1. INTRODUCCIÓN
Antes de nada, se ha de justificar la elección del tema del discurso [La magia de la Farmacobiotecnología: De las magistrales a los virus recombinantes en terapia génica]. En la historia de las sociedades, en general, se produce una evolución progresiva, que va en paralelo precisamente a los avances de la ciencia y en concreto de la Medicina. Quizás nos podamos plantear sin miedo a equívoco, que la farmacología ha permitido que se viva más y mejor. Pero, además, nuestras propias vidas, a veces, siguen, como puede ser en este caso, un paralelismo con estos avances.
Partiendo de estas premisas, la elección del argumento de mi discurso se ha potenciado más si cabe, por la circunstancia en mi caso de tener un origen, un transcurso y unos fines íntimamente relacionados con los avances en la Biomedicina. Historia, enfermedad y vocación investigadora, serían las palabras clave definitorias.
Y… ¿por qué lo de “magia”? Estamos en una realidad en que la farmacología es extraordinariamente novedosa, y pareciera que nos movemos en el mundo de la magia y la sorpresa. Porque, los fines terapéuticos que se persiguen en el momento presente, eran hace décadas inimaginables, y menos a través de las técnicas, formas farmacéuticas y procedimientos actuales que son una combinación de la actividad multidisciplinar de muchos especialistas (1). En este camino que va desde las fórmulas magistrales, de práctica muchas veces empírica, hasta las nuevas terapias avanzadas, han nacido áreas que son la combinación de distintos saberes como así es la Farmacobiotecnología o Biotecnología Farmacéutica. Somos capaces de controlar, en parte, a la Biología en su “alma” más íntima que es el genoma y somos capaces de crear modelos animales con una determinada patología para estudiar nuevos fármacos. ¡Si nuestros antepasados levantaran la cabeza! ¿No pareciera que todo esto tiene algo de mágico? Curar modificando el genoma de nuestras células mediante terapia génica. Pareciera que se cerrara un círculo desde las prácticas mágicas de la Prehistoria hasta la “magia” de la biotecnología de nuestro siglo XXI.
2. EL PRINCIPIO DE LA HISTORIA
El principio de la historia podría comenzar y de hecho así fue, en una botica de las muy antiguas de finales del S. XIX (2) (como la de Fortunata y Jacinta) en que un farmacéutico, D. Gabriel Herrero Tejedor (el bisabuelo del que ahora habla) en un pueblo perdido de la antes Castilla La Vieja, Navas de Oro, preparaba fórmulas magistrales que dispensaba en forma de papelinas y de obleas. Estos momentos de finales del S. XIX y principios del S. XX fueron el núcleo de cristalización de los avances científicos y de la farmacología que después vendrían. Pero también lo fue aquel farmacéutico, en sus últimos años de su vida, como inductor de una vocación farmacéutica en el biznieto en cuestión que optó por seguir la tradición familiar de ser boticario, pero, además, docente universitario e investigador en Biotecnología.
El siglo XIX supuso un cambio, una transformación única y abismal en el medicamento y, por ende, para la profesión del boticario que pasaría a denominarse farmacéutico. Ese paso vino acompañado del avance de la química, de la enseñanza en universidades, del desarrollo de la botánica y de la obtención de los primeros principios activos (3).
Las formulaciones más o menos empíricas como fórmulas magistrales individuales dejaban atrás la célebre y conocida fórmula verbal “hágase según arte” utilizada en las reboticas, para dar paso a la especialidad farmacéutica industrial. El boticario trabajaba con los denominados productos simples y otros medicamentos compuestos que elaboraba con arte, ciencia y cierta dosis de misterio y magia. La Triaca Magna (4) (Figura 1), un anti venenos secreto, con un total de 60 ingredientes que se encontraba plasmada en las Concordias desde el S. XVI, dejaba de ser la estrella a finales de ese S. XIX. Aquellos tiempos lo eran de preparaciones sencillas como: mantecas, enjundias, médulas preparadas, los simples, infusiones…; y otras preparaciones como: aceites, jabones, aguas destiladas, gargarismos y lavativas, supositorios o calas…. A mediados de 1800 se incorporarían, además, formas farmacéuticas más novedosas como las cápsulas de gelatina, las grageas, los comprimidos o los inyectables.

Figura 1. Triaca Magna de los antiguos aprobada de los modernos y en justicia y conciencia defendida con autoridad, experiencia y razón, manifestado por D. Domingo Guillén, médico de cámara de su Majestad. Impresión de la Real Audiencia y del Santo Tribunal de la Inquisición, Año 1724.
Aquel farmacéutico (Figura 2), el de toda la vida, D. Gabriel Herrero Tejedor nacido en Aldea del Rey (hoy Aldea Real), provincia de Segovia, en 1873, se licenció en Farmacia por la Universidad Central de Madrid en 1894 y ejerció en la farmacia de calle Carretas 33 (ya desaparecida) y paradójicamente hoy establecimiento de cannabis; también en Villadiego (Burgos); Otero de Herreros y finalmente en Navas de Oro en Segovia desde 1917. Casi 30 años de farmacéutico en esta última localidad sanando a ricos y pobres (a los que regalaba los remedios). Recibió el reconocimiento de esta última localidad con una calle dedicada con su nombre a su labor farmacéutica y por el Colegio Oficial de Farmacéuticos de Segovia a sus 100 años de edad.

Figura 2. D. Gabriel Herrero Tejedor nacido en Aldea del Rey (Segovia) en 1873, licenciado en Farmacia por la Universidad Central de Madrid en 1894 y farmacéutico en Navas de Oro (Segovia) desde 1917.
3. HASTA AQUÍ LOS ORÍGENES
A partir de ahora, la historia de una enfermedad, de una vocación científica y la revolución biomédica. Para ello, es necesario concretar dentro del amplio espectro de la biomedicina. Nos vamos a centrar en un tipo de patologías que son las coagulopatías congénitas. Y nos encontramos ya en los albores del S. XX. Puestos así en contexto podemos decir, en general, que aquel siglo XX dio lugar a la primera revolución farmacéutica, aunque en sus primeras décadas las cosas iban algo despacio.
La hematología empezaba a florecer y ya se hablaba de personajes importantes en la materia (5). Estos son los momentos en que la hemofilia irrumpía en España, en la Casa Real Española (6) (Figura 3) a partir de la Reina Victoria de Inglaterra, portadora espontánea de la enfermedad, hemofilia B, por una mutación espermática en su padre Eduardo de Kent. Su hija Beatriz también portadora, introduce directamente la enfermedad a través de su hija Victoria Eugenia que desposa con Alfonso XIII Rey de España en ese momento. Dos hijos de este matrimonio, Alfonso y Gonzalo sufren la enfermedad.
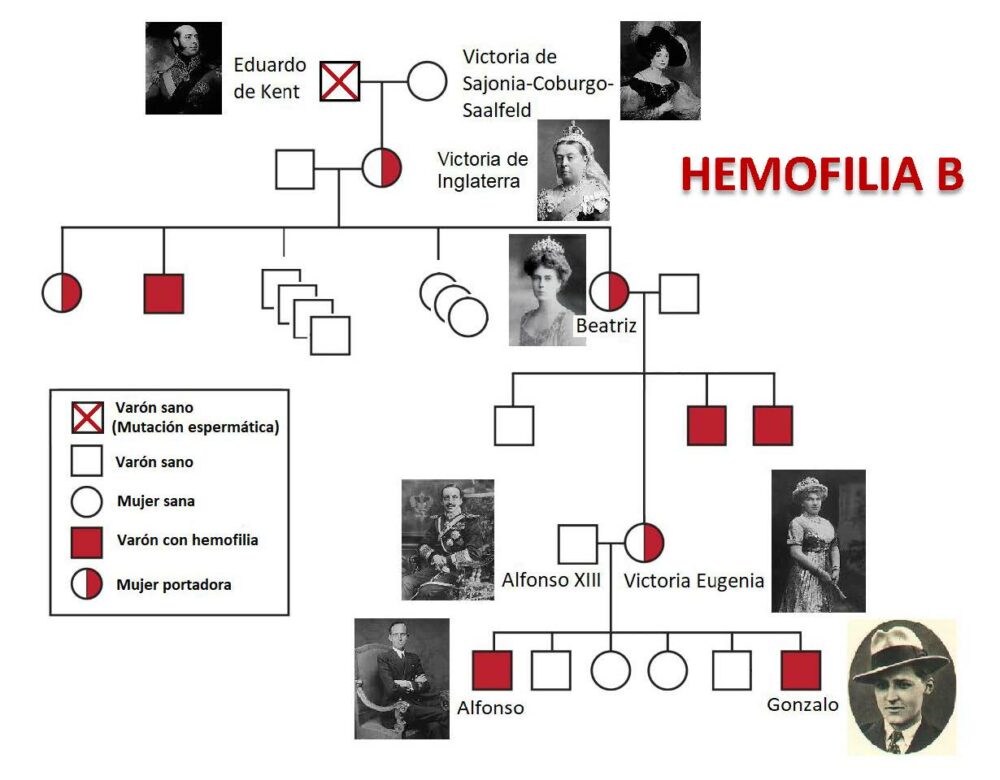
Figura 3. La hemofilia B en la Casa Real Española a partir de la Reina Victoria de Inglaterra, portadora espontánea de la enfermedad por una mutación espermática en su padre Eduardo de Kent. Su hija Beatriz también portadora, introduce directamente la enfermedad a través de su hija Victoria Eugenia que desposa con Alfonso XIII Rey de España. Dos hijos de este matrimonio, Alfonso y Gonzalo sufren la enfermedad.
Pero… ¿cuál era la situación médica en esos momentos entre 1900 y 1940? Con respecto a la hemofilia el desconocimiento era pleno, ni se conocían las causas y ni mucho menos el tratamiento. Un ilustre médico hematólogo italiano, Gustavo Pittaluga (7), fue nombrado médico personal de la Casa Real para hacerse cargo de la enfermedad. Los tratamientos: veneno de serpiente, clara de huevo, extracto de cacahuete, transfusiones de sangre, apósitos, inmovilización y aplicación de frio. Las consecuencias: mínima integración social, artropatía hemofílica invalidante y muerte a edad temprana. El Príncipe de Asturias, Alfonso, a los 31 años y el Infante Gonzalo a los 19.
Y ¿qué pasaba fuera de la Casa Real? Pues más o menos lo mismo, hemofilias por mutaciones espontáneas en muchas familias que padecerían la enfermedad. Vamos a tierras segovianas, por poner un ejemplo. Nuestro farmacéutico Gabriel se desposa con Mamerta en 1900; su primera hija Ana María nace en 1901; ésta desposa con Timoteo y su hija Carmen nace en 1930 (portadora de hemofilia en este caso hemofilia A por una mutación espontánea en los espermatozoides de su padre Timoteo), todo igual, pero sin ser Reina de Inglaterra, porque… las enfermedades no entienden de estirpes ni rangos. En 1955 nacería Antonio, aquí presente, con hemofilia A grave.
En la cronología temporal, un punto de inflexión se dio en 1937 con los estudios de Patek y Taylor (8) en donde se describía la fracción que llamaron globulina antihemofílica que, obtenida a partir de plasma humano de personas sanas, podía neutralizar los procesos hemorrágicos. Más tarde en 1946 Jessica Lewis y sus colaboradores (9) ensayaron los primeros derivados concentrados de proteínas de plasma para el tratamiento de los pacientes con trastornos hemorrágicos.
Y.. estamos ya sin darnos cuenta a mediados del S. XX en el inicio de la revolución farmacéutica (10) y de la medicina, una revolución en los diagnósticos, en la definición y clasificación de las enfermedades y además en la Farmacología. Se produce el paso de las drogas químicas y de origen vegetal a los fármacos que se denominan biológicos, o lo que es lo mismo aquellas moléculas grandes proteicas que se obtienen de un organismo, o se adecúan en un laboratorio, para que tengan efectos beneficiosos. Así, en la década de los 60´ se comenzaban a utilizar los llamados crioprecipitados de muy baja eficacia pero que solventaban en parte los problemas de sangrado.
Aun así, en aquellos años (y volvemos a Segovia como ejemplo) el diagnóstico de un niño hemofílico se solía realizar por los síntomas clínicos de sangrado a los pocos días desde el nacimiento hasta los dos años de edad con las primeras denticiones. Además, los crioprecipitados no tenían la suficiente potencia terapéutica como para evitar una artropatía hemofílica en los pacientes.
Pero la verdadera revolución en coagulopatías congénitas se dio en la segunda mitad del S. XX. Los genes codificantes de los factores de la coagulación se empezaron a clonar en especial el del factor VIII y el del factor IX cuya alteración da lugar a hemofilia A y B respectivamente. El diagnóstico molecular de las mutaciones comenzaba a dar sus primeros pasos y aparecían en los años 70´ los factores plasmáticos de pureza intermedia y después los de alta eficacia.
Mientras, en Segovia, 1973, D. Gabriel Herrero recibía el homenaje a toda una vida de farmacéutico por el Colegio Oficial de Farmacéuticos de Segovia. Un año más tarde casi coincidiendo con el final de su vida, 1974, su biznieto (cosas del azar o vete tu a saber), iniciaba en la Facultad de Farmacia de la Universidad Complutense de Madrid sus estudios de farmacéutico, por vocación y por consejo de su padre Antonio que pensaba en una profesión tranquila en la rebotica para su hijo con hemofilia (nada más lejos de la realidad que fue).
Los tiempos prometían. Aparecían noticias alentadoras sobre los nuevos factores plasmáticos, que padres y madres de pacientes hemofílicos buscaban y leían en la prensa. Ese mismo año de 1974 los pacientes aprendíamos la autoadministración intravenosa de aquellos factores plasmáticos de alta eficacia que nos devolverían la esperanza de vivir de otra forma, más y mejor.
Pero, cómo no siempre todo es de color de rosa, esos mismos factores plasmáticos que nos habían dado la esperanza de vivir, arrebataban la vida en los años 80´. La infección por el VIH (11) entró en la vida de los pacientes hemofílicos y casi el 80% de los pacientes no vivió para disfrutarlo.
Los años 90´ ofrecían un momento clave de revolución biomédica para las coagulopatías congénitas y para otras enfermedades, que fue el paso de los medicamentos biológicos a los biotecnológicos que son aquellos que, en base a los biológicos, se obtienen por técnicas generalmente de ingeniería genética. La era recombinante estaba servida (12-14) y los pacientes empezaban a disfrutar de medicamentos altamente eficaces y seguros. Los pacientes en su domicilio tenían su producto recombinante a su disposición para el autotratamiento.
Coincidiendo precisamente con esta revolución farmacológica, en las últimas décadas del S. XX, se dio un cambio en la forma de relacionarse los médicos y los enfermos desde un modelo paternalista a uno autonomista. Se empezó a introducir el concepto de tratar, no una enfermedad, sino a una persona que padecía su enfermedad. El sujeto tomaba un papel relevante en el arte de curar. Los aspectos sociales, psicológicos y personales tomaban peso en la práctica médica. Así, en 1973 la Asociación Americana de Hospitales aprobó la primera Carta de Derechos del Paciente (15), que suponía el reconocimiento oficial del derecho del enfermo a recibir una completa información sobre su situación clínica y a decidir entre las distintas opciones terapéuticas, como adulto autónomo y libre que era. Esto abrió las puertas al movimiento asociativo, y las asociaciones de pacientes fueron un hito en la historia de la medicina y la terapéutica.
Derivado de la posibilidad de diagnosticar un mayor número de enfermedades, aparece el concepto, a mediados de los años 80´, de enfermedad rara y ultra rara (16, 17) en base a la casuística de frecuencia en la incidencia (inferior a 1 de cada 2000 individuos o inferior a 1 de cada 50000). En coagulopatías congénitas hablamos de enfermedad rara, como, por ejemplo, la hemofilia A o la B y de ultra rara como el déficit de factor V. Esto que pareciera algo irrelevante a simple vista, se convierte en un inconveniente inesperado, como más adelante comentaré.
Pero cambiamos de siglo, al XXI, el que iba a ser el del fin del Mundo, aunque todavía estamos a tiempo. Nos seguimos sorprendiendo con los avances biomédicos, y si pensábamos que todo estaba ya conseguido o medio conseguido, surgen los medicamentos biotecnológicos mejorados de mayor vida media; surgen las llamadas nuevas terapias avanzadas (18), la terapia celular basada en células, la medicina regenerativa basada en trasplantes de órganos o tejidos y la terapia génica basada en corregir o sustituir genes alterados. Y si esto pareciera poco, surge la inmunoterapia (19), las terapias basadas en ARN mensajero y el CAR-T (20). En el diagnóstico, la biopsia líquida (21) por la que se pueden detectar y analizar distintas biomoléculas circulantes en fluidos biológicos como enzimas, proteínas, ácidos nucleicos o factores angiogénicos, representa una gran novedad ya que se trata de una técnica de alta especificidad y mínimamente invasiva porque se puede realizar en muestras, fundamentalmente en sangre, con aplicación en el diagnóstico prenatal y en el diagnóstico precoz del cáncer. La secuenciación masiva (22) de última generación por su parte complementa esta técnica y permite la secuenciación rápida y precisa del genoma.
4. ¿A DÓNDE NOS PUEDE LLEVAR TODO ESTO?
En general, seremos capaces, por ejemplo, de corregir un defecto hereditario en los genes; reproducir una enfermedad genética en un organismo; curar el cáncer mediante inmunoterapia y CAR-T; lograr, por ejemplo, que los niños con inmunodeficiencia combinada severa salgan de sus burbujas o que se devuelva el color de la vida a aquellas personas que padecen acromatopsia. También, que podamos controlar a largo plazo el sangrado en aquellos pacientes con coagulopatías congénitas mediante las terapias avanzadas, terapia celular o terapia génica.
El gran avance en el tratamiento de las coagulopatías congénitas y en concreto en hemofilia A y B ha sido la incursión de los fármacos biotecnológicos de origen recombinante en base a los plasmáticos. Presentan una larga vida media, gracias a la modificación en su estructura, por pegilación por ejemplo o por coexpresión con albúmina o inmunoglobulinas; son más eficaces y con menos efectos adversos; son el tratamiento óptimo y el de elección para hemofilia A y B; permiten una mejor inserción laboral y social; una alta calidad de vida; una muy buena adherencia y no tienen efectos secundarios ni riesgos de infección por patógenos. Además, presentan una muy baja inmunogenicidad ya que se obtienen y se modifican postraduccionalmente en células de origen humano.
Pero realmente la novedad farmacológica de este siglo XXI para la hemofilia ha sido el establecimiento de los nuevos protocolos de terapias avanzadas. Son procedimientos factibles tanto para enfermedades monogénicas como poligénicas y se justifican claramente en patologías graves o crónicas, cuando no existe un tratamiento o si lo hay este es tedioso, incómodo o con graves efectos secundarios adversos.
En hemofilia las células pluripotentes inducibles (iPSCs) son las células estrella para terapia celular, y en terapia génica, los vectores adenoasociados y lentivirales son los de mayor potencial clínico, en protocolos de terapia génica in vivo en que se inyecta en el torrente sanguíneo el propio vector que porta el gen terapéutico de factor VIII o de factor IX.
Así, el año pasado se aprobaron el Roctavian para hemofilia A (23) y el Hemgenix (24) para hemofilia B, basados ambos en vectores virales adenoasociados que se administran por vía intravenosa.
La pregunta clave es si mediante estos procedimientos se curará la hemofilia. La respuesta, hoy por hoy, es que no. No se corrige la causa endógena mutacional y en consecuencia se produce la expresión de la proteína durante unos pocos años y con fenotipos leves o moderados. Hay estudios muy interesantes, entre los que se encuentra la colaboración de nuestro grupo de investigación con la Dra. Follenzi de la Universidad de Piamonte en Novara (Italia) y la Dra. Mª José Sánchez de la Universidad Pablo de Olavide de Sevilla, en que intentamos corregir precisamente mediante técnicas de terapia celular y edición génica con CRISPR la mutación en hemofilia A en los estadios embrionario y fetal (25, 26).
¿Cuáles son los inconvenientes actuales de estos protocolos de terapia génica que hay que solventar? En primer lugar, la alta variabilidad individual en la respuesta. Esto hace que al existir ya un tratamiento recombinante actual muy eficaz y seguro para los pacientes con hemofilia, la terapia génica no representa, hoy por hoy, un tratamiento de primera línea. La segunda cuestión es que se alcanzan fenotipos moderados o leves (39 al 50% del valor de referencia). Tanto el decaimiento de la expresión de la proteína con el tiempo como los niveles alcanzados, se pueden explicar primero porque el virus no es integrativo de genoma, pero fundamentalmente por la alta inmunogenicidad que presenta la cápside de este tipo de virus adenoasociados que se deriva en una importante hepatotoxicidad con un aumento en las transaminasas. Esto obliga al uso concomitante de inmunosupresores, como son los corticoides, con sus bien conocidos efectos adversos. Y, por último, y no menos importante, es su coste que ronda los 3 millones de euros por inyección, lo que hace desconfiar de su eficiencia en la relación coste-beneficio, teniendo en cuenta que tampoco representan estos protocolos la cura de la enfermedad.
Las terapias avanzadas si funcionan bien serán un hito, pero atención porque vamos, hoy por hoy, por caminos empíricos en lo que respecta a sus efectos adversos y desconocemos muchas cosas, y los resultados pueden ser fatales, como así se notificó a finales del año pasado (27).
Otra desconfianza que nos puede surgir es la relativa a si estos procedimientos incrementarán aún más, si cabe, las brechas de inequidad que ya se dan en el momento presente en el Mundo con los tratamientos convencionales. Pensemos que, de los 400.000 pacientes con hemofilia en el Mundo, tan solo 100.000 se tratan adecuadamente. No se nos puede olvidar que la justicia y la equidad son pilares básicos para la bioética de la asistencia sanitaria.
En otro orden de cosas, otra cuestión puede ser si todas las enfermedades tienen la misma prioridad en sus tratamientos. Aquí inevitablemente tenemos que volver al concepto de enfermedad rara y ultra rara. No hace falta indagar demasiado para comprender que las prioridades de una enfermedad común, rara y ultra rara son obviamente muy diferentes. Si estudiamos, por ejemplo, la cadena de valor de traslación clínica entre la hemofilia A que es una enfermedad rara y el déficit de factor V que es una ultra rara (Figura 4), existen importantes diferencias por no decir que son dos mundos diferentes. En el caso de la hemofilia el proceso, desde los incipientes y empíricos procedimientos ya comentados, pasando por los medicamentos biológicos como el plasma y derivados, hasta llegar a los fármacos biotecnológicos y ahora a la terapia génica, ha seguido una evolución casi natural en base al avance de los conocimientos en biomedicina. Si nos fijamos en el déficit de factor V se puede decir que no ha habido ningún avance en su tratamiento que sigue siendo a base de plasma fresco congelado, como en aquel niño segoviano de 1960.
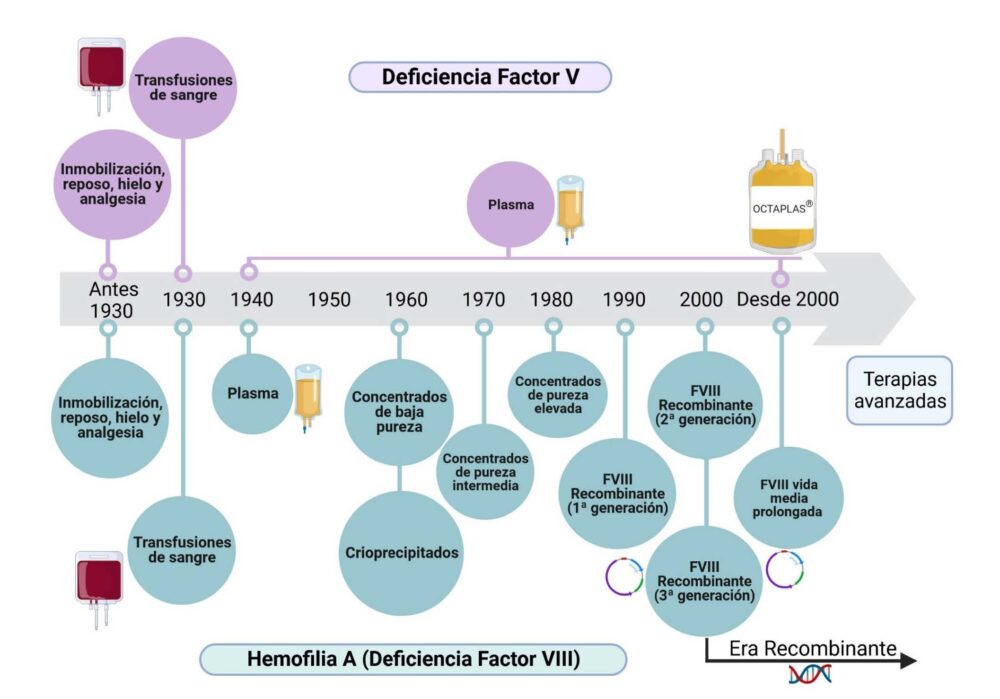
Figura 4. Cadena de valor de traslación clínica en hemofilia A y en el déficit de factor V, desde los incipientes y empíricos procedimientos de tratamiento, pasando por los medicamentos biológicos como el plasma y derivados, hasta llegar a los fármacos biotecnológicos (recombinantes) y la terapia génica.
Pero no es que no se haya investigado sobre factor V, más bien todo lo contrario. En los años 70´y 80´se purificó y caracterizó; en 2014 se obtuvo el primer factor V recombinante (28); en 2016 se estudió su estabilidad, seguridad farmacológica y sus propiedades farmacocinéticas (29), y en 2022, antes de ayer, se estableció su indicación no solo para el déficit de factor V sino también para otras situaciones como la coagulopatía traumática aguda (30). Pero… todavía no hay un factor V recombinante comercializado. Las razones económicas, en base a la mayor o menor incidencia de una enfermedad ¿pueden condicionar las prioridades?
5. NUESTRO GRUPO DE INVESTIGACIÓN: APORTACIONES AL CONOCIMIENTO
5.1. Los comienzos, circunstancias y desafíos
Hasta aquí la historia, ahora toca “arremangarnos” para contribuir a la generación del conocimiento y al desarrollo científico. Hace ahora 10 años, en 2013, nuestro grupo de terapias avanzadas: génica y celular del Campus de Excelencia Internacional de la Universidad Complutense de Madrid, para el tratamiento de las coagulopatías congénitas, se encontraba desarrollando un ambicioso proyecto sobre terapia génica no viral para la hemofilia B (31). Nuestra metodología era muy novedosa y, sobre todo, muy segura para los pacientes, ya que utilizábamos vectores no virales, como la nucleofección, para introducir el gen codificante de factor IX de la coagulación en células madre derivadas de tejido adiposo. Estas células se aislaban a partir de liposucciones y en gradientes de densidad y se caracterizaban por inmunocitometría de flujo y por diferenciación celular: adipogénica, osteogénica y condrogénica. Aunque los niveles de expresión eran muy bajos, estos estudios representaban el inicio de un protocolo seguro de terapia génica no viral. Pero la falta de financiación en ese momento, que nos impedía competir con grandes grupos de investigación en terapia génica viral para hemofilia B, hizo que el proyecto no pudiera continuar y se tuviera que cambiar el rumbo de las investigaciones.
Los momentos de cambio, en general, se producen por distintas circunstancias. En nuestro caso fueron las descritas de vía muerta, pero también se dieron otras que tienen que ver, precisamente, con las enfermedades ultra raras.
En 2013, en Jaén, Mª José y Manolo, aquí presentes, padres de Celia que presenta un déficit grave de factor V de la coagulación, buscaban ayuda y acudieron a la Federación Mundial y a la Española de Hemofilia para solicitar información sobre qué grupos investigaban en esa patología. No se encontraron grupos. Se afilian entonces a la Asociación Andaluza de Hemofilia, cuya presidenta en esos momentos, Lola Camero (aquí presente), se interesa por el tema y se convierte en la impulsora del proyecto. Contacta con el entonces presidente de la Comisión Científica de la Real Fundación Victoria Eugenia que era el que ahora habla, e iniciamos la búsqueda de información sobre investigaciones en factor V y tan solo un grupo italiano y otro de La Jolla hacían algo sobre ello. En 2014 se toma una decisión relevante de reorientación del grupo de investigación hacia factor V. En 2015, se organiza el primer evento de crowdfunding para el proyecto de factor V: RETO “Por la investigación en el déficit de factor V, con V de Victoria”, y en noviembre de ese mismo año se registra el proyecto en Orphanet. 2016, se consigue para Celia, como medicamento extranjero, el Octaplas® (32), que permite una mejor dosificación de los factores de la coagulación. 2017, se obtiene la primera ayuda de financiación por una empresa farmacéutica, y el que ahora os narra, después de 30 años en la investigación, y por primera vez, ponía cara y la máxima relevancia a su labor investigadora.
Pero aparte de las circunstancias que dieron lugar a este ambicioso proyecto, nos encontrábamos frente a muchos desafíos. Era un proyecto de gran envergadura en el que partíamos de cero (porque no había nada descrito) e íbamos hacia lo desconocido. Había que afrontar la muy alta responsabilidad que suponía, y se necesitaba para ello una colaboración multidisciplinar para abordarlo. A favor teníamos el apoyo y la esperanza de la sociedad que era de forma directa, de la propia paciente, de sus familiares, y de otras personas que creían en la causa. Todo esto canalizado a través de la creación de una muy joven asociación, la Asociación para la Investigación y Cura del Déficit de Factor V, Una Esperanza para Celia (Figura 5), cuya presidenta es la propia madre de Celia.

Figura 5. Asociación para la Investigación y Cura del Déficit de Factor V. Una Esperanza para Celia, que financia el proyecto de investigación sobre terapias avanzadas para el déficit de factor V. Disponible en: https://unaesperanzaparacelia.org/.
5.2. La consolidación y los apoyos. Objetivos
Nuestro grupo se fue consolidando con la colaboración de muchos investigadores y centros de investigación que apostaban por el proyecto porque…, las enfermedades olvidadas también debían ser investigadas.
Así, el objetivo general de nuestro proyecto es encontrar un tratamiento para el déficit de factor V mediante terapias avanzadas, terapia celular y terapia génica. Los objetivos específicos han sido y son la producción de factor V en hepatocitos obtenidos a partir de células madre mesenquimales de decidua de placenta humana; el estudio mutacional completo de la paciente y de sus progenitores, y la obtención de un modelo animal patológico en ratón y de un modelo celular, ambos deficitarios en factor V. Con la consiguiente estandarización de la medida de los niveles de factor V en ratón. Todos estos objetivos se han cumplido.
Son tres todavía las pruebas de inicio (de concepto) que nos hemos planteado: una la corrección de la mutación en el modelo celular in vitro mediante edición génica con CRISPR que ha sido exitosa, y mediante vectores lentivirales en que se han obtenido resultados positivos preliminares. Tenemos pendientes las otras dos, una de terapia celular mediante microtrasplante de hepatocitos funcionales mediante andamiajes o scaffolds que se encuentra en progreso, y la segunda de terapia génica mediante vectores lentivirales, con resultados preliminares positivos, ambas en el modelo patológico animal in vivo. Recientemente (ahora mismo se encuentra Juan Andrés de Pablo en Estados Unidos) hemos iniciado una colaboración con el grupo de la Dra. Drygalski del Centro de Tratamiento de Hemofilia y Trombosis de la Universidad de California en San Diego, con el objeto de ensayar su factor V recombinante en nuestro modelo patológico de ratón, mediante perfusión intravenosa.
5.3. La difusión de los resultados
Los resultados obtenidos hasta ahora por el grupo de investigación han dado lugar a distintas publicaciones, todas ellas en revistas de alto impacto y Q1.
Respecto a los estudios de terapia celular, se publicaron en la revista Biomedicine & Pharmacotherapy (33), donde se describió la obtención de células madre mesenquimales a partir de decidua de placenta humana y su diferenciación a hepatocitos que expresaban factor V.
Los estudios genéticos y hematológicos de la paciente y de sus progenitores se publicaron en la revista International Journal of Molecular Sciences (34), donde se describieron las mutaciones y su segregación, referenciándose, además, una nueva mutación en factor V, en el padre, que nunca se había descrito, la c.3279G.
La estandarización de los niveles de factor V, tiempo de protrombina y de tromboplastina parcial activada en ratón, que hasta ese momento no estaba descrita, se publicó en la prestigiosa revista Frontiers in Veterinary Science (35).
La obtención del modelo celular deficitario en factor V mediante CRISPR, se publicó también en la revista International Journal of Molecular Sciences (36) donde se describió la producción del modelo knock-out y la propia corrección de la mutación (que emulaba la misma que presenta nuestra paciente) mediante la misma técnica de edición génica. Se obtuvo, además, la patente del método de recuperación de la mutación mediante edición génica (37).
Respecto al modelo patológico animal en ratón deficitario en factor V, en este caso knock-in, obtenido también mediante CRISPR y técnicas de reproducción asistida in vitro, se ha obtenido la patente (38) y la publicación, enviada a la revista Thrombosis Research, se encuentra actualmente en revisión editorial.
6. CONCLUSIONES
Para finalizar, hay que decir que estamos, en general y también en nuestro grupo de investigación en particular, en un momento de euforia, gracias a las nuevas terapias avanzadas que permitirán una farmacología individualizada y personalizada.
Llegaremos a curar algunas enfermedades; a paliar otras, que no curar, durante un largo periodo de tiempo, que pudiera ser toda una vida para algunos pacientes; y en definitiva incrementaremos la esperanza y calidad de vida de las personas.
Todo ello gracias a la “magia” de la Farmacobiotecnología, porque somos capaces de actuar sobre el genoma y sobre los mecanismos más íntimos de la etiopatogenia.
Esperemos que la “magia” consiga también superar ese gran escollo que es la óptima traslación clínica desde los modelos animales al humano, y ya puestos a pedir, a que no veamos noticias como las que a veces se leen en la prensa (39).
Agradecimientos
Excelentísimos señores presidente, vicepresidente y secretario de la Real Academia Nacional de Farmacia, Excelentísimas señoras y Excelentísimos señores Académicos, señoras y señores, y otros más jóvenes y muy jóvenes. El inicio de mi disertación debe ser de profundo agradecimiento y admiración hacia la Real Academia Nacional de Farmacia y a quienes han depositado en mí su confianza al haberme acogido como Académico Correspondiente electo de esta noble corporación. Es un altísimo honor para mí. Mi especial gratitud a la Excma. Sra. Dña. María Molina Martín por haber accedido gustosamente a mi presentación en este acto.
Quiero recordar, hoy y aquí, a mis maestros de aquellos inolvidables años de promoción 1974-79 en la Facultad de Farmacia de la Universidad Complutense de Madrid, profesores todos ellos de una muy alta categoría humana y académica, y, en especial, a aquel profesor de Bioquímica, el Dr. Ángel Jiménez Solves, a veces exigente, pero que me inculcó la vocación por la Bioquímica y el poder de la fuerza de voluntad, y los que lo conocieron entenderán por qué digo esto. También a aquellos maestros en el “arte” de la Bioquímica y Biología Molecular de los que me nutrí de su sabiduría, en los primeros momentos de mi carrera investigadora predoctoral en la Universidad Autónoma de Madrid. El Dr. Alberto Sols y la Dra. Gertrudis de la Fuente, ya fallecidos, que lograron en mí el “enamoramiento” por hacer ciencia, por la enzimología, por la patología molecular, por la bioquímica y la biología molecular. De mi etapa postdoctoral en el Instituto Neurológico “Carlo Besta” de Milán, guardo recuerdo muy especial y nostálgico. Allí, en la maravillosa Milán de la Italia, el Dr. Stefano DiDonato, también fallecido hace unos años, y el Dr. Gaetano Finocchiaro, me abrieron los ojos a nuevas perspectivas y mundos en la investigación biomédica, y siempre reconocieron mis estudios y logros allí en tierras lejanas.
También, hoy y aquí, recordar y agradecer a esas otras personas que de forma indirecta han hecho posible esta extraordinaria circunstancia del día de hoy, mis padres Carmen y Antonio, sin los cuales no estaría aquí; al equipo médico de la Unidad de Hemofilia y al Servicio de Farmacia del Hospital Universitario La Paz de Madrid, sin los cuales tampoco estaría hoy aquí; a D. Antonio Camero y a Dña. Carmen Melero que, desde su extensa experiencia de vida y su interés por la cultura, siempre han creído en mi actividad profesional y siempre la han valorado como los que más; a la familia de Celia, nuestra paciente en estudio que padece deficiencia severa de factor V de la coagulación, que ha dejado en nuestras manos, con confianza plena, el llegar a una futura terapia avanzada para su enfermedad y que ha dado sentido y una nueva perspectiva a nuestra labor investigadora, y también, por qué no decirlo, a una muy alta responsabilidad; a nuestros hijos que son el relevo generacional, en base a lo que mejor o peor les hemos inculcado, y, en fin, y en especial, a Lola, mi mujer y compañera, aquí presente, que ha sido mi “motor” durante los últimos años, en momentos duros y especiales, y que revitalizó mi persona y mi actividad profesional. Desde el punto de vista profesional e investigador, agradecer a la Real Fundación Victoria Eugenia por concedernos dos premios de investigación que sirvieron de estímulo para proseguir con el proyecto en terapias avanzadas para el tratamiento de coagulopatías congénitas, y también como no, a aquellos investigadores que han colaborado y colaboran en nuestro proyecto de investigación, los Dres. Damián García-Olmo y Mariano García-Arranz de la Fundación Jiménez Díaz; Ana Isabel Flores del Instituto de Investigación 12 de Octubre; Sara Bernal del Hospital Sant Pau; José Carlos Segovia y Luis Javier Serrano del Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas; Pablo Bermejo del Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria; Luis Revuelta de la Facultad de Veterinaria de la Universidad Complutense de Madrid, y a los incansables predoctorales Juan Andrés de Pablo y Andrea Miguel, de la Facultad de Ciencias Biológicas de la Universidad Complutense de Madrid.
7. REFERENCIAS
- Pérez-Porto J, Gardey A. (2022) Multidisciplinariedad. Disponible en: https://definicion.de/multidisciplinariedad/.
- Muñoz-González MR. (2021). Momentos estelares de la historia de la farmacia durante el siglo XIX. (Trabajo Fin de Grado Inédito). Universidad de Sevilla, Sevilla. Disponible en: https://idus.us.es/bitstream/handle/11441/133052/MU%c3%91OZ%20GONZALEZ%20MARIA%20ROSA.pdf?sequence=1&isAllowed=y.
- Carmel Ferragud (2021). El nacimiento del medicamento industrial. Disponible en: https://sabersenaccio.iec.cat/es/el-nacimiento-del-medicamento-industrial/.
- Francisco J Puerto Sarmiento (2009). La Triaca Magna. Disponible en: https://ranf.com/wp-content/uploads/academicos/discursos/numero/puerto.pdf.
- Burgaleta-Alonso de Ozalla C. La especialidad de Hematología-Hemoterapia: Antecedentes. Desarrollo asistencial y científico y Perspectivas futuras. Rev Inv Edu Ciencias Salud (RIECS) 2023; 8: 1; doi: https://doi.org/10.37536/RIECS.2023.8.1.366.
- Ojeda-Thies C, Rodriguez-Merchan EC. Historical and political implications of haemophilia in the Spanish royal family. Haemophilia 2003; 9(2): 153-6; doi: https://doi.org/10.1046/j.1365-2516.2003.00732.x.
- Tomás F, Tamaro E. Biografia de Gustavo Pittaluga Fattorini. In: Biografías y Vidas. La enciclopedia biográfica en línea 2004. Disponible en: https://www.biografiasyvidas.com/biografia/p/pittaluga_fattorini.htm.
- Patek AJ Jr., Taylor FHL. Hemophilia. II. Some Properties of a Substance Obtained from Normal Human Plasma Effective in Accelerating the Coagulation of Hemophilic Blood. J Clin Invest 1937; 16(1): 113-24; doi: https://doi.org/10.1172/JCI100829.
- Lewis JH, Soulier JP, Taylor FHL. Chemical, Clinical and Immunological Studies on the Products of Human Plasma Fractionation. XXXIII. The Coagulation Defect in Hemophilia: The Effect in Vitro and in Vivo on the Coagulation Time in Hemophilia of a Prothrombin and Fibrinogen-Free Normal Plasma and its Derived Protein Fractions. J Clin Invest 1946; 25(6): 876-9; doi: https://doi.org/10.1172/JCI101775.
- Zhang YHP, Sun J, Ma Y. Biomanufacturing: history and perspective. J Ind Microbiol Biotechnol 2017; 44(4-5): 773-84; doi: https://doi.org/10.1007/s10295-016-1863-2.
- Levine PH. The acquired immunodeficiency syndrome in persons with hemophilia. Ann Intern Med 1985; 103(5): 723-6; doi: https://doi.org/10.7326/0003-4819-103-5-723.
- Stryjewska A, Kiepura K, Librowski T, Lochyński S. Biotechnology and genetic engineering in the new drug development. Part I. DNA technology and recombinant proteins. Pharmacol Rep 2013; 65(5): 1075-85; doi: https:// https://doi.org/10.1016/s1734-1140(13)71466-x.
- Adrio JL, Demain AL. Recombinant organisms for production of industrial products. Bioeng Bugs 2010; 1(2): 116-31; doi: https://doi.org/10.4161/bbug.1.2.10484.
- Ropers HH, Wieringa B. The recombinant DNA revolution: implications for diagnosis and prevention of inherited disease. Eur J Obstet Gynecol Reprod Biol 1989; 32(1): 15-23; doi: https://doi.org/10.1016/0028-2243(89)90119-6.
- Declaración de los Derechos del Paciente Asociación Norteamericana de Hospitales; 6 de febrero de 1973. Asociación Americana de Hospitales. Disponible en: https://www.codem.es/Adjuntos/CODEM/Documentos/Informaciones/Publico/f044efef-58a7-40e4-bb7b-91605df12553/B4694157-ADF7-4DD2-A221-229B18C518C7/18fb7dc6-1d99-4c69-a0f9-7d09b99f12de/18fb7dc6-1d99-4c69-a0f9-7d09b99f12de.pdf.
- Smith CIE, Bergman P, Hagey DW. Estimating the number of diseases – the concept of rare, ultra-rare, and hyper-rare. iScience 2022; 25(8): 104698; doi: https://doi.org/10.1016/j.isci.2022.104698.
- Klimova B, Storek M, Valis M, Kuca K. Global View on Rare Diseases: A Mini Review. Curr Med Chem 2017; 24(29): 3153-8; doi: https://doi.org/10.2174/0929867324666170511111803.
- Liras A. Biological therapies for inherited diseases: social and bioethical considerations. Hemophilia as an example. Expert Opin Biol Ther 2015; 15(5): 713-22; doi: https://doi.org/10.1517/14712598.2015.102945.
- Bergman PJ. Cancer Immunotherapy. Vet Clin North Am Small Anim Pract 2024; 54(3): 441-68; doi: https://doi.org/10.1016/j.cvsm.2023.12.002.
- Mitra A, Barua A, Huang L, et al. From bench to bedside: the history and progress of CAR T cell therapy. Front Immunol 2023; 14: 1188049; doi: https://doi.org/10.3389/fimmu.2023.1188049.
- Nikanjam M, Kato S, Kurzrock R. Liquid biopsy: current technology and clinical applications. J Hematol Oncol 2022; 15(1): 131; doi: https://doi.org/10.1186/s13045-022-01351-y.
- Hu T, Chitnis N, Monos D, Dinh A. Next-generation sequencing technologies: An overview. Hum Immunol 2021; 82(11): 801-11; doi: https://doi.org/10.1016/j.humimm.2021.02.012
- Blair HA. Valoctocogene Roxaparvovec: First Approval. Drugs 2022; 82(14): 1505-1510; doi: https://doi.org/10.1007/s40265-022-01788-y.
- Heo YA. Etranacogene Dezaparvovec: First Approval. Drugs 2023; 83(4): 347-352; doi: https://doi.org/10.1007/s40265-023-01845-0.
- Merlin S, Akula S, Cottonaro A, et al. Therapeutic potential of fetal liver cell transplantation in hemophilia A mice. Haematologica 2023; 108(6): 1544-54; doi: https://doi.org/10.3324/haematol.2022.282001.
- Serrano LJ, Cañete A, Garcia-Leal T, et al. Searching for a Cell-Based Therapeutic Tool for Haemophilia A within the Embryonic/Foetal Liver and the Aorta-Gonads-Mesonephros Region. Thromb Haemost 2018; 118(8): 1370-81; doi: https://doi.org/10.1055/s-0038-1661351.
- Lek A, Wong B, Allison Keeler A, et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. N Engl J Med 2023; 389(13): 1203-10; doi: https://doi.org/10.1056/NEJMoa2307798.
- von Drygalski A, Bhat V, Gale AJ, et al. An engineered factor Va prevents bleeding induced by anticoagulant wt activated protein C. PLoS One 2014; 9(8): e104304; doi: https://doi.org/10.1371/journal.pone.0104304.
- Gale AJ, Bhat V, Pellequer JL, et al. Safety, Stability and Pharmacokinetic Properties of (super)Factor Va, a Novel Engineered Coagulation Factor V for Treatment of Severe Bleeding. Pharm Res 2016; 33(6): 1517-26; doi: https://doi.org/10.1007/s11095-016-1895-3.
- Joseph BC, Miyazawa BY, Esmon CT, et al. An engineered activated factor V for the prevention and treatment of acute traumatic coagulopathy and bleeding in mice. Blood Adv 2022; 6(3): 959-69; doi: https://doi.org/10.1182/bloodadvances.2021005257.
- Olmedillas S, Garcia-Arranz M, Garcia-Olmo D, Liras A. Preliminary study on non-viral transfection of F9 (factor IX) gene by nucleofection in human adipose-derived mesenchymal stem cells. PeerJ 2016: 4: e1907; doi: https://doi.org/10.7717/peerj.1907.
- Biesert L, Suhartono H. Solvent/detergent treatment of human plasma—a very robust method for virus inactivation. Validated virus safety of OCTAPLAS. Vox Sang 1998; 74 (Suppl 1): 207-12; doi: https://doi.org/10.1111/j.1423-0410.1998.tb05474.x.
- Serrano LJ, de la Torre P, Liras A, Flores AI. Cell therapy for factor V deficiency: An approach based on human decidua mesenchymal stem cells. Biomed Pharmacother 2021; 142: 112059; doi: https://doi.org/10.1016/j.biopha.2021.112059.
- Bernal S, Pelaez I, Alias L, et al. High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. Int J Mol Sci 2021; 22(18): 9705; doi: https://doi.org/10.3390/ijms22189705.
- De Pablo-Moreno JA, Liras A, Revuelta L. Standardization of Coagulation Factor V Reference Intervals, Prothrombin Time, and Activated Partial Thromboplastin Time in Mice for Use in Factor V Deficiency Pathological Models. Front Vet Sci 2022; 9: 846216; doi: https://doi.org/10.3389/fvets.2022.846216.
- Serrano LJ, Garcia-Arranz M, De Pablo-Moreno JA, et al. Development and Characterization of a Factor V-Deficient CRISPR Cell Model for the Correction of Mutations. Int J Mol Sci 2022; 23(10): 5802; doi: https://doi.org/10.3390/ijms23105802.
- Patente “Método in vitro para recuperar la expresión del gen F5 que codifica el factor V de la coagulación”. Ref. ES2785323B2. Titulares: Universidad Complutense de Madrid y Fundació Institut de Recerca de L’hospital de La Santa Creu I Sant Pau. Inventores: Liras Martín, Antonio; Serrano Ramos, Luis Javier; Bernal Noguera, Sara. Boletín Oficial de la Propiedad Industrial (18-12-2020) Vol. 2: Invenciones. p. 9. Disponible en: https://consultas2.oepm.es/pdf/ES/0000/000/02/78/53/ES-2785323_B2.pdf.
- Patente “Mouse model, deficient in factor V”. Ref. ES2948817B2. Titulares: Universidad Complutense de Madrid y Consejo Superior de Investigaciones Científicas. Inventores: Liras Martín, Antonio; De Pablo Moreno, Juan Andrés; Revuelta Rueda, Luis; Miguel Batuecas, Andrea; Bermejo Alvarez, Pablo y González Brusi, Leopoldo. Boletín Oficial de la Propiedad Industrial (21-02-2024) Vol. 2: Invenciones. p. 8. Disponible en: https://consultas2.oepm.es/pdf/ES/0000/000/02/94/88/ES-2948817_B2.pdf.
- La falta de impulso a los biosimilares impide el ahorro de 431 millones de gasto farmacéutico en España. Diario Público (sep 2023). Disponible en: https://www.publico.es/sociedad/falta-impulso-biosimilares-impide-ahorro-431-millones-gasto-farmaceutico-espana.html.
