasemia, hipoalbuminemia, hipofosfatemia, aumento de fosfatasa alcalina, hipomagnesemiao, neutropenia febril, diarrea, mucositis, náuseas , hipocalcemia, dolor abdominal, sepsis, neutropenia, dolor de cabeza, aumento de la creatina fosfocinasa, vómitos, infecciones del tracto respiratorio superior, hipertransaminasemia, trombocitopenia, disminución del apetito, infecciones fúngicas, epistaxis, hiperpotasemia, infecciones por herpesvirus, insomnio, prolongación del intervalo QT del electrocardiograma, hipermagnesemia, hipernatremia, anemia e irritación ocular. La reacción adversa grave más común (≥5%) fue la neutropenia febril (11 %). Se produjeron reacciones adversas mortales en el 10 % de los pacientes, como sepsis (5 %), infecciones fúngicas (0,8 %) y edema cerebral (0,8 %). La discontinuación permanente debido a una reacción adversa ocurrió en el 20% de los pacientes.
Talquetamab (Talvey®) Janssen Cilag (EMA; European Medicines Agency/FDA; Food & Drug Administration, USA)
Indicación: Tratamiento en monoterapia de pacientes adultos con mieloma múltiple en recaída y refractario, que han recibido al menos 3 tratamientos previos, incluyendo un agente inmunomodulador, un inhibidor del proteasoma y un anticuerpo anti-CD38 y han presentado progresión de la enfermedad tras el último tratamiento.
Tipo: Medicamento biológico constituido por un anticuerpo biespecífico humanizado de inmunoglobulina G4-prolina, alanina, alanina (IgG4-PAA) dirigido contra el receptor acoplado a proteínas G de la familia C, grupo 5 y miembro D (GPRC5D) y los receptores CD3, producido mediante tecnología de ADN recombinante. Autorizado por la Unión Europea (EMA) el 21-8-2023 como medicamento huérfano (Orphan drug) y condicionalmente (Conditional marketing authorisation); autorizado también en Estados Unidos (FDA) el 9-8-2023.
Mecanismo: Talquetamab promueve una mayor citotoxicidad mediada por los linfocitos T mediante el reclutamiento de linfocitos T que expresan CD3 a células que expresan GPRC5D. Esto produce la activación de los linfocitos T e induce la posterior lisis de las células que expresan GPRC5D mediada por la perforina secretada y varias granzimas almacenadas en las vesículas secretoras de los linfocitos T citotóxicos. Talquetamab se dirige específicamente a las células del mieloma múltiple.
Eficacia clínica: Un ensayo de un solo grupo, abierto y multicéntrico en 288 pacientes con mieloma múltiple en recaída o refractario que habían recibido al menos tres tratamientos previos, incluyendo un inhibidor del proteasoma, un agente inmunomodulador y un anticuerpo monoclonal anti-CD38. Los pacientes recibieron una dosis 0,4 mg/kg por vía subcutánea semanalmente, tras dos dosis de escalada (0,01 y 0,06 mg/kg) en la primera semana de tratamiento, o de 8 mg/kg por vía subcutánea quincenalmente, tras tres dosis de escalada (0,01, 0,06 y 0,3 mg/kg), hasta la progresión de la enfermedad o una toxicidad inaceptable. Los resultados de la eficacia se basaron en la tasa de respuesta global, según lo determinado por la evaluación del Comité de Revisión Independiente utilizando los criterios del IMWG. La mediana de seguimiento de los pacientes tratados con la dosis de 0,4mg/kg semanales fue de 18,8 meses; se estima que el 51,5% de pacientes que respondieron al tratamiento mantuvieron la respuesta durante al menos 9 meses. La tasa de respuesta global fue del 74,1% (336,6% completa) con la dosis de 0,4 mg/kg/semanal y del 71,7% (38,7% completa) con la de 0,8 mg/kg/quincenal; la mediana de la duración de la respuesta fue de 9,5 meses con la dosis de 0,4 (no pudo determinarse con la de 0,8).
Eventos adversos: Los más comunes son síndrome de liberación de citocinas (SLC; 77%), disgeusia (72%), hipogammaglobulinemia (67%), trastorno de las uñas (56%), dolor musculoesquelético (48%), anemia (47%), trastorno de la piel (43%), fatiga (43%), pérdida de peso(40%), erupción (39%), boca seca (36%), neutropenia (35%), pirexia (33%), xerosis (32%), trombocitopenia (30%), infección del tracto respiratorio superior(29%), linfopenia (27%), disfagia (24%), diarrea (25%), prurito (23%), tos (23%), dolor (22%), apetito disminuido (22%) y cefalea (20%).
Elranatamab (Elrexfio®) Pfizer (FDA; Food & Drug Administration, USA)
Indicación: Tratamiento de pacientes adultos con mieloma múltiple en recaída o refractario que hayan recibido al menos cuatro líneas de terapia previas, incluido un inhibidor del proteasoma, un agente inmunomodulador y un anticuerpo monoclonal anti-CD38.
Tipo: Medicamento biológico constituido por un anticuerpo de tipo inmunoglobulina 2 kappa -alanina humanizado biespecífico, derivado de dos anticuerpos monoclonales, un anti-BCMA y un anti-CD3, cada uno de los cuales aporta una cadena pesada (H) distinta y una cadena ligera (L) distinta al biespecífico elranatamab; el anticuerpo biespecífico de 4 cadenas resultante está unido covalentemente mediante cinco enlaces disulfuro intercatenarios. Autorizado en Estados Unidos (FDA) el 14-8-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo biespecífico de maduración de células B (BCMA) dirigido a células T que se une a BCMA en células plasmáticas, plasmablastos y células de mieloma múltiple y a CD3 en células T, lo que conduce a la citolisis de las células que expresan BCMA. Elranatamab activa las células T, provocando la liberación de citocinas proinflamatorias (IL-2, IL-6, IL-8, IL-10, TNF-a y IFN-g) y la lisis de las células de mieloma múltiple.
Eficacia clínica: Un estudio multicéntrico, de un solo brazo y abierto que incluyó a 97 pacientes refractarios a al menos un inhibidor del proteasoma (IP), un agente inmunomodulador (IMiD) y un anticuerpo monoclonal anti-CD38. La eficacia se basó en la tasa de respuesta (57,7%, completa 25,8%) y la duración de la respuesta (DOR), según criterios del IMWG. La mediana del tiempo hasta la primera respuesta fue de 1,22 (0,9 a 6,5) meses. Con una mediana de seguimiento de 11,1 meses entre los que respondieron, la tasa de DOR a los 6 meses fue del 90,4 % y a los 9 meses fue del 82,3 %.
Eventos adversos: Los más comunes (≥20%) son síndrome de liberación de citocinas (RSC), fatiga, reacción en el lugar de la inyección, diarrea, infección del tracto respiratorio superior, dolor musculoesquelético, neumonía, disminución del apetito, erupción cutánea, tos, náuseas y pirexia. Las anomalías de laboratorio de grado 3 a 4 más comunes (≥30%) fueron disminución de linfocitos, disminución de neutrófilos, disminución de hemoglobina, disminución de glóbulos blancos y disminución de plaquetas. Se produjeron interrupciones permanentes del tratamiento debido a una reacción adversa en el 17% de los pacientes, siendo la causa más común el shock séptico (2,2%).
Momelotinib (Ojjaara®) GlaxoSmithKline (FDA; Food & Drug Administration, USA)
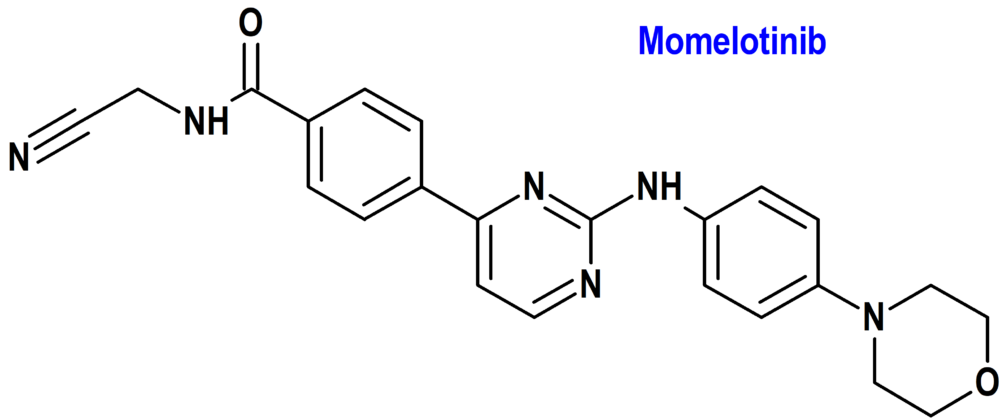
Indicación: Tratamiento de la mielofibrosis (MF) de riesgo intermedio o alto, incluida la MF primaria o secundaria [post-policitemia vera (PV) y post-trombocitemia esencial (ET)], en adultos con anemia.
Tipo: Medicamento sintético estándar derivado de un núcleo de aminopirimidina. Autorizado en Estados Unidos (FDA) el 15-9-2023 como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de Janus Kinase 1 y 2 de tipo salvaje (JAK1/JAK2) y JAK2V617F mutante, que contribuyen a la señalización de una serie de citocinas y factores de crecimiento que son importantes para la hematopoyesis y la función inmune. Momelotinib y su principal metabolito circulante humano, M21, tienen una mayor actividad inhibidora de JAK2 en comparación con JAK3 y la tirosina cinasa 2 (TYK2). Momelotinib y M21 inhiben adicionalmente el receptor de activina A tipo 1 (ACVR1), también conocido como receptor de activina tipo cinasa 2 (ALK2), que produce una inhibición posterior de la expresión de hepcidina hepática y una mayor disponibilidad de hierro, lo que resulta en una mayor producción de glóbulos rojos. La mielofibrosis es una neoplasia mieloproliferativa asociada con activación constitutiva y señalización JAK desregulada que contribuye a la inflamación y la hiperactivación de ACVR1. La señalización JAK recluta y activa proteínas STAT (transductores de señal y activación de la transcripción), lo que da como resultado la localización nuclear y la posterior regulación de la transcripción genética.
Eficacia clínica: Un ensayo doble ciego, aleatorizado y controlado con un comparador activo (danazol) en 195 adultos sintomáticos y anémicos con mielofibrosis que habían recibido previamente una terapia aprobada con inhibidores de JAK. La eficacia se estableció basándose en el porcentaje de pacientes tratados con momelotinib en comparación con danazol que lograron un MFSAF (formulario de evaluación de síntomas de mielofibrosis versión 4.0) con una reducción de la puntuación total de síntomas del 50 % o más en la semana 24 en comparación con su propia puntuación inicial (25 vs. 9%). Otros criterios de valoración incluyeron independencia de transfusión (30 vs. 20%), pacientes con reducción del volumen del bazo en un 35% o más (39 vs. 6%), cambio en la puntuación total de síntomas de MFSAF v4.0 desde el inicio (-9,4 vs. -3,1) y porcentaje de pacientes sin transfusiones (35 vs 17%).
Eventos adversos: Los más comunes son trombocitopenia (28%), diarrea (22%), hemorragia (22%), fatiga (21%), náuseas (16%), infección bacteriana (15%), dolor abdominal (13%), infección viral (12%), prurito (11%), enzimas hepáticas elevadas (10%) y pirexia (10%). Se produjeron reacciones adversas graves en el 35% de los pacientes y es precisa la interrupción permanente del tratamiento debido a una reacción adversa en el 18%. El 34% de los pacientes requieren una reducción de la dosis o la interrupción del tratamiento debido a una reacción adversa.
Rozanolixizumab (Rystiggo®) UCB (FDA; Food & Drug Administration, USA)
Indicación: Tratamiento de miastenia grave generalizada en pacientes adultos con anticuerpos anti-receptor de acetilcolina (AChR) o anti-tirosina cinasa específica de músculo (MuSK) positivos.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal IgG4 humanizado que se une al receptor Fc neonatal (FcRn). Autorizado en Estados Unidos (FDA) el 26-6-2023 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Se une al receptor Fc neonatal (FcRn), lo que resulta en la reducción de la IgG circulante. En pacientes con autoanticuerpos AChR y MuSK positivos que recibieron tratamiento, hubo una reducción en los niveles totales de IgG en relación con el valor inicial; las disminuciones en los niveles de autoanticuerpos AChR y autoanticuerpos MuSK siguieron un patrón similar.
Eficacia clínica: Estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, que incluyó a 200 pacientes, con un período de selección de 4 semanas y un período de tratamiento de 6 semanas seguido de 8 semanas de observación. La eficacia se midió mediante la escala MG-ADL, que evalúa el impacto de la miastenia grave generalizada en las funciones diarias de 8 signos o síntomas que normalmente se ven afectados; la puntuación total varía de 0 a 24, y la más alta indica un mayor deterioro. La variable principal de eficacia fue la comparación del cambio desde el inicio entre los grupos de tratamiento en la puntuación total de MG-ADL en el día 43: -3,4 (dosis de 7 mg/kg), -3,4 (dosis de 10 mg/kg) y -0,8 (placebo). La variable secundaria fue el cambio entre los grupos de tratamiento desde el inicio hasta el día 43 en la QMG, un sistema de calificación de 13 ítems que evalúa la debilidad muscular, cuya puntuación total posible oscila entre 0 y 39, donde la más alta indica un deterioro más grave: -5,4 (dosis de 7 mg/kg), -6,7 (dosis de 10 mg/kg) y -1,9 (placebo).
Eventos adversos: Los más comunes son (>5%) cefalea (44%), diarrea (20%), fiebre (17%), reacciones de hipersensibilidad (11%), náusea (10%), reacciones en el punto de administración (8%), infección del tracto respiratorio superior (8%), dolor abdominal (8%) y artralgia (7%).
Ritlecitinib (Litfulo®) Pfizer (EMA; European Medicines Agency/FDA; Food & Drug Administration, USA)
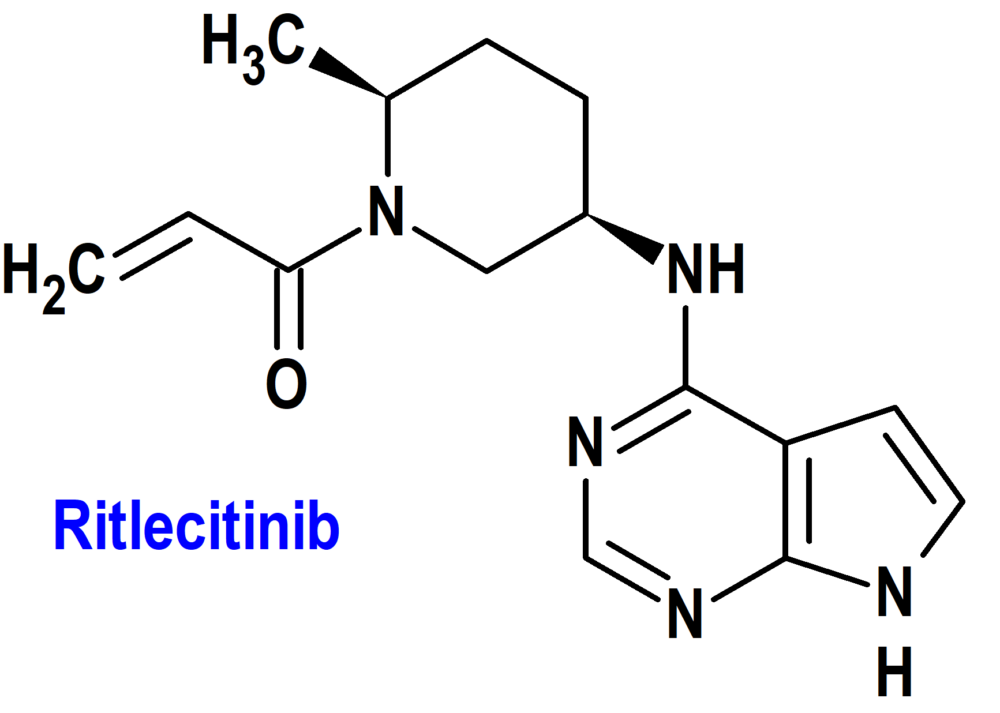
Indicación: Tratamiento de alopecia areata grave en adultos y adolescentes a partir de los 12 años.
Tipo: Medicamento sintético estándar derivado del núcleo de pirrolopirimidina; autorizado por la Unión Europea (EMA) el 15-9-2023; autorizado también en Estados Unidos (FDA) el 23-6-2023.
Mecanismo: Inhibe irreversiblemente la cinasa Janus tipo 3 (JAK3) y la tirosina cinasa expresada en la familia de cinasas del carcinoma hepatocelular (TEC) al bloquear el sitio de unión del trifosfato de adenosina (ATP). En entornos celulares, ritlecitinib inhibe la fosforilación de la familia de proteínas STAT (Signal Transducer and Activator of Transcription) inducida por citocinas mediada por receptores dependientes de JAK3. Además, ritlecitinib inhibe la señalización de los receptores inmunitarios dependientes de los miembros de la familia de cinasas TEC, aunque la relevancia de la inhibición de enzimas específicas de la familia JAK o TEC para la eficacia terapéutica no se conoce actualmente. El tratamiento fue asociado con una disminución temprana dependiente de la dosis en los niveles absolutos de linfocitos, linfocitos T (CD3) y subconjuntos de linfocitos T (CD4 y CD8); además, hubo una disminución temprana dependiente de la dosis en las células NK (CD16/56) que se mantuvo estable en el nivel más bajo hasta la semana 48.
Eficacia clínica: Un ensayo aleatorizado, doble ciego, controlado con placebo en 718 sujetos de 12 años de edad y mayores con alopecia areata con ≥50% de pérdida de cabello del cuero cabelludo, incluida la alopecia totalis y la alopecia universalis. La evaluación de la pérdida de cabello del cuero cabelludo se basó en la puntuación SALT (Severity of Alopecia Tool). En la semana 24, la proporción de sujetos tuvo una respuesta SALT ≤20 (20% o menos de pérdida de cabello del cuero cabelludo) fue del 23,0% (ritlecitinib) vs. 1,6% (placebo), mientras que la respuesta SALT ≤10 (10% o menos de pérdida de cabello del cuero cabelludo) fue del 13,4 vs. 1,5%.
Eventos adversos: Los más comunes (≥5%) son cefalea (10,8 %), diarrea (10,0 %), acné (6,2 %) y erupción (5,4 %).
Motixafortida (Aphexda®) BioLineRx (FDA; Food & Drug Administration, USA)
Indicación: En combinación con filgrastim (G-CSF) para movilizar células madre hematopoyéticas a la sangre periférica para su recolección y posterior trasplante autólogo en pacientes con mieloma múltiple.
Tipo: Medicamento sintético estándar constituido por un péptido cíclico formado por 14 aminoácidos: [N-(4-fluoro-benzoil)-L-arginil]-L-arginil-[(3-naftil)-L-alanil]-L-cisteinil-L-tirosil-L-citrulinil-L-lisil-D-lisil-L-prolil-L-tirosil-L-arginil-L-citrulinil-L-cisteinil-L-arginineamida, (4-13)-disulfuro cíclico. Autorizado en Estados Unidos (FDA) el 8-9-2023 como medicamento huérfano (Orphan drug); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor del receptor de quimiocina 4 con motivo C-X-C (CXCR4) y bloquea la unión de su ligando afín, el factor 1a derivado del estroma (SDF-1a)/ligando de quimiocina 12 con motivo C-X-C (CXCL12). SDF-1a y CXCR4 desempeñan un papel en el tráfico y la localización de células madre hematopoyéticas humanas en la médula. Una vez en la médula, la célula madre CXCR4 puede ayudar a anclar estas células a la matriz de la médula, ya sea directamente a través de SDF-1a o mediante la inducción de otras moléculas de adhesión. El tratamiento con motixafortida provoca leucocitosis y elevaciones de las células madre y progenitoras hematopoyéticas circulantes en la circulación periférica. Las células madre movilizadas por motixafortida son capaces de injertarse con capacidad de repoblación a largo plazo.
Eficacia clínica: Un estudio aleatorizado, doble ciego y controlado con placebo en 122 pacientes con mieloma múltiple. Antes de recibir motixafortida o placebo, los pacientes recibieron dosis matutinas diarias de filgrastim durante 4 días. La efectividad clínica midió según la proporción de pacientes que alcanzaron un objetivo de recolección de células de ≥6 × 10(6) células CD34+/kg en hasta 2 aféresis después de la administración de filgrastim y una sola administración de motixafortida o de placebo: 67,5 vs 9,5%.
Eventos adversos: Los más comunes son reacciones en el lugar de la inyección (73%), prurito (38%), enrojecimiento (33%), erupción (16%), urticaria (14%), eritema (12%), dolor de espalda (21%), parestesia (19%) , hipopotasemia (15%), náuseas (14%). Se produjeron reacciones adversas graves en el 5,4% de los pacientes.
(M) SISTEMA MÚSCULO-ESQUELÉTICO
Delandistrogene Moxeparvovec (Elevidys®) Sarepta (FDA; Food & Drug Administration, USA)
Indicación: Tratamiento de pacientes pediátricos ambulatorios de 4 a 5 años con distrofia muscular de Duchenne (DMD) con una mutación confirmada en el gen DMD.
Tipo: Medicamento de terapia avanzada (génica), constituido por un vector basado en el serotipo rh74 (AAVrh74) del virus adenoasociado, recombinante y no replicante que contiene el transgén de la microdistrofina bajo el control del promotor MHCK7; la proteína microdistrofina expresada es una versión abreviada (138 kDa, en comparación con el tamaño de 427 kDa de la distrofina expresada en células musculares normales) que contiene dominios seleccionados de la distrofina natural). Autorizado en Estados Unidos (FDA) el 22-6-2023 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Producto de terapia génica recombinante que consta de una cápside del serotipo rh74 (AAVrh74) del virus adenoasociado (AAV) recombinante no replicante y un casete de expresión de ssDNA flanqueado por repeticiones terminales invertidas (ITR) derivadas de AAV2. El casete contiene: un componente regulador del gen MHCK7, que comprende un promotor de la creatina cinasa 7 y un potenciador de la cadena pesada de la a-miosina, y el transgén de ADN que codifica la proteína de microdistrofina modificada.
Eficacia clínica: Dos ensayos clínicos multicéntricos, en progreso actualmente. El primero tuvo dos parte, la primera consistente en un período de 48 semanas, aleatorizado, doble ciego, controlado con placebo, mientras que la segunda tuvo un período de 48 semanas que comenzó después de completar la primera parte, donde los pacientes que recibieron placebo durante la Parte 1 fueron tratados con el fármaco y los pacientes tratados con el fármaco durante la Parte 1 recibieron placebo. La población del estudio consistió en pacientes masculinos ambulatorios con DMD (N = 41) de 4 a 7 años de edad con una mutación de marco de lectura confirmada o una mutación prematura del codón de terminación entre los exones 18 a 58 en el gen DMD. Los objetivos principales fueron evaluar la expresión de microdistrofina en el músculo esquelético y evaluar el efecto del fármaco en la puntuación total de la Evaluación ambulatoria de North Star (NSAA). El estudio 2 es un estudio abierto que incluye una cohorte de 20 sujetos masculinos ambulatorios con DMD de 4 a 7 años de edad. Los 20 sujetos tienen una mutación de cambio de marco confirmada, una mutación del sitio de empalme canónico o una mutación prematura del codón de parada en el gen DMD. El cambio en la puntuación total de la NSAA se evaluó desde el inicio hasta la semana 48 después de la infusión de ELEVIDYS o placebo, siendo la mediana del 40,8% en el estudio 1 y del 50,6% en el 2.
Eventos adversos: Los más comunes son vómitos (61%), náuseas (40%), aumento de valores en pruebas de función hepática (37%), pirexia (24%) y trombocitopenia (12%).
Palovaroteno (Sohonos®) Ipsen (FDA; Food & Drug Administration, USA)

Indicación: Para la reducción del volumen de nueva osificación heterotópica en adultos y pacientes pediátricos de 8 años o más para mujeres y de 10 años o más para hombres con fibrodisplasia osificante progresiva (FOP).
Tipo: Medicamento sintético estándar con estructura retinoide aromática. Autorizado en Estados Unidos (FDA) el 16-8-2023 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); por su parte, en la Unión Europea la Agencia Europea de Medicamentos (EMA) confirmó el 25-5-2023 su recomendación inicial del 26-1-2023 de denegar la autorización de comercialización de Sohonos, por considerar que no se podían extraer conclusiones firmes sobre los beneficios del medicamento, ya que la conclusión del solicitante se basaba en un análisis post hoc que no estaba ni científica ni clínicamente justificado y los objetivos de estudio preespecificados no se cumplieron, además los resultados de otros estudios y los limitados datos clínicos a largo plazo disponibles no respaldaron la eficacia. En cuanto a la seguridad, el riesgo de cierre fisario prematuro, que es un riesgo conocido con el tratamiento con retinoides en pacientes en crecimiento, no pudo ser adecuadamente mitigado con las medidas de minimización de riesgos propuestas por la empresa. Además, la EMA consideró que algunas cuestiones relativas a la calidad de la sustancia activa no se habían resuelto.
Mecanismo: En pacientes con FOP, la formación ósea anormal, incluida la osificación heterótrofa (HO), es impulsada por una mutación de ganancia de función en el receptor ALK2 (ACVR1) de tipo I de la proteína morfogenética ósea (BMP). El palovaroteno es un agonista del receptor del ácido retinoico, con particular selectividad en el subtipo gamma de RAR. A través de la unión a RARg, el palovaroteno disminuye la vía de señalización de BMP/ALK2 al inhibir la fosforilación de SMAD1/5/8, lo que reduce la condrogénesis dependiente de ALK2/SMAD y la diferenciación de osteocitos, lo que resulta en una reducción de la formación de hueso endocondral.
Eficacia clínica: Un estudio de un solo grupo en 97 sujetos con FOP con mutación R206H de 4 años o más. El criterio de valoración principal de eficacia fue el volumen anualizado de nueva osificación heterotópica (HO), evaluado mediante imágenes por TC de cuerpo entero de dosis baja (excluyendo la cabeza). La nueva HO media anualizada fue de 9,4 cm3/año en los sujetos que recibieron el tratamiento y de 20,3 cm3/año en los sujetos no tratados.
Eventos adversos: Los más comunes en uso crónico son piel seca (61%), labio seco (47%), artralgia (36%), prurito (34%), dolor en extremidades (29%), erupción (28%), alopecia (24%), eritema (19%), dolor de cabeza (19%), dolor de espalda (17%), exfoliación de la piel (15%), náuseas (15% ), dolor musculoesquelético (14%), mialgia (12%), ojo seco (10%), hipersensibilidad (10%), edema periférico (9%) y fatiga (5%). Se produjeron reacciones adversas graves en el 15% en la población de 8/10 años o más, siendo la reacción adversa grave más común el cierre epifisario prematuro; se produjeron reacciones adversas que condujeron a la interrupción permanente en el 8%, siendo la piel seca la causa más común (1%).
(N) SISTEMA NERVIOSO
Zuranolona (Zurzuvae®) Sage (FDA; Food & Drug Administration, USA)
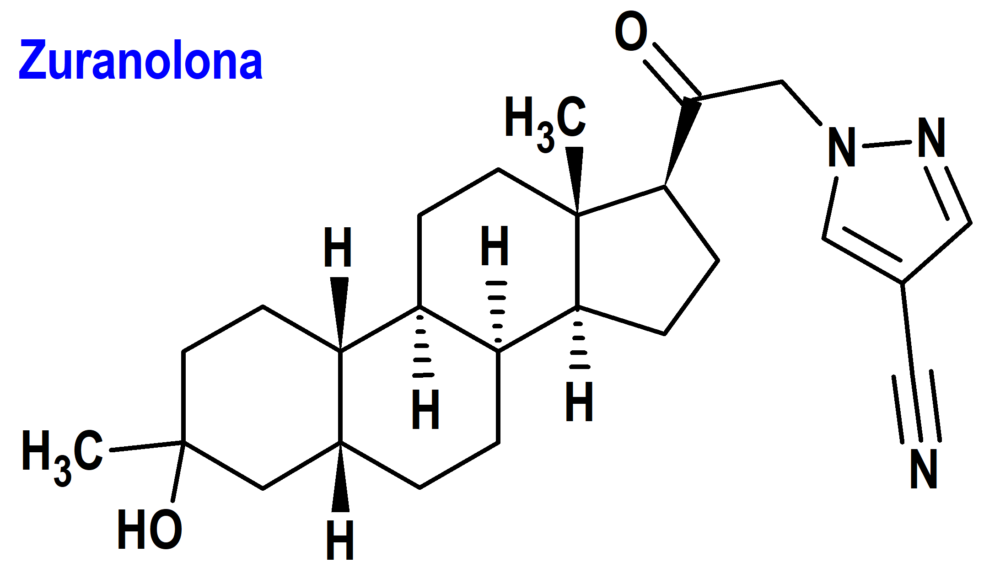
Indicación: Tratamiento de depresión posparto.
Tipo: Medicamento sintético estándar derivado de un núcleo esteroídico. Autorizado en Estados Unidos (FDA) el 4-4-2023 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Modulación alostérica de los receptores GABA (A).
Eficacia clínica: Dos estudios aleatorizados, controlados con placebo, doble ciego y multicéntricos, en mujeres con depresión posparto que cumplieron con los criterios del Manual Diagnóstico y Estadístico de Trastornos Mentales para un episodio depresivo mayor, con inicio de síntomas en el tercer trimestre o dentro de las 4 semanas posteriores al parto. El criterio principal de valoración para ambos estudios fue el cambio desde el inicio en los síntomas depresivos con relación al placebo, medido por la puntuación total de la escala de depresión de Hamilton de 17 ítems (HAMD-17) en el día 15. La diferencia con el placebo fue de 4,2 y 4,0 puntos en los estudios.
Eventos adversos: Los más comunes son somnolencia (36%), mareos (13%), diarrea (6%), fatiga (5%), infección del tracto urinario (5%), deterioro de la memoria (3%) y dolor abdominal (3%).
Atogepant (Aquipta®) AbbVie (EMA; European Medicines Agency)
Indicación: Profilaxis de la migraña en adultos que tengan al menos 4 días de migraña al mes.
Tipo: Medicamento sintético estándar derivado de un núcleo de espiropirrolpiridina-ciclopentapiridina. Autorizado por la Unión Europea (EMA) el 11-8-2023 con revisión prioritaria (Priority Medicines; PRIME); autorizado previamente en Estados Unidos (FDA) el 28-9-2021.
Mecanismo: Atogepant muestra afinidad, como inhibidor, hacia varios receptores de la familia de los receptores de la calcitonina/CGRP, que se considera que están implicados en la fisiopatología de la migraña. Sin embargo, aún no se ha establecido el mecanismo de acción preciso de atogepant en la profilaxis de la migraña.
Eficacia clínica: Un estudio aleatorizado, multicéntrico, doble ciego y controlado con placebo sobre 658 pacientes con migraña episódica (4 -14 días con migraña al mes), durante 12 semanas. La variable primaria de eficacia fue el cambio desde el inicio en la media de días con migraña al mes durante el periodo de tratamiento: -4,1 vs. -2,5.
Eventos adversos: Los más comunes son náuseas (9%), estreñimiento (8%) y fatiga/somnolencia (5%). La mayoría de las reacciones son de intensidad leve o moderada.
(R) SISTEMA RESPIRATORIO
Gefapixant (Lyfnua®) Merck Sharp Dohme (EMA; European Medicines Agency)
Indicación: Tratamiento de la tos crónica refractaria o idiopática.
Tipo: Medicamento sintético estándar derivado de un núcleo de diaminopirimidina; no autorizado previamente en Estados Unidos.
Mecanismo: Antagonista selectivo del receptor P2X3. Gefapixant también tiene actividad frente al subtipo de receptor P2X2/3. Los receptores P2X3 son canales de iones regulados por el ATP que se encuentran en las fibras C sensitivas del nervio vago en las vías respiratorias, que se activan en respuesta a la inflamación o a irritantes químicos. El ATP se libera de las células de la mucosa de las vías respiratorias en condiciones de inflamación. La unión del ATP extracelular a los receptores P2X3 se detecta como una señal de daño por parte de las fibras C. La activación de las fibras C, que el paciente siente como una necesidad de toser, inicia un reflejo de tos. El bloqueo de la señal que el ATP produce a través de los receptores P2X3 reduce la activación excesiva de los nervios sensitivos y la tos excesiva inducida por el ATP extracelular.
Eficacia clínica: Dos estudios de fase 3 de 52 semanas, multicéntricos, aleatorizados, doble ciego, controlados con placebo, en adultos con tos crónica refractaria (asociada a una enfermedad comórbida, tal como asma, enfermedad por reflujo gastroesofágico o síndrome de tos de las vías respiratorias superiores) o idiopática (no asociada a ninguna enfermedad comórbida). La variable primaria de eficacia fue la reducción de la frecuencia de la tos durante 24 horas (toses por hora): 61-63% (gefapixant) vs. 55-57% (placebo).
Eventos adversos: Los más comunes (>10%) son trastornos del gusto (disgeusia, ageusia, hipogeusia). La mayoría fueron de intensidad leve (65%) a moderada (32%).
(S) ÓRGANOS SENSORIALES
Avacincaptad Pegol (Izervay®) Iveric Bio (FDA; Food & Drug Administration, USA)
Indicación: Tratamiento de la atrofia geográfica (AG) secundaria a degeneración macular asociada con la edad (DMAE).
Tipo: Medicamento sintético biológico constituido por un aptámero de ácido ribonucleico (ARN), un oligonucleótido de 39 bases, que está unido covalentemente a una molécula ramificada de polietilenglicol (PEG, Pegol) de aproximadamente 970 unidades de etilenglicol. Autorizado en Estados Unidos (FDA) el 4-8-2023 mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Avacincaptad pegol es un aptámero de ARN, que se une e inhibe la proteína del complemento C5. evitando su escisión en C5a y C5b y disminuyendo así la formación del complejo de ataque a la membrana (MAC), el cual actúa como esquirlas y penetra las membranas de las células epiteliales del pigmento retiniano o fotorreceptor, destruyéndolas y provocando la atrofia regional de dicho epitelio.
Eficacia clínica: Dos estudios aleatorizados, multicéntricos, doble ciego, con control simulado, de 18 y 12 meses de duración en pacientes con AG por DMAE, con edades comprendidas entre 51 y 97 años con una media de 77 años; en total, 292 pacientes fueron tratados con avacincaptad pegol 2 mg y 332 pacientes recibieron tratamiento simulado. En ambos, se evaluó la tasa media de crecimiento de la AG (pendiente) desde el inicio hasta el mes 12, medido por fluorescencia automática (FAF) en 3 puntos temporales: el inicio, el mes 6 y el mes 12 (y en el mes 18 en uno de los estudios). La diferencia de la tasa de crecimiento de la atrofia geográfica (AG) a los 12 meses registrada con avacincaptad pegol con relación a la terapia simulada fue de 0,38-0,67 mm(2), lo que supone un 18-35% menos. Los pacientes que desarrollaron neovascularización coroidea fueron tratados concomitantemente con terapia anti-VEGF.
Eventos adversos: Los más comunes son hemorragia conjuntival (13%), aumento de la presión intraocular – PIO (9%), visión borrosa (8%), neovascularización coroidea (7%), dolor ocular (4%), moscas volantes (2%), blefaritis (2%).
(V) VARIOS
Piflufolastat 18F (Pyclari®) Curium PET (EMA; European Medicines Agency)

Indicación: Detección de lesiones que expresan el antígeno prostático específico de membrana (PSMA) mediante tomografía por emisión de positrones (PET) en adultos con cáncer de próstata (CaP) en estadificación primaria de pacientes con CaP de alto riesgo antes del tratamiento curativo inicial y en localización de recurrencias de CaP en pacientes con sospecha de recidiva basada en el aumento de los niveles séricos de antígeno prostático específico (PSA) después del tratamiento primario con intención curativa.
Tipo: Medicamento sintético estándar derivado del ácido pentanodioico, conteniendo un radionúclido constituido por un átomo de flúor-18 (F 18); autorizado por la Unión Europea (EMA) el 24-7-2023; autorizado previamente en Estados Unidos (FDA) el 26-5-2021.
Mecanismo: El antígeno prostático específico de membrana (PSMA) es una glucoproteína transmembrana que se expresa principalmente en el epitelio prostático humano normal a niveles bajos, pero que está sobreexpresada en tejidos tumorales, particularmente en células de cáncer de próstata, incluyendo metástasis. El flúor-18 (18F) es un radionúclido emisor de positrones que hace posible la tomografía por emisión de positrones (PET). El piflufolastato (18F) es un inhibidor selectivo del PSMA, marcado con flúor 18. Basándose en la intensidad de las señales, las imágenes PET obtenidas usando piflufolastato (18F) indican la presencia de tejidos que expresan PSMA.
Eficacia clínica: Tres ensayos clínicos multicéntricos, prospectivos y abiertos en varones con cáncer de próstata. En el primero (N=242). La sensibilidad fue del 28-39%, la especificidad del 95-98%, el valor predictivo positivo (VPP) del 72-81% y el valor predictivo negativo (VPN) del 81-84%). En el segundo estudio (N=208), el VPP fue del 85-87% y en el tercer estudio la sensibilidad fue del 58%.
Eventos adversos: Los más comunes son dolor de cabeza (1,4%), disgeusia (1,0%) y fatiga (0,5%).
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA) como la Food & Drug Administration (FDA) de Estados Unidos disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
