Premio Real Academia Nacional de Farmacia del Concurso Científico 2018 de la RANF
1. INTRODUCCIÓN
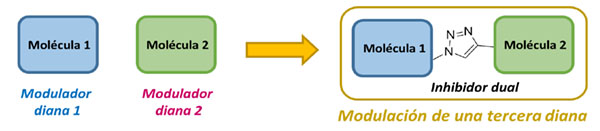
Un compuesto multidiana se caracteriza por presentar actividad biológica en más de una diana terapéutica. Tradicionalmente, los fármacos son diseñados para modular una diana específica de manera selectiva, considerando la modulación múltiple de otras dianas una característica indeseada. Sin embargo, esta estrategia de mono-modulación se ha visto insuficiente en el tratamiento de algunas enfermedades tales como enfermedades infecciosas, cáncer y desórdenes neurológicos (1). Para atacar estas enfermedades, la estrategia de modular varios mecanismos patológicos al mismo tiempo se ha venido usando en ensayos clínicos en los últimos años. Esta polifarmacología consiste en la combinación de diferentes moléculas que independientemente actúan en distintas dianas biológicas y ya se ha usado en enfermedades de tipo cardiovascular, cáncer o el VIH (2). Sin embargo, la combinación de varios fármacos de esta manera implica algunos riesgos y provoca ciertas dificultades tales como interacciones fármaco-fármaco no deseadas o múltiples efectos secundarios. Con el objetivo de modular varios mecanismos moleculares al mismo tiempo y reducir los riesgos asociados a la polifarmacología, los fármacos multidiana están adquiriendo gran importancia como una estrategia ideal y novedosa para tratar enfermedades complejas.
Las enfermedades neurodegenerativas, y más en concreto la enfermedad de Alzheimer (EA), se verían ampliamente beneficiadas por una aproximación de este tipo (3). Estas patologías complejas se caracterizan por su etiología idiopática, sus intrincados mecanismos moleculares y su naturaleza multifactorial. La EA es la enfermedad neurodegenerativa de mayor incidencia y presenta un elevado fracaso clínico a pesar de los grandes esfuerzos realizados a lo largo de los años para encontrar un fármaco efectivo. La complejidad de los mecanismos patológicos sugiere que las drogas tradicionales no serán capaces de producir un efecto terapéutico adecuado (4). Debido a estos hechos, los fármacos multidiana y las enfermedades neurodegenerativas han estado relacionados cada vez de manera más habitual en los últimos años, lo que ha hecho que varios compuestos multidiana hayan alcanzado ensayos clínicos (5). En el caso de estas enfermedades el uso de un compuesto multidiana resulta idóneo debido a que en el momento en el que son diagnosticadas, los pacientes ya presentan diversas patologías que necesitan tratamiento inmediato. El diseño específico de las moléculas necesita ser estudiado con detenimiento, persiguiendo unas características óptimas tales como: a) presentar una potencia similar en todas las dianas terapéuticas a modular, b) modular proteínas que presenten efectos sinérgicos o aditivos de manera moderada para obtener efectos secundarios menos acusados (6) y c) mantener las propiedades tipo fármaco.
En la EA los procesos patológicos principales son: inflamación cerebral, agregación proteica y neurodegeneración. Existen diferentes mecanismos implicados en la acumulación de proteínas celulares como el equilibro de fosforilación-defosforilación en el cual están implicadas las proteínas quinasas y las fosfatasas. En la EA hay dos tipos de agregados proteicos principales: las placas β-amiloides y los ovillos neurofibrilares. Las placas β-amiloides son depósitos neurotóxicos del péptido precursor del beta amiloide que se encuentran en la matriz extracelular debido a un procesamiento anormal realizado por la proteína beta-secretasa, BACE1 (7). Los ovillos neurofibrilares son depósitos intracelulares compuestos por proteína tau hiperfosforilada. Se han descrito varias quinasas implicadas en esta agregación patológica, tales como GSK3β, LLRK2 y CK1δ (8,9). Nuestro objetivo es diseñar compuestos multidiana que tengan al menos tres actividades biológicas: capaces de modular al mismo tiempo BACE1 y dos quinasas diferentes implicadas en la proteinopatía del Alzheimer. Además, debido a la implicación de estas quinasas en la neuroinflamación, la combinación de todas las actividades biológicas en una sola molécula podría dar lugar a un fármaco efectivo contra el Alzheimer.
El diseño racional de moléculas multidiana se basa en tres estrategias principales: unión, fusión y combinación (Figura 1).
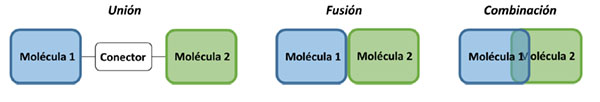
Figura 1. Estrategias principales para el diseño de ligandos multidiana
Para mantener las propiedades tipo fármaco, el incremento del tamaño y del peso molecular implicados en el diseño de compuestos multidiana constituye una de las principales desventajas (10). El aumento del peso molecular afecta directamente a las propiedades farmacocinéticas como la penetración de la barrera hematoencefálica o la biodisponibilidad oral. Por tanto, la estrategia de combinación, seguida de la fusión son las más deseables para mantener un peso molecular moderado. La estrategia de unión implica la adición de grupos funcionales que permitan conectar ambos esqueletos moleculares, aumentando el peso molecular del compuesto resultante. Sin embargo, esta estrategia ofrece la posibilidad de conectar los fragmentos de las moléculas originales de forma que mantengan su actividad inicial y sus interacciones con la diana. Esta tarea resulta muy complicada en el caso de la fusión y la combinación. Por este motivo, la estrategia de unión resulta la más apropiada para mantener la funcionalidad original de los esqueletos combinados. La posición del conector resulta de extrema importancia y se debe prestar especial atención al diseño para evitar disminuir las interacciones entre el ligando y la diana. Además, siguiendo esta estrategia se esperan variaciones mínimas de la actividad biológica del ligando multidiana respecto a la actividad inicial de los fragmentos, de manera que serán necesarios menos ciclos de optimización para obtener el fármaco final deseado.
Así, para el diseño de una molécula capaz de modular el progreso de la neurodegeneración presente en la EA utilizaremos la estrategia de unión, eligiendo un conector que además de realizar la función de unión de fragmentos nos proporcionará una tercera actividad biológica. El conector serviría entonces de nexo, así como de motivo de interacción con una tercera entidad biológica.
Finalmente, y para facilitar el proceso de selección de la mejor molécula multidiana de manera eficiente, hemos implementado la metodología de química click in situ. Esta metodología utiliza una de las enzimas diana, BACE1, como molde de reacción, de manera que se pueden combinar diferentes fragmentos y que la propia enzima catalice la reacción de formación de la molécula multidiana que se una de manera preferente al centro activo.
En este trabajo se describen el diseño, la síntesis y la evaluación biológica de cinco nuevos compuestos multidiana diseñados de manera innovadora con la estrategia de unión, así como una nueva metodología de selección de los mejores candidatos mediante la química click in situ.
2. MATERIALES Y MÉTODOS
2.1. General
Los reactivos se obtuvieron de diferentes casas comerciales y se usaron sin realizarles ningún tratamiento posterior. La purificación de los crudos de reacción se llevó a cabo mediante columnas cromatográficas, bien a presión media usando silica gel E. Merck, Grado 60, tamaño de partícula 0.040–0.063 mm, malla 230–240 ASTM o bien utilizando el Sistema de IsoleraOne de Biotage. El eluyente utilizado se especifica en cada caso. Los datos espectroscópicos de 1 RMN and 13C RMN se obtuvieron en un espectrómetro Bruker AV300 MHz. Los desplazamientos químicos se expresan en ppm. Las señales de multiplicidad (bs: singlete ancho, s: singlete, d: doblete, dd: doble doblete, ddd:doble doble doblete t: triplete, td: triple doblete m: multiplete, q: cuadruplete) y las constantes de acoplamiento (J=Hz) se indican para cada molécula. Los datos espectroscópicos se analizaron con el software MestreNova 10.0. Para los experimentos llevados a cabo con microondas se utilizó el equipo Biotage Initiator. Dichos experimentos se realizaron en el modo de control de temperatura y en viales sellados y especiales para dicho equipo. La temperatura se mide mediante un sensor de IR colocado en el reactor del microondas. La agitación de dichas reacciones se llevó a cabo mediante la presencia de una barrita magnética. Los datos de HPLCMS se obtuvieron con el equipo FinniganTM LXQ TM procedente de Termofisher. Los valores de punto de fusión de los diferentes compuestos fueron determinados en un equipo Büchi Melting Point M-560.
2.2. Síntesis de los fragmentos alquino y azida
Síntesis de 4-(azidometil)-N-(benzo[d]tiazol-2-il)benzamida (1)
Se disolvieron en DMF (10 mL) 354 mg de ácido 4-(azidometil)benzoico (2.0 mmol), 383 mg de EDCI (4.0 mmol) y 488 mg de DMAP (2.0 mmol). A continuación, tras 5 min de agitación a t.a., se añadieron 300 mg de 2-amino-benzotiazol (2.0 mmol). La mezcla resultante se dejó en agitación durante toda una noche a la misma temperatura. Se añadió agua (35 mL) y AcOEt (35 mL). La fase orgánica resultante se lavó consecutivamente con disoluciones saturadas de NaHCO3 y NaCl. Se evaporó el disolvente de la fase orgánica bajo presión reducida y el crudo de reacción obtenido se purificó mediante cromatografía en columna usando hexano/acetato de etilo 9:1 como eluyente, obteniéndose finalmente un sólido blanco (426 mg, 69% de rendimiento).

1H RMN (300 MHz, DMSO-d6): δ 12.95 (s, 1H), 8.19 (d, J = 8.2 Hz, 2H), 8.03 (d, J = 7.6, 1.2 Hz, 1H), 7.80 (d, J = 8.2 Hz, 1H), 7.56 (d, J = 8.2 Hz, 2H), 7.48 (dd, J = 8.2, 1.3 Hz, 1H), 7.35 (td, J = 7.6, 1.2 Hz, 1H), 4.60 (s, 2H). 13C RMN (75 MHz, DMSO-d6): δ 166.1, 159.3, 148.9, 141.1, 131.2 (2C), 129.2 (2C), 128.8 (2C), 126.7, 124.2, 122.2, 120.8, 53.5.
ESI calc para C15H11N5OS [M + H]+ 310,0757; encontrada: 310,0765.
Síntesis de 2-(4-(azidometil)fenil)-N-(benzo[d]tiazol-2-il)acetamida (4)
Preparación de cloruro de 2-(4-(bromometil)fenil)acetilo
Sobre una disolución de 2.35 g (10 mmol) de ácido 2-(4-(bromometil)fenil)acético en 15 mL de cloroformo se añadieron 3.85 g (32 mmol) de cloruro de tionilo. La mezcla se calentó a 70 ºC durante 6 horas obteniéndose una disolución amarilla. Se enfrió a temperatura ambiente y se eliminó el disolvente a presión reducida proporcionando 2.37 g (0.96 mmol, 96% de rendimiento) del cloruro deseado como un sólido amarillo que se usó sin posterior tratamiento. 1H RMN (400 MHz, CDCl3): δ 7.44–7.38 (m, 2H), 7.28–7.24 (m, 2H), 4.48 (s, 2H), 4.14 (s, 2H). 13C RMN (100.6 MHz, CDCl3): δ 171.6, 137.8, 131.3 (2C), 129.9, 129.6 (2C), 52.6, 32.6.
Preparación de N-(benzo[d]tiazol-2-il)-2-(4-(azidometil)fenil)acetamida
Sobre una disolución de aminobenzotiazol (600 mg, 4.0 mmol) en THF (7.5 mL) se añadió el cloruro de ácido obtenido anteriormente (990 mg, 4.0 mmol) a temperatura ambiente. A continuación, la muestra se calentó bajo radiación microondas a 100 ºC durante 15 min. Una vez enfriada la mezcla se añadió agua (35 mL) y DCM (65 mL). Tras la separación de las fases, el disolvente de la fase orgánica se eliminó a presión reducida y el sólido obtenido se purificó mediante cromatografía en columna usando hexano/acetato de etilo 9:1 como eluyente, obteniéndose finalmente un sólido blanco (732 mg, 53% de rendimiento).
Sobre una disolución de N-(benzo[d]tiazol-2-il)-2-(4-(bromometil)fenyl)acetamida (0.150 g, 0.43 mmol) en DMSO (2 mL), se añadió azida sódica (0.042 g, 0.65 mmol) y la mezcla resultante se dejó en agitación durante 18 h a temperatura ambiente. A continuación se añadió agua (5 mL) y la mezcla de reacción se extrajo con éter dietílico (20 mL x 3). La fase orgánica se secó sobre MgSO4, se filtró y concentró a presión reducida. El sólido obtenido se purificó mediante cromatografía en columna usando hexano/acetato de etilo 9:1 como eluyente, obteniéndose finalmente el producto deseado como un sólido blanco (109 mg, 79% rendimiento).

1H RMN (300 MHz, CDCl3): δ 9.92 (bs, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.37 (ddd, J = 8.2, 7.3, 1.3 Hz, 1H), 7.26 (td, J = 7.7, 1.2 Hz, 1H), 7.22 – 7.12 (m, 4H), 4.26 (s, 2H), 3.77 (s, 2H). 13C RMN (75 MHz, CDCl3): δ 169.5, 159.0, 148.3, 135.7, 133.2, 132.4, 130.3 (2C), 129.3 (2C), 126.9, 124.6, 122.0, 121.1, 54.7, 43.5.
ESI calc para C16H13N5OS [M + H]+ 324,0814; encontrada: 324,0814.
Síntesis de 3-etil-5-(prop-2-in-1-il)tiazolidin-2,4-diona (5)
Se enfrió a 0 ºC una disolución de isocianato de etilo (163 μL, 2.0 mmol) e isocianato de propargilo (189 μL, 2.0 mmol) en THF (5 mL). A continuación se añadió lentamente cloruro de sulfurilo (167 μL, 2 mmol) y la mezcla se dejó en agitación durante toda la noche a temperatura ambiente. Tras 30 min de agitación al aire se elimina el disolvente a presión reducida. La mezcla de reacción se purificó mediante cromatografía en columna usando un gradiente de eluyente (0-20% de acetato de etilo en hexano) obteniéndose un aceite amarillo (243 mg, 66% rendimiento).

1H RMN (300 MHz, CDCl3): δ 4.37 (d, J = 2.5 Hz, 2H), 3.65 (q, J = 7.2 Hz, 2H), 2.23 (t, J = 2.7 Hz, 1H), 1.22 (t, J = 7.2 Hz, 3H). 13C RMN (75 MHz, CDCl3): δ 165.5, 152.0, 76.5, 72.8, 40.6, 31.8, 14.3.
ESI calc para C7H9N2O2S [M + H]+ 185,0379; encontrada: 185,0385.
Síntesis de (Z)-3-(4-(prop-2-in-1-iloxi)benciliden)indolin-2-ona (6)
Una disolución de isatina (294 mg, 2 mmol) y 4-(prop-2-in-1-iloxi)anilina (294 mg, 2 mmol) en etanol (3 mL) se calentó a reflujo durante 1 h y a continuación se dejó en agitación durante 16 h. La mezcla obtenida se filtró y el sólido obtenido se purificó mediante cromatografía en columna usando una mezcla 1:1 de hexano y acetato de etilo, obteniendo un sólido naranja (110 mg, 20% rendimiento). Pf: 230 – 232 ºC.
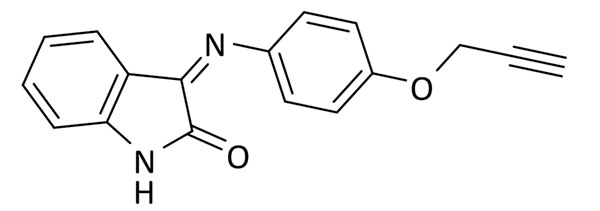
1H RMN (300 MHz, DMSO-d6): δ 10.97 (s, 1H), 7.35 (td, J = 7.7, 1.3 Hz, 1H), 7.13 – 7.08 (m, 2H), 7.03 – 6.97 (m, 2H), 6.90 (d, J = 7.8 Hz, 1H), 6.76 (td, J = 7.7, 1.0 Hz, 1H), 6.59 (dd, J = 7.8, 1.2 Hz, 1H), 4.85 (d, J = 2.4 Hz, 2H), 3.62 (t, J = 2.4 Hz, 1H). 13C RMN (75 MHz, DMSOd6): δ 164.1, 155.4, 155.3, 147.3, 144.3, 134.7, 125.5, 122.2, 119.7 (2C), 116.3 (2C), 116.2, 111.9, 79.7, 78.8, 56.3.
ESI calc para C17H13N2O2 [M + H]+ 277,0972; encontrada: 277,0979.
Síntesis de N-(benzo[d]tiazol-2-il)-4-(prop-2-in-1-iloxi)benzamida (2)
Síntesis de ácido 4-(prop-2-in-1-iloxi)benzoico:
Se añadió 3-bromopropino (600 μL, 7.67 mmol) a una mezcla de 4-hidroxibenzoato de metilo (991 mg, 6.52 mmol) y carbonato potásico (3.47 g, 25.1 mmol) en acetonitrilo (32 mL). La mezcla resultante se dejó en agitación y a reflujo durante toda la noche. Después de enfriar la mezcla a temperatura ambiente y adicionar agua, se hizo una extracción con acetato de etilo (20 mL × 3). La fase orgánica se lavó con una disolución saturada de NaCl, se secó con Na2SO4 y se concentró hasta sequedad. El residuo se purificó en cromatografía en columna (gradiente: 0-20% de MeOH en DCM), obtiéndose un aceite incoloro (929 mg, 75% rendimiento). 1H RMN (600 MHz, CDCl3): δ 8.08 (d, J = 9.0 Hz, 2H), 7.00 (d, J = 9.0 Hz, 2H), 4.75 (s, 2H), 3.89 (s, 3H), 2.55 (s, 1H). 13C RMN (75 MHz, CDCl3): δ 172.6, 157.1, 130.7, 127.4, 115.4, 78.9, 75.9, 56.2, 52.4, 40.7.
3.8 g (20 mmol) de 4-propiniloxibenzoato de metilo se disolvieron en 30 mL de metanol y THF (1:1 v/v). A continuación se adicionaron 5 mL de una disolución acuosa de NaOH (4 g, 100 mmol) y la mezcla de reacción se dejó agitando a temperatura ambiente durante 16 h. Después de haber ajustado el pH ≈ 3 con HCl 5M, el disolvente se eliminó a presión reducida. Posteriormente se añadieron 200 mL de agua y la mezcla se dejó agitando 0.5 h, se filtró y el precipitado se lavó con agua fría. Finalmente se obtuvo un sólido blanco (3.48 g, 98 % rendimiento). 1H RMN (300 MHz, DMSO-d6): δ 7.86 (d, J = 8.3 Hz, 2H), 7.01 (d, J = 8.3 Hz, 2H), 4.84 (s, 2H), 3.55 (t, 1H). 13C RMN (75 MHz, DMSO-d6): δ 173.0, 155.9, 130.4, 127.8, 114.6, 79.4, 78.2, 55.3, 39.8
Se disolvieron 704 mg de ácido 4-(prop-2-in-1-iloxi)benzoico (4.0 mmol), 766 mg de EDCI (4.0 mmol) y 976 mg de DMAP (8.0 mmol) en DMF. A continuación, tras 5 min de agitación, se adicionaron 600 mg de 2-amino-benzotiazol (4.0 mmol) y la mezcla resultante se dejó con agitación durante 16 h. Posteriormente se añadió acetato de etilo (35 mL) y la fase insoluble se filtró y lavó con agua fría, obteniéndose un sólido blanco (852 mg, 69% rendimiento). Pf: 150-152 ºC.
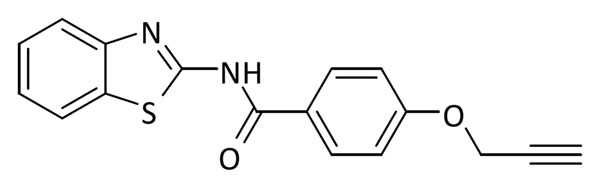
1H RMN (300 MHz, DMSO-d6): δ 12.77 (bs, 1H), 8.17 (d, J = 8.9 Hz, 2H), 8.02 (d, J = 7.6, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.47 (ddd, J = 8.2, 7.3, 1.3 Hz, 1H), 7.34 (td, J = 7.6, 1.2 Hz, 1H), 7.22 – 7.09 (m, 2H), 4.94 (d, J = 2.4 Hz, 2H), 3.66 (s, 1H). 13C RMN (75 MHz, DMSO-d6): δ 165.6, 161.2, 159.3, 148.9, 132.0, 130.8 (2C), 126.6, 125.1, 124.05, 122.2, 120.7, 115.2 (2C), 79.2 (2C), 56.2.
ESI calc para C17H13N2O2S [M + H]+ 309,0692; encontrado: 309,0699.
Procedimiento general para la síntesis de los triazoles (3, 7-10):
Se dejó reaccionando una mezcla de alquino (0.3 mmol, 1 eq) y azida (0.3 mmol, 1 eq) en DMF (3.0 mL) toda una noche a temperatura ambiente, en presencia de sulfato de cobre (CuSO4·5H2O) (7.5 mg, 10 mol%) y ascorbato sódico (11.9 mg, 20 mol%). A continuación, sobre la mezcla de reacción se añadió agua y se extrajo con una mezcla DCM:MeOH (9:1). La fase orgánica se lavó con una disolución 5% de hidróxido amónico, se secó sobre sulfato magnésico, se concentró a presión reducida y el residuo se purificó con cromatografía flash de silica gel
(eluyente: mezcla DCM – MeOH).
N-(benzo[d]tiazol-2-il)-4-((1-(4-(benzo[d]tiazol-2-ilcarbamoil)bencil)-1H-1,2,3-triazol-4-il)metoxi)benzamida (3)
Se utilizó N-(benzo[d]tiazol-2-il)-4-(prop-2-in-1-iloxi)benzamida (58.8 mg, 0.2 mmol) y 4-(azidometil)-N-(benzo[d]tiazol-2-il)benzamida (59.0 mg, 0.2 mmol). Rendimiento: 71 mg, 57%. Sólido blanco; pf: 280 – 282 ºC (descomp.).
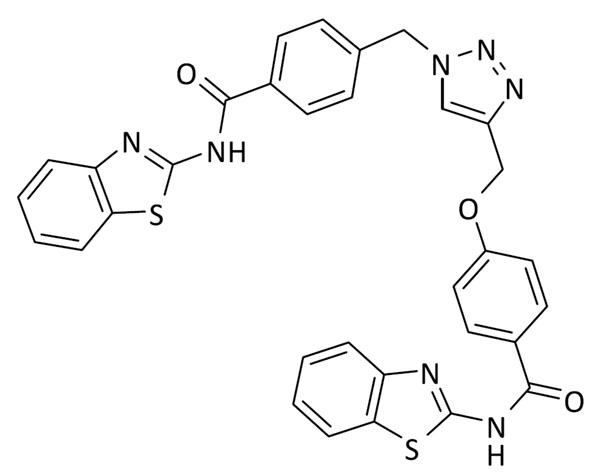
1H RMN (300 MHz, DMSO-d6): δ 12.5 (bs, 2H), 8.42 (s, 1H), 8.23 – 8.09 (m, 4H), 8.06 – 7.95 (m, 2H), 7.86 – 7.71 (m, 2H), 7.52 – 7.43 (m, 4H), 7.39 – 7.28 (m, 2H), 7.22 (d, J = 8.9 Hz, 2H), 5.77 (s, 2H), 5.30 (s, 2H). 13C RMN (75 MHz, DMSO-d6): δ 166.0, 165.5, 162.0, 159.30, 159.29, 148.8 (2C), 143.0, 141.1, 132.1, 131.9, 130.8 (2C), 129.2 (2C), 128.4 (2C), 126.6, 126.5, 125.6, 124.6, 124.6, 124.1, 123.9, 122.1, 122.0, 120.7 (2C), 115.0 (2C), 61.7, 52.8.
ESI calc para C32H24N7O3S2 [M + H]+ 618,1377; encontrado: 618,1385.
N-(benzo[d]tiazol-2-il)-4-((4-((2-etil-3,5-dioxo-1,2,4-tiadiazolidin-4-il)metil)-1H-1,2,3-triazol-1-il)metil)benzamida (7)
Se utilizó 4-(azidometil)-N-(benzo[d]tiazol-2-il)benzamida (59 mg, 0.2 mmol) y 3-etil-5-(prop-2-in-1-il)tiazolidin-2,4-diona (37 mg, 0.2 mmol). Rendimiento: 66 mg, 67%. Sólido blanco; pf: 204 – 206 ºC.
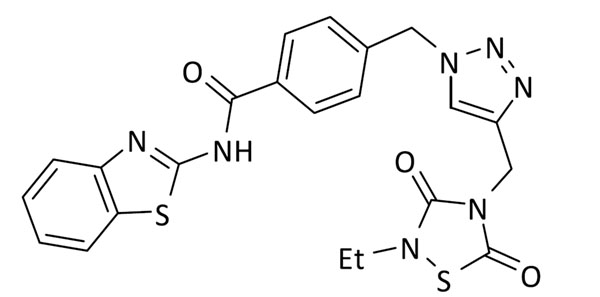
1H RMN (300 MHz, DMSO-d6): δ 12.92 (s, 1H), 8.23 (s, 1H), 8.14 (d, J = 8.3 Hz, 2H,), 8.03 (d, J = 7.6, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.56 – 7.41 (m, 3H), 7.35 (td, J = 7.6, 1.2 Hz, 1H), 5.71 (s, 2H), 4.83 (s, 2H), 3.64 (q, J = 7.1 Hz, 2H), 1.16 (t, J = 7.1 Hz, 3H). 13C RMN (75 MHz, DMSOd6): δ 166.2 (2C), 152.5, 148.9 (2C), 142.1, 141.2, 132.1, 131.9, 129.3 (2C), 128.5 (2C), 126.7, 124.6, 124.2, 122.3, 120.8, 52.8, 40.1, 37.5, 14.0.
ESI calc para C22H20N7O3S2 [M + H]+ 494,1064; encontrado: 494,1072.
N-(benzo[d]tiazol-2-il)-2-(4-((4-((2-etil-3,5-dioxo-1,2,4-tiadiazolidin-4-il)metil)-1H-1,2,3-triazol-1-il)metil)fenil)acetamida (8)
Se utilizó 2-(4-(azidometil)fenil)-N-(benzo[d]tiazol-2-il)acetamida (64 mg, 0.2 mmol) y 3-etil-5-(prop-2-in-1-il)tiazolidin-2,4-diona (37 mg, 0.2 mmol). Rendimiento: 62 mg, 61%. Sólido blanco; pf: 226 – 228 ºC (descomp.).
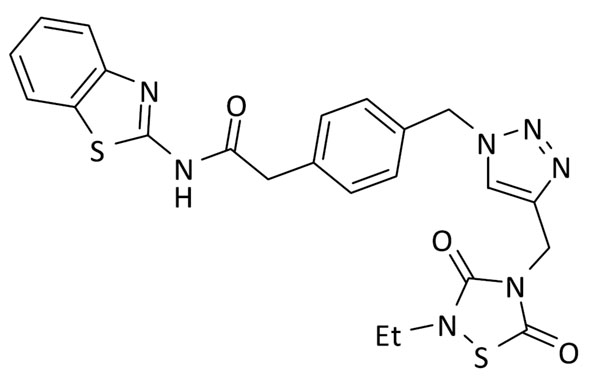
1H RMN (300 MHz, DMSO-d6): δ 12.62 (s, 1H), 8.14 (s, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.43 (td, J = 7.6, 1.2 Hz, 1H), 7.36 (d, J = 8.3 Hz, 2H), 7.35 – 7.23 (m, 3H), 5.57 (s, 2H), 4.81 (s, 2H), 3.85 (s, 2H), 3.61 (q, J = 7.1 Hz, 2H), 1.13 (t, J = 7.2 Hz, 3H). 13C RMN (75 MHz, DMSO-d6): δ 170.5, 166.1, 158.4, 152.5, 149.0, 142.0, 135.1, 135.1, 131.9, 130.2 (2C), 128.6 (2C), 126.6, 124.2, 124.0, 122.2, 121.0, 53.0, 41.9, 40.0, 37.5, 14.0.
ESI calc para C23H22N7O3S2 [M + H]+ 507,1147; encontrado: 507,1156.
(Z)-N-(benzo[d]tiazol-2-il)-2-(4-((4-((4-((2-oxoindolin-3-iliden)amino)fenoxi)metil)-1H-1,2,3-triazol-1-il)metil)fenil)acetamida (9)
Se utilizó 2-(4-(azidometil)fenil)-N-(benzo[d]tiazol-2-il)acetamida (64 mg, 0.2 mmol) y (Z)-3-(4-(prop-2-in-1-iloxi)benciliden)indolin-2-ona (62 mg, 0.2 mmol). Rendimiento: 55 mg, 46%. Sólido naranja; pf: 166 – 168ºC.

1H RMN (300 MHz, DMSO-d6): δ 12.5 (s, 1H), 10.89 (s, 1H), 8.25 (s, 1H), 7.95 – 7.78 (m, 2H), 7.67 (d, J = 8.0 Hz, 1H), 7.42 – 7.17 (m, 6H), 7.06 (d, J = 8.8 Hz, 2H), 6.94 – 6.86 (m, 2H), 6.82 (d, J = 7.8 Hz, 1H), 6.68 (t, J = 7.6 Hz, 1H), 6.54 (d, J = 7.6 Hz, 1H), 5.54 (s, 2H), 5.10 (s, 2H), 3.77 (s, 2H). 13C RMN (75 MHz, DMSO-d6): δ 170.4, 162.7, 158.2, 156.2, 155.0, 148.8 (2C), 147.2, 143.8, 135.0 (2C), 134.6, 131.8, 130.2 (3C), 128.5 (2C), 126.5, 125.4, 125.1, 123.9, 122.1 (3C), 119.7 (2C), 115.9 (2C), 111.8, 61.71, 53.0, 41.8.
ESI calc para C23H26N7O3S [M + H]+ 600,6765; encontrado: 600,6774.
N-(benzo[d]tiazol-2-il)-4-((1-(4-(2-(benzo[d]tiazol-2-ilamino)-2-oxoetil)bencil)-1H-1,2,3-triazol-4-il)metoxi)benzamida (10)
Se utilizó 2-(4-(azidometil)fenil)-N-(benzo[d]tiazol-2-il)acetamida (64 mg, 0.2 mmol) y 4-(azidometil)-N-(benzo[d]tiazol-2-il)benzamida (59.0 mg, 0.2 mmol). Rendimiento: 69 mg, 55%. Sólido blanco; pf: 198 – 200 ºC (descomp.).
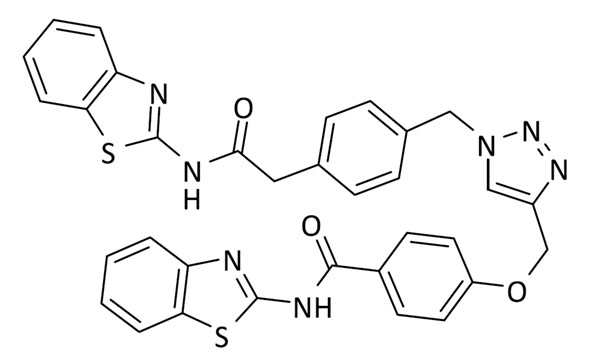
1H RMN (300 MHz, DMSO-d6): δ 12.57 (s, 2H), 8.33 (s, 1H), 8.15 (d, J = 8.4 Hz, 2H), 8.05 – 7.91 (m, 2H), 7.80 – 7.65 (m, 2H), 7.49 – 7.24 (m, 8H), 7.22 (d, J = 8.4 Hz, 2H), 5.61 (s, 2H), 5.25 (s, 2H), 3.84 (s, 2H). 13C RMN (75 MHz, DMSO-d6): δ 170.5, 165.7, 162.8 (2C), 162.1, 158.3, 148.9, 143.0, 135.2, 135.1, 131.9, 130.9 (2C), 130.3 (2C), 128.6 (2C), 126.6 (2C), 125.3, 124.7, 124.0 (2C), 122.2 (2C), 121.0 (2C), 120.7, 115.1 (2C), 61.8, 53.1, 41.9.
ESI calc para C33H26N7O3S [M + H]+ 632,1533; encontrado: 632,1542.
2.3. Estudios de actividad
La actividad de los compuestos frente a las diferentes quinasas se evaluó como se detalla a continuación.
Ensayo enzimático LRRK2
Los inhibidores de LRRK2 fueron evaluados por la compañía ThermoFisher bajo el siguiente ensayo. El ensayo en sí se puede dividir en dos fases: una fase de reacción de la quinasa y una fase de detección de ADP. En la fase de reacción de la quinasa, todos los componentes requeridos para la reacción se agregan al pocillo, y la reacción se deja incubar durante 60 minutos. Pasado este tiempo, se agrega una solución de detección que consiste en un anticuerpo anti-ADP marcado con europio, un marcador ADP marcado con Alexa Fluor ™ 647 y AEDT (para detener la reacción de la quinasa). El ADP formado por la reacción de la quinasa (en ausencia de un inhibidor) desplazará el marcador ADP marcado con Alexa Fluor 647 del anticuerpo, dando como resultado una disminución en la señal TR-FRET. En presencia de un inhibidor, la cantidad de ADP formada por la reacción de la quinasa se reduce, y la interacción resultante del anticuerpo traza da como resultado una señal alta de TR-FRET. La formación de ADP se determina calculando la relación de emisión del pocillo de ensayo.
La mezcla 2X LRK2 / ERM (Lrrktide) se prepara en 50 mM Tris pH 8.5, 0.01% Brij-35, 10 mM MgCl2, 1 mM EGTA, 0.02% NaN3. La reacción final de la quinasa con un volumen de 10 μL consiste en 3,75 – 70 ng LRRK2 y 200 μM ERM (Lrrktide) en 25 mM de Tris / 7,5 mM HEPES pH 8,2, 0,005% Brij-35,5 mM MgCl2, 0,5 mM EGTA, 0.01% NaN3. Después de 1 hora de reacción de la quinasa, se añaden 5 μL de mezcla de detección.
Ensayo enzimático CK1δ
Se utilizó el Kit de Quinasa-Glo de Promega para detectar la actividad de los compuestos frente a los ensayos tamponados de CK1δ Kinase-Glo utilizando placas de 96 pocillos negras. En un ensayo típico, se añadieron 10 μl de compuesto (disuelto en DMSO a una concentración de 1 mM y diluido de antemano en tampón a la concentración deseada) y 10 μl (16 ng) de enzima a cada pocillo seguido de 20 μl de tampón con 0,1 % de caseína como sustrato y 4 μM de ATP. La concentración final de DMSO en la mezcla de reacción no excedió del 1%. Después de una incubación de 60 minutos a 30 ºC, la reacción enzimática se detuvo con 40 μl de reactivo Kinase-Glo. La luminiscencia se registró después de 10 minutos utilizando un lector multimodo Fluostar Optima (BMG Labtechnologies Gmbh, Offenburg, Alemania). La actividad es proporcional a la diferencia del ATP total y consumido. Las actividades inhibitorias se calcularon sobre la base de las actividades máximas medidas en ausencia de inhibidor. La CI50 se definió como la concentración de cada compuesto que reduce un 50% la actividad enzimática con respecto a la que se produce sin inhibidor.
2.3.1. Ensayo enzimático GSK-3β
La enzima GSK-3β recombinante humana y el sustrato polipéptido prefosforilado se compraron a Millipore (Millipore Ibérica SAU). El ensayo de quinasa Kinase-Glo se obtuvo de Promega (Promega Biotech Ibérica, SL). El ATP y todos los demás reactivos proceden de Sigma Aldrich (St. Louis, MO). El tampón utilizado en los ensayos contenía 50 mM de HEPES (pH 7.5), 1 mM de AEDT, 1 mM de EGTA y 15 mM de acetato de magnesio.
Los ensayos de Kinase-Glo se realizaron en medio tamponado utilizando placas de 96 pocillos negros. En un ensayo típico, 10 μL (10 μM) de compuesto (disuelto en DMSO a una concentración de 1 mM y diluido previamente en el tampón a la concentración deseada) y 10 μL (20 ng) de enzima se añadieron a cada pocillo seguido de 20 μL de disolución tampón de concentración 25 μM sustrato y 1 μM ATP. La concentración final de DMSO en la mezcla de reacción no excedió del 1%. Después de 30 minutos de incubación a 30 ºC, la reacción enzimática se detuvo con 40 μL de reactivo Kinase-Glo. La luminiscencia se registró después de 10 minutos utilizando un lector multimodo Fluostar Optima (BMG Labtechnologies Gmbh, Offenburg, Alemania). La actividad es proporcional a la diferencia del ATP total y consumido. La actividad inhibitoria de cada compuesto se calculó en base a las actividades máximas medidas en ausencia de inhibidor. La CI50 se definió como la concentración de cada compuesto que reduce un 50 % la actividad enzimática con respecto a la que se produce sin inhibidores.
2.3.2. Ensayo enzimático BACE1
Los ensayos con la enzima BACE1 se realizaron in vitro utilizando FRET. Se utilizó un sustrato peptídico a base de APP (rodamina-EVNLDAEFK-quencher, Km de 20 μM) portador de la mutación sueca y que contiene una rodamina como donante de fluorescencia y un atenuador en cada extremo. El sustrato intacto es débilmente fluorescente y se vuelve altamente fluorescente al sufrir la escisión enzimática. Los ensayos se realizaron en un tampón de acetato de sodio de 50 mM, pH 4,5 y una concentración enzimática final (1 U/mL). El cribado enzimático se realizó a una concentración 10 μM del inhibidor. La mezcla fue incubada durante 60 minutos a 25ºC en ausencia de luz y luego se detuvo añadiendo una disolución de acetato de sodio 2,5 M. La fluorescencia se midió con un lector de microplacas Fluostar Optima (BMG Labtechnologies Gmbh, Offenburg, Alemania) a 545 nm de excitación y 585 nm de emisión.
2.4. Metodología química click in situ
Los fragmentos 1 y 2 se incubaron en 50 μL de una disolución tampón de acetato sódico (50 mM y pH 4,5) a concentraciones de 30 μM y 60 μM. Se añadió la enzima BACE1 a una concentración final de 0,5 μM. Las soluciones se dejaron a temperatura ambiente durante 24 horas y se analizaron con HPLC-MS mediante la detección de modo iónico único centrado en la masa de los triazoles esperados.
3. RESULTADOS Y DISCUSIÓN
El principal objetivo del presente trabajo es el diseño y síntesis de moléculas multidiana capaces de modular al mismo tiempo diferentes mecanismos patológicos, tales como la inflamación, la neurodegeneración y la agregación del péptido beta amiloide (Aβ), implicados en la enfermedad de Alzheimer. Las proteínas quinasas juegan un papel importante en enfermedades neurodegenerativas ya que muchas de ellas son capaces de fosforilar de manera anormal la proteína tau provocando su agregación en forma de depósitos insolubles y tóxicos para las neuronas (11,12).
Dada la amplia experiencia del grupo de investigación en el diseño y síntesis de inhibidores de estas proteínas quinasas, el primer paso para el diseño racional de las moléculas multidiana se centró en la búsqueda de fragmentos en nuestra quimioteca MBC (13). Los fragmentos seleccionados debían ser fácilmente modificables en alguna posición donde no se viera afectada la actividad biológica de la molécula por la introducción de una nueva funcionalidad que pudiera conectar varios fragmentos. En este caso se planteó que el conector elegido podría proporcionar al compuesto final una tercera actividad biológica no presente en los fragmentos aislados. BACE1 es una enzima ácido aspártico proteasa localizada en la membrana celular y altamente expresada en el cerebro, especialmente en el tejido neuronal implicado en la agregación del péptido beta amiloide. El péptido Aβ se produce por la proteólisis de la proteína precursora del amiloide (PPA), catalizada tanto por la gama secretasa como por la beta secretasa o BACE1. Se ha demostrado que tanto mutaciones en el gen de BACE1 que afectan al sitio de unión de la PPA, como la inhibición de BACE1, provocan una disminución en la formación de péptido beta amiloide (14, 15). Estos hechos validan la enzima BACE1 como una buena diana terapéutica en el tratamiento de la EA.
Los inhibidores de proteínas quinasas son, habitualmente, moléculas de bajo peso molecular (16, 17), por debajo de los 280 Da. Por el contrario, los inhibidores de BACE1 se caracterizan por ser moléculas grandes, como por ejemplo péptidos, debido a la amplia cavidad catalítica que posee esta proteasa. Basándonos por tanto en los fragmentos inhibidores de quinasa, se introdujeron los conectores adecuados en las posiciones más favorables para la obtención de una molécula final con actividad inhibidora de BACE1. Conviene señalar que el bolsillo de unión de BACE1 tiene preferencia por alojar grupos amida debido a la fijación de sustratos de tipo peptídico que debe ocurrir previamente para la correcta actividad catalítica de la enzima. Por tanto, el conector elegido fue el motivo 1,2,3-triazol, debido a su característica de grupo bioisóstero de amida (18), de manera que el conector pudiera aportar un fragmento de unión a la proteasa. Además este heterociclo es sintéticamente accesible por medio de la química click, a través de una cicloadición 1,3 dipolar de azidas y alquinos catalizada por cobre ampliamente utilizada en química médica (19).
Se seleccionaron diferentes inhibidores de quinasa sintetizados previamente en el laboratorio para llevar a cabo un estudio de relación estructura química-actividad biológica que nos permitiera razonar las posiciones más favorables de introducción de los grupos funcionales necesarios para la síntesis de los triazoles. Estos sitios se señalan en la siguiente figura (Figura 2).
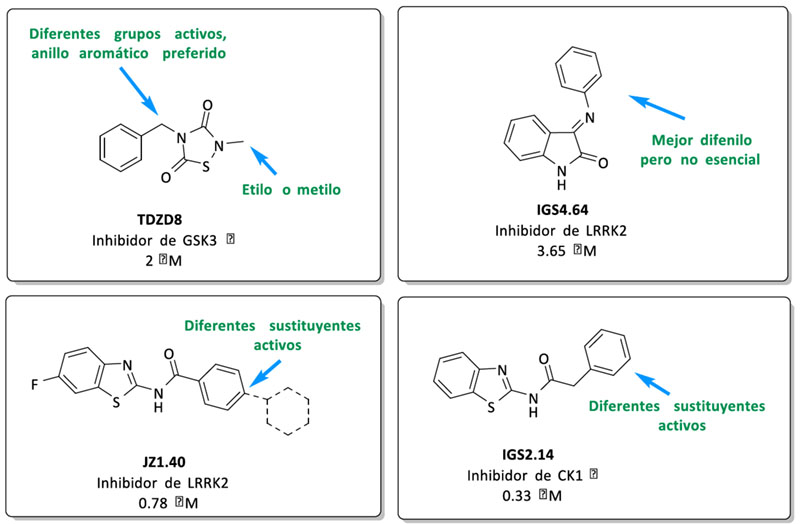
Figura 2. Modificaciones estructurales realizadas en los inhibidores de quinasa iniciales
El inhibidor de GSK-3β seleccionado fue TDZD-8. Este compuesto de la familia de las tiadiazolidindionas, fue el primer compuesto ATP no-competitivo descrito para esta enzima (20). La incorporación del grupo funcional se efectuó en el nitrógeno en posición 4, donde diferentes sustituyentes aportan actividad biológica al compuesto. Para los fragmentos inhibidores de LRRK2 se eligieron dos familias diferentes: la primera es la familia de las indolinonas (21), cuyos compuestos presentan valores de CI50 del orden micromolar cuando las moléculas tienen un sustituyente de tipo fenilo en la posición para-. La segunda familia es la de los benzotiazoles, donde la sustitución en posición para– en el anillo de fenilo es compatible con la actividad biológica de los compuestos y fue la seleccionada para la incorporación tanto de azidas como de alquinos. Finalmente, como inhibidor de CK1δ, se eligieron compuestos también de la familia de los benzotiazoles pero esta vez con un grupo metileno entre la amida y en anillo aromático (22). Estos compuestos presentan menores restricciones en la relación estructuraactividad y debido a razones sintéticas los grupos funcionales fueron introducidos en posición para-.
3.1. Síntesis y evaluación biológica in vitro.
Una vez se seleccionaron las mejores posiciones para la introducción de los grupos funcionales, se diseñó una ruta sintética basándonos en los reactivos comerciales disponibles y en la eficiencia sintética que nos proporcionara los fragmentos alquino y azida necesarios.
Se sintetizaron cinco fragmentos: dos azidas derivadas de inhibidores de LRRK2 y CK1δ y tres alquinos derivados de GSK3β, LRRK2 y CK1δ.
La síntesis del inhibidor de GSK3β presentó algunos retos sintéticos debido a que los reactivos necesarios para la formación del anillo de tiadiazolinonas son isocianatos e isotiocianatos. Ambos son incompatibles con múltiples reacciones químicas. En este caso, la estrategia sintética se cambió ligeramente para obtener el fragmento necesario de manera rápida y eficiente. El anillo aromático necesario para la actividad de GSK3β se eliminó con la hipótesis de que el anillo de triazol formado tras la química click se posicionaría de una manera similar en el compuesto final. De esta manera el conector se incorporó en la estructura inicial del inhibidor de GSK3β.
Después de la síntesis de todos los fragmentos, y para testar nuestra hipótesis, primero se sintetizó el triazol 3 que posee dos fragmentos inhibidores de LRRK2 de tipo benzotiazol. El compuesto 3 sirvió como prueba de concepto, tanto del mantenimiento de la actividad en LRRK2 como de la nueva actividad biológica del triazol en BACE1. Tras la evaluación biológica, se observó actividad en el rango de bajo micromolar para ambas enzimas lo que confirma nuestra hipótesis inicial (Figura 3, Tabla 1).
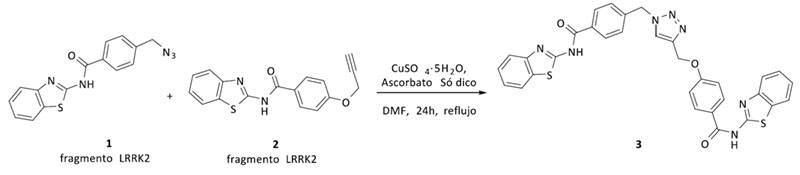
Figura 3. Síntesis del primer compuesto multidiana mediante química click catalizada por cobre
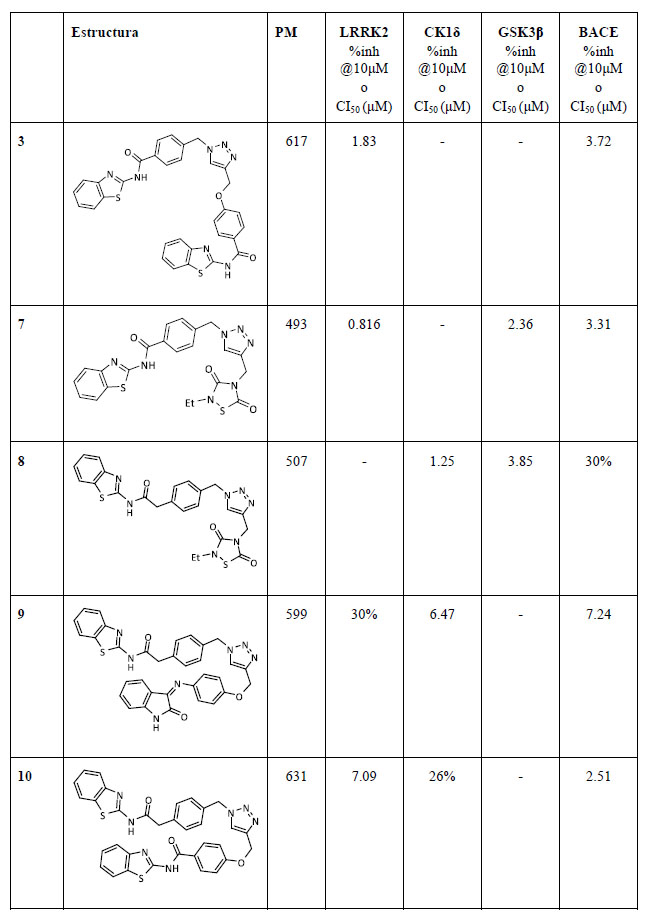
Tabla 1. Estructura química y actividad biológica de los triazoles en sus enzimas respectivas
A continuación, diferentes parejas de fragmentos inhibidores de quinasas se combinaron para obtener otros cuatro triazoles multidiana, que poseyeran actividad en BACE1 y en LRRK2-GSK3β (7), CK1δ-GSK3β (8), o CK1δ -LRRK2 (9, 10) respectivamente (Figura 4).
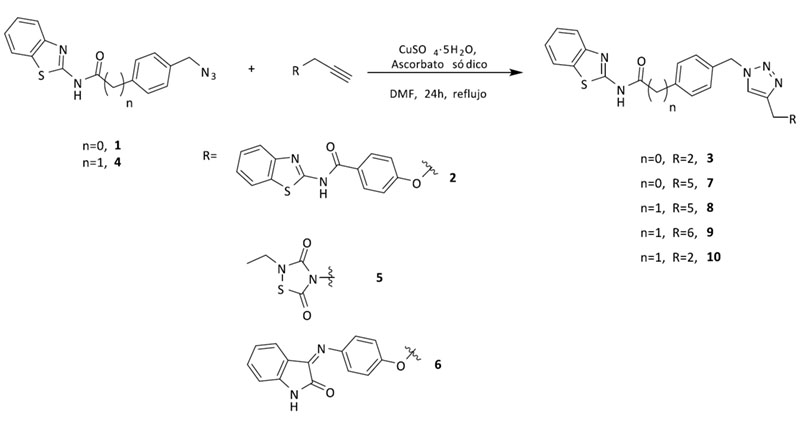
Figura 4. Estructura y síntesis de los triazoles 1,2,3-disustituídos
La actividad inhibitoria de los compuestos sintetizados se evaluó en sus enzimascorrespondientes así como en BACE1, para establecer si las nuevas estructuras químicas mantenían su potencial inhibitorio (Tabla 1).
Los resultados mostraron que todos los triazoles sintetizados eran capaces de inhibir BACE1, cuatro de ellos en el rango de bajo micromolar y solamente el compuesto 8 presentaba una inhibición del 30% a 10 μM.
Además, la actividad inicial de los fragmentos se mantuvo en general en todos los triazoles a excepción de los compuestos de CK1δ. La capacidad inhibitoria se vio reducida en un orden de magnitud para el compuesto 9 y dos órdenes para el compuesto 10.
La actividad inhibitoria de los compuestos iniciales frente a BACE1 también se evaluó, quedando demostrado que ninguna de las estructuras en las que se basaron los fragmentos poseía la capacidad de interaccionar con la proteasa (Tabla 2).
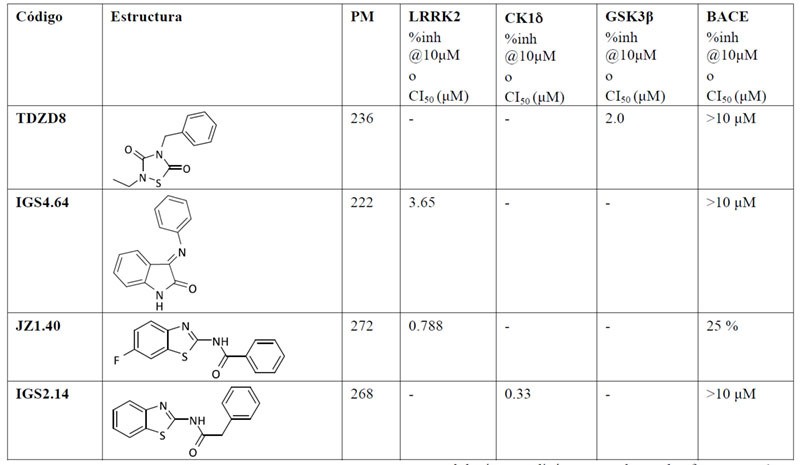
Tabla 2. Estructura química y actividad biológica de los precursores de los fragmentos en sus quinasas correspondientes y en BACE1
3.2. Química click in-situ
Finalmente, tras los resultados exitosos obtenidos con los triazoles multidiana se ha implementado una metodología para seleccionar de manera efectiva nuevos compuestos multidiana. Para ello, se utilizó la química click in situ que, en lugar de utilizar un catalizador metálico para la formación de los triazoles, emplea la enzima como molde de reacción para construir el inhibidor si los fragmentos se colocan de manera adecuada en el centro activo de la proteasa (23). La puesta a punto de la metodología se realizó empezando por los fragmentos 1 y 2, incubados 24h a concentraciones de 30 y 60 μM, en presencia y ausencia de BACE1. Tras analizar las mezclas por HPLC-MS se observaron picos cuya masa se correspondía al triazol correspondiente, solo en los casos en los que los fragmentos se incubaron con la proteasa (Figura 5). Este nuevo procedimiento permitirá una selección más eficiente de los mejores candidatos sin la necesidad de sintetizar un gran número de moléculas, ahorrando tiempo y costes en el proceso de descubrimiento de estos compuestos multidiana.

Figura 5. Los fragmentos que dieron lugar al triazol 3 se incubaron en distintas condiciones (a, c) con BACE1 y (b, d) sin la enzima. Los fragmentos se encontraban a una concentración de 30 μM (a, b) y 60 μM (c, d)
4. CONCLUSIONES Y TRABAJO FUTURO
En este trabajo se han diseñado, sintetizado y evaluado cinco moléculas multidiana que han demostrado mantener la actividad inhibitoria inicial de los fragmentos de partida en sus quinasas respectivas así como presentado una actividad inhibitoria adicional en BACE1. Este hecho deriva de la combinación adecuada de fragmentos inhibidores de quinasa con un conector de tipo triazol. Los resultados aquí presentados implican el descubrimiento no sólo de cinco nuevos prometedores candidatos para el tratamiento o estudio de la EA sino también la introducción de una manera novedosa de optimizar el diseño de compuestos multidiana. Con esta nueva metodología, los compuestos multidiana diseñados con la metodología de unión podrían incluir ahora una actividad adicional si se selecciona el conector adecuado y se coloca en la posición más beneficiosa para la actividad. Además, se han obtenido resultados preliminares que indican que la química click in situ podría ser una estrategia eficiente para el descubrimiento de entidades multidiana haciendo que la enzima seleccione los mejores candidatos.
5. ABREVIATURAS
Aβ- beta amiloide
ADP- adenosín difosfato
AEDT- ácido etilendiaminotetraacético
AEGTA- ácido etilenglicoltetraacético
ATP- adenosín trifosfato
BACE1- beta secretasa
CI50– concentración que inhibe la actividad enzimática a la mitad
CK1δ- casein quinasa 1 isoforma δ
DCM- diclorometano
DMAP- 4-dimetilaminopiridina
DMF- dimetilformamida
DMSO- dimetilsulfóxido
EA- enfermedad de Alzheimer
EDCI- hidrocloruro de N-(3-Dimetilaminopropil)-N′-etilcarbodiimida
ERM- ezrina/radixina/moesina
ESI- ionización por electrospray
FRET- transferencia de energía por resonancia de fluorescencia
GSK3β- quinasa de la glucógeno sintasa 3 isoforma β
HEPES- ácido 4-(2-hidroxietil)piperazin-1-iletanosulfónico
HPLC- cromatografía líquida de alta precisión
IR- radiación infrarroja
LRRK2- quinasa rica en repeticiones de leucina isoforma 2
MS- espectrometría de masas
RMN- resonancia magnética nuclear t.a.- temperatura ambiente
THF- tetrahidrofurano
TR-FRET- Transferencia de energía por resonancia de fluorescencia resuelta en el tiempo
VIH- virus de la inmunodeficiencia humana
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
6. AGRADECIMIENTOS
Este trabajo ha sido financiado por el MINECO (SAF2016-76693-R). V.N. poseee un contrato pre-doctoral FPU (FPU16/04466). V.P. quiere agradecer a la Caixa por su beca Junior Leader.
7. REFERENCIAS
- Ramsay RR, Popovic-Nikolic MR, Nikolic K, Uliassi E, Bolognesi ML. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018; 7: 3.
- Rutherford GW, Sangani PR, Kennedy GE. Three- or four- versus two-drug antiretroviral maintenance regimens for HIV infection. Cochrane Database Syst. Rev.4. 2003. Frishman WH, Zuckerman AL. Amlodipine/atorvastatin: the first cross risk factor polypill for the prevention and treatment of cardiovascular disease. Expert Rev. Cardiovasc. Ther. 2004; 2(5), 675–681.
- Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M, Melchiorre C. Multitarget-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008; 51: 347–372
- Van der Schyf CJ. The use of multi-target drugs in the treatment of neurodegenerative diseases, Expert. Rev. Clin. Pharmacol., 2011;4:3, 293-298,
- Bachurin SO, Bovina EV, Ustyugov AA. Drugs in Clinical Trials for Alzheimer’s Disease: The Major Trends. Med. Res. Rev. 2017; 37: 1186-1225.
- Bottegoni G, Favia AD, Recanatini M, Cavalli A. The role of fragment-based and computational methods in polypharmacology. Drug Discov Today. 2012;17: 23–34.
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016; 8:(6), 595-608.
- Martin L, Latypovac X, Wilsona CM, Magnaudeixa A, Perrina ML, Yardina C, Terroa F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res Rev. 2013; 12: 289-309.
- Bailey, R.M.; Covy, J.P.; Melrose, H.L.; Rouseau, L.; Watkinson, R.; Knoght, J.; Miles, S.; Farrer, M. J.; Dickson, D.W.; Giasson, B. I.; Lewis, J., LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol 2013; 126: (6), 809-827.
- Savelieff MG, Nam G, Kang J, Lee HJ, Lee M, Lim MH. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem Rev. 2018; Aug 10. doi: 10.1021/acs.chemrev.8b00138.
- Morales-Garcia, J.; Salado, I. G.; Sanz-San Cristobal, M.; Gil, C.; Pérez-Castillo, A.; Martínez, A.; Pérez, D. I. Biological and pharmacological characterization of Benzothiazole based CK-1δ inhibitors in models of Parkinson’s disease. ACS Omega 2017; 2: 5215-5220.
- Palomo, V.; Perez, D. I.; Perez, C.; Morales-Garcia, J. A.; Soteras, I.; Alonso-Gil, S.; Encinas, A.; Castro, A.; Campillo, N. E.; Perez-Castillo, A.; Gil, C.; Martinez, A. 5-Imino-1,2,4-thiadiazoles: first small molecules as substrate competitive inhibitors of glycogen synthase kinase 3. J. Med. Chem. 2012; 55: 1645-1661
- Sebastián-Pérez, V.; Roca, C.; Awale, M.; Reymond, J. L.; Martínez, A.; Gil, C.; Campillo, N. E. The Medicinal and Biological Chemistry (MBC) library: an efficient source on new hits. J. Chem. Inf. Model. 2017; 57: 2143-2151
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999; 286: 735-41.
- Nishitomi K, Sakaguchi G, Horikoshi Y, Gray AJ, Maeda M, Hirata-Fukae C, Becker AG, Hosono M, Sakaguchi I, Minami SS, Nakajima Y, Li HF, Takeyama C, Kihara T, Ota A, Wong PC, Aisen PS, Kato A, Kinoshita N, Matsuoka Y. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem. 2006; 99:1555-63.
- Morales-Garcia, J.; Salado, I. G.; Sanz-San Cristobal, M.; Gil, C.; Pérez-Castillo, A.; Martínez, A.; Pérez, D. I. Biological and pharmacological characterization of Benzothiazole based CK-1δ inhibitors in models of Parkinson’s disease. ACS Omega 2017; 2: 5215-5220.
- Palomo, V.; Martinez, A. Glycogen synthase kinase 3 (GSK-3) inhibitors: a patent update (2014-2015). Expert Opin. Ther. Pat. 2017; 27: 657-666.
- Mohammed I, Kummetha IR, Singh G, Sharova N, Lichinchi G, Dang J, Stevenson M, Rana TM. 1,2,3-Triazoles as Amide Bioisosteres: Discovery of a New Class of Potent HIV-1 Vif Antagonists J Med Chem. 2016;59(16):7677-82.
- Kolb HC1, Sharpless KB The growing impact of click chemistry on drug discovery. Drug Discov Today. 2003; 8:(24), 1128-37
- Martinez A, Alonso M, Castro A, Pérez C, Moreno FJ. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002; 45: 1292-1299.
- Salado IG, Zaldivar-Diez J, Sebastián-Pérez V, Li L, Geiger L, González S, Campillo NE, Gil C, Morales AV, Perez DI, Martinez A. Leucine rich repeat kinase 2 (LRRK2) inhibitors based on indolinone scaffold: Potential pro-neurogenic agents. Eur J Med Chem. 2017; 138: 328-342.
- Salado IG, Redondo M, Bello ML, Perez C, Liachko NF, Kraemer BC, Miguel L, Lecourtois M, Gil C, Martinez A, Perez DI. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. J Med Chem. 2014;57: 2755-72.
- Lewis, W. G., Green, L. G., Grynszpan, F., Radić, Z., Carlier, P. R., Taylor, P., Finn, M. G. and Sharpless, K. B. Click Chemistry In Situ: Acetylcholinesterase as a Reaction Vessel for the Selective Assembly of a Femtomolar Inhibitor from an Array of Building Blocks. Angew. Chem. Int. Ed., 2002; 41: 1053-1057.
