Premio Real Academia Nacional de Farmacia del Concurso Científico 2018 de la RANF
1. INTRODUCTION
Microglial cells constitute the resident immune cell population of the mammalian central nervous system (CNS). These cells monitor environmental changes and serve as damage sensors within the brain. Any type of injury or pathological process leads to activation of the microglia from their surveillant/resting state. In response to injury, microglial cells change their morphology taking on an ameboid appearance and then start to migrate to the site of injury, proliferate, produce/release several substances that affect pathological processes, such as proinflammatory cytokines, tumor necrosis factor (TNF)-α and interleukin (IL)-6 or IL-12, or even phagocytose damaged cells or debris to maintain the brain homeostasis (1).
However, excessive activation of the microglia could contribute to the progression of chronic neurodegenerative diseases, though the underlying mechanisms are still unclear. Regarding Alzheimer’s disease (AD), this disorder is characterized by complex molecular and cellular alterations, including the development of extracellular amyloid-beta (Aβ) deposits, intracellular aggregated phosphorylated tau, dystrophic neurites, loss of synapses and neurons, and a prominent gliosis. The reactive gliosis involves alterations in morphology and function of microglia and astrocytes. It has long been recognized that Aβ neuritic plaques are surrounded by activated microglial cells that could contribute to Aβ phagocytosis and/or compaction (2, 3). In this sense, microglial activation could improve the AD pathology reducing the Aβ levels (4). However, the release of proinflamatory mediators, such as cytokines or chemokines, from reactive microglia is thought to contribute to the chronic inflammatory response associated to AD, which has a prominent role in the progression and severity of the disease (5, 6).
Nucleotides act as widespread extracellular signaling molecules. The physiological effects of these compounds are mediated through an extended family of purinoceptors that are classified into ionotropic P2X or metabotropic P2Y receptors (7, 8). Microglia express multiple P2 receptors that differentially sense extracellular nucleotides and, therefore, P2 receptor-mediated microglial responses have received much attention (9-12). Among these purinoceptors, the P2X7 receptor has gained prominent recognition as a regulator of inflammatory responses (13). Microglial cells express P2X7 receptors (14, 15) which drive microglial activation and proliferation (16). P2X7 receptors are overexpressed in microglial cells in the neuroinflammatory foci of numerous neurodegenerative conditions (17) and the activation of these P2X7 receptors is coupled to the maturation and secretion of the key proinflammatory cytokine IL-1β (18), thereby triggering or potentiating neuroinflammation. These results lead to propose the use of P2X7 receptor antagonists for the treatment of CNS inflammation (19, 20). Interestingly, the P2X7 receptor seems to be also involved in the regulation of the processing of the amyloid precursor protein (APP) (21) and the in vivo inhibition of this receptor conduced to a reduction of the amyloid plaques in a mouse model of AD (22).
Microglia sense ATP or other nucleotides by multiple P2 receptors after which they change in different phenotypes. However, it is necessary to investigate what is the origin of extracellular ATP in the brain under both physiological and pathological conditions. Microglia have been shown to release ATP when the cells are exposed to different stimuli, such as ATP or glutamate (23, 24). The signaling pathways that mediate the ATP release from microglia remain unclear, although Connexin 43 (Cx43) hemichannels have been shown to be involved (25). However, a recent work showed that microglial cells express the nucleotide vesicular transporter (VNUT), which transports cytosolic nucleotides into vesicles using a proton-mediated membrane potential as a driving force, and these cells are able to release ATP by VNUTdependent exocytotic mechanisms (26).
Due to the close relationship that exists among ATP, the P2X7 receptor and microglial cells in inflammatory and neurodegenerative processes, where the extracellular nucleotide can have a vesicular origin, we evaluated whether the expression of VNUT and the P2X7 receptor, as well as the ATP release, could be modified in the reactive microglia. To achieve microglia activation we use the lipopolysaccharide (LPS), one of the main components of gram-negative bacterial cell wall, that induces microglial cells activation through interaction with the Toll-like receptor 4 (TLR4) (27). Additionally, we analyzed the effect of the β-amyloid peptide β1-42, which is able to activate and stimulate the phagocytic activity of the microglial cells (28), on the expression of VNUT and the ATP release in the microglia.
2. MATERIALS AND METHODS
2.1. Cell Culture
Primary cultures of microglial cells were performed according to the procedure described by (29), with some modifications. Briefly, the whole brain was removed aseptically from postnatal day 5 C57BL/6 mice and submitted to digestion with papain 100 U/ml (Worthington, Lake Wood, NJ) (previously activated in Earle’s Balanced Salt Solution (EBSS) buffer containing 5 mM l-Cys and 2 mM EDTA), in the presence of 100 U/ml of DNAse (Worthington, Lake Wood, NJ), 1 mM CaCl2 and 1 mM MgCl2. Then, cells were resuspended in Dulbecco’s Modified Eagle’s medium (DMEM) containing 10% (v/v) fetal calf serum, 25 mM glucose, 2 mM glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin and 2.5 μg/ml amphotericin, and they were plated in culture flasks at a density of 70,000 cells/cm2. Cells were maintained in culture until they reached confluence (approximately 10–12 days), replacing the medium every 3–4 days. This procedure allows obtaining an astroglial culture, as neurons do not survive under such conditions. To separate astrocytes and microglia, flasks were shaken at 250 rpm for 2 h at 37°C in an orbital shaker, and after that, the supernatant contains microglia whereas astrocytes remains adhered to the culture surface. Culture medium was then centrifuged at 200 x g for 10 min and microglial cells were resuspended and seeded onto culture plates.
Treatment with the lipopolysaccharide (LPS) was initiated 48h after seeding and the compound was added at 1μg/ml concentration for 24h (26). In the case of the treatment with β1-42, the β-amyloid peptide was prepared according to (30, 31) and added at a final concentration of 5 μM for 18h. Control non-amyloidogenic β42-1 peptide was prepared and added under the same conditions.
2.2. Reverse Transcription PCR and Quantitative Real-Time PCR
Total RNA was obtained from cultured microglia using a SpeedTools Total RNA Extraction kit (Biotools), following the manufacturer’s instructions. After reversed transcribed using M-MLV reverse transcriptase, 6 μg of random primers and 350 μM deoxynucleotide triphosphates (dNTPs) (all from Thermo Fisher Scientific). Quantitative real-time PCR (qPCR) reactions were performed in final volume of 25 μl using the LuminoCt qPCR Ready Mix (Sigma–Aldrich), 5 μl of the cDNA synthesized previously and 1.25 μl of specific commercial TaqMan® gene expression assays for mouse: VNUT (Mm00805914_m1), P2X7 (Mm00440582_m1), P2Y2 (Mm02619978_s1) and the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Mm99999915_g1), all from Applied Biosystems. The qPCR assays were carried out using a StepOnePlus Real-Time PCR System (Applied Biosystems) as follows: denaturation at 95°C for 20 s, followed by 40 cycles of 95°C for 1 s and 60°C for 20 s. The expression of each gene was normalized to that of the endogenous housekeeping gene GAPDH amplified in parallel.
2.3. Western Blotting
Microglial cells were lysed and homogenized for 1 h at 4°C in lysis buffer (pH 7.4) containing 50 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P40 and CompleteTM Protease Inhibitor Cocktail Tablets (Roche Diagnostics GmbH). The cell lysate was then centrifuged at 16,000 × g for 9 min and the supernatants collected. Protein extracts were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes, which were then blocked for 1h at room temperature (∼25°C) with 3% bovine serum albumin (BSA) in Tris buffered saline (TBS) (10 mM Tris, 137 mM NaCl, 2.7 mM KCl, 5 mM Na2HPO4, 1.4 mM KH2PO4 [pH 7.5]) with 0.1% Tween 20. The membranes were probed with primary antibodies raised against VNUT (1:500, MBL) or P2X7 (1:1000, Alomone Labs). Antibody binding was detected by ECL chemiluminescence (Amersham GE Healthcare), and images were captured with ImageQuant LAS 500 (GE Healthcare Life Sciences) and analyzed using ImageQuant TL.
2.4. Immunofluorescence
Microglial cells cultured on coverslips in 35 mm dishes were fixed for 15 min with 4% paraformaldehyde and then rinsed with phosphate buffered saline (PBS) twice for 10 min. Afterward, coverslips were blocked and permeabilized for 1h at room temperature with blocking solution (0.1% Triton X-100, 5% fetal bovine serum and 10% bovine serum albumin in PBS). The coverslips were then incubated overnight at 4°C with the corresponding primary antibodies: rabbit anti-VNUT (1:100, MBL), mouse anti-Iba1 (1:300, Wako) or rat anti-CD11b (1:500, ThermoFisher Scientific). The following day, the cells were washed three times with PBS and incubated with the corresponding fluorescent-tagged secondary antibodies (Thermo Fisher Scientific) for 1h at room temperature. The nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI, Thermo Fisher Scientific) and the coverslips were finally mounted on glass slides using FluoroSaveTM Reagent (Calbiochem). Images were acquired on a Leica TCS SPE confocal microscope.
2.5. ATP release
Adenosine triphosphate release was measured using the ENLITEN® rLuciferase/Luciferin reagent (Promega). Microglial cells were maintained for 30 min at 37°C in Mg2+-free Locke’s buffer (140 mM NaCl, 4.5 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 5.5 mM glucose and 10 mM HEPES [pH 7.4]) containing 100 μM ARL67156, a competitive inhibitor of ecto-ATPases (32), and subsequently, 50 μl of extracellular medium was collected to measure the ATP levels. The samples were centrifuged at 600 × g for 5 min at 4°C and 10 μl of supernatant were transferred onto 96-well plate placed on ice. The plate was then situated in a FLUOstar OPTIMA Microplate Luminometer (BMG LABTECH GmbH) and 100 μl of rLuciferase/Luciferin reagent was automatically injected into each well at room temperature.
2.6. Statistical Analysis
The data shown are mean values ± standard error of the mean (s.e.m) and all independent experiments shown were reproduced 3–6 times. The figures were generated and the statistical analyses performed using GraphPad Prism 6 (GraphPad Software). The results were analyzed using an unpaired Student’s t-tests. A p-value ≤ 0.05 was considered statistically significant.
3. RESULTS
3.1. Effect of LPS treatment in the expression of purinergic receptors and VNUT in the microglia
Microglial cells were stimulated with 1μg/ml LPS for 24h and mRNA levels of the P2X7 receptor and VNUT were analyzed. In order to confirm the effectiveness of the treatment, mRNA levels of the purinergic P2Y2 receptor, which expression increases after stimulation with the lipopolysaccharide (33), were also measured.
As can be observed in Figure 1, LPS treatment induced a significant increase in metabotropic P2Y2 receptor expression, whereas P2X7 mRNA levels were not affected. On the other hand, VNUT mRNA levels were significantly reduced after LPS stimulation (Figure 1A).
The protein levels of the nucleotide vesicular transporter and the P2X7 receptor were also measured by Western blot analyses and a marked reduction in the amount of both proteins was observed after LPS treatment (Figures 1D-F).
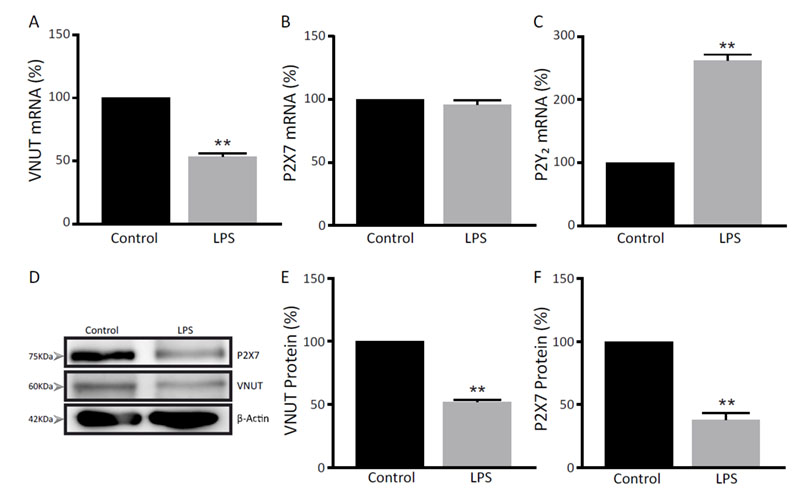
Figure 1. Effect of LPS treatment in the expression of purinergic receptors and VNUT in microglia cultures. Q-PCR analysis of mRNA levels of VNUT (A), P2X7 receptor (B) and P2Y2 receptor (C) after stimulation with LPS (1μg/ml) for 24h. Values are represented as a percentage of those obtained in untreated (control) cells, which were set as a 100%. (D) Western blot detection of VNUT and P2X7 proteins in cellular lysates of both LPS treated and untreated microglial cells. VNUT (E) and P2X7 receptor (F) protein levels were normalized with respect to β-actin. Values are represented as a percentage of those obtained in untreated cells, which were set as a 100%. Data are the mean ± SEM of three independent experiments. **p < 0.01 (Student’s t-test).
3.2. Effect of LPS treatment in the ATP release by the microglia
The effect of LPS in the microglial culture was visualized by immunofluorescence techniques, using a specific marker of microglial cells, such as the Iba1 (ionized calcium-binding adapter molecule 1) protein (34). As can be observed in figure 2A, microglial cells acquire an amoeboid morphology that is characteristic of the reactive microglia after 24h of treatment with the lipopolysaccharide, which confirmed that the LPS treatment was effective. Regarding the distribution of the nucleotide vesicular transporter, this was mainly localized in the proximal zone to the cell nucleus (Figure 2A).
The release of ATP after the treatment with LPS was measured by luminometric techniques. As can be observed in Figure 2B, the treatment with the lipopolysaccharide for 5 min induced a significant increase in the luminescence values, which means a higher liberation of the nucleotide to the extracellular medium. These results are in agreement with those obtained by other research groups (35). However, the stimulation with LPS for 24h did not significantly modify the recorded luminescence values (Figure 2C).
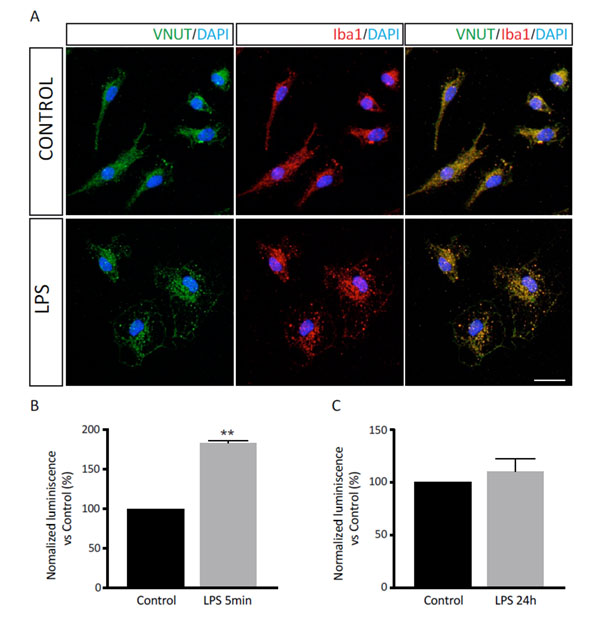
Figure 2. Effect of LPS treatment in the ATP release by the microglia. (A) Representative photomicrographs of quiescent (upper panels) and LPS treated (lower panels) microglial cells showing VNUT (green) and Iba1 (red) immunodetection. Nuclei are counterstained with DAPI (blue). A merge image is shown. Scale bar = 15 μm. (B, C) Luminometric measure of the extracellular ATP in microglial cells treated with LPS for 5 min (B) or 24 h (C). Luminiscence values were normalized with respect to those obtained in untreated (control) cells and are represented as a percentage. Data are the mean ± SEM of three independent experiments. **p < 0.01 (Student’s t-test).
3.3. Effect of the amyloid peptide β1-42 in VNUT expression in microglial cells. Release of ATP
Stimulation of the microglia with the amyloid peptide β1-42 at a 5 μM concentration for 18h resulted in a significant increase in the ATP release to the extracellular medium when compared with the effect of the nonamyloidogenic (control) β42-1 peptide (Figure 3A). However, VNUT expression was unaffected by the treatment with the amyloidogenic peptide as both the mRNA and protein levels of the transporter remained constant in β1-42 or β42-1 treated cells (Figura 3B).
Moreover, the treatment with the amyloidogenic peptide induced a morphological change in the microglial cells which acquired the amoeboid morphology that is characteristic of the reactive microglia (Figure 3C).
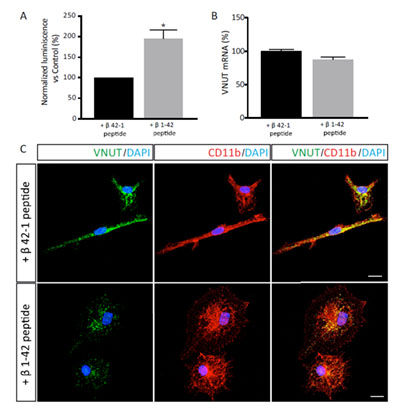
Figure 3. Effect of the amyloid peptide β1-42 in VNUT expression in microglial cells. Release of ATP. Luminometric measure of extracellular ATP (A) and Q-PCR analysis of the VNUT mRNA levels (B) in microglial cells treated with the control peptide β42-1 or the amyloidogenic peptide β1-42 for 18h. Values were normalized with respect to those obtained in cells treated with the control peptide and are represented as a percentage. Data are the mean ± SEM of three independent experiments. *p < 0.05 (Student’s t-test). (C) Representative photomicrographs of microglial cells treated with the control peptide β42-1 (upper panels) or the amyloidogenic peptide β1-42 (lower panels) showing immunodetection for VNUT (green) and the microglial cell marker CD11b (red). Nuclei are counterstained with DAPI (blue). Scale bar = 10 μm.
4. DISCUSSION
A common characteristic of the CNS diseases that courses with inflammation is that the activation of microglial cells occurs at the initial stages (36, 37). Then, cells migrate to the damaged area where they carry out phagocytic processes and mediate the inflammatory responses (1). Purinergic system plays an important role in the regulation of such processes, ATP being a key molecule in the development of microglial functions. Due to the relevance of the presence of extracellular ATP in the physiopathological context of the microglia, we asked whether such presence could be mediated by a VNUTdependent mechanism.
Our first methodological approach was the activation of the microglia through the use of the lipopolysaccharide LPS. Stimulation with this bacterial endotoxin has been used as a study model for neuroinflammatory processes, which are present in neurodegenerative diseases (38, 39). The conditions of the treatment of the microglia with LPS were based on previous studies carried out in a microglial cell line (26). The effectiveness of this stimulation was confirmed by the morphological change observed, microglial cells acquiring an amoeboid morphology, as well as by the increase in the expression of the P2Y2 receptor (33). LPS treatment did not modify the mRNA levels of the P2X7 receptor, although a significant reduction in the amount of the protein was observed. These results could reflect a change in the turnover or degradation of the P2X7 protein induced by the lipopolysaccharide. In the case of VNUT, the 24-hour treatment with LPS produced a significant decrease in its expression both at the mRNA and protein levels. These data does not agree to those described in a previous study in which the vesicular transporter was identified in the microglia (26). In this case, although the conditions tested were the same, the authors of the work used a cell line instead of a primary microglial culture. Although it is a widely used study model, it is important to consider the origin of this microglial cell line, obtained from the brain of a transgenic mouse deficient in the p53 gene (40). The absence of this gene, known as the guardian of the genome, enhances the proliferative potential of the cells (41), so that the pattern of gene expression can differ considerably from that of the primary microglia from wild type animals. Likewise, in other microglial cell lines, discrepancies have been observed in the responses to the same treatment, not only with respect to the primary microglial culture, but also in the same cell line used (42). Therefore, this could explain the differences observed regarding the expression of VNUT.
On the other hand, LPS caused a significant increase in the amount of ATP released to the extracellular medium in a five-minute interval, which is coincident with previous data obtained by other research groups (35). These results confirm that LPS induces the release of ATP in the microglia, with the initial response being very pronounced. However, the luminescence values recorded in LPS treated cells for 24 hours did not significantly differ from the control values. We cannot define the mechanism involved in LPS-induced ATP release from microglial cells. Despite this, the robust expression of VNUT in both LPS and untreated cells make possible that such release could be mediated at least in part by VNUT-dependent exocytotic mechanism. However, the decrease in transporter expression by treatment of the microglial cells with LPS suggests that another mechanism other than exocytosis, such as release trough connexins or panexins (25, 43), could mediates the ATP release from these cells.
Our second methodological approach was based in the use of the amyloid peptide β1-42. Again, the microglial cells acquired an amoeboid morphology, similar to observed after LPS treatment and typical of reactive microglia, following stimulation with the β1-42 peptide. In this case, treatment with the amyloid peptide β1-42 for 18 hours did not alter the expression of VNUT, but produced a significant increase in the extracellular levels of ATP. β1-42-induced ATP release from microglial cells has been described in a previous work (44). However, in this case acute (less than one hour) stimulation periods were used. Our results, however, showed long-term potentiation of ATP release by β1-42 peptide treatment in microglial cells. Although it cannot be confirmed that the release of ATP is mediated by a VNUT-dependent exocytotic mechanism, this idea cannot be ruled out, taking into account the expression of the transporter in the microglial cells that, unlike the treatment with LPS, remains unchanged in β1-42 stimulated cells.
It has been previously shown that microglial cells express the amyloid precursor protein (APP) and hence these cells are able to produce and release the β-amyloid peptide (45-47). The ATP released by the microglia in response to β1-42 peptide stimulation could activate microglial P2 receptors that in turn regulate β-amyloid peptide formation. It has been described that P2X7 and P2Y2 receptors modulate the α-secretase-dependent processing of the APP protein, these receptors having opposite effects on β-amyloid peptide generation (21). Activation of the P2X7 receptor leads to a reduction of α-secretase activity which enhances the amyloidogenic processing of the APP and the formation of β-amyloid peptide. However, activation of the P2Y2 receptor increases α-secretase activity, thus precluding the generation of the β-amyloid peptide (21). As activation of the microglial cells lead to reduction in the expression of the amyloidogenic P2X7 receptor and to an increase in the expression of the non-amyloidogenic P2Y2 receptor, this could represent a negative feedback mechanism by which the β-amyloid peptide limits or reduces its own formation and secretion by the microglia. Additionally, it has been previously described that the nucleotides released by the microglia in response to β1-42 peptide stimulation enhance cell migration and promote β-amyloid peptide phagocytosis through activation of the P2Y2 receptor (44). However, to have a complete image of this process it is necessary to take also into account the effect of the microglial ATP release on purinergic receptors expressed in cells other than microglia, such as surrounding neurons and astrocytes.
5. CONCLUSIONS
ATP and other nucleotides are important signal molecules that regulate a wide diversity of microglial functions. One of the possible sources of extracellular nucleotides in the CNS is their release from microglial cells. It has been demonstrated that microglia are able to release ATP and other nucleotides in response to a variety of stimuli.
Our results demonstrate that activation of microglial cells by LPS or the β-amyloid peptide β1-42 induces an increase in the ATP release, measured by luminometric techniques. This release could be mediated, at least in part, by a VNUT-dependent exocytotic mechanism, as VNUT appear to be robustly expressed by both resting and reactive microglia. However, in the case of the activation by LPS, the reduction in the expression of the transporter in activated cells, suggests that a different mechanisms, other than exocytosis, could be also involved in the ATP release from these cells.
Moreover, activation of microglial cells induces a reduction in the expression of the purinergic P2X7 receptor and an increase in P2Y2 receptor expression. In the case of the activation by β1-42 peptide, it can be hypothesized that this could represent a negative feedback mechanism by which the β-amyloid peptide limits their own formation and release by microglial cells.
Taken together, our results highlight the relevance of a new element, the nucleotide vesicular transporter (VNUT), and its role in the ATP release, in the complex interplay among extracellular ATP, purinergic receptors and microglial cells in the context of neuroinflammatory responses in neurodegenerative diseases.
6. ACKNOWLEDGEMENTS
This work was funded by the Spanish Ministerio de Economía y Competitividad (BFU 2014-53654-P), by the Comunidad de Madrid (BRADE-CM S2013/ICE-2958) and by Fundación Ramón Areces research funding (PR2018/16-02). FO acknowledges the Ramon y Cajal Program of the Spanish Ministry of Economy and Competitiveness (RYC-2013-13290).
7. REFERENCES
- Kettenmann H, Hanisch UK, Noda M, et al. Physiology of Microglia. Physiological Reviews 2011; 91: 461-553.
- Frenkel D, Wilkinson K, Zhao LZ, et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nature Communications 2013; 4.
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. Journal of Neuroscience 2008; 28: 8354-60.
- Michaud JP, Halle M, Lampron A, et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proceedings of the National Academy of Sciences of the United States of America 2013; 110: 1941-6.
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nature Reviews Neuroscience 2015; 16: 358-72.
- Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurology 2015; 14: 388-405.
- Abbracchio MP, Burnstock G, Boeynaems JM, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 2006; 58: 281-341.
- North RA. Molecular physiology of P2X receptors. Physiological Reviews 2002; 82: 1013-67.
- Koizumi S, Ohsawa K, Inoue K, et al. Purinergic receptors in microglia: Functional modal shifts of microglia mediated by P2 and P1 receptors. Glia 2013; 61: 47-54.
- Domercq M, Vazquez-Villoldo N, Matute C. Neurotransmitter signaling in the pathophysiology of microglia. Frontiers in Cellular Neuroscience 2013; 7.
- Ohsawa K, Kohsaka S. Dynamic Motility of Microglia: Purinergic Modulation of Microglial Movement in the Normal and Pathological Brain. Glia 2011; 59: 1793-9.
- Bianco F, Fumagalli M, Pravettoni E, et al. Pathophysiological roles of extracellular nucleotides in glial cells: differential expression of purinergic receptors in resting and activated microglia. Brain Research Reviews 2005; 48: 144-56.
- Lister MF, Sharkey J, Sawatzky DA, et al. The role of the purinergic P2X7 receptor in inflammation. J Inflamm (Lond) 2007; 4: 5.
- Matute C, Torre I, Perez-Cerda F, et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J Neurosci 2007; 27: 9525-33.
- Xiang Z, Burnstock G. Changes in expression of P2X purinoceptors in rat cerebellum during postnatal development. Brain Res Dev Brain Res 2005; 156: 147-57.
- Monif M, Reid CA, Powell KL, et al. The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J Neurosci 2009; 29: 3781-91.
- Weisman GA, Camden JM, Peterson TS, et al. P2 receptors for extracellular nucleotides in the central nervous system: role of P2X7 and P2Y(2) receptor interactions in neuroinflammation. Mol Neurobiol 2012; 46: 96-113.
- Ferrari D, Pizzirani C, Adinolfi E, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 2006; 176: 3877-83.
- Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs 2011; 20: 897-915.
- Friedle SA, Curet MA, Watters JJ. Recent patents on novel P2X(7) receptor antagonists and their potential for reducing central nervous system inflammation. Recent Pat CNS Drug Discov 2010; 5: 35-45.
- Leon-Otegui M, Gomez-Villafuertes R, Diaz-Hernandez JI, et al. Opposite effects of P2X7 and P2Y2 nucleotide receptors on alpha-secretasedependent APP processing in Neuro-2a cells. FEBS Lett 2011; 585: 2255-62.
- Diaz-Hernandez JI, Gomez-Villafuertes R, Leon-Otegui M, et al. In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3beta and secretases. Neurobiol Aging 2012; 33: 1816-28.
- Dou Y, Wu HJ, Li HQ, et al. Microglial migration mediated by ATP-induced ATP release from lysosomes. Cell Res 2012; 22: 1022-33.
- Liu GJ, Kalous A, Werry EL, et al. Purine release from spinal cord microglia after elevation of calcium by glutamate. Mol Pharmacol 2006; 70: 851-9.
- Ma Y, Cao W, Wang L, et al. Basal CD38/cyclic ADP-ribose-dependent signaling mediates ATP release and survival of microglia by modulating connexin 43 hemichannels. Glia 2014; 62: 943-55.
- Imura Y, Morizawa Y, Komatsu R, et al. Microglia release ATP by exocytosis. Glia 2013; 61: 1320-30.
- Zielasek J, Hartung HP. Molecular mechanisms of microglial activation. Adv Neuroimmunol 1996; 6: 191-22.
- Kopec KK, Carroll RT. Alzheimer’s beta-amyloid peptide 1-42 induces a phagocytic response in murine microglia. J Neurochem 1998; 71: 2123-31.
- Pons S, Trejo JL, Martinez-Morales JR, et al. Vitronectin regulates Sonic hedgehog activity during cerebellum development through CREB phosphorylation. Development 2001; 128: 1481-92.
- McLarnon JG, Ryu JK, Walker DG, et al. Upregulated expression of purinergic P2X(7) receptor in Alzheimer disease and amyloid-beta peptide-treated microglia and in peptide-injected rat hippocampus. J Neuropathol Exp Neurol 2006; 65: 1090-7.
- Webster S, Bradt B, Rogers J, et al. Aggregation statedependent activation of the classical complement pathway by the amyloid beta peptide. J Neurochem 1997; 69: 388-98.
- Levesque SA, Lavoie EG, Lecka J, et al. Specificity of the ecto-ATPase inhibitor ARL 67156 on human and mouse ectonucleotidases. Br J Pharmacol 2007; 152: 141-50.
- Eun SY, Seo J, Park SW, et al. LPS potentiates nucleotide-induced inflammatory gene expression in macrophages via the upregulation of P2Y2 receptor. Int Immunopharmacol 2014; 18: 270-6.
- Ito D, Imai Y, Ohsawa K, et al. Microglia-specific localisation of a novel calcium binding protein, Iba1. Molecular Brain Research 1998; 57: 1-9.
- Pascual O, Ben Achour S, Rostaing P, et al. Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proceedings of the National Academy of Sciences of the United States of America 2012; 109: E197-E205.
- Iannaccone S, Cerami C, Alessio M, et al. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat Disord 2013; 19: 47-52.
- Bachstetter AD, Norris CM, Sompol P, et al. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer’s disease-related pathology. J Neurosci 2012; 32: 10201-10.
- Guerriero F, Sgarlata C, Francis M, et al. Neuroinflammation, immune system and Alzheimer disease: searching for the missing link. Aging Clin Exp Res 2017; 29: 821-31.
- Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 2016; 12: 719-32.
- Ohsawa K, Imai Y, Nakajima K, et al. Generation and characterization of a microglial cell line, MG5, derived from a p53-deficient mouse. Glia 1997; 21: 285-98.
- Tsukada T, Tomooka Y, Takai S, et al. Enhanced proliferative potential in culture of cells from p53-deficient mice. Oncogene 1993; 8: 3313-22.
- Stansley B, Post J, Hensley K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J Neuroinflammation 2012; 9: 115.
- Gajardo-Gomez R, Labra VC, Orellana JA. Connexins and Pannexins: New Insights into Microglial Functions and Dysfunctions. Front Mol Neurosci 2016; 9: 86.
- Kim HJ, Ajit D, Peterson TS, et al. Nucleotides released from Abeta(1)(-)(4)(2) -treated microglial cells increase cell migration and Abeta(1)(-)(4)(2) uptake through P2Y(2) receptor activation. J Neurochem 2012; 121: 228-38.
- Haass C, Hung AY, Selkoe DJ. Processing of betaamyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci 1991; 11: 3783-93.
- Sondag CM, Combs CK. Amyloid precursor protein mediates proinflammatory activation of monocytic lineage cells. J Biol Chem 2004; 279: 14456-63.
- Manocha GD, Floden AM, Rausch K, et al. APP Regulates Microglial Phenotype in a Mouse Model of Alzheimer’s Disease. J Neurosci 2016; 36: 8471-86.