1. INTRODUCCIÓN
La Organización Mundial de la Salud (OMS) define la diabetes mellitus como: “una enfermedad crónica que aparece cuando el páncreas no produce insulina suficiente o cuando el organismo no utiliza eficazmente la insulina que produce”. Ésta se clasifica principalmente en dos tipos atendiendo a su origen etiopatológico. Por un lado, la diabetes mellitus tipo I, también conocida como insulinodependiente, afecta principalmente a personas jóvenes e individuos que son incapaces de producir insulina de forma endógena. Por otro lado, la diabetes mellitus tipo II (DM II), es una afección más propia de personas en edad adulta y/o avanzada, las cuales han desarrollado resistencia a la acción de esta hormona en los tejidos periféricos de su cuerpo. A estas dos variantes se le la diabetes gestacional que se manifiesta en mujeres durante el embarazo y que puede suponer un aumento del riesgo de padecer DM II en etapas posteriores de la vida.
Si bien todas las formas presentan síntomas similares, destacando: poliuria, polidipsia y polifagia (todos ellos patognomónicos de la enfermedad), los trastornos visuales, complicaciones cardiovasculares y la afección renal son las consecuencias que hacen de la DM II una de las mayores causas de morbi-mortalidad en los países desarrollados (1) actualmente. Del mismo modo, la diabetes no sólo se ha relacionado con las complicaciones mencionadas anteriormente sino también con un aumento en la prevalencia de ciertas neoplasias, así como de deterioros cognitivos, como la enfermedad de Alzheimer (2). Las elevadas cifras de prevalencia de este trastorno lo convierten en una pandemia de la era moderna, estimándose que en 2012 la carga total de mortalidad asociada a la DM II fue de 3,7 millones de muertes. Además, en 2014 se estimó una prevalencia total de la enfermedad de 422 millones de personas (mayores de 18 años), siendo Europa la tercera región con mayor prevalencia (64 millones de afectados) según el Informe Mundial sobre la Diabetes publicado en el 2016 por la OMS. Debido al cambio en los hábitos de vida y al aumento de la esperanza de vida, se estima que estas cifras seguirán elevándose en los años venideros, constituyendo un auténtico problema no sólo para las personas afectadas, sino también para los sistemas de salud encargados de hacer frente a este reto económico y asistencial para la salud pública.
En la actualidad, el arsenal farmacológico disponible para el manejo en pacientes de la DM II lo constituyen los hipoglucemiantes no insulínicos (sulfonilureas, meglitinidas, metformina, tiazolidinonas, inhibidores de α -glucosidasa, etc) (3). Sin embargo, estas terapias, focalizadas en el aumento de la insulinemia plasmática, no son totalmente óptimas. Independientemente del número y tipo de efectos adversos, estas aproximaciones no combaten aspectos importantes de la DM II, como la reducción de la sensibilidad de las células b pancreáticas a la glucosa o el incremento de glucosa vía gluconeogénesis. Por ello, la búsqueda de nuevas dianas y el desarrollo de nuevos antidiabéticos orales continúa siendo una de las principales líneas de investigación, tanto de industrias farmacéuticas como de grupos académicos de todo el mundo.
Hacia finales del siglo XX se clonó el primer gen de una proteína tirosina fosfatasa, identificada como Proteína Tirosina Fosfatasa 1B (PTP1B) o Proteína Tirosina Fosfatasa de tipo No-receptor (PTPN1) (4). Esta es una enzima de 435 aminoácidos que es capaz de catalizar la hidrólisis de los residuos de fosfotirosina del receptor de insulina activado, y otros sustratos relacionados con la misma, como el sustrato I del receptor de insulina y el receptor de leptina. Esta proteína se ha asociado a los fenómenos de resistencia a la insulina y a la obesidad, al tratarse de un regulador negativo de las vías de señalización de la insulina y la leptina (5). No sólo eso, sino que PTP1B ha sido ampliamente validada como una prometedora diana terapéutica para el tratamiento de la DM II (6): los ratones knock-out para el gen que codifica esta proteína presentan una mayor sensibilidad a la insulina y a la pérdida de sobrepeso (7). Estos hechos han motivado el desarrollo de numerosas moléculas potenciales candidatas a fármaco capaces de inhibir la PTP1B por parte de la industria farmacéutica (8). Se pueden distinguir dos grandes grupos atendiendo a la zona específica de unión de estas moléculas en la enzima: inhibidores dirigidos al centro activo e inhibidores dirigidos a sitios alostéricos (8), (9). Los primeros constituyen sin ninguna duda la familia más numerosa, con numerosas moléculas de distinta naturaleza, aunque casi todas ellas con la característica común de presentar grupos cargados negativamente a pH fisiológico, con el fin de mimetizar los restos de fosfotirosina presentes en el segmento intracelular del receptor de insulina. Esta característica compromete la biodisponibilidad de todas estas moléculas para su administración por vía oral, dada su baja permeabilidad a través de membranas biológicas. Por otro lado, la alta homología del centro activo de la PTP1B con respecto a otras tirosina fosfatasas, supone un gran inconveniente a la hora de desarrollar inhibidores lo suficientemente selectivos (10). Estos hechos han tenido como resultado que ningún inhibidor dirigido al centro activo haya alcanzado los ensayos clínicos. Por otro lado, existen inhibidores alostéricos de PTP1B, la mayoría de ellos con un carácter mucho más lipófilo, como el ertiprotafib, que llegó a ensayos clínicos de fase I, o la trodusquemina (un derivado de fitoesterol), que alcanzó ensayos clínicos de fase II (8). Esto se debe a su mejor biodisponibilidad y mayor selectividad, puesto que los sitios alostéricos de las proteínas no se encuentran tan conservados dentro de una misma familia y suponen una fuente de selectividad (11), (12). Dentro de este grupo de inhibidores destaca una familia de benzbromaronas descrita por Wiesmann en 2004 (Figura 1) (13).
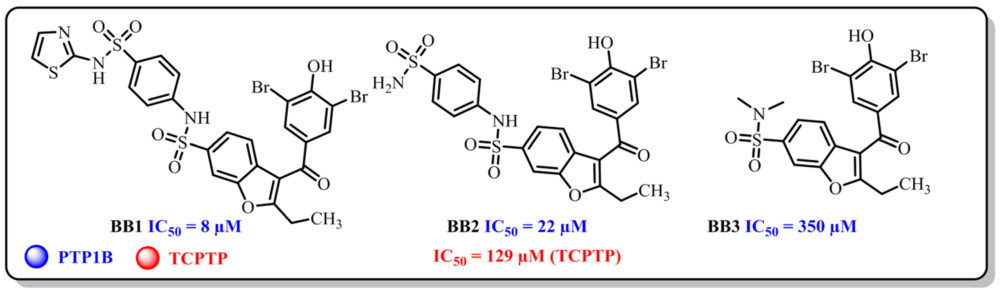
Figura 1. Inhibidores alostéricos de PTP1B descritos por Wiesman y colaboradores y su actividad frente a PTP1B y TCPTP
Estos compuestos demostraron unirse a una región no antes explorada de la PTP1B que se encuentra a 20 Å del centro activo tal y como se puedo determinar en los estudios de cristalografía de rayos X (PD: 1T48, 1T49 y 1T4J). Esta región está compuesta por las a hélices 3, 6 y 7 situadas cerca del extremo C-terminal de la proteína, tal y como se puede observar en la Figura 2.
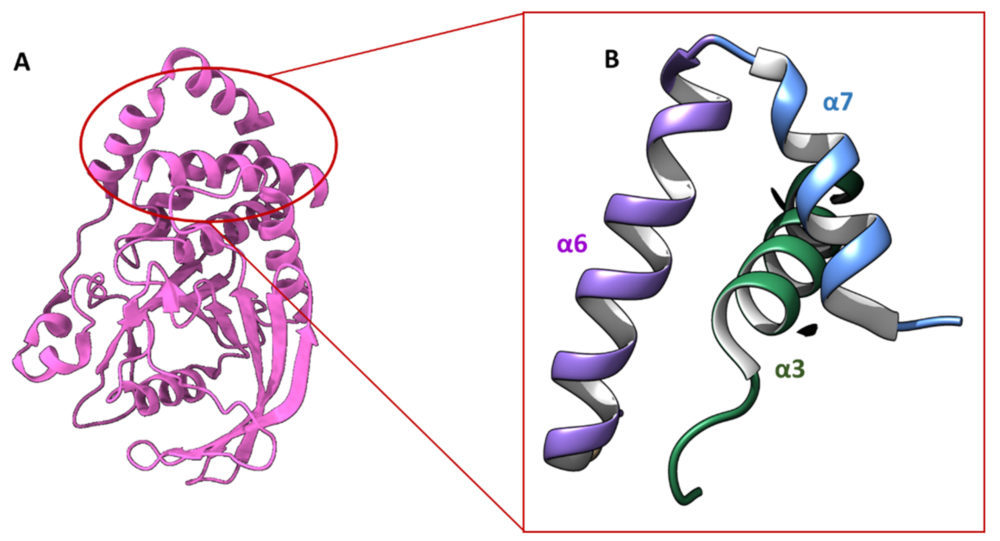
Figura 2. A) Estructura tridimensional de PTP1B (Entrada del PDB:1OEM) B) Ampliación de la estructura sitio alostérico descrito por Wiesman y colaboradores cuando no presenta inhibidor
Este hecho supuso el inicio de nuevos programas de búsqueda de inhibidores de PTP1B partiendo del concepto de “diseño basado en estructura” a comienzos del siglo XXI. Esta nueva familia de inhibidores alostéricos es capaz de desplazar el residuo de Trp291 situado en la hélice α7 de PTP1B con el fin de acomodarse en su interior generando una nueva cavidad y desestructurando la hélice α7. La unión del inhibidor desencadena una serie de movimientos en el esqueleto proteico que se traducen en el bloqueo del bucle catalítico de la PTP1B (14). Este bucle, conocido entre todas las tirosina fosfatasas de la familia como WPD-loop, se encuentra en una posición abierta antes de alojar el sustrato en su interior. Una vez los residuos de fosfotirosina se acomodan en el centro catalítico, es necesario el cierre del bucle para que tenga lugar el ciclo catalítico de la enzima. Sin embargo, tras la unión de las benzbromaronas al correspondiente sitio alostérico, el WPD-loop queda bloqueado en su conformación abierta de tal manera que, aunque permite que el sustrato se aloje en su interior, la enzima es catalíticamente inactiva y por lo tanto, queda inhibida (15).
Durante la última década, numerosos estudios computacionales han pretendido arrojar luz al mecanismo de inhibición alostérica, a las bases del diseño de inhibidores o a la dinámica del bolsillo alostérico de PTP1B (16), (17), (14). Sin embargo, no existe ningún tipo de estudio que trate las bases de selectividad entre PTP1B y su homóloga más cercana, la Proteína Tirosina Fosfatasa de Linfocitos T (TCPTP). La inhibición de esta última, con la que PTP1B comparte más de un 71% de identidad, conduce a efectos adversos indeseables como la destrucción de la médula ósea, y procesos de eritropoyesis y linfopoyesis aberrantes (18). Además, los modelos murinos knock-out para el gen de TCPTP demostraron ser inviables a las pocas semanas del nacimiento (19).
Desde su inicio hasta nuestros días, las técnicas de modelado molecular han experimentado un desarrollo exponencial y se han convertido en herramientas indispensables en campos tan variados como la farmacología, biología y bioquímica estructural, el diseño de fármacos y la química biológica (20). Más en concreto, los estudios de dinámica molecular y el cálculo de energía libre han sido aplicados con éxito al estudio de la catálisis enzimática (21), la resistencia a fármacos (22), (23), el diseño de fármacos (24), (25), o la más novedosa farmacología de sistemas (26).
Teniendo en cuenta todos los antecedentes y la falta de conocimiento acerca de las bases estructurales que rigen la selectividad entre ambas enzimas, clave para la obtención de nuevos fármacos y mejor comprensión de la función de ambas proteínas a nivel molecular, se planteó un estudio computacional que permitiese arrojar luz sobre el mecanismo. El objetivo de este trabajo ha sido el estudio de la benzbromarona BB2, descrita por Wiesmann, mediante técnicas computacionales de dinámica molecular clásica y cálculo de energía libre con el fin de explicar su selectividad y asentar las bases para el futuro el diseño de inhibidores selectivos hacia PTP1B.
2. MATERIAL Y MÉTODOS
2.1. Modelado por homología
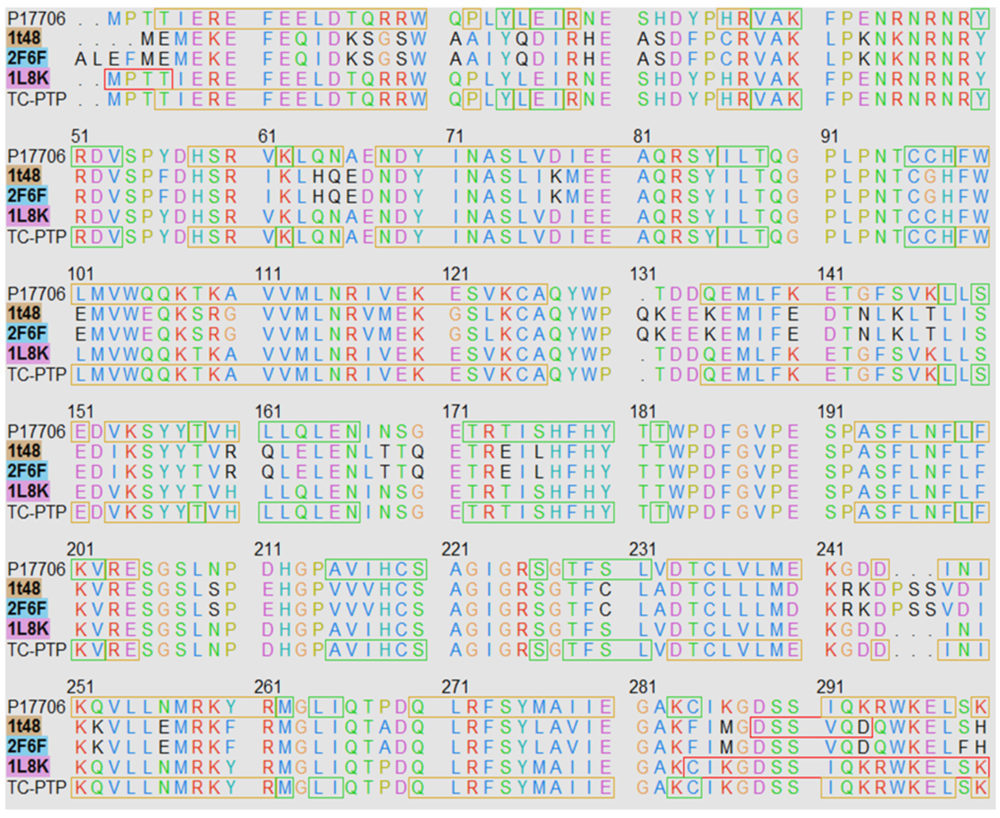
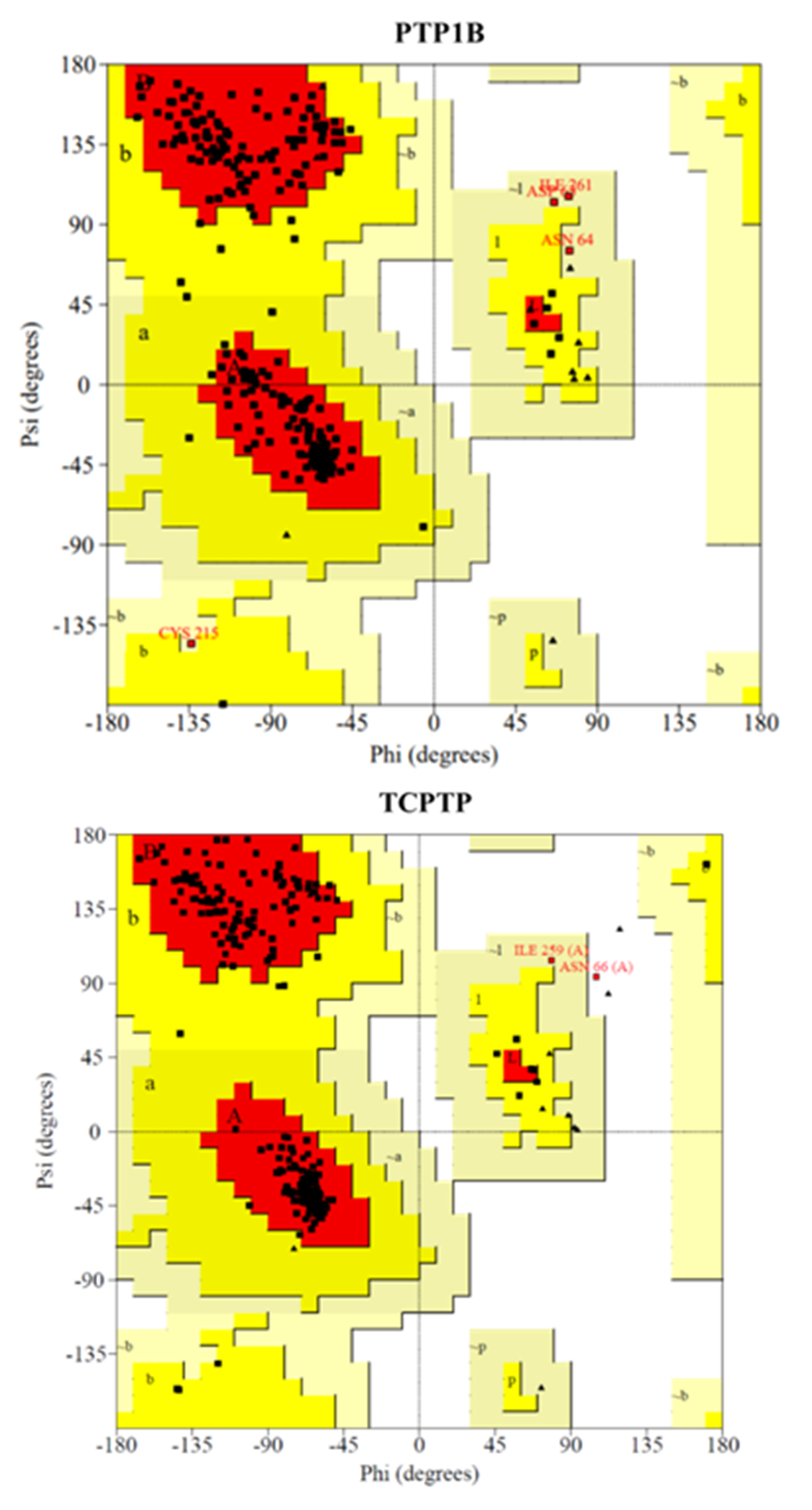
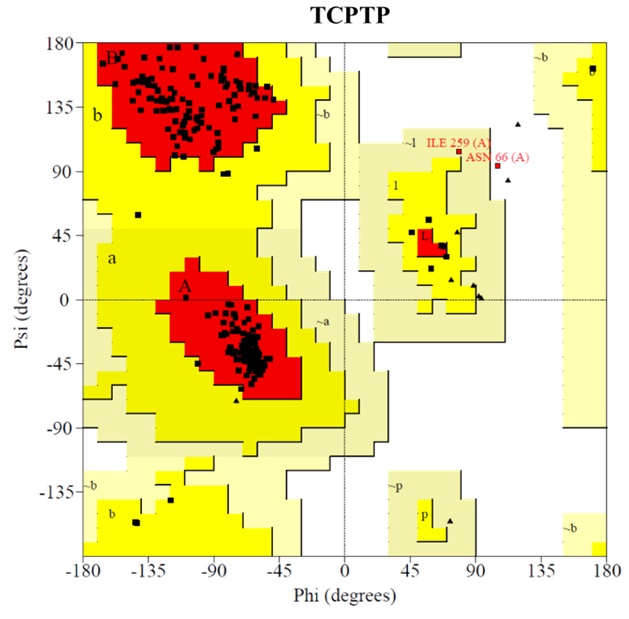
Dado que no existen estructuras tridimensionales de inhibidores alostéricos con la hélice α7 resuelta se hizo necesario utilizar técnicas de modelado por homología. Para la construcción de los modelos se emplearon las estructuras depositadas en el Protein Data Bank (PDB) como 1T48 y 1L8K para PTP1B y TCPTP, respectivamente. Para incorporar las estructuras no resueltas de la hélice α7 en ambas proteínas y el loop a6 en 1L8K, se empleó un método descrito en la bibliografía previamente con el fin de crearlas con el WPD-loop abierto (27). Junto a estas estructuras se utilizó la PTP1B completa y con el WPD loop cerrado 2F6F. Del mismo modo, se utilizó el ligando BB2 presente en 1T48 para introducir restricciones en ambos modelos. Los moldes se alinearon con las secuencias de la base de datos génica Uniprot para ambas enzimas (P18031 para PTP1B y P17706 para TCPTP) a través de la interfaz gráfica de usuario del programa Modeller 9.2 en UCSF Chimera 1.13 (ver material complementario) (28) . Los modelos mejor posicionados según su puntuación en la función Z-DOPE de Modeller fueron seleccionados y sometidos a una inspección visual para ajustar aquellos rotámetros peor posicionados. El estado de protonación de la proteína se investigó empleando el servidor H++ (29), a continuación, tanto el modelo como la red de enlaces de hidrógeno se comprobó y optimizó con el servidor WHAT-IF (30). Tras esto se comprobaron los mapas de Ramachandran con el fin de validar la calidad de los modelos (ver material complementario).
2.2. Simulaciones de dinámica molecular
Las simulaciones de dinámica molecular se llevaron a cabo empleando la suite de programas Amber 16 junto con AmberTools 16 (31). La proteína se describió empleando el campo de fuerzas ff14SB, mientras que para el ligando se utilizó GAFF. Las cargas de este últimos se asignaron empleando el módulo Antechamber y ajustándolas a un nivel teórico AM1-BCC. Ambos complejos se solvataron en un cubo de moléculas de aguas TIP3P de 12 Å de tamaño de lado y se adicionaron 5 iones de sodio con el fin de neutralizar el sistema empleando el módulo Leap de AmberTools. A continuación, se pasó a la fase de preparación del sistema por pasos: minimización de los hidrógenos, minimización de las moléculas de disolvente, minimización del sistema completo (aplicando un potencial de restricciones de 5 kcal/mol en los Cα de la proteína), calentamiento hasta los 300 K durante 20 ps con las mismas restricciones, equilibrado durante 100 ps a 300 K (manteniendo constante el número de partículas, temperatura y volumen) y equilibrado, liberando las restricciones posicionales gradualmente durante 100 ps (manteniendo constante temperatura, volumen y presión). Tras esta última fase de equilibrado, se pasó a la producción de dos réplicas para cada sistema de dinámica molecular clásica durante 25 ns, empleando un intervalo de integración de 2 fs. La producción se llevó a cabo a través de GPUs utilizando el módulo PMEMD.CUDA implementado en Amber. Los cálculos requirieron 7 días para completar cada una de las réplicas. Todos los sistemas se simularon por duplicado empleando las condiciones de límite periódico, y aplicando el método de Ewald para tratar las interacciones electrostáticas de corto alcance con un límite de 8 Å. Así mismo, se empleó el algoritmo SHAKE (32) en todos los enlaces en los que participan átomos de hidrógeno con el fin de simplificar los cálculos durante la dinámica. Las trayectorias de la dinámica molecular se analizaron empleando el software Cpptraj, VMD y Chimera.
2.3. Cálculos con WaterSwap
Para el cálculo de la energía libre se empleó el programa basado en el método de Montecarlo, WaterSwap (33). Las estructuras empleadas para llevar a cabo estos cálculos procedían de los 15 ns de dinámica cuando el sistema se encontraba en equilibrio termodinámico. Los cálculos se llevaron a cabo en el módulo implementado en el paquete de programas Sire, utilizando los mismos datos de topología y parámetros generados para la dinámica molecular (Amber ff14SB). La energía libre de unión absoluta se calculó mediante el método de integración termodinámica de réplica de intercambio, a lo largo de 16 ventanas de transformación ligando-moléculas de agua. Todos los cálculos requirieron de 7 días para ser completados. Las energías finales se obtuvieron utilizando el script de Python freenrg, también presente en la suite Sire.
3. DISCUSIÓN Y RESULTADOS
3.1. Modelado por homología
Para llevar a cabo el estudio con la benzbromarona BB2, se hizo necesario el uso de técnicas de modelado por homología que permitiesen predecir las estructuras tridimensionales de ambas enzimas. Las entradas depositadas en el PDB co-cristalizadas con inhibidores alostéricos, carecen de la hélice α 7 como consecuencia de la alta movilidad que presenta esta hélice tras la unión del inhibidor y que imposibilita su resolución por técnicas convencionales de cristalografía de rayos X. Por otro lado, la única estructura disponible de la TCPTP (1L8K) carece también de esta hélice y presenta una baja resolución. Es por esto por lo que se hizo necesario recurrir al modelado por homología para obtener estructuras tridimensionales completas y de buena calidad de ambas enzimas. Para generar los modelos se utilizó el método descrito por Shinde y colaboradores (27), empleando como moldes para el modelado las estructuras cristalinas de PTP1B con el bucle catalítico abierto, 1T48, y con la hélice α7 resuelta, 2F6F. En el caso de la TCPTP se utilizó 1L8K. Superponiendo los moldes con las correspondientes secuencias de ambas proteínas extraídas de la base de datos Uniprot se generaron modelos en base al alineamiento para ambas enzimas que posteriormente fueron refinados e inspeccionados visualmente de forma cuidadosa. Las estructuras generadas obtuvieron una puntuación Z-DOPE de -1,27 para PTP1B y -1,07 para TCPTP, valores que corresponden a modelos fiables y bien resueltos.
Una vez construidos, se pasó a llevar a cabo una inspección visual de las estructuras generadas. En primer lugar, la observación del sitio alostérico modelado para ambas enzimas permitió tener una idea de la geometría de este. Como se puede comprobar en la Figura 3, el sitio alostérico está constituido por una cavidad comprendida entre las hélices α3,α6 y α7, con un carácter altamente hidrofóbico debido a la naturaleza de los aminoácidos que lo constituyen.
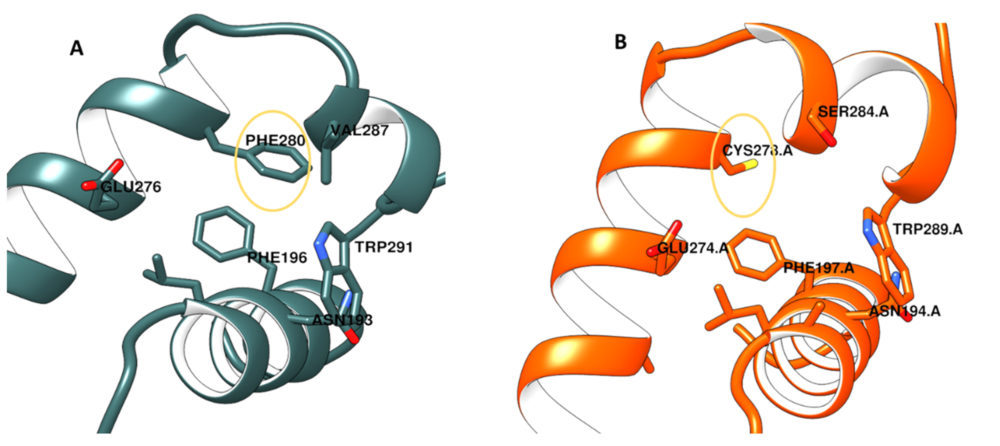
Figura 3. A) Bolsillo alostérico de PTP1B modelada. B) Bolsillo alostérico de TCPTP modelada
A pesar de la gran similitud entre los bolsillos alostéricos de ambas enzimas, hay algunas diferencias bastante reseñables: por un lado, el residuo de Phe280 de la PTP1B se encuentra sustituido por una cisteína que en la secuencia de TCPTP ocupa la posición 278. Esto es una diferencia bastante notable puesto que la naturaleza y tamaño de estos aminoácidos es bastante distinta. Algo similar ocurre con la Val287 que pasa a ser una ocupada por la Ile284 de TCPTP. Así mismo existen algunas diferencias también en el extremo C-terminal, pero estas no fueron contempladas por tratarse de una zona alejada del bolsillo y con alta movilidad. Estos primeros resultados parecen indicar que, si bien ambos son bolsillos hidrófobos, el de la TCPTP presentaría un carácter más polar y mayor tamaño por la presencia del residuo de cisteína en lugar de fenilalanina.
3.2. Dinámica molecular
Con el fin de profundizar en el proceso de interacción entre BB2 y ambas fosfatasas, se llevaron a cabo experimentos de dinámica molecular entre los complejos ligando-enzima. La geometría inicial de los complejos se obtuvo, en el caso de la PTP1B, directamente del cristal, y en el caso de la TCPTP por superposición del ligando BB2 durante la fase de modelado. Un primer vistazo a las geometrías iniciales permite revelar las claves de la interacción, como se puede comprobar en la Figura 4.
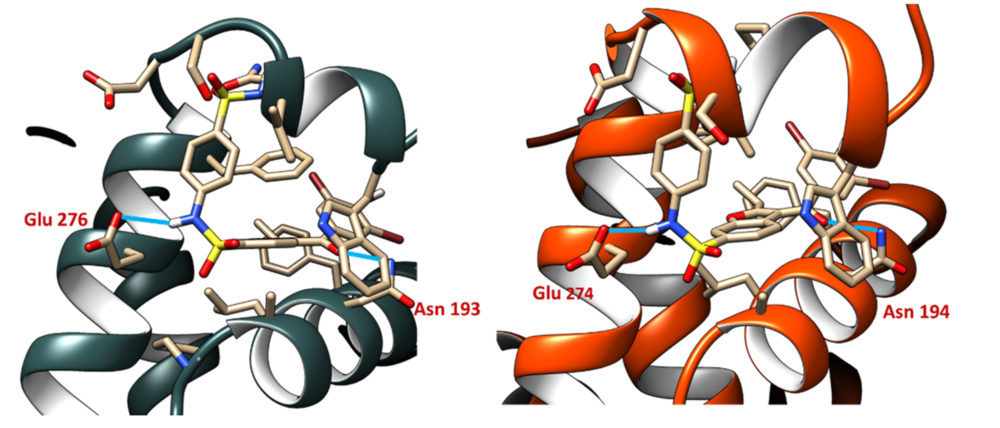
Figura 4. Geometrías iniciales de los complejos BB2-enzima para la dinámica molecular (izquierda PTP1B y derecha TCPTP). Los enlaces de hidrógeno entre la Asn193/194 y el Glu 276/274 se encuentran marcados como líneas azules
Las benzbromaronas se asientan en la cavidad alostérica estableciendo numerosos contactos hidrofóbicos con residuos de glicina, isoleucina y el esqueleto carbonado de la proteína. Se pueden diferenciar dos enlaces de hidrógeno iniciales: uno por parte del grupo carbonilo de BB2 que interacciona con los residuos Asn193 y Asn194 de PTP1B y TCPTP, respectivamente gracias a su –NH2; y otro por parte del grupo sulfonamida, a través de su –NH, que interacciona con el COOH del Glu276 de PTP1B y el Glu274 en el caso de la TCPTP. Hay que destacar que a los contactos hidrofóbicos se le unen fuertes interacciones de tipo apilamiento-π que establece el esqueleto de benzofurano de BB2 con los anillos aromáticos de los residuos Phe196, Phe280 y Trp291 de la PTP1B. En el caso de la TCPTP, estos contactos se mantienen a excepción del apilamiento-π paralelo de la Phe280, ya que en esta posición se encuentra la Cys278. La pérdida de este apilamiento podría ser una de las causas que contribuyese negativamente a su unión a la TCPTP (IC50 =129 µM) favoreciendo por tanto la selectividad del compuesto (6 veces más selectivo sobre la PTP1B, IC50 = 22 µM) (13).
Las técnicas de dinámica molecular convencional permiten simular con un alto grado de exactitud los complejos ligando-proteína en condiciones que simulen el ambiente biológico (presencia explícita de disolvente y de iones para fijar el pH y fuerza iónica) a lo largo del tiempo y de este modo poder estudiar los eventos que tengan lugar a nivel molecular. Una vez minimizados los distintos átomos del sistema (para eliminar geometrías forzadas y contactos demasiado cercanos), éste se calentó gradualmente hasta 25 ºC, se hizo una pequeña fase de equilibrado para reestablecer la densidad y se pasó a la fase de producción, donde se generan los resultados de la dinámica que se analizan y comentan durante la discusión.
Uno de los parámetros más empleados para evaluar la estabilidad de los complejos es el cálculo de la desviación cuadrática media (RMSD) a lo largo del tiempo de las trayectorias generadas. Este parámetro representa las fluctuaciones de la posición de los átomos de distintos elementos, como el esqueleto proteico, comparándola con la conformación inicial a lo largo de la simulación. Su monitorización nos permite comprobar si el sistema ha alcanzado el equilibrio (se mantiene estable), o, por el contrario, continúa evolucionando en el espacio conformacional en búsqueda de estados termodinámicamente más favorecidos para el sistema.
Como se puede observar en la Figura 5, ambos sistemas parecen haber alcanzado el equilibrio, especialmente a partir de los 5 ns de dinámica, cuando los valores de RMSD presentan menor fluctuación y se mantienen sin grandes incrementos. No obstante, se pueden encontrar pequeñas diferencias si se estudia el valor medio a lo largo de toda la dinámica para ciertas partes de la proteína.
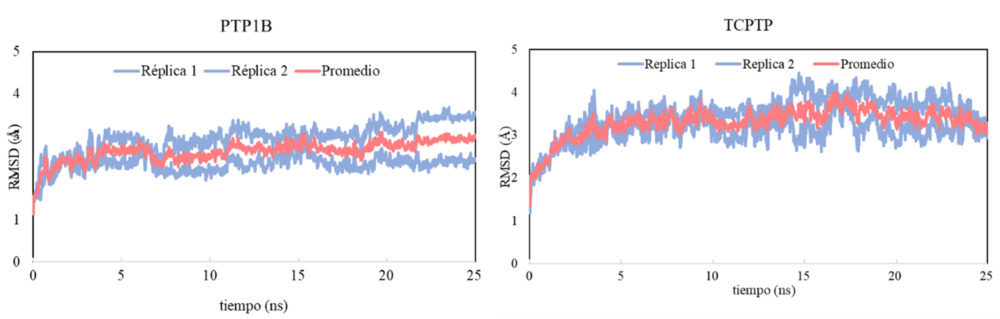
Figura 5. Evolución del RMSD a lo largo de la dinámica molecular para el complejo BB2-PTP1B (izquierda) y BB2-TCPTP (derecha)
Tal y como recoge la Tabla 1, los valores de RMSD correspondientes a los átomos del soluto (proteína-ligando) son superiores en ambos pares de réplicas para la TCPTP, lo que sugiere una menor estabilidad del complejo y además mayor flexibilidad. Lo mismo se puede deducir de los valores obtenidos para los carbonos alfa del esqueleto peptídico. Además, atendiendo a la movilidad del bucle catalítico de ambas fosfatasas (WPD-loop), se puede observar que en el caso de la TCPTP estos valores son ligeramente superiores comparados con su homóloga. Este hecho sugiere que el bucle no se encuentra estabilizado en la conformación abierta (catalíticamente inactiva) de la que se parte en ambas dinámicas. Esta posición queda fijada tras la unión del inhibidor alostérico, sin embargo, en ausencia de éste, el bucle tiende a moverse entre la conformación abierta y la cerrada (34) (catalíticamente activa) lo que se traduce en una movilidad mayor y por tanto en valores superiores de RMSD.
Tabla 1. Valores medios de RMSD a lo largo de toda la simulación de dinámica molecular
PTP1B TCPTP
Réplica 1 Réplica 2 Réplica 1 Réplica 2
Complejo 2,93 ± 0,3 2,32 ± 0,21 3,12 ± 0,33 3,47 ± 0,48
Cα 2,4 ± 0,42 1,66 ± 0,22 2,28 ± 0,35 2,78 ± 0,51
WPD-loop 1,29 ± 0,29 1,33 ± 0,25 1,44 ± 0,27 1,57 ± 0,27
Dado que el estudio general del RMSD del complejo parece sugerir una menor estabilidad en el caso de la TCPTP, se decidió analizar por separado este valor para los átomos pesados (distintos de hidrógeno) que forman la benzbromarona BB2.
Como se puede observar en la Figura 6, el ligando no solo presenta un valor bastante más alto en el caso de la TCPTP (2,18 Å de media en ambas réplicas frente a 0,63 de PTP1B), sino que además fluctúa más en el interior del bolsillo, lo que se observó durante la visualización de las trayectorias de la dinámica. Este fenómeno se debe a la ausencia de la interacción correspondiente a la Phe280 (sustituida por un residuo de cisteína en la TCPTP), y a un apilamiento-π paralelo que mantiene mucho más fijo el ligando en el caso de PTP1B. El análisis de estos valores de RMSD parece estar de acuerdo con la actividad experimental observada, lo cual supone una validación del método teórico.

Figura 6. Evolución de los valores de RMSD de BB2 a lo largo de la dinámica molecular tanto para la PTP1B (azul) como TCPTP (rojo)
La pérdida de estabilidad del complejo TCPTP también se puede constatar midiendo las distancias entre los átomos que participan en el enlace de hidrógeno entre el carbonilo de BB2 y el residuo de asparagina presente en ambas proteínas.
Para el análisis de este enlace de hidrógeno se tomaron como límites una distancia de 3.5 Å y un ángulo de relajación entre los pares de átomos participantes de 30 ºC.
Tal y como se puede observar en la Figura 7, a pesar de que el enlace de hidrógeno se mantiene dentro de la distancia estándar durante la mayor parte de la dinámica existen diferencias significativas en ambas las simulaciones para ambas enzimas. En el caso de la TCPTP las fluctuaciones en la distancia de este enlace son más abundantes y frecuentes alcanzando distancias superiores a 3,5 Å y por tanto incompatibles con la existencia de un enlace de hidrógeno. Este hecho se debe a que BB2 presenta una mayor movilidad dentro de la cavidad de TCPTP por la falta de la interacción de apilamiento extra con la fenilalanina que lo mantendría más fijo tal y como previamente fue observado. Este hecho da lugar a una distorsión en la geometría y posición carbonilo de BB2 con respecto a la asparagina, dificultando así la formación de un enlace de hidrógeno.
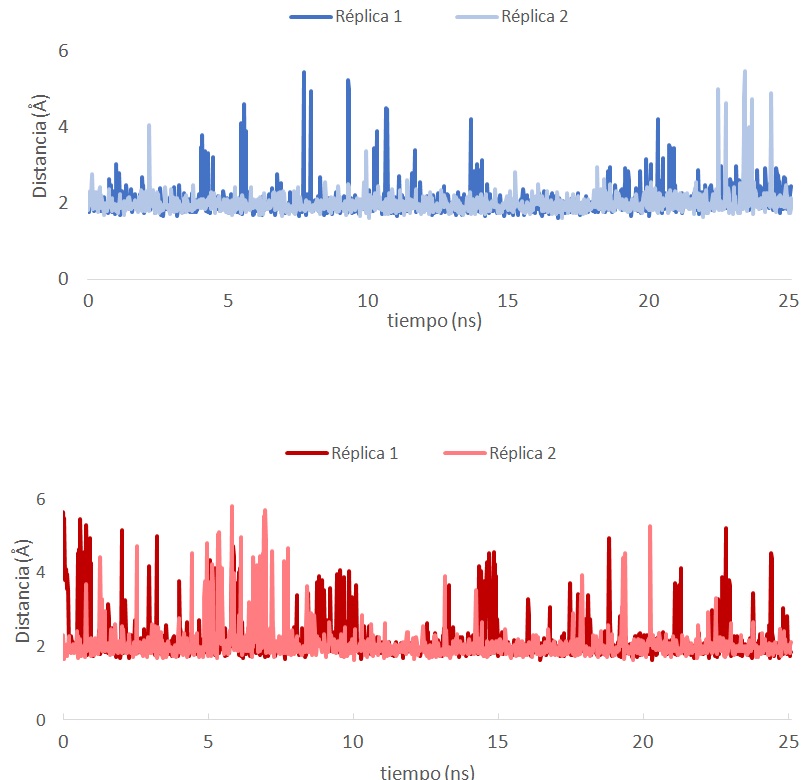
Figura 7. Monitorización a lo largo del tiempo de la distancia entre el oxígeno carbonílico de BB2 y el NH de la asparagina: Arriba PTP1B, abajo TCPTP
Si bien, durante el transcurso de la simulación de dinámica molecular estos fueron los enlaces de hidrógeno más estables, no fueron los únicos. El grupo carbonilo de BB2 es capaz de establecer eventualmente interacciones con el NH del Trp291PTP1B o Trp279TCPTP. Sin embargo, esta interacción es mucho menos estable, y a ella se le une un enlace de hidrógeno temporal entre el grupo SO2 de la sulfonamida (a través del átomo de oxígeno) que se puede establecer con los NH del esqueleto peptídico de la proteína. Junto a estos, se les unen enlaces de hidrógeno con una persistencia menor al 2% que ocurren en momentos concretos de la dinámica molecular. No obstante, el número total de enlaces de hidrógenos establecidos a lo largo del tiempo por el ligando en ambos complejos también presenta diferencias.
En la Figura 8 se muestra el número total de enlaces de hidrógenos que es capaz de establecer BB2 con su proteína a lo largo del tiempo en una replicada de cada sistema. Como se puede observar existen dos enlaces de hidrógeno que se mantienen relativamente estables a lo largo del tiempo que son los estudiados previamente. Sin embargo, en el diagrama se refleja la posibilidad de nuevos enlaces de hidrógeno con otros grupos funcionales. Se puede observar que existe una mayor población de enlaces de hidrógeno en el caso del complejo BB2-PTP1B. Este hecho refleja la mayor estabilidad del ligando dentro de esta proteína a diferencia de TCPTP, lo que a su vez se traduce en una menor afinidad por este último, debida en gran parte a la menor estabilidad del complejo formado.

Figura 8. Diagramas de número total de enlaces de hidrógenos establecidos a lo largo de la dinámica entre el ligando y PTP1B (A) y TCPTP (B)
3.3. Cálculo de energía libre
La afinidad de un fármaco por su diana está íntimamente relacionada con la energía libre de unión a la misma (ΔG) (35). Durante los últimos años, los químicos y bioquímicos computacionales han desarrollado numerosos algoritmos con el fin de determinar este parámetro, de gran utilidad durante el proceso de desarrollo de fármacos, especialmente en las campañas hit-to-lead (36). Del mismo modo, el cálculo de la energía libre de unión puede ser empleado para discernir la afinidad y, por tanto, la selectividad de un mismo ligando por dos dianas distintas (37). Para determinar la energía libre de unión de BB2 hacia la PTP1B y TCPTP se decidió emplear un método bastante novedoso conocido como WaterSwap (33). Este algoritmo emplea coordenadas de reacción para intercambiar el ligando en el interior del sitio de unión por un número de moléculas de agua equivalente a su volumen (Figura 9).

Figura 9. A) Aspecto tridimensional de BB2 durante la simulación con WaterSwap. B) Moléculas de agua intercambiadas por BB2 durante la simulación de WaterSwap
Una vez intercambiados, se calcula la energía libre absoluta a través de tres métodos distintos a lo largo del gradiente de coordenadas de reacción: algoritmo de Bennets, integración termodinámica (IT) y perturbación de energía libre (PEL). A lo largo de las coordenadas de reacción se emplean además movimientos de Montecarlo que permitan tener en cuenta la flexibilidad del sistema y promediar los gradientes de energía libre. Cuando los tres valores son similares, la simulación de WaterSwap ha encontrado la convergencia en las coordenadas de reacción y se puede tomar la media de ambos como valor de energía libre absoluta.
Tabla 2. Resultados WaterSwap para el cálculo de energía libre (kcal/mol) entre BB2 y PTP1B o TCPTP
WATERSWAP PTP1B TCPTP
Bennetts -15,65 -3,64
PEL -13,9 -4,99
IT -15,65 -3,29
ΔG Media -15,06 -3,97
Como se puede observar en la Tabla 2, la simulación ha alcanzado la convergencia puesto que las desviaciones de los valores promedio para la PTP1B y la TCPTP son inferiores a 1 (0,82 y 0,47 kcal/mol, respectivamente). Resultó muy interesante comprobar la capacidad que tiene este método para discernir entre la afinidad de BB2 por una su diana PTP1B, y sin embargo no mostrar a penas afinidad por TCPTP. Para la PTP1B los valores de ΔG están cercanos a las -15 kcal/mol, siendo valores acordes a un proceso de unión favorable y de un ligando de potencia media. En el caso de la TCPTP, los valores son mucho peores (-3,97 kcal/mol). Esto quiere decir que el proceso de unión de BB2 hacia la TCPTP no es favorable termodinámicamente por lo que la afinidad por esta es muy baja, lo cual es acorde a los valores experimentales de IC50. Este hecho lo explica la falta del contacto hidrofóbico y del apilamiento π que proporciona el residuo Phe280 de la PTP1B. La presencia de cisteína y serina en TCPTP da lugar a un aumento en el tamaño de la cavidad (343 Å3 en comparación con los 227 Å3 de la PTP1B) que en condiciones normales está ocupada por moléculas de agua. La interacción con la Phe280 de la PTP1B juega un papel clave, ya que permite compensar la pérdida de entropía por desolvatación del receptor con la contribución entálpica a la energía libre de unión que proporciona el apilamiento-π de los anillos aromáticos e interacciones de Van der Waals.
Si se estudia la contribución energética de los residuos que rodean el ligando se pueden observar cómo se cumple esta hipótesis.
Tal y como se muestra en la Figura 10 el cambio del residuo Phe280 por Cys278 supone una pérdida de la energía de unión de casi 2 kcal/mol completas, confirmándose una vez más la importancia de este residuo. Además, la falta de esta interacción supone, como se comentó anteriormente, una pérdida de la estabilidad del ligando en el bolsillo de unión. Este efecto se observa en cómo algunos aminoácidos contribuyen en menor medida a la energía de unión en el caso de la TCPTP como por ejemplo la serina o incluso el triptófano. La mayor movilidad que presentan ahora estos aminoácidos tiene como consecuencia una mayor dificultad para la desolvatación del sitio de unión y reemplazar las moléculas de agua por BB2.
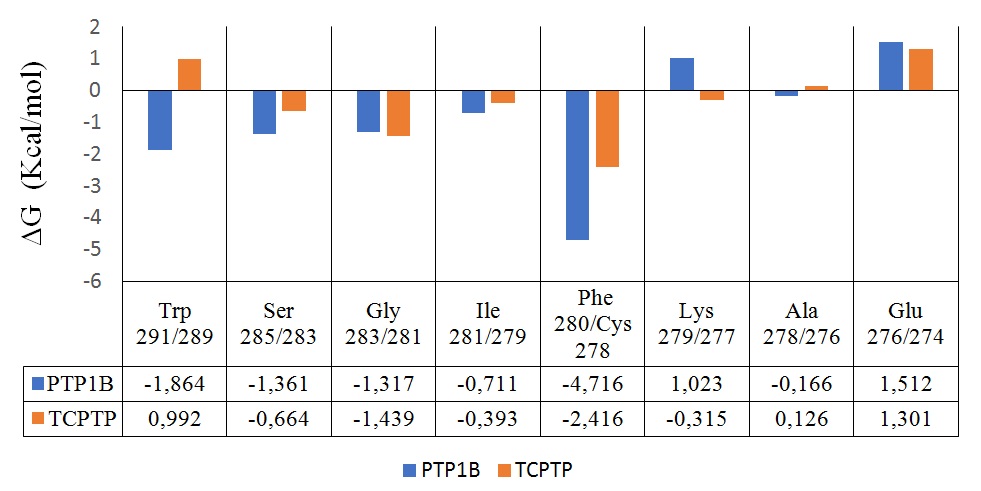
Figura 10. Contribución energética por residuo a la unión de BB2 en aminoácidos seleccionados de PTP1B y TCPTP
Cabe destacar que el residuo de Glu a pesar de presentar un enlace de hidrógeno estable durante la dinámica no presenta una energía libre de unió favorable. Esto puede deberse a que se encuentra muy cercano al anillo de benceno del ligando y la desolvatación del mismo no está muy favorecida a pesar de que luego se establezca un enlace de hidrógeno.
Teniendo en cuenta todos estos resultados, se podría formular un modelo farmacofórico que permitiese diseñar inhibidores potentes y selectivos de PTP1B (Figura 11). Básicamente este farmacóforo lo constituiría un núcleo de anillos aromáticos bien carbo o heterocíclicos responsables del apilamiento-π paralelo con la Phe280. Estos deberían estar unidos en la disposición adecuada al aceptor de enlace de hidrógeno necesario para interactuar con el residuo de Asn193. A estos puntos se le uniría un donador de enlace de hidrógeno situado fuera de este plano a 4 Å y 34º capaz de interaccionar con el residuo de Glu274, preferiblemente un grupo sulfonamida por su geometría. Finalmente, este debería estar unido a otro anillo aromático capaz de interactuar por apilamiento-π en forma de T con el residuo de Phe280.
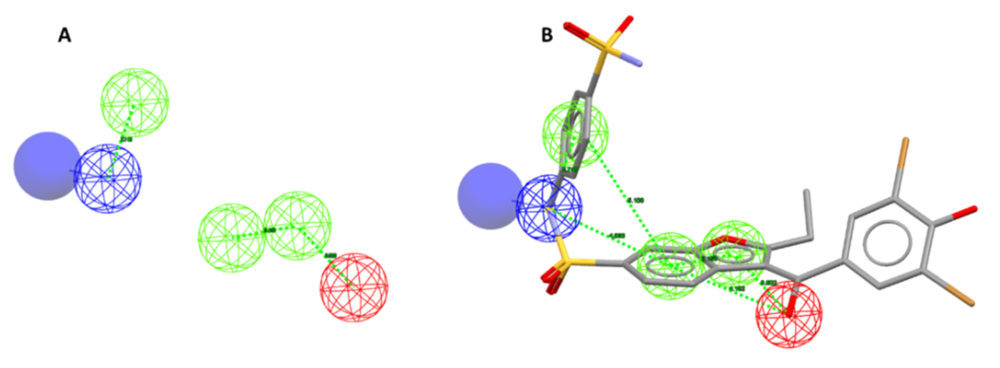
Figura 11. A) Modelo farmacofórico para inhibidores alostéricos y selectivos de PTP1B. B) Superposión del modelo y BB2, en la que se muestran las interacciones aromáticas (verde), aceptoras (roja) y donadoras (azul) de enlace de hidrógeno y su proyección geométrica
4. Conclusiones
Desde su descubrimiento hasta nuestros días, numerosos grupos de investigación han intentado desarrollar fármacos inhibidores de PTP1B al tratarse de una de las dianas más prometedoras para el tratamiento de la DM II. El fracaso de la mayoría de estos fármacos se debe a su baja biodisponibilidad y a la falta de selectividad frente a otras fosfatasas, especialmente la TCPTP. Si bien se ha profundizado tanto experimental como computacionalmente en el desarrollo de inhibidores del sitio activo, el bolsillo alostérico no ha sido tan explorado hasta el momento. Dado que el diseño racional de fármacos y más en concreto las técnicas computacionales hoy día son cruciales en la identificación de nuevas moléculas con potencia farmacológico se ha planteado un estudio teórico sobre la selectividad de los inhibidores alostéricos entre PTP1B y TCPTP. Las técnicas de dinámica molecular y cálculo de energía libre han permitido proponer una hipótesis sólida acerca de la selectividad entre ambas proteínas. Los modelos así generados han demostrado una mayor estabilidad de la PTP1B frente a la TCPTP, lo cual está en acorde a los valores experimentales de IC50 y valida esta aproximación.
La afinidad de un fármaco por su diana viene determinada por el número de interacciones totales que establece con la misma. Este estudio demuestra cómo a pesar de que en ambos sistemas las interacciones son similares a priori, no lo es su comportamiento. En el caso de la TCPTP los enlaces de hidrógenos resultaron ser menos estables y mantenidos a lo largo del tiempo, teniendo un efecto negativo en la estabilidad del sistema y por tanto en la afinidad total de BB2 por TCPTP. Más en concreto, nuestras simulaciones han revelado cómo el papel clave lo juega la falta de un residuo de fenilalanina en la TCPTP que estabiliza el complejo enzima-receptor a través de interacciones de apilamiento-π y además contribuye a la energía libre de unión y por tanto a la afinidad de forma decisiva. Si bien, los enlaces de hidrógenos son de las interacciones intermoleculares más fuertes, el anormal entorno hidrofóbico del bolsillo alostérico parece favorecer mucho más las interacciones de carácter apolar y aromáticas, acusando la repercusión que tiene el residuo de Phe280 en PTP1B.
La combinación de estas dos aproximaciones computacionales no sólo ha servido de base para hacer una propuesta sólida acerca de esta selectividad y generar un modelo cualitativo, sino que podrá utilizarse para el diseño de nuevos inhibidores puesto que ha demostrado, y especialmente los cálculos de energía libre, ser capaz de discernir las diferencias y los determinantes que hacen que esta unión sea favorable hacia PTP1B, en acorde a los valores experimentales.
Del mismo modo, se ha puesto de manifiesto como estas técnicas computacionales constituyen una herramienta de gran utilidad para comprender procesos bioquímicos a nivel molecular. Así pues, este estudio también abre una puerta para el diseño racional de fármacos dirigidos a esta diana y que presenten la selectividad suficiente para progresar en ensayos clínicos.
5. Listado de abreviaturas
Organización Mundial de la Salud (OMS), diabetes mellitus tipo II (DMII), Proteína tirosina fosfatasa 1B (PTP1B), proteína tirosina fostasa de linfocitos T (TCPTP), Protein Data Bank (PDB), raíz cuadrada de la desviación cuadrática media (RMSD), integración termodinámica (IT) y perturbación de energía libre (PEL).
6. Agradecimientos
El autor agradece a la Universidad de Bristol los medios y recursos computacionales de alto rendimiento para poder llevar a cabo todas las simulaciones del trabajo, así como la financiación del EPSRC. Así mismo, se agradece al profesor Adrian Mulholland la aceptación en su grupo de investigación y a la doctora Kara E. Ranaghan por sus consejos. Del mismo modo, el autor agradece al doctor Ramón Alajarín su ayuda en la supervisión y confección del manuscrito y al doctor Juan José Vaquero por la aceptación en su grupo de investigación. Finalmente, se agradece al Ministerio de Economía y Competitividad la concesión de una ayuda FPU (FPU16/01647) y a la Universidad de Alcalá por la concesión de una Ayuda de Movilidad en el año 2018 (Bolsa de Viajes 2018).
7. Referencias
- Chatterjee S b, Khunti K, Davies MJ. Type 2 diabetes. The Lancet. 2017;389(10085):2239-51.
- Wong E, Backholer K, Gearon E, Harding J, Freak-Poli R, Stevenson C, et al. Diabetes and risk of physical disability in adults: a systematic review and meta-analysis. The Lancet Diabetes & Endocrinology. 2013;1(2):106-14.
- Gómez-Huelgas R, Martínez-Castelao A, Artola S, Górriz JL, Menéndez E. Tratamiento de la diabetes tipo 2 en el paciente con enfermedad renal crónica. Medicina Clínica. 2014;142(2):85.e1-.e10.
- Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell. 1992;68(3):545-60.
- Feldhammer M, Uetani N, Miranda-Saavedra D, Tremblay ML. PTP1B: A simple enzyme for a complex world. Critical Reviews in Biochemistry and Molecular Biology. 2013;48(5):430-45.
- Barr AJ. Protein tyrosine phosphatases as drug targets: strategies and challenges of inhibitor development. Future Medicinal Chemistry. 2010;2(10):1563-76.
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased Insulin Sensitivity and Obesity Resistance in Mice Lacking the Protein Tyrosine Phosphatase-1B Gene. Science. 1999;283(5407):1544-8.
- Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nature Reviews Drug Discovery. 2002;1:696.
- Hussain H, Green IR, Abbas G, Adekenov SM, Hussain W, Ali I. Protein tyrosine phosphatase 1B (PTP1B) inhibitors as potential anti-diabetes agents: patent review (2015-2018). Expert Opinion on Therapeutic Patents. 2019;29(9):689-702.
- Low J-L, Chai CLL, Yao SQ. Bidentate Inhibitors of Protein Tyrosine Phosphatases. Antioxidants & Redox Signaling. 2013;20(14):2225-50.
- Grover AK. Use of Allosteric Targets in the Discovery of Safer Drugs. Medical Principles and Practice. 2013;22(5):418-26.
- Rocheville M, Garland SL. An industrial perspective on positive allosteric modulation as a means to discover safe and selective drugs. Drug Discovery Today: Technologies. 2010;7(1):e87-e94.
- Wiesmann C, Barr KJ, Kung J, Zhu J, Erlanson DA, Shen W, et al. Allosteric inhibition of protein tyrosine phosphatase 1B. Nature Structural &Amp; Molecular Biology. 2004;11:730.
- Li S, Zhang J, Lu S, Huang W, Geng L, Shen Q, et al. The Mechanism of Allosteric Inhibition of Protein Tyrosine Phosphatase PLOS ONE. 2014;9(5):e97668.
- Baskaran SK, Goswami N, Selvaraj S, Muthusamy VS, Lakshmi BS. Molecular Dynamics Approach to Probe the Allosteric Inhibition of PTP1B by Chlorogenic and Cichoric Acid. Journal of Chemical Information and Modeling. 2012;52(8):2004-12.
- Shinde RN, Sobhia ME. Binding and discerning interactions of PTP1B allosteric inhibitors: Novel insights from molecular dynamics simulations. Journal of Molecular Graphics and Modelling. 2013;45:98-110
- Cui W, Cheng Y-H, Geng L-L, Liang D-S, Hou T-J, Ji M-J. Unraveling the Allosteric Inhibition Mechanism of PTP1B by Free Energy Calculation Based on Umbrella Sampling. Journal of Chemical Information and Modeling. 2013;53(5):1157-67.
- Zhang S, Chen L, Luo Y, Gunawan A, Lawrence DS, Zhang Z-Y. Acquisition of a Potent and Selective TC-PTP Inhibitor via a Stepwise Fluorophore-Tagged Combinatorial Synthesis and Screening Strategy. Journal of the American Chemical Society. 2009;131(36):13072-9.
- Li X, Wang L, Shi D. The design strategy of selective PTP1B inhibitors over TCPTP. Bioorganic & Medicinal Chemistry. 2016;24(16):3343-52.
- Wade RC, Salo-Ahen OMH. Molecular Modeling in Drug Design. Molecules (Basel, Switzerland). 2019;24(2):321.
- Mendoza F, Gómez H, Lluch JM, Masgrau L. α1,4-N-Acetylhexosaminyltransferase EXTL2: The Missing Link for Understanding Glycosidic Bond Biosynthesis with Retention of Configuration. ACS Catalysis. 2016;6(4):2577-89.
- Woods CJ, Malaisree M, Long B, McIntosh-Smith S, Mulholland AJ. Computational Assay of H7N9 Influenza Neuraminidase Reveals R292K Mutation Reduces Drug Binding Affinity. Scientific Reports. 2013;3:3561.
- Callegari D, Ranaghan KE, Woods CJ, Minari R, Tiseo M, Mor M, et al. L718Q mutant EGFR escapes covalent inhibition by stabilizing a non-reactive conformation of the lung cancer drug osimertinib. Chemical Science. 2018;9(10):2740-9.
- Liu X, Shi D, Zhou S, Liu H, Liu H, Yao X. Molecular dynamics simulations and novel drug discovery. Expert Opinion on Drug Discovery. 2018;13(1):23-37.
- Rudling A, Gustafsson R, Almlöf I, Homan E, Scobie M, Warpman Berglund U, et al. Fragment-Based Discovery and Optimization of Enzyme Inhibitors by Docking of Commercial Chemical Space. Journal of Medicinal Chemistry. 2017;60(19):8160-9.
- Wang T, Wu Z, Sun L, Li W, Liu G, Tang Y. A Computational Systems Pharmacology Approach to Investigate Molecular Mechanisms of Herbal Formula Tian-Ma-Gou-Teng-Yin for Treatment of Alzheimer’s Disease. Frontiers in Pharmacology. 2018;9(668).
- Shinde RN, Kumar GS, Eqbal S, Sobhia ME. Screening and identification of potential PTP1B allosteric inhibitors using in silico and in vitro approaches. PLOS ONE. 2018;13(6):e0199020
- Šali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. Journal of Molecular Biology. 1993;234(3):779-815.
- Anandakrishnan R, Aguilar B, Onufriev AV. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Research. 2012;40(W1):W537-W41.
- Rodríguez R, Chinea G, Lopez N, Pons T, Vriend G. Homology modeling, model and software evaluation: three related resources. Bioinformatics. 1998;14(6):523-8.
- Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr., et al. The Amber biomolecular simulation programs. Journal of computational chemistry. 2005;26(16):1668-88.
- Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics. 1977;23(3):327-41.
- Woods CJ, Malaisree M, Hannongbua S, Mulholland AJ. A water-swap reaction coordinate for the calculation of absolute protein–ligand binding free energies. The Journal of Chemical Physics. 2011;134(5):054114.
- Cui DS, Lipchock JM, Brookner D, Loria JP. Uncovering the Molecular Interactions in the Catalytic Loop That Modulate the Conformational Dynamics in Protein Tyrosine Phosphatase 1B. Journal of the American Chemical Society. 2019;141(32):12634-47.
- Lamoree B, Hubbard RE. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017;61(5):453-64.
8. Material Complementario
Alineamiento de las secuencias generado con el programa Modeller durante el proceso de modelado por homología.