1. INTRODUCCIÓN
La microbiota, el conjunto de los microorganismos que colonizan nuestros epitelios (la piel, las cavidades orofaríngea, respiratoria y, en términos cuantitativos, especialmente la digestiva), ha sido contemplada por largo tiempo con sospecha por su potencial como foco de enfermedades infecciosas. Sin embargo, aportaciones provenientes de la ecología microbiana y de la genómica, en particular de las tecnologías que permiten la secuenciación masiva del DNA en entornos naturales complejos (metagenómica) y de la caracterización mediante espectrometría de masas de las moléculas presentes en dichos entornos (metabolómica), nos describen en la actualidad un escenario en el que la inmensa mayoría de los microorganismos que componen la microbiota participan en una red de interacciones beneficiosas para el organismo hospedador (1,2). Los resultados de las aproximaciones experimentales “ómicas”, integrados por la biología de sistemas, a la par nos abruman y fascinan con grandes cifras que parecen propias de la cosmología: las alrededor de 10000 especies de nuestra microbiota (principalmente bacterias, pero también arqueas, hongos y protistas) comprenden un número total de células (≈3,8 x 1013) parejo a las del cuerpo humano, mientras que los genes que aquéllas portan (el microbioma) excede en una razón 100:1 a los aproximadamente 22000 descritos en nuestro genoma (3).
La relación entre microbiota y enfermedades neurodegenerativas arranca de estudios epidemiológicos que relacionan eventos de infección respiratoria o intestinal, con capacidad proinflamatoria, y un agravamiento o aceleración de los síntomas neurológicos en pacientes ya diagnosticados de neurodegeneración (4). Durante los últimos años, tanto las infecciones como las perturbaciones en la composición de la microbiota (disbiosis) están siendo estudiadas en relación con la fase silente asintomática de las enfermedades neurodegenerativas, o asociadas con sus estadios iniciales (pródomos) (5-10). Está comúnmente aceptado que cuando las enfermedades neurodegenerativas pasan a una fase sintomática el margen para una intervención terapéutica es ya escaso, por lo que hay un interés renovado por caracterizar aquellos factores, entre ellos las infecciones y la disbiosis, que pudiesen participar en primera instancia como desencadenantes de dichas enfermedades (11).
En este artículo contemplaré primero el estado de la cuestión concerniente a las relaciones entre microbiota bacteriana y SNC, en particular lo relativo a los metabolitos con actividad neurotrópica asociados a disbiosis y la evidencia disponible, superando la mera correlación que no implica necesariamente causalidad (12-13), sobre la neurotoxicidad de las proteínas y péptidos de naturaleza amiloide generados por la microbiota (Figura 1). Por último, se presentará nuestro trabajo en el desarrollo de un sistema modelo mínimo, totalmente bacteriano, de una proteinopatía amiloide intracelular que delinea aspectos esenciales de la rutas mitocondriales en neurodegeneración (Figura 2).
2. INTERACCIONES ENTRE LOS MICROORGANISMOS Y EL SNC
2.1. Metabolitos neurotrópicos asociados a la microbiota y su disbiosis
Los estudios que han abordado la microbiota humana mediante aproximaciones de biología integrativa (metagenómica, proteómica, metabolómica y análisis de sistemas) han puesto de manifiesto, en las heces y en el mucus que recubre el intestino grueso, la presencia de moléculas orgánicas con un origen claramente microbiano: ácidos grasos de cadena corta (SCFA, por sus siglas en inglés: acetato, propionato y butirato, principalmente), vitaminas y derivados de aminoácidos, así como subproductos de la alteración por parte de los microorganismos de biomoléculas del hospedador (14-16). Dichos metabolitos pueden ser reconocidos por receptores específicos en las células del epitelio intestinal, tales como los del tipo Toll (TLRs) o los acoplados a proteínas-G (GPCRs), que transmiten señales a factores transcripcionales específicos con el resultado de un fortalecimiento de la integridad de la barrera intestinal, tanto de las uniones apicales estrechas como de las que delimitan la lamina propria basal. Sin embargo, la interacción con metabolitos como poliaminas o histamina resultan en el epitelio en una respuesta mediada por el inflamasoma, lo que produce el debilitamiento, e incluso la disrupción, de dicha barrera (15). Un correcto balance de la microbiota prima la primera de las respuestas mencionadas, mientras que su disbiosis da lugar a la segunda de ellas.
Una ruta alternativa por la que los metabolitos generados por la microbiota podrían alcanzar el SNC es el denominado “eje intestino-cerebro”. La capa tisular subyacente al epitelio intestinal presenta enervaciones, tanto aferentes (sensoras) como eferentes (motoras), del sistema parasimpático. En concreto, el nervio vago es el canal por el que, mediante transporte axonal retrógrado, diversas moléculas con origen microbiano y propiedades neurotransmisoras y moduladoras (SCFAs, GABA, oxitocina, entre otras) ejercerían una influencia neta sobre el SNC (17,18). Esta hipótesis ha sido confirmada por diversos estudios conductuales llevados a cabo con roedores, vinculando dichos metabolitos con una reducción sintomática de estados de estrés, ansiedad y depresión. En lo que concierne a los procesos inflamatorios locales en el intestino, los SCFAs ejercen una actividad antiinflamatoria, al modular la diferenciación de los linfocitos T reguladores (mediada por un aumento de IL-10), reducir la actividad de macrófagos y células dendríticas y disminuir la proliferación de linfocitos T citotóxicos (15). Dichos efectos locales pueden ejercerse también a distancia sobre el SNC, pues algunas de esas pequeñas moléculas, dispersadas por el torrente sanguíneo, son capaces de atravesar la barrera hematoencefálica y de ejercer su efecto sobre neuronas y células gliales (9). Así, en el caso de enfermedades neurales de naturaleza autoinmune e inflamatoria, como la esclerosis múltiple, se comprobó un efecto beneficioso de derivados del aminoácido triptófano, vía la acción de la triptofanasa bacteriana y modificaciones enzimáticas adicionales que generan ligandos reconocidos por los receptores de arilos hidrocarbonados (AHR) en los astrocitos, con el resultado de una reducción de la neuroinflamación asociada a la enfermedad en un modelo animal (19).
En los últimos años hay un interés creciente en la capacidad de vesículas extracelulares (ECVs) de membrana, generadas naturalmente en todos los organismos vivos, para actuar como vehículos de la señalización intercelular, tanto en condiciones fisiológicas como patológicas. La microbiota intestinal es una de las fuentes más abundantes de ECVs, pudiendo éstas transportar tanto metabolitos como pequeños RNAs reguladores y proteínas (20-22). Como se comentará más adelante, las ECVs son consideradas también potenciales vehículos para la propagación de los agregados amiloides.
Los efectos beneficiosos de los metabolitos arriba indicados se ven alterados de manera significativa en la disbiosis que suele estar clínicamente asociada a las enfermedades neurodegenerativas, pudiendo por el contrario tener un efecto promotor de la progresión de éstas (15). Paso a continuación a pormenorizar esa relación ambivalente entre metabolitos microbianos y neurodegeneración en el contexto de tres de las enfermedades con mayor prevalencia e impacto sociosanitario.
2.1.1. Enfermedad de Parkinson (EP)
Esta enfermedad neurodegenerativa, descrita por vez primera en 1817 por el médico inglés que le da nombre, James Parkinson (1755-1824), comprende un conjunto de patologías relacionadas con la agregación de la proteína sináptica α-sinucleína en diversas áreas cerebrales, en especial en zonas del mesencéfalo como la substantia nigra (pars compacta), formando los denominados cuerpos de Lewy en neuronas dopaminérgicas. Sus síntomas implican disfunciones motoras y, en fases avanzadas, cognitivas. Es la segunda enfermedad neurodegenerativa con mayor prevalencia (hasta un 1% de los mayores de 60 años) (11).
La enfermedad de Parkinson fue la primera neurodegeneración para la que se obtuvo evidencia experimental sobre su vinculación con la microbiota. En 2016, el grupo de Sarkis Mazmanian (23), utilizando un modelo murino de EP que sobreexpresaba un transgén de la α-sinucleína humana, descubrió que ratones que habían sido sometidos desde su nacimiento a condiciones de esterilidad (ausencia de microbiota), o a un tratamiento antibiótico intensivo, manifestaban en diversas pruebas de motricidad una respuesta similar a la observada, tras el mismo tratamiento, en una cohorte de ratones control con genotipo silvestre. Cuando ambos grupos de animales se criaban en condiciones en las que su microbiota era la adquirida espontáneamente en las jaulas y bajo un régimen de alimentación estándar, los ratones transgénicos para la α-sinucleína mostraban dificultades claras para alcanzar en las pruebas de motricidad una puntuación similar a la lograda por los controles. Estudios de inmunohistoquímica revelaron, en el caso de los ratones transgénicos, un aumento neto de α-sinucleína agregada y fosforilada en la Ser129 (marcadores característicos de la EP) en las áreas cerebrales habitualmente afectadas en dicha patología. El análisis metagenómico de las heces de los roedores indicó una presencia mayor de Proteobacterias en los ratones transgénicos y una disminución correlativa de la de Firmicutes, que podría estar detrás del aumento, según se desprendía de estudios metabolómicos, de SCFAs en las heces de los ratones transgénicos. Esta observación tiene su paralelo en los resultados similares de análisis realizados con heces humanas de individuos control y de pacientes de la EP. Al realizar una transferencia de esas heces humanas a los ratones, los transgénicos para α-sinucleína manifestaban una exacerbación de los síntomas sólo cuando el material fecal procedía de pacientes de EP. Lo más sugestivo es que bastaba suplementar con SCFAs la dieta de los ratones criados en condiciones de esterilidad o tratados con antibióticos para que los transgénicos para la α-sinucleína mostrasen la plena sintomatología motora y la histopatología asociadas a la enfermedad. Con las limitaciones inherentes a tratarse de un modelo murino en un contexto de sobreexpresión de la α-sinucleína, este estudio representó la primera evidencia científica de la vinculación entre la microbiota y sus metabolitos en el desarrollo de una enfermedad neurodegenerativa (23), posteriormente extendida a otras moléculas (24).
2.1.2. Enfermedad de Alzheimer (EA)
Esta dolencia neurodegenerativa, la de mayor prevalencia (hasta el 6% de la población mayor de 60 años), lleva el nombre de Alois Alzheimer (1864-1915) quien la describió en una publicación fechada en 1907. La enfermedad, que cursa con un deterioro cognitivo progresivo y severo, afecta primero a la corteza entorrinal y al hipocampo para extenderse después a la corteza temporal, la amígdala y finalmente a otras áreas corticales. Se vincula con la aparición de agregados amiloides fibrilares de dos proteínas, Tau (factor regulador del ensamblaje de los microtúbulos) que, relacionada con su hiperfosforilación, forma ovillos intracelulares en las neuronas, y los péptidos Aβ1-42 y Aβ1-40 (provenientes de la proteólisis de APP, una proteína de la membrana sináptica), que constituyen densas placas extracelulares (11).
Existe evidencia de una actividad antimicrobiana del péptido Aβ1-42, lo que podría indicar una posible respuesta innata ante infecciones bacterianas (25,26). Una relación directa entre la EA y la microbiota se reportó en 2019 al describirse que una bacteria responsable de infecciones periodontales, Porphyromonas gingivalis, estaba presente en muestras de anatomía patólogica postmortem en pacientes de esa neurodegeneración (27). Estudios de inmunocitoquímica empleando anticuerpos contra las gingipaínas, proteasas características de la bacteria que son secretadas para la desorganización tisular del hospedador durante la colonización de éste, así como análisis de PCR cuantitativa, mostraron la huella de P. gingivalis en los tejidos analizados. Dada la extraordinaria capacidad de esta bacteria para generar ECVs, lo más probable es que los marcadores moleculares mencionados sean transportados por ECVs que hayan atravesado la barrera hematoencefálica, no debido a una infección in situ (28). Se desconocen los detalles sobre cómo la bacteria podría ser un factor relevante en la etiología y el desarrollo de la EA en el nivel molecular, pero tanto los fibras de Tau como las placas formadas por péptidos Aβ colocalizaban con las gingipaínas en un modelo murino de infección. Estudios realizados con ratones mostraron que el tratamiento con un inhibidor específico de las gingipaínas reducía la carga bacteriana y los niveles de marcadores de la enfermedad como Aβ1-42 y TNFα (27).
Un estudio reciente (2023) realizado por un consorcio liderado por David Holtzman (29), ha arrojado información adicional sobre cómo la microbiota intestinal puede impactar en el desarrollo de la enfermedad de Alzheimer, dependiendo del alelo del gen APOE, un factor clave en la neurodegeneración mediada por Tau, expresado en ratones modelo de la enfermedad, con efectos más marcados para las isoformas ApoE3 y ApoE4. El haber sido criados en ambiente estéril, al igual que el tratamiento con antibióticos (30), reducía la huella histopatológica de la enfermedad, en términos de atrofia cerebral y detección de Tau fosforilada en el hipocampo, y (sobre todo en fondos genéticos que expresan el alelo ApoE3) conllevaba una reducción significativa de la extensión y ramificación de las terminaciones en las células de la microglía (29). Dichos resultados fueron desiguales en machos y hembras, siendo las segundas mucho menos sensibles al tratamiento antibiótico, en consonancia con estudios metagenómicos que mostraron que los ratones de diferente sexo presentaban microbiotas con composiciones marcadamente distintas. El análisis metabolómico de las heces de los ratones mostraba que la ingesta de antibióticos correlacionaba con una disminución significativa de los niveles de SCFAs en las muestras (29), en el mismo sentido de lo apuntado en la sección anterior para el modelo murino de la EP (23). Un segundo trabajo, en el que se realizaron transferencias fecales a ratas desde cohortes de donantes humanos diagnosticados de Alzheimer o controles sanos, mostró la capacidad de la microbiota de los pacientes, enriquecida en bacterias del género Desulfovibrio, para interferir con la neurogénesis en el hipocampo, correlacionándose con síntomas tanto histológicos (epitelio intestinal) como conductuales y de aprendizaje (31).
2.1.3. Esclerosis lateral amiotrófica (ELA)
La ELA es una enfermedad que afecta a las neuronas motoras y que cursa de manera relativamente rápida, conduciendo a la parálisis total del paciente y a su fallecimiento por insuficiencia respiratoria. Fue descrita por vez primera en 1873 por Jean-Martin Charcot (1825-1893). Al contrario de lo que sucede con la EP y la EA, donde una o dos proteínas forman las inclusiones fibrilares cerebrales características de esas enfermedades, en el caso de la ELA son diversas las proteínas potencialmente implicadas (Sod1, TDP-43, Fus y C9orf72, entre otras). En el SNC, las áreas principalmente afectadas son el córtex motor y el tronco cerebral. Su prevalencia es menor que las de la EA y la EP (hasta 8 casos por cada 100000 habitantes), siendo hasta un 15% de dicha cifra de origen genético, ligado principalmente a mutaciones en el gen de la superóxido dismutasa 1 (SOD1) (11).
Quizás el hallazgo más significativo relacionado con una vinculación entre microbiota y ELA sea el reportado en 2019 por los laboratorios de Eran Elinav y Eran Segal (32), nuevamente en un modelo transgénico murino de la enfermedad que, en este caso, expresaba una variante alélica mutante humana del gen SOD1. En ratones criados en condiciones de esterilidad, o sometidos a un tratamiento antibiótico, diversos ensayos de motricidad mostraron que la composición de la microbiota es un factor agravante y acelerador de los síntomas propios de la ELA. Estudios paralelos de metagenómica indicaron que los ratones transgénicos presentaban la pérdida casi absoluta de Akkermasia muciniphila, una bacteria presente por el contrario en la microbiota intestinal de los animales control. Aproximaciones metabolómicas mostraban que esa disbiosis estaba correlacionada con una disminución significativa del metabolito nicotinamida (una de las formas de la vitamina B3). Bastaba con inocular A. muciniphila en los ratones para que en éstos mejoraran sensiblemente los síntomas de la ELA, mientras que componentes de la microbiota de los ratones transgénicos (como Ruminococcus y Parabacteroides) los exacerbaban. Lo que resultó decisivo fue que el suplementar nicotinamida en la dieta de los ratones afectados era suficiente para que éstos recuperaran cierta motricidad, sin que llegase a ser pareja a la estimada para los controles. No obstante, esa mejora sintomática no tenía reflejo en un aumento significativo de la supervivencia de los ratones modelo de ELA. Por último, estudios realizados con heces de pacientes de ELA y controles sanos mostraron que, efectivamente, los niveles de la vitamina B3 se hallaban reducidos en los primeros.
En resumen (Figura 1), la evidencia acumulada durante los últimos años permite afirmar que la microbiota tiene un impacto neto significativo en el desarrollo, y quizás también en la etiología, de las enfermedades neurodegenerativas que, en primera instancia, parece ser ejercido por metabolitos secretados o modificados por los microorganismos, lo que abre una ventana al desarrollo futuro de intervenciones propiamente farmacológicas (33,34). Sin embargo, es necesaria una doble nota de cautela pues las evidencias anteriormente comentadas provienen de sistemas modelo animales y su extrapolación a pacientes humanos es incierta. Por otro lado, en todos los casos mencionados las actuaciones sobre la microbiota tienen efectos de alivio sintomático, no propiamente terapéutico. En este sentido, cabe señalar que la crisis actual producida por las multirresistencias bacterianas a los antibióticos acarrea una amenaza adicional al restringir también la utilidad de estos fármacos para la modulación de la microbiota como parte del tratamiento de enfermedades neurodegenerativas. En la siguiente sección se tratará sobre una segunda vía, conceptualmente más compleja, a través de la cual los microorganismos actúan sobre el curso de las enfermedades neurodegenerativas.
2.2. Disbiosis y amiloides neurotóxicos
Rudolf Virchow (1821-1902), uno de los padres de la histología, describió en 1854 la presencia de inclusiones en secciones fijadas de cerebros de pacientes con edad avanzada cuando aquéllas se trataban con una tinción específica del almidón, por lo que las denominó amiloides. Poco después, en 1859, el químico Friedrich Kekulé (1829-1896) identificó correctamente el material componente de dichas inclusiones como “albuminoide”, es decir, proteico, pese a lo cual la denominación que ha prevalecido es la primera (35).
En la actualidad se ha determinado que cada una de las enfermedades neurodegenerativas (y algunas de las sistémicas, como diversas amiloidosis renales, hepáticas y articulares) estudiadas hasta fecha se correlaciona con la presencia de depósitos amiloides de una proteína específica distinta, propia de la patología concreta, localizados en regiones anatómicamente diferentes, características también de cada enfermedad (36). En el nivel molecular, los amiloides son agregados de una proteína dada en los que cada una de las moléculas de ésta transforma su estructura incrementando su composición en hebras-β, que se apilan perpendicularmente constituyendo el eje de una estructura fibrilar (denominada lámina-β cruzada) (37). Las proteínas amiloidogénicas presentan pues al menos dos plegamientos alternativos (desordenado o nativo de partida y amiloide final), siendo el primero el único que puede ser predicho por los algoritmos basados en inteligencia artificial, como AlphaFold, que por el momento son incapaces de identificar la estructura amiloide alternativa (38-39). El amiloide es el estado termodinámicamente más estable que puede adoptar una proteína y, debido a las complejidades de su trayectoria de plegamiento y ensamblaje (control cinético), por influencia de las condiciones físico-químicas y de la presencia de ligandos (cofactores), puede dar lugar a estructuras fibrilares alternativas (polimorfos) que suelen correlacionarse con diversos fenotipos (síntomas) de la enfermedad con la que se relacionan (37,40). Otra característica particular de los amiloides es el fenómeno de polimerización nucleada, por el que agregados discretos de una proteína amiloidogénica (usualmente oligómeros prefibrilares de la misma) son capaces de transferir por contacto su conformación amiloide a moléculas solubles (nativas) de esa misma proteína, e incluso a otras con un grado de identidad de secuencia elevado, en lo que se denomina agregación o “siembra” cruzada (40). Esta agregación cruzada es la responsable de la capacidad de propagación intercelular de los amiloides, tanto por la vía retrovagal como, una vez en el propio SNC, entre áreas cerebrales distales o, en el caso de los amiloides infectivos (cuyo paradigma es el prion PrP) (41), entre organismos. Dicha nucleación se detecta como una aceleración de la progresión de la enfermedad en el organismo receptor in vivo, o como un adelanto de la fase exponencial (por reducción del tiempo de latencia necesario para generar el núcleo amiloide) en las curvas sigmoideas características de la cinética de agregación in vitro (40).
2.2.1. Biopelículas bacterianas y enfermedad de Parkinson
Las biopelículas son consorcios microbianos que constituyen formas de persistencia ante condiciones ambientales no favorables, incluido el tratamiento con antibióticos, lo que supone un problema sanitario de primer orden en el actual contexto de expansión incontrolada de las multirresistencias. En las biopelículas, los microorganismos en sí entretejen una compleja trama extracelular formada por polisacáridos, DNA y proteínas de naturaleza amiloide, siendo estas últimas de particular importancia para dotar de resistencia mecánica al conjunto del biomaterial resultante (42,43). La primera proteína amiloidogénica caracterizada como parte de una biopelícula fue CsgA, que es el constituyente principal de unos pili denominados curli en la bacteria Gram-negativa Escherichia coli (44). La secreción y ensamblaje como amiloide de CsgA es un proceso complejo que requiere múltiples cofactores, entre ellos un canal específico en la membrana externa, chaperonas periplasmáticas y una proteína nucleadora de la amiloidogénesis (45). Estructuras fibrilares amiloides análogas a CsgA/curli se han descrito posteriormente en otras bacterias Gram-negativas (FapC en Pseudomonas) (46) y Gram-positivas (TasA en Bacillus, Bap y PSMs en Staphylococcus) (47-49).
La primera evidencia sobre una posible relación con la neurodegeneración de los amiloides extracelulares secretados por la microbiota se obtuvo en estudios in vitro realizados por el grupo de Matthew Chapman (2015), en los que una chaperona (CsgC) encargada de contribuir al ensamblaje de curli era capaz de inhibir la agregación de la α-sinucleína humana recombinante (50). In vivo, el equipo de Robert Friedland (2016) mostró en ratas y en el nematodo Caenorhabditis elegans, un hospedador versátil para estudiar la microbiota por la facilidad para controlar la composición de las bacterias con las que se le alimenta, que curli generaba depósitos amiloides de la α-sinucleína en células neurales, tanto en ganglios entéricos como cerebrales (51). En otro estudio realizado en gusanos transgénicos para la α-sinucleína humana, expresada en el tejido muscular o en diversas neuronas, ésta era inducida a agregar (resultando en una reducción de la motilidad) cuando la dieta estaba constituida por E. coli silvestre, mientras que dicho efecto no se observaba en la misma medida cuando el genotipo de las bacterias suministradas como alimento era ∆csgA. Si en la dieta se incluía una molécula polifenólica natural con propiedades anti-agregativas de los amiloides (epigalocatequina-3-galato, EGCG) la agregación de la α-sinucleína se reducía significativamente (52). En un tercer trabajo, el cambio a una dieta constituida por B. subtilis era capaz de revertir ese efecto proagregativo de E coli, lo que se debía a un aumento en los gusanos del metabolismo de esfingolípidos inducido por Bacillus (53).
Utilizando el mismo modelo murino transgénico de EP mencionado anteriormente (23), los grupos de M. Chapman y S. Mazmanian mostraron que la colonización de ratones criados en esterilidad con E. coli secretora de curli o con un control en el que se habían delecionado varios genes de curli, entre ellos csgA, tenía efecto dispar en los animales (54): en cuanto a habilidades motoras, ratones a los que se habían transferido con la dieta bacterias ∆csgA presentaban una respuesta mejor que los que habían recibido bacterias silvestres y, en lo que se refiere a marcadores histológicos, se observaba una menor acumulación de la forma más amiloidogénica de la α-sinucleína (fosforilada en la Ser129). La inclusión de EGCG en la dieta, como se ha apuntado anteriormente para C. elegans (52), producía una mejora significativa en las habilidades motoras de los ratones inoculados con las bacterias silvestres (54).
Dirigiendo la mirada a otros componentes de la microbiota distintos de las bacterias, un trabajo reciente ha mostrado que la proteína priónica Sup35 de la levadura Saccharomyces cerevisiae también es capaz de inducir la agregación citotóxica de la α-sinucleína, tanto in vitro como en un modelo murino de infección por vía nasal, siendo ésta bloqueada por un tratamiento con el antifúngico fluconazol (55). Estas observaciones apoyan que el impacto de la microbiota sobre las enfermedades neurodegenerativas, al menos en el caso de la EP, podría no deberse sólo a su componente mayoritario procariótico.
Es importante considerar otros factores que colateralmente contribuyan a la acción de la microbiota sobre el SNC. Un estudio reciente, abordó el efecto de la dieta sobre la agregación de la α-sinucleína en el modelo transgénico murino, midiendo su efecto sobre distintas habilidades motoras (SNC) y, localmente, en el intestino (56). El resultado, cuando la microbiota intestinal secretaba curli, fue una exacerbación de los síntomas de la EP si los ratones además estaban alimentados con una dieta pobre en fibra, mientras que una alimentación rica en fibra tenía un efecto protector. Estos resultados van en la línea trazada por el popular dicho: de alguna manera, “somos lo que comemos”.
2.2.2. Perspectivas sobre el impacto de los amiloides bacterianos en las enfermedades neurodegenerativas
Cabe la posibilidad de que amiloides de origen bacteriano dispares a CsgA/curli (o sus análogos FapC, TasA, Bap y PSMs) puedan actuar como nucleadores de la agregación de diversas proteínas en el organismo hospedador. Búsquedas computacionales en las secuencias completas de los genomas bacterianos presentes en el tracto gastrointestinal, dirigidas a localizar genes codificadores de proteínas con potencial amiloide, han identificado, sólo en la subfamilia de proteínas intrínsecamente desordenadas con dominios del tipo prion, unas 138000 proteínas probablemente amiloidogénicas, distribuidas entre más del 60% de las bacterias del microbioma (57). Nuevamente, los modelos basados en C. elegans han abierto camino a la validación experimental con alto rendimiento de todas esas secuencias, seleccionando aquellas que son agregativas, citotóxicas y con efectos sobre el comportamiento de los gusanos, complementada con diversas aproximaciones de biología molecular y biofísicas (58). El fenotipo (relativo al aprendizaje y condicionamiento conductual) que expresaban los gusanos era similar al descrito en modelos más complejos de EA. En un estudio anterior, gusanos que expresaban Aβ1-42 o Sod1 presentaban también neurodegeneración compatible con EA y ELA, respectivamente, tras la ingesta de una estirpe de E. coli que expresaba CsgA (52).
Si los amiloides con origen en la microbiota intestinal son potencialmente causantes de neurodegeneración, por su capacidad para nuclear la agregación cruzada de proteínas amiloidogénicas humanas, su neutralización es un objetivo de indudable interés biomédico. En nuestro laboratorio estamos desarrollando mediante aproximaciones de biología sintética dispositivos basados en porinas, proteínas intrínsecas de la membrana externa bacteriana con estructura de barril-β, a los que hemos dotado de la capacidad para unirse a péptidos y proteínas amiloides a través de su reconocimiento homotípico (el ensamblaje amiloide de secuencias idénticas) (59). Para ello, hemos insertado en uno de los lazos extracelulares de la porina de E. coli OmpF un péptido con la misma secuencia que la de un segmento amiloidogénico presente en una proteína bacteriana diana. El ensamblaje amiloide del péptido, marcado con una sonda fluorescente, sobre el lazo que incorpora su secuencia en OmpF es detectable en la superficie de las células de E. coli mediante microscopía de fluorescencia (59). Las bacterias podrían actuar así como atractores de los amiloides generados por la microbiota intestinal, evitando que pudieran alcanzar por la vía retrovagal el SNC.
El ensayista y matemático norteamericano de origen libanés Nassim Taleb (n. 1960) acuñó el término “cisne negro” para referirse a los sucesos que ocurren por sorpresa, pues ningún analista los había previsto ni tenido en cuenta porque, a priori, eran improbables, y que terminan teniendo un gran impacto. Paradójicamente, a posteriori, esos acontecimientos se racionalizan haciéndolos predecibles o explicables, dando así la impresión de que se esperaba que ocurrieran. En este sentido, la microbiota fue un cisne negro en el campo de las enfermedades neurodegenerativas, si bien en la actualidad está ya en la fase de su plena aceptación, en la que caben esperar nuevos hallazgos que consoliden y extiendan el papel desempeñado por los metabolitos y los amiloides bacterianos (Figura 1).
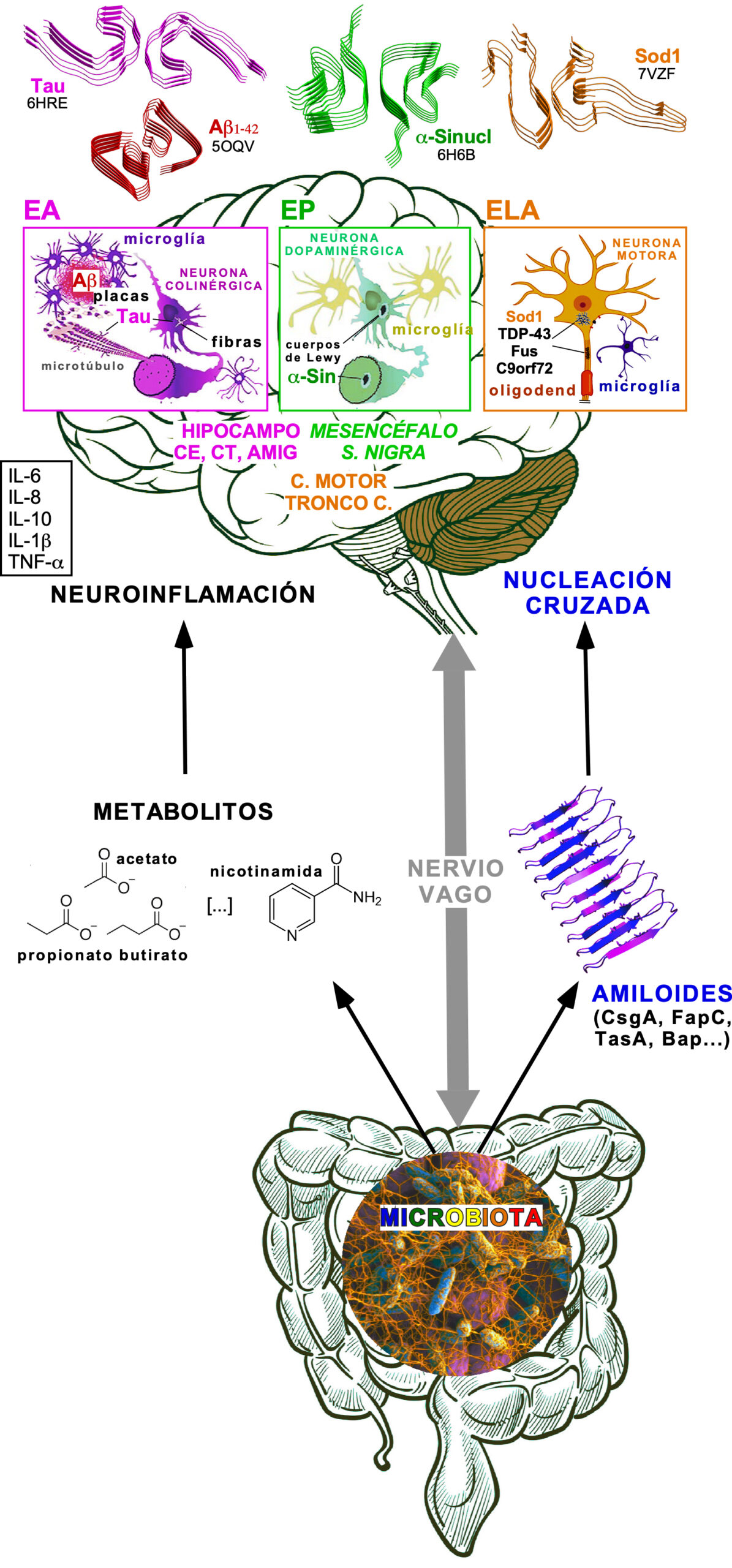
Figura 1. Una visión esquemática integrada de las interacciones entre microbiota y neurodegeneración, mediadas a través del “eje intestino-cerebro”. El dibujo recoge algunos de los metabolitos microbianos (izquierda) y de las proteínas bacterianas amiloidogénicas (derecha), que son relevantes en las tres enfermedades neurodegenerativas revisadas en este trabajo (arriba). Secciones fibrilares de algunas proteínas cuyas estructuras son moldeadas sobre los amiloides secretados por la microbiota se muestran por los paneles específicos de cada enfermedad
3. RepA-WH1, EL PRIMER MODELO BACTERIANO DE AMILOIDOSIS INTRACELULAR
Uno de los protagonistas de la creación de la biología molecular, Jacques Monod (1910-1976), en una apuesta por la universalidad de esa disciplina desarrollada fundamentalmente sobre modelos procarióticos, afirmó que (en biología) “lo que es válido para E. coli es válido para el elefante”, aforismo que fue perdiendo fuerza al ir desentrañándose la extraordinaria complejidad de los organismos eucariotas multicelulares. Cabe plantearse si sería posible invertir los términos de la sentencia de Monod, adaptando a las bacterias un proceso exclusivo de los organismos más complejos; así ¿podría desarrollarse a partir de éstas un modelo minimalista para el estudio de las proteinopatías amiloides? Con ese objetivo, en el laboratorio del autor se adoptó a finales de la década de 2000 una aproximación propia de la biología sintética para el estudio de los amiloides bacterianos, tanto en cuanto a su caracterización estructural y funcional como para explorar sus posibles aplicaciones en biotecnología y biomedicina.
3.1. RepA-WH1, un amiloide funcional en la replicación del DNA plasmídico
Las proteínas Rep son factores esenciales para la replicación de muchos plásmidos en las bacterias Gram negativas. En el laboratorio del autor se estudia, desde hace 30 años, la proteína RepA del plásmido pPS10 de Pseudomonas (Figura 2A). Como dímero, RepA reprime la transcripción de su propio promotor mediante la unión a una secuencia operadora repetida inversamente, mientras que su monomerización al unirse a una secuencia de 22 pb repetida en tándem (iterones, en número de cuatro, ubicados en el origen de replicación, oriV) activa a RepA para la iniciación de la replicación del DNA plasmídico (61). Una vez que se completa la replicación del DNA, RepA modula el ensamblaje de un amiloide funcional que acopla físicamente las dos copias del plásmido resultantes, inhibiendo así rondas adicionales de replicación (62). Clave en esa conmutación funcional en RepA es un cambio conformacional que afecta al dominio N-terminal (WH1) en la proteína, incrementando su contenido en lámina-β, y que es desencadenado por su unión específica, pero con baja afinidad, a uno de los extremos de cada iterón (63-65). El reconocimiento con mayor afinidad del otro extremo por el dominio C-terminal en RepA (WH2) (66), limita la unión de la proteína a la extensión precisa abarcada por los iterones (88 pb) (62). Así, la restricción topológica de su agregación por la secuencia diana en el DNA es el mecanismo a través del cual RepA ensambla un amiloide funcional, evitando su polimerización indefinida que podría llevar a una agregación amiloide citotóxica (67).
3.2. Perspectivas estructurales sobre los amiloides de RepA-WH1
El primer hallazgo sobre las bases moleculares de la amiloidogénesis del dominio RepA-WH1 (Figura 2B) fue que, una vez separado de WH2, su unión transitoria a distintas secuencias cortas (11 pares de bases) de DNA del plásmido promovía la formación de diferentes ensamblajes proteicos, incluyendo fibras (68). Es interesante que la mutación más frecuente encontrada en cribados genéticos dirigidos a modular la actividad de RepA en la replicación del plásmido, el cambio de Ala31 por Val en una región propensa a la amiloidogénesis en WH1 (L26VLCAVSLI34) (61), mejoraba sustancialmente el ensamblaje de RepA-WH1 en fibras amiloides in vitro. Éstas tenían unas dimensiones de 25 nm de grosor y una longitud variable, según reveló la microscopía electrónica de transmisión (TEM), estando constituidas por la asociación lateral de varios filamentos más finos (4 nm), en los cuales la proteína se ensamblaba como hebras helicoidales simples o dobles con una sección axial tubular. Los filamentos se torsionaban en las fibras dando lugar a una superhélice levógira, de acuerdo con observaciones de microscopía de fuerza atómica (AFM) (69).
Estudios recientes de resonancia magnética nuclear (NMR) revelaron que la unión del DNA a RepA-WH1 desestabilizaría su interfaz de dimerización como lámina-β, al romper la red de interacciones catión-π entre cadenas laterales de aminoácidos que contribuye a mantener unidas las dos subunidades de la proteína, aumentando así la inestabilidad intrínseca de susextremos N y C-terminales (70). Esta desestabilización liberaría el lazo que incluye la secuencia amiloidogénica arriba mencionada para iniciar el autoensamblaje de RepA-WH1.
Siendo el DNA un inductor de la amiloidogénesis de RepA-WH1 (68), buscamos mediante un cribado computacional inhibidores de dicha interacción. Identificamos una variante tetrasulfonada de la molécula de índigo (S4-índigo) como ligando con capacidad para unirse a una superficie rica en residuos de arginina, centrados en torno al eje de simetría binario del dímero RepA-WH1 y en posición distal al segmento amiloidogénico, que estaría implicada en el reconocimiento del DNA (71) (Figura 2B). Ensayos in vitro demostraron que esta molécula, efectivamente, se unía a la proteína con afinidad submicromolar y era capaz de bloquear el ensamblaje de las fibras RepA-WH1 (71). Estudios de NMR revelaron que el S4-índigo también previene la amiloidogénesis a través de un segundo mecanismo: la unión a un bolsillo entre la inestable hélice-α N-terminal y una horquilla-β implicada en la dimerización, bloqueando así la disociación promovida por el DNA de los dímeros RepA-WH1 (70). El S4-índigo fue el primer inhibidor alostérico descrito de una amiloidogénesis.
3.3. Interruptores sintéticos de la amiloidogénesis de RepA-WH1
Hemos explorado formas alternativas de controlar la amiloidogénesis de RepA-WH1, además de la unión al DNA (Figura 2B). Entre ellas, destaca que vesículas lipídicas que incluyen fosfolípidos ácidos (cardiolipina, fosfatidil-glicerol) promovían tanto la agregación de RepA-WH1 como su inserción en membranas modelo, formando poros oligoméricos anulares con un diámetro similar al descrito para los túbulos fibrilares amiloides (72). La disposición espacial en las membranas de las cabezas de fosfolípidos cargadas negativamente podría proporcionar un patrón bidimensional reconocido por RepA-WH1, de forma mimética al de los fosfatos en el DNA efector. Explorando otros efectos de superficies en la amiloidogénesis, RepA-WH1 se funcionalizó sobre nanopartículas de oro, formando oligómeros resistentes a la desnaturalización que nuclearon eficientemente el crecimiento de fibras amiloides, lo que generó una firma espectral en ensayos de dispersión Raman mejorada en superficie (SERS) que puede utilizarse para monitorizar la polimerización amiloide (73) (Figura 2B).
Otra herramienta generada para conseguir controlar la amiloidogénesis de RepA-WH1 fue un anticuerpo monoclonal (B3h7) que reconocía los oligómeros de la proteína en la vía que conduce a las fibras amiloides, inhibiendo así el ensamblaje de éstas (74) (Figura 2B). B3h7 fue decisivo para identificar el nucleoide bacteriano como el lugar del ensamblaje inicial de los amiloides RepA-WH1 (74), y el puente transplasmídico entre moléculas de RepA unidas a los iterones como un oligómero amiloide (véase más arriba) (67) (Figura 2A).
La fusión optogenética de un dominio fotorreceptor vegetal (LOV2, que responde a la luz azul) con RepA-WH1 ha permitido recientemente canalizar el plegamiento de esta última proteína hacia la amiloidogénesis (75). En una quimera LOV2-WH1, la absorción de fotones de luz azul por el cofactor mononucleótido de flavina (FMN) sito en el dominio LOV2 despliega la hélice-a C-terminal en éste (Jα) y, por tanto, también la hélice-α N-terminal de WH1 (α1) que estaba fusionada a ella, lo que desestabiliza el dominio WH1 completo (Figura 2B). Cuando se utilizó para nuclear la amiloidogénesis in vitro, al ser iluminada con luz azul la quimera LOV2-WH1 moldeó el ensamblaje de oligómeros RepA-WH1, mientras que en la oscuridad dio lugar a haces de fibras y láminas de RepA-WH1. A nivel macroscópico, la oscuridad produjo una transición de fase irreversible de líquido a hidrogel en LOV2-WH1. La expresión bajo iluminación con luz azul de LOV2-WH1 en E. coli condujo a la generación de agregados oligoméricos citotóxicos que redujeron la viabilidad de las bacterias, allanando el camino a una nueva clase de potenciales agentes antimicrobianos (que denominamos “optobióticos”) (75).
El segmento amiloidogénico hidrofóbico en RepA-WH1 es, por sí mismo, una pieza adecuada para el desarrollo de dispositivos sintéticos cuando se incluye en otros amiloides funcionales conocidos (Figura 2C). Al sustituir las repeticiones polares oligopeptídicas ricas en glutamina y asparagina de la proteína de levadura Sup35 por varias copias en tándem del péptido amiloidogénico R25LVLCAVSLID35G de RepA-WH1, se construyó una serie de nuevos priones híbridos, [REP-PSI+] (76), que imitaban el fenotipo del prion natural de levadura [PSI+] al causar la lectura corrida de codones de terminación de la traducción (77). Este diseño se aplicó después para fusionar las mismas repeticiones de RepA-WH1 a RF1, uno de los dos factores de terminación de la traducción de E. coli, de tal manera que la agregación amiloide de la quimera daba lugar a que los ribosomas tradujeran, sin detenerse, el mRNA a través de un codón de parada prematuro insertado en el gen reportero lacZ. Se implementó así en bacterias un concepto de ingeniería inspirado en un prion de levadura, lo que permitió además el ensayo in vivo de moléculas naturales anti-amiloidogénicas, como la EGCG y el resveratrol (78).
3.4. RepA-WH1 genera una proteinopatía priónica sintética en bacterias
La presión evolutiva para el plegamiento eficiente de las proteínas parece haber seleccionado negativamente de forma natural cualquier amiloidosis proteotóxica intracelular en bacterias (40). Por lo tanto, para abordar la generación de una amiloidosis bacteriana sintética, expresamos RepA-WH1 en E. coli con una proteína fluorescente fusionada a su extremo C-terminal, para poder visualizarla y sustituir al dominio WH2, liberando así a WH1 de su papel como amiloide funcional (79) (Figura 2D). Este enfoque permitió rastrear la generación de agregados citosólicos que inhibían la proliferación bacteriana, comprobar la capacidad de estos agregados, una vez purificados, de nuclear el crecimiento de fibras amiloides de RepA-WH1 in vitro, y explorar la dominancia en la coagregación de distintas variantes mutantes de la proteína (79,80).
La segregación (vertical) entre célula madre y células hijas de agregados proteicos suele ser asimétrica en E. coli para garantizar que una de las dos células resultantes no herede proteínas dañadas (81). Sin embargo, al estudiar la proliferación de E. coli dentro de los canales de un dispositivo microfluídico, los agregados de RepA-WH1 se segregaron por igual entre las células hijas, reduciendo así la probabilidad de conseguir bacterias “curadas” de partículas amiloides y permitiendo la propagación epigenética estable de éstas en los linajes celulares (82). Esas células bacterianas, siendo isogénicas, mostraron dos fenotipos distintos en relación con los agregados: o bien múltiples y globulares, con citotoxicidad aguda, provocando la filamentación y la muerte celulares; o bien partículas únicas y alargadas, en forma de cometa y con apariencia fluida, que eran levemente tóxicas y se fragmentaban en dos con la formación del septo en la división celular (Figura 2D). Ambos tipos de agregados eran reactivos a una sonda química amiloidotrópica (BTA-1), aunque ésta mostraba una mayor afinidad por los agregados globulares (82). Por lo tanto, estos dos tipos de partículas responden a estirpes o variantes amiloides distintas, características de los priones: secuencias proteicas únicas que pueden adoptar al menos dos conformaciones agregadas alternativas, cada una de las cuales determina un fenotipo diferente (41,77). Mientras que la variante amiloide elongada de RepA-WH1 tiende a convertirse espontáneamente en la globular, la chaperona bacteriana DnaK (Hsp70), pero no ClpB (Hsp104), es capaz de revertir dicha conversión en el citoplasma bacteriano, proporcionando una demostración convincente de la capacidad de una Hsp70 para detoxificar amiloides in vivo (82) (Figura 2D). La síntesis in vitro, mediante un sistema de transcripción-traducción reconstituido (PURE), de RepA-WH1 dentro de vesículas lipídicas en presencia de chaperonas (Hsp70 y Hsp40) reproduce los patrones de agregación observados en las bacterias (83).
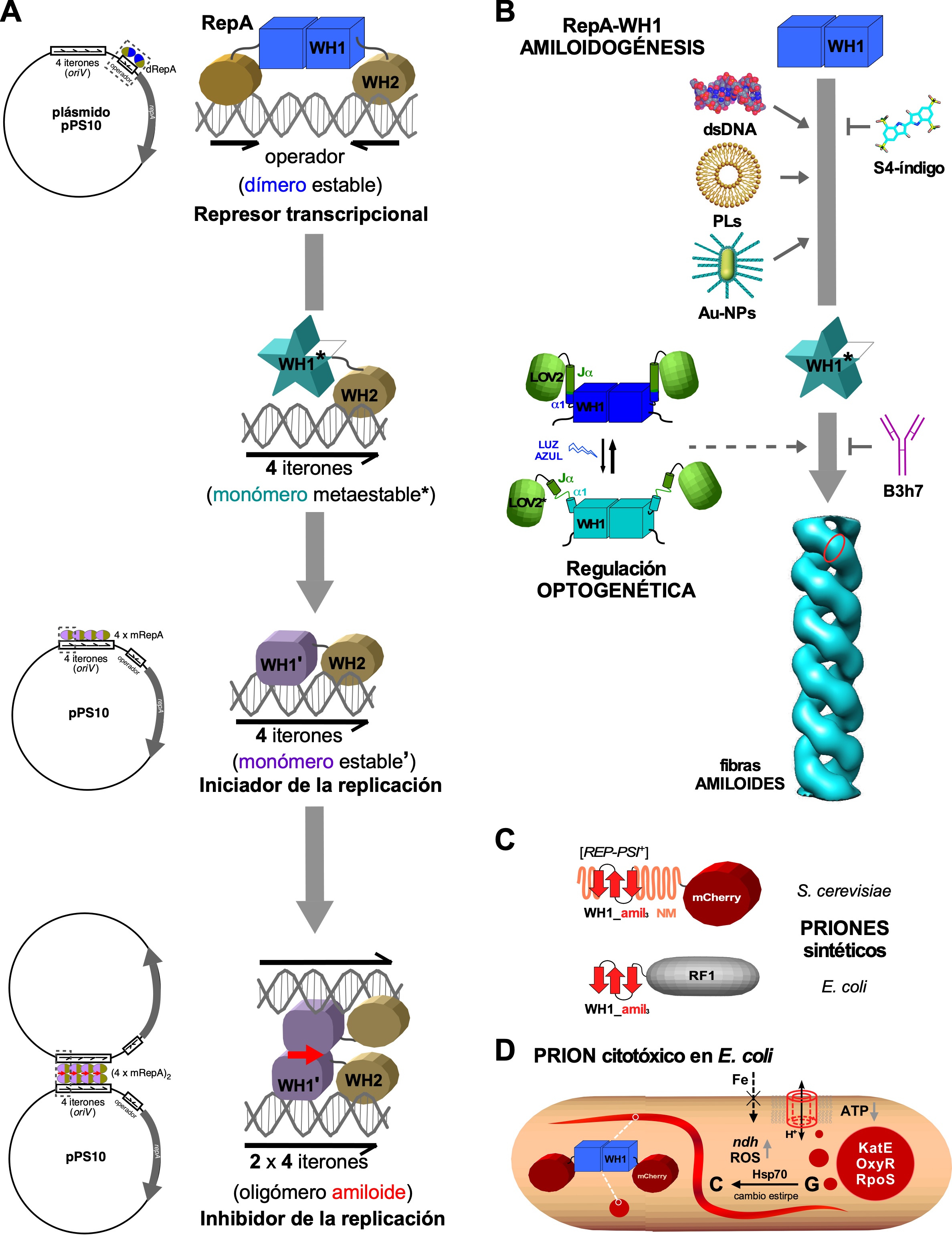
Figura 2. La proteína multifuncional RepA del plásmido de Pseudomonas pPS10 y sus aplicaciones en biología sintética. A) Cambios conformacionales que experimenta el dominio WH1 son responsables de la transición de la proteína RepA desde un represor transcripcional de su propio gen (unión al operador repA) a iniciar la replicación del plásmido pPS10 (unión a los iterones en oriV) y, finalmente, a inhibir ésta al asociar moléculas del plásmido a través de un puente de naturaleza amiloide (flechas rojas). B) Diversos ligandos naturales y sintéticos promueven o inhiben la transformación del dominio WH1 en fibras amiloides, permitiendo así controlar ese proceso. C) La secuencia amiloidogénica en RepA-WH1 puede ser utilizada para promover la agregación de otras proteínas, generando así priones sintéticos en levaduras y bacterias. D) RepA-WH1, fusionada a un marcador fluorescente (mCherry), permite caracterizar su agregación amiloide y propagación epigenética como prion en E. coli, así como trazar las vías responsables de su citotoxicidad, comunes a las descritas en las mitocondrias en las enfermedades neurodegenerativas
Tras la caracterización de RepA-WH1 como una proteína priónica, otros investigadores identificaron in silico en los proteomas bacterianos proteínas adicionales como potenciales priones (57), dos de las cuales se han caracterizado además in vivo: los dominios intrínsecamente desordenados del terminador transcripcional Rho en Clostridium botulinum (84) y de la proteína de unión al DNA monocatenario SSB en Campylobacter hominis (85).
Para estudiar las bases moleculares de la toxicidad de RepA-WH1 en E. coli, la transcriptómica diferencial y el análisis proteómico de las proteínas atrapadas con los agregados de RepA-WH1, validados por diversos ensayos funcionales, nos permitieron trazar las rutas sistémicas de la toxicidad del amiloide (86). Como se había observado in vitro (véase más arriba) (72), RepA-WH1 construye poros en la membrana bacteriana interna, drenando así la fuerza protón-motriz, lo que reduce la síntesis de ATP y cualquier transporte acoplado (en particular, la captación de hierro mediada por sideróforos). Como último recurso, la expresión de una deshidrogenasa alternativa (NdhII), menos eficiente en la respiración que el complejo I, genera especies reactivas del oxígeno (ROS) a través de su autooxidación. Una fracción del proteoma de E. coli, que incluye enzimas (KatE) y factores de transcripción (OxyR, RpoS) que forman parte de la respuesta bacteriana al estrés oxidativo o de la envuelta celular, coagrega con RepA-WH1, contribuyendo así a la muerte de las bacterias (86) (Figura 2D). En su conjunto, este cuadro se asemeja a algunas de las vías de toxicidad descritas para las mitocondrias, cuyo origen evolutivo es bacteriano, en la neurodegeneración amiloide (87).
Para cumplir todas las características de un prion, una proteína amiloide también debe ser transmisible horizontalmente, es decir, entre células donantes y receptoras de distinto linaje como lo hace un agente infeccioso (41). La transmisibilidad horizontal de RepA-WH1 se probó en células de mamífero cultivadas (88). En primer lugar, las mismas variantes de RepA-WH1 que se habían ensayado en bacterias (véase más arriba) se expresaron en el citosol de células murinas: mientras que las que incluían la mutación hiperamiloidogénica A31V formaron agregados amiloides citotóxicos, la proteína silvestre (wt) permaneció soluble y las células permanecieron viables. A continuación, las fibras RepA-WH1(A31V) ensambladas in vitro, o las células murinas que las expresaban, se cocultivaron, como donantes de agregados amiloides, con células receptoras humanas que expresaban de forma estable y soluble RepA-WH1(wt), fusionada a una segunda proteína fluorescente distinta. En ambos casos, las partículas RepA-WH1 fueron captadas fácilmente por las células receptoras pero, a diferencia de CsgA/curli con la α-sinucleína (véase más arriba) (51-54), la agregación intracelular sólo se produjo en las receptoras que expresaban RepA-WH1(wt) soluble, que podía moldearse como amiloide al unirse a los agregados una vez internalizados (88). Cabe destacar que la mera captación de agregados de RepA-WH1 por las células de mamífero no era tóxica: puesto que esa proteína no tiene similitud de secuencia significativa con ninguna proteína del proteoma humano, se requirió modificar genéticamente las células receptoras para que éstas expresaran heterólogamente RepA-WH1. Esto constituye una prueba de la bioseguridad del prion bacteriano sintético.
4. CONCLUSIONES
Los microorganismos que componen la microbiota son una fuente virtualmente inagotable de moléculas moduladoras de la fisiología de nuestro organismo, a través de cauces de comunicación e integración que han evolucionado conjuntamente, siendo determinantes tanto para una correcta homeostasis como, en condiciones de disbiosis, para el desarrollo de enfermedades, entre las que aquí se han glosado las neurodegenerativas. Las proteínas con potencial amiloidogénico generadas por la microbiota constituyen una segunda familia de factores que impactan directamente sobre la neurodegeneración, por su contrastada capacidad para moldear en las terminaciones nerviosas intestinales y en el SNC la agregación amiloide de proteínas cuyo plegamiento anómalo está implicado en las enfermedades neurodegenerativas. Nuestros estudios de biología sintética sobre la proteína amiloidogénica intracelular plasmídica RepA-WH1 ilustran el potencial de un amiloide bacteriano sintético como modelo mínimo “genérico” para comprender la biología de los priones y desarrollar nuevas estrategias para su utilización en biotecnología y biomedicina.
El novelista escocés Robert L. Stevenson (1850-1894), en su clásica novela “El extraño caso del Dr. Jekyll y Mr. Hyde” (1886), primera recreación literaria de una patología causada por la adicción a las drogas, describía cómo una misma persona (en nuestro caso, digamos proteína) podía tener dos personalidades (v.g., conformaciones) alternativas, una sana y sociable (esto es, funcional) y otra enferma y asocial (amiloide disfuncional), siendo la transformación de la primera en la segunda lenta e irreversible. Esperemos que, en el caso de las enfermedades neurodegenerativas, ese destino trágico pueda ser revertido gracias a la investigación sobre los amiloides en la que, a buen seguro, los microorganismos desempeñarán un papel fundamental.
Agradecimientos
El trabajo en el grupo del autor es parte del proyecto de I+D+i con referencia PID2021-124866OB-I00, financiado por MICIU/AEI/10.13039/501100011033, “FEDER, una manera de hacer Europa”. El autor está en deuda de gratitud con sus maestros científicos, y con sus compañeros de laboratorio (estudiantes, doctorandos, posdoctorales, y técnicos) y colaboradores, tanto en el CSIC como fuera del mismo, por la excepcional labor científica de éstos a lo largo de casi cuatro décadas (1987-2024). Este artículo está dedicado a la familia del autor por su afecto, estímulo, apoyo y comprensión.
5. REFERENCIAS
- Proctor LM, Creasy HH, Fettweis JM, Lloyd-Price J, Mahurkar A, Zhou W, et al. The integrative human microbiome project. Nature. 2019;569:641-8.
- Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017;20:145–55.
- Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e10002533.
- Lotz SK, Blackhurst BM, Reagin KL, Funk KE. Microbial infections are a risk factor for neurodegenerative diseases. Front Cell Neurosci. 2021;15:691136.
- Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. 2016;167:915–32.
- De la Fuente-Nuez C, Torres-Meneguetti B, Franco OL, Lu TK. Neuromicrobiology: How microbes influence the brain. ACS Chem Neurosci. 2018;9:141–50.
- Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P. Triggers, facilitators, and aggravators: Redefining Parkinson’s disease pathogenesis. Trends Neurosci. 2019;42:4-13.
- Bana B, Cabreiro F. The microbiome and aging. Annu Rev Genet. 2019;53:239–61.
- Zhang H, Chen Y, Wang Z, Xie G, Liu M, Yuan B, et al. Implications of gut microbiota in neurodegenerative diseases. Front Immunol. 2022;13:785644.
- Wilson DM III, Cookson MR, Van den Bosch L, Zetterberg H, Holtzman DM, Dewachter I. Hallmarks of neurodegenerative diseases. Cell. 2023;186:693–714
- Clerigué-Louzado J, Martín-Cámara O. La neurodegeneración desde un punto de vista holístico. An R Acad Nac Farm. 2023;89:327-64.
- Schmidt TSB, Raes J, Bork P. The human gut microbiome: From association to modulation. Cell. 2018;172:1198–215.
- Cannon T, Gruenheld S. Microbes and Parkinson’s disease: from associations to mechanisms. Trends Microbiol. 2022;30:749-60.
- Friedland RP. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J Alzheimers Dis. 2015;45:349–62.
- Nicolas GR, Chang PV. Deciphering the chemical lexicon of host–gut microbiota interactions. Trends Pharmacol Sci. 2019;40:430–5.
- Choi H, Mook-Jung I. Functional effects of gut microbiota-derived metabolites in Alzheimer’s disease. Curr Op Neurobiol. 2023;81:102730
- Fülling C, Dinan TG, Cryan JF. Gut microbe to brain signaling: What happens in vagus. Neuron. 2019;101:998–1002.
- Morais LH, Schreiber HL IV, Mazmanian SK. The gut microbiota–brain axis in behaviour and brain disorders. Nat Rev Microbiol. 2021;19:241-55.
- Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med. 2016;22:586–97.
- Toyofuku M, Nomura N, Eberl L. Types and origins of bacterial membrane vesicles. Nat Rev Microbiol. 2019;17:13-24.
- Ñahui Palomino RA, Vanpouille C, Costantini PE, Margolis L. Microbiota–host communications: Bacterial extracellular vesicles as a common language. PLoS Pathog. 2021;17:e1009508.
- Xie J, Haesebrouck F, Van Hoecke L, Vandenbroucke RE. Bacterial extracellular vesicles: An emerging avenue to tackle diseases. Trends Microbiol. 2023;31:1206-24.
- Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469–80.
- Rosario D, Bidkhori G, Lee S, Bedarf J, Hildebrand F, Le Chatelier E. et al. Systematic analysis of gut microbiome reveals the role of bacterial folate and homocysteine metabolism in Parkinson’s disease. Cell Rep. 2021;34:108807.
- J. Soscia, J. E. Kirby, K. J. Washicosky, S. M. Tucker, M. Ingelsson, B. Hyman, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505.
- Spitzer P, Condic M, Herrmann M, Oberstein TJ, Scharin-Mehlmann M, Gilbert DF, et al. Amyloidogenic amyloid-beta-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci Rep. 2016;6:32228.
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5:eaau3333.
- Liu S, Butler CA, Ayton S, Reynolds EC, Dashper SG. Porphyromonas gingivalis and the pathogenesis of Alzheimer’s disease. Crit Rev Microbiol. 2023;doi: 10.1080/1040841X.2022.2163613.
- Seo D, O’Donnell D, Jain N, Ulrich JD, Herz J, Li Y, et al. ApoE isoform– and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science. 2023;379:eadd1236.
- Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci Rep. 2016;6:30028.
- Grabrucker S, Marizzoni M, Silajdžić E, Lopizzo N, Mombelli E, Nicolas S, et al. Microbiota from Alzheimer’s patients induce deficits in cognition and hippocampal neurogenesis. Brain. 2023; 1-19.
- Blacher E, Bashiardes S, Shapiro H, Rothschild D, Mor U, Dori-Bachash M, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature. 2019:572:474–80.
- Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. 2018;174:1368–405.
- Suez J, Elinav E. The path towards microbiome-based metabolite treatment. Nat Microbiol. 2017;2:17075.
- Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE. A new era for understanding amyloid structures and disease. Nat Rev Mol Cell Biol. 2018;19:755–73.
- Walker LC, Jucker M. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci. 2018;21:1341–9.
- Sawaya MR, Hughes MP, Rodriguez JA, Riek R, Eisenberg DS. The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell. 2021;184:4857–73.
- Pinheiro F, Santos J, Ventura S. AlphaFold and the amyloid landscape. J Mol Biol. 2021;433:167059.
- Ragonis-Bachar P, Axel G, Blau S, Ben-Tal N, Kolodny R, Landau M. What can AlphaFold do for antimicrobial amyloids? Proteins. 2024;92:265-81.
- Louros N, Schymkowitz J, Rousseau F. Mechanisms and pathology of protein misfolding and aggregation. Nat Rev Mol Cell Biol. 2023;24:912-33.
- Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nat Rev Genet. 2018;19:405–18.
- Taglialegna A, Lasa I, Valle J. Amyloid structures as biofilm matrix scaffolds. J Bacteriol. 2016;198:2579-88.
- Van Gerven N, Van der Verren SE, Reiter DM, Remaut H. The role of functional amyloids in bacterial virulence. J Mol Biol. 2018;430:3657–84
- Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851-5.
- Deshmukh M, Evans ML, Chapman MR. Amyloid by design: Intrinsic regulation of microbial amyloid assembly. J Mol Biol. 2018;430:3631–41.
- Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, Hein KL, et al. Functional amyloid in Pseudomonas. Mol Microbiol. 2010;77:1009-20.
- Taglialegna A, Navarro S, Ventura S, Garnett JA, Matthews S, Penades JR, et al. Staphylococcal Bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathog. 2016;12:e1005711.
- El Mammeri N, Hierrezuelo J, Tolchard J, Cámara-Almirón J, Caro-Astorga J, Álvarez-Mena A, et al. Molecular architecture of bacterial amyloids in Bacillus biofilms. FASEB J. 2019;33:12146–63.
- Rayan B, Barnea E, Khokhlov A, Upcher A, Landau M. Differential fibril morphologies and thermostability determine functional roles of Staphylococcus aureus PSMα1 and PSMα3. Front Mol Biosci. 2023;10:1184785.
- Evans ML, Chorell E, Taylor JD, Åden J, Götheson A, Li F, et al. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol Cell. 2015;57:445-55.
- Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep. 2016;6:34477.
- Wang C, Lau CY, Ma F, Zheng C. Genome-wide screen identifies curli amyloid fibril as a bacterial component promoting host neurodegeneration. Proc Natl Acad Sci USA. 2021;118:e2106504118.
- Goya ME, Xue F, Sampedro-Torres-Quevedo C, Arnaouteli S, Riquelme-Dominguez L, Romanowski A, et al. Probiotic Bacillus subtilis protects against alpha-synuclein aggregation in C. elegans. Cell Rep. 2020;30:367–80.
- Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, Shastri GG, et al. A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. eLife. 2020;9:e53111.
- Meng L, Liu C, Li Y, Chen G, Xiong M, Yu T, et al. The yeast prion protein Sup35 initiates α-synuclein pathology in mouse models of Parkinson’s disease. Sci Adv. 2023;9:eadj1092.
- Schmit KJ, Garcia P, Sciortino A, Aho VTE, Pardo-Rodriguez B, Thomas MH, et al. Fiber deprivation and microbiome-borne curli shift gut bacterial populations and accelerate disease in a mouse model of Parkinson’s disease. Cell Rep. 2023;42:113071.
- Iglesias V, de Groot NS, Ventura S. Computational analysis of candidate prion-like proteins in bacteria and their role. Front Microbiol. 2015;6:1123.
- Seira Curto J, Dominguez Martinez A, Sotillo Sotillo P, Serrat Garcia M, Fernandez MR, Sanchez de Groot N. Microbiome-derived prion-like proteins and their potential to trigger cognitive dysfunction. bioRxiv. 2023;doi:10.1101/2023.10.19.563052.
- Vendrell-Fernández S, Lozano-Picazo P, Cuadros-Sánchez P, Tejero-Ojeda MM, Giraldo R. Conversion of the OmpF porin into a device to gather amyloids on the E. coli outer membrane. ACS Synth Biol. 2022;11:655-67.
- Taleb NN. El cisne negro: El impacto de lo altamente improbable. Ed. Paidós Ibérica. 2011; ISBN: 9788449326622.
- Giraldo R, Fernández-Tresguerres ME. Twenty years of the pPS10 replicon: Insights on the molecular mechanism for the activation of DNA replication in iteron-containing bacterial plasmids. Plasmid. 2004;52:69-83.
- Gasset-Rosa F, Díaz-López T, Lurz R, Prieto A, Fernández-Tresguerres ME, Giraldo R. Negative regulation of pPS10 plasmid replication: Origin pairing by zipping-up DNA-bound RepA monomers. Mol Microbiol. 2008;68:560-72.
- Giraldo R, Fernández-Tornero C, Evans PR, Díaz-Orejas R, Romero A. A conformational switch between transcriptional repression and replication initiation in the RepA dimerization domain. Nat Struct Biol. 2003;10:565–71.
- Díaz-López T, Lages-Gonzalo M, Serrano-López A, Alfonso C, Rivas G, Díaz-Orejas R, et al. Structural changes in RepA, a plasmid replication initiator, upon binding to origin DNA. J Biol Chem. 2003;278:18606-16
- Díaz-López T, Dávila-Fajardo C, Blaesing F, Lillo MP, Giraldo R. Early events in the binding of the pPS10 replication protein RepA to single iteron and operator DNA sequences. J Mol Biol. 2006;364:909-20.
- Giraldo R, Andreu JM, Díaz-Orejas R. Protein domains and conformational changes in the activation of RepA, a DNA replication initiator. EMBO J. 1998;17:4511-26.
- Molina-García L, Gasset-Rosa F, Moreno-del Álamo M, Fernández-Tresguerres ME, Moreno-Díaz de la Espina S, Lurz R, et al. Functional amyloids as inhibitors of plasmid DNA replication. Sci Rep. 2016;6:25425.
- Giraldo R. Defined DNA sequences promote the assembly of a bacterial protein into distinct amyloid nanostructures. Proc Natl Acad Sci USA. 2007;104:17388–93.
- Torreira E, Moreno-del Álamo M, Fuentes-Perez ME, Fernández C, Martín-Benito J, Moreno-Herrero F, et al. Amyloidogenesis of bacterial prionoid RepA-WH1 recapitulates dimer to monomer transitions of RepA in DNA replication initiation. Structure. 2015;23:183–9.
- Pantoja-Uceda D, Oroz J, Fernández C, de Alba E, Giraldo R, Laurents DV. Conformational priming of RepA-WH1 for functional amyloid conversion detected by NMR spectroscopy. Structure. 2020;28:336–47.
- Gasset-Rosa F, Maté MJ, Dávila-Fajardo C, Bravo J, Giraldo R. Binding of sulphonated indigo derivatives to RepA-WH1 inhibits DNA-induced protein amyloidogenesis. Nucleic Acids Res. 2008;36:2249¬56.
- Fernández C, Núñez-Ramírez R, Jiménez M, Rivas G, Giraldo R. RepA-WH1, the agent of an amyloid proteinopathy in bacteria, builds oligomeric pores through lipid vesicles. Sci Rep. 2016;6:23144.
- Fernández C, González-Rubio G, Langer J, Tardajos G, Liz-Marzán LM, Giraldo R, et al. Nucleation of amyloid oligomers by RepA-WH1 prionoid-functionalized gold nanorods. Angew Chem Int Ed. 2016;55:11237–41.
- Moreno-del Álamo M, Moreno-Díaz de la Espina S, Fernández-Tresguerres ME, Giraldo R. Pre-amyloid oligomers of the proteotoxic RepA-WH1 prionoid assemble at the bacterial nucleoid. Sci Rep. 2015;5:14669.
- Giraldo R. Optogenetic navigation of routes leading to protein amyloidogenesis in bacteria. J Mol Biol. 2019;431:1186–202.
- Gasset-Rosa F, Giraldo R. Engineered bacterial hydrophobic oligopeptide repeats in a synthetic yeast prion, [REP-PSI+]. Front Microbiol. 2015;6:311.
- Harvey ZH, Chen Y, Jarosz DF. Protein-based inheritance: Epigenetics beyond the chromosome, Mol Cell. 2018;69:195-202.
- Molina-García L, Giraldo R. Enabling stop codon read-through translation in bacteria as a probe for amyloid aggregation. Sci Rep. 2017;7:11908.
- Fernández-Tresguerres ME, Moreno-Díaz de la Espina S, Gasset-Rosa F, Giraldo R. A DNA-promoted amyloid proteinopathy in Escherichia coli. Mol Microbiol. 2010;77:1456–69.
- Molina-García L, Giraldo R. Aggregation interplay between variants of the RepA-WH1 prionoid in Escherichia coli. J Bacteriol. 2014;196:2536–42.
- Schramm FD, Schroeder K, Jonas K. Protein aggregation in bacteria. FEMS Microbiol Rev. 2020;44:54–72.
- Gasset-Rosa F, Coquel AS, Moreno-del Álamo M, Chen P, Song X, Serrano AM, et al. Direct assessment in bacteria of prionoid propagation and phenotype selection by Hsp70 chaperone. Mol Microbiol. 2014;91:1070–87.
- Fernández C, Giraldo R. Modulation of the aggregation of the prion-like protein RepA-WH1 by chaperones in a cell-free expression system and in cytomimetic lipid vesicles. ACS Synth Biol. 2018;7:2087–93.
- Yuan AH, Hochschild A. A bacterial global regulator forms a prion. Science. 2017;355:198–201.
- Jager K, Orozco-Hidalgo MT, Springstein BL, Joly-Smith E, Papazotos F, McDonough EK, et al. Measuring prion propagation in single bacteria elucidates a mechanism of loss. Proc Natl Acad Sci USA. 2023;120:e2221539120.
- Molina-García L, Moreno-del Álamo M, Botias P, Martín-Moldes Z, Fernández M, Sánchez-Gorostiaga A, et al. Outlining core pathways of amyloid toxicity in bacteria with the RepA-WH1 prionoid. Front Microbiol. 2017;8:539.
- Calabrese G, Molzahn C, Mayor T. Protein interaction networks in neurodegenerative diseases: From physiological function to aggregation. J Biol Chem. 2022;298:102062.
- Revilla-García A, Fernández C, Moreno-del Álamo M, de los Ríos V, Vorberg IM, Giraldo R. Intercellular transmission of a synthetic bacterial cytotoxic prion-like protein in mammalian cells. mBio. 2020;11:e02937-19.
