1. INTRODUCCIÓN
Las enfermedades infecciosas han supuesto un gran reto en cuanto a morbilidad, mortalidad y secuelas para la humanidad a través de los siglos. A pesar de los enormes avances científicos y tecnológicos logrados en materia de prevención y tratamiento, la lucha frente a estas enfermedades continúa hoy en día más viva que nunca. La emergencia o reemergencia de ciertas enfermedades infecciosas constituye un hecho de gran relevancia en la actualidad. El elevado crecimiento demográfico, el comercio internacional, una educación en materia sanitaria insuficiente, los desplazamientos poblacionales como consecuencia de conflictos, pobreza y guerras locales, el cambio climático y las inestabilidades sociales, configuran diferentes caras de la globalización creando las condiciones óptimas para que enfermedades infecciosas que estaban confinadas en determinadas zonas geográficas, se puedan encontrar en la actualidad en cualquier punto del planeta y transmitirse rápidamente provocando una epidemia global.
Desde comienzos del siglo XXI, la salud pública a nivel mundial se ha visto profundamente amenazada no solo por la emergencia de nuevos patógenos, entre los que destacan el coronavirus asociado al síndrome respiratorio grave y agudo (SARS-Co), el virus de la gripe A H1N1, las cepas de gripe aviar A H5N1 y H7N9 o el coronavirus causante del síndrome respiratorio de Oriente Medio (MERS-CoV), sino también por la emergencia de patógenos conocidos como el virus de Zica, el virus del Dengue, el virus de Chikungunya, el virus de Marburgo o el virus del Ebola tanto en zonas endémicas como no endémicas. Además, la aparición de brotes de enfermedades prevenibles como el cólera, la poliomelitis o el sarampión como consecuencia del descenso en las coberturas de inmunización en países que llevaban años libres de ellas, junto con la presencia de enfermedades históricas como el Síndrome de Inmunodeficiencia Adquirida (SIDA), la Hepatitis C, la tuberculosis o la malaria, complican aún más el panorama sanitario internacional. Dada la relevancia de estas enfermedades, el Fondo Mundial (Global Fund) se marcó el objetivo de conseguir 14.000 millones de dólares para luchar contra el VIH, la malaria y la tuberculosis, habiendo alcanzado en octubre de 2019 la cifra de 13.000 millones de dólares, lo que constituye un logro de cooperación internacional. España aportará 100 millones de euros.
Las estadísticas reportadas por la Organización Mundial de la Salud (OMS) referente al número de afectados por los diferentes brotes y su expansión territorial reflejan la vulnerabilidad que existe frente a los patógenos emergentes a nivel global y representan un motivo de preocupación tanto para los organismos de salud pública como para la comunidad científica, ya que muchas de las enfermedades infecciosas a las que nos enfrentamos actualmente presentan una patogénesis desconocida y carecen de tratamientos efectivos tanto para su prevención como para su cura. Unido a esto, tampoco puede descartarse el peligro que supone la elevada tasa de mutación y recombinación que poseen estos agentes patógenos, lo que les permite derivar en nuevas variantes capaces de franquear la barrera inter-específica y trasladarse de su reservorio habitual a otros reservorios, modificar su modo de contagio o aumentar su grado de virulencia, pudiendo provocar consecuencias nefastas sobre la salud humana. No cabe duda que hoy en día los problemas sanitarios están mejor definidos que hace dos décadas, que existe una colaboración nacional e internacional más estrecha a la hora de tratar cuestiones claves para la salud y que se dispone de un mayor conocimiento, medios y capacidad para abordar problemas emergentes. Sin embargo, queda aún mucho camino por recorrer.
La innovación científica en los últimos años ha transformado nuestra capacidad para enfrentar los retos que suponen las infecciones emergentes y reemergentes. Hoy en día contamos con un amplio abanico de nuevas y sofisticadas tecnologías que permiten grandes progresos en la detección y control potencial de enfermedades infecciosas entre las que destacan la secuenciación genómica avanzada, la proteómica, la bioinformática, la biología estructural, la epigenómica y la biología de sistemas. A pesar de que el diagnóstico y el tratamiento temprano de las infecciones tienen una influencia directa sobre la incidencia de transmisión, la prevención basada en vacunas representa la estrategia más efectiva a largo plazo. Debido a esto, se han ido introduciendo en este campo las nuevas tecnologías con el fin de ir modificando los conceptos aplicados hasta ahora con las vacunas disponibles y adaptarlos a los requerimientos de los patógenos a los que nos enfrentamos actualmente y para los cuales no existe una vacuna preventiva eficaz.
El grupo de poxvirus y vacunas liderado por el Dr. Mariano Esteban en el Centro Nacional de Biotecnología (CNB), perteneciente al Consejo Superior de Investigaciones Científicas (CSIC), representa un pilar importante en España dentro del campo de la investigación y generación de candidatos vacunales frente a enfermedades infecciosas utilizando como plataforma cepas altamente atenuadas del virus vaccinia (VACV). Con más de 300 publicaciones originales en revistas arbitradas, este grupo ha realizado importantes aportaciones en los campos de la virología, la inmunología y la vacunología. Desde mi incorporación en el año 1998 hasta la fecha, mi trabajo se ha centrado en el estudio comparativo del comportamiento y la patogénesis de las cepas altamente atenuadas del virus vaccinia MVA (virus modificado de Ankara) y NYVAC (virus vaccinia de Nueva York) con el propósito final de trasladar el conocimiento básico a la clínica a través del desarrollo de candidatos vacunales frente a enfermedades prevalentes, con énfasis en la infección causada por el Virus de la Inmunodeficiencia Humana (VIH).
En esta revisión llevaré a cabo una descripción temporal de las principales aportaciones que hemos realizado desde el conocimiento básico de la biología del vector MVA, tanto in vitro como in vivo, en comparación con la cepa atenuada NYVAC, hasta su evaluación como candidato vacunal frente al VIH/SIDA en ensayos clínicos. Detallaré la generación y caracterización del vector recombinante MVA que expresa los antígenos Env, Gag, Pol y Nef del subtipo B del VIH-1 (denominado MVA-B) y revisaré los datos preclínicos que respaldaron la evaluación de MVA-B como la primera vacuna profiláctica y terapéutica frente al VIH-1 ensayada en humanos en España. Además, valoraré los resultados de los ensayos clínicos y discutiré las líneas de investigación en las que estamos trabajando actualmente teniendo como premisa los últimos avances científicos realizados en el campo de vacunas frente al VIH/SIDA.
Lógicamente, mi contribución científica se ha visto beneficiada por el liderazgo del Dr. Mariano Esteban y por la colaboración con miembros del laboratorio y con otros investigadores europeos y americanos con los que trabajamos conjuntamente en pro de la consecución de una vacuna frente al VIH. Además, la aportación económica de la Fundación Bill & Melinda Gates, Fundación Botín, Unión Europea, proyectos del Plan Nacional, Fundación para la Investigación y Prevención del SIDA (FIPSE) y Red Española de Investigación en SIDA (RIS), ha contribuido sustancialmente a los avances que hemos llevado a cabo en el desarrollo de candidatos vacunales frente al VIH-1, permitiendo realizar una traslación desde la investigación básica hasta los ensayos preclínicos y clínicos.
A continuación, esbozaré como hemos ido avanzando a lo largo de los años desde el entendimiento de la biología de los virus utilizados hasta su aplicación como vacunas.
2. BIOLOGÍA DE LAS CEPAS ATENUADAS MVA Y NYVAC. SIMILITUDES Y DIFERENCIAS.
2.1. Poxvirus. Aspectos generales
Los poxvirus son una familia de virus ADN de gran tamaño y complejidad, capaces de infectar tanto vertebrados como invertebrados. El miembro más estudiado de esta familia es el virus vaccinia (VACV) (1); sin embargo, el más conocido es el virus de la viruela humana, causante de una de las enfermedades más devastadoras que ha conocido el hombre a lo largo de su historia y, a su vez, la única enfermedad hasta el momento combatida mediante vacunación a escala mundial y declarada erradicada oficialmente por la OMS en el año 1980.
Una de las características que distingue a los poxvirus de otros virus ADN es que permanece en el citoplasma de la célula infectada durante todo el ciclo infeccioso (Figura 1).
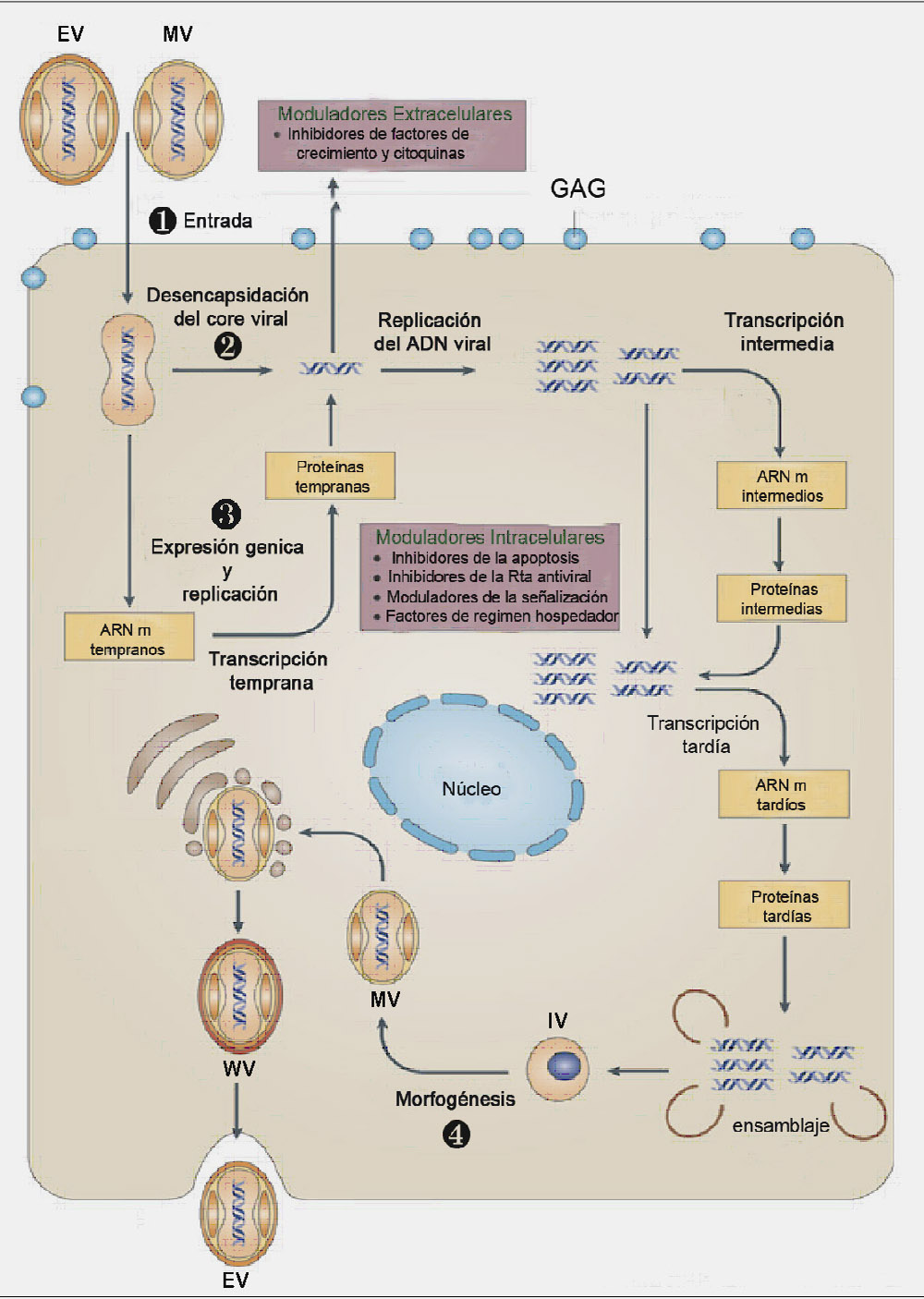
Figura 1. Ciclo infectivo del virus vaccinia (VACV). En el esquema se muestra el ciclo completo de infección del virus, que se encuentra dividido en: Entrada (1), desensamblaje (2), transcripción y replicación (3) y morfogénesis y salida de la progenie viral (4). IV: virus inmaduro; MV: virus intracelular maduro; WV: virus intracelular con envuelta; EV: virus extracelular.
La replicación viral es un proceso complejo, altamente conservado y en cascada en el que se distinguen 3 fases de síntesis de ARN mensajeros y de proteínas virales que se diferencian temporalmente en tempranas, intermedias y tardías (2). Después de la entrada y desencapsidación del virus, se produce la transcripción de los genes tempranos que codifican proteínas que intervienen en la modulación de los mecanismos de defensa de la célula, en la replicación del ADN y en la transcripción de genes intermedios. La replicación del ADN tiene lugar en áreas del citoplasma denominadas factorías virales y ocurre durante las dos primeras horas tras la entrada del virus. Como consecuencia de dicha replicación, se generan unas 10000 copias del genoma viral por célula de las que sólo la mitad llega a empaquetarse. En la etapa intermedia se transcriben proteínas implicadas en la transcripción de genes tardíos que, a su vez, codifican proteínas estructurales del virión y enzimas necesarias para la trascripción de los genes tempranos que también se incorporan en la partícula viral. Una vez replicado el ADN y sintetizadas todas las proteínas virales, comienza el proceso de morfogénesis que ocurre en varias etapas e involucra la producción de diferentes formas virales. Este proceso comienza con la formación de crestas membranosas que rodean un material electrodenso que contiene el ADN viral y las proteínas del core, dando lugar a la formación de estructuras esféricas denominadas virus inmaduros (IVs, “Inmature Virus”). A continuación, tiene lugar la proteólisis y condensación del core viral, transformando los IVs en viriones intracelulares maduros (MVs, “Mature Virus”) que representan las primeras formas infectivas del VACV. Una proporción de los MVs son transportados a través de microtúbulos desde las factorías virales hasta el trans-golgi, donde adquieren una segunda membrana transformándose en virus intracelulares con envuelta (WVs, “Wrapped Virus”). Estos WVs son también transportados a través de la red de microtúbulos hasta la membrana plasmática, fusionándose con esta última y liberando al exterior virus con envuelta (EVs, “Enveloped Virus”) que pueden permanecer retenidos en la superficie de la célula infectada o liberarse al medio. Los EVs retenidos en la superficie inducen la formación de colas de actina que proyectan las partículas virales fuera de la célula, permitiendo la infección célula a célula, mientras que los EVs liberados al medio facilitan la diseminación del virus a larga distancia (3).
El VACV ha contribuido al desarrollo de una parte importante de la virología moderna, ya que además de ser el primer virus animal visualizado al microscopio electrónico, crecido en cultivos celulares, purificado físicamente y analizado químicamente, fue uno de los primeros en ser utilizado como vector de expresión de antígenos heterólogos o como vacunas vivas gracias a los avances en la tecnología del ADN recombinante (4), lo que supuso una revolución en el campo de las vacunas.
Existen una serie de características únicas que hacen de los poxvirus una excelente plataforma para la generación de candidatos vacunales. Entre ellas destacan: (i) la flexibilidad de su genoma, que permite la inserción de grandes fragmentos de ADN exógeno (hasta 25 Kb) sin pérdida de infectividad; (ii) la ausencia de persistencia o integración genómica debido a su replicación citoplasmática; (iii) la facilidad para generar virus recombinantes altamente estables que expresen niveles elevados del transgen; (iv) la capacidad de inducir una potente respuesta inmune tanto humoral como celular con inmunidad duradera tras una única inoculación; (v) la baja prevalencia en la población mundial de inmunidad frente al vector por la interrupción de la vacunación frente a la viruela en los años 70; (vi) la estabilidad del producto liofilizado, su bajo coste, facilidad de producción, distribución y administración a nivel global; y (vii) la disponibilidad de cepas altamente atenuadas incapaces de replicar en células humanas como MVA y NYVAC.
El virus modificado de Ankara se derivó de la cepa vaccinia Ankara corioalantoidea (CVA), utilizada en Turquía como vacuna frente a la viruela, tras más de 570 pases sucesivos en cultivos primarios de fibroblastos embrionarios de pollo (CEFs, “Chicken Embryo Fibroblasts”). Durante el curso de su generación, perdió un 15 % del genoma parental y, como consecuencia, su capacidad para replicar en células humanas y en la mayoría de células de mamífero. En 1998, Antoine y colaboradores publicaron la secuencia completa de MVA. Su genoma contiene 178 Kb frente a las 208 Kb de su cepa parental. Se han mapeado 193 ORFs (fases de lectura abierta) que corresponden a 177 genes, 25 de los cuales están parcialmente delecionados y/o han sufrido mutaciones dando lugar a proteínas truncadas (5). Estos genes codifican mayoritariamente proteínas inmunomoduladoras responsables de contrarrestar la respuesta antiviral de la célula hospedadora, proteínas que determinan el rango de hospedador y proteínas estructurales que controlan la morfogénesis viral. Esta cepa se utilizó como vacuna profiláctica durante las últimas décadas de la campaña de erradicación de la viruela en Alemania en más de 120000 individuos sin presentar ninguna complicación, incluso en pacientes inmunodeprimidos, avalando su seguridad como vector vacunal.
A diferencia de MVA que sufrió un proceso de atenuación clásica, la cepa NYVAC fue generada mediante la deleción selectiva de genes. En la década de los 90, Tartaglia y colaboradores eliminaron del genoma de la cepa Copenhagen del virus vaccinia (VACV-COP) 18 genes no esenciales mediante ingeniería genética. Los genes delecionados codifican proteínas implicadas en virulencia, patogenicidad e interacción con la célula hospedadora. El resultado de este proceso fue la obtención de un virus atenuado, con una capacidad reducida de replicación en un amplio rango de células de mamífero, entre las que se incluyen las de origen humano, y altamente seguro cuando se administró en ratones inmunodeprimidos (6).
Debido al interés progresivo que existe en la aplicación clínica de estas cepas altamente atenuadas de poxvirus como candidatos vacunales frente a un abanico de enfermedades infecciosas y cáncer, la primera parte de esta revisión se centrará en describir los principales hallazgos que hemos realizado durante los últimos años para ampliar el conocimiento sobre el comportamiento y la patogénesis de las cepas MVA y NYVAC tanto in vitro como in vivo, y para definir el perfil de seguridad e inmunogenicidad que inducen los virus recombinantes basados en ellas en diferentes modelos animales. A su vez, resaltaremos las principales estrategias que hemos explorado, basándonos en el conocimiento básico generado, para optimizar la capacidad inmunogénica de ambos vectores.
Estos estudios se han desarrollado en el CNB en el marco de múltiples proyectos de I+D+i financiados en convocatorias competitivas de administraciones o entidades públicas y privadas tanto nacionales como internacionales. Los resultados obtenidos se han publicado en forma de artículos originales, revisiones o capítulos de libros en revistas científicas relacionadas con virología, inmunología o desarrollo de vacunas, como se detalla en la bibliografía. Esta investigación también formó parte de la memoria titulada “Caracterización in vitro e in vivo de los vectores atenuados de poxvirus MVA y NYVAC como candidatos vacunales frente al VIH/SIDA” presentada bajo mi codirección por el estudiante José Luis Nájera García para optar al grado de Doctor en Ciencias por la Universidad Autónoma de Madrid en el año 2007. La tesis fue calificada con sobresaliente cum laude y obtuvo el premio Juan Abelló Pascual, concedido por la Real Academia de Doctores de España.
2.2. Comportamiento in vitro de las cepas MVA y NYVAC
Si bien algunos aspectos del comportamiento de MVA en diferentes tipos celulares habían sido reportados con anterioridad a nuestros estudios (7), poco se sabía sobre la biología o el comportamiento de NYVAC en células en cultivo. Para ampliar este conocimiento realizamos un estudio comparativo de los efectos que producía la infección por MVA o NYVAC en cultivos primarios de CEF o células de riñón de hámster recién nacido (BHK-21) (líneas celulares permisivas) o en cultivos de células de origen humano o murino (no permisivas).
Los resultados de este trabajo, publicados en la revista “J. Virol.” (8), confirmaron que MVA y NYVAC eran incapaces de replicar de forma productiva en células no permisivas, mientras que exhibían cinéticas de replicación similares en condiciones permisivas. No obstante, observamos un comportamiento diferencial entre ambas cepas que podría influir sobre la capacidad inmunogénica de ambos vectores vacunales (Figuras 2 y 3).
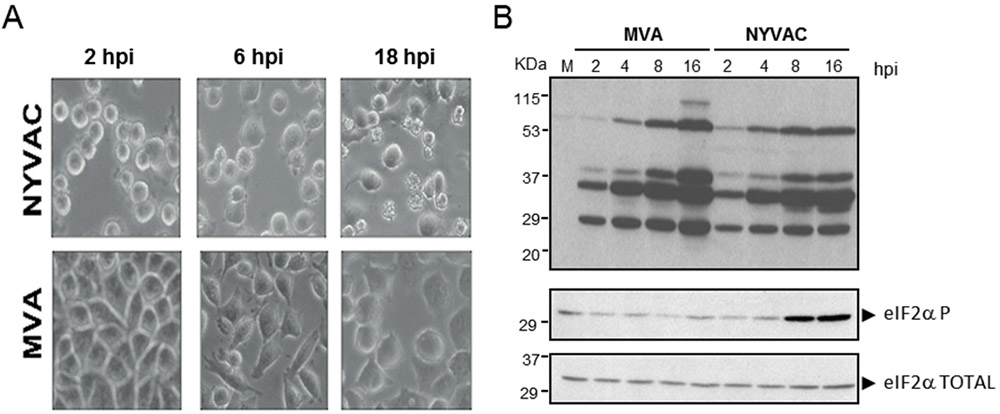
Figuras 2. Comportamiento diferencial de MVA y NYVAC en células de origen humano (HeLa) a diferentes tiempos post-infección. A: Efecto citopático analizado mediante microscopía de contraste. B: Análisis por western-blot de la acumulación de proteínas virales (arriba) y de los niveles de fosforilación de eIF2α con respecto al total (abajo).

Figura 3. Efecto de la expresión de proteínas virales sobre la morfogénesis de MVA y NYVAC. A: Análisis por western-blot de la expresión de proteínas virales tempranas y tardías en condiciones permisivas (BHK-21) y no permisivas (HeLa). A la derecha se esquematizan los diferentes estadíos del ciclo viral en los que están implicadas algunas de las proteínas virales tardías que participan en el proceso de morfogénesis de VACV. B: Microscopía electrónica de células HeLa infectadas con MVA o NYVAC. La barra situada en la esquina superior derecha de cada panel indica la magnificación de cada fotografía (500 nm). N: núcleo; M: mitocondria; FV: factoría viral. Las flechas indican los viriones inmaduros (IVs).
Entre las principales diferencias encontradas destacan: (i) NYVAC produce un efecto citopático (EC) más severo que el producido por MVA desde tiempos muy tempranos y en un amplio rango de células, tanto permisivas como no permisivas, lo que indica que es un efecto independiente de la restricción del hospedador (Figura 2A); (ii) en condiciones permisivas el rendimiento de virus asociado a células obtenido tras la infección por NYVAC es inferior al obtenido en células infectadas por MVA. Esta reducción podría estar relacionada con el marcado efecto citopático asociado a la infección por NYVAC y podría explicar las dificultades encontradas por algunos grupos para producir altos títulos de recombinantes basados en este vector; (iii) la infección por NYVAC en condiciones no permisivas produce un bloqueo traduccional que se encuentra asociado a un incremento significativo en los niveles de fosforilación de la subunidad alfa del factor eIF2 (eIF2) (Figura 2B) y que afecta a la expresión de ciertas proteínas virales tardías, algunas de ellas implicadas en el proceso de maduración de los viriones, como las proteínas estructurales A27, A17 o L1 (Figura 3A). Como consecuencia de esto, el bloqueo en la morfogénesis ocurre antes o durante la formación de los viriones inmaduros (IVs) (Figura 3B).
Esto difiere de lo que nuestro grupo había reportado previamente en relación a la infección por MVA, donde la expresión de proteínas virales tempranas y tardías ocurre de forma similar a como se produce en células permisivas, pero existe un bloqueo en el proceso de morfogénesis, posterior a la formación de los IVs (9, 10); y (iv) a diferencia de MVA, NYVAC induce en las células infectadas potentes cambios morfológicos y bioquímicos característicos de la muerte por apoptosis, un fenómeno que demostramos depende de la activación de las caspasas (cisteín-proteasas) y de la ARNasa L (Endorribonucleasa L). La activación de las caspasas se demostró mediante la detección por western-blot de la forma procesada de la proteína celular PARP (Poly (ADP-Ribosa) Polimerasa) (Figura 4A), mientras que la activación de la ARNasa L se verificó por el patrón de degradación de las subunidades 28S y 18S del ARN ribosómico (Figura 4B). La magnitud de la apoptosis fue cuantificada por citometría de flujo, detectándose un 42% de células con fenotipo apoptótico tras 24 horas de infección por NYVAC frente al 17% detectado tras la infección por MVA (Figura 4C).

Figura 4. Inducción de muerte celular por apoptosis tras la infección viral. A: Análisis por western-blot del procesamiento de la proteína celular PARP. B: Degradación del ARN ribosomal. Las flechas indican los productos de degradación de las subunidades 28S y 18S del ARN ribosómico. C: Cuantificación de la apoptosis mediante citometría de flujo en presencia (+) o ausencia (-) de zVAD. El porcentaje de células apoptóticas para cada condición se muestra en la parte superior de cada barra.
Muchas de las diferencias observadas in vitro entre MVA y NYVAC se vieron reflejadas en el patrón de expresión génica que inducían ambas cepas en células dendríticas derivadas de monocitos humanos inmaduros (MDDC) y que fue analizado utilizando la tecnología de microarrays (11). De los 544 genes diferencialmente regulados por la infección, 283 eran regulados específicamente tras la infección por MVA. Ambos vectores regulan al alza genes que codifican para citoquinas, quimioquinas y moléculas implicadas en la captación y procesamiento de antígenos, incluidos los genes del complejo principal de histocompatibilidad, lo que resalta la capacidad que poseen ambos vectores para activar los mecanismos de inmunidad innata. No obstante, la activación que promueve MVA, tanto en número de genes como en niveles de expresión, es significativamente superior a la que promueve NYVAC. Por ejemplo, los niveles de ARNm que codifican las proteínas IL-12β, IFN-β y TNF-a aumentaron cinco veces o más tras la infección por MVA en comparación con la infección por NYVAC, mientras que los niveles de expresión de IL-1a e IL-1β aumentaron sólo tras la infección por NYVAC. Del mismo modo, aunque ambos virus regularon de manera similar los perfiles de expresión de factores de transcripción tales como NF-κB y STAT, el aumento en la expresión de los genes OASL, MDA5 y RIG-I, implicados en la respuesta antiviral, fue exclusivo de la infección por MVA.
Estas diferencias observadas en el comportamiento de MVA y NYVAC en células en cultivo se atribuyen fundamentalmente a las numerosas deleciones y mutaciones que se introdujeron en sus respectivos genomas durante el curso de su atenuación (Figura 5). Por ello, decidimos realizar un estudio comparativo de los genomas de MVA y NYVAC con la finalidad de identificar algún factor viral que pudiera ser el responsable de las diferencias observadas entre ambos vectores. Existen 5 genes ausentes en el genoma de NYVAC que están intactos en el de MVA: C6L, C7L, N2L, I4L y J2R. Entre ellos seleccionamos el gen C7L como uno de los posibles genes responsables del comportamiento diferencial entre ambas cepas. Este gen se encuentra altamente conservado dentro del género orthopoxvirus, tiene expresión temprano-tardía y se ha definido como un gen de rango de hospedador necesario para la replicación del virus en células humanas (12, 13). Para determinar si el gen C7L podría ser el responsable del comportamiento diferencial observado, decidimos reintroducirlo en el genoma de NYVAC generando para ello el virus recombinante NYVAC-C7L. La reinserción del gen C7L es capaz de prevenir la fosforilación del factor eIF2 y la degradación del ARN ribosomal, lo que se traduce en un rescate de la expresión de las proteínas virales tardías cuya síntesis se encontraba inhibida en la infección por NYVAC (A17, A27 o L1). Como consecuencia de esto, el nuevo vector recupera la capacidad para crecer en células humanas y murinas. Del mismo modo, la expresión de la proteína C7 inhibe la apoptosis que se induce tras la infección con la cepa parental.
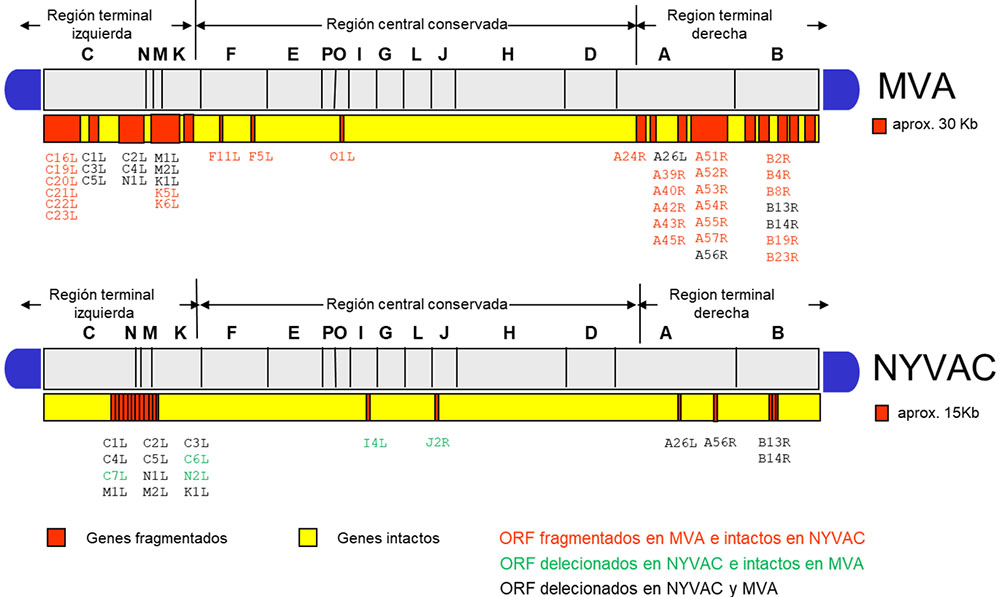
Figura 5. Análisis comparativo de los genomas de MVA y NYVAC. Los genes han sido nombrados según la nomenclatura correspondiente a la cepa Copenhagen propuesta.
En estudios in vivo el recombinante NYVAC-C7L incrementó su eficiencia de replicación; sin embargo, mantuvo un fenotipo atenuado. Todos estos resultados evidenciaron que el gen C7L posee propiedades anti-apoptóticas y desempeña un papel importante en el control de la síntesis de proteínas virales, dos funciones no descritas hasta el momento y que avalaron la solicitud de la patente “Vectores en los que se inserta el gen C7L y uso de los mismos en la fabricación de vacunas y de composiciones para terapia génica”, inscrita en España con número de solicitud P2006601240 y cuya entidad titular de derechos es el Consejo Superior de Investigaciones Científicas (CSIC).
2.3. Interacción virus-célula hospedadora
Los virus son parásitos intracelulares obligados que requieren de la maquinaria metabólica celular para replicarse. El reconocimiento del virus por la célula hospedadora desencadena una serie de eventos inmediatos y no-específicos que promueven la producción de interferones (IFNs), genes estimulados por IFN (ISGs, “Interferon Stimulated Genes”) y citoquinas pro-inflamatorias, y cuyo objetivo es resolver la infección viral. Este reconocimiento viral se lleva a cabo por los receptores de reconocimiento de patrones (PRRs, “Pattern recognition receptors”), que son proteínas codificadas en la línea germinal y que participan en el sistema inmunitario innato identificando patrones moleculares asociados a patógenos (PAMPs, “Pathogen-Associated Molecular Patterns”) y patrones moleculares asociados a daño (DAMPs, “Damage-Associated Molecular Patterns”). Entre los PRRs mejor descritos y con implicaciones en infecciones virales se encuentran los receptores tipo toll (TLRs, “Toll-Like Receptors”), los receptores tipo RIG-I (proteína inducible por ácido retinoico) (RLRs, “Retinoic acid-inducible gene I (RIG-I)-Like Receptors”) y los receptores tipo NOD (proteínas con dominio de oligomerización y unión a nucleótidos) (NLRs, “Nucleotide binding oligomerization domain (NOD)-like receptors”). Estas moléculas se expresan en los macrófagos, en las células del epitelio pulmonar y en las células reclutadas del sistema inmune, aunque también se han encontrado en células endoteliales, estromales, neuronales y fibroblastos. La detección de determinados componentes virales por los receptores TLR se produce en la superficie celular y en el compartimento endosómico. En la superficie, TLR2 o TLR4 reconocen virus con genoma ADN o ARN mientras que en el compartimento endosomal, TLR7, TLR3 o TLR9 detectan moléculas de ARN monocatenario, ARN bicatenario o secuencias de ADN, respectivamente. Dentro de los receptores RLR se ha demostrado que RIG-I y MDA5 (proteína codificada por el gen asociado con la diferenciación del melanoma 5) funcionan como sensores de ARN viral citosólico. Por otra parte, se ha descrito que NALP3, un miembro de la familia de receptores NLR, está involucrado en la detección tanto de virus ADN como ARN. NALP3, ASC y pro-caspasa 1 forman un complejo molecular citosólico multimérico, conocido como inflamasoma NALP3, que controla el procesamiento de la pro-interleuquina 1 (pro-IL-1) y la pro-interleuquina 18 (pro-IL-18) en sus formas activas: IL-1 e IL-18, respectivamente (14).
La mayoría de los estudios realizados sobre el proceso de reconocimiento de los poxvirus por parte de la célula hospedadora se habían llevado a cabo con cepas competentes en replicación que codifican inmunomoduladores que actúan de forma coordinada y a diferentes niveles para contrarrestar la respuesta antiviral inducida por la célula. Debido a este hecho, nos pareció muy interesante y novedoso averiguar cómo ocurría este proceso con cepas atenuadas de VACV.
En colaboración con el grupo del Dr. Thierry Calandra nos propusimos identificar las bases moleculares del reconocimiento innato de las cepas atenuadas MVA y NYVAC, así como evaluar de forma comparativa el perfil inflamatorio que inducían ambos vectores al infectar macrófagos de origen humano y murino. El estudio mostró que la infección de macrófagos humanos por MVA induce altos niveles de IFN-β quimioquinas (IL-8, MIP-1 α, MIP-1, MCP-1, RANTES e IP-10) e IL-1ra, mientras que los niveles de citoquinas pro-inflamatorias (TNF, IL-1 α IL-1β, IL-6 e IL-12 p40) fueron menores. Por su parte, la infección por NYVAC indujo la secreción de niveles bajos de IL-8, IL-1β e IFN-b y no se hallaron niveles detectables de TNF, IL-6, MIP-1α, RANTES o IP-10. El análisis de la producción de quimioquinas independientes de IFN-βen macrófagos aislados de ratones deficientes en receptores TLRs o en las moléculas adaptadoras MyD88 y TRIF revelaron un papel fundamental del complejo heterodimérico TLR2-TLR6 y de la proteína adaptadora MyD88 en la detección tanto de MVA como de NYVAC. Del mismo modo, sólo MDA5 e IPS-1 estuvieron involucrados en la detección intracelular de ambas cepas y en la producción de IFN-β y de las quimioquinas dependientes de IFN-β en macrófagosTanto en MVA como en NYVAC la interacción entre TLR2-MyD88 y el inflamasoma NALP3 fue esencial para la expresión y el procesamiento de la pro-IL-1b en IL-1β madura. La detección de MVA por los diferentes receptores de reconocimiento de patrones descritos (TLR2-TLR-6-MyD88, MDA5-IPS-1 y el inflamasoma NALP3) activó de forma eficiente múltiples vías de señalización intracelular, incluyendo NFkB, ERK-1/2, JNK, IRF3, IRF7 y STAT-1, mientras que la detección de NYVAC indujo una activación muy débil de dichas rutas (Figura 6).
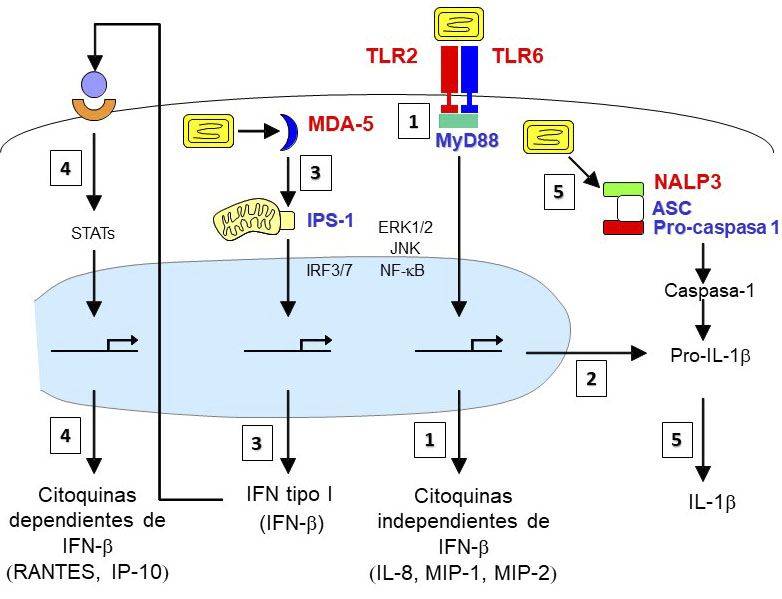
Figura 6. Rutas activadas por MVA en macrófagos. En la superficie celular, el heterodímero TLR2-TLR6 detecta al virus MVA e induce por un lado la producción de quimioquinas independientes de IFNb (IL-8, MIP-1 y MIP-2) (1) y por otro la producción de pro-IL-1b (2). Tras la entrada del virus en la célula, los componentes virales citosólicos (posiblemente ARNm tempranos o ADN viral) activan la vía MDA-5-IPS-1 para liberar IFN-b (3) y la inducción posterior de quimioquinas dependientes de IFN-b(tales como RANTES e IP-10) después de la activación del receptor de IFN tipo I (4). Finalmente, la infección por MVA conduce a la activación del inflamasoma NALP3 (compuesto por NALP3, ASC y pro-caspasa 1) que permite el procesamiento de la caspasa-1, la maduración de la pro-IL-1β y la secreción de IL-1β (5). La partícula viral de MVA aparece indicada en amarillo.
Este trabajo, publicado en la revista “PLoS Pathogens” (15), además de definir los componentes moleculares involucrados en el reconocimiento de las cepas atenuadas MVA y NYVAC, demostró por primera vez la implicación directa de MDA-5 en la detección innata de un virus ADN y la interacción entre las vías TLR y NLR en el contexto de una infección viral.
2.4. Distribución in vivo
Los poxvirus, y en particular el VACV, pueden diseminarse dentro del hospedador mediante: (i) propagación directa de célula a célula utilizando colas de actina; (ii) como virus libre; (iii) leucocitos infectados; y/o (iv) motilidad celular inducida por virus. Se cree que los EVs son particularmente importantes para una rápida diseminación célula a célula in vivo, mientras que los MVs probablemente contribuyan a la propagación a distancia del virus tras la muerte celular o ruptura de la membrana (16).
En contraste con las cepas del VACV competentes en replicación, los virus atenuados MVA y NYVAC no producen progenie viral en la mayoría de las células de mamíferos; sin embargo, como definimos anteriormente, poseen un comportamiento diferencial in vitro que afecta tanto a procesos de replicación y morfogénesis viral como al perfil inflamatorio que desencadenan en la célula hospedadora.
Con estos antecedentes, era importante caracterizar la diseminación in vivo de ambos virus y analizar la cinética de expresión de genes heterólogos codificados por recombinantes basados en ellos en diferentes tejidos. Para abordar este objetivo generamos vectores recombinantes basados en MVA o NYVAC que expresan como marcador reportero la proteína luciferasa. Los virus recombinantes obtenidos fueron inoculados en ratones BALB/c empleando diferentes rutas sistémicas o de mucosas y su capacidad de diseminación fue evaluada mediante imágenes de bioluminiscencia (BLI, “Bioluminescence Imaging”). Esta técnica ofrece la posibilidad de estudiar procesos biológicos en organismos vivos y se basa en la detección de luz visible emitida tras la descarboxilación oxidativa de la luciferina, una reacción que es catalizada por la enzima luciferasa en presencia de ATP y oxígeno (17). Además de monitorizar la expresión de un determinado gen, esta tecnología nos permite cuantificar en un mismo animal la progresión de la infección viral en el espacio y en el tiempo, identificando las variaciones en replicación y diseminación del virus. De hecho, dada la gran utilidad que ha tenido en el campo de la poxvirología, hemos escrito un capítulo dedicado a ella en el libro “Vaccinia Virus. Methods and Protocols” por invitación de su editor, el Dr. Jason Mercer (18).
Este estudio, publicado en la revista “Journal of General Virology”, con mi contribución como primera autora (19), demostró que existen diferencias en el comportamiento in vivo entre las cepas MVA y NYVAC que afectan tanto a los niveles de expresión del gen heterólogo como a su permanencia dentro del animal. En la mayoría de las rutas analizadas la expresión del gen reportero es transitoria, limitándose a las 24 horas post-inoculación en el caso de los animales inmunizados con el recombinante basado en MVA y permaneciendo hasta las 72 horas post-inoculación en el recombinante basado en NYVAC. En ninguno de los órganos analizados (cavidad peritoneal, bazo, nódulos linfáticos y ovarios) se detectaron partículas infecciosas, confirmando la capacidad de replicación restringida de ambos vectores y avalando su perfil de seguridad. Las rutas de administración sistémicas, en particular la ruta intraperitoneal (i.p.) y la intramuscular (i.m.), resultaron más efectivas que las rutas de mucosas. Ambos virus tienen la capacidad de alcanzar e infectar tejidos diana distintos a los del sitio de inoculación; sin embargo, los resultados obtenidos al evaluar la cinética de expresión de luciferasa mediante ensayos bioquímicos indicaron que la eficiencia de expresión génica de MVA es mayor que la de NYVAC a tiempos tempranos post-infección, lo que podría estar relacionado con la susceptibilidad de determinados tipos celulares a la infección por ambas cepas (Figura 7).
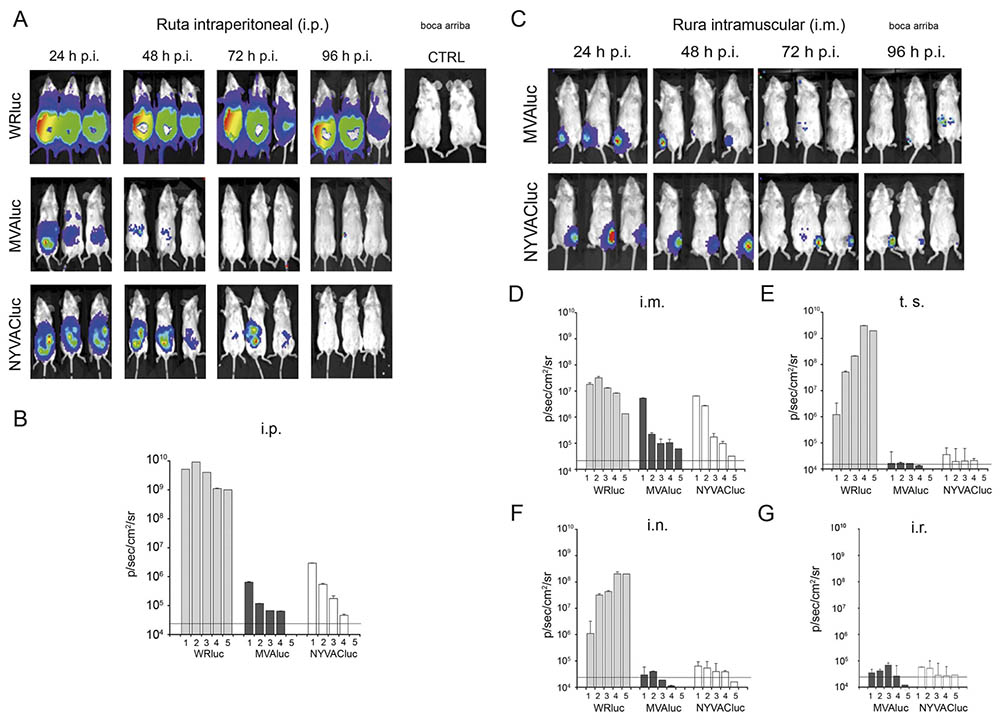
Figura 7: Distribución de los virus WRluc, MVAluc y NYVACluc en ratones inmunizados por diferentes rutas. A: Localización de la señal de luciferasa en ratones inoculados por ruta intraperitoneal (i.p.) a distintos tiempos post-infección. En la esquina superior derecha se muestra la señal procedente de los ratones inoculados con PBS. B: Cuantificación de la emisión de fotones a lo largo del tiempo en la región de interés (RI) tras la inoculación por ruta intraperitoneal. C: Localización de la señal de luciferasa en ratones inoculados por ruta intramuscular (i.m.) a distintos tiempos post-infección. D: Cuantificación de la emisión de fotones a lo largo del tiempo en RI tras la inoculación por ruta intramuscular. E: Cuantificación de la emisión de fotones a lo largo del tiempo en RI tras la inoculación por escarificación en la base de la cola (t.s.). F: Cuantificación de la emisión de fotones a lo largo del tiempo en RI tras la inoculación por ruta intranasal (i.n.). G: Cuantificación de la emisión de fotones a lo largo del tiempo en RI tras la inoculación por ruta intrarectal (i.r.). En las gráficas se representan los valores obtenidos a lo largo del tiempo (días) junto con la desviación estándar. La línea horizontal representa el nivel basal de luminiscencia.
Para extender estos resultados a otro modelo animal realizamos un estudio, como parte de un equipo multidisciplinario liderado por el Dr. Max Corbett, que evaluó la distribución, seguridad e inmunogenicidad de virus recombinantes basados en MVA y NYVAC cuando se administran mediante aerosoles a primates no humanos. Esta ruta de inoculación ofrece ventajas potenciales de seguridad, logística y ahorro de costes sobre las rutas tradicionales de vacunación y representa una alternativa en el contexto de patógenos transmitidos por ruta de mucosas.
Los resultados obtenidos demostraron la viabilidad y el potencial del uso del aerosol para administrar las cepas atenuadas MVA y NYVAC. Las imágenes de gammagrafía in vivo realizadas a tiempo real en las regiones de la cabeza y el tórax de los animales que inhalaron los vectores radiomarcados revelaron que tanto MVA como NYVAC se depositan de forma eficiente en varias regiones de la mucosa, incluyendo los pulmones, los senos paranasales, la boca, la región orofaríngea, el estómago y el duodeno superior. De forma intermitente se detectaron acumulaciones en el esófago y la tráquea, pero en ningún caso se detectó radiactividad en cerebro u ojos. A pesar de que la cantidad total de deposición en la superficie de mucosas fue equivalente para los dos vectores, se observaron diferencias regionales, siendo mayor la deposición de MVA en el tracto respiratorio y la de NYVAC en el tracto gastrointestinal superior. Los animales permanecieron sanos durante todo el período de experimentación y no presentaron ninguna patología cerebral ni cambios histológicos en las regiones de deposición de los vectores asociados a la vacunación, acreditando el perfil de seguridad de ambas cepas atenuadas en primates. A pesar de la baja dosis de vacuna administrada, la absorción eficiente de los antígenos virales por parte de las células que conforman los tejidos de las mucosas garantizó la generación de respuestas inmunes específicas, tanto celulares como humorales, que fueron detectables hasta 5 meses después de la inmunización. Este estudio, publicado en la revista “PNAS” (20), fue el primero que exploró y demostró el perfil de seguridad e inmunogenicidad de los vectores basados en las cepas atenuadas MVA y NYVAC cuando se administran mediante aerosol.
En conjunto, todos estos resultados in vivo confirmaron que tanto las rutas sistémicas como las de mucosas son seguras y efectivas para la administración de recombinantes basados en las cepas atenuadas MVA y NYVAC.
2.5. Inmunogenicidad de recombinantes basados en MVA y NYVAC que expresan antígenos del VIH
Los resultados descritos hasta el momento demuestran que ambas cepas atenuadas poseen un comportamiento diferencial tanto in vitro como in vivo. Para definir si estas diferencias condicionaban la capacidad inmunogénica de ambos vectores, decidimos llevar a cabo un estudio comparativo entre virus recombinantes basados en MVA o NYVAC que expresaban antígenos del VIH.
En este sentido, nuestro laboratorio fue seleccionado para participar en un proyecto cooperativo y multidisciplinario denominado “EuroVacc” (de las siglas en inglés “European Vaccine Effort Against HIV/AIDS”), financiado por la comisión europea de control de enfermedades infecciosas, y cuyo objetivo principal era el desarrollo de candidatos vacunales frente al VIH/SIDA explorando diferentes plataformas. Nuestra aportación se centraba en la generación y caracterización de virus recombinantes basados en la cepa atenuada MVA, así como en la evaluación de su inmunogenicidad en protocolos homólogos o heterólogos que incluían otros vectores desarrollados dentro del consorcio. Todos los vectores producidos dentro del marco del proyecto expresaban los antígenos Env, Gag, Pol y Nef del VIH-1 que incluyen los principales determinantes antigénicos reconocidos durante la infección viral. Los genes sintéticos que codificaban dichos antígenos fueron diseñados y optimizados para eliminar regiones antigénicas no deseadas que pudieran comprometer la seguridad del vector, así como para mejorar su expresión en células humanas. Teniendo en cuenta la diversidad del VIH-1, se propuso que los genes a incluir procedieran de aislados virales de los subtipos B y C. El subtipo B del VIH-1 es predominante en América, Europa occidental, Australia y Japón, y aunque representa sólo el 12% de las infecciones a nivel mundial, es el más utilizado en el desarrollo de prototipos de vacunas. El subtipo C, sin embargo, es el más abundante a nivel global, representando más del 50% de las infecciones, y predomina especialmente en el África subsahariana, India, China y Nepal (21).
El primer virus recombinante que generamos en el laboratorio fue el denominado MVA-B. Este virus tiene insertado dentro del mismo locus viral (TK, timidina quinasa) los genes que codifican la proteína de la envuelta gp120, procedente del aislado primario BX08, y la poliproteína de fusión Gag-Pol-Nef (GPN), procedente del aislado IIIB, ambos pertenecientes al subtipo B. Los genes env y gpn se encuentran en posición opuesta bajo el control transcripcional del promotor viral sintético temprano-tardío pE/L. Esta misma aproximación se empleó para la generación del virus recombinante MVA-C, que expresa los mismos antígenos del VIH-1 pero procedentes del aislado asiático 97CN54 perteneciente al subtipo C. Por razones de seguridad y teniendo en cuenta su posible aplicación clínica, ambos recombinantes se generaron utilizando un plásmido de transferencia, diseñado por nosotros, que contiene un gen marcador de selección flanqueado por 2 repeticiones de la región izquierda del locus TK, de manera que tras los primeros pases de purificación este gen marcador se pierde mediante un proceso de recombinación homóloga entre ambas repeticiones. Los virus recombinantes MVA-B y MVA-C son equivalentes a los generados por la empresa Sanofi-Aventis sobre la cepa parental NYVAC y que han sido referidos en los trabajos publicados como NYVAC-B y NYVAC-C. Tanto los vectores del subtipo B (MVA-B y NYVAC-B) como los del subtipo C (MVA-C y NYVAC-C) expresan la proteína gp120 monomérica como un producto que se libera al sobrenadante y GPN como una poliproteína de fusión intracelular. Cuando se caracterizaron en células en cultivo observamos que todos ellos (i) habían incorporado en su genoma los genes heterólogos sin ninguna modificación genética y no presentaban contaminación con la cepa parental; (ii) presentaban cinéticas de expresión de los antígenos heterólogos similares; (iii) eran capaces de replicar eficientemente en cultivos primarios de CEF, obteniéndose altos rendimientos virales; y (iv) eran muy estables, conservando la integridad del inserto tras someterse a pases sucesivos en cultivos celulares. En la Figura 8 se muestra un ejemplo representativo de la caracterización in vitro de los virus recombinantes del subtipo B.
Figura 8: Caracterización in vitro de MVA-B y NYVAC-B. A: Representación esquemática del locus TK de los virus recombinantes MVA-B y NYVAC-B donde se encuentran insertados los genes que codifican para las proteínas gp120BX08 y GPN del VIH-1 bajo el control transcripcional del promotor viral sintético temprano-tardío (sE/L). B: Análisis por PCR del locus viral TK. C: Cinética de expresión de las proteínas gp120BX08 y GPN en células infectadas con MVA-B o NYVAC-B analizada por western-blot. D: Cinética de crecimiento de los virus MVA-B y NYVAC-B en cultivos primarios de CEF. La gráfica de la izquierda representa el virus liberado al sobrenadante y la de la derecha el virus que permanece asociado a las células. E: Expresión de las proteínas gp120BX08 y GPN tras pases sucesivos de MVA-B en cultivos primarios de CEF.
Cabe destacar que la inserción de los antígenos del VIH-1 en el genoma viral no modifica las características intrínsecas de sus respectivos vectores parentales, como la inducción de apoptosis o la inhibición de la síntesis de proteínas a tiempos tardíos descritas para la cepa NYVAC.
A pesar de que los poxvirus han demostrado ser unos excelentes inmunógenos, la experiencia obtenida tanto en los ensayos preclínicos como en los ensayos clínicos indica que su utilización en sistemas combinados de inmunización, preferentemente en la dosis de refuerzo, representa la vía más efectiva para lograr una potenciación de la respuesta inmune específica (22). En este sentido, nuestro laboratorio ha sido pionero en el desarrollo y optimización del protocolo de inmunización denominado “prime-boost” (inmunización-refuerzo) empleando vectores basados en poxvirus en el “boost”. Dicho protocolo ha logrado inducir respuestas inmunes celulares que correlacionaron con protección en el modelo de malaria (23).
Para evaluar de forma comparativa la inmunogenicidad inducida por los vectores generados utilizamos diferentes protocolos de inmunización, tanto homólogos como heterólogos, utilizando como “prime” vectores de ADN o recombinantes basados en el virus del bosque Semliki (SFV), seguido de un “boost” con los vectores basados en MVA o NYVAC, todos expresando los mismos antígenos. Empleamos dos modelos de ratón, la cepa BALB/c y los ratones transgénicos humanizados HHD, que solamente disponen de la quimera humana HLA-A2.1 como molécula de histocompatibilidad de clase I. De modo general, observamos que tanto los vectores basados en MVA (MVA-B y MVA-C) como los basados en NYVAC (NYVAC-B y NYVAC-C) eran capaces de inducir una potente respuesta inmune celular específica frente a los antígenos Env, Gag, Pol y Nef del VIH-1, detectada por la técnica de ELISPOT, tanto en ratones BALB/c como en ratones humanizados HHD. Sin embargo, detectamos diferencias en la magnitud y la amplitud de la respuesta generada que dependían del protocolo de inmunización empleado en cada modelo. En protocolos de inmunización homólogos, los recombinantes basados en MVA estimularon una respuesta celular VIH-1-específica superior a la inducida por los recombinantes basados en NYVAC, mientras que la respuesta específica frente al vector presentaba un comportamiento contrario (NYVAC > MVA). Las combinaciones heterólogas (ADN/Pox o SFV/Pox) demostraron ser más inmunogénicas que el protocolo homólogo (Pox/Pox). En ratones BALB/c, los recombinantes basados en NYVAC expandieron de forma más efectiva los clones de células T específicos frente al VIH-1 que habían sido inicialmente estimulados por los vectores de ADN o SFV administrados en el “prime”, obteniéndose una magnitud de la respuesta de células T superior a la inducida por los recombinantes basados en MVA. En este modelo animal, la respuesta celular específica se distribuyó de forma equitativa entre los 4 antígenos del VIH-1 expresados por los diferentes candidatos vacunales. Sin embargo, en ratones transgénicos HHD la magnitud de la respuesta celular VIH-1-específica inducida por ambos vectores fue similar y estuvo dirigida esencialmente frente a los grupos de péptidos representativos del antígeno de la envuelta (Env). El patrón de citoquinas detectado en los sobrenadantes de los cultivos celulares de linfocitos del bazo tras la re-estimulación con las diferentes mezclas de péptidos del VIH-1 demostró una clara polarización de la respuesta celular hacia un fenotipo Th1, independientemente del protocolo o del modelo animal empleado. Además de la respuesta celular inducida, tanto los recombinantes basados en MVA como los basados en NYVAC estimularon la producción de anticuerpos de unión específicos frente al antígeno de la envuelta del VIH-1.
Estos resultados fueron los primeros que describieron comparativamente el potencial inmunogénico de los vectores basados en MVA y NYVAC in vivo. El trabajo fue publicado en dos artículos originales, con mi contribución como primera autora, en la revista “Vaccine” (24, 25).
A pesar de que en ambas cepas de ratones obtuvimos resultados prometedores, el modelo murino no permitía evaluar la eficacia de estos recombinantes a la hora de conferir protección frente a la infección por el VIH. Debido a esto, y en colaboración con el grupo del Dr. Jonathan Heeney en el marco del proyecto EuroVacc, decidimos realizar un estudio en primates no humanos para evaluar la inmunogenicidad de los recombinantes basados en MVA y NYVAC, así como determinar su capacidad para conferir protección frente a un desafío intravenoso con la cepa altamente patogénica SHIV89.6p, un virus híbrido entre el VIH-1 y el virus de la inmunodeficiencia de simio (VIS) denominado SHIV (del inglés “Simian-HumanImmunodeficiency Virus) capaz no sólo de infectar macacos sino de producir un síndrome similar al SIDA. Este virus híbrido contiene los genes env, tat, vpu y rev del aislado primario 89.6 del VIH-1 y los genes restantes del VISmac239 (26) y, a través de pases sucesivos en macacos y en cultivo, se ha adaptado a multiplicarse y provocar la depleción de los linfocitos CD4 y la muerte de los macacos en menos de un año después de la inoculación.
Para poder llevar a cabo el estudio generamos recombinantes basados en MVA y NYVAC, equivalentes a MVA-B y NYVAC-B, pero que expresan la proteína monomérica gp120 del virus SHIV89.6p y la poliproteína de fusión GPN procedente del virus VISmac239. Ambos vectores fueron caracterizados in vitro, confirmándose un comportamiento similar al observado con los recombinantes que expresan los antígenos del VIH-1 de los subtipos B y C. Una vez generados dichos recombinantes, los macacos fueron inmunizados utilizando el protocolo heterólogo ADN/Pox. Este estudio reveló que tanto el recombinante basado en MVA como el basado en NYVAC eran capaces de conferir una protección similar frente a la infección con el virus altamente patogénico SHIV89.6p (Figura 9A). Tras el desafío, todos los animales resultaron infectados; sin embargo, los animales inmunizados con los recombinantes MVA y NYVAC mantuvieron niveles estables de células T CD4 durante un periodo de tiempo superior a un año y fueron capaces de controlar la viremia, manteniendo la carga viral o disminuyéndola hasta 2 logaritmos por debajo de la detectada en los animales del grupo control durante todo el periodo de análisis (Figura 9B). La protección observada se correlacionó con la inducción de una respuesta de células T específicas frente a los diferentes antígenos ya que los niveles de anticuerpos neutralizantes antes del desafío fueron insuficientes para prevenir la infección de los animales inmunizados. En ambos grupos, la respuesta de células T específica inducida antes del desafío estaba dirigida fundamentalmente frente al antígeno de la envuelta, como ocurría en los ratones transgénicos HHD, mientras que después del desafío se dirigió mayoritariamente frente a los antígenos Gag-Pol-Nef. A pesar de que mediante la técnica de ELISPOT no se observaron diferencias en la magnitud de la respuesta de células T específicas entre ambos vectores, el análisis por citometría de flujo reveló que la respuesta celular específica inducida por MVA estaba mediada por linfocitos T CD8+, mientras que la inducida por NYVAC estaba mediada por linfocitos T CD4+ (Figura 9C). Esta activación diferencial de células T CD4 o CD8 por parte de los vectores se mantuvo tras el desafío y durante todo el período que duró el experimento. Ambos subtipos de células T activadas presentaban un fenotipo polifuncional y capacidad proliferativa.

Figura 9: Eficacia de los recombinantes basados en MVA y NYVAC frente a la infección por el virus híbrido SHIV89.6P en primates no humanos. A: Porcentaje de supervivencia después del desafío. B: Niveles de carga viral (arriba) y número absoluto de células T CD4 (abajo) en los animales individuales de cada grupo de inmunización. C: Contribución de las poblaciones de células T CD4+ y T CD8+ en la respuesta específica frente al VIH-1 antes del desafío (izquierda) y después del desafío (derecha).
Estos resultados demostraron por primera vez que recombinantes basados en MVA y NYVAC eran capaces de conferir una protección similar frente a la infección por SHIV en primates no humanos a través de la potenciación de respuestas celulares diferentes: en el caso de MVA mediada por células T CD8+ y en el caso de NYVAC mediada por células T CD4+. Este estudio, publicado en la revista “J. Virol.” (27), permitió la traslación de los vectores basados en las cepas atenuadas MVA y NYVAC generados en el marco del proyecto EuroVacc de la evaluación preclínica a la fase clínica.
Los vectores MVA-B y MVA-C fueron objeto de la patente europea “Vectores recombinantes basados en el virus modificado de Ankara (MVA) como vacunas preventivas y terapéuticas contra el SIDA” (EP1921146B1), cuya entidad titular de derechos es el Consejo Superior de Investigaciones Científicas (CSIC), y que fue transferida a la compañía farmacéutica Laboratorios Esteve S.A.
2.6. Optimización de vectores recombinantes basados en MVA y NYVAC
La capacidad restringida de replicación de las cepas atenuadas MVA y NYVAC en diferentes líneas celulares de mamífero y en células humanas sin duda confiere a estos vectores un perfil de seguridad óptimo para su utilización en clínica, pero también representa una limitación si consideramos que la cantidad de antígeno que se expresa está limitada al tiempo que permanece el virus en el organismo (48-72h). Sin embargo, a pesar de esta limitación, ambos vectores han demostrado ser inmunogénicos en los diferentes modelos animales ensayados, debido, en gran medida, a la respuesta inflamatoria que inducen en las células infectadas y que, a su vez, está correlacionada con las deleciones en genes inmunomoduladores que han sufrido ambos genomas virales durante su generación.
Como parte de un consorcio internacional financiado por la Fundación Bill & Melinda Gates con el fin de desarrollar una vacuna contra el VIH/SIDA basado en la optimización de los vectores de poxvirus MVA y NYVAC, nuestro grupo se dedicó a explorar nuevas estrategias que permitieran mejorar el perfil inmunogénico de ambas cepas. Para lograr este propósito nos plantemos abordar diferentes aproximaciones que incluían: (i) eliminar genes inmunomoduladores virales todavía presentes en el genoma viral; (ii) mejorar la capacidad replicativa del vector; (iii) utilizar moléculas coestimuladoras; (iv) mejorar la fortaleza del promotor viral; y (v) optimizar los insertos. Las diferentes estrategias se ensayaron sobre los recombinantes de MVA y NYVAC que expresan los antígenos del VIH-1 de los subtipos B y C, administrados utilizando el protocolo heterólogo ADN/Pox.
2.6.a. Deleción selectiva de genes inmunomoduladores
Como mencionamos con anterioridad, las cepas atenuadas MVA y NYVAC exhibían comportamientos distintos tanto in vitro como in vivo debido a las diferencias que existen en sus genomas como resultado del proceso de generación al que fueron sometidas. La infección con la cepa MVA induce un perfil inflamatorio mucho más agudo que NYVAC, lo que garantiza el reclutamiento de mediadores inmunológicos que son capaces de activar una respuesta innata efectiva que clarifica de forma eficiente el virus poco después de su entrada en el organismo. Esta activación se produce debido a la cantidad de genes inmunomoduladores que aparecen delecionados o mutados en su genoma, que son mayores que los que se encuentran delecionados en la cepa NYVAC, y que actúan en rutas de señalización claves en la evasión de la respuesta antiviral. Sin embargo, ambos virus conservan intactos muchos otros genes con funciones similares. Con estos antecedentes, decidimos llevar a cabo deleciones selectivas de genes que estuvieran implicados en una misma ruta de señalización, o incluso en rutas diferentes. El hecho de que NYVAC conserve un número mayor de genes relacionados con la evasión de la respuesta antiviral nos permitía un margen más amplio de experimentación para evaluar el efecto sobre la respuesta inmune específica de una deleción individual o de múltiples combinaciones.
De forma general, la estrategia de deleción selectiva de genes inmunomoduladores produjo un aumento de la inmunogenicidad inducida por los vectores generados. Por ejemplo, la deleción de los genes B19R y/o B8R que codifican receptores solubles de IFN tipo I y tipo II, respectivamente, mejoró la magnitud y la calidad de la respuesta de células T CD8 específicas frente al VIH-1 durante las fases adaptativa y de memoria en el modelo murino (28). Asimismo, el análisis transcriptómico realizado en monocitos humanos infectados con estos mutantes demostró que ambas deleciones activan la expresión de los factores IRF3, IRF7 y STAT1 y estimulan la producción de altos niveles de IFN tipo I y de genes estimulados por IFNs (ISGs); sin embargo, sólo la doble deleción B8R/B19R logró inducir la máxima respuesta inflamatoria (29). La combinación de deleciones en genes relacionados con la ruta de señalización del IFN (B19R, B8R) junto con regiones genómicas (B6R-B10R) que incluyen genes que codifican el receptor soluble de TNF-a (B7) o proteínas con funciones desconocidas (B6, B9 y B10) no logró mejorar la inmunogenicidad inducida por el doble mutante de deleción ΔB19R/B8R (30). Un efecto similar se observó cuando delecionamos genes que bloquean la ruta de activación del factor de transcripción NFκB a diferentes niveles. La deleción individual del gen A46R, que actúa a un nivel superior en la ruta de señalización de los TLRs interfiriendo con la interacción TLR-molécula adaptadora, aumenta la secreción de las citoquinas TNF, IL-6 e IL-8 en macrófagos humanos in vitro y mejora la magnitud de la respuesta inmune de las células T CD4 y CD8 específicas del VIH-1 in vivo (31); sin embargo, cuando realizamos deleciones secuenciales sobre este mutante de otros genes virales siguiendo el nivel en el que actúan bloqueando la ruta de activación de NFκB (A52R-K7R-B15), observamos que sólo la doble deleción ΔA46R/A52R logró inducir un beneficio inmunológico con respecto al mutante simple (30). Curiosamente, cuando abordamos la estrategia de deleción de esta ruta manteniendo intacto el gen A46R pero delecionando A52R-K7R-B15R, descubrimos que sólo la deleción triple era capaz de restaurar la función de NFκB en macrófagos. De este modo, la infección con NYVAC-C Δ3 activa la vía de señalización de NFκB produciendo un aumento en la expresión de citoquinas/quimioquinas (IL-6, MCP-1, MIP-1β, KC) que incrementa la migración de neutrófilos (Na y Nβ) al sitio de inoculación y su activación. Una vez activados, la infección con NYVAC-C Δ3 promueve una mayor migración de neutrófilos, preferentemente de los neutrófilos Nβ, a los órganos linfoides secundarios. Estos neutrófilos Nβ sobre-expresan en su superficie marcadores específicos de células presentadoras de antígeno que les permiten activar a las células T CD8 específicas de forma directa (Figura 10).

Figura 10: Mecanismo de activación de neutrófilos y su implicación en la activación de células T CD8+ tras la infección por NYVAC-C o NYVAC-C Δ3.
El aumento en el tráfico de neutrófilos al sitio de infección correlacionó con una mayor respuesta de células T específicas frente al VIH-1. Estos resultados, publicados en la revista “PNAS”, identificaron por primera vez a los neutrófilos como un mediador celular involucrado en la respuesta inmune inducida por VACV, un aspecto novedoso que sin duda resulta de gran importancia de cara a la utilización de estos vectores como candidatos vacunales (32).
La deleción selectiva de genes inmunomoduladores todavía presentes en el genoma de MVA también demostró tener un beneficio inmunológico. Por ejemplo, la simple deleción de genes que codifican inhibidores de la vía de señalización de IFN tipo I (C6L) (33), apoptosis (F1L) (34), proteína de unión a IL-18 (C12L) (35) o un inhibidor de IRF-3 (N2L) (36), indujo un aumento en la magnitud, polifuncionalidad y durabilidad de las células T CD4 y CD8 específicas frente al VIH-1. Estos resultados se potenciaron aún más con los dobles mutantes de deleción ΔA41L/B16R (37) o ΔC6L/K7R (38).
Los datos de inmunogenicidad derivados de la deleción del gen C6L en el genoma de MVA formaron parte de la patente “Vectores recombinantes basados en el virus modificado de Ankara (MVA) con deleciones en el gen C6L como vacunas frente al VIH/SIDA y otras enfermedades” (ES2401904B1) cuya entidad titular de derechos es el Consejo Superior de Investigaciones Científicas (CSIC).Estos estudios en su conjunto demostraron que la eliminación selectiva de genes inmunomoduladores representa una estrategia adecuada para mejorar la inmunogenicidad de recombinantes basados en las cepas MVA y NYVAC. Sin embargo, es necesario llevar a cabo un diseño racional de la misma ya que dependiendo de la combinación de deleciones que se realice podemos conseguir una modulación positiva o negativa de la respuesta inmune específica. Tal y como hemos corroborado en nuestros estudios, un mayor número de deleciones no conlleva necesariamente un aumento en la inmunogenicidad.
2.6.b. Aumento de la capacidad replicativa del vector
Esta estrategia consiste en la generación de nuevos vectores basados en poxvirus que han recuperado la capacidad de replicación en células humanas, aumentando así el tiempo y el nivel de expresión del antígeno heterólogo en el hospedador, pero sin perder el fenotipo atenuado.
Como habíamos observado con anterioridad, la re-inserción del gen de rango de hospedador C7L en el genoma de NYVAC confirió al virus la capacidad de replicar en células humanas y murinas, manteniendo un fenotipo atenuado in vivo. Cuando re-insertamos el gen C7L en el genoma del virus recombinante NYVAC-B observamos un aumento relevante en los niveles de expresión del antígeno heterólogo in vitro que correlacionó con un incremento significativo en la magnitud y amplitud de la respuesta de células T específicas frente al VIH-1 in vivo (39).
Igualmente, demostramos que la reinserción simultánea de C7L y K1L, otro gen de rango de hospedador, en el genoma de NYVAC-C, o la combinación de las estrategias de restauración de la competencia de replicación con la deleción de genes inmunomoduladores (B19R), confieren al vector una serie de características biológicas in vitro entre las que podemos destacar: mayores niveles de expresión del antígeno heterólogo, competencia de replicación en queratinocitos humanos y fibroblastos dérmicos, inducción de la expresión de IFN-y genes inducidos por IFN y activación de genes implicados en el procesamiento y presentación de antígenos. Esto correlaciona con una mejora significativa en la presentación cruzada del antígeno por parte de las células diana infectadas a las células T CD8 específicas del VIH, así como una mayor proliferación de los clones de memoria. Además, estos vectores poseen una propagación limitada en tejidos y mantienen un fenotipo altamente atenuado in vivo (40, 41).
2.6.c. Uso de moléculas coestimuladoras
El objetivo de esta estrategia consiste en mejorar la inmunogenicidad de los vectores atenuados mediante el suministro simultáneo de una molécula estimuladora junto con el antígeno heterólogo. En nuestro caso, esta aproximación fue abordada mediante la utilización de la forma hexamérica soluble de CD40L (sCD40L). Nuestro estudio reveló que la administración de sCD40L era necesaria tanto en el “prime” junto con el vector de ADN como en el “boost” junto con MVA-B o NYVAC-B para lograr un aumento en la magnitud y amplitud de la respuesta de células T específicas frente a los antígenos del VIH-1. La administración de sCD40L un día después de cada inmunógeno también resultó efectiva, pero no representó una mejora en comparación con la administración conjunta. Este trabajo puso de manifiesto el beneficio inmunoestimulador de la molécula sCD40L (42).
2.6.d. Mejora de la fortaleza del promotor viral
Todos los virus recombinantes descritos en este trabajo contienen los genes heterólogos insertados bajo el control transcripcional de un promotor viral sintético temprano-tardío que garantiza su expresión durante ambas fases de la transcripción. Sin embargo, es posible mejorar el promotor viral mediante el intercambio de los elementos centrales para lograr maximizar la expresión del antígeno recombinante a tiempos muy tempranos después de la infección. En este sentido, se ha descrito que la eficiencia con la que se procesa y presenta un antígeno en la superficie de las células infectadas influye en su reconocimiento y en la potenciación de la respuesta inmune específica generada (43).
A partir de las secuencias promotoras (200 nt aguas arriba del codón de inicio) de 37 genes clasificados como genes inmediatos-tempranos mediante el análisis transcriptómico de VACV, se realizó un análisis bioinformático que definió el elemento central o secuencia consenso temprana que compartían estos genes, así como la distancia existente entre este núcleo de secuencia y el codón de iniciación. Una vez definidos ambos elementos, se diseñó un nuevo promotor sintético tardío-temprano optimizado al que denominamos promotor LEO (“Late Early Optimized”) que, con respecto al promotor sintético tradicional, compartía el núcleo de la parte tardía, pero intercambiado de posición, seguido de la nueva secuencia consenso temprana definida y de un espaciador de 38 nucleótidos. El nuevo promotor se incorporó en un plásmido de transferencia controlando la transcripción del gen reportero GFP (proteína verde fluorescente) y su fortaleza se estudió en el contexto del recombinante basado en MVA generado (MVA-LEO-GFP). En comparación con el promotor tradicional, LEO mejoró significativamente los niveles de expresión de GFP en la primera hora después de la infección, lo que se correlacionó con una mejora en la respuesta inmune de células T CD8+ GFP-específicas (44). Un alargamiento de la región espaciadora de 38 a 160 nucleótidos (LEO160) mejoró la expresión temprana y la respuesta de células T CD8+ y CD4+ específicas de memoria frente al antígeno LACK de Leishmania (45).
2.6.e. Optimización del inserto
Además de las modificaciones del vector que acabamos de describir, otro enfoque que hemos explorado con el objetivo de mejorar la inmunogenicidad de los recombinantes basados en poxvirus ha sido la optimización del antígeno heterólogo. Una de las estrategias que hemos ensayado para superar el desafío que supone la diversidad global del VIH-1 ha sido la utilización de secuencias consenso o mosaicos de la envuelta (Env) en lugar de secuencias derivadas de un determinado aislado viral. Los mosaicos son pequeños conjuntos de antígenos vacunales diseñados computacionalmente, utilizando un algoritmo genético, para aumentar la presentación de los epítopos potenciales de células T (PTE) presentes en las proteínas naturales. Una vacuna de mosaico es un «cóctel» que contiene varias secuencias complementarias que combinadas proporcionan una cobertura casi óptima de los PTE que circulan en una determinada población diana.
Cuando estos antígenos se ensayaron de forma comparativa en primates no humanos utilizando el protocolo heterólogo ADN/Pox/proteína se observó que tanto los inmunógenos de consenso como los mosaicos indujeron respuestas celulares y humorales específicas y cross-reactivas significativamente mayores que las potenciadas por el inmunógeno clásico derivado de un aislado viral (46).
También ensayamos nuevos formatos de antígenos Env y GPN optimizados que diferían de los que expresan los vectores recombinantes MVA y NYVAC de los subtipos B y C, en los que la proteína Env gp120 se secreta en forma monomérica y GPN se produce como una poliproteína de fusión intracelular. Los nuevos recombinantes generados expresaban la proteína Env gp140 que se secretaba en una conformación trimérica más parecida a la conformación nativa que adopta en el virus (NYVAC-gp140) o la proteína GPN que fue optimizada para sufrir procesamiento proteolítico y liberar partículas como virus (VLP) inducidas por Gag (NYVAC-Gag-Pol-Nef). Ambos vectores estimulan respuestas innatas específicas en células humanas superiores a su antecesor (NYVAC-C) y presentan un fenotipo atenuado tras su inoculación intracraneal en ratones BALB/c adultos inmunocomprometidos o en ratones CD1 recién nacidos. En protocolo homólogo Pox/Pox, ambos virus recombinantes indujeron respuestas de células T CD4 polifuncionales específicas frente a Env o CD8 específicas frente a Gag (47). Estos vectores también se han ensayado en combinación con vectores de ADN que expresan los mismos antígenos y con la proteína gp120 monomérica en un ensayo clínico fase 1b que involucró a 96 voluntarios sanos con bajo riesgo de infección. Los resultados, publicados recientemente, demostraron la seguridad y tolerabilidad de los vectores y revelaron que la administración conjunta de la proteína Env gp120 con los vectores de ADN o NYVAC durante el “priming” induce una respuesta temprana y potente de anticuerpos IgG de unión frente a los lazos variables V1/V2 de la proteína de la envuelta del VIH-1 sin afectar la respuesta celular inducida por los diferentes regímenes de inmunización. Los autores de dicho trabajo proponen considerar este enfoque para la inducción de anticuerpos preventivos en futuros ensayos de eficacia (48).
3. MVA-B como candidato vacunal frente al VIH-1
Las cepas atenuadas de poxvirus MVA y NYVAC representan una plataforma formidable para el desarrollo de candidatos vacunales frente a un amplio abanico de enfermedades infecciosas, parasitarias e incluso en cáncer, como ha quedado demostrado en los diferentes ensayos preclínicos y clínicos que se han llevado a cabo (49-51). Sin embargo, como hemos descrito en la primera parte de esta revisión, ambas cepas poseen características intrínsecas propias que determinan comportamientos diferenciales tanto in vitro como in vivo. Considerando que el perfil inflamatorio que induce MVA es superior al inducido por NYVAC, lo que garantiza una respuesta innata más potente que va a determinar la calidad de la respuesta adaptativa que se desarrolle contra al patógeno y teniendo en cuenta que produce una activación preferencial de células T CD8, decidimos apostar por ella para dar el salto a la clínica y evaluarla como candidato vacunal frente al VIH/SIDA.
En esta segunda parte del trabajo haremos una revisión de las estadísticas de la infección por VIH/SIDA a nivel global y en España, detallaremos los principales retos a los que nos enfrentamos a la hora de desarrollar candidatos vacunales frente al VIH/SIDA y discutiremos los principales resultados que hemos obtenido de los ensayos clínicos fase I realizados en España con el vector MVA-B, generado por nosotros, como vacuna profiláctica en pacientes sanos y como vacuna terapéutica en pacientes infectados por VIH-1.
Indudablemente la investigación clínica genera el conocimiento que permite mejorar las intervenciones futuras para incidir de forma más efectiva en problemas que afectan a la salud pública. Sin embargo, abordar proyectos de ensayos clínicos requiere de un fuerte apoyo económico y del trabajo coordinado de grupos multidisciplinares que garanticen el bienestar y la seguridad de los voluntarios que se presentan al estudio y la solidez de los resultados que se derivan de ellos. Gracias al soporte económico de entidades públicas y privadas como la Red Española de Investigación en SIDA (RIS), el Fondo de Investigaciones Sanitarias (FIS), la Fundación para la Investigación y Prevención del SIDA (FIPSE) y la Fundación Marcelino Botín se pudieron abordar en España los ensayos clínicos RISVAC02 y RISVAC03 que detallaremos más adelante. Los resultados de estos estudios han tenido gran repercusión mediática en nuestro país y, aunque sólo se han realizado en fase I como estudios exploratorios de seguridad, tolerabilidad e inmunogenicidad en un número reducido de pacientes, han marcado las pautas y el camino a seguir en ensayos futuros.
3.1. La infección por VIH/SIDA en cifras
El virus de la inmunodeficiencia humana (VIH), identificado hace ya casi 40 años como agente causal del Síndrome de la Inmunodeficiencia Adquirida (SIDA), continúa siendo letal para la humanidad, acumulando ya más de 39 millones de muertes en todo el mundo. Las estadísticas reportadas por la Organización Mundial de la Salud (OMS) estiman que hoy en día un total de 37.9 millones de personas conviven con la infección a nivel mundial.
La introducción de la terapia antirretroviral (TAR) en el tratamiento de la infección por VIH ha constituido uno de los éxitos más espectaculares en medicina en el siglo pasado, transformando la infección por VIH de una sentencia de muerte a una enfermedad crónica manejable. A nivel mundial ha tenido lugar una reducción en la morbilidad y mortalidad relacionadas con el VIH y en la actualidad un total de 23.3 millones de personas tienen acceso al tratamiento, lo que representa el 62% de los infectados. Sin embargo, a pesar de estos grandes avances, alrededor de un millón de personas mueren de enfermedades relacionadas con el VIH cada año y se producen 1,7 millones de nuevas infecciones (www.unaids.org).
En España la infección por VIH mantiene su expansión a pesar de todos los esfuerzos destinados para frenarla. Según los datos del último informe sobre vigilancia epidemiológica del VIH/SIDA (2017), entre 140.000 y 170.000 personas viven con VIH y se producen 3381 nuevas infecciones al año, dejando en evidencia que los proyectos en materia de prevención no están teniendo el efecto deseado en la sociedad. La mayoría de nuevos casos se producen en hombres (84,6%) y la media de edad se sitúa en los 35 años. Además, tanto en Europa como en España, 1 de cada 5 personas infectadas desconocen que lo están. Aparte del efecto deletéreo que tiene este desconocimiento para los enfermos, su impacto sobre la salud pública es demoledor. Se ha estimado que el 20% de infectados sin diagnosticar es responsable del 60% de las nuevas infecciones. Por otra parte, el diagnóstico tardío de la infección conlleva el deterioro de la inmunidad del paciente encareciendo enormemente su tratamiento.
El programa conjunto de las naciones unidas sobre VIH/SIDA (ONUSIDA) se ha planteado como meta para el año 2020 el objetivo 90-90-90 para controlar y erradicar el virus. Este objetivo persigue que el 90% de las personas con VIH estén diagnosticadas, que el 90% de las personas diagnosticadas reciban tratamiento y que el 90% de las que reciban tratamiento presenten una carga viral indetectable. A finales del año 2018 las cifras globales eran 79-62-53 (79% diagnosticados, 62% con acceso al tratamiento y 53% con carga viral indetectable) lo que nos hace pensar que alcanzar la meta propuesta va a resultar difícil; no obstante, es necesario seguir aunando esfuerzos para erradicar esta enfermedad.
En España, la prevención y el diagnóstico precoz son los principales ‘talones de Aquiles’ para alcanzar el objetivo 90-90-90 establecido por ONUSIDA. A pesar de que nuestras cifras son más esperanzadoras que las globales (82-92-88) aún queda camino por recorrer.
Conseguir este objetivo en materia de tratamiento implica una reducción en más del 80% del número de muertes y nuevas infecciones a largo plazo. Sin embargo, es necesario contemplar otras intervenciones que puedan implementarse para contrarrestar los efectos adversos asociados con una terapia antirretrovial prolongada y el elevado coste que dicha terapia representa.
En ausencia de vacunas, poner fin a la pandemia del VIH/SIDA tendrá un coste sin precedentes. En el año 2015, el coste global destinado a la atención del VIH/SIDA fue de 49 billones de dólares. En el año 2018, ONUSIDA informó que se invirtieron 21.3 billones de dólares en países en desarrollo para luchar contra el VIH/SIDA, datos que se sitúan un 21% por debajo de los 26.2 billones de dólares que se estima serán necesarios para dar “una respuesta más eficaz” en el año 2020. Sin embargo, existen estudios de modelajes matemáticos que estiman que la introducción de una vacuna parcialmente eficaz, pero con una alta cobertura, podría reducir sustancialmente el número de nuevas infecciones por VIH (52). Se plantea que la implementación de una estrategia integral que combine una vacuna parcialmente efectiva con el tratamiento como prevención junto a la profilaxis previa a la exposición podría representar una alternativa eficaz y mucho más rentable, considerando incluso un coste de $105 por serie de 5 dosis (53).
3.2. Retos en el desarrollo de vacunas frente al VIH/SIDA
Las vacunas son consideradas una de las intervenciones de salud pública más rentables, pero el desarrollo de una vacuna eficaz contra el VIH/SIDA ha demostrado ser uno de los desafíos científicos más complejos debido a la naturaleza del virus y su interacción con el sistema inmunitario humano. Ninguna persona se ha recuperado naturalmente de una infección por el VIH, y hasta la fecha se desconocen los correlatos que indiquen qué respuestas inmunitarias se necesitan para bloquear o eliminar el virus. Además, existen otros muchos obstáculos que, aunque a lo largo de los años se han ido esclareciendo en cierta medida gracias a las diferentes investigaciones en el tema, continúan siendo retos que dificultan el desarrollo de una vacuna. Entre estos obstáculos podemos destacar la elevada variabilidad genética del VIH, el todavía escaso conocimiento sobre la patogénesis de la infección, la alta tasa de mutación que permite al virus evadir rápidamente la respuesta del hospedador, la infección latente que produce en células poco accesible al sistema inmune y donde crea sus reservorios naturales, la destrucción de células dianas del sistema inmune como consecuencia de la infección, el enmascaramiento de epítopos que están directamente implicados en la entrada viral y la ausencia de un modelo animal que reproduzca la infección que ocurre en los humanos.
Además de los problemas meramente científicos, el avance en el desarrollo de vacunas frente al VIH-1 también se enfrenta a problemas logísticos. Existe todavía una escasa coordinación internacional, una inversión insuficiente (cerca del 1% del esfuerzo global), un interés marginal por parte de la industria farmacéutica cuya prioridad se centra en las terapias antirretrovirales y la percepción de que el problema del VIH/SIDA se puede resolver con las intervenciones existentes (circuncisión, microbicidas, profilaxis pre-exposición, tratamiento como prevención) sin necesidad de una vacuna.
No obstante, y a pesar de estos obstáculos, se han hecho y se siguen haciendo grandes progresos en el desarrollo de candidatos y plataformas vacunales destinados a prevenir y tratar la infección.
El único ensayo clínico en fase III que ha demostrado hasta el momento algún grado de eficacia frente a la infección por VIH-1 fue el denominado RV144, llevado a cabo en Tailandia entre los años 2003-2009 y en el que se involucraron más de 16000 voluntarios heterosexuales sanos con riesgo moderado de infección por VIH. El estudio, supervisado y financiado por el Ministerio Tailandés de Salud Pública, el Instituto estadounidense de Alergias y Enfermedades Infecciosas (NIAID) y el Ejército de los Estados Unidos, evaluó la combinación de un poxvirus recombinante basado en la cepa ALVAC, denominado vCP1521, y la proteína AIDSVAX® B/E co-formulada con Alúmina utilizando la estrategia “prime-boost”. El vector vCP1521 expresa los antígenos Gag-Pol del aislado LAI del VIH-1 perteneciente al subtipo B y la proteína monomérica gp120 de la forma recombinante circulante CRF01_AE unida a la región transmembrana gp41 del aislado LAI, mientras que AIDSVAX® B/E es una vacuna de subunidades de gp120 bivalente que contiene la proteína de la envuelta de la forma recombinante circulante CRF01_AE y la del aislado MN del subtipo B (54).
Los resultados de este ensayo clínico reflejaron que el protocolo de vacunación ensayado proporcionó una eficacia del 31.2% en la prevención de la infección por VIH-1 en el transcurso de 42 meses a pesar de la ausencia de anticuerpos neutralizantes. La presencia de anticuerpos IgG no neutralizantes frente a los lazos variables V1/V2 de la proteína de la envuelta viral y de anticuerpos que median la citotoxicidad celular dependiente de anticuerpos (ADCC) en presencia de bajos niveles de IgA en plasma correlacionaron con un menor riesgo de infección por el virus (55). Además, desde el punto de vista de la respuesta celular, se detectó la presencia de células T CD4+ efectoras y polifuncionales en individuos vacunados protegidos (56).
A pesar de que la eficacia obtenida fue modesta, este ensayo clínico puso de manifiesto el hecho de que conseguir una vacuna preventiva frente al VIH/SIDA era posible, algo que se había puesto en duda tras los fracasos registrados en anteriores ensayos de eficacia con recombinantes de adenovirus y proteína gp120, y marcó el camino en el diseño y evaluación de nuevos candidatos vacunales. Además, demostró que se dispone actualmente de las infraestructuras necesarias para desarrollar ensayos de eficacia en países con alta incidencia de la infección, y que es necesario llevar a cabo grandes mejoras en los candidatos vacunales con el fin de incrementar su grado de protección.
El hecho de que una cepa altamente atenuada como ALVAC fuera uno de los componentes de este ensayo ha reforzado la utilización de los poxvirus como plataforma a incluir en ensayos futuros.
3.3. Efecto de MVA-B sobre células dendríticas
Las células dendríticas (DCs) son los centinelas naturales del sistema inmune, derivan de precursores hematopoyéticos y proceden principalmente de linaje mieloide. Al ser las principales células profesionales presentadoras de antígeno (APCs) son importantes tanto en la estimulación/activación como en la regulación de la intensidad de la respuesta de linfocitos T y linfocitos B. La exposición de las DCs a agentes infecciosos induce cambios funcionales y de expresión génica los cuales, de manera coordinada, activan su capacidad para interactuar con los linfocitos T y promover tanto su proliferación como su diferenciación funcional.
Se ha demostrado tanto in vitro como in vivo que recombinantes basados en la cepa atenuada MVA infectan de forma efectiva las células presentadoras de antígeno, fundamentalmente las células mieloides MHC clase II+ (DCs y macrófagos) (57), que representan el eslabón que conecta la inmunidad innata con la inmunidad adaptativa. Sin embargo, se conocía poco sobre el impacto de estos vectores en las DCs humanas y su capacidad para presentar los antígenos heterólogos y activar células T específicas. Debido a estas razones decidimos, en primer lugar, evaluar el efecto que tiene la expresión de los antígenos del VIH-1 desde el candidato vacunal MVA-B sobre el patrón de expresión génica de estas células; y, en segundo lugar, estudiar ex vivo la capacidad de las células dendríticas derivadas de monocitos humanos (MDDCs) infectadas por MVA-B para presentar los antígenos del VIH-1 y activar a células T humanas autólogas de pacientes infectados con el virus.
Mediante la tecnología de microarrays realizamos un análisis de los cambios transcripcionales que ocurrían en las MDDCs aisladas de varios donantes sanos tras la infección por MVA-B en comparación con la infección con la cepa parental MVA. Identificamos 442 genes regulados diferencialmente relacionados con la expresión de los antígenos del VIH-1, de los cuales 387 fueron regulados positivamente y 55 lo hicieron negativamente. Por categorías funcionales los genes regulados estaban implicados fundamentalmente en el tráfico de células inmunes, hematopoyesis, presentación antigénica y respuestas de células T y B. Un análisis más detallado de los datos reveló que la expresión de los genes del VIH-1 desde el virus MVA-B activa de forma eficiente la expresión de quimioquinas (CCL3, CCL4, CCL20, CXCL10, CXCL13 y CCR8), citoquinas (IFN-α, TNF-α e IFN-β) y proteínas quinasas implicadas en rutas de señalización (JAK1 e IRAK2). También se expresaron de forma diferencial genes involucrados en la ruta de procesamiento y presentación antigénica: el gen que codifica para la proteína MICA se reguló positivamente, mientras que los genes HLA-DRA y HSPA5 fueron regulados negativamente. Demostramos que la regulación de la expresión de MICA se debía al antígeno Env del VIH-1 expresado desde MVA-B y no de GPN. Los ensayos funcionales indicaron que esta proteína mejora la interacción entre las células diana infectadas por MVA-B y los linfocitos citotóxicos (58). Resultados similares fueron obtenidos cuando se analizó el perfil de expresión génica inducido por los antígenos del VIH-1 del subtipo C expresados por MVA-C en células dendríticas mieloides (mDCs) y plasmacitoides (pDCs) (59).
En un modelo ex vivo investigamos el impacto de la infección por el candidato vacunal MVA-B en la biología de MDDCs de pacientes asintomáticos infectados por VIH-1 y evaluamos la capacidad de estas MDDCs infectadas para activar respuestas de células T específicas de VIH-1 en linfocitos T autólogos. Cuando analizamos el efecto de la infección in vitro de MDDCs por MVA-B observamos que las células infectadas presentan un fenotipo maduro, secretan altos niveles de citoquinas y quimioquinas (IL1-a, IL-7, TNF-a, IL-6, IL-12, IL-15, IL-8, MCP-1, MIP-1a, MIP-1b, RANTES, IP-10, MIG e IFN-a) y adquieren capacidad para migrar hacia gradientes de quimioquinas linfoides (CCL19 o CCL21). Además, la infección por MVA-B indujo la apoptosis de las células infectadas y los cuerpos apoptóticos resultantes fueron engullidos por MDDCs no infectadas que presentaron de forma cruzada los antígenos del VIH-1 a los linfocitos T CD8 autólogos. El co-cultivo de MDDCs infectadas por MVA-B con linfocitos T autólogos indujo una respuesta de células T CD8 específicas del VIH-1 altamente funcional caracterizada por un aumento en su capacidad proliferativa, la secreción de IFN-g, IL-2, TNF-a, MIP-1a, MIP-1a, RANTES e IL-6 y una fuerte actividad citotóxica contra linfocitos T CD4+ autólogos infectados por VIH-1 (reducción del 69.8%) (Figura 11) (60).

Figura 11: Proliferación de células T específicas frente al VIH-1 y secreción de citoquinas inducidas por células dendríticas derivadas de monocitos (MDDCs) autólogas infectadas con MVA-B. A: Gráfico representativo de la dilución del marcador de proliferación CFSE mediante citometría de flujo en células T CD3+ CD8+ estimuladas con MDDCs autólogas infectadas con la cepa parental MVA o con el recombinante MVA-B. B: Porcentaje de proliferación de células T CD8+ y T CD4+ estimuladas con MDDCs autólogas infectadas con la cepa parental MVA, con el recombinante MVA-B o pulsadas con la proteína p24 soluble del VIH-1. C: Secreción de citoquinas después del co-cultivo de linfocitos T CD8+ y MDDCs infectadas con MVA o MVA-B. (***) p<0.005, (**) p<0.01, (*) p< 0.05.
Estos estudios demostraron ex vivo la capacidad del candidato vacunal MVA-B para estimular la maduración y activación de las células dendríticas derivadas de monocitos humanos e inducir la activación de células T CD8 específicas frente al VIH-1 polifuncionales y con capacidad citotóxica para eliminar de forma efectiva células T CD4 infectadas por VIH-1.
3.4. MVA-B como vacuna profiláctica
Los resultados derivados de la evaluación preclínica en diferentes modelos animales y en experimentación ex vivo demostraron que el candidato vacunal MVA-B cumplía las exigencias de seguridad e inmunogenicidad necesarias para ser evaluado en ensayos clínicos. Debido a esto, se presentó al Ministerio de Sanidad Español una propuesta para evaluar MVA-B como vacuna profiláctica frente al VIH/SIDA en un ensayo clínico fase I. Después de recabar toda la información solicitada y cumplir con las exigencias requeridas, las autoridades regulatorias aprobaron el ensayo clínico, denominado RISVAC02, con identificador NCT00679497.
El estudio RISVAC02 tenía como objetivo evaluar la seguridad e inmunogenicidad del candidato vacunal MVA-B con fines profilácticos. El ensayo aleatorizado, multicéntrico, doble ciego y controlado con placebo, incluyó un total de 30 individuos sanos, que fueron seleccionados de un total de 356 que se ofrecieron a participar en esta investigación pionera. Los voluntarios, en su mayoría hombres (80%), con rangos de edad entre 19 y 48 años, estaban sanos, tenían bajo riesgo de infección por VIH-1 y no presentaban inmunidad pre-existente frente al virus de la viruela. Se distribuyeron entre los hospitales universitarios Gregorio Marañón, en Madrid, y Clinic, en Barcelona, y fueron asignados al azar (con aleatorización equilibrada [1:1] a través de una lista de aleatorización generada por ordenador sin ninguna restricción) para recibir 3 inyecciones intramusculares (1 × 108 ufp/dosis) de MVA-B (n = 24) o placebo (n = 6) a las semanas 0, 4 y 16. El seguimiento se realizó hasta la semana 48 (1 año).
Desde el punto de vista de seguridad podemos decir que la vacuna MVA-B fue segura y bien tolerada. Durante todo el período de seguimiento se registraron un total de 169 efectos adversos (EA) (158 en el grupo de vacunados y 11 en el grupo placebo), la mayoría de grado 1-2. Los EA más frecuentes que se reportaron fueron cefaleas, dolor en la zona de la inyección o malestar general.
Desde el punto de vista inmunológico detectamos respuestas celulares específicas frente al VIH-1 en el 75% de los voluntarios vacunados por la técnica de ELISPOT, que cuantifica el número de células T productoras de IFN-γ específicas frente a grupos de péptidos representativos de los 4 antígenos del VIH-1 expresados por MVA-B (Env, Gag, Pol y Nef). La frecuencia de respondedores aumentó significativamente después de la segunda dosis de la vacuna, del 33% al 67%, y se mantuvo después de la tercera dosis y hasta el final del estudio. La respuesta detectada estaba dirigida fundamentalmente frente a los grupos de péptidos representativos del antígeno Env, aunque después de la tercera dosis la respuesta fue más balanceada.
En cuanto a la respuesta humoral, el 96% de los vacunados desarrollaron anticuerpos de unión específicos frente a la proteína de la envuelta gp160 del subtipo B después de tres dosis de MVA-B, que se mantuvo en el 73% de los vacunados al cabo del año (Figura 12A). Del mismo modo, el 100% de los vacunados desarrollaron anticuerpos de unión específicos frente al vector después de tres dosis de MVA-B que se mantuvo en el 77% de los vacunados al cabo del año (Figura 12B). Interesantemente, el 54% de los vacunados desarrollaron anticuerpos neutralizantes frente al aislado homólogo BX08 del VIH-1, aunque los títulos detectados eran bajos (Figura 12C) (61).

Figura 12: Respuesta humoral específica frente al VIH-1 y VACV en voluntarios del ensayo clínico RISVAC02. A: Frecuencia de respondedores y niveles de anticuerpos IgG de unión frente a la proteína Env del VIH-1. B: Frecuencia de respondedores y niveles de anticuerpos IgG de unión frente a los antígenos de VACV. C: Niveles de anticuerpos neutralizantes frente al aislado autólogo BX08 del VIH-1. La línea discontinua representa el umbral considerado como respuesta positiva. V: vacunados; P: placebo.
Aunque el ensayo de ELISPOT es la técnica estandarizada que se utiliza internacionalmente para medir la respuesta inmune celular inducida por los diferentes candidatos vacunales frente al VIH-1 que se ensayan en clínica, proporciona una información muy limitada. Debido a ello se han ido implementando otros métodos analíticos más complejos, entre los que destaca sin lugar a dudas la citometría de flujo multiparamétrica que permite caracterizar una célula por su fenotipo de superficie y, al mismo tiempo, conocer el patrón de secreción de citoquinas, quimioquinas, capacidad proliferativa o capacidad citotóxica entre otras. La disponibilidad de amplios paneles de reactivos de gran calidad facilita la aplicación de esta técnica en la rutina clínica. Por estas razones decidimos implementarla para llevar a cabo una caracterización más detallada de la respuesta inmune celular inducida por el candidato vacunal MVA-B. El estudio se realizó en 16 voluntarios de los 30 que se incluyeron en el ensayo RISVAC02 (13 vacunados y 3 placebos) que habían sido reclutados en el Hospital Gregorio Marañón (Madrid) y utilizamos linfocitos de sangre periférica frescos aislados el mismo día de la toma de muestra de los pacientes. La caracterización de la magnitud, polifuncionalidad y fenotipo de memoria de las células T CD4 y CD8 específicas frente al VIH-1 inducidas tras la vacunación se realizó empleando la técnica de tinción de citoquinas intracelulares y evaluando específicamente los niveles de secreción de IFN-γ, IL-2, TNF-α y MIP-1β. De forma general podemos destacar que la vacunación con MVA-B estimuló respuestas de células T CD4 (69,2%) y CD8 (92.3%) específicas frente al VIH-1 en los individuos vacunados. La frecuencia de respondedores después de dos dosis de MVA-B fue similar a la obtenida después de la tercera dosis de la vacuna (75% versus 69.2%). Las células T CD4+ estaban dirigidas frente al antígeno Env, mientras que las células T CD8+ reconocían a los antígenos Env, Gag, Pol y Nef de manera similar (Figura 13).
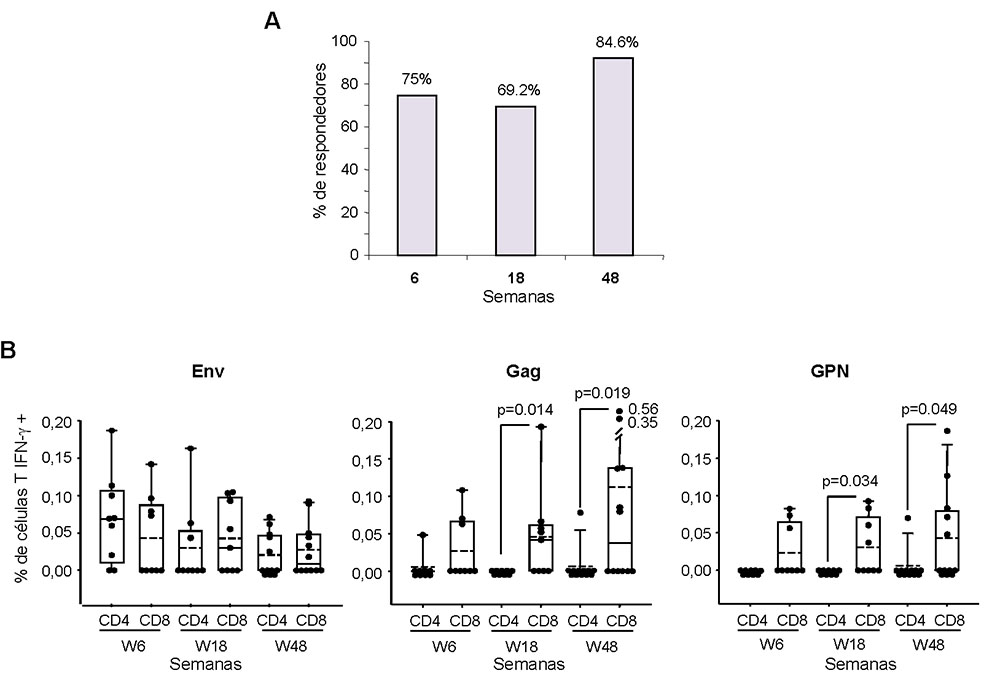
Figura 13: Respuesta celular específica frente al VIH-1 en voluntarios del ensayo clínico RISVAC02. A: Frecuencia de respondedores a los diferentes tiempos. B: Porcentajes de células T CD4 y T CD8 que producen IFN-g frente a los antígenos Env, Gag o GPN del VIH-1.
Entre el 25%-45% de las células T específicas mostraban un perfil polifuncional, caracterizado por la secreción simultánea de dos o más citoquinas. Al año de haber comenzado el ensayo con MVA-B analizamos la magnitud, frecuencia y fenotipo de las células T específicas de memoria incluyendo en el panel de reactivos los anticuerpos que reconocen las moléculas de superficie CD45RA y CCR7 que definen las siguientes poblaciones de células T de memoria: centrales de memoria (TCM; CD45RA-CCR7+, efectoras de memoria (TEM; CD45RA- CCR7-) o efectoras de memoria diferenciadas (TEMRA; CD45RA+ CCR7-). Observamos que la magnitud y la frecuencia de la respuesta de células T específicas se mantuvieron sin diferencias significativas con respecto a la detectada después de tres dosis de la vacuna. Las células T CD4 específicas de memoria se distribuían entre los fenotipos TEM y TCM, mientras que las células T CD8 específicas de memoria se distribuían principalmente entre los fenotipos TEM y TEMRA (Figura 14) (62).

Figura 14: Fenotipo de la respuesta de células T específicas de memoria frente al VIH-1. A: Distribución de las células T CD4+ y T CD8+ de memoria según la expresión de los marcadores CCR7 en combinación con CD45RA. B: Perfiles fenotípicos representativos de las células T CD4 y CD8 específicas de memoria frente al VIH-1. (*) p< 0.05.
La presencia de células T con fenotipo TEM o TEMRA se ha correlacionado con el control de la infección por VIH-1 (63); del mismo modo, estas poblaciones han sido relevantes en el control temprano de la infección por el virus de simio altamente patogénico (64), de ahí la importancia de que sean estimuladas por el candidato vacunal MVA-B.
Después de 4 años de haber comenzado el ensayo clínico RISVAC02, tratamos de contactar con los 24 voluntarios vacunados con MVA-B para ver si accedían a recibir una cuarta dosis del vector. El estudio se explicó en detalle a todos los participantes que se pudieron localizar y que cumplían los criterios de elegibilidad. De ellos, 13 dieron su consentimiento informado por escrito para recibir una inyección intramuscular (1 × 108 ufp/dosis) de MVA-B y someterse a un seguimiento por 12 semanas para evaluar la seguridad, tolerabilidad e inmunogenicidad de la dosis tardía administrada. El estudio denominado RISVAC02 boost fue aprobado por los comités éticos institucionales de los Hospitales Clinic y Gregorio Marañón y por las autoridades regulatorias españolas. Los voluntarios fueron en su mayoría hombres (92.3%), con rangos de edad entre 24 y 53 años. Después de la vacunación se registraron un total de 64 efectos adversos, la mayoría de grado 1 (96%), y no se informó ninguno de grados 3 ó 4, confirmando el perfil de seguridad de MVA-B. Al analizar la respuesta celular mediante la técnica de ELISPOT, observamos que previamente a la administración de la cuarta dosis de la vacuna el 20% de los participantes mantenían respuestas celulares específicas frente al VIH-1, aunque de baja magnitud. Esta frecuencia experimentó un pequeño incremento tras la cuarta dosis de MVA-B (38%) y se mantuvo hasta la semana 12. El análisis por citometría de flujo multiparamétrica confirmó estos resultados. Después de 4 años, sólo un 12.5% de los voluntarios mantenían respuestas de células T CD4+ y T CD8+ específicas. Esta frecuencia se incrementó hasta el 45% a las semanas 2 y 4 después del “boost” tardío con MVA-B, volviendo a los niveles basales a la semana 12. La respuesta T CD4+ estaba dirigida frente a Env, mientras que la respuesta CD8+ se dirigía de forma similar frente a los 4 antígenos. El 20% de las células T CD4+ y el 35% de las células T CD8+ secretaban simultáneamente dos o más citoquinas. La respuesta frente al vector por su parte se mantuvo en el 12.5% de los vacunados al cabo de los 4 años; sin embargo, a la semana 4 después de la cuarta dosis de MVA-B, se detectó en el 80% de los vacunados una respuesta T CD8+.
En cuanto a la respuesta humoral, sí logramos detectar un efecto positivo tras la cuarta inmunización. El porcentaje de voluntarios con niveles de anticuerpos de unión específicos frente a la proteína gp120 homóloga se incrementó del 21.3% detectado al cabo de los 4 años, hasta el 92.3% a la semana 2 después del “boost” tardío (Figura 15A). Los títulos de anticuerpos neutralizantes también se incrementaron significativamente, llegando a valores en el orden de 104 a este tiempo (Figura 15B). Este título medio fue 10 veces mayor que el detectado después de 3 dosis de MVA-B en el ensayo RISVAC02 y se encontraba en el mismo rango que el reportado en el ensayo de eficacia RV144 (54, 55) o en otros estudios que emplean recombinantes basados en MVA (65, 66). Estos datos resaltan la capacidad del vector MVA-B para inducir respuestas de anticuerpos de larga duración frente a la proteína Env del VIH-1, probablemente proporcionando señales de supervivencia a las células B que les permiten expandirse eficientemente después de un refuerzo tardío. Además, la cuarta dosis de MVA-B fue capaz de inducir anticuerpos neutralizantes en suero frente al aislado homólogo del VIH-1 en más del 75% de los voluntarios, que se correlacionaron de forma positiva con los niveles de anticuerpos de unión frente a gp120 (Figura 15C) (67).

Figura 15: Respuesta humoral específica frente al VIH-1 después de una cuarta dosis de MVA-B administrada a los cuatros años de haber comenzado el estudio RISVAC02. A: Niveles de anticuerpos IgG de unión frente a la proteína Env del VIH-1. B: Niveles de anticuerpos neutralizantes frente al aislado autólogo BX08 del VIH-1. C: Correlación entre títulos de anticuerpos IgG de unión frente a la proteína Env y títulos de anticuerpos neutralizantes frente al aislado autólogo BX08 del VIH-1.
Los datos derivados de este ensayo clínico fueron publicados en las revistas “Vaccine”, “J. Virol.” y “PLOSone”, con mi contribución como primera autora en 2 de ellos. En conjunto, este estudio demostró que la inoculación de tres dosis del candidato vacunal MVA-B en voluntarios sanos siguiendo un protocolo homólogo es segura, tolerable e inmunogénica, induciendo respuestas inmunes celulares y humorales específicas y duraderas frente al VIH-1 en el 75% y 95% de los vacunados, respectivamente, e involucrando a subpoblaciones de células T CD4+ y CD8+ polifuncionales. Por otra parte, una cuarta dosis de la vacuna al cabo de los 4 años indujo aumentos moderados en las respuestas de células T específicas frente al VIH-1, pero aumentó significativamente las respuestas de anticuerpos frente a la proteína Env del VIH-1, así como la generación de anticuerpos neutralizantes frente al aislado homólogo.
El ensayo clínico RISVAC02 fue el primer estudio fase I en humanos realizado en España con un candidato vacunal frente al VIH/SIDA. Se realizó con fondos de organizaciones públicas y fundaciones privadas españolas en un marco colaborativo y multidisciplinario en el que los voluntarios representaron el pilar fundamental. Representó un desafío para todos los centros que estuvimos involucrados, pero el trabajo mereció la pena. Demostramos que tenemos las infraestructuras necesarias para afrontar con rigor científico proyectos de gran envergadura y hacer posible la traslación de candidatos vacunales desde la preclínica a los ensayos clínicos, una tarea que sólo unos pocos afortunados logran ver cumplir. Además, estos estudios de vacunación frente al VIH tuvieron una gran difusión en los medios (prensa, radio y televisión) tanto nacionales como internacionales.
3.5. MVA-B como vacuna terapéutica
Aunque la morbilidad y la mortalidad de la infección por VIH-1 han disminuido drásticamente debido a la introducción de la terapia antirretroviral combinada (TAR), el tratamiento a largo plazo puede ocasionar resistencia a la medicación y toxicidad, sin olvidar el elevado coste económico que representa su implementación, sobre todo para países en desarrollo que poseen altas tasas de individuos infectados. Debido a ello, resulta necesario explorar nuevas estrategias terapéuticas que restauren y/o potencien respuestas inmunes específicas frente al VIH-1 que permitan controlar la replicación viral en ausencia de TAR durante períodos prolongados de tiempo. Sin embargo, el desarrollo racional y seguro de estas intervenciones se dificulta por la falta de parámetros bien definidos relacionados con el control inmune del VIH-1 y la incertidumbre con respecto a las evaluaciones más adecuadas que deben aplicarse en los ensayos de vacunas clínicas.
Los resultados de seguridad e inmunogenicidad obtenidos en el estudio RISVAC02 avalaron la propuesta presentada al Ministerio de Sanidad Español para evaluar MVA-B como vacuna terapéutica frente al VIH/SIDA. El ensayo clínico fase I, denominado RISVAC03, fue aprobado por las autoridades regulatorias con identificador NCT01571466 y tenía como objetivos, por un lado, evaluar la seguridad e inmunogenicidad del candidato vacunal MVA-B en pacientes crónicos infectados con VIH-1 que estaban en tratamiento antirretroviral (TAR) y, por otro lado, determinar su capacidad de actuar sobre el reservorio de virus latente y el rebote de la carga viral tras la interrupción controlada de la terapia cuando se administra sólo o en combinación con disulfiram, un medicamento utilizado para reactivar el reservorio viral.
Previamente a la realización de este estudio, nuestro grupo evaluó en qué medida los diferentes inhibidores virales que conforman la terapia antiretroviral en las unidades de VIH de los hospitales españoles podrían afectar a la replicación del candidato vacunal MVA-B en cultivos celulares establecidos y en células dendríticas humanas. Observamos que los inhibidores de VIH-1 ensayados no ejercían ningún efecto sobre la cinética de crecimiento del virus MVA-B in vitro, incluso a concentraciones más altas que las empleadas normalmente en el tratamiento. Además, tampoco se vieron afectadas ni la cinética ni los niveles de expresión de las proteínas gp120 y GPN expresadas por MVA-B (68).
RISVAC03 fue un ensayo aleatorizado, multicéntrico y doble ciego en el que participaron 30 voluntarios con infección crónica por VIH-1 controlados por TAR y con recuentos de células T CD4 >450 células/mm3. Todos los pacientes dieron su consentimiento informado por escrito y fueron asignados al azar [aleatorización equilibrada (2:1)] para recibir tres inyecciones intramusculares (1 × 108 ufp/dosis) de MVA-B (n=20) o placebo (n=10) a las semanas 0, 4 y 16. A las 8 semanas después de la última dosis de MVA-B (semana 24) se interrumpió la terapia antirretroviral y se evaluó la dinámica del rebote viral durante las primeras 12 semanas tras la interrupción. A un subconjunto de pacientes (8 vacunados y 4 placebos) no se les interrumpió la TAR en la semana 24 y recibieron una cuarta dosis de MVA-B en la semana 36 combinada con disulfiram (250 mg una vez al día) durante 3 meses (desde la semana 36 a la semana 48). En esto grupo, la interrupción de la terapia se realizó en la semana 48 y se evaluó la dinámica del rebote viral igual que en el grupo principal. En el estudio estuvieron implicados los Hospitales Germans Trias i Pujol y Clinic de Barcelona y el Hospital Gregorio Marañón de Madrid.
Durante todo el seguimiento del estudio se reportaron un total de 326 efectos adversos (257 en el grupo de los vacunados y 69 en el grupo placebo). El 90% de los EA en el grupo vacunado fueron de grado 1 y el 8% de grado 2. Los clasificados como grado 3 y 4 no estuvieron relacionados con la vacunación. Los EA más comunes que se registraron fueron dolor y malestar general. Estos datos confirman que el vector MVA-B es seguro y bien tolerado, incluso en pacientes seropositivos.
Cuando analizamos la respuesta celular inducida por MVA-B mediante la técnica de ELISPOT observamos que en comparación con los receptores de placebo, los voluntarios vacunados con MVA-B mostraron un aumento tanto en la magnitud de la respuesta de células T específicas frente al VIH-1, alcanzando los máximos valores después de la segunda inmunización (semana 6), como en su amplitud, llegando a reconocer 7 grupos de péptidos representativos de los antígenos incluidos en el vector MVA-B después de tres dosis de la vacuna (semana 18). MVA-B logró expandir fundamentalmente las respuestas pre-existentes frente a Gag (Figura 16) (69).
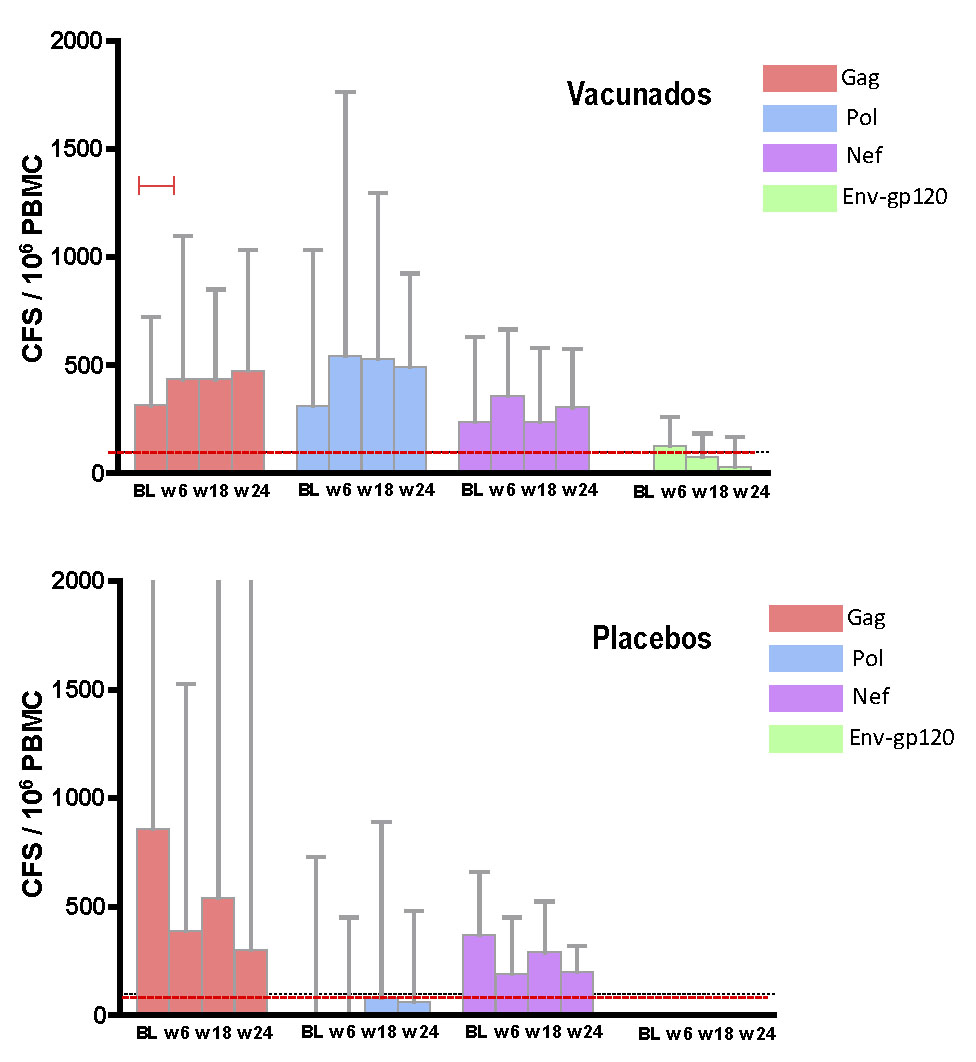
Figura 16: Respuesta celular específica frente a los antígenos Env, Gag, Pol y Nef del VIH-1 en voluntarios del ensayo clínico RISVAC03. Los valores representan el número de células secretoras de IFN- / 106 PBMCs frente a los antígenos del VIH-1 presentes en la vacuna MVA-B en los individuos vacunados (panel superior) y en los receptores de placebo (panel inferior) antes de la vacunación (BL), después de dos (W6) o tres (W18) dosis de MVA-B o en el momento de la interrupción de la terapia (W24). La línea discontinua representa el umbral considerado como respuesta positiva.
Para ampliar estos resultados, evaluamos por citometría de flujo multiparamétrica la magnitud, amplitud y polifuncionalidad de las respuestas celulares específicas frente al VIH-1 inducidas y/o expandidas por el vector MVA-B en comparación con el grupo placebo. El estudio se realizó en 22 de los 30 voluntarios reclutados (14 vacunados y 8 placebos) debido a la disponibilidad de muestras. A diferencia del grupo receptor de placebo, la vacunación con MVA-B mejoró significativamente la frecuencia, magnitud y amplitud de las respuestas de células T CD4 específicas frente a los antígenos Gag y GPN del VIH-1. El número total de respuestas T CD4+ detectadas en el grupo de vacunados se incrementó desde 5 (semana 0) hasta 22 tras tres dosis de MVA-B, y se mantuvo en 16 en el momento de la interrupción de la terapia antirretroviral. No obstante, es difícil distinguir si las nuevas respuestas que se detectaron provenían de células T CD4 vírgenes o de células T CD4+ preexistentes y cuyos niveles se encontraban por debajo del nivel umbral de detección de la técnica. Por el contrario, no hubo ninguna variación temporal significativa en las respuestas de células T CD8+ asociada con la administración de la vacuna (Figura 17).
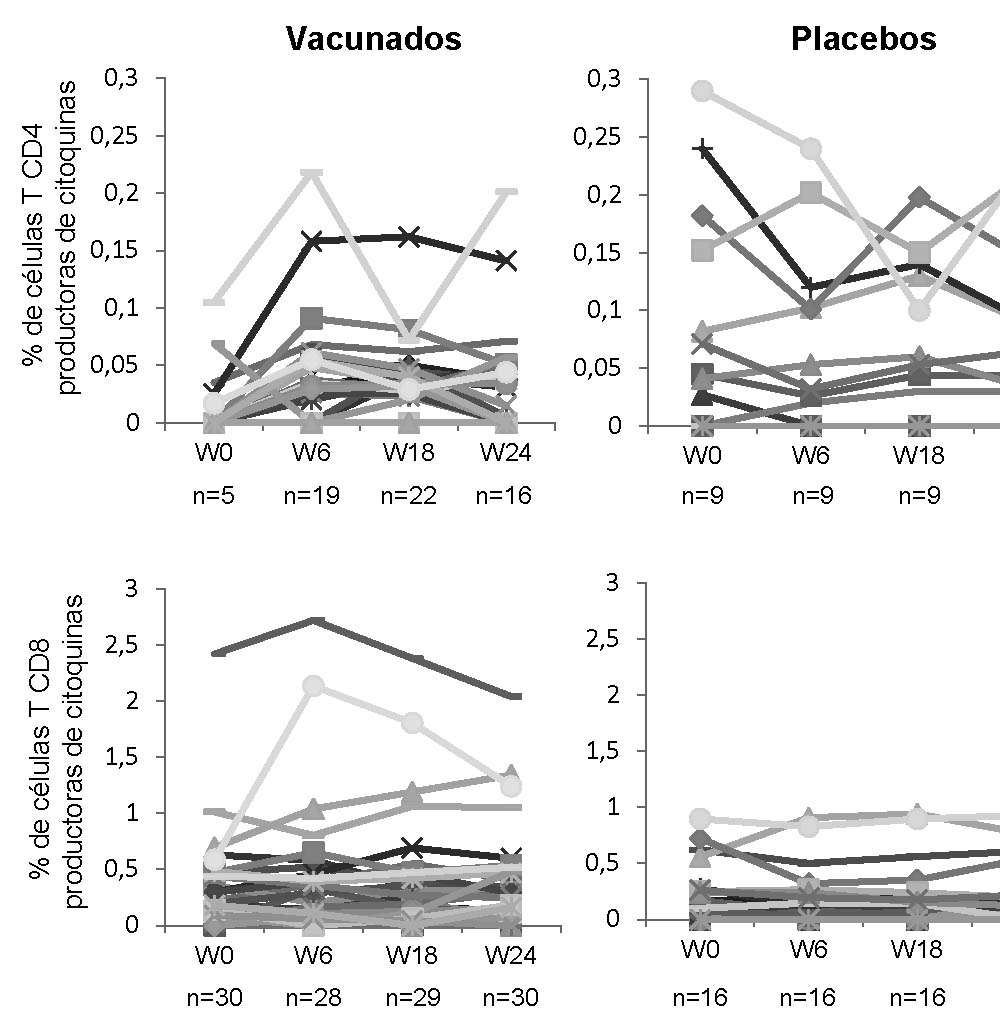
Figura 17: Respuesta de células T CD4 y T CD8 específicas frente al VIH-1 en voluntarios del ensayo clínico RISVAC03 determinada por citometría de flujo multiparamétrica. Los gráficos representan la cinética de la respuesta de células T CD4 (panel superior) y CD8 (panel inferior) específicas frente al VIH-1 (Env+Gag+GPN) para cada uno de los individuos vacunados o receptores de placebo incluidos en el ensayo a lo largo del tiempo. n representa el número de respuestas positivas frente a Env, Gag y GPN a cada tiempo analizado.
Cuando analizamos el perfil funcional de la respuesta frente al VIH-1 observamos que tanto en el grupo de vacunados como en el grupo placebo las células T específicas preexistentes eran altamente polifuncionales. En el grupo de voluntarios vacunados, el 40% de las células T CD4+ y el 60% de las células T CD8+ exhibían al menos dos funciones efectoras. La inmunización con MVA-B indujo un aumento de la frecuencia tanto de las células T CD4+ que secretan simultáneamente IFN-γ + IL-2 + TNF-α, como de las células T CD8+ que secretan simultáneamente CD107a + IFN-γ + TNF-α. La vacunación también mejoró las frecuencias de células T CD4+ con fenotipo efector de memoria (70).
Cuando se analizó el efecto de la vacunación sobre la respuesta humoral observamos que no hubo ninguna variación temporal significativa ni en los niveles de anticuerpos de unión a Env ni en su capacidad neutralizante que pudiera asociarse con la administración de MVA-B. La gran mayoría de los participantes presentaban niveles altos y variables de anticuerpos de unión a gp120 (100% en ambos grupos) y a la región V1/V2 de gp120 (75% y 67% en vacunados y placebo, respectivamente) al inicio del estudio, que se mantuvieron sin cambios a lo largo del mismo. Igualmente, la actividad neutralizante de los anticuerpos frente al aislado homólogo fue muy variable entre los diferentes pacientes, pero no hubo cambios significativos después de la vacunación.
Después de la interrupción de la terapia se cuantificaron niveles detectables de carga viral en el plasma (pVL) de todos los pacientes. Sin embargo, hubo un retraso moderado, pero significativo, en la cinética de rebote viral en los voluntarios que recibieron MVA-B, pero no el disulfiram, durante las primeras 4 semanas después de la interrupción (Figura 18).
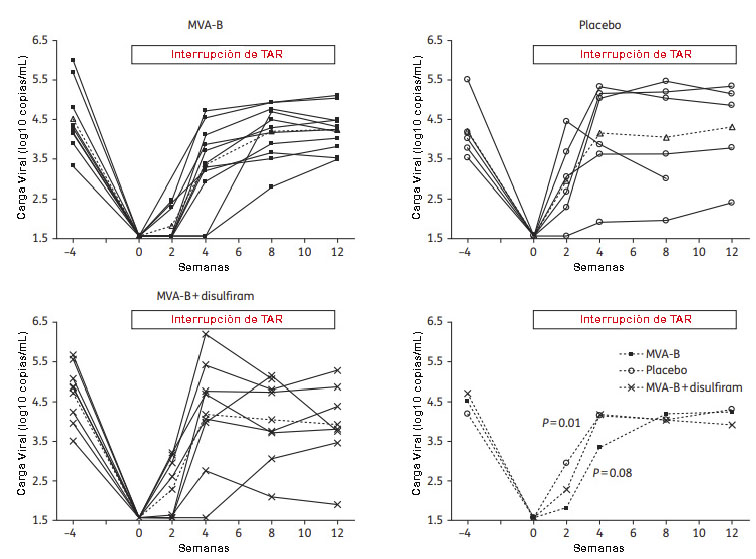
Figura 18: Rebote de la carga viral del VIH-1 después de la interrupción de la terapia antirretroviral (TAR). Se representan los valores individuales de carga viral plsmática (pVL) antes del inicio de TAR (–4) y durante el período de interrupción controlada de TAR en los diferentes grupos.
El tratamiento con disulfiram no demostró tener ningún efecto en términos de reactivación del VIH-1 latente. El tamaño del reservorio proviral latente en células T CD4+ circulantes en la semana 48 (12 semanas después del tratamiento con disulfiram) no difirió significativamente del detectado en la semana 36. A pesar de que no hubo correlación debido al tamaño muestral, observamos que en el momento en que se interrumpió la terapia antiretroviral (semana 24), los individuos vacunados que tenían un retraso en la cinética de rebote viral presentaban frecuencias más elevadas de células T CD8 específicas frente a Gag, porcentajes más altos de células T CD4 específicas frente al VIH-1 con fenotipo efector de memoria y títulos más altos de anticuerpos de unión anti-V1/V2 de gp120. Sin embargo, estos parámetros inmunológicos no fueron suficientes para el control del VIH-1 en los voluntarios vacunados. Esta observación podría estar relacionada con el aumento moderado que se induce tras la vacunación de los niveles del marcador de agotamiento celular PD-1 detectados en la población de células T CD8+ (71).
Si bien los resultados de cohortes transversales de infección natural por VIH apuntan a varios marcadores inmunes asociados con la carga viral, no se han identificado parámetros inmunes sólidos que puedan servir como predictores confiables del control viral en pacientes que reciben vacunas terapéuticas e interrumpen el tratamiento antirretroviral (72). En este sentido, el ensayo RISVAC03 permitió integrar parámetros virológicos e inmunológicos que podrían pronosticar el retraso clínicamente modesto, pero estadísticamente significativo, que se produjo en la cinética de rebote viral de los individuos vacunados con MVA-B y así definir predictores del control viral. Para ello, se analizaron las respuestas inmunes de células T y B específicas que se desarrollaron tras la interrupción del tratamiento, se realizaron estudios filogenéticos de la población viral antes y después de la interrupción, determinando el número de mutaciones de escape presentes en Gag asociadas al antígeno leucocitario humano-I (HLA-I) y se midió de forma precisa los niveles de ADN proviral del VIH-1 en células T CD4 de los voluntarios antes de la vacunación y después de la interrupción de la terapia. Todos estos parámetros se integraron en un modelo de regresión lineal que también tenía en consideración la presencia de alelos protectores de HLA-I (B57, B270 y B51), si los voluntarios habían sido receptores de vacuna o de placebo, la administración de disulfiram, los conteos de células T CD4 y la carga viral en plasma antes del tratamiento. De todos ellos, la única variable que se asoció independientemente con el tiempo de retraso del rebote viral fue el nivel de ADN proviral del VIH-1 presente antes de la vacunación. Un nivel bajo de ADN proviral del VIH-1 antes de la vacunación se asoció de forma independiente con un retraso en el rebote viral y un pico de viremia menor después de la interrupción del tratamiento. A su vez, un pico de viremia pequeño también se correlacionó positivamente con un número reducido de polimorfismos en la secuencia de Gag asociados a alelos HLA-I presentes en la población de virus de rebote. Estos resultados sugieren que tanto los niveles de ADN proviral del VIH-1 como la cantidad de polimorfismos en Gag asociados a HLA-I pueden ser considerados como parámetros virológicos que influyen en el resultado clínico de las interrupciones estructuradas del tratamiento. A pesar de que estos datos deberán ser validados en ensayos clínicos más extensos, su incorporación en futuros ensayos de vacunas terapéuticas podría contribuir a la implementación de interrupciones del tratamiento mucho más seguras (73).
En conjunto, los resultados del ensayo clínico RISVAC03 demostraron que el vector MVA-B es seguro, tolerado e inmunogénico en pacientes con infección crónica por VIH-1 controlados por tratamiento antirretroviral. La vacunación por MVA-B expandió y también indujo nuevas respuestas de células T CD4 específicas, polifuncionales y de fenotipo TEM dirigidas principalmente frente a los antígenos Gag y GPN y, aunque mantuvo la especificidad de antígeno, frecuencia, amplitud, polifuncionalidad y perfil fenotípico de las respuestas de células T CD8 específicas pre-existentes, también provocó un aumento moderado de su agotamiento. Independientemente del beneficio inmunológico, la respuesta inducida por MVA-B no es capaz de controlar el rebote viral que se produce tras la interrupción del tratamiento antirretroviral, lo que indica que es necesario optimizar su uso empleando estrategias combinadas de vacunación en protocolos heterólogos junto a otros vectores e incluso utilizando inmunomoduladores que reviertan el estado de agotamiento que presentan de base las células T CD8+ en pacientes crónicos y que de forma moderada se incrementa tras la vacunación.
4. PERSPECTIVA ACTUAL Y EXPERIMENTACIÓN FUTURA
Existe un consenso a nivel científico de que el candidato vacunal ideal frente al VIH debe ser capaz de estimular tanto una respuesta de anticuerpos ampliamente neutralizante que proteja frente a la infección viral como una respuesta celular potente que garantice la eliminación de las células infectadas. Sin embargo, el desafío para obtener este candidato ideal radica en que los antígenos virales, los sistemas de administración, los adyuvantes y el entorno de citoquinas y quimioquinas que promueven la respuesta humoral deseada no son los mismos que garantizan una respuesta celular potente. Por ello, la opción más realista para superar este desafío consiste en la combinación de candidatos vacunales que de forma independiente estimulen una de estas ramas del sistema inmune, empleando protocolos heterólogos de inmunización.
El ensayo clínico RV144 ha ido marcando el ritmo sobre los parámetros inmunológicos que deben tenerse en consideración a la hora de evaluar nuevas estrategias vacunales frente al VIH-1; de ahí que la mayoría de los ensayos determinen el nivel de anticuerpos no neutralizantes frente a la región V1/V2 de gp120 o la función efectora ADCC de los anticuerpos IgG generados frente a la envuelta como una estrategia para identificar un régimen de vacuna eficaz. De hecho, cada vez se investiga más en la identificación de correlatos inmunes asociados a riesgo de infección utilizando muestras procedentes de los ensayos de eficacia que se han realizado en humanos y de estudios en primates no humanos. Recientemente se ha publicado un trabajo en el que se sugiere que en próximos ensayos de eficacia se tengan en consideración además de los parámetros descritos en el ensayo RV144, la función efectora ADCP (fagocitosis celular dependiente de anticuerpos) de los anticuerpos IgG generados frente a la envuelta, la presencia de anticuerpos de isotipo IgG3 con reactividad cruzada y el nivel de células T CD8+ polifuncionales como correlatos inmunes mecanísticos de protección, ya que todos ellos estuvieron asociados a un menor riesgo de infección por VIH-1 en los ensayos evaluados (74).
Por otra parte, además de los correlatos empíricos derivados de los diferentes ensayos, se ha determinado el tipo de respuesta inmune que se genera en un grupo de pacientes seropositivos que controlan de forma natural la infección sin necesidad de terapia antirretrovial llamados controladores de élite. En ellos se ha evidenciado: (i) la presencia de anticuerpos neutralizantes de amplio espectro, incluso dirigidos frente a diferentes determinantes antigénicos del trímero de gp120; (ii) la existencia de respuestas celulares mediadas por linfocitos T CD8+ con perfil polifuncional, principalmente frente al antígeno Gag del VIH-1; y (iii) la detección de anticuerpos de unión frente a la proteína de la envuelta con isotipos más funcionales (IgG3), con mayor afinidad por receptores de Fc y mejores funciones efectoras (ADCC). Estos hallazgos también han dirigido el curso de la generación de nuevas estrategias vacunales hacia la búsqueda de candidatos que mimeticen esta inmunidad. Por un lado, se desarrollan inmunógenos que generen anticuerpos neutralizantes de amplio espectro, por ejemplo, mediante el diseño de vacunas guiadas por estructuras moleculares o usando proteínas de la envuelta en conformación nativa; y, por otro lado, se ha seguido explorando en la búsqueda de inmunógenos potenciadores de la respuesta celular también de amplio espectro. En este sentido, las construcciones sintéticas que emplean regiones conservadas del proteoma viral implicadas funcionalmente con protección o el desarrollo de proteínas tipo Mosaico constituyen las aproximaciones más prometedoras.
Los ensayos clínicos RISVAC02 y RISVAC03 evidenciaron que la administración de MVA-B en un sistema homólogo de vacunación con fines profilácticos o terapéuticos, a pesar de ser inmunogénica, no logra controlar el rebote viral una vez interrumpida la terapia, evidenciando la necesidad de realizar cambios significativos que permitan mejorar estos resultados. En este sentido, y desde la perspectiva de los avances más relevantes en campo, hemos optado tanto por optimizar los antígenos incluidos en el vector, como por explorar estrategias heterólogas que combinen varios vectores para así potenciar la calidad de la respuesta inmune generada.
Para evitar la inmunodominancia que ejerce el antígeno de la envuelta del VIH-1 sobre los antígenos Gag-Pol-Nef que se co-expresan desde el vector vacunal MVA-B y detectada tanto en el ensayo profiláctico como en el terapéutico, hemos diseñado un inmunógeno multi-epitópico destinado a activar células T, denominado TMEP-B. Este antígeno sintético incluye aquellas regiones de las proteínas Gag, Pol y Nef del VIH-1 pertenecientes al subtipo B que están ampliamente conservadas entre los diferentes aislados virales circulantes y que se han asociado funcionalmente con el control viral. Este tipo de aproximación basada en el uso de epítopos en vez de en antígenos completos ofrece una serie de ventajas entre las que destacan su seguridad, inmunogenicidad y una mayor cobertura poblacional ya que permite tanto la inclusión de aquellas regiones antigénicas relevantes para la protección que pueden unirse a un número máximo de moléculas de HLA) cubriendo toda la diversidad poblacional, como la exclusión de aquellas secuencias que se han relacionado con virulencia. Los inmunógenos basados en elementos altamente conservados del VIH-1 se desarrollan bajo la premisa de que las mutaciones que se generan en las regiones conservadas como consecuencia de un mecanismo viral de evasión de la respuesta inmune del hospedador tendrían un elevado coste para la replicación del virus, generándose variantes más susceptibles de ser eliminadas por el sistema inmune. Además, este tipo de inmunógenos modifican las jerarquías de inmunodominancia observadas en la infección por VIH-1 y en las vacunas que codifican antígenos virales nativos, ya que dirigen la respuesta de las células T a regiones que son subdominantes y poco inmunogénicas en el contexto de la proteína completa.
El nuevo inmunógeno TMEP-B ha sido introducido en un vector de ADN (ADN-TMEP) y también expresado desde MVA (MVA-TMEP) para en ambos casos ensayarlo en protocolos heterólogos combinado con MVA-B. Ambos vectores han sido caracterizados in vitro y evaluados in vivo de forma comparativa con el sistema homólogo MVA-B/MVA-B que representa el protocolo empleado en clínica. El uso de una primera dosis o “prime” con ADN-TMEP seguido de una dosis refuerzo o “boost” con MVA-B indujo respuestas de células T CD8 específicas frente al VIH-1 significativamente superiores, con un reconocimiento de epítopos más amplio y un perfil polifuncional mayor que la combinación homóloga MVA-B/MVA-B. Además, también se detectaron respuestas más elevadas de células T CD4 y T cooperadoras foliculares (Tfh) específicas frente al VIH-1 usando este régimen (75). Del mismo modo, la combinación ADN-TMEP/MVA-TMEP también fue significativamente superior a la combinación MVA-B/MVA-B expandiendo de forma eficiente la respuesta específica frente al antígeno Gag (76). Estos resultados, aunque aún preliminares, demuestran un cambio en el patrón de inmunodominancia estimulado por MVA-B, expandiendo la respuesta específica frente a epítopos funcionalmente relacionados con protección frente a la infección. Actualmente estamos involucrados en desarrollar vectores de ARNm que producen la proteína TMEP-B para su posterior evaluación como plataforma alternativa a los vectores de ADN y para ensayarlos en combinación con los vectores recombinantes basados en MVA.
Otro factor limitante del vector MVA-B es que expresa la proteína gp120 de la envuelta del VIH-1 en forma monomérica y, por lo tanto, logrando estimular solamente anticuerpos que neutralizan el aislado autólogo.
Durante la última década, la implementación de técnicas de aislamiento, cultivo y clonación de células B individuales aplicada a muestras infectadas por VIH-1 ha permitido la identificación de una nueva generación de anticuerpos neutralizantes de amplio espectro (bNAbs) que reconocen un gran número de cepas del VIH-1 con gran potencia. Estos anticuerpos, presentes en un porcentaje muy bajo de pacientes (1%), se desarrollan después de varios años de infección (2-4 años) y requieren extensos procesos de hipermutación somática. Su descubrimiento ha abierto nuevas vías para la investigación básica en inmunología, biología estructural y vacunología. Gracias a ellos se ha podido avanzar enormemente en el diseño de proteínas de la envuelta que adopten estructuras triméricas con una conformación similar a la envuelta “nativa” expresada en la partícula viral.
En este sentido, también estamos trabajando en el desarrollo de nuevos vectores recombinantes basados en MVA que expresen distintas formas de la proteína trimérica gp140 del VIH-1 con capacidad para inducir anticuerpos de amplio espectro de acción frente al VIH-1 en modelos animales. En una primera aproximación hemos generado vectores recombinantes que expresan la proteína trimérica gp145 anclada a membrana sola (MVA-gp145), o conjuntamente con la poliproteína GPN que fue optimizada para sufrir procesamiento proteolítico y liberar partículas similares a virus (VLPs) inducidas por Gag (MVA-gp145-GPN). Estos vectores son capaces de expresar la proteína gp145 en la membrana de las células infectadas y exponer epítopos críticos que son reconocidos por un amplio panel de bNAbs, indicando una conformación similar a la nativa. Además, en el doble recombinante la proteína de la envuelta también se incorpora en las VLPs inducidas por Gag. En estudios in vivo ambos recombinantes administrados en sistema homólogo generan respuestas de células T específicas frente al VIH-1 y activan células B específicas frente a Env del centro germinal que correlacionan con altos niveles de anticuerpos de unión específicos frente a Env (77). Los próximos pasos con estas construcciones implican su evaluación en conejos para poder determinar la capacidad neutralizante de los anticuerpos que logran inducir.
Estos nuevos estudios en los que estamos implicados, junto con otros dirigidos a la eliminación de los reservorios virales en personas infectadas y financiados por la Unión Europea, proyectos HIVACAR y EHVA, nos permitirán definir con qué antígeno optimizado y con qué régimen de inmunización procederemos en un futuro para conseguir respuestas celulares y humorales óptimas que nos permitan controlar la infección por VIH. La lucha contra el VIH es una prioridad sanitaria en la que están implicados muchos países, entre ellos España, y constituye una gran satisfacción personal poder colaborar estrechamente con los mejores equipos de investigación mundiales, destacando la perseverancia que nos debe de conducir a alcanzar ese logro final de la eliminación de uno de los virus con mayor tasa de mortalidad global.
5. REFERENCIAS
- Moss B. Poxviridae: the viruses and their replication. 4ª ed. Philadelphia, PA 19106 USA: Knipe, D. M. ; Howley, P. M.; 2001. 1249-83 p.
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344(1):48-54.
- Blasco R, Moss B. Extracellular vaccinia virus formation and cell-to-cell virus transmission are prevented by deletion of the gene encoding the 37,000-Dalton outer envelope protein. J. Virol. 1991;65(11):5910-20.
- Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. USA. 1996;93(21):11341-8.
- Antoine G, Scheiflinger F, Dorner F, Falkner FG. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244(2):365-96.
- Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188(1):217-32.
- Drexler I, Heller K, Wahren B, Erfle V, Sutter G. Highly attenuated modified vaccinia virus Ankara replicates in baby hamster kidney cells, a potential host for virus propagation, but not in various human transformed and primary cells. J Gen Virol. 1998;79 ( Pt 2):347-52.
- Nájera JL, Gómez CE, Domingo-Gil E, Gherardi MM, Esteban M. Cellular and biochemical differences between two attenuated poxvirus vaccine candidates (MVA and NYVAC) and role of the C7L gene. J. Virol.. 2006;80(12):6033-47.
- Sancho MC, Schleich S, Griffiths G, Krijnse-Locker J. The block in assembly of modified vaccinia virus Ankara in HeLa cells reveals new insights into vaccinia virus morphogenesis. J Virol. 2002;76(16):8318-34.
- Gallego-Gómez JC, Risco C, Rodríguez D, Cabezas P, Guerra S, Carrascosa JL, et al. Differences in virus-induced cell morphology and in virus maturation between MVA and other strains (WR, Ankara, and NYCBH) of vaccinia virus in infected human cells. J. Virol. 2003;77(19):10606-22.
- Guerra S, Nájera JL, González JM, López-Fernández LA, Climent N, Gatell JM, et al. Distinct gene expression profiling after infection of immature human monocyte-derived dendritic cells by the attenuated poxvirus vectors MVA and NYVAC. J. Virol. 2007;81(16):8707-21.
- Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, et al. Vaccinia virus host range genes. Virology. 1990;179(1):276-86.
- Oguiura N, Spehner D, Drillien R. Detection of a protein encoded by the vaccinia virus C7L open reading frame and study of its effect on virus multiplication in different cell lines. J. Gen. Virol. 1993;74 ( Pt 7):1409-13.
- Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3(6):920-40.
- Delaloye J, Roger T, Steiner-Tardivel QG, Le Roy D, Knaup Reymond M, Akira S, et al. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathogens. 2009;5(6):e1000480.
- Smith GL, Law M. The exit of vaccinia virus from infected cells. Virus Res. 2004;106(2):189-97.
- Sadikot RT, Blackwell TS. Bioluminescence imaging. Proc Am Thorac Soc. 2005;2(6):537-40, 11-2.
- Perdiguero B, Gómez CE, Esteban M. Bioluminescence Imaging as a Tool for Poxvirus Biology. Methods in molecular biology (Clifton, NJ). 2019;2023:269-85.
- Gómez CE, Nájera JL, Domingo-Gil E, Ochoa-Callejero L, González-Aseguinolaza G, Esteban M. Virus distribution of the attenuated MVA and NYVAC poxvirus strains in mice. J. gen- Virol. 2007;88(Pt 9):2473-8.
- Corbett M, Bogers WM, Heeney JL, Gerber S, Genin C, Didierlaurent A, et al. Aerosol immunization with NYVAC and MVA vectored vaccines is safe, simple, and immunogenic. Proc. Natl. Acad. Sci. USA. 2008;105(6):2046-51.
- Bbosa N, Kaleebu P, Ssemwanga D. HIV subtype diversity worldwide. Curr Opin HIV AIDS. 2019;14(3):153-60.
- Excler JL, Kim JH. Novel prime-boost vaccine strategies against HIV-1. Expert Rev. Vaccines. 2019;18(8):765-79.
- Li S, Rodrigues M, Rodríguez D, Rodríguez JR, Esteban M, Palese P, et al. Priming with recombinant influenza virus followed by administration of recombinant vaccinia virus induces CD8+ T-cell-mediated protective immunity against malaria. Proc. Natl. Acad. Sci. USA. 1993;90(11):5214-8.
- Gómez CE, Nájera JL, Jiménez EP, Jiménez V, Wagner R, Graf M, et al. Head-to-head comparison on the immunogenicity of two HIV/AIDS vaccine candidates based on the attenuated poxvirus strains MVA and NYVAC co-expressing in a single locus the HIV-1BX08 gp120 and HIV-1(IIIB) Gag-Pol-Nef proteins of clade B. Vaccine. 2007;25(15):2863-85
- Gómez CE, Nájera JL, Jiménez V, Bieler K, Wild J, Kostic L, et al. Generation and immunogenicity of novel HIV/AIDS vaccine candidates targeting HIV-1 Env/Gag-Pol-Nef antigens of clade C. Vaccine. 2007;25(11):1969-92
- Reimann KA, Li JT, Voss G, Lekutis C, Tenner-Racz K, Racz P, et al. An env gene derived from a primary human immunodeficiency virus type 1 isolate confers high in vivo replicative capacity to a chimeric simian/human immunodeficiency virus in rhesus monkeys. J. Virol. 1996;70(5):3198-206.
- Mooij P, Balla-Jhagjhoorsingh SS, Koopman G, Beenhakker N, van Haaften P, Baak I, et al. Differential CD4+ versus CD8+ T-cell responses elicited by different poxvirus-based human immunodeficiency virus type 1 vaccine candidates provide comparable efficacies in primates. J. Virol. 2008;82(6):2975-88.
- Gómez CE, Perdiguero B, Nájera JL, Sorzano CO, Jiménez V, González-Sanz R, et al. Removal of vaccinia virus genes that block interferon type I and II pathways improves adaptive and memory responses of the HIV/AIDS vaccine candidate NYVAC-C in mice. J. Virol. 2012;86(9):5026-38.
- Delaloye J, Filali-Mouhim A, Cameron MJ, Haddad EK, Harari A, Goulet JP, et al. Interleukin-1- and type I interferon-dependent enhanced immunogenicity of an NYVAC-HIV-1 Env-Gag-Pol-Nef vaccine vector with dual deletions of type I and type II interferon-binding proteins. J. Virol. 2015;89(7):3819-32.
- Gómez CE, Perdiguero B, Sánchez-Corzo C, Sorzano COS, Esteban M. Immune Modulation of NYVAC-Based HIV Vaccines by Combined Deletion of Viral Genes that Act on Several Signalling Pathways. Viruses. 2017;10(1).
- Perdiguero B, Gómez CE, Di Pilato M, Sorzano CO, Delaloye J, Roger T, et al. Deletion of the vaccinia virus gene A46R, encoding for an inhibitor of TLR signalling, is an effective approach to enhance the immunogenicity in mice of the HIV/AIDS vaccine candidate NYVAC-C. PloS one. 2013;8(9):e74831.
- Di Pilato M, Mejías-Pérez E, Zonca M, Perdiguero B, Gómez CE, Trakala M, et al. NFkappaB activation by modified vaccinia virus as a novel strategy to enhance neutrophil migration and HIV-specific T-cell responses. Proc. Natl. Acad. Sci. USA. 2015;112(11):E1333-42.
- García-Arriaza J, Nájera JL, Gómez CE, Tewabe N, Sorzano CO, Calandra T, et al. A candidate HIV/AIDS vaccine (MVA-B) lacking vaccinia virus gene C6L enhances memory HIV-1-specific T-cell responses. PloS one. 2011;6(8):e24244.
- Perdiguero B, Gómez CE, Nájera JL, Sorzano CO, Delaloye J, González-Sanz R, et al. Deletion of the viral anti-apoptotic gene F1L in the HIV/AIDS vaccine candidate MVA-C enhances immune responses against HIV-1 antigens. PloS one. 2012;7(10):e48524.
- Falivene J, Del Medico Zajac MP, Pascutti MF, Rodríguez AM, Maeto C, Perdiguero B, et al. Improving the MVA vaccine potential by deleting the viral gene coding for the IL-18 binding protein. PloS one. 2012;7(2):e32220.
- García-Arriaza J, Gómez CE, Sorzano CO, Esteban M. Deletion of the vaccinia virus N2L gene encoding an inhibitor of IRF3 improves the immunogenicity of modified vaccinia virus Ankara expressing HIV-1 antigens. J. Virol.. 2014;88(6):3392-410.
- García-Arriaza J, Nájera JL, Gómez CE, Sorzano CO, Esteban M. Immunogenic profiling in mice of a HIV/AIDS vaccine candidate (MVA-B) expressing four HIV-1 antigens and potentiation by specific gene deletions. PloS one. 2010;5(8):e12395.
- García-Arriaza J, Arnaez P, Gómez CE, Sorzano CO, Esteban M. Improving Adaptive and Memory Immune Responses of an HIV/AIDS Vaccine Candidate MVA-B by Deletion of Vaccinia Virus Genes (C6L and K7R) Blocking Interferon Signaling Pathways. PloS one. 2013;8(6):e66894.
- Nájera JL, Gómez CE, García-Arriaza J, Sorzano CO, Esteban M. Insertion of vaccinia virus C7L host range gene into NYVAC-B genome potentiates immune responses against HIV-1 antigens. PloS one. 2010;5(6):e11406.
- Kibler KV, Gómez CE, Perdiguero B, Wong S, Huynh T, Holechek S, et al. Improved NYVAC-based vaccine vectors. PloS one. 2011;6(11):e25674.
- Quakkelaar ED, Redeker A, Haddad EK, Harari A, McCaughey SM, Duhen T, et al. Improved innate and adaptive immunostimulation by genetically modified HIV-1 protein expressing NYVAC vectors. PloS one. 2011;6(2):e16819.
- Gómez CE, Nájera JL, Sánchez R, Jiménez V, Esteban M. Multimeric soluble CD40 ligand (sCD40L) efficiently enhances HIV specific cellular immune responses during DNA prime and boost with attenuated poxvirus vectors MVA and NYVAC expressing HIV antigens. Vaccine. 2009;27(24):3165-74.
- Moutaftsi M, Salek-Ardakani S, Croft M, Peters B, Sidney J, Grey H, et al. Correlates of protection efficacy induced by vaccinia virus-specific CD8+ T-cell epitopes in the murine intranasal challenge model. Eur J Immunol. 2009;39(3):717-22.
- Di Pilato M, Mejías-Pérez E, Gómez CE, Perdiguero B, Sorzano CO, Esteban M. New vaccinia virus promoter as a potential candidate for future vaccines. J. Gen. Virol.. 2013;94(Pt 12):2771-6.
- Di Pilato M, Sánchez-Sampedro L, Mejías-Pérez E, Sorzano CO, Esteban M. Modification of promoter spacer length in vaccinia virus as a strategy to control the antigen expression. J. Gen. Virol.. 2015;96(8):2360-71
- Hulot SL, Korber B, Giorgi EE, Vandergrift N, Saunders KO, Balachandran H, et al. Comparison of Immunogenicity in Rhesus Macaques of Transmitted-Founder, HIV-1 Group M Consensus, and Trivalent Mosaic Envelope Vaccines Formulated as a DNA Prime, NYVAC, and Envelope Protein Boost. J. Virol.. 2015;89(12):6462-80.
- Perdiguero B, Gómez CE, Cepeda V, Sánchez-Sampedro L, García-Arriaza J, Mejías-Pérez E, et al. Virological and immunological characterization of novel NYVAC-based HIV/AIDS vaccine candidates expressing clade C trimeric soluble gp140(ZM96) and Gag(ZM96)-Pol-Nef(CN54) as virus-like particles. J. Virol.. 2015;89(2):970-88.
- Pantaleo G, Janes H, Karuna S, Grant S, Ouedraogo GL, Allen M, et al. Safety and immunogenicity of a multivalent HIV vaccine comprising envelope protein with either DNA or NYVAC vectors (HVTN 096): a phase 1b, double-blind, placebo-controlled trial. Lancet HIV. 2019.
- Gómez CE, Nájera JL, Krupa M, Esteban M. The poxvirus vectors MVA and NYVAC as gene delivery systems for vaccination against infectious diseases and cancer. Curr. Gen. Ther. 2008;8(2):97-120.
- Gómez CE, Nájera JL, Krupa M, Perdiguero B, Esteban M. MVA and NYVAC as vaccines against emergent infectious diseases and cancer. Curr. Gen. Ther. 2011;11(3):189-217
- Gómez CE, Perdiguero B, García-Arriaza J, Esteban M. Clinical applications of attenuated MVA poxvirus strain. Expert review of vaccines. 2013;12(12):1395-416.
- Harmon TM, Fisher KA, McGlynn MG, Stover J, Warren MJ, Teng Y, et al. Exploring the Potential Health Impact and Cost-Effectiveness of AIDS Vaccine within a Comprehensive HIV/AIDS Response in Low- and Middle-Income Countries. PloS one. 2016;11(1):e0146387.
- de Montigny S, Adamson BJS, Masse BR, Garrison LP, Jr., Kublin JG, Gilbert PB, et al. Projected effectiveness and added value of HIV vaccination campaigns in South Africa: A modeling study. Sci Rep. 2018;8(1):6066.
- Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361(23):2209-20.
- Kim JH, Excler JL, Michael NL. Lessons from the RV144 Thai phase III HIV-1 vaccine trial and the search for correlates of protection. Annu Rev Med. 2015;66:423-37.
- Lin L, Finak G, Ushey K, Seshadri C, Hawn TR, Frahm N, et al. COMPASS identifies T-cell subsets correlated with clinical outcomes. Nat Biotechnol. 2015;33(6):610-6.
- Altenburg AF, van de Sandt CE, Li BWS, MacLoughlin RJ, Fouchier RAM, van Amerongen G, et al. Modified Vaccinia Virus Ankara Preferentially Targets Antigen Presenting Cells In Vitro, Ex Vivo and In Vivo. Sci Rep. 2017;7(1):8580.
- Guerra S, González JM, Climent N, Reyburn H, López-Fernández LA, Nájera JL, et al. Selective induction of host genes by MVA-B, a candidate vaccine against HIV/AIDS. J. Virol.. 2010;84(16):8141-52.
- Gómez CE, Perdiguero B, Jiménez V, Filali-Mouhim A, Ghneim K, Haddad EK, et al. Systems analysis of MVA-C induced immune response reveals its significance as a vaccine candidate against HIV/AIDS of clade C. PloS one. 2012;7(4):e35485.
- Climent N, Guerra S, García F, Rovira C, Miralles L, Gómez CE, et al. Dendritic cells exposed to MVA-based HIV-1 vaccine induce highly functional HIV-1-specific CD8(+) T cell responses in HIV-1-infected individuals. PloS one. 2011;6(5):e19644.
- García F, Bernaldo de Quiros JC, Gómez CE, Perdiguero B, Nájera JL, Jiménez V, et al. Safety and immunogenicity of a modified pox vector-based HIV/AIDS vaccine candidate expressing Env, Gag, Pol and Nef proteins of HIV-1 subtype B (MVA-B) in healthy HIV-1-uninfected volunteers: A phase I clinical trial (RISVAC02). Vaccine. 2011;29(46):8309-16.
- Gómez CE, Nájera JL, Perdiguero B, García-Arriaza J, Sorzano CO, Jiménez V, et al. The HIV/AIDS vaccine candidate MVA-B administered as a single immunogen in humans triggers robust, polyfunctional, and selective effector memory T cell responses to HIV-1 antigens. J. Virol. 2011;85(21):11468-78.
- Pantaleo G, Koup RA. Correlates of immune protection in HIV-1 infection: what we know, what we don’t know, what we should know. Nat Med. 2004;10(8):806-10.
- Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473(7348):523-7.
- Goepfert PA, Elizaga ML, Sato A, Qin L, Cardinali M, Hay CM, et al. Phase 1 safety and immunogenicity testing of DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J Infect Dis. 2011;203(5):610-9.
- Goepfert PA, Elizaga ML, Seaton K, Tomaras GD, Montefiori DC, Sato A, et al. Specificity and 6-month durability of immune responses induced by DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J Infect Dis. 2014;210(1):99-110.
- Guardo AC, Gómez CE, Díaz-Brito V, Pich J, Arnaiz JA, Perdiguero B, et al. Correction: Safety and vaccine-induced HIV-1 immune responses in healthy volunteers following a late MVA-B boost 4 years after the last immunization. PloS one. 2018;13(4):e0195915.
- García-Arriaza J, Arnaez P, Jiménez JL, Gómez CE, Muñoz-Fernández MA, Esteban M. Vector replication and expression of HIV-1 antigens by the HIV/AIDS vaccine candidate MVA-B is not affected by HIV-1 protease inhibitors. Virus Res. 2012;167(2):391-6.
- Mothe B, Climent N, Plana M, Rosas M, Jiménez JL, Muñoz-Fernández MA, et al. Safety and immunogenicity of a modified vaccinia Ankara-based HIV-1 vaccine (MVA-B) in HIV-1-infected patients alone or in combination with a drug to reactivate latent HIV-1. J. Antimicrob. Ther. 2015;70(6):1833-42.
- Gómez CE, Perdiguero B, García-Arriaza J, Cepeda V, Sánchez-Sorzano CO, Mothe B, et al. A Phase I Randomized Therapeutic MVA-B Vaccination Improves the Magnitude and Quality of the T Cell Immune Responses in HIV-1-Infected Subjects on HAART. PloS one. 2015;10(11):e0141456.
- Rallon N, Mothe B, López Bernaldo de Quiros JC, Plana M, Ligos JM, Montoya M, et al. Balance between activation and regulation of HIV-specific CD8+ T-cell response after modified vaccinia Ankara B therapeutic vaccination. AIDS. 2016;30(4):553-62.
- Hurst J, Hoffmann M, Pace M, Williams JP, Thornhill J, Hamlyn E, et al. Immunological biomarkers predict HIV-1 viral rebound after treatment interruption. Nat Commun. 2015;6:8495.
- Rosas-Umbert M, Mothe B, Noguera-Julian M, Bellido R, Puertas MC, Carrillo J, et al. Virological and immunological outcome of treatment interruption in HIV-1-infected subjects vaccinated with MVA-B. PloS one. 2017;12(9):e0184929.
- Neidich SD, Fong Y, Li SS, Geraghty DE, Williamson BD, Young WC, et al. Antibody Fc effector functions and IgG3 associate with decreased HIV-1 risk. J Clin Invest. 2019.
- Perdiguero B, Raman SC, Sánchez-Corzo C, Sorzano COS, Valverde JR, Esteban M, et al. Potent HIV-1-Specific CD8 T Cell Responses Induced in Mice after Priming with a Multiepitopic DNA-TMEP and Boosting with the HIV Vaccine MVA-B. Viruses. 2018;10(8).
- Perdiguero B, Sánchez-Corzo C, CO SS, Mediavilla P, Saiz L, Esteban M, et al. Induction of Broad and Polyfunctional HIV-1-Specific T Cell Responses by the Multiepitopic Protein TMEP-B Vectored by MVA Virus. Vaccines. 2019;7(3).
- Perdiguero B, Sánchez-Corzo C, Sorzano COS, Saiz L, Mediavilla P, Esteban M, et al. A Novel MVA-Based HIV Vaccine Candidate (MVA-gp145-GPN) Co-Expressing Clade C Membrane-Bound Trimeric gp145 Env and Gag-Induced Virus-Like Particles (VLPs) Triggered Broad and Multifunctional HIV-1-Specific T Cell and Antibody Responses. Viruses. 2019;11(2).
- Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E. The complete DNA sequence of vaccinia virus. Virology. 1990;179(1):247-66, 517-63.
