1. INTRODUCCIÓN
Los organismos inferiores y superiores constan de una maquinaria celular específica con la que mantienen un equilibrio entre la expresión y la degradación de proteínas para el correcto funcionamiento celular. En células eucariotas, los principales sistemas son el Sistema Ubiquitina-Proteasoma (UPS) y el sistema endo-lisosomal (1).
El UPS está formado por un conjunto de enzimas que se encarga del marcaje de proteínas intracelulares con moléculas de ubiquitina, y por la proteasoma 26S cuya función es el reconocimiento de estas proteínas ubiquitinadas y su posterior degradación (2). Por otro lado, el papel del sistema endo-lisosomal es la degradación de proteínas extracelulares o de membrana que han sido previamente glicosiladas (3). La disfunción de ambos sistemas se ha visto relacionada con el origen de procesos patológicos tales como el cáncer y enfermedades neurodegenerativas (especialmente en la enfermedad de Alzheimer), por lo que tanto el estudio de su funcionamiento, como la búsqueda de ligandos que puedan modular su actividad, se ha convertido en uno de los campos que más interés está despertando en los últimos años en el ámbito del descubrimiento de fármacos (2).
1.1 Sistema endo-lisosomal para la degradación de proteínas
En las células eucariotas, las vías endocítica y secretora están organizadas en varios compartimentos celulares conectados a través de pequeñas vesículas. Entre sus funciones fisiológicas se encuentran el mantenimiento de la composición lipídica y proteica de la célula, el transporte de componentes a/desde la membrana plasmática, así como el control del tamaño y composición de distintos orgánulos. Ambas vías presentan un componente común, el lisosoma. Estos son orgánulos ácidos encargados del metabolismo degradativo de macromoléculas extra e intracelulares en procesos endocíticos, fagocíticos y autofágicos, respectivamente (4). Además, están formados por un conjunto de enzimas hidrolasas, proteasas, glicosidasas y lipasas en las que recae su capacidad degradativa (5).
La vía endo-lisosomal (Figura 1) comienza con la invaginación de la membrana plasmática, ya sea a través de un mecanismo mediado por clatrina o por receptores de membrana, y la consecuente formación de una pequeña vesícula en el interior de la célula. Esta vesícula se fusiona con una endosoma temprano, cuyo interior ligeramente ácido propicia la disociación de la macromolécula extracelular endocitada y su receptor. En este punto, el receptor puede ser devuelto a la membrana o incorporarse a la red trans-Golgi (TGN; “trans-Golgi Network”). Mientras tanto, la endosoma temprano madura a endosoma tardío donde comienza la degradación de la carga endosomal, que terminará en los lisosomas (6).
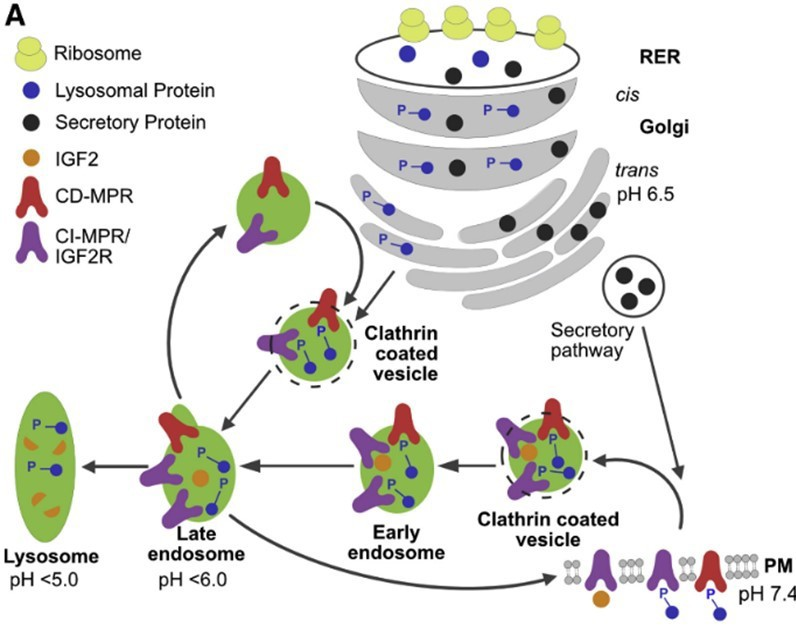
Figura 1. Representación gráfica de la vía endo-lisosomal mediada por los receptores CI-MPR y CD-MPR. Imagen extraída del artículo de L. J. Olson et al. (5)
Las enzimas lisosomales se sintetizan en el retículo endoplasmático y el complejo de Golgi y, a continuación, entran a formar parte de la TGN. Para que estas enzimas sean transportadas hasta los lisosomas deben estar glicosiladas con residuos de manosa-6- fosfato (M6P), o bien, con fosfodiésteres de manosa-6-fosfato N-acetilglucosamina (Man-P-GlcNAc),(7) que harán que sean reconocidas y transportadas por el receptor de manosa-6-fosfato independiente de cationes (CI-MPR) o por el receptor de manosa-6- fosfato dependiente de cationes (CD-MPR).(8) Además, a pH fisiológico, CI-MPR también puede encontrarse en la membrana plasmática y mediar la endocitosis tanto de hidrolasas lisosomales extracelulares como de otras proteínas no glicosiladas (Figura 1) (6).
1.2 Receptor de manosa-6-fosfato independiente de cationes (CI-MPR)
CI-MPR es un receptor transmembrana de 300 kDa implicado en diversos procesos fisiológicos relacionados con la génesis lisosomal, el tráfico intracelular y extracelular de proteínas, así como la regulación del crecimiento y movilidad celular (5). Asimismo, ha sido estudiado como diana para el tratamiento del cáncer y enfermedades lisosomales (9).
Las principales localizaciones subcelulares de este receptor son el aparato de Golgi (90%) y la membrana plasmática (10%), en esta última cumple una función fundamentalmente endocítica (10). Además de internalizar hidrolasas extracelulares glicosiladas, presenta como sustratos al factor de crecimiento de insulina II (IGF2), plasminógeno y fibronectina, entre otros (11). IGF2 es un pequeño polipéptido similar estructuralmente a la insulina que lleva a cabo funciones relacionadas con la migración, diferenciación, crecimiento y supervivencia celular debido a su interacción con el receptor de factor de crecimiento de insulina I (IGF1R) (12). Dada la relevancia biológica de IGF2, CI-MPR también puede recibir el nombre de receptor del factor de crecimiento de insulina II (IGF2R), puesto que es capaz de disminuir la concentración extracelular de este a nivel local y circulante (10).
CI-MPR (Figura 2a) se compone principalmente de 15 dominios extracelulares de unos 150 aminoácidos cada uno que presentan una estructura conservada de barril b y a los que se les denomina dominios homólogos del receptor de manosa-6-fosfato (MRH), puesto que presentan entre un 15 y 38% de identidad de secuencia (13). A pesar de esto, tan solo los dominios MRH3, 5, 9 y 15 reconocen azúcares derivados de manosa.(14) Su sitio de unión está altamente conservado y se compone principalmente por la secuencia “QREY” (Glutamina, Arginina, ácido Glutámico, Tirosina), clave para el reconocimiento de este azúcar. Los aminoácidos glutamina (Q), arginina (R) y tirosina (Y) están localizados en las láminas b3, b8 y b9, respectivamente, mientras que el ácido glutámico (E) lo está en el Loop D (entre las láminas b8 y b9) (Figura 2b). No obstante, como se puede observar en el alineamiento de secuencias mostrado en la Figura 3a, los dominios MRH3, 5 y 9 presentan la secuencia “QREY” completa, mientras que MRH15 posee un residuo de valina en vez de arginina (15).
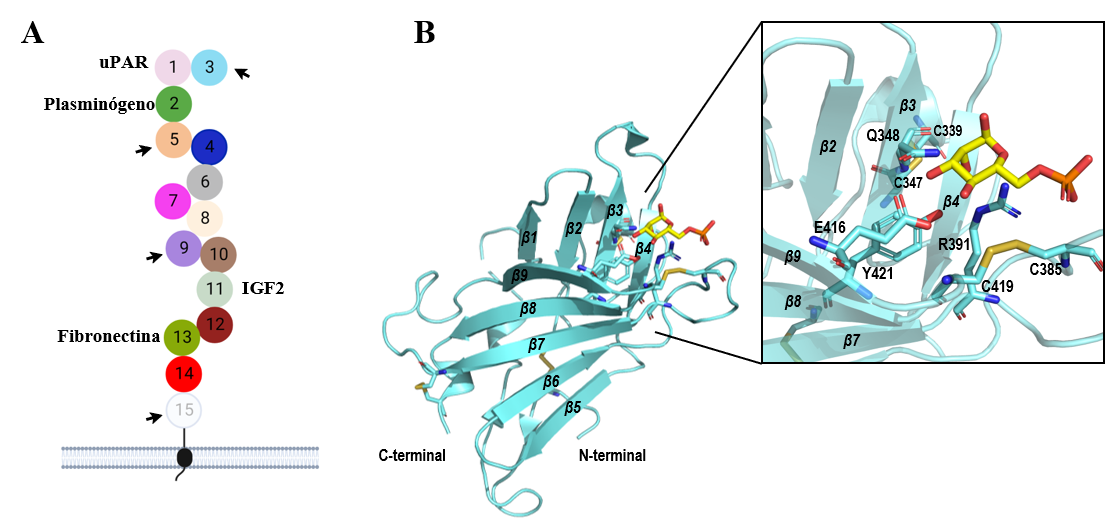
Figura 2. (A) Esquema del receptor CI-MPR adaptado del trabajo de L. J. Olson et al. (5) Las flechas indican la posición de los dominios de reconocimiento de azúcares. (B) Estructura general de un dominio de reconocimiento de azúcares (dominio MRH3, extraído del PDB código 1SZ0). Esta imagen ha sido generada con PyMOL (16)
A pesar de su elevada homología, ligeros cambios estructurales en el sitio de unión a manosa hacen que estos dominios difieran en cuanto a su especificidad por el sustrato. Los dominios MRH3 y MRH9 reconocen específicamente a monoésteres de M6P,(14) (17) mientras que el dominio MRH5 es exclusivo de fosfodiésteres de Man-P-GlcNAc,(7) y MRH15 reconoce tanto a monoésteres como a fosfodiésteres (15). Los dominios MRH5 y MRH15 no presentan un enlace disulfuro adyacente al sitio de unión de manosa, mientras que MRH3 y MRH9 sí, por lo que el bolsillo es de mayor tamaño y puede abarcar ligandos más voluminosos como los fosfodiésteres de Man-P-GlcNAc.(7, 15) Por otro lado, los dominios MRH3 y MRH9 contienen aminoácidos clave para la interacción con monoésteres de M6P. El dominio MRH3 presenta un residuo de S386 (código PDB 1SZ0) que interacciona con el grupo fosfato de M6P, cuya posición ocupa la H1320 (código PDB 6Z32) en el dominio MRH9 (Figura 3b) (17). El residuo H1320 ha sido descrito como un aminoácido esencial para la especificidad de este dominio por monoésteres de M6P.(18) Asimismo, en el caso de MRH3, la cooperación entre los dominios MRH1-3 hace que incremente en 1000 veces la afinidad por este sustrato.(7) En el caso de MRH15, aunque no se dispone de estructura cristalográfica, ha sido estudiada la naturaleza de la interacción de este dominio con M6P. En 2015, L. J. Olson et al. (15) construyeron un modelo del dominio MRH15 y mediante estudios de mutagénesis establecieron que el residuo R2170, que se encontraba cerca del sitio de unión a manosa, era clave para el reconocimiento de M6P.
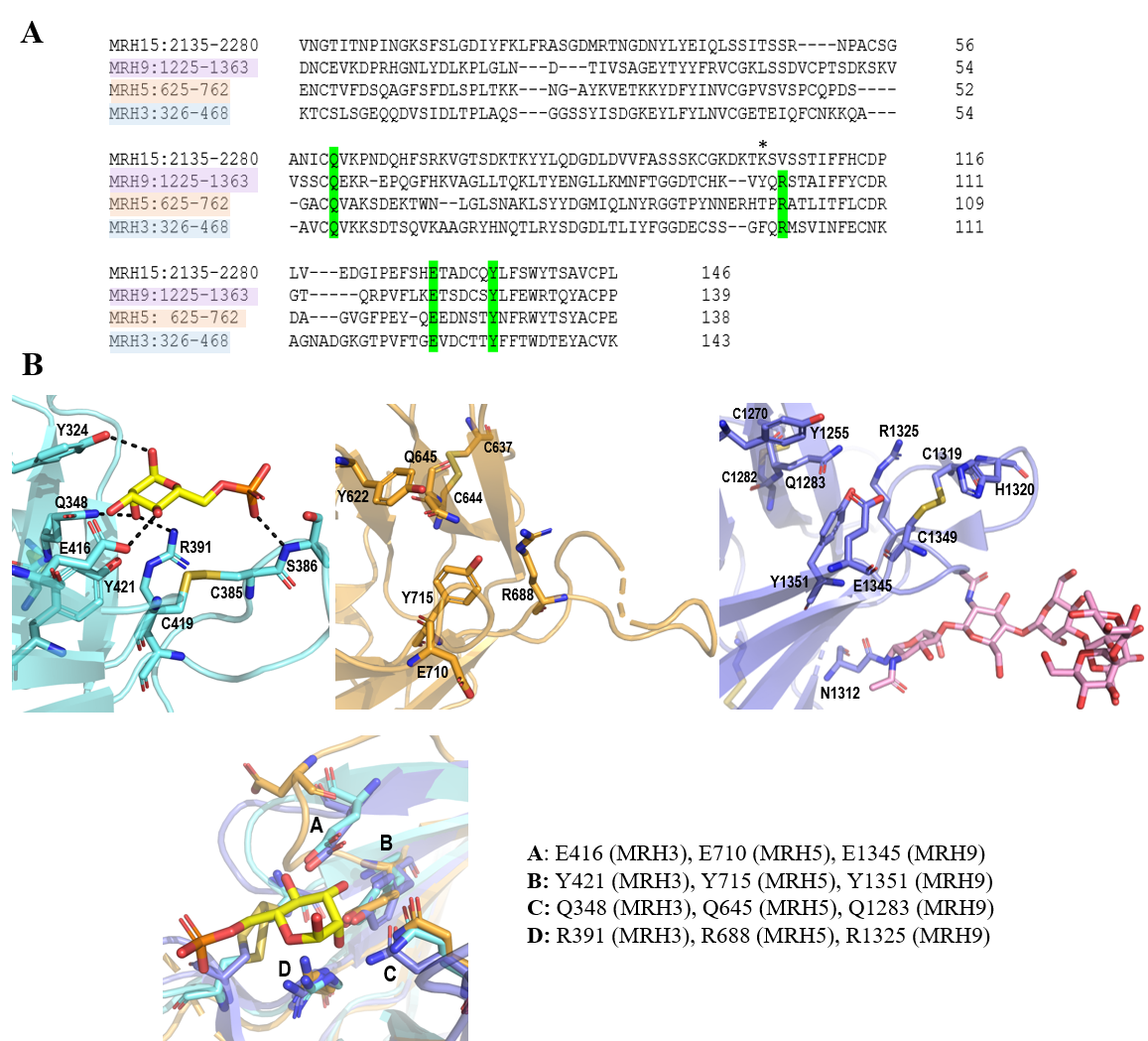
Figura 3. (A) Alineamiento de las secuencias de los dominios MRH3, MRH5, MRH9 y MRH15 llevada a cabo con Clustal Omega (19). (B) Estructuras tridimensionales de los dominios MRH3 (izquierda, azul), MRH5 (centro, naranja) y MRH9 (derecha, violeta) depositadas en el PDB bajo los códigos 1SZ0, 6V02 y 6Z32, respectivamente. El ligando M6P se muestra en color amarillo (código PDB 1SZ0), mientras que la glicosilación del dominio MRH9 (código PDB 6Z32) aparece en rosa. En la parte inferior, se muestra una superposición del sitio de unión a azúcares donde se destaca la secuencia consenso “QREY”. El dominio MRH15 ha sido omitido porque no se dispone de estructura cristalográfica. Estas imágenes han sido generadas con PyMOL (16).
El resto de los dominios extracelulares de CI-MPR no reconocen azúcares a pesar de su elevada homología. El dominio MRH1 tiene como ligando endógeno al receptor activador de plasminógeno (uPAR), MRH2 al plasminógeno, MRH11 a IGF2 y MRH13 a fibronectina. La función específica que desempeñan los dominios no mencionados es aún desconocida (5, 20).
Existen varias hipótesis en torno a la disposición tridimensional que debe adquirir el receptor para que este pueda ser internalizado en la célula. En 2020, el grupo de investigación de A. Bassim Hassan y M. P. Crump (17) resolvió por primera vez la estructura de los dominios MRH7 a MRH11 humanos utilizando difracción de rayos X (código PDB, 6Z32). En este cristal, el dominio MRH9 se encontraba glicosilado en el residuo D1312 con un oligosacárido de manosa y glucosa, lo que provocaba la formación de un dímero estabilizado a través de las interacciones establecidas entre la glicosilación del dominio MRH9 de un monómero y el sitio de unión a M6P del dominio MRH9 del otro (Figura 4a). Por otro lado, en ese mismo año, X. Li et al. resolvieron por crio-microscopía electrónica (Cryo-EM) la estructura de los dominios MRH4 a MRH14 del receptor CI-MPR bovino (código PDB, 6UM2) unido a IGF2. (21) En este caso, el domino MRH9 no se encuentra dimerizado puesto que en la estructura no se aprecian glicosilaciones, sino que adquiere una “conformación de pistola”. Adicionalmente, se observa que la interacción de IGF2 con el dominio MRH11 podría estar estabilizada por los dominios MRH6 y MRH8 (Figura 4b).
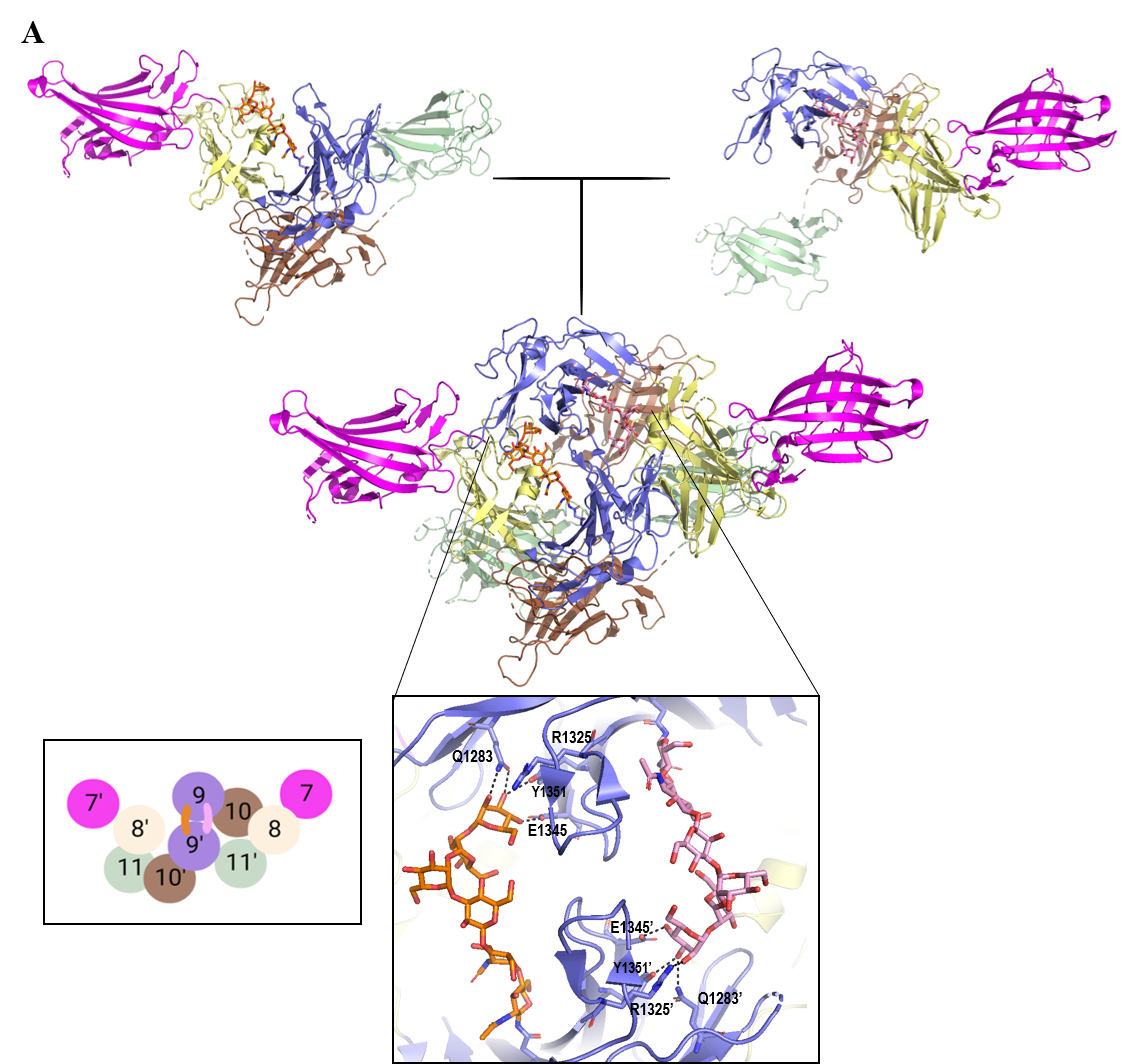
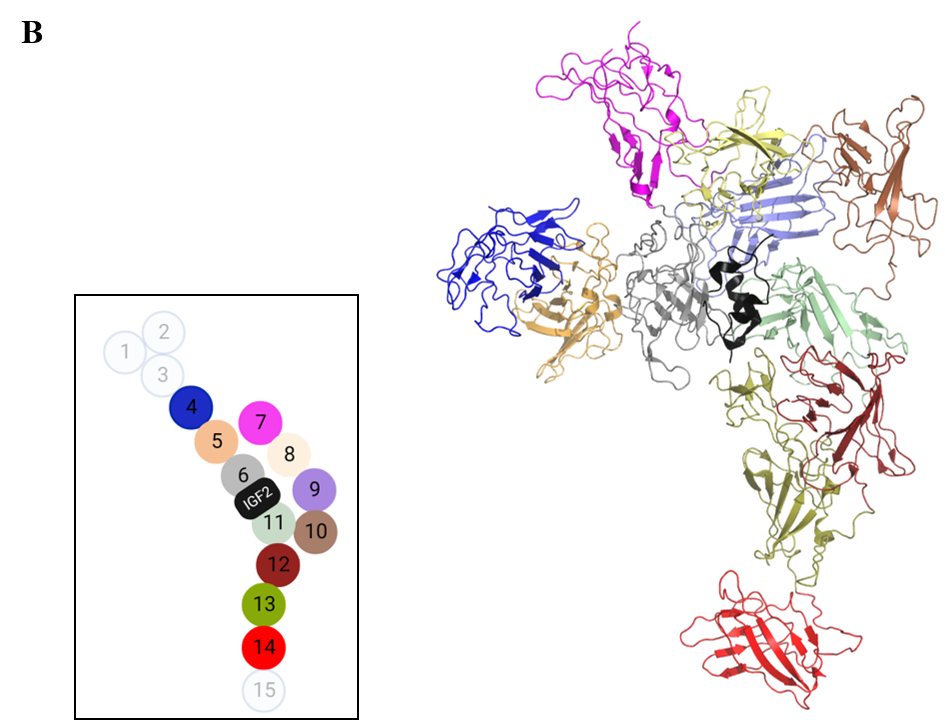
Figura 4. Principales hipótesis de conformación del receptor CI-MPR. (A) “Hipótesis de dimerización (17). Cada monómero contiene los dominios MRH7-MRH11 (código PDB 6Z32). La dimerización se establece mediante el dominio MRH9 de un monómero y el dominio MRH9’ del otro. Su formación es posible gracias a las glicosilaciones presentes en ambos monómeros, el oligosacárido de color rosa corresponde al monómero MRH9, mientras que el naranja a MRH9’. Los enlaces de hidrógeno están representados por líneas negras. (B) Hipótesis de “conformación pistola”. Basada en la estructura tridimensional de los dominios MRH4-14 unidos a IGF2 de origen bovino resueltos por difracción de rayos X (código PDB 6UM2). El esquema es una adaptación del descrito en el trabajo de X. Li et al.(21). Los esquemas se han creado en Biorender.com. Las estructuras en 3D se han obtenido con PyMOL (16).
Debido a la alta complejidad estructural del receptor CI-MPR, su gran tamaño y la gran cantidad de ligandos endógenos con los que interacciona, no se ha llegado a un consenso sobre su mecanismo de internalización. Asimismo, se dispone de escasa información estructural. Un artículo publicado por R. G. MacDonald et al. (11) apoya la hipótesis de dimerización del receptor, demostrando experimentalmente que una serie de ligandos multidentados con residuos de manosa eran capaces de promover la dimerización del receptor y, con ello, la internalización y degradación de IGF2. De este modo, catalogaron a IGF2 como “passenger ligand” o ligando polizón. En esta misma línea, L. M. Ali et al. (22). diseñaron un ligando de este receptor capaz de aumentar su internalización y además estudiaron su comportamiento dinámico en disolución e in silico mediante técnicas de espectroscopía NOESY (“Nuclear Overhauser Effect Spectroscopy”) y dinámica molecular, respectivamente. Este ligando consta de un grupo fluoróforo, seis unidades de γ-péptido y tres colas acabadas en manosa-6-fosfonato (M6Pn), que se unirán a los dominios de reconocimiento de azúcares del receptor (Figura 5). El cambio de M6P por M6Pn no compromete el modo de unión del ligando al receptor CI-MPR y dicha modificación se llevó a cabo principalmente para impedir la degradación del grupo fosfato por las fosfatasas plasmáticas.
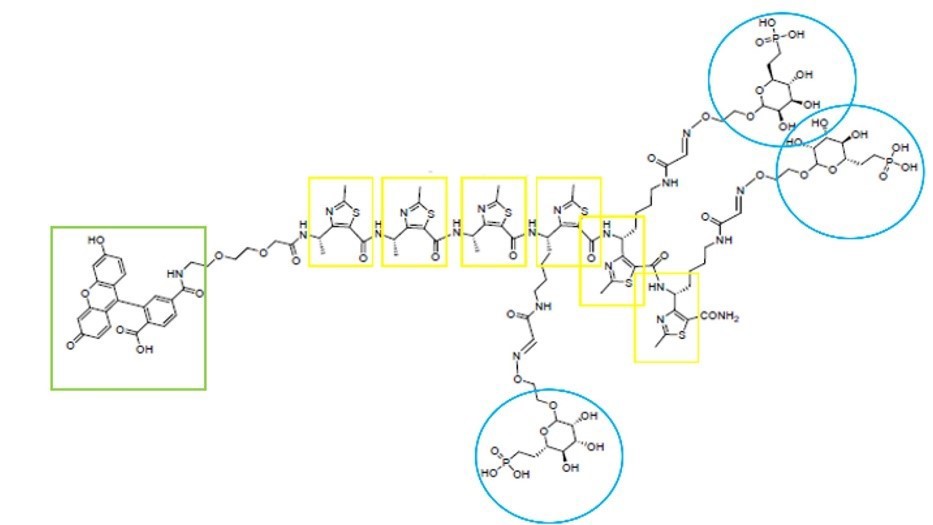
Figura 5. Estructura del ligando del receptor CI-MPR diseñado por L. M. Ali et al. (22). Los residuos de M6Pn se encuentran representados en azul, unidos a ellos a través de un espaciador, se encuentran los monómeros del γ-péptido (amarillo); y, por último, en verde, el grupo fluorescente.
1.3 Estrategias de degradación dirigida a proteínas: LYTACS
El enfoque tradicional para el tratamiento de enfermedades consiste en la modulación de la actividad de una proteína con una molécula pequeña, que puede actuar como agonista o como antagonista. Sin embargo, alrededor del 75% del proteoma humano no es susceptible de ser modulado con moléculas pequeñas, lo que se conoce como “undruggable proteome”, y por ello surgió la estrategia de degradación dirigida de proteínas (“Targeted Protein Degradation”, TPD) (23). Ésta consiste en aprovechar la maquinaria degradativa de las células con el fin de disminuir la concentración de una proteína de interés, ya sea intracelular, extracelular o de membrana (24). Hoy en día, se hace un uso más amplio de la estrategia TPD, pues no se limita al “undruggable proteome”, sino a cualquier proteína terapéuticamente relevante.
Uno de los métodos de degradación selectiva de proteínas más utilizado consiste en el reclutamiento del UPS mediante unas moléculas hereobifuncionales llamadas “PROteolysis TArgeting Chimeras” (PROTACs). Estos compuestos median la ubiquitinación de proteínas citosólicas y su posterior degradación por la proteasoma 26S (25). Por otro lado, recientemente se ha descrito una nueva tecnología con la que se recluta al sistema endo-lisosomal para la degradación de proteínas extracelulares mediante unas moléculas a las que se ha denominado “LYsosome TArgeting Chimeras” (LYTACs). Los LYTACs son capaces de unirse simultáneamente a un receptor transmembrana y a una proteína de interés y así mediar su internalización celular y su posterior degradación lisosomal (26).
Estos receptores pertenecen a la familia de receptores dirigidos al lisosoma (“Lysosome Targeting Receptors”, LTRs) y facilitan el transporte de proteínas a este orgánulo celular. Hasta la fecha, solo el receptor CI-MPR y el “Asialoglycoprotein receptor” (ASPGR) han sido utilizados en la tecnología LYTAC. (9, 27) Ambos son receptores que poseen dominios de reconocimiento a azúcares, no obstante, mientras CI-MPR tiene como sustratos azúcares derivados de M6P (como ya se ha mencionado en el apartado anterior), los sustratos de ASGPR son azúcares derivados de N-acetilgalactosamina (GalNAc). Otra diferencia entre ambos receptores es su distribución tisular, mientras que CI-MPR es un receptor ubicuo, ASGPR es exclusivamente hepático (9). En este trabajo nos centramos en el estudio del receptor CI-MPR.
En el año 2020, C. Bertozzi et al. (28) desarrollaron una prueba de concepto de la tecnología LYTAC. Carloyn Bertozzi ha obtenido el Premio Nobel de Química en el año 2022 por el desarrollo de la química click y la química bioortogonal y sus aplicaciones para el estudio de diferentes sistemas biológicos, entre otros, los LYTACs (29). Para ello, diseñaron una serie de moléculas capaces de unirse simultáneamente a la porción extracelular del receptor CI-MPR y a varias proteínas tanto extracelulares, como de membrana (Figura 6). En primer lugar, diseñaron un LYTAC basado en un polímero de M6P (20-90 unidades) y biotina como ligandos de unión a CI-MPR y NeutrAvidina-647 (proteína fluorescente), respectivamente (Figura 6a). De este modo demostraron la posibilidad de mediar la degradación lisosomal de una proteína usando una molécula pequeña como ligando. Adicionalmente, consiguieron degradar ApoE4, una proteína extracelular implicada en el desarrollo de la enfermedad de Alzheimer, acoplando el polímero de M6P a un anticuerpo anti-apoE4. Además de proteínas extracelulares, lograron la degradación lisosomal del receptor del factor de crecimiento endotelial (EGFR) que se ubica en la membrana plasmática (Figura 6b).
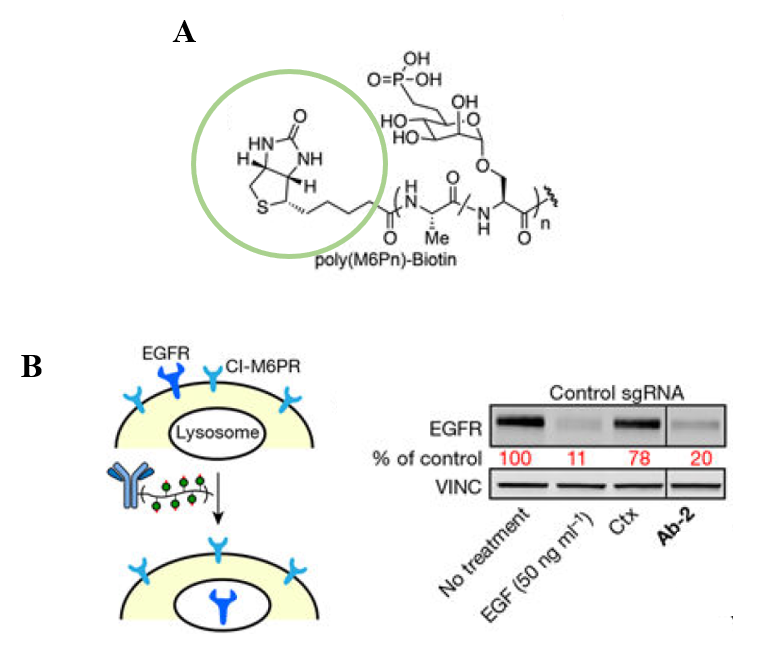
Figura 6. Ejemplos de los primeros LYTACs diseñados por C. Bertozzi et al. (28). (A) Estructura de un LYTAC con un polímero de M6P (poly(M6Pn)) como ligando de unión a CI-MPR y con biotina (verde) como ligando de unión a la proteína fluorescente NeutrAvidina-647. (B) Representación esquemática del mecanismo de acción de un LYTAC que media la degradación lisosomal del receptor de membrana EGFR (izquierda). A la derecha, los resultados de un ensayo Western Blot, en el que se observa degradación de EGFR en presencia del LYTAC (Ab-2). Las imágenes han sido extraídas del artículo de C. Bertozzi et al. (28).
1.4 MMP-2 como posible diana terapéutica de LYTACs
Las Metaloproteasas de la Matriz (MMPs) son una familia de endopeptidasas dependientes de zinc responsables de la remodelación de la matriz extracelular. (30) Estas enzimas juegan un papel fundamental a nivel fisiológico y fisiopatológico, ya que están involucradas en la proliferación, diferenciación, apoptosis celular y angiogénesis, por lo que una desregulación de sus niveles se ve relacionada con la aparición de procesos metastásicos, inflamatorios y de crecimiento tumoral (31).
Esta familia se puede dividir en seis subfamilias en función de la especificidad de sustrato: colagenasas (MMP-1, 8, 13 y 18), gelatinasas (MMP-2 y 9), estromelisinas (MMP-3, 10, 11, 27), matrilisinas (MMP-7 y 26), asociadas a membrana (MMP-14, 15, 16 y 24), metaloelastasas (MMP-12) y otras (MMP-19, 20, 21, 23 y 28) (32, 33).
Las enzimas MMP-2 y MMP-9 degradan el colágeno tipo IV, que es el componente principal de la matriz extracelular (32). MMP-2 es una diana validada para el tratamiento de muchos tipos de cáncer, pues su sobreexpresión está relacionada con la invasión tumoral, metástasis y angiogénesis. (33) Por el contrario, a pesar de su similitud estructural y funcional, MMP-9 está involucrada tanto en procesos oncogénicos como antitumorales por lo que su eficacia como diana contra el cáncer está en entredicho. Por esta razón, diferentes grupos de investigación se centran en el diseño y síntesis de inhibidores selectivos de MMP-2 para el tratamiento del cáncer (34-36).
Este trabajo tiene como finalidad el diseño computacional de un LYTAC para la degradación selectiva de MMP-2. Esta molécula deberá poseer la capacidad de unirse simultáneamente a MMP-2 y al receptor transmembrana CI-MPR, que será el encargado de su internalización y de su transporte hasta los lisosomas, donde se degradará. En este trabajo se han llevado a cabo estudios computacionales y se han alcanzado los siguientes objetivos:
Análisis detallado de las estructuras cristalográficas depositadas en el Protein Data Bank (PDB) (37) para estudiar las diferentes conformaciones que puede adoptar CI-MPR en presencia y ausencia de ligandos endógenos.
Estudio del modo de unión de M6P a los dominios de reconocimiento de azúcares de CI-MPR (MRH3, 5, 9 y 15).
Diseño de un LYTAC basado en la estructura del ligando propuesto por L. M. Ali et al. (22) y un inhibidor de MMP-2 desarrollado previamente descrito (36).
Estudio del comportamiento dinámico y análisis conformacional del LYTAC- MMP2 y posterior comparación con el ligando de CI-MPR descrito por L. M. Ali et al. (22).
2. MATERIALES Y MÉTODOLOGÍA
2.1 Análisis Cristalográfico
En primer lugar, se realizó una búsqueda de las estructuras cristalográficas del receptor CI-MPR en la base de datos PDB (Tabla 1), (37) con el objetivo de analizar los modos de unión de sus ligandos endógenos utilizando PyMOL (16).
Tabla 1. Estructuras del receptor CI-MPR, depositadas en el PDB, que han sido utilizadas para el desarrollo de este trabajo.
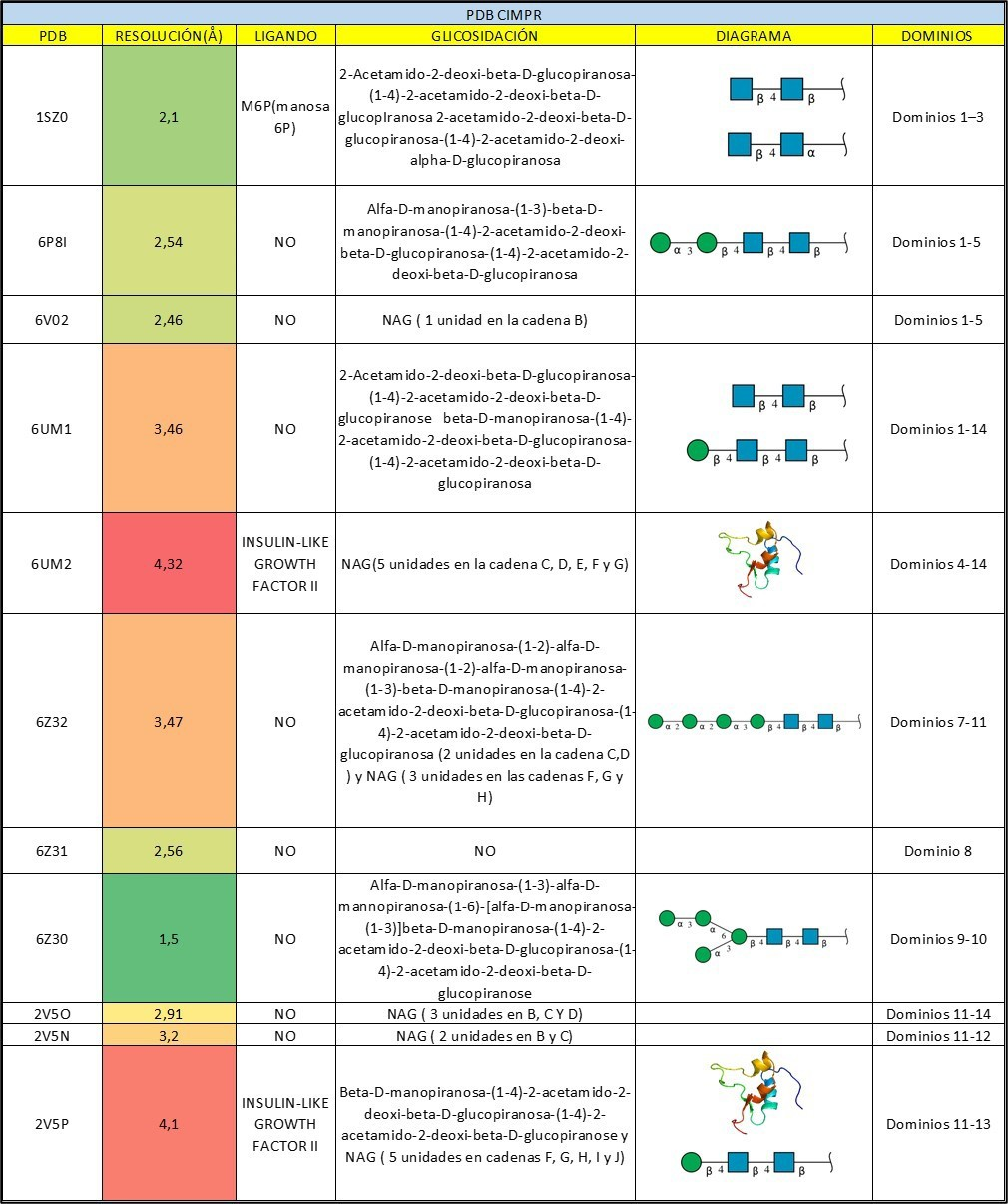
En general, se seleccionaron aquellas estructuras que presentaban mejor resolución, una secuencia de aminoácidos más completa y que, además, incluyeran los ligandos de interés: M6P e IGF2. No obstante, también se analizaron estructuras con una peor resolución pero que aportaban información clave acerca de la disposición espacial de los dominios del receptor, como por ejemplo la estructura con el código 6UM2. Esta estructura fue obtenida por Cryo-EM a pH fisiológico con una resolución de 4.32 Å e incluye los dominios MRH4-14 adoptando una “conformación de pistola”, además, el ligando IGF2 se encuentra unido al dominio MRH11, por lo que se utilizó para analizar su modo de unión.
Con el fin de estudiar el reconocimiento de azúcares por parte del receptor, se analizaron las estructuras :1SZ0, 6V02 y 6Z32. El cristal 1SZ0, de origen bovino, fue obtenido por difracción de rayos X con una resolución de 2.10 Å. Este comprende los dominios N- terminales del receptor (MRH1-3) y se utilizó para estudiar el modo de unión de M6P al dominio MRH3. Por otro lado, el cristal 6V02 (resolución 2.46 Å) engloba a los dominios MRH1-5, pero no presenta al ligando M6P unido al dominio MRH5, por lo que para analizar su modo de unión se tuvo que recurrir a técnicas de docking molecular (Apartado 2.3). Los dominios MRH7-11 están cristalizados y depositados en PDB: 6Z32 (3.47 Å), en el que se observa una dimerización del receptor a través de glicosilaciones pertenecientes al dominio MRH9. Esta estructura apoya la hipótesis de la necesidad de dimerización de CI-MPR para que este pueda ser internalizado por la célula. Por último, no hay depositada ninguna estructura en el PDB del último dominio de reconocimiento de azúcares, dominio MRH15. Por esta razón y por su poca accesibilidad al encontrarse muy próximo a la membrana, se concluyó no estudiarlo como sitio de unión al LYTAC.
2.2 Modelado por homología
El único cristal de un dominio de reconocimiento de azúcares unido a M6P depositado en el PDB es 1SZ0 y este es de origen bovino. Puesto que los dominios MRH3 bovino y humano presentan una homología del 78.9%, se realizó un modelado por homología con el servidor SwissModel (38) utilizando el cristal 1SZ0 como molde. Posteriormente, se comparó el modo de unión de M6P en el cristal y en el molde mediante superposición en PyMOL.
2.3 Docking molecular
Este es un estudio computacional que pretende predecir el modo de unión más favorable de un ligando a su proteína diana, mediante la exploración de las diferentes conformaciones que el ligando puede adoptar con el objetivo final de determinar la afinidad y actividad de estos. Para ello, se prepara una “grid” (39) de un tamaño preestablecido en la proteína diana, la cual consiste en energías de interacción pre- calculadas entre la proteína y una serie de sondas atómicas que exploran el sitio de unión del ligando. Para llevar a cabo este estudio se empleó la interfaz gráfica de la Suite de Schrödinger, MAESTRO (40).
Esta técnica se utilizó para predecir el modo de unión de M6P a los dominios MRH5 y MRH9, puesto que este ligando ya se encuentra unido a MRH3 en el cristal 1SZ0. No obstante, se decidió llevar a cabo el docking de M6P en el dominio MRH3 (1SZ0) para validar el uso de la técnica. Por un lado, el ligando se preparó usando la herramienta LigPrep, (41) en la que se tuvieron en cuenta los dos posibles estados de protonación del grupo fosfato del residuo de manosa (cargas -1 y -2). Por otro lado, se usó la herramienta Protein Preparation Wizard (42) para preparar los dominios MRH3 (1SZ0), MRH5 (6V02) y MRH9 (6Z32), es decir, se añadieron protones, se modelaron las cadenas laterales de aminoácidos que faltaban y se corrigieron los estados de protonación de los residuos de las proteínas a pH 7. Posteriormente, se construyó una grid de tamaño suficiente para abarcar el sitio de unión a manosa de los tres dominios y se centró en la secuencia consenso “QREY”. Como último paso, se realizó el docking con la herramienta Glide (43).
Los resultados obtenidos fueron analizados cualitativamente utilizando PyMOL y cuantitativamente mediante los valores de Docking Score obtenidos para cada una de las poses del ligando en la proteína.
2.4 Dinámica molecular (MD)
La dinámica molecular se basa en cálculos teóricos de mecánica molecular para obtener una trayectoria en la que la posición y velocidad de los diferentes átomos del sistema varían en función del tiempo de simulación. El campo de fuerzas utilizado es AMBER 20 (44). Esta metodología permitió determinar la estabilidad del modo de unión del ligando M6P en el dominio MRH3 (código PDB 1SZ0), así como estudiar el comportamiento dinámico libre en agua del ligando de CI-MPR diseñado por L. M. Ali et al. (22) y el LYTAC-MMP2.
Antes de llevar a cabo las simulaciones de dinámica molecular, los ligandos fueron parametrizados con mecánica cuántica utilizando el programa Gaussian 09 (45). Se optimizó su geometría y, en un segundo paso, se les calculó la distribución de cargas puntuales ajustando el potencial electrostático molecular (MEP) obtenido a un modelo con ayuda de un método intrínseco de este programa.
A continuación, los sistemas (ligando libre o complejo proteína-ligando) fueron preparados utilizando el software de AMBER 20 (44). Puesto que los ligandos contienen un azúcar fosfatado y que el dominio MRH3 de CI-MPR presenta glicosilaciones, se emplearon simultáneamente campos de fuerza para glúcidos, GLYCAM_06j-1 (46) y para proteínas, Protein.ff19SB (47).
En un primer paso, los sistemas se minimizaron en vacío utilizando Sander (44) con el fin de eliminar posibles anomalías estructurales de las moléculas. A continuación, se solvataron las estructuras en octaedros truncados de agua TIP3P (“Three-site Tranferrable Intermolecular Potencial”) con un número total de moléculas de agua que dependerá del tamaño del sistema con la consiguiente adición de contraiones para neutralizar su carga. Tras la obtención de los sistemas solvatados, estos se minimizaron de nuevo con Sander para evitar problemas derivados de posibles interacciones desfavorables entre los iones y las moléculas de agua. Posteriormente se llevó a cabo una fase de calentamiento en la cual los sistemas pasaron de una temperatura de 100K a 300K en 20 picosegundos (ps). En esta fase solo se permitió el movimiento de las moléculas de agua, pues se aplicó una fuerza de 50 Kcal/mol para mantener inmóvil a todos los átomos del soluto. El último paso, aunque previo a la producción de la dinámica, consiste en una fase de equilibrado en cinco pasos de 100 ps. Durante estos cinco pasos, la restricción inicial del soluto se fue reduciendo lo que permitió una mejor interacción entre éste y las moléculas de agua, las cuales se fueron disponiendo sobre la superficie accesible al disolvente del soluto. Para finalizar, se ejecutó la simulación de dinámica molecular de diferentes periodos de duración según el sistema molecular.
Las dinámicas moleculares fueron analizadas con el módulo cpptraj de AMBER 20 (44) con el que se extrajeron los confórmeros mayoritarios de cada sistema.
Dinámica molecular del complejo MRH3-M6P
Se llevó a cabo una simulación de 100 nanosegundos (ns) del cristal 1SZ0 para analizar la estabilidad del modo de unión de M6P al dominio MRH3. Para el ligando M6P se seleccionó como estado de ionización -2 preferente, puesto que es el que mejor resultado dio en el docking y el mayoritario a pH fisiológico (Figura 7b). Se mantuvieron los dominios MRH1 y MRH2, ya que ha sido descrito su importante papel en la estabilización del modo de unión (7).
Dinámica molecular del ligando de CI-MPR y del LYTAC- MMP2
Las simulaciones de dinámica molecular de ambos ligandos libres en agua fueron de 200 ns. El ligando de CI-MPR fue simulado con carga neutra puesto que es la carga que se le asignó en las simulaciones de dinámica molecular mostradas en el artículo de referencia, con el objetivo de comparar los resultados (22). Sin embargo, tras un cálculo de pKa con Marvin Sketch (48). se determinó que los estados de protonación más probables en agua para M6Pn eran -1 y -2 (Figura 7a). Teniendo en cuenta esta predicción, el LYTAC- MMP2 se simuló tanto con carga -3 como con carga -6, ya que presenta tres residuos de M6Pn.
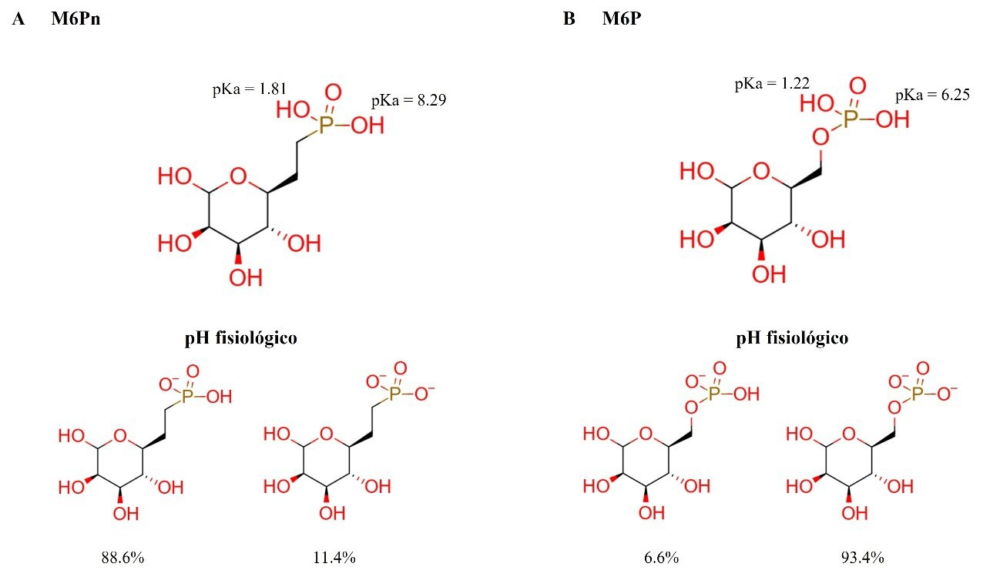
Figura 7. Valores de pKa y distribución de microespecies a pH fisiológico (7.4) de un residuo de M6Pn (A) y uno de M6P (B) calculados con Marvin Sketch (48).
3. RESULTADOS Y DISCUSIÓN
3.1 Análisis de los dominios de reconocimiento de azúcares de CI-MPR
Este análisis tiene como objetivo determinar cuál de los cuatro dominios de reconocimiento de azúcares presentes en el receptor CI-MPR es el más plausible para la interacción con un LYTAC.
En primer lugar, como la única estructura depositada en el PDB de un dominio de reconocimiento de azúcares unido a M6P era de origen bovino (código PDB 1SZ0), se decidió realizar un modelado por homología de la proteína humana usando este cristal como molde. El modelo obtenido con Swissmodel (38) presentaba una alta homología (78.9%) con los dominios MRH1, 2 y 3 de origen bovino, asimismo, los aminoácidos claves para la interacción con M6P (secuencia “QREY”) se encontraban en la misma disposición espacial (Figura 8). Por esta razón, se decidió emplear la estructura del dominio MRH3 depositada en el cristal 1SZ0 para el resto del estudio computacional, pues no se esperaban diferencias significativas en el modo de unión a M6P o en su comportamiento en simulaciones de dinámica molecular.
Figura 8. (A) Modelo de los dominios MRH1, 2 y 3 humanos obtenidos con Swissmodel, (38) usando el cristal 1SZ0 como molde. Las zonas más conservadas están representadas en azul, mientras las de menor homología en naranja. (B) Superposición con PyMOL (16) del dominio MRH3 del cristal 1SZ0 (cian) y el dominio MRH3 humano obtenido (gris). Se muestra el modo de unión del ligando M6P (amarillo) y la secuencia “QREY”. Los enlaces de hidrógeno están representados con líneas negras.
3.1.a Estudio del modo de unión de M6P a los dominios MRH3, MRH5 y MRH9
El modo de unión de M6P a los dominios MRH5 y MRH9 se estudió mediante técnicas de docking molecular. Con el fin de validar este método para este tipo de macromoléculas, se empleó el cristal 1SZ0 como control.
Los cristales empleados para la realización del docking fueron: 1SZ0 (dominios MRH1, MRH2, MRH3), 6V02 (dominio MRH5) y 6Z32 (dominio MRH9). En el caso del ligando, se tuvieron en cuenta sus estados de protonación más probables, que son -1 y -2. En la siguiente tabla (Tabla 2) se muestran los mejores resultados obtenidos del docking en función del estado de protonación de M6P y el dominio de reconocimiento de azúcares.
Tabla 2. Resultados del docking molecular del ligando M6P con diferentes estados de protonación en los dominios MRH3, MRH5 y MRH9. Los datos de “Docking Score” hacen referencia a la energía de interacción ligando-proteína y está en Kcal/mol.

El resultado de docking del ligando M6P en el dominio MRH3 reproduce la pose encontrada en el cristal 1SZ0 (Figura 9b). La carga del ligando parece no influir en la conformación que adopta éste en el sitio de unión, no obstante, cuando presenta carga -2 la unión es ligeramente más favorable energéticamente (Tabla 2). En ambos casos, se establecen cuatro enlaces de hidrógeno intermoleculares entre el anillo de manosa y las cadenas laterales de E416, Y324 y Q348. Además, el grupo fosfato interacciona con la R391 a través de un enlace iónico reforzado con enlace de hidrógeno y con el grupo amino del esqueleto de S386 (Figura 9a).
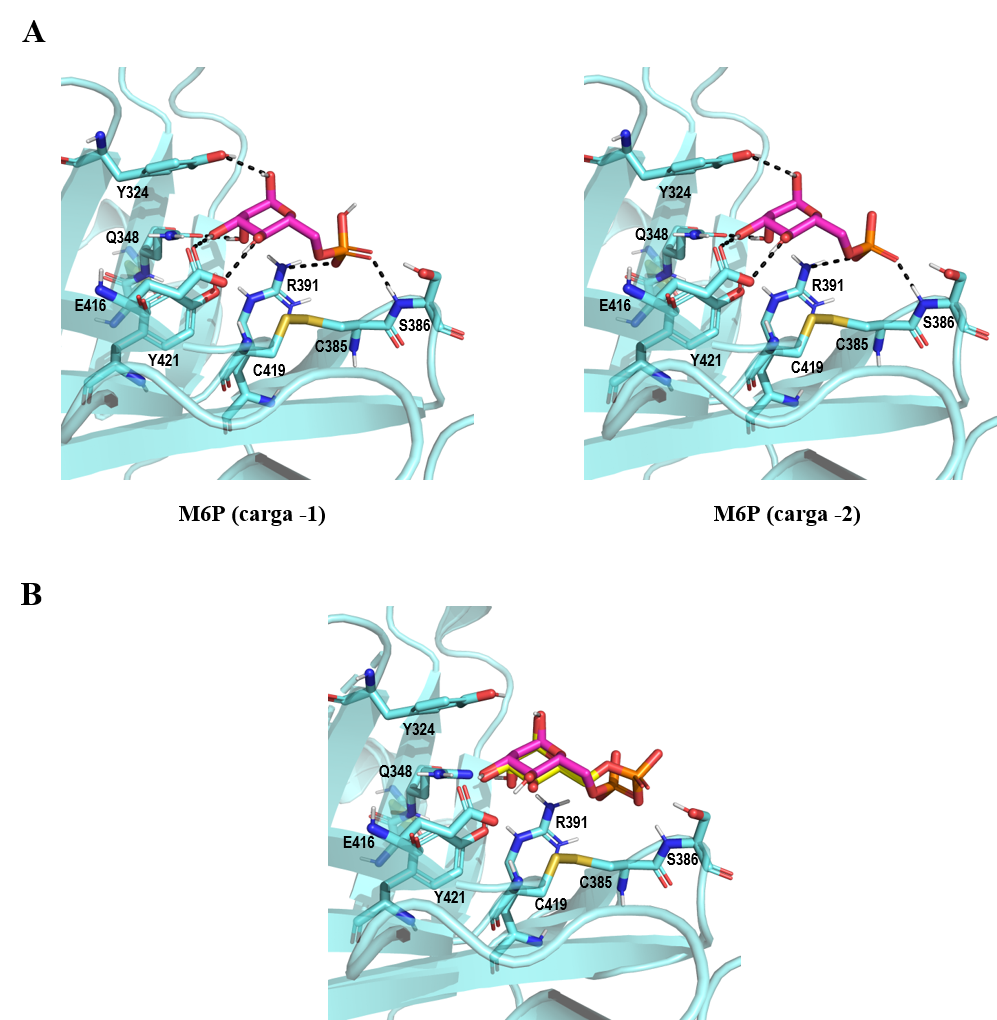
Figura 9. (A) Resultados de docking del ligando M6P (magenta) con cargas -1 y -2 en el dominio MRH3 (cian, código PDB 1SZ0). (B) Superposición del mejor resultado de docking de M6P (magenta) en el dominio MRH3 (cian) y el cristal 1SZ0 (M6P en amarillo). Los enlaces de hidrógeno están representados con líneas negras. Las imágenes han sido generadas con PyMOL (16)
En el caso del dominio MRH5, a pesar de que energéticamente el modo de unión del ligando es favorable (Tabla 2), éste no explora el sitio de unión a azúcares del dominio (Figura 10). Este resultado podría ser debido a que MRH5 es específico de fosfodiésteres de Man-P-GlcNAc y a que el bolsillo es ligeramente más grande por la ausencia de un puente disulfuro.
Figura 10. Resultados de docking del ligando M6P (magenta) con cargas -1 y -2 en el dominio MRH5 (naranja, código PDB 6V02). Además, se muestra el ligando M6P (amarillo) extraído del cristal 1SZ0 y superpuesto en este dominio. Los enlaces de hidrógeno están representados con líneas negras. Las imágenes han sido generadas con PyMOL (16)
Por último, los resultados de docking en el dominio MRH9 muestran que la unión de M6P con carga -2 es más favorable, puesto que establece un mayor número de interacciones (Tabla 2). M6P con carga -1 establece dos enlaces de hidrógeno entre el anillo de manosa y las cadenas laterales de Y1255 y E1345, además de un enlace iónico reforzado con enlace de hidrógeno entre R1325 y el grupo fosfato. Por otro lado, la M6P con carga -2 establece tres enlaces de hidrógeno entre el anillo de manosa y las cadenas laterales de Q1283 y E1345, además de otro entre el grupo fosfato y el grupo amino del esqueleto de H1320 (Figura 11).
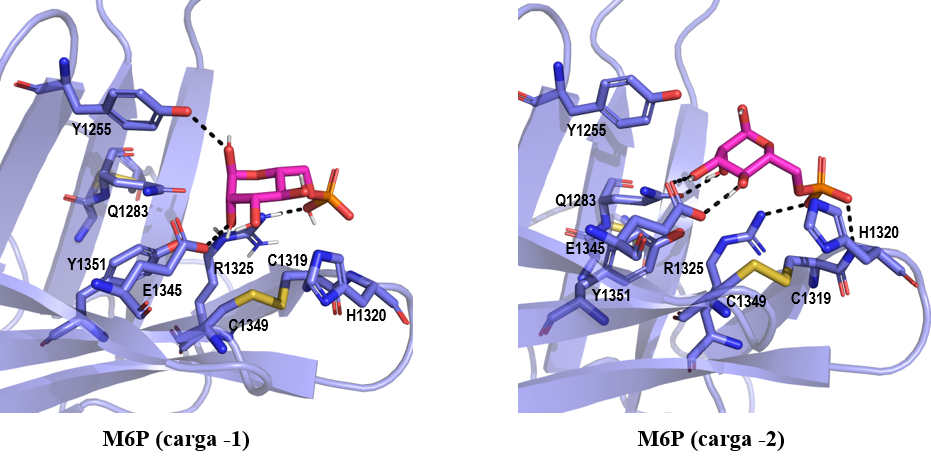
Figura 11. Resultados de docking del ligando M6P (magenta) con cargas -1 y -2 en el dominio MRH9 (violeta, código PDB 6Z32). Los enlaces de hidrógeno están representados con líneas negras. Las imágenes han sido generadas con PyMOL (16)
Adicionalmente a este estudio, se consideraron las dos hipótesis de internalización del receptor CI-MPR (17, 21) para concretar cuál sería la más plausible para la interacción con un LYTAC. La hipótesis de dimerización del receptor se sustenta en el aislamiento de los dominios MRH7-MRH11 dimerizados a través de una glicosilación en el dominio MRH9 (código PDB 6Z32) de dos monómeros adyacentes (Figura 4a). Por otro lado, los dominios MRH4-MRH14 de origen bovino se resolvieron por Cryo-EM (código PDB 6UM2) unidos a IGF2 y se encontraban en “conformación pistola” (Figura 4b). Aunque ambos cristales fueron resueltos a pH fisiológico, el dominio MRH9 en la estructura 6Z32 presentaba una glicosilación completa acabada en un residuo de manosa en N1312 (Figura 3b) que hacía posible la dimerización, mientras que en la estructura encontrada bajo el código 6UM2 estas glicosilaciones se reducían a un solo monosacárido de 2- acetamido-2-deoxi-b-D-glucopiranosa (NAG). En este último caso, la ausencia de glicosilaciones completas podría imposibilitar la formación de un dímero, lo que podría dar lugar a una interpretación sesgada del proceso fisiológico.
Además, se encontró en el dominio MRH3 (código PDB 1SZ0) una glicosilación de NAG en el residuo N365, muy próximo al sitio de unión de la M6P, lo que abriría la posibilidad de observar una dimerización análoga a la encontraba en la estructura 6Z32 si la glicosilación fuese completa.
Dados los resultados de docking y el estudio de dimerización de CI-MPR, se concluyó que la interacción LYTAC-receptor era más probable a través del dominio MRH3, tanto por su especificidad por monoésteres de manosa como por su accesibilidad. No obstante, no se debe descartar la posibilidad de que el ligando de CI-MPR descrito por L. M. Ali et al. (22) (Figura 5), al poseer tres residuos derivados de manosa, interaccione con más de un dominio del mismo monómero del receptor.
A continuación, se decidió estudiar la estabilidad del modo de unión de un único residuo de M6P en el dominio MRH3 mediante simulaciones de dinámica molecular libre en agua usando el software AMBER20 (44). Para ello, se seleccionó la estructura 1SZ0 que incluía además los dominios MRH1 y MRH2, que se mantuvieron durante la simulación ya que tienen un papel cooperativo en el modo de unión del ligando (7). En cuanto a M6P, solo se tuvo en cuenta el estado de protonación -2, puesto que fue el que mejor energía de interacción obtuvo en los resultados de docking (Tabla 2). Con el fin de evaluar la estabilidad del complejo se extrajeron ocho confórmeros y se analizó la desviación cuadrática media (“Root-Mean-Square Desviation”, RMSD) entre la proteína y el ligando. El porcentaje de distribución de los confórmeros a lo largo de los 20 ns de simulación es de 33.9% para el confórmero 1 (c1), 29.3% para c2, 17.8% para c3, 7.4% para c4, 5.6% para c5, 5.2% para c6, 0.7% para c7 y 0.2% para c8. Se seleccionaron cuatro de los ocho confórmeros obtenidos, puesto que representaban diferentes modos de unión del ligando M6P al dominio MRH3 (Figura 12).
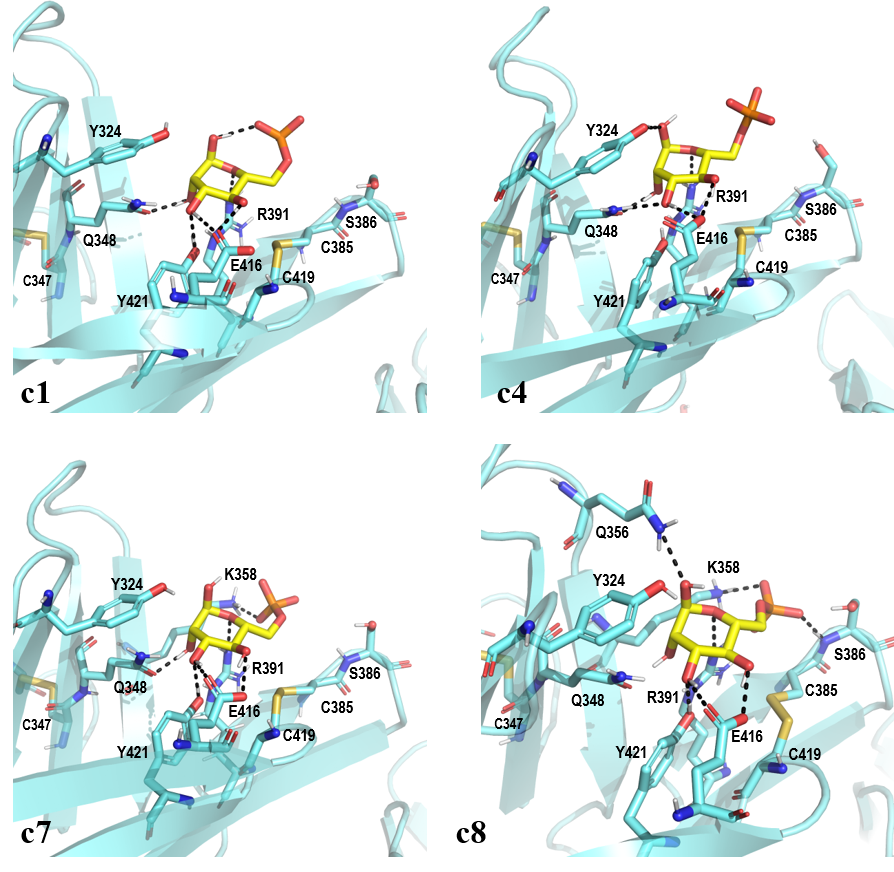
Figura 12. Confórmeros representativos del modo de unión del ligando M6P (amarillo) en el dominio MRH3 (cian, código PDB 1SZ0) extraídos de una simulación de 20 ns de dinámica molecular. Los enlaces de hidrógeno están representados con líneas negras. Las imágenes han sido generadas con PyMOL (16).
En el confórmero mayoritario (c1) destaca la formación de un enlace de hidrógeno intramolecular entre uno de los oxígenos del grupo fosfato y un grupo hidroxilo del anillo de manosa. Esta interacción impide que el grupo fosfato interaccione con el esqueleto de S386, tal y como está descrito en la bibliografía, (17) sin embargo, se pierde en el resto de confórmeros. Además, se establecen cinco enlaces de hidrógeno intermoleculares entre el anillo de manosa y las cadenas laterales de los residuos Q348, Y421, E416 y R391. En el confórmero c4, el enlace de hidrógeno intramolecular desaparece y en su lugar se forma un enlace de hidrógeno entre el grupo hidroxilo de Y421 y el anillo de manosa (Figura 12).
Aunque los confórmeros c7 y c8 no sean cuantitativamente representativos en la simulación, se observa una serie de interacciones interesantes para estabilizar el modo de unión del ligando (Figura 12). En ambos casos, el grupo fosfato establece un enlace iónico reforzado con enlace de hidrógeno con la cadena lateral del residuo K358. Además, en el confórmero c8 se observan dos enlaces de hidrógeno adicionales entre el grupo fosfato y la cadena lateral del residuo Q356 y el grupo amino del esqueleto de S386.
El RMSD mide la variación en la posición de los átomos de una molécula empleando una conformación dada como referencia. En este caso, la estructura de referencia es la encontrada en el cristal 1SZ0. La representación del RMSD (Å) frente al tiempo de simulación (ns) (Gráfica 1) muestra que el modo de unión de la M6P al dominio MRH3 es estable, puesto que tan solo se desvía 1.5 Å de media respecto a la estructura de referencia (línea verde). Debido al mayor tamaño y flexibilidad de los dominios MRH1, MRH2 y MRH3 en comparación con M6P, la gráfica (línea azul) sufre más fluctuaciones y desviación respecto a la estructura de referencia en este caso.
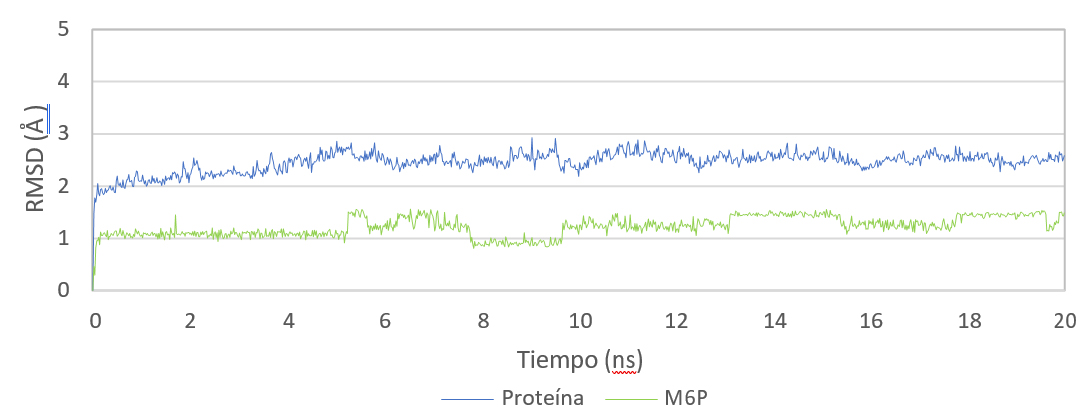
Gráfica 1. Representación gráfica de la variación del RMSD (Å) en función del tiempo de simulación (20 ns) del complejo formado entre los dominios N-terminales de CI-MPR (MRH1, MRH2 y MRH3) y M6P.
3.2 Diseño de un LYTAC para la degradación selectiva de MMP-2 (LYTAC – MMP2)
Con la información estructural disponible, se procedió al diseño de un LYTAC combinando la estructura del ligando de CI-MPR propuesto por L. M. Ali et al. (22) y la de un inhibidor selectivo de MMP-2 previamente descrito (Figura 13) (36). Para ello, se sustituyó el grupo fluoróforo (señalado en verde, Figura 5) del ligando de CI-MPR por el inhibidor de MMP-2. El punto de anclaje del inhibidor se seleccionó cuidadosamente para que no influyese en su modo de unión a la enzima.
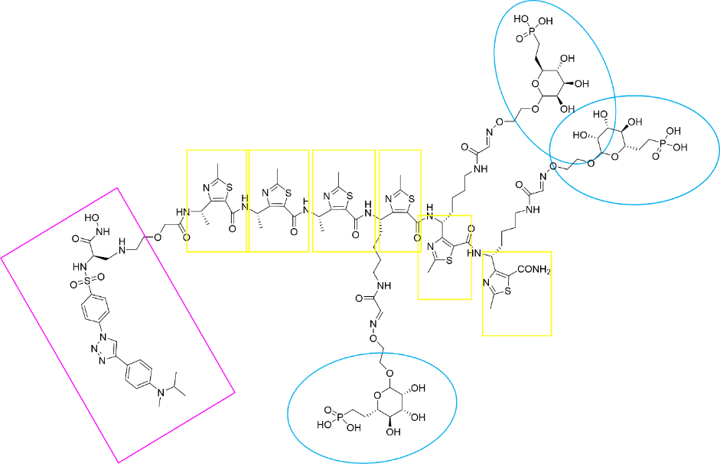
Figura 13. Estructura del LYTAC-MMP2. Los residuos de M6Pn se encuentran representados en azul, unidos a ellos a través de un espaciador, se encuentran los monómeros del γ-péptido (amarillo); y, por último, en morado, el inhibidor selectivo de MMP-2
3.3 Comportamiento dinámico del ligando de CI-MPR y del LYTAC propuesto
L. M. Ali et al. (22) estudiaron el comportamiento del ligando de CI-MPR (Figura 5) en una disolución de tampón fosfato 50 mM mediante técnicas de espectroscopía NOESY. Posteriormente, utilizaron los resultados obtenidos como restricciones para las simulaciones de dinámica molecular del ligando, usando agua como solvente implícito y el software AMBER16 (49). Por el contrario, en este trabajo no se aplicaron restricciones en las simulaciones de dinámica molecular, se empleó agua como solvente explícito y se utilizó una versión más actualizada del software AMBER (AMBER 20) (44).
La finalidad del estudio fue la de estudiar el comportamiento dinámico libre en agua del ligando de CI-MPR y compararlo con el del LYTAC-MMP2.
3.3.a Diámica molecular del ligando de CI-MPR
En el artículo publicado por L. M. Ali et al. (22). se ejecutó la dinámica molecular del ligando de CI-MPR con carga neutra, por lo que se decidió mantener este estado de ionización a pesar de no ser el mayoritario a pH fisiológico (Figura 7) de acuerdo con los resultados obtenidos con el programa Marvin Sketch (48).
Se llevó a cabo una simulación de dinámica molecular lo suficientemente larga para observar grandes cambios conformacionales en la molécula, concretamente, su duración fue de 200 ns (10000 pasos). Para su análisis se extrajeron doce confórmeros con diferentes porcentajes de distribución: 64.8% para c1, 13.2% para c2, 12.3% para c3, 6.3% para c4, 1.81% para c5, 0.74% para c6, 0.33% para c7, 0.21% para c8, 0.11% para c9, 0.05% para c10, 0.04% para c11 y 0.02% para c12. De acuerdo con estos resultados, el ligando puede adoptar conformaciones a lo largo de la simulación de dinámica molecular, en las que se establezcan un mayor número de interacciones intramoleculares o “conformación cerrada” o conformaciones en las que sus partes constituyentes se encuentren más distanciadas entre sí o “conformación abierta”. La conformación abierta deja accesibles los residuos de manosa para su interacción con el receptor, por lo que se considera la conformación bioactiva.
La mayoría de los confórmeros extraídos presentan una disposición cerrada, caracterizada por apilamientos π-π entre los monómeros del γ-péptido, así como por interacciones de enlace de hidrógeno entre los grupos polares de la molécula. En la Figura 14a se muestran el confórmero mayoritario (c1) en una conformación cerrada y el “confórmero bioactivo” (c8). Como se puede observar, la conformación bioactiva tan solo se explora en un 0.21% de la dinámica.
Adicionalmente, se representó la variación del RMSD del ligando a lo largo del tiempo de simulación (Figura 14b). Se utilizó una conformación abierta del ligando como estructura de referencia, con el objetivo de que fuese similar a la conformación bioactiva. Se considera que dos conformaciones son lo suficientemente similares si la variación del RMSD es igual o inferior a 2 Å. En este caso, la posición media de los átomos del ligando se desvía entre 2.5 Å y 4.5 Å respecto a la conformación inicial, por lo que ésta no se vuelve a explorar a lo largo de la dinámica.
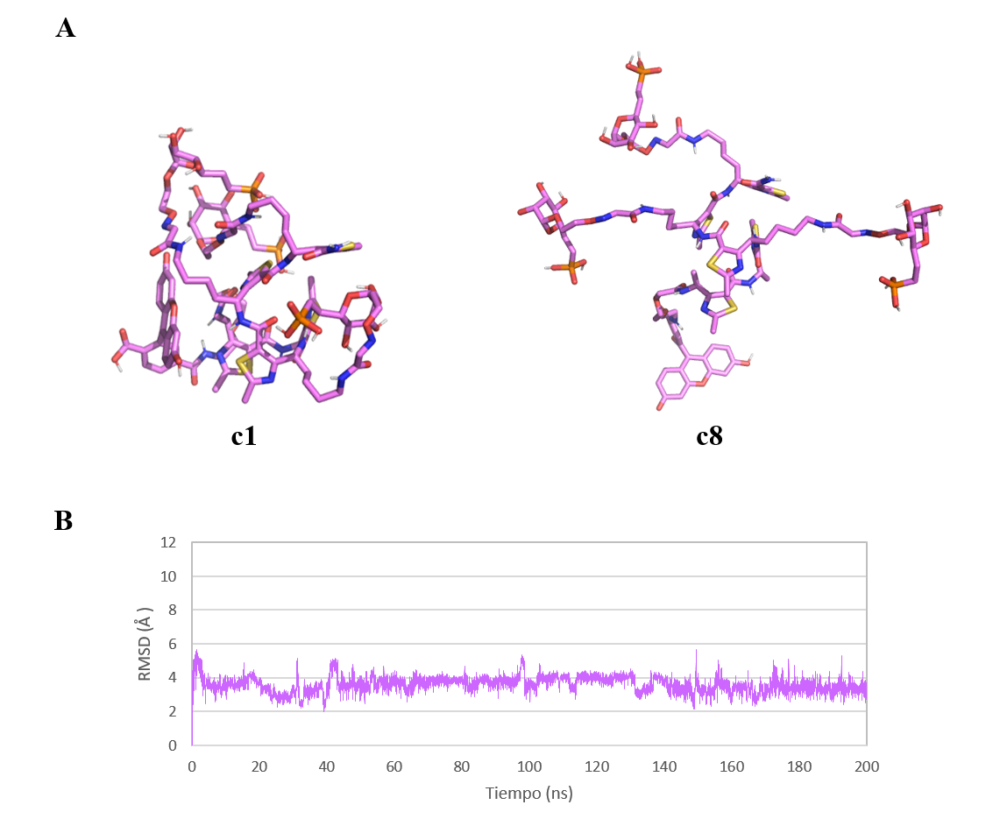
Figura 14. (A) Confórmero mayoritario (c1) y confórmero bioactivo (c8) extraídos de 200 ns de dinámica molecular del ligando de CI-MPR. Las imágenes han sido generadas con PyMOL (16). (B) Representación gráfica de la variación de RMSD (Å) frente al tiempo de simulación (200 ns) del ligando de CI-MPR (22) en agua.
3.3.b Dinámica molecular del LYTAC-MMP2
La molécula propuesta está formada por tres extensiones en su estructura acabadas en residuos de M6Pn como ligando de unión al receptor CI-MPR, una cadena de γ-péptido como estructura base y un inhibidor para reclutar a la proteína de interés, MMP-2 (Figura 13).
La producción de la dinámica molecular, así como el análisis de los resultados se llevó a cabo de forma análoga al ligando de CI-MPR, no obstante, al contrario que en el caso anterior, para las simulaciones de dinámica molecular del LYTAC se tuvo en cuenta el estado de protonación del grupo fosfonato. Como ya se indicó en el apartado de Materiales y Métodos, a pH fisiológico el grupo fosfonato se encuentra en un 88.6% con carga -1 y en un 11.4% con carga -2 (Figura 7). A pesar de esto y con el objetivo de poder comparar el comportamiento dinámico del ligando de CI-MPR y el LYTAC, se decidió modelar también con carga neutra. Desafortunadamente, la dinámica del compuesto con carga neutra falló por problemas en el término electrostático y, por lo tanto, los datos no se muestran en este trabajo.
Dinámica molecular del LYTAC-MMP2 (δ= -3)
El estado de protonación -3 sería el mayoritario para el LYTAC a pH fisiológico, puesto que dispone de tres residuos terminales de M6Pn.
Al igual que en el caso anterior, se extrajeron doce confórmeros con los siguientes porcentajes de distribución: 35.7% para c1, 16.51% para c2, 13.9% para c3, 12.83% para c4, 10.06% para c5, 3.3% para c6, 2.43% para c7, 1.71% para c8, 1.37% para c9, 1.07% para c10, 1.05% para c11 y 0.06% para c12. En este caso, la dispersión de los resultados es mayor, lo que indicaría un mayor número de cambios conformacionales representativos a lo largo de la dinámica. El confórmero mayoritario (c1) (Figura 15a) se encuentra en una conformación cerrada estabilizada principalmente por apilamientos π-π entre el γ-péptido y ligando. En cambio, los confórmeros c3 y c11 presentan una disposición espacial más cercana a la bioactiva. Por un lado, aunque la disposición de los residuos de M6Pn del confórmero c3 pudiesen permitir la unión al receptor, el inhibidor de MMP-2 se encuentra inaccesible debido al establecimiento de interacciones intramoleculares con el γ-péptido. Por otro lado, el confórmero c11, aunque es minoritario, adopta la conformación extendida necesaria para que el compuesto pueda unirse a ambas dianas.
La desviación conformacional del LYTAC a lo largo de la dinámica respecto a la conformación bioactiva de referencia oscila entre 3 y 6 Å (Figura 15b). Sin embargo, en ocasiones llega a ser menor a 2 Å, por lo que se podría concluir que el LYTAC sí es capaz de explorar ocasionalmente la conformación bioactiva a lo largo de la simulación.
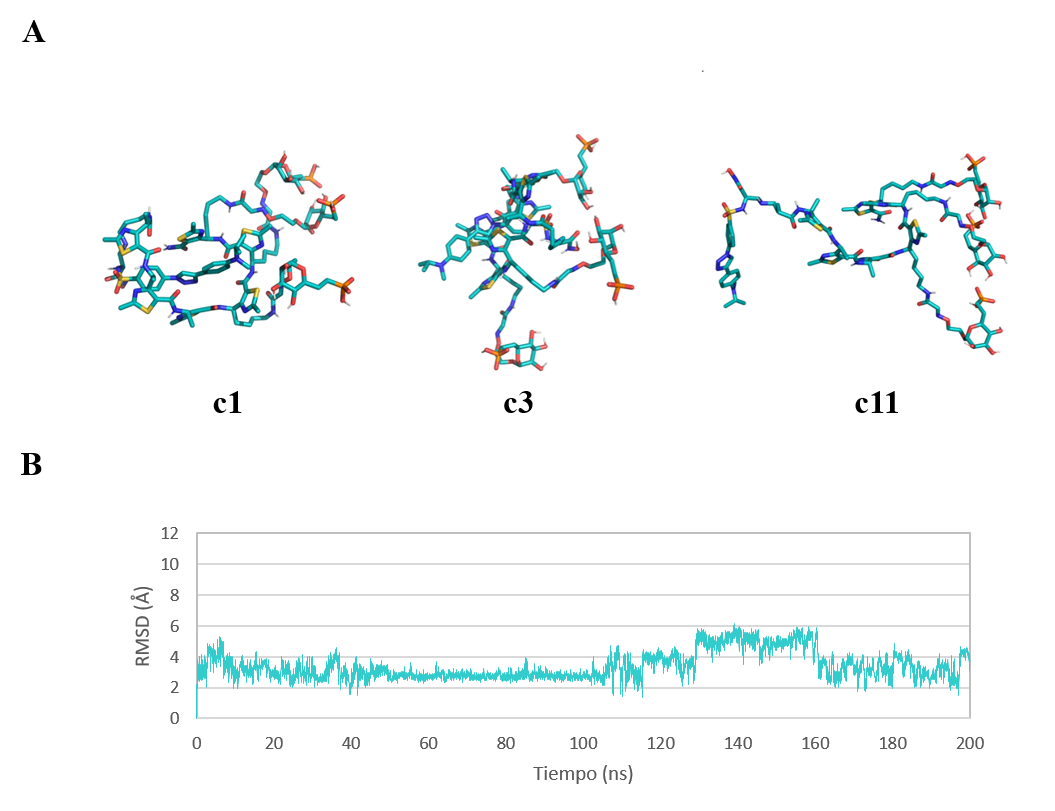
Figura 15. (A) Confórmeros representativos extraídos de 200 ns de dinámica molecular del LYTAC-MMP2 con carga -3. Las imágenes han sido generadas con PyMOL.(16) (B) Representación
Dinámicaica molecular del LYTAC-MMP2 (δ= -6)
El estado de ionización -2 para los residuos de M6Pn dan como resultado una microespecie minoritaria del LYTAC a pH fisiológico. No obstante, ya que en el organismo existen regiones con pH superior al fisiológico y que el porcentaje de distribución de la M6Pn a este estado de ionización pasa de 11.4% (pH 7.4) a 34% (pH 8), se decidió que el estudio de su comportamiento dinámico era biológicamente relevante.
En este caso, los doce confórmeros extraídos tienen una distribución de: 71.17% para c1, 11.37% para c2, 4.88% para c3, 3.8% para c4, 2.13% para c5, 1.61% para c6, 1.45% para c7, 1.01% para c8, 0.87% para c9, 0.71% para c10, 0.54% para c11 y 0.45% para c12. Teniendo en cuenta estos datos y el análisis visual del confórmero mayoritario (c1) (Figura 16a), el LYTAC se mantiene en una conformación cerrada durante prácticamente toda la dinámica.
Aunque no se vea representado en la Figura 16b, el LYTAC es capaz de explorar conformaciones más abiertas, como las representadas por los confórmeros c6 y c11 (Figura 16a). Además, curiosamente, se aprecia una disminución paulatina del valor de RMSD a lo largo de la simulación lo que llevaría a pensar que, si se alargara el tiempo de simulación, el LYTAC podría llegar a explorar la conformación inicial o bioactiva.
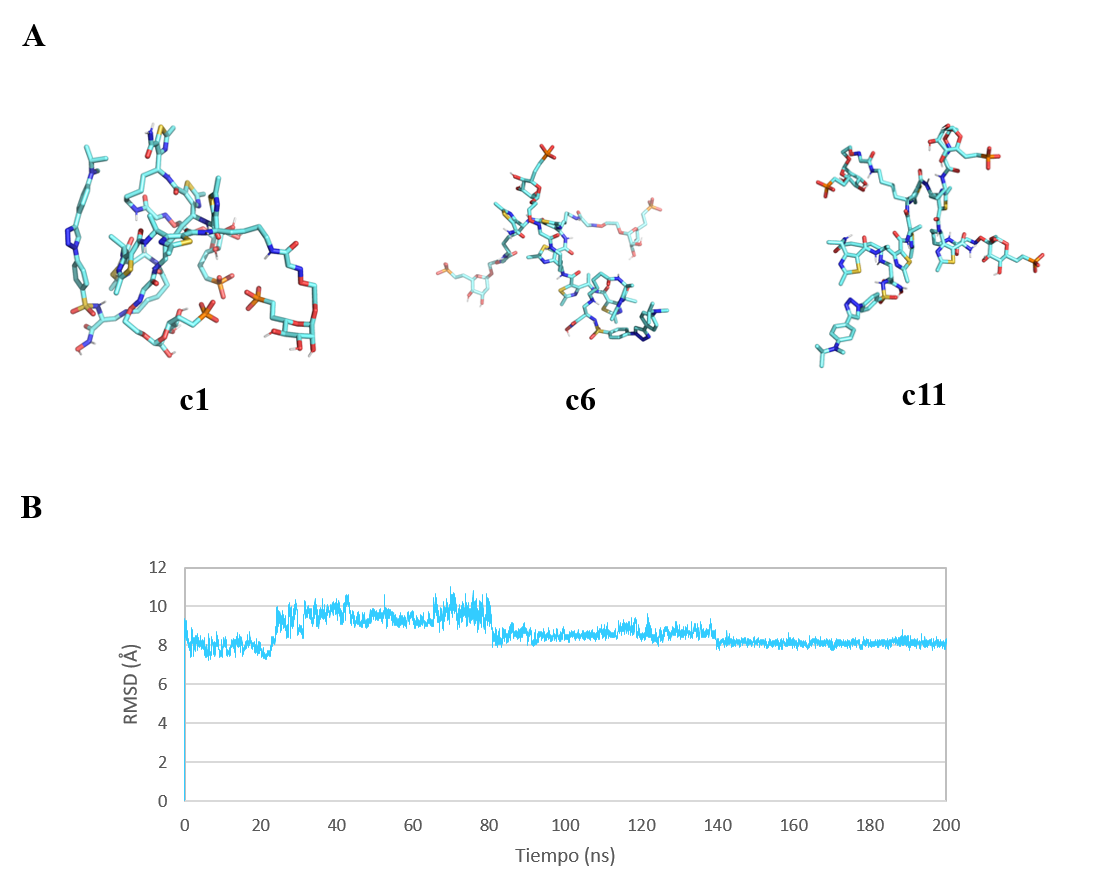
Figura 16. (A) Confórmeros representativos extraídos de 200 ns de dinámica molecular del LYTAC-MMP2 con carga -6. Las imágenes han sido generadas con PyMOL (16). (B) Representación gráfica de la variación de RMSD (Å) frente al tiempo de simulación (200 ns) del LYTAC-MMP2 con carga -6 en agua.
4. CONCLUSIONES
Mediante el estudio estructural del receptor CI-MPR se han propuesto argumentos para apoyar la hipótesis de la dimerización del receptor a través de glicosilaciones terminadas en residuos de M6P. Asimismo, se ha propuesto al dominio MRH3 como dominio de reconocimiento de azúcares más plausible para la interacción del receptor con un LYTAC. Se ha demostrado mediante técnicas de docking que la interacción entre éste y la M6P está favorecida energéticamente en comparación con los resultados obtenidos para los dominios MRH5 y MRH9. Del mismo modo, mediante simulaciones de dinámica molecular se ha propuesto un modo de unión de M6P al dominio MRH3 que permanece estable a lo largo del tiempo se simulación.
Por otro lado, se ha diseñado un LYTAC basado es un ligando de CI-MPR previamente descrito en la bibliografía, que razonadamente podría tener utilidad para la degradación de MMP-2 (LYTAC-MMP2). El análisis conformacional del LYTAC-MMP2 muestra un comportamiento similar al del ligando de CI-MPR de referencia, sin embargo, no se han reproducido los resultados descritos en la bibliografía. La razón podría ser la falta de datos de los NOE que L. M. Ali et al. (22) aplicaron como restricciones para llevar a cabo las simulaciones de dinámica molecular del ligando de CI-MPR. Las principales diferencias en el comportamiento dinámico entre el LYTAC-MMP2 y el ligando de referencia han surgido debido a la presencia del inhibidor de MMP-2 y su capacidad de interaccionar con el resto de la molécula mediante interacciones de tipo π-π entre sus grupos aromáticos. Para continuar este trabajo, se llevará a cabo la simulación de dinámica molecular del LYTAC-MMP2 con carga cero, para poder así compararla con las simulaciones realizadas en este trabajo y tras la optimización de la estructura, se planteará su síntesis y posterior evaluación biológica.
5. REFERENCIAS
- Chen B, Retzlaff M, Roos T, Frydman J. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol. 2011;3(8):a004374.
- Marshall RS, Vierstra RD. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front Mol Biosci. 2019;6:40.
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13(7):448-462.
- Kim JJ, Olson LJ, Dahms NM. Carbohydrate recognition by the mannose-6-phosphate receptors. Curr Opin Struct Biol. 2009;19(5):534-542.
- Olson LJ, Misra SK, Ishihara M, Battaile KP, Grant OC, Sood A, et al. Allosteric regulation of lysosomal enzyme recognition by the cation-independent mannose 6-phosphate receptor. Commun Biol. 2020;3(1):498.
- van Meel E, Klumperman J. Imaging and imagination: understanding the endo-lysosomal system. Histochem Cell Biol. 2008;129(3):253-266.
- Olson LJ, Peterson FC, Castonguay A, Bohnsack RN, Kudo M, Gotschall RR, et al. Structural basis for recognition of phosphodiester-containing lysosomal enzymes by the cation-independent mannose 6-phosphate receptor. Proc Natl Acad Sci U S A. 2010;107(28):12493-12498.
- Le Borgne R, Hoflack B. Protein transport from the secretory to the endocytic pathway in mammalian cells. Biochim Biophys Acta. 1998;1404(1-2):195-209.
- Li J, Sahagian GG. Demonstration of tumor suppression by mannose 6-phosphate/insulin-like growth factor 2 receptor. Oncogene. 2004;23(58):9359-9368.
- Brown J, Jones EY, Forbes BE. Interactions of IGF-II with the IGF2R/cation-independent mannose-6-phosphate receptor mechanism and biological outcomes. Vitam Horm. 2009;80:699-719.
- Zavorka ME, Connelly CM, Grosely R, MacDonald RG. Inhibition of insulin-like growth factor II (IGF-II)-dependent cell growth by multidentate pentamannosyl 6-phosphate-based ligands targeting the mannose 6-phosphate/IGF-II receptor. Oncotarget. 2016;7(38):62386-62410.
- Chao W, D’Amore PA. IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev. 2008;19(2):111-120.
- Castonguay AC, Olson LJ, Dahms NM. Mannose 6-phosphate receptor homology (MRH) domain-containing lectins in the secretory pathway. Biochim Biophys Acta. 2011;1810(9):815-826.
- Bohnsack RN, Song X, Olson LJ, Kudo M, Gotschall RR, Canfield WM, et al. Cation-independent mannose 6-phosphate receptor: a composite of distinct phosphomannosyl binding sites. J Biol Chem. 2009;284(50):35215-35226.
- Olson LJ, Castonguay AC, Lasanajak Y, Peterson FC, Cummings RD, Smith DF, et al. Identification of a fourth mannose 6-phosphate binding site in the cation-independent mannose 6-phosphate receptor. Glycobiology. 2015;25(6):591-606.
- DeLano WL. Pymol: An open-source molecular graphics tool. CCP4 Newsletter On Protein Crystallography, 40, 82-92.2002.
- Bochel AJ, Williams C, McCoy AJ, Hoppe HJ, Winter AJ, Nicholls RD, et al. Structure of the Human Cation-Independent Mannose 6-Phosphate/IGF2 Receptor Domains 7-11 Uncovers the Mannose 6-Phosphate Binding Site of Domain 9. Structure. 2020;28(12):1300-1312.e5.
- Marron-Terada PG, Hancock MK, Haskins DJ, Dahms NM. Recognition of Dictyostelium discoideum lysosomal enzymes is conferred by the amino-terminal carbohydrate binding site of the insulin-like growth factor II/mannose 6-phosphate receptor. Biochemistry. 2000;39(9):2243-2253.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology [Internet]. 2011 2011/10//; 7:[539 p.]. Available from: http://europepmc.org/abstract/MED/21988835Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/21988835/?tool=EBIAvailable from: https://www.ncbi.nlm.nih.gov/pmc/articles/pmid/21988835/pdf/?tool=EBIAvailable from: https://doi.org/10.1038/msb.2011.75Available from: https://europepmc.org/articles/PMC3261699Available from: https://europepmc.org/articles/PMC3261699?pdf=render.
- Brown J, Jones EY, Forbes BE. Keeping IGF-II under control: lessons from the IGF-II-IGF2R crystal structure. Trends Biochem Sci. 2009;34(12):612-619.
- Wang R, Qi X, Schmiege P, Coutavas E, Li X. Marked structural rearrangement of mannose 6-phosphate/IGF2 receptor at different pH environments. Sci Adv. 2020;6(7):eaaz1466.
- Ali LMA, Simon M, El Cheikh K, Aguesseau-Kondrotas J, Godefroy A, Nguyen C, et al. Topological Requirements for CI-M6PR-Mediated Cell Uptake. Bioconjug Chem. 2019;30(10):2533-2538.
- Ottis P, Crews CM. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem Biol. 2017;12(4):892-898.
- Zorca CE, Fallahi A, Luo S, Eldeeb MA. Multifaceted targeted protein degradation systems for different cellular compartments. Bioessays. 2022;44(6):e2200008.
- Zhang X, Liu H, He J, Ou C, Donahue TC, Muthana MM, et al. Site-Specific Chemoenzymatic Conjugation of High-Affinity M6P Glycan Ligands to Antibodies for Targeted Protein Degradation. ACS Chemical Biology. 2022;17(11):3013-3023.
- Alabi SB, Crews CM. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem. 2021;296:100647.
- Ahn G, Banik SM, Miller CL, Riley NM, Cochran JR, Bertozzi CR. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat Chem Biol. 2021;17(9):937-946.
- Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 2020;584(7820):291-297.
- Nobel Prize [Available from: https://www.nobelprize.org/prizes/chemistry/.
- Verma RP, Hansch C. Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem. 2007;15(6):2223-2268.
- Bassiouni W, Ali MAM, Schulz R. Multifunctional intracellular matrix metalloproteinases: implications in disease. The FEBS Journal. 2021;288(24):7162-7182.
- Morgunova E, Tuuttila A, Bergmann U, Isupov M, Lindqvist Y, Schneider G, et al. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science. 1999;284(5420):1667-1670.
- Gimeno A, Beltrán-Debón R, Mulero M, Pujadas G, Garcia-Vallvé S. Understanding the variability of the S1′ pocket to improve matrix metalloproteinase inhibitor selectivity profiles. Drug Discov Today. 2020;25(1):38-57.
- Zapico JM, Puckowska A, Filipiak K, Coderch C, de Pascual-Teresa B, Ramos A. Design and synthesis of potent hydroxamate inhibitors with increased selectivity within the gelatinase family. Org Biomol Chem. 2015;13(1):142-156.
- Zapico JM, Acosta L, Pastor M, Rangasamy L, Marquez-Cantudo L, Coderch C, et al. Design and Synthesis of Water-Soluble and Potent MMP-13 Inhibitors with Activity in Human Osteosarcoma Cells. Int J Mol Sci. 2021;22(18):9976.
- Fabre B, Ramos A, de Pascual-Teresa B. Targeting matrix metalloproteinases: exploring the dynamics of the s1′ pocket in the design of selective, small molecule inhibitors. J Med Chem. 2014;57(24):10205-10219.
- The Protein Data Bank [Internet]. 2000. Available from: https://www.rcsb.org/.
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296-w303.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J Med Chem. 2006;49(21):6177-6196.
- Schrödinger Release 2022-3 : Maestro, Schrödinger, LLC, New York, NY, 2021.
- Schrödinger Release 2020-1 : LigPrep, Schrödinger, LLC, New York, NY, 2020.
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221-234.
- Schrödinger Release 2018-1: Glide, Schrödinger, LLC, New York, N. (2018). www.schrodinger.com/Glide.
- J. Swails, R.C. Walker, R.C. Walker, X. Wu, L. Xiao, P.A. Kollman. Amber 20. San Francisco: University of California; 2020.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, R. Cheeseman, et al. Gaussian 09. Wallingford CT: Gaussian Inc.; 2016.
- Kirschner KN, Yongye AB, Tschampel SM, González-Outeiriño J, Daniels CR, Foley BL, et al. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Comput Chem. 2008;29(4):622-655.
- Tian C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J Chem Theory Comput. 2020;16(1):528-552.
- Cherinka B, Andrews BH, Sánchez-Gallego J, Brownstein J, Argudo-Fernández M, Blanton M, et al. Marvin: A Tool Kit for Streamlined Access and Visualization of the SDSS-IV MaNGA Data Set. Astron J. 2019;158:74.
- D.A. Case, R.M. Betz, D.S. Cerutti, T.E. Cheatham, III, T.A. Darden, R.E. Duke, T.J. Giese, H. Gohlke, A.W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T.S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K.M. Merz, G. Monard, H. Nguyen, H.T. Nguyen, I. Omelyan, A. Onufriev, D.R. Roe, A. Roitberg, C. Sagui, C.L. Simmerling, W.M. Botello-Smith, J. Swails, R.C. Walker, J. Wang, R.M. Wolf, X. Wu, L. Xiao and P.A. Kollman (2016), AMBER 2016, University of California, San Francisco.
