1. INTRODUCCIÓN
No cabe duda de que lo que nos ha ocurrido a todos con la pandemia y lo que ocurre en el día a día del trabajo analítico está gobernado por la sorpresa. En el ámbito analítico es la sorpresa de obtener resultados no esperados ya sea dentro de especificaciones o fuera, ya sea dentro de expectativas o fuera.
Y esa sorpresa necesariamente lleva a la necesidad de obtener soluciones lo antes posible. El tiempo es fundamental y debe además llevar implícita la seguridad de que la solución es la adecuada.
Desde que empecé a trabajar en el Laboratorio de Técnicas instrumentales y a lo largo de los años transcurridos en mi actividad profesional, he ido obteniendo resultados analíticos que, como digo, no siempre son los esperados. Esta situación conlleva una serie de preguntas como las siguientes:
- ¿Cuáles son los resultados analíticos que se acercan más a la verdad? ¿Los esperados o los obtenidos?
- Si hay discrepancia entre resultados obtenidos en ensayos repetidos siguiendo los mismos pasos analíticos, ¿cuál es la causa de dicha discrepancia?
- ¿Qué hacer ante valores analíticos fuera de especificaciones o fuera de expectativas?
- En general, ¿podemos estar seguros de la validez de los resultados analíticos incluso cuando están dentro de los límites de normalidad?
Desgraciadamente, la mayoría de esas preguntas no tienen respuesta o, por lo menos, una respuesta inequívoca, pero, en su momento, me estimularon a entrar en el mundo de la validación analítica como herramienta para reducir a límites razonables los errores en los laboratorios analíticos y a estar permanentemente en contacto con la evolución de la normativa que se ha ido generando a este respecto.
2. LA ICH Y SUS GUÍAS DE CALIDAD
La armonización de los requisitos reglamentarios, tan necesaria en el ámbito de la industria farmacéutica, fue iniciada por la Comunidad Europea (CE), en la década de 1980 (1), ya que la CE (actualmente Unión Europea) se movió hacia el desarrollo de un mercado único de los productos farmacéuticos. El éxito obtenido en Europa demostró que la armonización era factible por lo que se empezaron a mantener conversaciones multilaterales entre Europa, Japón y los EE.UU. sobre las posibilidades de armonización.
Sin embargo, hasta 1989 no empezó a materializarse ningún plan de acción específico, lo que ocurrió en la Conferencia de la Organización Mundial de la Salud (OMS) de Autoridades Reguladoras de Medicamentos (ICDRA) de París. Poco después, las autoridades se reunieron en el seno de la International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) para discutir una iniciativa conjunta de regulación internacional armonizada de la industria farmacéutica.
El nacimiento de la ICH (actualmente International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use) tuvo lugar en una reunión organizada por la European Federation of Pharmaceutical Industries and Associations (EFPIA) en Bruselas en abril de 1990 en la que los representantes de las agencias reguladoras y las asociaciones industriales de Europa, Japón y los EE.UU. discutieron las implicaciones y los términos de referencia más amplios de la ICH. En la primera reunión del Comité Directivo de la ICH se decidió que los temas seleccionados para la armonización se dividirían en Seguridad, Calidad y Eficacia para reflejar los tres criterios que sirven de base para la aprobación y autorización nuevos medicamentos, siendo las guías encuadradas en el campo de la Calidad (codificadas con la letra Q) las que más interesan desde el punto de vista del análisis de los medicamentos.
2.1. La guía ICH Q2
Dentro de la categoría Q, una de las guías ICH con mayor solera es la Q2-Validación Analítica (2), finalizada y emitida en octubre de 1994 como “Guía Q2A-Texto”, y que identifica los parámetros necesarios para la validación de métodos analíticos, incluyendo también las características que deben ser tenidas en cuenta durante la validación de los procedimientos analíticos que vayan a formar parte de las solicitudes de registro.
De forma análoga, en noviembre de 1996 se emitió la “Guía Q2B-Metodología”, en la que se incluyeron las pruebas experimentales requeridas, junto con su interpretación estadística, para la validación de los procedimientos analíticos. En noviembre de 2005 se fusionaron ambas guías en un único texto rebautizado como Q2 (R1), sin ningún cambio en el contenido de las dos directrices originales.
En la figura 1 se muestran las características o parámetros que deben ser evaluados durante la validación de diferentes tipos de métodos analíticos, de acuerdo con la guía Q2 (R1).

– esta característica no se evalúa normalmente + esta característica se evalúa normalmente * en caso de que hayan realizado las pruebas de reproductibilidad, no es necesaria la precisión ** la falta de ESPECIFICIDAD podrá compensarse con otro procedimiento *** puede necesitarse en algunos casos[/caption]
Figura 1: parámetros evaluados según la guía ICH Q2(R1)
Esta lista no es exhaustiva ya que es de destacar que otras características fundamentales, como la Robustez, no aparecen en la tabla, si bien la guía indica claramente que este tipo de parámetros deben ser tenidos en cuenta durante el desarrollo de un método analítico.
A pesar de este tipo de carencias, el contenido de la guía está plenamente incorporado a las Farmacopeas y guías oficiales más importantes, lo que hace que la validación de un método analítico sea de facto y de iure, un requisito legal imprescindible en cualquier solicitud de registro.
Esta situación tiene, como principal ventaja, la homogenización global de prácticas de validación, con el consiguiente reconocimiento por parte de las diferentes Autoridades Sanitarias. Sin embargo, la presencia en las guías de una lista de características como las de la figura anterior, agravada por la extendida costumbre de utilizar como definitivos unos criterios de aceptación generales recomendados para cada uno de dichas características, en muchos casos ha convertido el ejercicio de validación basada en la guía ICH Q2 (R1) en una especie de “formulario” que hay que rellenar, sin que con ello se llegue a un conocimiento profundo del funcionamiento de los métodos analíticos y de los factores internos y externos que pueden afectar a los resultados obtenidos.
Esto resulta especialmente importante cuando se obtienen resultados analíticos atípicos o fuera de especificaciones (OOS) ya que, en el entorno de las Normas de Correcta Fabricación, estos resultados no se pueden descartar sin haber efectuado una investigación de laboratorio cuyo objeto es conocer su “causa raíz”, es decir, intentando identificar y corregir las causas profundas de los acontecimientos, en lugar de tratar simplemente sus síntomas.
Es evidente que la investigación de laboratorio puede llegar a indicar que, desde el punto de vista analítico, el resultado sea correcto y que, por lo tanto, se haya puesto de manifiesto un problema en la muestra analizada. Pero también puede darse el caso de que la “causa raíz” esté en la propia metodología analítica y que, como consecuencia, haya que poner en marcha acciones correctivas y preventivas que impidan, o reduzcan de forma significativa, que el error analítico vuelva a producirse. En muchos casos, estas acciones pasan por una modificación del método lo que resulta especialmente grave si se ven afectados parámetros operativos validados según la ICH e incluidos en los correspondientes dosieres de registros lo que, consecuentemente, obliga a presentar variaciones ante las Autoridades Sanitarias, con la consiguiente pérdida de tiempo y de confianza en los resultados analíticos obtenidos hasta ese momento.
2.2. Nuevas guías ICH y la calidad por diseño
A lo largo del tiempo, los trabajos de la ICH en el ámbito de la calidad han dado como resultado más guías que han proporcionado una nueva dimensión tanto al proceso de desarrollo de los medicamentos como al de la producción. Estas guías son las siguientes:
- Q8 (R2): PHARMACEUTICAL DEVELOPMENT (Step 5), de agosto de 2009 (3)
- Q9: QUALITY RISK MANAGEMENT (Step 5) de noviembre de 2005 (4)
- Q10: PHARMACEUTICAL QUALITY SYSTEM (Step 5) de junio de 2008 (5)
- Q11: DEVELOPMENT AND MANUFACTURE OF DRUG SUBSTANCES (CHEMICAL ENTITIES AND BIOTECHNOLOGICAL/BIOLOGICAL ENTITIES) (Step 5) de mayo de 2012 (6)
- Q12: TECHNICAL AND REGULATORY CONSIDERATIONS FOR PHARMACEUTICAL PRODUCT LIFECYCLE MANAGEMENT (Step 5) de noviembre de 2019 (7)
- Q13: CONTINUOUS MANUFACTURING OF DRUG SUBSTANCES AND DRUG PRODUCTS (Step 1) de noviembre de 2018 (8)
- Q14: ANALYTICAL PROCEDURE DEVELOPMENT GUIDELINE (Step1) de noviembre de 2018 (9).
Llamo la atención sobre estas dos últimas guías por estar aún en fase de desarrollo y sobre la revisión de la Q2 (R1) que se está llevando en paralelo.
Según indica el Expert Working Group encargado de estas tareas “El alcance de la revisión de la guía Q2 (R1) incluirá principios de validación que cubren el uso analítico de datos espectroscópicos o de espectrometría (por ejemplo, NIR, Raman, NMR o MS) (…). Estas pautas propuestas (Q2 (R2) y Q14) están destinadas a complementar las guías ICH de la Q8 a la Q12, así como la guía Q13 para la fabricación continua, actualmente en Step 1”(9).
Con respecto a la guía Q14 (9), el mencionado grupo indica que “Esta nueva directriz está destinada a mejorar la comunicación regulatoria entre la industria y los reguladores y facilitar una aprobación más eficiente, sólida basada en los riesgos y en la ciencia, así como la gestión de cambios analíticos posteriores a la aprobación”.
Es importante destacar que estamos en un momento muy interesante ya que, según el plan de trabajo publicado, está previsto que se abra un periodo de consulta pública a partir de enero de 2021 que permitirá no sólo verificar si se ratifica el proceso de desarrollo y validación de métodos analíticos que se describe a continuación, sino poder participar directamente en la redacción final de las guías.
En estas guías, la Calidad por Diseño (QbD) viene definida como un “enfoque sistemático que comienza con unos objetivos previamente definidos y que enfatiza la comprensión de los productos y de los procesos, y el control del proceso basándose en el rigor científico y en la gestión de riesgos para la calidad”. Esta definición ya está incluida en el Anexo 15 de las Normas de Correcta Fabricación (NCF) (10).
Como se puede observar, hasta ahora la QbD ha ido claramente enfocada a los procesos productivos desde la fase de desarrollo, pero en ningún caso las guías actualmente en vigor mencionan expresamente a los métodos analíticos, explicando así la necesidad de la guía Q14 y la actualización de la Q2(R1).
Sin embargo, ya en el año 2007, Borman et al. (11) indicaban que existía la posibilidad de aplicar los principios de la QbD al desarrollo y validación de métodos analíticos para asegurar su precisión intermedia y su robustez (incluyendo las de la tecnología analítica de procesos – PAT), tal como se define en la guía ICH Q2(R1) y en la USP, y proponían una metodología que se está aplicando cada vez con mayor frecuencia. Hay que indicar que los métodos oficiales de Farmacopeas y los métodos microbiológicos quedan excluidos de esta metodología.
En cualquier caso, cabe destacar que hay autores (12) que hablan de Validación de Métodos por Diseño (Method Validation by Design – MVbD) y que citan iniciativas presentadas a la FDA en el ámbito del proyecto “Pharmaceutical Quality for the 21st Century. A Risk-Based Approach” (13), como puede ser el documento “Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification and Procedure Performance Verification” emitido en septiembre de 2013, por un comité de la USP creado al efecto.
En definitiva, se trata de una herramienta de desarrollo cuyo interés en la comunidad científica va incrementándose con el tiempo. Como aportación personal, la figura 2 muestra una pequeña revisión bibliográfica que cubre desde el año 2005 al año 2019, utilizando bases de datos habituales como Embase, Medline, Pubmed o American Chemical Society (14-25).
Publicaciones sobre QbD en métodos analíticos
Fuentes: ACS, Embase, Medline, Pubmed y propias
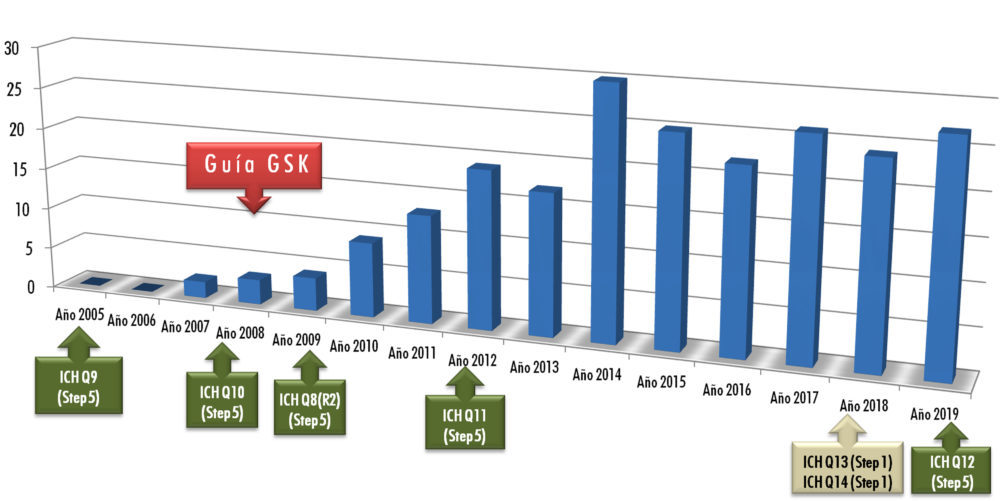
Figura 2: número de publicaciones sobre QbD en métodos analíticos (2005-2019)
De su observación resulta evidente el incremento del número de trabajos científicos posterior a la publicación de las Guías ICH Q8, Q10 y Q11, aunque resultan llamativas las publicaciones pioneras del año 2007, de autores pertenecientes al laboratorio en el que desarrollo mi actividad profesional, y que coinciden con la puesta en marcha de una guía interna de manejo de la QbD en el desarrollo y validación de métodos analíticos.
Queda pendiente por ver si la metodología que se describa en la nueva guía Q14 y la actualización de la Q2(R1) recoge los enfoques científicos del desarrollo de procedimientos analítico utilizados hasta ahora, uno de los cuales, quizá el más empleado, es el que se describe a continuación.
3. APLICACIÓN DE LA QbD. EL MÉTODO ANALÍTICO COMO PROCESO PRODUCTIVO
El primer paso para la aplicación de la QbD en el desarrollo y validación de un método analítico es considerarlo como un conjunto de operaciones secuenciales (preparación de la muestra objeto de análisis, metodología instrumental, calibración, definición de resultados y de especificaciones, etc.) que, partiendo de una “entradas” (inputs) permiten la obtención de unas “salidas” (outputs) o, en otros términos, la obtención de unos “productos” – los resultados analíticos – que deben cumplir unas especificaciones para considerarlos válidos per se, independientemente de las consecuencias a las que puedan dar lugar (por ejemplo, la aceptación o el rechazo de un lote de un medicamento).
En este sentido, como cualquier proceso productivo, la ejecución de un método analítico está sujeto a la influencia de factores externos que pueden dar lugar a que los resultados no sean válidos per se y que su utilización conlleve una decisión errónea.
La esencia de la QbD es el conocimiento de esos factores externos y de sus efectos e interacciones, lo que, en principio se plantea como una tarea casi inabarcable si se intentan conocer todos los posibles factores y todas las posibles interacciones. Es lo que se denomina el Espacio de Conocimiento que, en una primera aproximación, puede ayudar a definir qué partes de ese espacio tienen un efecto negativo sobre un determinado método analítico.
Sin embargo, desde un punto de vista de desarrollo y tomando como base el conocimiento existente, es mucho más útil definir el Espacio de Diseño, es decir, el que contiene la suma de todos los factores que dan lugar a unas condiciones aceptables en cuanto a sus efectos sobre el método analítico.
Finalmente, desde un punto de vista pragmático, el objetivo final es el de definir el Espacio de Control, que se puede mover dentro del Espacio de Diseño, y que es el que delimita las condiciones operativas normales en la ejecución de un método analítico.
Aunque, en general, los Espacios de Conocimiento, Diseño y Control son combinaciones e interacciones multidimensionales de los factores considerados, la figura 3 describe, de manera gráfica y en tres dimensiones, las relaciones entre estos tres Espacios.
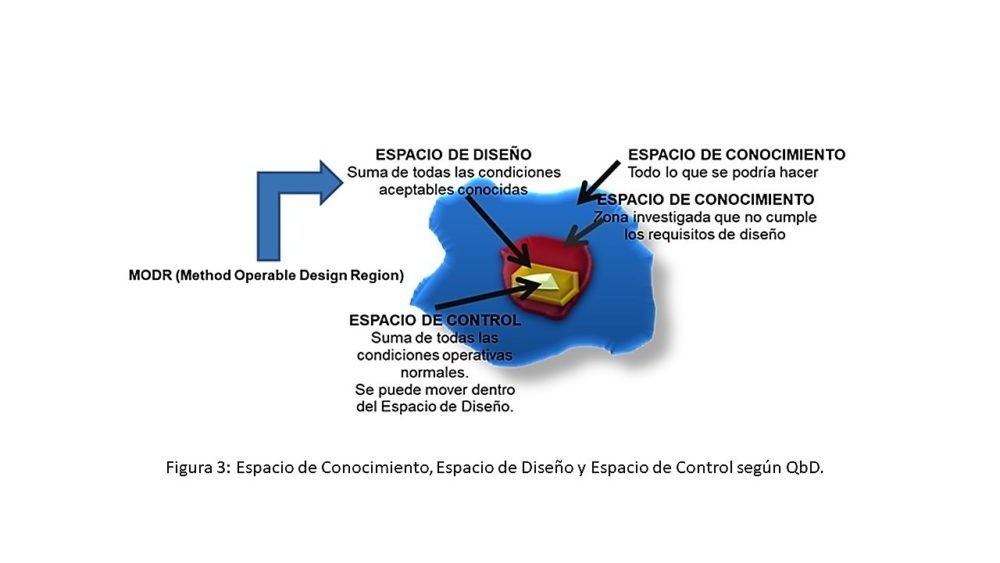
Figura 3: Espacio de Conocimiento, Espacio de Diseño y Espacio de Control según QbD
La aplicación de la QbD, por lo tanto, permite obtener evidencias científicas del desarrollo de métodos robustos, capaces de resistir la influencia de esos factores externos, es decir, de producir datos de calidad de forma consistente, siempre que el Espacio de Control se mueva dentro del Espacio de Diseño.
Para ello, la metodología de la QbD se basa en los siguientes cuatro pasos secuenciales:
a) Definición del diseño
b) Selección del diseño
c) Definición de control
d) Verificación del control
3.1. Definición del diseño
Es evidente que el primer paso debe ser el propósito del método analítico (14), distinguiendo, por ejemplo, si se trata de un método que se va a utilizar para la liberación de lotes o para caracterizar un producto, y teniendo claro cuál es el perfil de calidad del objeto de análisis (QTPP) y sus atributos críticos de calidad (CQA) según la guía ICH Q8 (R2), incluyendo las especificaciones del producto ya que son éstas las que definen el perfil analítico del método (Analytical Target Profile o ATP, análogo al QTPP en terminología ICH) y no al contrario (17).
En términos generales, la terminología QbD en el ámbito analítico puede resumirse conforme a lo indicado en la figura 4.
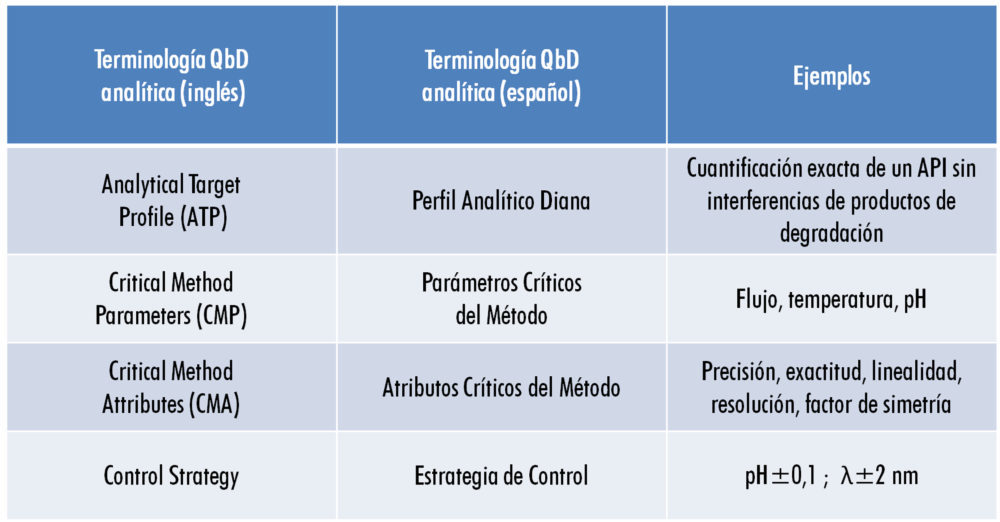
Figura 4: terminología QbD analítica
Para construir el ATP es necesario determinar los indicadores del comportamiento del método analítico (sus CMA), que no son otros que los parámetros tradicionalmente utilizados en la validación analítica (precisión, exactitud, linealidad, etc.), pero, sobre todo, hay que determinar los criterios de aceptación aplicables a cada uno de los indicadores en función del objeto de análisis (18) para que el método desarrollado y validado se ajuste a su propósito (fit-to-purpose).
Si se estudian con detalle los CMA con el fin de asignarles criterios de aceptación, se observa que hay dos grupos claramente diferenciados:
Atributos cuya naturaleza matemático-estadística o cuyo origen instrumental permite la asignación de criterios de aceptación a priori (y en cierto modo, independiente del objeto de análisis);
Atributos para los que es necesario conocer las características del producto objeto de análisis antes de asignar criterios de aceptación.
En la tabla 1 se detallan los componentes de los dos grupos.
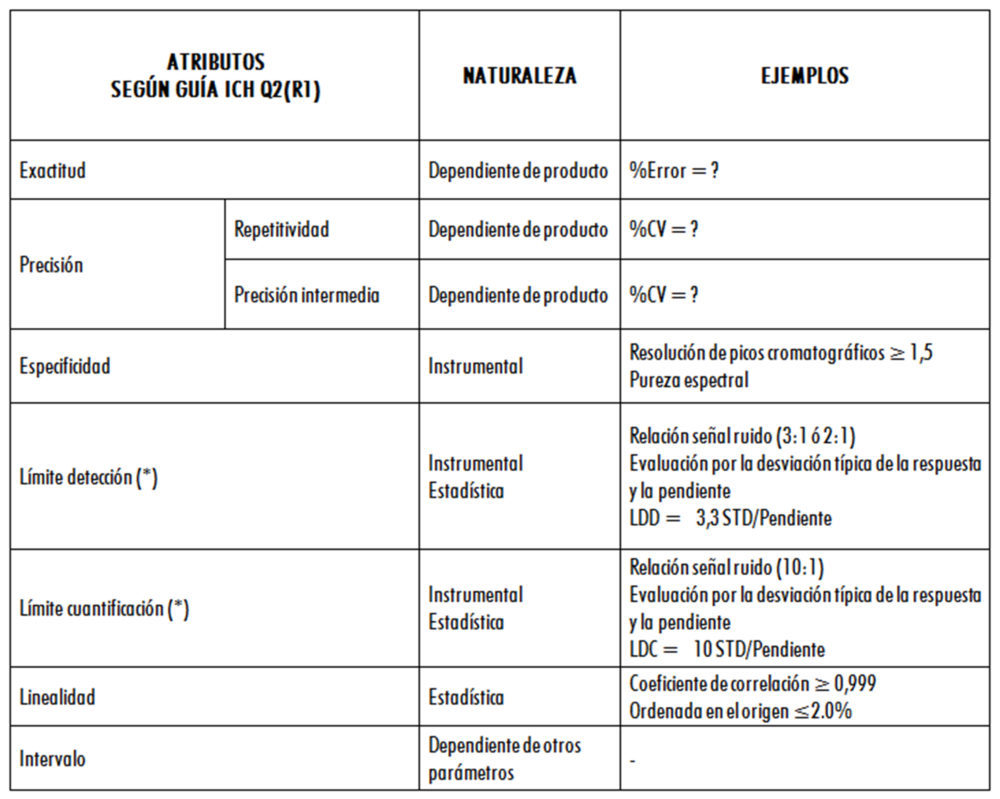
(*) A su vez, se tiene que demostrar la precisión y la exactitud en el nivel de concentración determinado mediante este atributo.
Por citar un ejemplo, supongamos que se quiere establecer el criterio de aceptación de un ensayo de repetitividad, lo que implica asignar un valor numérico al Coeficiente de Variación (%CV). Para ello, es posible utilizar la relación precisión-tolerancia (P/TOL), definida por la siguiente fórmula:
P/TOL = 6σmétodo/(Límite superior – Límite inferior) donde σ es la desviación estándar del método analítico.
Generalmente se considera que, si P/TOL es menor o igual a 0,3, la precisión del método es aceptable, pero si P/TOL es mayor que 0,3, dicha precisión es inaceptable.
Sin embargo, no es este el punto final del proceso ya que sus implicaciones se extienden también al equipo instrumental que se pretenda utilizar, toda vez que no será adecuado el empleo de un equipo cuya precisión sea peor que el mencionado Coeficiente de Variación.
En definitiva, la gran revolución de la definición del diseño, como primer paso en la aplicación de la QbD en el desarrollo y validación de métodos analíticos, consiste en condicionar todos los elementos que intervienen en el análisis a las características del medicamento que se pretende analizar, partiendo de los límites superior e inferior obtenidos a partir de los ensayos clínicos.
3.2. Selección del diseño
Describir la selección del diseño es equivalente a describir el desarrollo del proceso analítico en sí.
Es evidente que, al definir el diseño conforme a lo indicado en el apartado anterior, el desarrollador tiene en mente una técnica instrumental concreta, pero no la forma de realizar cada uno de los pasos del análisis, empezando por la preparación de las muestras y acabando con la expresión de los resultados finales. Por ello, es conveniente utilizar esquemas de trabajo secuenciales que cubran los siguientes puntos, siempre que sean aplicables:
- Tipo de equipo instrumental
- Condiciones ambientales
- Materiales y reactivos
- Preparación de las muestras
- Sistema de introducción de las muestras preparadas en el equipo instrumental
- Condiciones operativas del equipo instrumental
- Sistema de detección
- Sistema de obtención y proceso de datos
Obviamente, no se trata sólo de realizar un ejercicio teó-rico ya que cada uno de los puntos anteriores debe ir acompañado del correspondiente trabajo experimental, como tradicionalmente se ha realizado a lo largo de la historia. La diferencia con la aplicación de la QbD estriba en que se debe tener siempre presente el ATP para aceptar o descartar los resultados obtenidos durante el desarrollo analítico, si bien, dependiendo del estadio de dicho desarrollo y del tipo de análisis, no es necesario tener en cuenta todos los parámetros que definen el ATP. En este sentido, si se trata de un ensayo de valoración del componente principal de un medicamento, se puede obviar la precisión intermedia, la reproducibilidad, el intervalo y los límites de detección y cuantificación, además de la robustez, en las fases iniciales de desarrollo, incorporando estos parámetros en las fases finales. Algo parecido ocurre en las fases iniciales del desarrollo de un método de análisis cuantitativo de impurezas, para el que, además de los anteriores, se debe incorporar el límite de cuantificación en las fases finales (19).
3.3. Definición de control
Describir la definición de control es equivalente a realizar una evaluación de riesgos durante el desarrollo del proceso analítico, evaluación que necesariamente debe comenzar con la enumeración de todos los posibles factores que pueden afectar al ATP, tanto en un sentido positivo como negativo. Estos son los denominados “Parámetros Críticos del Método” o CMP, análogos a los “Parámetros Críticos del Proceso” o CPP descritos en la guía ICH Q8.
La utilización de experiencias previas en el desarrollo de otros métodos analíticos, la bibliografía disponible y, especialmente, las opiniones de analistas experimentados (voice of the customer) deben ser las fuentes utilizadas para la obtención de una lista bruta de los CMP anteriormente mencionados. Esa lista bruta define el Espacio de Conocimiento que, como ya se ha indicado anteriormente, no es viable como tal salvo como punto de partida para la definición del Espacio de Diseño, mediante una labor de clasificación razonada y sistemática de los parámetros considerados.
Para ello, el primer paso consiste en tener en cuenta los puntos descritos en el apartado anterior como componentes de un esquema de trabajo secuencial para el desarrollo de un proceso analítico, ya que no son casuales. Responden a una estrategia necesaria para la definición de control ya que en cada uno de ellos deben encuadrarse los CMP que pueden influir en el comportamiento de un método analítico en desarrollo. Así habrá parámetros relacionados las condiciones ambientales, otros relacionados con la preparación de las muestras, u otros relacionados con las condiciones operativas del equipo instrumental.
En todo caso, la lista de CMP considerados en cada apartado debe ser lo más exhaustiva posible, huyendo de listados prefabricados similares a formularios, que podrían llevar a no tomar en consideración algún parámetro verdaderamente importante. Además, la cantidad y variedad de CMP hace necesario el uso de herramientas de permitan ordenarlos y clasificarlos, por lo que es frecuente el uso de un diagrama de Ishikawa.
Sin embargo, la mera enumeración de CMP no es suficiente para definir el control, toda vez que la influencia de cada uno de ellos no es uniforme. Así habrá fundamentalmente tres tipos de parámetros, denominados factores en esta clasificación:
a) Factores que generan un cierto “ruido” en los resultados analíticos, denominados factores N, y que están relacionados con la precisión intermedia;
b) Factores que producen un efecto sustancial sobre los resultados analíticos, pero cuya cuantificación en términos de efectos y de los intervalos aceptables es desconocida. Se denominan factores X y están relacionados con la robustez;
c) Factores que, durante la ejecución del análisis, deben estar controlados para no producir ningún efecto. Se denominan factores C.
Cada uno de estos tipos de factores tiene un tratamiento distinto, tanto desde el punto de vista del análisis de riesgos y determinación de los más influyentes, como desde el punto de vista estadístico, tal como puede observarse en la figura 5.
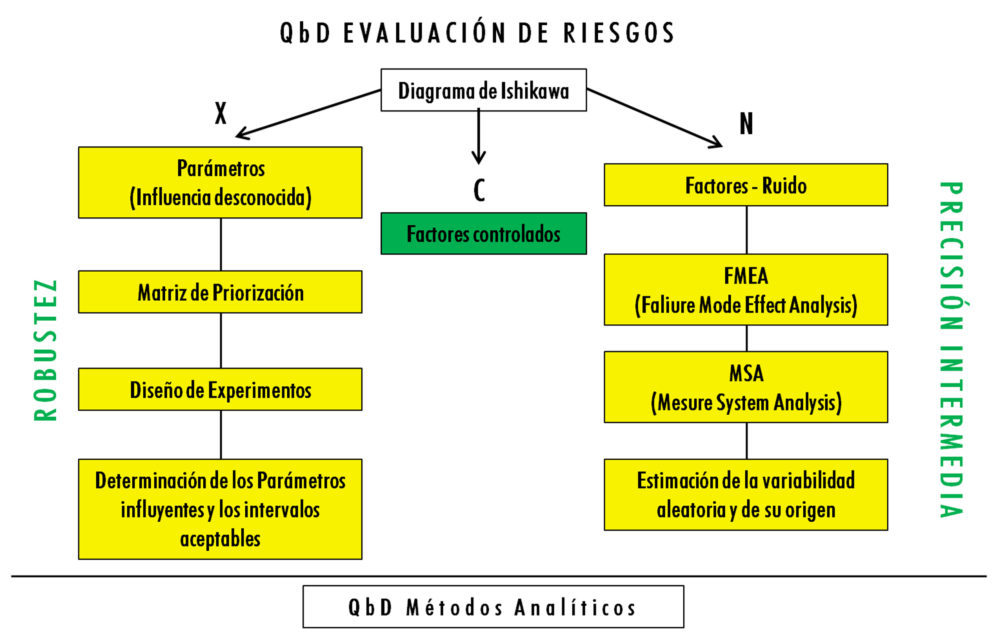
Figura 5: evaluación de riesgos en el desarrollo de métodos analíticos según QbD
FACTORES X – ROBUSTEZ
Son los primeros factores que hay que estudiar, ya que de su estudio se desprenden las condiciones operativas más adecuadas en primera instancia para ejecutar el procedimiento analítico.
La herramienta más útil para el análisis de riesgos es la denominada Matriz de Priorización, basada en la asignación de un valor numérico previamente definido al efecto o impacto de cada factor sobre cada uno de los CMA.
Una vez finalizado el análisis de riesgos, la herramienta estadística más adecuada para su análisis es el Diseño de Experimentos (DoE), que es un modelo estadístico clásico cuyo objetivo es averiguar si unos determinados factores influyen en una respuesta de interés y, en caso de existir, cuantificar dicha influencia.
FACTORES N – PRECISIÓN INTERMEDIA
El estudio de los factores N debe realizarse tras finalizar el estudio de los factores X y, al igual que con estos, debe comenzar con una tarea de clasificación de aquellos que presentan un mayor riesgo para la precisión intermedia en caso de fallo.
La herramienta más útil es el Análisis Modal de Fallos y Efectos, más conocida por sus siglas en inglés FMEA (Failure Mode Effect Analysis). La metodología para la realización de un FMEA establece las siguientes etapas:
1. Identificación de Riesgos: Identificación de fallos (hazards) y determinación de consecuencias para cada uno de los factores N.
2. Estimación del riesgo para cada uno de los fallos detectados:
- Gravedad
- Probabilidad del fallo
- Clasificación del riesgo
- Prioridad del riesgo
3. Evaluación del riesgo y establecimiento de las medidas de control necesarias para reducirlo.
La gravedad evalúa el impacto, en caso de que el fallo suceda, sobre la calidad de los resultados analíticos, en este caso en cuanto a la precisión intermedia (o indirectamente sobre la calidad/seguridad/eficacia del producto), de manera que se establecen cinco niveles:
- CATASTRÓFICO (nivel 5): Impacto directo sobre la calidad de los resultados analíticos, que suponga una amenaza grave y no evitable para las decisiones sobre la calidad/seguridad/eficacia del producto.
- ALTO (nivel 4): Impacto directo sobre la calidad de los resultados analíticos, que suponga una amenaza grave y evitable para las decisiones sobre la calidad/seguridad/eficacia del producto.
- MEDIO (nivel 3): Impacto indirecto sobre la calidad de los resultados analíticos, que suponga una amenaza grave y no evitable para las decisiones sobre la calidad/seguridad/eficacia del producto.
- BAJO (nivel 2): Impacto indirecto sobre la calidad de los resultados analíticos, que suponga una amenaza grave y evitable para las decisiones sobre la calidad/seguridad/eficacia del producto.
- INSIGNIFICANTE (nivel 1): Ausencia de impacto.
Por otra parte, se evalúa la probabilidad de que el fallo suceda con criterios cualitativos, estableciendo los siguientes cinco niveles:
- CASI SEGURA (nivel 5)
- PROBABLE (nivel 4)
- POSIBLE (nivel 3)
- IMPROBABLE (nivel 2)
- CASI NULA (nivel 1)
A partir de la probabilidad de que suceda el fallo y su impacto, se determina el riesgo asociado a cada uno de los fallos posibles.
Una vez finalizado el análisis de riesgos, y utilizando aquellos con riesgos altos y medios, la herramienta estadística más adecuada para su análisis es el Análisis del Sistema de Medida, más conocido por sus siglas MSA (Measurement System Analysis) que, partiendo de un diseño de ANOVA, permite no sólo conocer el grado de dispersión de los valores analíticos, sino también el porcentaje relativo de aportación de cada uno de los factores a la variabilidad total. Al analizar dichos valores, se pueden dar dos circunstancias:
Que la mayor contribución corresponda a las muestras analizadas lo que indica que el método analítico es adecuado y permite la discriminación de muestras de diferente origen;
Que la mayor contribución corresponda a los factores analíticos (analista o equipo instrumental), lo que obliga a poner en marcha acciones correctivas conforme al FMEA.
ESTABLECIMIENTO FINAL DE LA DEFINICIÓN DE CONTROL
A lo largo del estudio de los factores X y de los factores N, puede darse la circunstancia de que los resultados obtenidos no sean todo lo satisfactorios que cabría esperar.
Así puede ocurrir que el DoE no muestre cambios significativos en determinadas respuestas (por ejemplo, obteniendo superficies de respuesta planas) o que el MSA muestre que, a pesar de poner en marcha acciones correctivas se siga observando que los factores analíticos tienen porcentajes de aportación elevados.
Normalmente esto significa que muy probablemente se ha considerado como factor N uno que debía haber sido clasificado como factor X y viceversa, por lo que será conveniente rehacer la clasificación y repetir algunos de los ensayos en las nuevas condiciones.
En cualquier caso, es absolutamente imprescindible que todos los factores enumerados en el primer paso de la definición de control hayan tenido algún tratamiento, bien en el estudio de la robustez, bien en el estudio de la precisión intermedia. Una vez realizada la verificación correspondiente, denominada también “Procedure Performance Qualification” y que requiere un protocolo de trabajo previamente definido, se podrá pasar a expresar de manera completa el Espacio de Diseño que, en el caso de métodos analíticos, se denomina “Method Operable Design Region” o MODR.
4. VERIFICACIÓN DEL CONTROL
Una vez completadas las fases anteriores, e incluido el método en el dossier de registro, hay que mantener el estado de validación de forma permanente en su utilización habitual. Para ello, lo más frecuente es hacer ensayos de idoneidad del sistema (system suitability) de forma periódica utilizando patrones analíticos o muestras completamente caracterizadas, es decir, con todas sus características conocidas en cuanto a identidad, composición o pureza. En este sentido, la frecuencia de realización podrá depender del uso al que esté destinado el método analítico y, por lo tanto, la criticidad de las decisiones que se puedan tomar a partir de los resultados obtenidos en un análisis con dicho método (por ejemplo, la liberación o el rechazo de un lote de medicamento para uso humano), pero en ningún caso se podrán obviar los resultados de los ensayos de idoneidad que deberán ser significativamente parecidos a los obtenidos durante la fase de desarrollo y validación, desde el punto de vista estadístico.
Por lo tanto, cualquier desviación implicará una investigación de laboratorio y las correspondientes acciones correctivas y, en su caso, preventivas. Si estas requieren la modificación de las condiciones operativas, habrá que verificar si dichas condiciones se mantienen dentro del Espacio de Diseño, en cuyo caso no será necesario llevar a cabo ninguna revalidación ni modificación del registro original. Obviamente, si las nuevas condiciones operativas se salen del Espacio de Diseño, habrá que repetir los pasos anteriores para definir uno nuevo con la consiguiente presentación de una variación del registro ante las Autoridades Sanitarias.
5. APLICACIÓN DE LA QbD FUERA DEL ÁMBITO DEL ANÁLISIS DE MEDICAMENTOS
Llegados a este punto en la descripción de la aplicación de la QbD en el desarrollo y validación de métodos analíticos, cabe plantearse dos preguntas:
¿Es posible aplicar la QbD fuera del ámbito del análisis de medicamentos?
¿Qué aporta un desarrollo analítico siguiendo QbD frente a la metodología tradicional?
La respuesta a la primera pregunta es obvia ya que no existe ningún impedimento regulatorio para la aplicación de QbD, siempre que se trate del desarrollo de un método analítico físico o físico-químico.
Por otra parte, en nuestra experiencia, podemos afirmar que el ámbito real de aplicación de la QbD para métodos analíticos puede abarcar no sólo el análisis cuantitativo de medicamentos, sino también otros métodos analíticos en los que se determinan otras propiedades. Tal es el caso de la citoferometría, modalidad de electroforesis de células y partículas en suspensión, con la que es posible determinar la movilidad electroforética de eritrocitos normales y patológicos, entre otras muestras.
Esta afirmación está basada en una serie de trabajos de validación de un programa informático desarrollado “ad hoc” en la fase PQ (Process Qualification o Cualificación de Proceso) según la Guía GAMP 5 (26). En estos trabajos se reprocesaron datos del desarrollo original de un método citoferométrico realizado a partir del año 1986 como parte de mi tesis doctoral, de acuerdo con los principios QbD actuales de manera que fuera posible verificar el correcto funcionamiento del programa informático.

Figura 6: citoferómetro
Los datos analíticos originales se obtuvieron con un citoferómetro Zeiss, equipo instrumental que se muestra en la figura 6 y que consta de las siguientes partes:
Un microscopio de eje óptico horizontal, uno de cuyos oculares presenta una lente reticulada que permite medir el tiempo que tardan los eritrocitos en recorrer una determinada distancia.
Una cámara de electroforesis de 1,4 cm de longitud y 7,6 x 10-2 cm de profundidad introducida en una cámara termostatizada que permite variar la temperatura de trabajo.
Una fuente de alimentación capaz de generar una diferencia de potencial entre 0 y 900 V.
Dos electrodos de platino.
Un conmutador que permite la inversión del campo eléctrico generado.
Para realizar las medidas de movilidad electroforética se aplicó un potencial determinado y se midió con un cronómetro de precisión el tiempo que tardan los eritrocitos en recorrer 0,0032 cm (dos intervalos de retículo) en las dos direcciones del campo eléctrico aplicado, cuando se encuentran en una solución salina isotónica cuya resistencia específica se conoce previamente.
Puesto que la movilidad electroforética µ se define mediante la expresión matemática,

siendo: l = distancia recorrida (cm)
T = tiempo empleado (s)
i = intensidad de corriente aplicada (A)
r = resistencia específica del medio de suspensión (Ω x cm)
h = longitud de la cámara de electroforesis (cm)
t = profundidad de la cámara de electroforesis (cm)
es evidente que se puede trabajar directamente con las medidas de tiempo (inversamente proporcionales a los valores de movilidad electroforética) para seguir la metodología QbD de una manera rápida y eficaz.
Adicionalmente, los nuevos resultados se compararon con los obtenidos mediante la metodología tradicional de manera que se pudo responder a la segunda pregunta planteada al comienzo de este apartado.
Como punto de partida, y según el desarrollo del método citoferométrico original (27), la metodología más conveniente consiste en la aplicación de un campo eléctrico de 10 mA a eritrocitos recién lavados (conservados a 4 ºC si es necesario), suspendidos en solución salina fisiológica (pH = 5,65, fuerza iónica resistencia = 0.1 M) y medidos dentro de una cámara termostatizada a 25-30 ºC, con el fin de obtener resultados analíticos con una precisión y una selectividad adecuadas.
Por su parte, para el desarrollo y validación del método analítico según la QbD se siguieron los siguientes pasos secuenciales (steps) descritos en las guías internas del laboratorio donde desarrollo mi actividad profesional:
Step 1: desarrollo del método (method development), en el que habitualmente se diseña el método, se establece el ATP, los CPM y los CMA basados en la guía ICH Q2(R1) (2), así como los correspondientes criterios de aceptación que deben ser comprobados experimentalmente.
Step 2: evaluación del riesgo (risk assessment), en el que se determinan todos los factores que potencialmente pueden tener un impacto en el comportamiento del método analítico, y se establece su nivel de prioridad y su posible efecto sobre la robustez (factores X) y la reproducibilidad (factores N).
Step 3: robustez del método (method robustness), en el que se emplea el Diseño de Experimentos para definir el Espacio de Diseño que es la zona en la que los factores X permiten el cumplimiento de los criterios de aceptación de los CMA (zona también denominada MODR o Method Operable Design Region) y el Espacio de Control, que es la zona en la que se obtienen los mejores resultados en el cumplimiento de dichos criterios y que, en principio, es la que define las condiciones normales de uso del método analítico.
Step 4: reproducibilidad del método (method ruggedness), en el que se emplean técnicas de MSA (Measurement System Analysis) para definir el % de contribución a la variabilidad total del método analítico por parte de factores relacionados con el sistema de medida, con los analistas y con las muestras objeto de análisis.
Otras etapas, como el Step 5 (definición del control del método), el Step 6 (validación del método) y el Step 7 (mejora continua y control de cambios) quedan fuera del ámbito del presente trabajo.
5.1 Step 1. Desarrollo del método
Los resultados se muestran en las tablas 2 y 3. En esta última se muestran los criterios de aceptación de los CMA, indicando su aplicabilidad, los valores propuestos en función de los trabajos iniciales de desarrollo por QbD, y los aplicados en el desarrollo original.
Tabla 2: terminología analítica según QbD
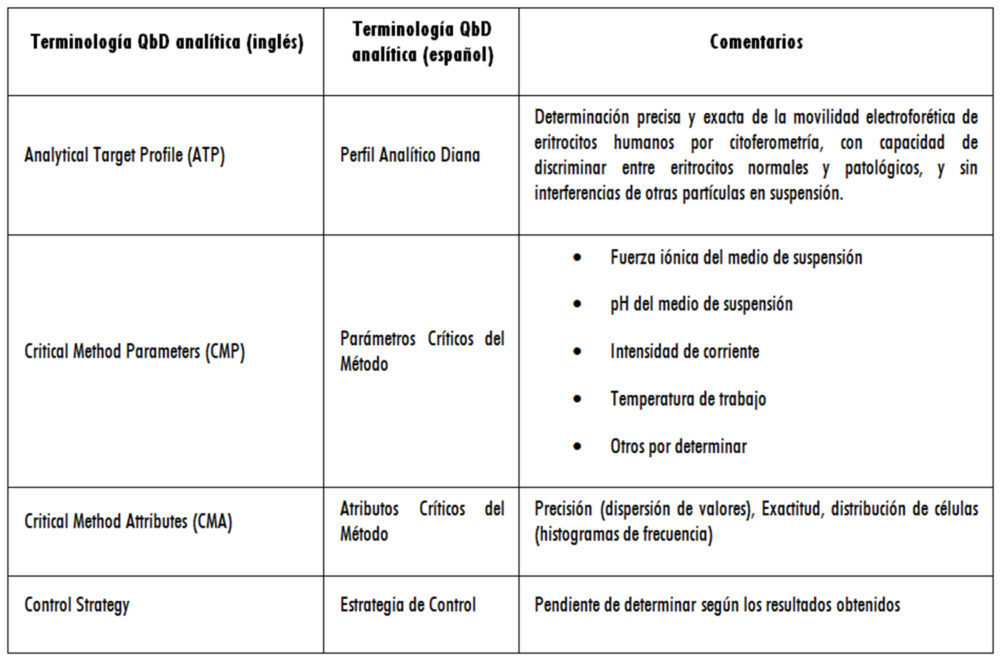
Tabla 3: comparación de criterios de aceptación según la metodología aplicada
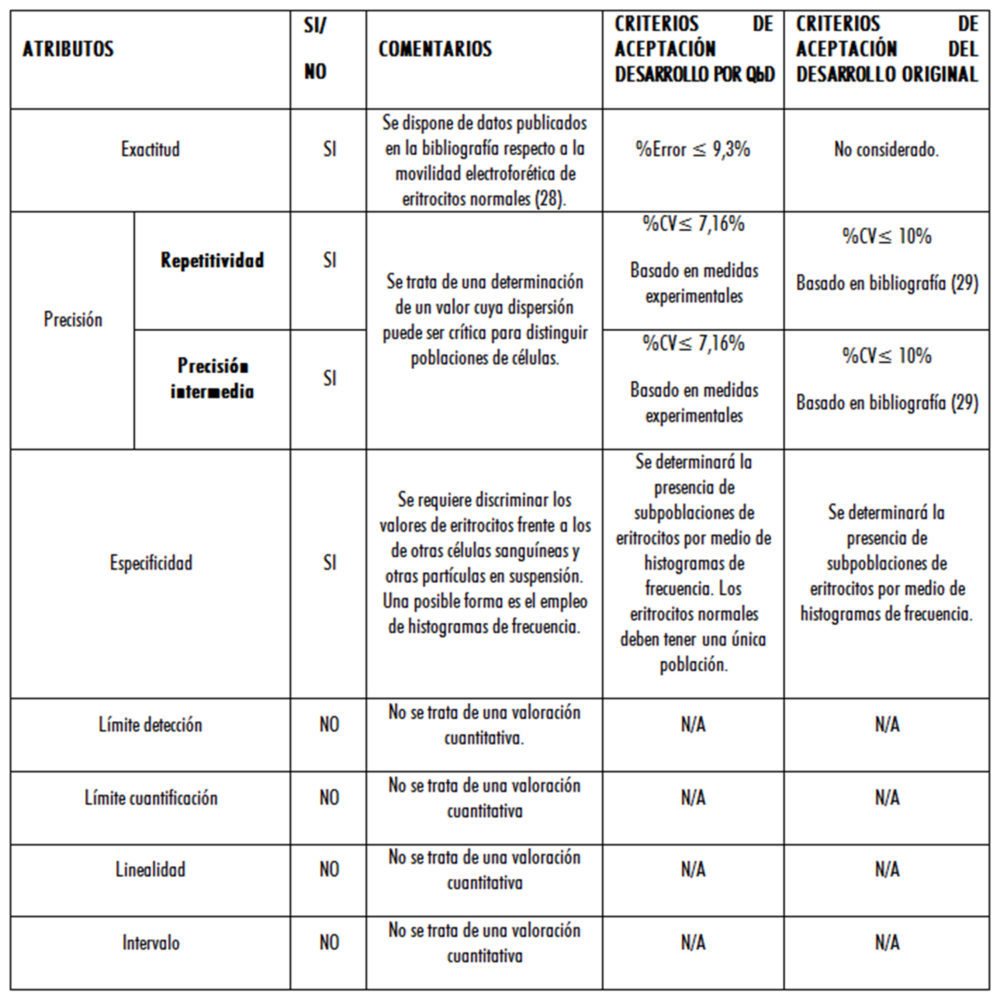
Con respecto al criterio de aceptación del error, se tuvieron en cuenta las pautas del documento CEA-ENAC-LC/02 de enero de 1998 (30), ya que, al disponer de 20 valores experimentales, se pudo realizar una evaluación de la incertidumbre Tipo A que dio como resultado UA = 0,149715055 = 0,15 s (para 19 grados de libertad y un nivel de confianza del 95,45 %).
Como dicha incertidumbre supone un 3,4 % del valor medio, inicialmente se consideró como % error admisible la diferencia entre el valor de tiempo para la movilidad electroforética de la bibliografía (28), que corresponde a 4,00 s y el experimental (4,37 s), lo que da 9,3 %, superior a la incertidumbre y, por lo tanto, detectable.
5.2 Step 2. Evaluación del riesgo
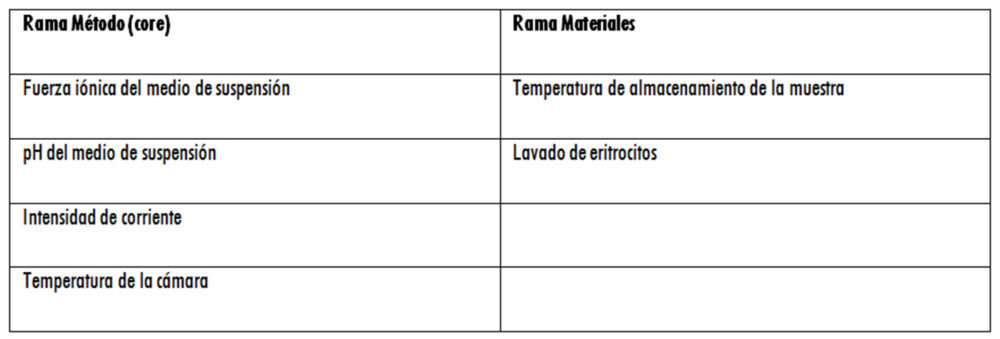
En cuanto al Step 2, el análisis de riesgo inicial, realizado mediante una matriz de priorización, permitió la construcción de un diagrama de Ishikawa y la determinación de los factores X susceptibles de ser incluidos en los diseños de experimentos correspondientes y que, tras una serie de ensayos realizados aplicando revisión bibliográfica, comparaciones de medias o análisis OFAT, quedaron definitivamente clasificados como se indica en la tabla 4.
Tabla 4: factores X
5.3 Step 3. Robustez del método
Como consecuencia de lo anterior, los resultados obtenidos en los experimentos diseñados, aplicando los correspondientes criterios de aceptación son los mostrados en la figura 7.
En el primer caso – Rama Método (core): dos factores numéricos (pH y Fuerza Iónica) y una respuesta medida (% Error) -, se considera que el MODR se define por la utilización de un medio isotónico con un pH entre 5,00 y 5,75, con una Fuerza Iónica entre 0,1 y 0,2 M.
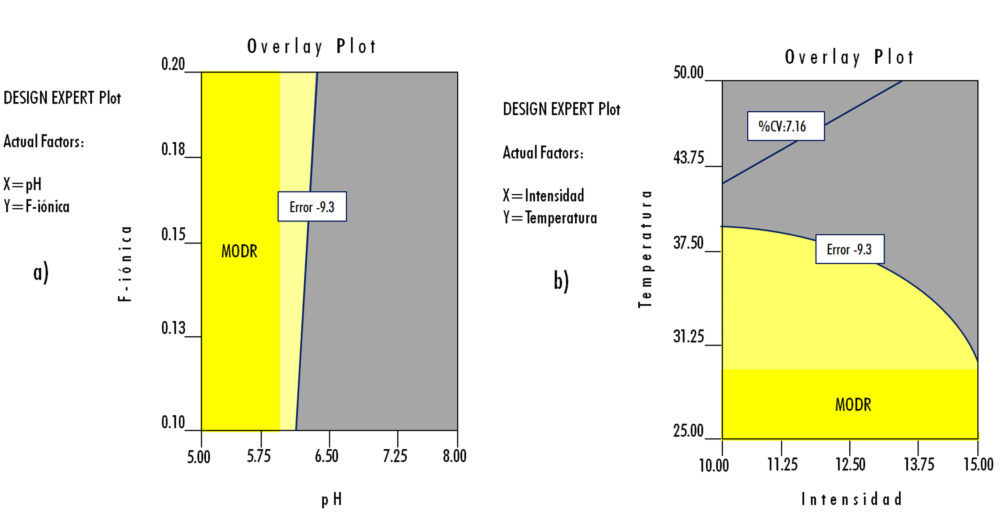
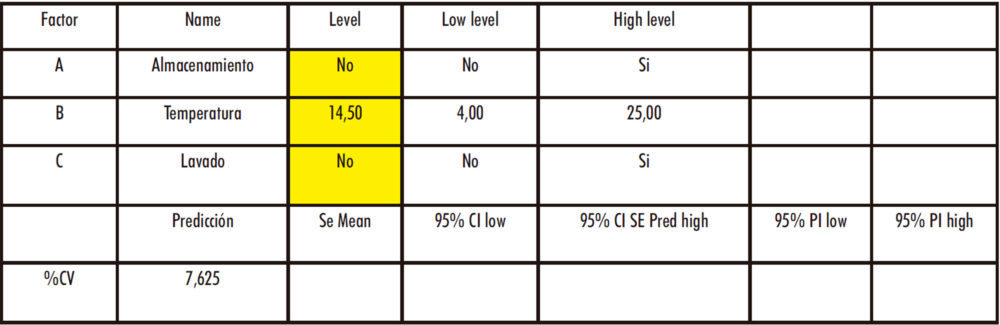
Figura 7: ensayos de robustez. a): dos factores numéricos (pH y Fuerza Iónica) y una respuesta medida (% Error); b) dos factores numéricos (temperatura e Intensidad de corriente) y dos respuestas (% Error y %CV); c) dos factores categóricos (almacenamiento y lavado), un factor numérico (temperatura de conservación) y una respuesta (% CV).
En el segundo caso – Rama Método (core): dos factores numéricos (Temperatura e Intensidad de corriente) y dos respuestas (% Error y %CV) -, se considera que el MODR que cumple los criterios de aceptación de ambas respuestas medidas se define con una T entre 25 ºC y 31 ºC y una intensidad de corriente entre 10 y 15 mA.
En el tercer caso – Rama Materiales: dos factores categóricos (almacenamiento y lavado), un factor numérico (temperatura de conservación) y una respuesta (% CV) -, se considera que las condiciones experimentales óptimas consisten en la utilización de eritrocitos frescos y no lavados. En cuanto a la temperatura de conservación, en caso de ser necesaria, el sistema informático indica una T = 14,50 oC pero, al ser esto inviable desde un punto de vista práctico (atendiendo a una posible climatización del laboratorio o a una posible cámara de conservación con ese punto de consigna), se considera más adecuado conservar los eritrocitos a una T =4 ºC. No es posible establecer un MODR al haber valores categóricos y no numéricos, por lo que estos pasan a ser factores C.
5.4 Step 4. Reproducibilidad del método
Una vez identificados los factores X, N y C en los pasos anteriores, y habiendo estudiado el efecto de los factores X, se centró la atención en los factores N para su estudio por MSA (Step 4).
Las causas potenciales de variabilidad pueden ser:
debidas a las muestras, para las que hay que determinar su estabilidad;
debidas al instrumento, que pueden afectar a la repetitividad y a la estabilidad;
debidas al operador.
Se estudiaron los factores N determinados en el Step 1 para determinar cuáles afectan a la repetitividad y cuáles afectan a la reproducibilidad. El resultado de ese estudio es el siguiente:
EQUIPO: mantenimiento, historia de uso, tipo de filtros utilizados. Estos afectan a la repetitividad.
AMBIENTE: temperatura del laboratorio, HR del laboratorio, localización del equipo. Estos afectan a la reproducibilidad.
INLET: introducción de muestras. Afecta a la repetitividad.
DETECTOR: estado de la lámpara. Afecta a la repetitividad.
PERSONAL: formación, competencia, precisión, patrones de trabajo, procedimientos. Estos afectan a la reproducibilidad.
MUESTRA: lípidos, TG, índices eritrocitarios, sexo del paciente, grupo sanguíneo. Estos afectan a la repetitividad y a la reproducibilidad.
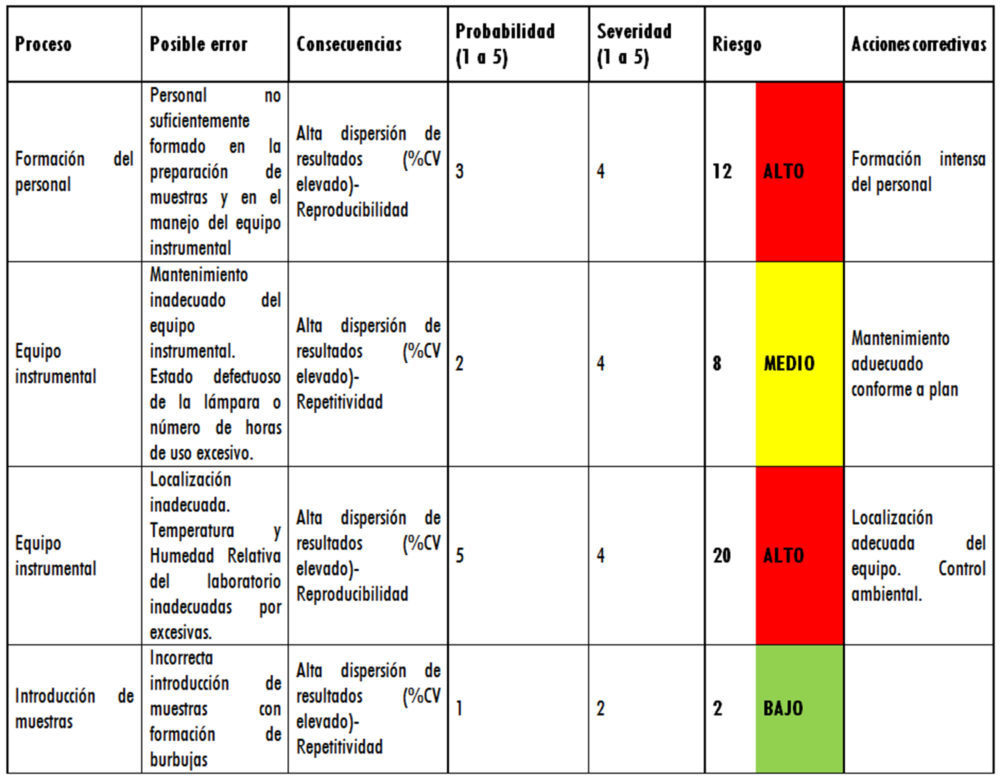
Con esto datos se procedió a la realización del FMEA agrupando los factores por niveles de riesgo. Los resultados se muestran en la tabla 5 y en ellos se observa que conviene estudiar personal y equipo por ser los de riesgo ALTO y MEDIO.
Tabla 5: tabla FMEA
Se planteó un protocolo estándar de trabajo de 5 muestras x 2 operadores x 2 replicados para estudiar el efecto de la formación y experiencia del operador y del cambio de ubicación de equipo y, con los resultados de este estudio (figura 8), se pudo constatar la poca capacidad de discriminación de muestras debido especialmente a la muy elevada influencia del personal (88% appraiser).
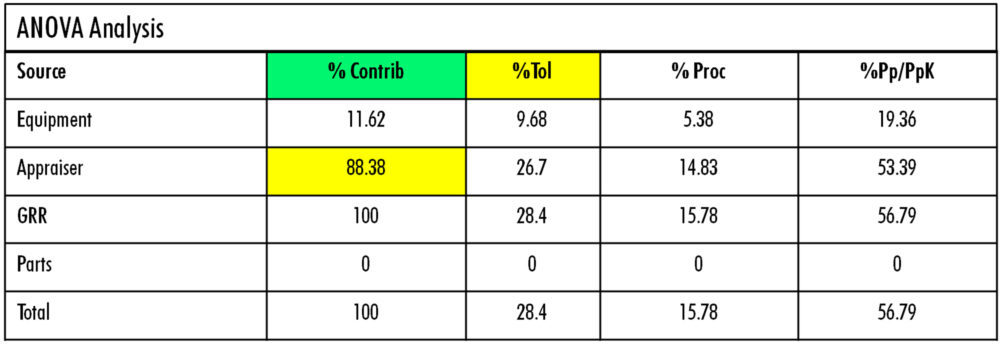
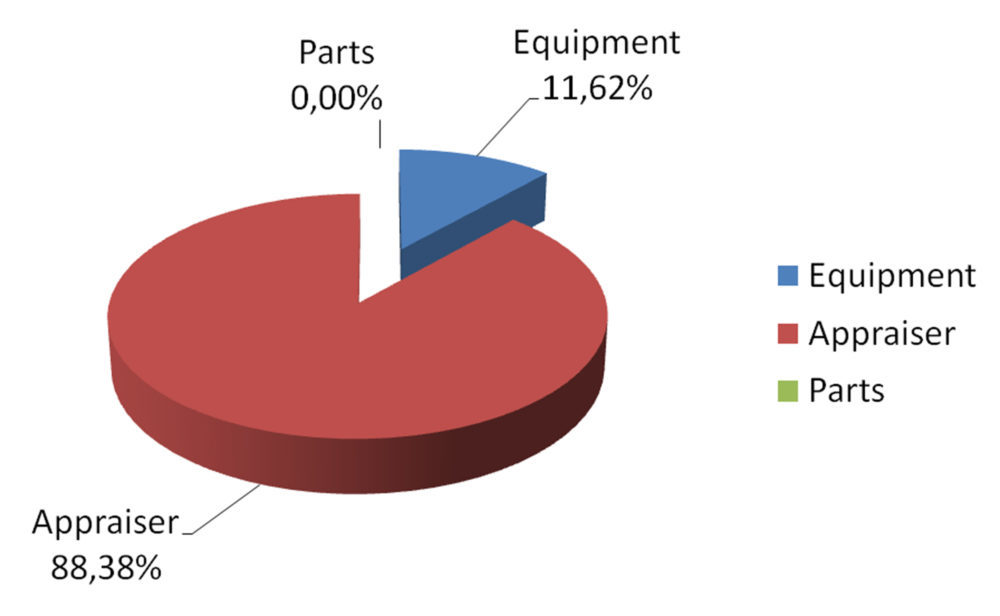
Figura 8: análisis MSA
Este aspecto merece una consideración especial ya que aporta información muy interesante que puede ser aplicada en el ámbito académico. Hay que partir del hecho de que se tomaron datos del trabajo experimental de una tesis doctoral, obtenidos en momentos diferentes, es decir, al principio del doctorado (aproximadamente tras un año de experiencia, con el equipo ubicado en un laboratorio) y al final del doctorado (aproximadamente tras más de tres años de experiencia, con el mismo equipo ubicado en otro laboratorio). En ambos casos las muestras consideradas fueron del mismo tipo (eritrocitos humanos del mismo donante) y sin aportación porcentual a la variabilidad total. Analizando específicamente la representación gráfica de la variación por “appraiser” o analista (figura 9) donde Appraiser 1 corresponde a datos iniciales y Appraiser 2 corresponde a datos obtenidos varios años después, resulta evidente que los resultados de Appraiser 2 presentan una dispersión menor que los de Appraiser 1 y que la experiencia adquirida no deja de ser el resultado de un proceso de formación análogo al mencionado en la tabla FMEA. A esto hay que añadir que, al revisar los datos de la propia tesis doctoral, los resultados sobre la evidencia de subpoblaciones eritrocitarias mediante histogramas de frecuencias en muestras patológicas están más definidos tras varios años de experiencia, lo que es un claro reflejo de este proceso formativo.
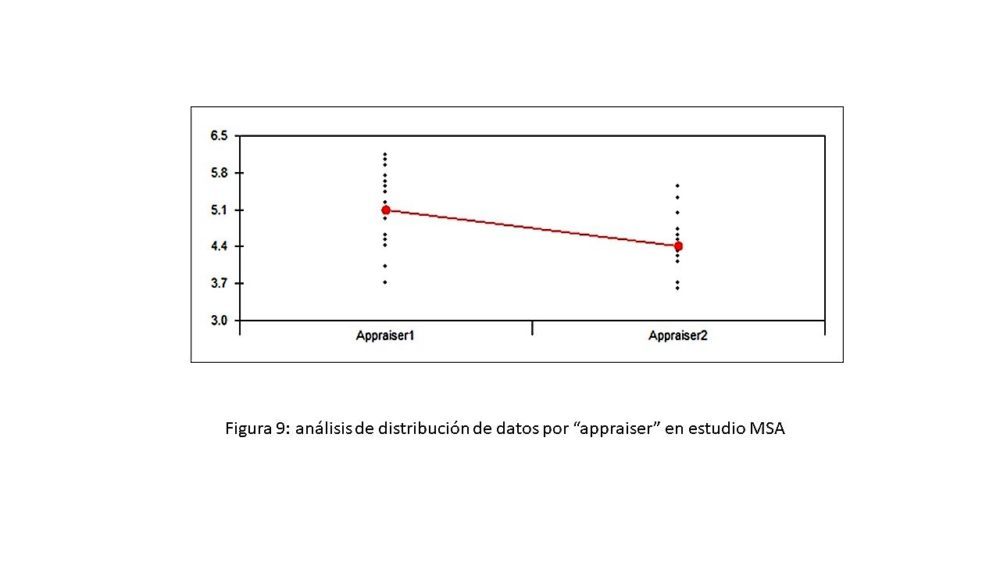
Figura 9: análisis de distribución de datos por “appraiser” en estudio MSA
Así pues, una importante aportación de la utilización de la metodología QbD en el ámbito académico es la de plantearse una repetición periódica y planificada de determinados ensayos a lo largo de la realización de un trabajo experimental prolongado en el tiempo, como puede ser una tesis doctoral, análogamente a lo que es una práctica habitual de la industria farmacéutica, como forma de verificación constante de cumplimento de las condiciones idóneas de formación para la realización de ensayos analíticos y, consecuentemente, para la obtención de resultados analíticos válidos.
5.5 Comparación del desarrollo analítico siguiendo qbd frente a la metodología tradicional
A modo de resumen de los datos presentados en los apartados anteriores, se muestran dos tablas (una para factores X – tabla 6 – y otra para factores N- tabla 7) en la que se comparan ambas metodologías.
Tabla 6: comparación de resultados para factores X
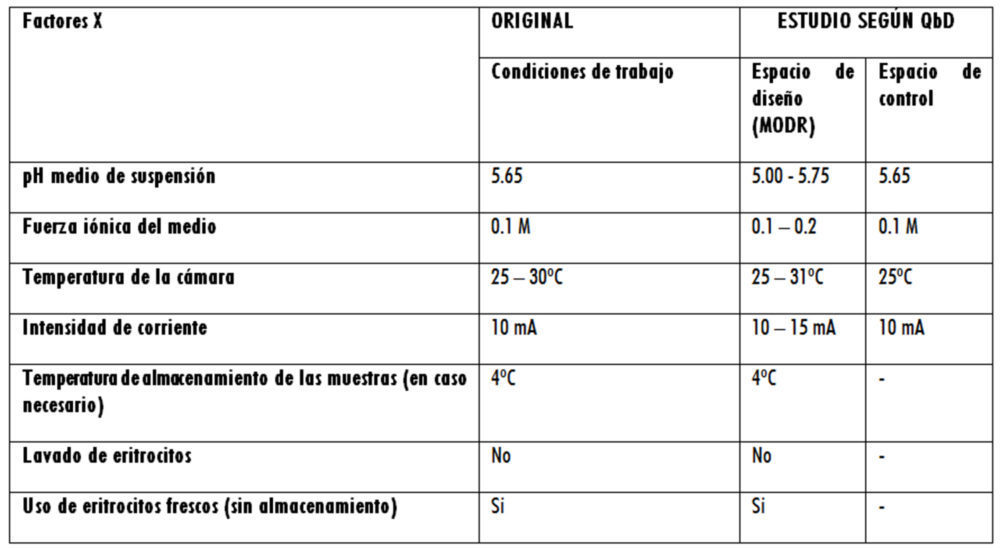
Tabla 7: comparación de resultados para factores N
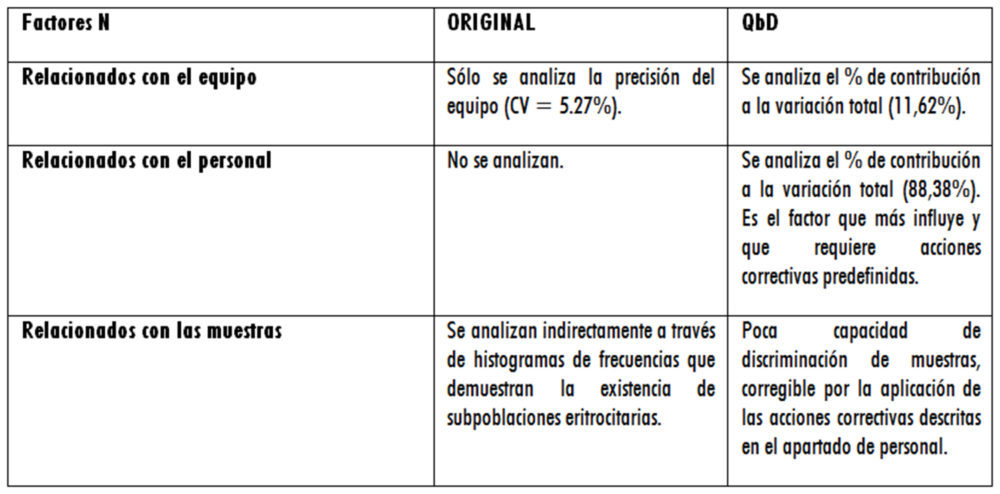
Se observa que los resultados obtenidos siguiendo QbD aportan un mayor conocimiento y control del método citoferométrico, así como una mayor flexibilidad en el manejo de los factores físicos y físico-químicos necesarios y unas herramientas adecuadas para controlar las principales fuentes de variabilidad analítica.
6. CONCLUSIONES
Independientemente de cómo quede redactada la nueva Guía ICH Q14 y de que la metodología descrita tenga cabida en dicha redacción, no cabe duda de que se han podido vislumbrar las ventajas de la aplicación de la QbD en el desarrollo y validación de métodos analíticos frente a las prácticas tradicionales, tanto en el ámbito del análisis de medicamentos como en otros ámbitos analíticos.
Para la industria farmacéutica, es evidente que poder demostrar cómo se han identificado los parámetros críticos de un método analítico y cómo estos están controlados en el trabajo habitual, constituyen el fundamento de una interacción necesaria entre analistas y estadísticos y de una relación productiva y bidireccional de la Industria con las Autoridades Sanitarias y con la Universidad, desde un punto de vista científico.
Sin embargo, como se ha pretendido demostrar en esta disertación, no hay que limitar esta visión a la actividad de la industria farmacéutica ya que, al tratarse la QbD de una metodología de trabajo muy rigurosa desde el punto de vista científico, su utilidad en el ámbito académico en general, y universitario en particular, es extremadamente grande.
La incorporación de nuevos métodos analíticos desarrollados con la metodología QbD conlleva un esfuerzo adicional, pero “a posteriori” aporta un significativo ahorro de tiempo, tanto en la investigación básica como en la I+D aplicada, lo que es especialmente importante cuando se trata de poner nuevos medicamentos a disposición de los pacientes y más en las circunstancias que nos está tocando vivir.
No olvidemos que los pacientes no pueden esperar. Muchas gracias.
7. REFERENCIAS
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). History. Disponible en: https://www.ich.org/page/history.
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Validation Of Analytical Procedures: Text And Methodology Q2(R1). Disponible en: https://database.ich.org/sites/default/files/Q2_R1__Guideline.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Pharmaceutical Development Q8(R2). Disponible en: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Quality Risk Management Q9. Disponible en: https://database.ich.org/sites/default/files/Q9_Guideline.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Pharmaceutical Quality System Q10. Disponible en: https://database.ich.org/sites/default/files/Q10_Guideline.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Development And Manufacture Of Drug Substances (Chemical Entities And Biotechnological/Biological Entities) Q11. Disponible en: https://database.ich.org/sites/default/files/Q11_Guideline.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Technical And Regulatory Considerations For Pharmaceutical Product Lifecycle Management Q12. Disponible en: https://database.ich.org/sites/default/files/Q12_Guideline_Step4_2019_1119.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH): Final Concept Paper ICH Q13: Continuous Manufacturing of Drug Substances and Drug Products dated 14 November 2018. Endorsed by the Management Committee on 15 November 2018. Disponible en: https://database.ich.org/sites/default/files/Q13_EWG_Concept_Paper.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Final Concept Paper ICH Q14: Analytical Procedure Development and Revision of Q2(R1) Analytical Validation dated 14 November 2018. Endorsed by the Management Committee on 15 November 2018 Disponible en: https://database.ich.org/sites/default/files/Q2R2-Q14_EWG_Concept_Paper.pdf
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Guía de Normas de Correcta Fabricación de Medicamentos de Uso Humano y Veterinario Anexo 15: Cualificación y validación. Disponible en: https://www.aemps.gob.es/industria/inspeccionNCF/guiaNCF/docs/anexos/anexo-15.pdf
- Borman P, Nethercote P, Chatfield M, Thompson D, Truman K. The Application of Quality by Design to Analytical Methods. Pharm Tech 2007; 31 (10): 142-152.
- Alassandro M, Little T, Fleitman J. Method Validation by Design to Support Formulation Development. Pharm Tech Europe 2013; 25(4): 48-54.
- Department of Health and Human Services. U.S Food and Drug Administration. Pharmaceutical cGMPS for the 21st Century — A risk-based approach. Final Report. Disponible en: https://www.fda.gov/media/77391/download
- Little T. Assay Development and Method Validation Essentials. BioPharm International 2012; 25(11): 48-51.
- LoBrutto R. An Insight into Analytical Quality by Design in the Pharmaceutical Industry. The Column 2013; 9(15): 14-7.
- Nethercote P, Ermer J. Quality by Design for Analytical Methods. Pharm Tech Europe 2012; 24(10): 52-6.
- Burgess C. Calibration of Instruments: Is Your UV Spectrometer Accurate Enough? Pharm Tech 2014; 38(1): 56-9.
- Burgess C. Evaluating Risk-Based Specifications for Pharmaceuticals. Pharm Tech Europe 2013; 25(7): 30-6.
- Chambers D, Guo G, Kleintop B, Rasmussen H, Deegan S, Nowak S, Patterson K, Spicuzza J, Szulc M, Tombaugh K, Trone M, Yuabova Z. GMPs for Method Validation in Early Development. An Industry Perspective (Part II). Pharm Tech Europe 2012; 24(8): 26-32.
- Rignall A, Christopher D, Crumpton A, Hawkins K, Lyapustina S, Memmesheimer H, Parkinson A, Smith MA, Wyka B, Kaerger S. Quality by Design for analytical methods for use with orally inhaled and nasal drug products. Pharm Tech Europe, 2008; 29(10): 24-31.
- Embase® (página de inicio de Internet). Disponible en: https://www.embase.com/
- National Library of Medicine. National Center for Biotechnology Information. Pubmed® (página de inicio de Internet). Disponible en: https://www.ncbi.nlm.nih.gov/pubmed
- American Chemical Society (ACS) Publications (página de inicio de Internet). Disponible en: https://pubs.acs.org/
- Ferrándiz F. Aplicación de la Calidad por Diseño (Quality by Design) en el desarrollo y validación de métodos analíticos. Discurso de ingreso. Academia de Farmacia de Castilla y León 2014.
- McDowall D. Life Cycle and Quality by Design for Chromatographic Methods. LC-GC Europe 2014; 27(2): 91-7.
- ISPE GAMP® 5 Guide: A Risk-Based Approach to Compliant GxP Computerized Systems (página de inicio de Internet). Disponible en: https://ispe.org/publications/guidance-documents/gamp-5.
- Ferrándiz F, Ródenas S, Del Castillo B. Análisis citoferométrico de eritrocitos humanos. Análisis Clínicos 1988; XIII: 117-120.
- Abramson HA, Moyer LS, Gorin MH. Electrophoresis of proteins and the chemistry of cell surfaces. New York: Reinhold Publishing Corporation 1942.
- Liteanu C, Rica I. Statistical theory and methodology of trace analysis. New York: John Wiley & sons 1980.
- Entidad Nacional de Acreditación – ENAC. Expresión de la incertidumbre de medida en las calibraciones. Disponible en: http://www.proycal.com/Imagenes/CEA-ENAC-LC-02%20Rev.%201.pdf
AGRADECIMIENTOS
Es de bien nacidos ser agradecidos. Por ello deseo que mis palabras sean de profundo agradecimiento a todos los Académicos que han depositado en mí su confianza al haberme acogido como Académico Correspondiente en esta Real Academia. Espero corresponder a dicha confianza con mi participación activa en la vida de la corporación. Ganas y entusiasmo no me van a faltar.
Permítanme que exprese además mi especial gratitud al Excmo. Prof. Dr. D Fidel Ortega por sus amables palabras en la presentación y, como no podía ser de otra manera, al Excmo. Prof. Dr. D Benito del Castillo García, mi maestro y amigo, desde que allá por el año 1984, tuvo la osadía de permitirme ser monitor de prácticas de Técnicas Instrumentales en la Facultad de Farmacia de la Universidad Complutense de Madrid y dejarme defender mi Tesina y mi Tesis Doctoral, algunos años más tarde y en el mismo laboratorio. Hago mías sus palabras: “amigos, en el acuerdo y en el desacuerdo, siempre”.
He de hacer mención también al no menos significativo hecho de que, al permitirme entrar en la Cátedra de Técnicas Instrumentales, D. Benito me puso en contacto directo con una de las personas que más han influido en mi desarrollo profesional, tanto en el ámbito académico como en la industria farmacéutica, y a la que estoy profundamente agradecido por ello. Me refiero a la Dra. Sofía Ródenas de la Rocha quien, mucho antes de que se publicase la Guía ICH Q2, tan importante para el análisis instrumental en la industria farmacéutica, ya aplicaba de manera práctica (y con 10 años de antelación respecto a la fecha de publicación de la mencionada Guía ICH) los conceptos de Calidad, GMP, GLP, precisión, exactitud, linealidad o validación, no sólo en el trabajo experimental sino en las mismas prácticas de la Facultad. Pocos conocen lo importante que es para la industria farmacéutica recibir alumnos que conocen estos términos y su contenido, especialmente si se dedican a tareas de I+D, Registros, Producción, Control de Calidad o Garantía de Calidad, que son el núcleo de la actividad farmacéutica industrial.
Es importante destacar también que la Dra. Ródenas me puso en contacto con la Dra. Ana Villegas del Servicio de Hematología del Hospital Clínico de Madrid, con la que codirigió mi tesis doctoral, y con la que tuve la oportunidad de ver la aplicación práctica de esos conceptos, que dejaban de ser meramente teóricos para convertirse en una forma de trabajo que, además, establecía puentes de colaboración con otros centros fuera de la Facultad.
