Discurso de ingreso como Académica Correspondiente en la Real Academia Nacional de Farmacia.
1. INTRODUCCIÓN
El estrés oxidativo, término que describe la alteración de la homeostasis redox en células y tejidos con un incremento de los niveles de especies reactivas de oxígeno (ROS) u oxidantes, es un mecanismo patogénico común que subyace a múltiples enfermedades como las enfermedades cardiovasculares, los desórdenes neurodegenerativos, la inflamación y el cáncer. El oxígeno no fue considerado una molécula tóxica hasta el reconocimiento de que los radicales libres de oxígeno eran utilizados en los mecanismos de defensa de huésped de células eucariotas y bacterias para defenderse de, atacar y eliminar potenciales invasores. En este sentido, durante las seis últimas décadas, el concepto de que las ROS son moléculas tóxicas que deben de ser suprimidas y eliminadas ha sido ampliamente aceptado en la comunidad científica.
El papel perjudicial de las ROS en la enfermedad cardiovascular está bien documentado y se sabe que juegan un papel clave en la patogénesis de hipertensión, aterosclerosis, fallo cardiaco, ictus, hipertrofia cardiaca y en las complicaciones vasculares de enfermedades metabólicas como la diabetes, la obesidad y el síndrome metabólico. Con motivo de una estancia sabática en el año 2009 en el Departamento de Fisiología del New York Medical College (EEUU), en el Grupo de Estrés Oxidativo Vascular liderado por el Profesor Michael Wolin, tuve la oportunidad de familiarizarme conceptual y experimentalmente con el campo de estudio de las ROS en el sistema vascular, iniciando así una línea de investigación enfocada al estudio el papel del estrés oxidativo en la disfunción endotelial asociada a enfermedad metabólica. En el camino, además de la patología, nos encontramos con hallazgos en el laboratorio que señalan el papel relevante de las ROS en la función endotelial de lechos vasculares claves como el coronario o renal. Estos hallazgos, así como investigaciones relevantes en este campo de otros autores, quiero compartir hoy con ustedes.
Las ROS y especies reactivas de N2 pueden ser consideradas moléculas inestables que contienen oxígeno o nitrógeno y que tienen un electrón reactivo, o bien son intermediarios que pueden generar moléculas reactivas. Son entidades químicas reactivas que comprenden dos grandes grupos:
Radicales libres del O2: anión superóxido O2.-, anión hidroxilo OH–, óxido nítrico NO.. Son especies que tienen un electrón no pareado y son altamente reactivas.
Derivados del O2 no radicales: peróxido de hidrógeno H2O2 y peroxinitrito (ONOO). Debido a que las membranas celulares son permeables al H2O2, este actúa como principal molécula señalizadora tanto en procesos patológicos como fisiológicos.
Las principales fuentes de generación de ROS en la pared vascular son (1,2):
La mitocondria, donde se genera O2.- y H2O2 a partir de los complexos I y II de la cadena respiratoria que representa un 80 % de la producción endógena de O2.- producida basalmente.
Las NADPH oxidasas, complejos multienzimáticos que producen O2.- o H2O2 de forma primaria y no como parte de otras reacciones.
La xantín oxidasa, que genera O2.- como intermediario en la reacción en la que cataliza la oxidación de la xantina para formar ácido úrico.
La NOS desacoplada, que en situaciones de estrés oxidativo que oxidan el sustrato de síntesis tetrahidrobiopterina, se desacopla y genera O2.- en vez de NO.
En los sistemas biológicos, incluido el sistema vascular, el O2.- es rápidamente reducido a H2O2 por los sistemas antioxidantes de la SOD: SOD1 o CuZnSOD citosólica, SOD2 o MnSOD mitocondrial y SOD3, extracelular. O bien por enzimas como la catalasa que transforma el H2O2 en agua, la glutation peroxidasa (GPx) y las peroxiredoxinas (Prx) que eliminan el H2O2 gracias al alto poder reductor del glutation o la tioredoxina (2).
2. ROS Y FUNCIÓN ENDOTELIAL
Las ROS y en particular el H2O2, que es más estable y es capaz de difundir por las membranas y alcanzar las dianas celulares, actúan en la señalización oxidativa celular. Tradicionalmente han sido consideradas perjudiciales por sus implicaciones patológicas en diversas enfermedades humanas. Así, cuando se altera el balance entre los sistemas generadores de ROS y la actividad de los sistemas antioxidantes, lo que conlleva a una situación de estrés oxidativo, los niveles de ROS elevados van a favorecer la oxidación de diversas moléculas como proteínas, ácidos nucleicos y fosfolípidos, alterando su función y originando daño celular y tisular. En la pared vascular van a estar implicados en procesos inflamación vascular, remodelado, calcificación del músculo liso vascular (MLV) y disfunción endotelial (3). Sin embargo, la evidencia experimental acumulada sostiene que las ROS juegan un papel fundamental como moléculas señalizadoras en los procesos fisiológicos. Si se mantiene el equilibrio entre los sistemas generadores de ROS y los sistemas antioxidantes, las especies reactivas de oxígeno actúan como señalizadores e interaccionan mediante reacciones redox con canales iónicos, enzimas o factores de transcripción, participando así en la proliferación, migración, diferenciación fenotipo del MLV y los procesos de contracción y dilatación en los vasos sanguíneos (3).
El endotelio es la capa interna de los vasos sanguíneos que actúa como interfase entre la sangre y la pared vascular. El endotelio sano es un órgano paracrino, autocrino y endocrino que juega un papel clave en la homeostasis, secretando activamente diversas moléculas vasoactivas y tróficas que afectan la vasomoción, la proliferación y el crecimiento de células endoteliales y del MLV, las interacciones células endotelial-leucocito, la adhesión plaquetaria, la coagulación, la permeabilidad y la inflamación (4,5). En respuesta a estímulos físicos como el shear stress o fuerzas de cizallamiento producidas por el flujo sanguíneo o los incrementos de presión, o en respuesta a la hipoxia u otros estímulos químicos, el endotelio libera diversos factores vasodilatadores, anticoagulantes y antinflamatorios como el óxido nítrico (NO) o la prostaciclina (PGI2), y factores vasoconstrictores, con acciones proliferativas y proagregantes como el tromboxano A2 (TXA2) y la endotelina (ET-1). Sin embargo, el endotelio controla el tono vascular no solamente mediante la liberación de NO y PGI2, sino que también es capaz de activar por otras vías una hiperpolarización del MLV adyacente, que fue inicialmente adscrita a factores liberados por el endotelio que difundían al MLV activando canales de K+ y produciendo hiperpolarización y relajación, y que fueron inicialmente denominados “factores hiperpolarizantes derivados del endotelio” o EDHF (endothelium-derived hyperpolarizing factor). Entre los posibles EDHF fueron incluidos metabolitos del ácido araquidónico derivados de las vías de las citocromo P450 (CYP) epoxigenasas y de la lipooxigenasa, péptidos, gases como el H2S y el CO, y también ROS como el H2O2. Posteriormente, el concepto EDHF se amplía a “hiperpolarización derivada del endotelio” o EDH (endothelium-derived hyperpolarization) al demostrarse que este tipo de respuesta vasodilatadora incluye no solamente la hiperpolarización de las células de MLV, sino que se inicia con la hiperpolarización de las células endoteliales que posteriormente se propaga al MLV (6,7).
La primera evidencia de la participación del H2O2 en las respuestas vasodilatadores tipo EDH fue proporcionada por el laboratorio de Shimokawa y col (8) de la División de Medicina Cardiovascular de la Universidad Tohoku (Japón), quienes demostraron que las relajaciones tipo EDH y las correspondientes hiperpolarizaciones de arterias de resistencia mesentéricas, resistentes al bloqueo de las enzimas NO sintasa (NOS) y ciclooxigenasa (COX) pero inhibids por bloqueantes de canales de K+, eran reducidas por la enzima catalasa o por la eliminación del gen de la SOD citosólica en ratones Cu,Zn-SOD-/- (8). Ese mismo año, este grupo también demuestra que el H2O2 es un EDHF endógeno en los microvasos coronarios que participa en los mecanismos de autorregulación del flujo sanguíneo coronario, predominando las respuestas vasodilatadoras mediadas por H2O2 en las arterias coronarias más pequeñas (9). Posteriormente, las investigaciones llevadas a cabo en el laboratorio de William Chillian en EEUU confirman que el H2O2 acopla el flujo sanguíneo coronario al metabolismo cardiaco, demostrando la existencia de una correlación entre el consumo de oxígeno de los miocitos cardiacos y la producción de H2O2, así como también entre la producción de H2O2 y el flujo sanguíneo coronario (10). En la circulación coronaria nuestro grupo de investigación ha demostrado también que el papel vasoactivo del H2O2 como molécula señalizadora varía dependiendo del diámetro arterial, actuando como vasodilatador en las arterias coronarios de menor calibre y otras arterias de resistencia, como las mesentéricas de tercer orden, mientras que en los segmentos proximales de la arteria coronaria descendente izquierda, el H2O2 es un vasoconstrictor que libera TXA2 como factor contráctil derivado del endotelio y activa la entrada de Ca2+ a través de canales dependientes de voltaje tipo L el MLV coronario (11).
En esta misma línea de investigación y siguiendo con el estudio del papel de las ROS en la fisiología y fisiopatología endotelial, investigamos si el H2O2 podría jugar también un papel en la vasodilatación dependiente del endotelio de las arterias renales de resistencia, al igual que ocurría en el lecho arterial coronario y en otros lechos vasculares periféricos. El riñón recibe un 25 % del gasto cardiaco y el sistema vascular es esencial para llevar a cabo la función renal de depuración del plasma y de mantenimiento de la homeostasis de los líquidos y electrolitos en el organismo. Las investigaciones del grupo de Ingrid Fleming y Rudi Busse, del Instituto de Fisiología Carrdiovascular de la Universidad de Frankfurt, habían descrito en las arterias intrarrenales humanas una relajación endotelial de tipo EDH resistente al bloqueo de la NOS y acompañada por una hiperpolarización del MLV renal, cuya naturaleza y origen permanecían por dilucidar (12). En estudios llevados a cabo en nuestro laboratorio en arterias interlobares de la rata, demostramos que el H2O2 es un vasodilatador dependiente del endotelio, ya que las relajaciones no mediadas por NO o prostanoides podían ser inhibidas por la catalasa y miméticos de la glutation peroxidasa como el ebselen. Además, el H2O2 activa canales de K+ dependientes de Ca2+ (KCa) en el endotelio e hiperpolariza la células endoteliales iniciando así una respuesta EDH que se propaga al MLV, donde se produce también una hiperpolarización y un descenso del Ca2+ intracelular que conduce a la relajación de las arterias renales (13).
Las enzimas citocromo P450 (CYP) epoxigenasas tienen la capacidad de metabolizar el ácido araquidónico y generar ácidos epoxieicosatrienóicos (EETs) e hidroxieicosatetraeonoicos (HETEs). El riñón tiene una expresión significativa de las enzimas CYP y tanto los EETs como los HETEs actúan en las células del epitelio tubular renal alterando el transporte de sodio. Sin embargo, también se ha atribuido a los derivados del ácido araquidónico por la vía de las CYP epoxigenasas y CYP hidroxilasas, los EETs y HETEs respectivamente, un papel en el control local del flujo sanguíneo renal. El 20-HETE contrae las arteriolas aferentes y contribuye a la autorregulación del flujo sanguíneo renal, mientras que los EETs han sido propuestos como factores hiperpolarizantes dependientes del endotelio (EDHFs) (14). En el endotelio vascular coronario, las enzimas CYP2C son una fuente de ROS, y el inhibidor selectivo de la CYP2C9, sulfafenazol, abole la generación de O2.- en células que sobreexpresan esta isoenzima o en células endoteliales estimuladas con bradicinina, si bien potencia pero no inhibe las relajaciones dependientes del endotelio de las arterias coronarias, indicando que las CYP2C epoxigenasas son una fuente de ROS vasoconstrictoras en las arterias coronarias (15). En las arterias intrarrenales de la rata, sin embargo, nuestro grupo ha demostrado que las CYP2C epoxigenasas se expresan en el endotelio renal colocalizadas con la eNOS (Figura 1) y las relajaciones endoteliales sensibles a catalasa y la producción de H2O2 por estimulación del endotelio en arterias intactas son reducidas por el inhibidor de la CYP2C epoxigenasa, lo que sugiere que esta enzima es una fuente de generación de H2O2 vasodilatador en arterias renales (13).
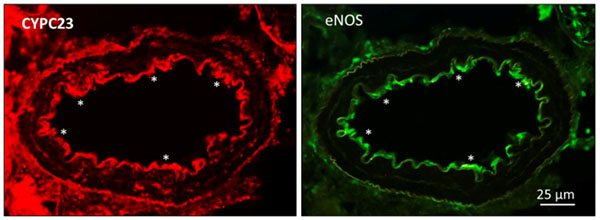
Figura 1. “Expresión de la enzima CYP2C23 colocalizada con eNOS en el endotelio de las arterias renales interlobares del riñón de ratas Wistar”.
2.1. NADPH oxidasas y sistema vascular
Las enzimas NADPH oxidasas de la familia Nox son las únicas enzimas que generan ROS de forma primaria y catalizan la conversión de O2 en O2.- usando NADPH como donante de electrones. Otras enzimas como las COX, las CYP epoxigenasas o las enzimas de la cadena de transporte mitocondrial pueden producir ROS pero como subproductos de su función normal. Las Nox representan una fuente importante de ROS en el sistema vascular y juegan un papel esencial tanto en la salud como en la patología vascular. Las Nox se expresan en abundancia en el plasma y en las membranas plasmática y lisosomal de los fagocitos, neutrófilos y macrófagos, y su activación es responsable del “estallido respiratorio u oxidativo” (Figura 2), proceso por el cual algunas células son capaces de producir y liberar ROS como O2.- y H2O2, con un aumento marcado de la demanda de O2 por parte de la célula, mecanismo utilizado por las células del sistema inmune para producir compuestos oxidantes -H2O2 y anión hipoclorito- con capacidad microbicida.
En función del número de grandes subunidades catalíticas transmembrana, se han identificado 7 homólogos Nox en el genoma humano: de Nox1 a Nox5, Duox1 y Duox2, que difieren en su nivel de expresión y control, en el tipo de ROS que generan y en el control de su activación (16). Todas las Nox son enzimas multisubunidad en las que las distintas subunidades necesitan formar un complejo para generar ROS. El control de la activación de estas enzimas se ejerce por calcio o por interacciones proteína-proteína, con la excepción de la Nox4 que es constitutivamente activa. La Nox5 depende de calcio y genera anión O2.- de forma primaria (17), mientras que el otro grupo de Nox, dependientes de calcio, son la Duox1 y Duox2 que producen H2O2, aunque su expresión es muy baja en el sistema vascular. El resto de las Nox (Nox1-4) requieren una subunidad, la p22phox, para la estabilización de la subunidad Nox. La Nox4 no requiere ninguna subunidad adicional además de la p22phox para la actividad constitutiva de la enzima y, al igual que las enzimas Duox, parece producir directamente H2O2. La Nox1, la Nox2 y la Nox3 requieren subunidades citosólicas para su activación. En el modelo clásico, el leucocito, la Nox2 es activada por la p67phox (y la Rac2), y el complejo es fijado a la Nox2 por la p47phox que actúa como adaptador. En el caso de la Nox1, la función activadora de la p67phox es llevada a cabo por NoxA1 (Nox-Activator1) y la función organizadora de la p47phox por la NoxO1 (Nox-Organizer1) (17).
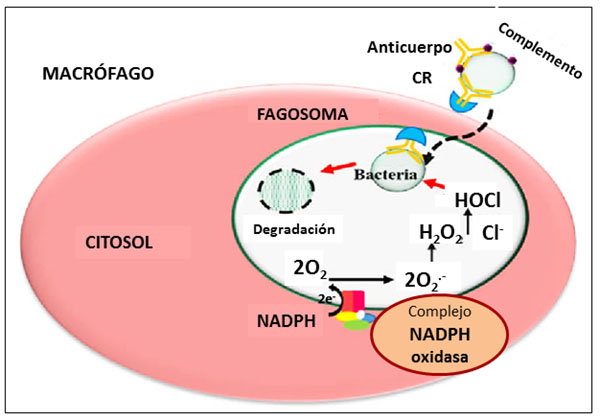
Figura 2. “Estallido respiratorio u oxidativo” de los fagocitos del sistema inmune”
Expresión de las Nox en la pared vascular
En células no fagocíticas, incluyendo las células de la pared vascular, las Nox se expresan a niveles mucho menores que en los fagocitos y se distribuyen no solamente en la membrana plasmática sino también en otros compartimentos subcelulares como la mitocondria, el retículo endoplásmico, el núcleo y los lisosomas. En estas células las Nox producen de manera controlada niveles no citotóxicos de ROS que participan en la señalización REDOX durante el metabolismo celular normal.
Cuatro de las isoformas Nox, Nox1, Nox2, Nox4 y Nox5 se expresan en el endotelio, aunque también están presentes en otras células vasculares como las del MLV, los fibroblastos adventiciales y diferentes subgrupos de leucocitos presentes en la pared vascular durante los estados de enfermedad. La Nox2, que es la isoforma predominante en los leucocitos, fue la primera isoforma identificada en las células endoteliales y se encuentra también en los fibroblastos y en los miocitos cardiacos, siendo, por otra parte, una de las Nox más importantes en el contexto de patología vascular. La Nox1, isoforma típica de las células epiteliales del colon, y sus proteínas organizadora y activadora NoxO1 y NoxA1, se expresan en las células endoteliales y han sido asociadas con la generación de ROS inducidas por lipoproteínas LDL oxidadas, y por tanto con el estrés oxidativo vascular en condiciones fisiopatológicas. La Nox1 se expresa fundamentalmente en el MLV de roedores, aunque su papel en el MLV humano parece ser llevado a cabo por Nox2 y Nox5 (1,16,18).
Dentro de la familia de proteínas Nox, la expresión de la Nox4 en las células endoteliales, y en menor medida en el MLV, es mucho mayor que la de cualquiera de las otras isoformas de la Nox. Tiene actividad constitutiva, se localiza en la mitocondria y en el retículo endoplásmico (RE) de las células endoteliales, y a diferencia Nox1 y Nox2 que producen primariamente O2.-, la Nox4 genera principalmente H2O2. La Nox5 es la única Nox endotelial que no es activada por subunidades citosólicas phox o Rac, sino que tiene un dominio N-terminal similar a la calmodulina, con cuatro lugares de unión para el Ca2+, y su actividad está modulada, por tanto, por cambios en la concentración intracelular de Ca2+ (16).
En el sistema cardiovascular se ha descrito fundamentalmente un incremento de la expresión de las isoformas Nox1 y Nox2 ligado al estrés oxidativo en situaciones de disfunción vascular asociada a enfermedades vasculares o complicaciones en el curso de enfermedad metabólica como hipertensión, hipercolesterolemia, diabetes, obesidad, síndrome metabólico y enfermedad arterial (18). En estas situaciones existe un estado proinflamatorio en la pared vascular y las proteínas citosólicas activadoras de Nox1 y Nox2 son, a su vez, inducidas por mediadores de la inflamación como el TNFɑ, trombina, angiotensina II (AII), hiperglucemia u oxilípidos (17). Por el contrario, mientras que los estudios con animales transgénicos o en los que se ha eliminado el gen de la Nox1 y Nox2 indican que estas isoformas participan en la patología vascular, los estudios con Nox4 indican que su actividad puede conferir protección vascular (19).
2.2. Papel de las Nox en el riñón
En el riñón todas las isoformas de Nox -Nox1, Nox2, Nox4 y Nox5- se expresan de forma específica dependiendo de la región de la nefrona y del tipo celular, y han sido localizadas en el mesangio, los túbulos, la mácula densa, el endotelio y el MLV renal. La Nox2 también ha sido implicada en diversas funciones renales como el feedback o retroalimentación túbulo glomerular y la respuesta de la mácula densa a las altas concentraciones de sal con el fin de regular el volumen de filtrado, la regulación del tono de la arteriola aferente y el manejo de glucosa y el transporte de electrolitos en el túbulo (20). De todas las isoformas Nox, la Nox4 es la que se expresa con mayor abundancia (túbulos, células mesangiales y podocitos) y, por ello, fue inicialmente denomina Renox, si bien ha sido asociada e implicada como isoforma principal responsable del estrés oxidativo y la lesión renal en la nefropatía diabética. Así, la regulación al alza de Nox4 producida por la hiperglucemia y otros factores incrementados en el medio diabético, incluyendo el sistema renina-angiotensina y el factor de crecimiento transformante (TGF), y el consiguiente estrés oxidativo derivado de Nox4 conduciría a la hipertrofia glomerular y al acúmulo de la matriz mesangial responsables de la lesión glomerular, la proteinuria y la fibrosis que subyacen a la nefropatía diabética (21).
A pesar de la abundancia de literatura científica que implica al estrés oxidativo de la Nox en las complicaciones vasculares de diabetes, incluyendo la nefropatía diabética, el papel funcional de estas enzimas en el sistema vascular y la hemodinámica renal ha permanecido durante tiempo sin clarificar, a pesar del papel atribuido a los ROS en la vasodilatación-dependiente del endotelio de las arterias renales. Por esta razón, en un estudio traslacional llevado a cabo en nuestro grupo de investigación de la UCM en colaboración con el Hospital Universitario Puerta de Hierro de Madrid, investigamos si las Nox, en particular la Nox2 y Nox4, constituían una fuente funcional de H2O2 en el endotelio renal y participaban, por tanto, en la vasodilatación dependiente del endotelio. Las relajaciones endoteliales resistentes al bloqueo de la eNOS y de la COX fueron inhibidas por el bloqueante no selectivo de Nox apocinina, que inhibió también la generación de O2.- tanto en arterias intrarrenales humanas como de la rata. Además, la Nox4 es una fuente de H2O2 endotelial vasodilatador en las arterias renales, se colocaliza con las eNOS en el endotelio renal (Figura 3) y los inhibidores de la Nox4 y los antioxidantes mitocondriales como el mitoTEMPO reducen las relajaciones dependientes del endotelio no-NO no-prostanoide de las arterias renales. También pudimos demostrar que la Nox2 es una fuente endotelial de H2O2 vasodilatador in las arterias renales (Figura 3). Nox4 y Nox2 son fuentes funcionalmente relevantes de generación H2O2 que contribuyen a la vasodilatación renal dependiente del endotelio y tienen, por tanto, un papel protector de la función vascular renal (22).
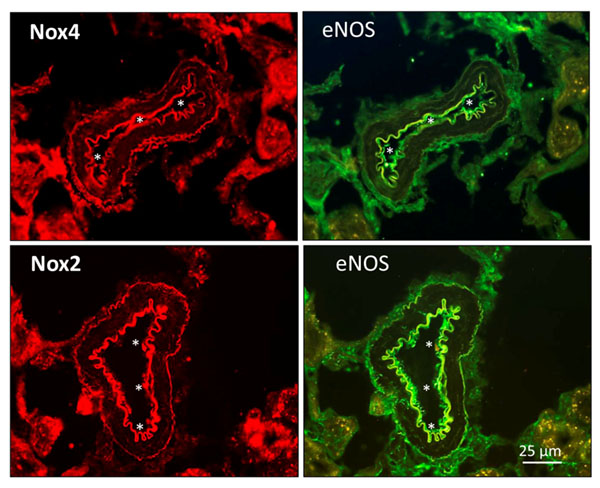
Figura 3. “Expresión de las enzimas Nox4 y Nox2 colocalizadas con eNOS en el endotelio de las arteriolas de la corteza renal del riñón de rata Wistar”
3. ROS y DISFUNCIÓN VASCULAR
El estrés oxidativo y la disfunción endotelial asociada son factores patogénicos clave que subyacen a las enfermedades cardiovasculares como hipertensión, aterosclerosis, fallo cardiaco, ictus, hipertrofia cardiaca, enfermedad arterial coronaria, y a las complicaciones vasculares de enfermedades metabólicas como la diabetes, la obesidad y el síndrome metabólico, incluyendo la nefropatía diabética. Los pacientes con enfermedad vascular o factores de riesgo cardiovascular (hipercolesterolemia, tabaquismo, diabetes mellitus) presentan disfunción endotelial caracterizada por el descenso de biodisponibilidad de NO asociada a la mayor generación de ROS.
La obesidad es un problema de salud pública cuya incidencia incrementa de forma alarmante y afecta en la actualidad a niños y adolescentes, constituyendo un factor de riesgo clave para el desarrollo de enfermedad metabólica y cardiovascular (23, 24). El sobrepeso y la obesidad se definen como una acumulación anormal o excesiva de grasa que puede ser perjudicial para la salud. Según el informe de la OMS de 2018, la obesidad ha alcanzado proporciones epidémicas a nivel mundial, y cada año mueren como mínimo 2,6 millones de personas a causa de la obesidad o el sobrepeso. Entre 1975 y 2016 la prevalencia mundial de la obesidad casi se ha triplicado. Por otra parte, en el mundo hay más de 42 millones de menores de cinco años con sobrepeso y la obesidad infantil es uno de los problemas de salud pública más graves del siglo XXI.
En condiciones de salud, los depósitos de grasa del organismo se mantienen en balance por medio de mecanismos homeostáticos que regulan los sustratos de los alimentos (glucosa, y ácidos grasos libres) y la acción de hormonas como la leptina y la insulina. La obesidad se caracteriza por un exceso en el acúmulo de grasas debido a una falta de balance crónico entre el ingreso y el gasto energético (el aumento del consumo de alimentos muy ricos en calorías sin un aumento proporcional de la actividad física produce un aumento de peso), lo cual va a afectar a diferentes tipos celulares incluyendo adipocitos, hepatocitos, células del músculo esquelético, células endoteliales y células del sistema inmune, originando estrés oxidativo, estrés del retículo endoplásmico, inflamación, resistencia a la insulina, para dar lugar en última instancia a disfunción metabólica. La relación entre obesidad y enfermedad metabólica y vascular está bien definida. Los estudios epidemiológicos de Bays y col (24), en los que se determina la distribución del índice de masa corporal (BMI) en la población de pacientes con 50 % son obesos; b) el 35 % de pacientes con dislipidemias tienen sobrepeso y casi un 35 % son obesos; c) el 35 % de pacientes con hipertensión tienen sobrepeso y casi un 40 % son obesos. La similar distribución del BMI en pacientes diabéticos, con dislipidemia e hipertensos se explica porque a estos desórdenes subyacen mecanismos patogénicos comunes (24).
3.1. Adiposopatía
Entre las respuestas celulares adversas al exceso de nutrientes (oxidación de ácidos grasos libres -AGL- y glucosa en la mitocondria y otros procesos en la célula) se produce un exceso de ROS o estrés oxidativo en asociación con una respuesta inflamatoria. La acumulación excesiva de tejido adiposo e hipertrofia de los adipocitos conduce a una situación de hipoxia que provoca la infiltración de macrófagos y otras células del sistema inmune y la inflamación del tejido adiposo o “adiposopatía” (Figura 4). Así, el tejido adiposo hipertrofiado adquiere un fenotipo pro-inflamatorio caracterizado por el exceso de generación de ROS y secreta mayores cantidades de citoquinas, adipoquinas y AGL, promoviendo una inflamación de bajo grado que conduce a resistencia a la insulina no solo en el músculo esquelético e hígado, sino también en las células endoteliales produciendo disfunción endotelial y vascular (5,25). Esta situación de inflamación del tejido adiposo en la obesidad se reproduce en el tejido adiposo perivascular, que adquiere también un fenotipo inflamatorio, con alteración en el perfil de secreción de adipoquinas y citoquinas inflamatorias, lipotoxicidad e incremento del estrés oxidativo que conduce a la disfunción endotelial. Así, a pesar de que se ha descrito que el estrés oxidativo sistémico está incrementado y se correlaciona con el BMI enfermedad metabólica y vascular, demuestran que: a) el 22 % de pacientes con diabetes tienen sobrepeso y casi un y la circunferencia de la cintura en individuos obesos y modelos animales de obesidad (26), los estudios experimentales revelan la ausencia de cambios en el estrés oxidativo sistémico asociados a una función endotelial anormal, lo que sugiere que el estrés oxidativo vascular es el mayor determinante de la disfunción endotelial en los estadíos tempranos de la obesidad (27).
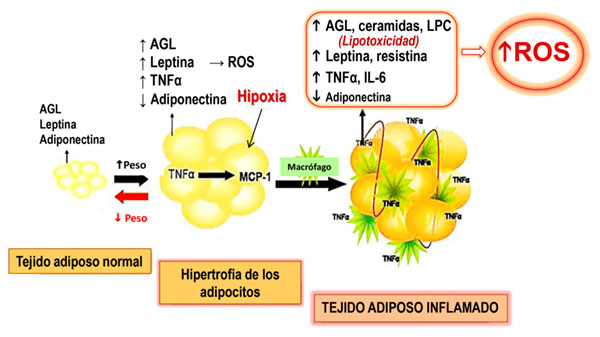
Figura 4. Adiposopatía. Inflamación del tejido adiposo en la obesidad. AGL: ácidos grasos libres; LPC: lipofosfatidil colina; TNFα: factor de necrosis tumoral α; MCP-1: proteína quimiotáctica de monocitos 1; ROS: especies reactivas de O2. Modificado de Baker y col. (25).
3.2. Estrés oxidativo, disfunción endotelial y obesidad
A pesar de que el endotelio vascular puede adaptarse a diversos estreses incluyendo el estrés mecánico, metabólico y oxidativo, la inflamación y la hipoxia, la disfunción endotelial, que es un indicador temprano de enfermedad vascular, representa un fenotipo endotelial mal adaptado caracterizado por la alteración de la vasodilatación, la angiogénesis y la función barrera del endotelio, junto con un incremento de la expresión de factores proinflamatorios y protrombóticos. Aunque diversos factores pueden comprometer la síntesis de NO, que normalmente protege la pared vascular de los eventos moleculares que conducen a la arteriosclerosis, se considera que la causa primaria de la disfunción endotelial en la obesidad es la reducción de la cantidad de NO biodisponible debido al estrés oxidativo. El NO es rápidamente inactivado por la reacción con el O2.- que genera anión peroxinitrito, un radical oxidante poderoso y altamente tóxico que causa daño en el DNA, proteínas y lípidos, produce desacoplamiento de la eNOS, lo que generaría más O2.-, aumenta la apoptosis, la lesión tisular y la inflamación. Además, el estrés oxidativo generado por la lipotoxicidad activa el factor de transcripción sensible a REDOX NFκ-B, que a su vez favorece la expresión de genes inflamatorios y el incremento de las moléculas de adhesión, COX-2, TNFɑ, IL6 y CRP, así como la regulación al alza de las subunidades de Nox y la generación de más estrés oxidativo. La NADPH oxidasa es una fuente principal de generación de estrés oxidativo a nivel vascular en la obesidad, y se ha descrito que la expresión y actividad de las subunidades Nox1 y Nox4, así como las subunidades reguladoras p22- y p47phox están incrementada en aorta, arterias cerebrales y coronarias de modelos de obesidad genética e inducida por dieta (5).
3.3. Estrés oxidativo y disfunción endotelial coronaria en la obesidad
La obesidad y el síndrome metabólico incrementan el riesgo de enfermad cardiaca y fallo cardiaco, siendo el estrés oxidativo en el corazón un factor patogénico común en el desarrollo de enfermedad cardiovascular en la obesidad, la diabetes y otros estados de resistencia a la insulina (28). Utilizando un modelo experimental de obesidad genética/síndrome metabólico, la rata Zucker obesa, en nuestro grupo de investigación pudimos demostrar que en las arterias coronarias de ratas obesas el estrés oxidativo basal y el derivado de Nox1, Nox2 and Nox4 está incrementado, a pesar de que la función endotelial está preservada en arterias coronarias, debido a un incremento compensador de la actividad de la enzima de síntesis de NO y de los sistemas antioxidantes (29,30).
La obesidad y la diabetes conducen a un estado proinflamatorio de la pared vascular, y los prostanoides derivados de la COX-2 son importantes mediadores de la repuesta inflamatoria. En los vasos sanguíneos sanos, la mayoría de los prostanoides son producidos por la isoforma constitutiva COX-1, localizada en el endotelio vascular y asociada principalmente a la producción de TXA2, mientras que la COX-2 se considera tradicionalmente una isoforma inducible que se expresa a niveles bajos o indetectables en tejidos normales, pero es rápidamente inducida por estímulos inflamatorios, mitogénicos y mecánicos. La expresión de COX-2 es rápidamente inducida por citoquinas, promotores de tumor y factores de crecimiento. La COX-2 se regula al alza en condiciones de inflamación de bajo grado en la pared vascular, en estados de resistencia a la insulina como la diabetes (31) y en la hipertensión (32,33), constituyendo una fuente importante de estrés oxidativo asociado a disfunción endotelial. Nuestro grupo ha demostrado que la COX-2 se encuentra también regulada al alza en las arterias coronarias en la obesidad (34), y su expresión incrementada también se asocia con el aumento del estrés oxidativo vascular (35). Además, hemos confirmado la existencia de un mecanismo de amplificación de la producción de ROS y un asa de retroalimentación positiva COX-2- ROS que contribuye al estrés oxidativo vascular en la obesidad (Figura 5), y previamente descrito en hipertensión (32,33). Así, el estrés oxidativo puede incrementar la expresión y la actividad de la COX-1 y de la COX-2, y específicamente, el H2O2 estimula la expresión de COX-2 tanto en células endoteliales como como de MLV. Los datos obtenidos por nosotros demuestran que el H2O2 regula al alza la expresión e incrementa la actividad de la COX-2 en las arterias coronarias. La COX-2 tiene una expresión baja en el endotelio coronario de animales control, pero la exposición aguda al oxidante H2O2 induce la expresión rápida de la enzima tanto en el endotelio como en el MLV y un incremento marcado en los niveles de contenido de la proteína COX-2 en animales control, similar al contenido en arterias coronarias de animales obesos. El incremento de la expresión de la COX-2 está acoplado a un incremento de la producción de O2.- y del estrés oxidativo derivado de COX-2 (35)
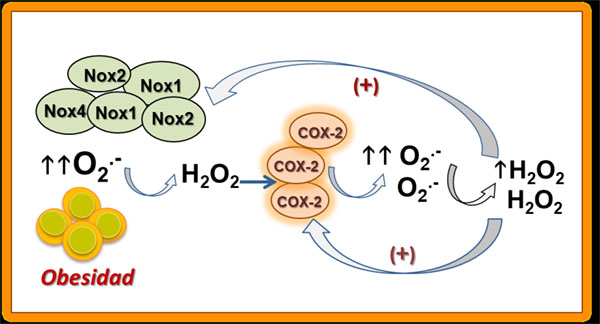
Figura 5. “Asa de retroalimentación positiva COX-2- ROS y contribución al estrés oxidativo vascular en la obesidad”.
Sin embargo, en las arterias coronarias, la expresión incrementada de COX‐2 y el consiguiente incremento del estrés oxidativo arterial en la obesidad no se encuentra asociado con una vasodilatación endotelial alterada sino que, por el contrario, se asocia con el incremento de la producción basal de una prostaglandina relajante (34), lo que también había sido descrito en arterias coronarias de pacientes diabéticos (36). Posteriormente, nuestro grupo identificó esta prostaglandina vasodilatadora como PGE2, que actuando sobre receptores EP4 probablemente juega un papel protector frente al daño oxidativo producido por los ROS en las arterias coronarias (35), consistente con el papel beneficioso recientemente atribuido a este receptor en el proceso inflamatorio asociado a la obesidad (37,38). La relevancia de estos hallazgos, que muestran por una parte que la COX-2 es una fuente importante de estrés oxidativo y riesgo cardiovascular en las arterias coronarias en la obesidad, pero, por otra, que esta isoenzima está implicada en efectos vasculares protectores mediante la liberación de PGE2 vasodilatadora del endotelio, sugiere que el enfoque de los esfuerzos terapéuticos para combatir el estrés oxidativo vascular y la disfunción endotelial en la obesidad debe dirigirse hacia dianas por debajo de la COX-2 en la vía de señalización de esta enzima, es decir, a nivel del receptor EP4, para así desviar el balance eficacia/riesgo cardiovascular descrito para los antinflamatorios no esteroides (AINES) (38,39).
3.4. Estrés oxidativo y disfunción endotelial renal en la obesidad
La obesidad y el Síndrome Metabólico, conjunto de alteraciones metabólicas y cardiovasculares que incluyen adiposidad visceral, resistencia a la insulina, dislipidemia e hipertensión y que predisponen a la diabetes tipo 2, se asocian con un incremento del riesgo de complicaciones diabéticas como son la nefropatía diabética y la enfermedad renal crónica (ERC). Sin embargo, investigaciones recientes sugieren que la enfermedad renal crónica puede desarrollarse en individuos obesos no diabéticos, y los estudios epidemiológicos indican que la obesidad constituye un factor de riesgo de enfermedad renal crónica independiente de la presencia de diabetes, hipertensión y otras comorbilidades (40). En realidad, el incremento global de la ERC se produce en paralelo con la epidemia global de obesidad. La microalbuminuria, que posteriormente progresa a albuminuria abierta, es la indicación más temprana de la disfunción vascular renal asociada (41).
Nuestros estudios iniciales sobre disfunción endotelial y estrés oxidativo se llevaron a cabo en el lecho vascular renal, donde el objetivo fue valorar la función endotelial y la produccción vascular de ROS con el fin de determinar si los ROS y sus fuentes (COX-2 y Nox) estaban implicados en la disfunción vascular y nefropatia asociada a la obesidad. En el modelo de obesidad genética de la rata Zucker, las arterias renales interlobares presentan disfunción endotelial como se deduce de las vasodilatación dependiente del endotelio, reducida y asociada a un incremento de los niveles basales y estimulados por ET-1 de O2.-., derivados de la actividad incrementada tanto de las Nox como de la xanthin oxidasa y de la NOS desacoplada en el riñón de ratas obesas. Además, la COX-2 está regulada al alza y asociada al incremento del estrés oxidativo arterial y a la disfunción endotelial renal en la obesidad. Así, la COX-2 es constitutiva y se expresa en el endotelio de las arterias renales de ratas control, estando acoplada a la producción de prostanoides contráctiles, como se deduce del incremento de las respuestas relajantes dependientes del endotelio al bloquear esta isoenzima. En las arterias renales de animales obesos, se produce un incremento marcado de la expresión de COX-2 que se extiende al MLV y se asocia con un aumento de la producción de prostanoides contráctiles y O2.-, ya que el bloqueo de las COX-2 normaliza las respuestas relajantes dependientes del endotelio y también la producción incrementada de ROS. Por tanto, la COX‐2 está regulada al alza y asociada al incremento del estrés oxidativo arterial y a la disfunción endotelial renal en la obesidad (42).
3.5. Estrés oxidativo y disfunción endotelial renal en la obesidad: papel de las Nox
En la última parte de esta conferencia explicaré nuestros hallazgos sobre el papel del estrés oxidativo dependiente de la NADPH oxidasa en la disfunción vascular renal en la obesidad, sobre todo, teniendo en cuenta la controversia existente sobre el papel específico de las distintas subunidades de Nox en la nefropatía asociada a la enfermedad metabólica, ya que por una parte, isoenzimas como la Nox4 han sido consideradas junto con la mitocondria como las fuentes de estrés oxidativo más importantes en la patogénesis de la nefropatía diabética (20,21), mientras que por otro lado, nuestros datos en las arterias renales humanas y datos de otros autores demuestran el papel protector vascular de Nox4 y Nox2 en la función endotelial bajo condiciones fisiológicas (19,22). En el curso de nuestras investigaciones nos encontramos en el laboratorio con esta paradoja que, en realidad, fue el motivo que nos hizo acudir a la fisiología para valorar el papel de los ROS en la función endotelial renal. Así, a pesar del estrés oxidativo vascular que ya habíamos descrito en las arterias renales de ratas obesas (42), la disfunción endotelial renal asociada no era revertida por antioxidantes clásicos como el mimético de la SOD tempol, antioxidantes mitocondriales o inhibidores no selectivos de las Nox como la apocinina. Por otra parte, el efecto protector a nivel vascular demostrado para la Nox4 en arterias renales de ratas obesas estuvo comprometido: su expresión en el endotelio vascular fue menor y los inhibidores de esta subunidad apenas redujeron ni las relajaciones mediadas por H2O2 ni la producción de H2O2 derivada de NADPH (43). Además, si bien los niveles totales de O2.- estuvieron incrementados en tejido vascular y cortical renal, la producción de H2O2 estuvo reducida coincidiendo con el menor contenido de Nox4, subunidad de localización mitocondrial, lo cual indicaría una disfunción mitocondrial con alteración del metabolismo de las ROS. En realidad, y a pesar del papel atribuido al estrés oxidativo derivado de Nox4 en la nefropatía diabética, nuestros hallazgos confirmarían el papel protector de esta enzima en la función endotelial renal, e indican que posiblemente la pérdida de este papel beneficioso es en realidad lo que contribuiría a la disfunción vascular asociada a la obesidad (43).
Por el contrario, otras subunidades de la familia de las NADPH oxidasas como la Nox1 parecen ser responsables del estrés oxidativo vascular que subyace a la disfunción endotelial renal en la obesidad y el síndrome metabólico. Tanto la Nox1 como la Nox2 han sido vinculadas a los procesos de inflamación vascular en estados de resistencia a la insulina como la diabetes y la hipertensión (44-46). La Nox1 es una fuente importante de generación de O2.- en células endoteliales, de MLV y fibroblastos, tanto en condiciones fisiológicas como fisiopatológicas. Participa en el remodelado vascular inducido por shear stress, mientras que en la patología ha sido implicada en procesos proliferativos e hipertensión (45). En el riñón de las ratas obesas, la Nox1 estuvo regulada al alza en arterias y corteza renal, e inhibidores selectivos de la Nox1 redujeron la formación de O2.- y mejoraron las relajaciones dependientes del endotelio en arterias renales. Estos datos demuestran un papel relevante de la producción de ROS derivada de Nox1 en el estrés oxidativo vascular y en la disfunción endotelial asociados a la obesidad, mientras que la expresión reducida de la Nox4 junto con una menor producción de H2O2 y una vasodilatación dependiente de H2O2 reducida pueden disminuir los efectos protectores de la Nox4 y contribuir a la lesión renal en la obesidad (43).
4. CONCLUSIONES
En resumen, los estudios llevados a cabo en los últimos años han confirmado el papel perjudicial de las ROS en la enfermedad cardiovascular y la implicación del estrés oxidativo en la disfunción endotelial que subyace a la arteriosclerosis, hipertensión, enfermedad arterial coronaria y complicaciones vasculares de diabetes, obesidad y otros estados de resistencia a la insulina. Sin embargo, la evidencia experimental acumulada en las dos últimas décadas ha permitido también un cambio en el paradigma que consideraba a las ROS únicamente como moléculas perjudiciales con una implicación clave en la patología cardiovascular, siendo en la actualidad consideradas moléculas de señalización cruciales en la fisiología endotelial y vascular. Las ROS juegan un papel esencial como factores EDH en la función endotelial de los lechos vascular renal y coronario, acoplando en este último el flujo sanguíneo al metabolismo. A pesar de actuar como fuentes de factores EDH (Nox) y factores endoteliales contráctiles (COX-2) en condiciones fisiológicas, ambas enzimas contribuyen de forma importante al estrés oxidativo vascular durante la obesidad. Esto podrían explicar el fracaso clínico de muchas de los ensayos con antioxidantes y sugiere que las estrategias terapéuticas destinadas a combatir el estrés oxidativo y las complicaciones vasculares en estados de resistencia a la insulina deben de tener en cuenta, para ser eficaces, las particularidades del metabolismo de las ROS en los distintos lechos vasculares.
5. FINANCIACIÓN
Trabajos financiados por los proyectos SAF2009-10448 y SAF2012-31631 del MICINN y SAF2016-77526 del MINECO, España
6. AGRADECIMIENTOS
Me gustaría expresar mi agradecimiento a una serie de personas que me han apoyado en mi vida profesional y personal a lo largo de estos años. En primer lugar, al Prof. Albino García Sacristán, Académico de número de esta institución y Catedrático de Fisiología de la Universidad Complutense de Madrid, por haberme abierto en su día, hace ya muchos años, las puertas del Departamento de Fisiología de la Facultad de Veterinaria, donde pude iniciarme y formarme en la investigación y docencia en Fisiología. Gracias por sus enseñanzas y apoyo durante todo este tiempo.
Mi agradecimiento también a los profesores Medardo Hernández, Sara Benedito y Luis Rivera, Catedráticos de Fisiología de esta Universidad, compañeros de ciencia y de docencia durante una larga trayectoria como profesores e investigadores en la Universidad Complutense. Y gracias a todos los profesores del Departamento de Fisiología de la Facultad de Farmacia de la Universidad Complutense, que funcionan como un grupo compacto que nos permite en la actualidad proporcionar a los estudiantes de Farmacia las enseñanzas de Fisiología, base de posteriores disciplinas esenciales en su carrera como profesionales de Ciencias de la Salud.
Me gustaría mencionar a las personas que han participado en las investigaciones que he mostrado a lo largo de esta conferencia, Dras. Mercedes Muñoz, María Pilar Martínez, Elvira López-Oliva, Cristina Contreras, Ana Sánchez, Belén Climent, Elvira Santiago, Dres. Medardo Hernández y Luis Rivera, y Sres. Claudia Rodríguez y Estéfano Pinilla, muchos de ellos profesores de la Universidad Complutense, algunos antiguos doctorandos y otros doctorandos actuales. También, al Dr. Javier Sáenz Medina del Servicio de Urología del Hospital Universitario Puerta de Hierro-Majadahonda de Madrid, por su valiosa colaboración en los estudios traslacionales de estrés oxidativo en riñón humano. A todos ellos, gracias por su aportación a la investigación y al trabajo científico que he presentado hoy, así como también a los Sres. Manuel Perales y Francisco Puente por su inestimable apoyo técnico.
Quiero mencionar y agradecer también a los investigadores excelentes con los que he tenido la oportunidad de colaborar en mis estudios sobre disfunción endotelial, estrés oxidativo y obesidad. A la Prof. Mercedes Salaices, Académica Numeraria de esta institución, y a la Dra. Ana Briones, ambas del Departamento de Farmacología de la Universidad Autónoma de Madrid. A los Dres. Carmen Martínez y Ramaranson Adriantsitohaina del laboratorio de “Stress oxydant et pathologies métaboliques l Sopam” del INSERM, Universidad de Angers (Francia). Al profesor Michael Wolin del Departamento de Fisiología del New York Medical College (EEUU) y al Dr. Miguel López, del Departamento de Fisiología y del CIMUS de la Universidad de Santiago de Compostela.
Me gustaría también recordar y agradecer de forma especial al Prof. Mike Mulvany, del Departamento de Farmacología de la Universidad de Arhus (Dinamarca), con quien tuve el privilegio de iniciar mis investigaciones en arterias de resistencia, y quien ha sido sin duda mi mayor mentor científico. También al Profesor Niels Nyborg, gran farmacólogo vascular de la misma universidad, con el que comencé las investigaciones sobre el endotelio arterial a finales de la década de los 80, y del que aprendí mucho de lo que después guiaría mis pasos en la investigación sobre función y disfunción endotelial.
Mi agradecimiento a los profesores Antonio Rodríguez Artalejo y María Teresa Miras Portugal, Académicos numerarios de esta institución, por su consejos y su apoyo en momentos importantes de mi carrera académica.
Y finalmente, last but not least, mi agradecimiento, de corazón, a mi familia; ellos han estado y siempre están ahí. A mi hija Kooki, que sin duda ha sido y es mi mejor proyecto y mi mayor estímulo vital. A las mujeres de mi familia, mis hermanas, mi madre y mi abuela. Y también a los amigos y gente querida que me han apoyado en mi trayectoria personal y vital. Gracias.
7. REFERENCIAS
- Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research, Antioxid. Redox Signal 2014; 20: 164-82.
- Satoh K, Godo S, Saito H, Enkhjargal B, Shimokawa H. Dual roles of vascular-derived reactive oxygen species–with a special reference to hydrogen peroxide and cyclophilin A. J Mol Cell Cardiol 2014; 73: 50-6:
- Byon CH, Heath JM, Chen Y. Redox signaling in cardiovascular pathophysiology: A focus on hydrogen peroxide and vascular smooth muscle cells. Redox Biol 2016; 9: 244-53.
- Flammer AJ, Lüscher TF. Human endothelial dysfunction: EDRFs. Pflugers Arch – Eur J Physiol 2010; 459: 1005-13.
- Prieto D, Contreras C, Sánchez A. Endothelial dysfunction, obesity and insulin resistance. Curr Vasc Pharmacol 2014; 12: 412-26.
- Félétou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond) 2009; 117: 139-55.
- Godo S, Shimokawa H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic Biol Med 2017; 109: 4-10.
- Morikawa K, Shimokawa H, Matoba T, Kubota H, Akaike T, Talukder MA, Hatanaka M, Fujiki T, Maeda H, Takahashi S, Takeshita A.. Pivotal role of Cu,Znsuperoxide dismutase in endothelium-dependent hyperpolarization J Clin Invest 2003; 112: 1871-9.
- Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, Ogasawara Y, Kajiya F. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation 2003; 107: 1040-5.
- Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, Rogers PA, Knudson JD, Dick GM, Swafford A, Chilian WM. Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 2006; 26: 2614-21.
- Santiago E, Contreras C, García-Sacristán A, Sánchez A, Rivera L, Climent B, Prieto D. Signaling pathways involved in the H2O2-induced vasoconstriction of rat coronary arteries. Free Radic Biol Med 2013; 60: 136-46.
- Büssemaker E, Popp R, Binder J, Busse R, Fleming I. Characterization of the endothelium-derived hyperpolarizing factor (EDHF) response in the human interlobar artery. Kidney Int 2003; 63: 1749-55.
- Muñoz M, López-Oliva ME, Pinilla E, Martínez MP, Sánchez A, Rodríguez C, García-Sacristán A, Hernández M, Rivera L, Prieto D. CYP epoxygenasederived H2O2 is involved in the endothelium-derived hyperpolarization (EDH) and relaxation of intrarenal arteries. Free Radic Biol Med 2017; 106: 168-83.
- Imig JD. Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function. Prostaglandins Other Lipid Mediat 2013; 104-105: 2-7.
- Fleming I, Michaelis UR, Bredenkötter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 2001; 88: 44-51
- Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic Biol Med 2014; 76: 208-26.
- Schröder K, Weissmann N, Brandes RP. Organizers and activators: Cytosolic Nox proteins impacting on vascular function. Free Radic Biol Med 2017; 109: 22-32.
- Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease?Trends Endocrinol Metab 2014; 25: 452-63.
- Schröder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 2012; 110: 1217-25.
- Sedeek M, Nasrallah R, Touyz RM, Hébert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 2013; 240): 1512-8.
- Gorin Y, Block K. Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med 2013; 61: 130-4.
- Muñoz M, Martínez MP, López-Oliva ME, Rodríguez C, Corbacho C, Carballido J, García-Sacristán A, Hernández M, Rivera L, Sáenz-Medina J, Prieto D. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. 2018; 19: 92-104.
- Wisse BE, Kim F, Schwartz MW. An integrative view of obesity. Science 2007; 318: 928-9.
- Bays HE, Toth PP, Kris-Etherton PM, Abate N, Aronne LJ, Brown WV, Gonzalez-Campoy JM, Jones SR, Kumar R, La Forge R, Samuel VT. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. Journal of Clinical Lipidology 2013; 7: 304-383
- Bakker W, Eringa EC, Sipkema P, van Hinsbergh VW. Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res 2009; 335: 165-89.
- Keaney JF Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ. Framingham Study. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 2003; 23: 434-9.
- Galili O, Versari D, Sattler KJ, Olson ML, Mannheim D, McConnell JP, Chade AR, Lerman LO, Lerman A. Early experimental obesity is associated with coronary endothelial dysfunction and oxidative stress. Am J Physiol Heart Circ Physiol 2007; 292: H904-1.
- Alexander CM, Landsman PB, Teutsch TM, Haffner SM. NCEP-defined metabolic syndrome, diabetes, and prevalence of coronary heart disease among NHANES III participants age 50 years and older. Diabetes 2003; 52: 1210-14.
- Villalba N, Martínez P, Bríones AM, Sánchez A, Salaices M, García-Sacristán A, Hernández M, Benedito S, Prieto D. Differential structural and functional changes in penile and coronary arteries from obese Zucker rats. Am J Physiol Heart Circ Physiol 2009; 297: H696-707.
- Contreras C, Sánchez A, García-Sacristán A, Martínez MC, Andriantsitohaina R, Prieto D. Preserved insulin vasorelaxation and up-regulation of the Akt/eNOS pathway in coronary arteries from insulin resistant obese Zucker rats. Atherosclerosis 2011; 217: 331-9.
- Shi Y, Vanhoutte PM. Oxidative stress and COX cause hyper-responsiveness in vascular smooth muscle of the femoral artery from diabetic rats. Br J Pharmacol 2008; 154: 639-51.
- Tian XY, Wong WT, Leung FP, Zhang Y, Wang YX, Lee HK, Ng CF, Chen ZY, Yao X, Au CL, Lau CW, Vanhoutte PM, Cooke JP, Huang Y. Oxidative stressdependent cyclooxygenase-2-derived prostaglandin f(2α) impairs endothelial function in renovascular hypertensive rats. Antioxid Redox Signal 2012; 16: 363-73.
- Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, García-Redondo L, Esteban V, Redondo JM, Alonso MJ, Briones AM, Salaices M. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal 2013; 18: 1851-65.
- Sánchez A, Contreras C, Martínez P, Villalba N, Benedito S, García-Sacristán A, Salaices M, Hernández M, Prieto D. Enhanced cyclooxygenase 2-mediated vasorelaxation in coronary arteries from insulin-resistant obese Zucker rats. Atherosclerosis 2010; 213: 392-9.
- Santiago E, Martínez MP, Climent B, Muñoz M, Briones AM, Salaices M, García-Sacristán A, Rivera L, Prieto D. Augmented oxidative stress and preserved vasoconstriction induced by hydrogen peroxide in coronary arteries in obesity: role of COX-2. Br J Pharmacol 2016; 173: 3176-95.
- Szerafin T, Erdei N, Fülöp T, Pasztor ET, Edes I, Koller A, Bagi Z. Increased cyclooxygenase-2 expression and prostaglandin-mediated dilation in coronary arterioles of patients with diabetes mellitus. Circ Res 2006; 99: e12-7
- Tang SY, Monslow J, Todd L, Lawson J, Puré E, Fitz Gerald GA. Cyclooxygenase-2 in endothelial and vascular smooth muscle cells restrains atherogenesis in hyperlipidemic mice. Circulation 2014; 129: 1761-1769.
- Yasui M, Tamura Y, Minami M, Higuchi S, Fujikawa R, Ikedo T et al. (2015). The Prostaglandin E2 receptor EP4 regulates obesity related inflammation and insulin sensitivity. PLoS One 10: e0136304.
- Bhala N, Emberson J, Merhi A, Abramson S, Arber N, Baron JA, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: metaanalyses of individual participant data from randomised trials. Lancet 2013; 382: 769-79.
- Abrass CK. Overview: obesity: what does it have to do with kidney disease? J Am Soc Nephrol 2004; 15: 2768-72.
- de Vries AP, Ruggenenti P, Ruan XZ, Praga M, Cruzado JM, Bajema IM, D’Agati VD, Lamb HJ, Pongrac Barlovic D, Hojs R, Abbate M, Rodriquez R, Mogensen CE, Porrini E. ERA-EDTA Working Group Diabesity. Fatty kidney: emerging role of ectopic lipid in obesityrelated renal disease. Lancet Diabetes Endocrinol 2014; 2: 417-22.
- Muñoz M, Sánchez A, Pilar Martínez M, Benedito S, López-Oliva ME, García-Sacristán A, Hernández M, Prieto D. COX-2 is involved in vascular oxidative stress and endothelial dysfunction of renal interlobar arteries from obese Zucker rats. Free Radic Biol Med 2015; 84:77-90.
- Muñoz M, López-Oliva ME, Rodríguez C, Martínez MP, Sáenz-Medina J, Sánchez A, Climent B, Benedito S, García-Sacristán A, Rivera L, Hernández M, Prieto D. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2019; 28: 101330.
- Thompson JA, Larion S, Mintz JD, Belin de Chantemèle EJ, Fulton DJ, Stepp DW. Genetic Deletion of NADPH Oxidase 1 Rescues Microvascular Function in Mice With Metabolic Disease. Circ Res 2017; 121: 502-11.
- Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ Res 2018; 122: 877-902.
- Rezende F, Moll F, Walter M, Helfinger V, Hahner F, Janetzko P, Ringel C, Weigert A, Fleming I, Weissmann N, Kuenne C, Looso M, Rieger MA, Nawroth P, Fleming T, Brandes RP, Schröder K. The NADPH organizers NoxO1 and p47phox are both mediators of diabetesinduced vascular dysfunction in mice. Redox Biol 2018; 15: 12-21.
