1. INTRODUCCIÓN
Las interacciones entre fármacos y alimentos, así como entre fármacos y plantas medicinales son frecuentes en la práctica clínica (1) y adquieren aún más relevancia cuando se trata de medicamentos de estrecho margen terapéutico, como los antineoplásicos, puesto que pueden dar lugar con mayor probabilidad a efectos adversos (2).
Además, la respuesta a las interacciones alimentomedicamento (iAM) no es la misma en todos los pacientes, siendo más grave en desnutridos, geriátricos, con cáncer o trasplantados (2). Por ello, es necesario implantar guías y protocolos de recomendaciones para la administración de fármacos de manera correcta. En este contexto, las nuevas tecnologías pueden suponer un apoyo a los profesionales sanitarios a la hora de realizar recomendaciones sobre la adecuada administración del fármaco. Por ejemplo, existe una aplicación llamada Medisonda desarrollada por el Servicio de Farmacia del Hospital de Vigo en la que a través de un buscador de principio activo y seleccionando la vía de administración enteral, ofrece recomendaciones sobre la administración de las distintas formas farmacéuticas (2). No obstante, existe controversia entre la recomendaciones ofrecidas por la EMA y las de la FDA en cuanto a la administración con o sin comidas de un medicamento, por lo que la información del etiquetado autorizada por las agencias regulatorias debería ser periódicamente revisada y estandarizada para minimizar las interacciones y mejorar el cumplimiento y adherencia del paciente (3).
La interacción con alimentos no siempre es similar en todos los fármacos del mismo grupo terapéutico, ya que debido a que pueden tener características químicas totalmente diferentes pueden presentar distinto comportamiento farmacocinético. En esta línea cabe destacar los inhibidores de tirosin-kinasa (TKI), cuyas interacciones con alimentos se discutirán en esta minirevisión. Además, se señalan algunos aspectos centrales de los TKI y su mecanismo de acción.
1.1. Inhibidores de tirosin kinasa
Los inhibidores de tirosin-kinasa constituyen un grupo de fármacos de administración oral diseñados para interferir en vías de señalización dependientes de receptores de tirosin-kinasa (4).
Como es sabido, el ATP es una molécula formada por una adenosina compuesta por un anillo de adenina, un azúcar de tipo ribosa y tres grupos fosfatos (figura 1). Por su parte, las kinasas son enzimas que catalizan la transferencia de grupos fosfatos del ATP (adenosin trifosfato) a proteínas implicadas en modular señales de crecimiento celular.
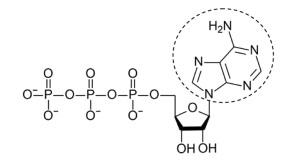
Figura 1. Estructura del ATP y lugar de unión al dominio intracelular del receptor . La unión del ATP a la proteína kinasa se realiza por puentes de hidrógeno entre la adenina (señalada con un círculo de puntos en la figura) y la hendidura de unión al ATP.
Los TKI pueden clasificarse según actúen sobre receptores de tirosin-kinasa o independientemente a ellos (no receptores de tirosin-kinasa). Los receptores de tirosinkinasa de tipo VEGFR (Vascular Endothelial Growth Factor o receptor de factor de crecimiento endotelial vascular) juegan un papel fundamental en rutas de señalización implicadas en la angiogénesis y se sobreexpresan en gran cantidad de tumores (4). Los receptores de tirosin-kinasa presentan un dominio extracelular de unión al ligando (N-terminal), un dominio transmembrana y un dominio intracelular con actividad tirosina kinasa (C-terminal). El dominio intracelular tiene una estructura bilobulada con una hendidura para la unión del ATP (adenosine triphosphate o trifosfato de adenosina) entre los dos lóbulos (figura 2).
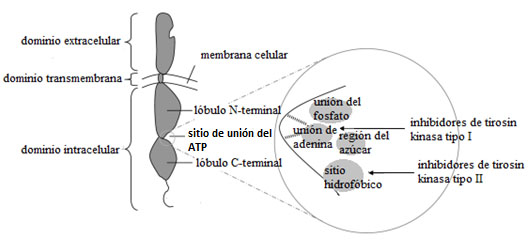
Figura 2. Estructura de un receptor de tirosin-kinasa. Se señalan las regiones de unión de los TKI I y II. Modificada de Gotink et al. (4).
Cuando el ligando se une al dominio extracelular del receptor se produce la dimerización de éste dando lugar a un cambio conformacional que activa el dominio catalítico y se produce una autofosforilación de los residuos de tirosina del dominio kinasa citoplasmático (4). Como parece lógico, la mayoría de los TKI compiten con el ATP por su sitio de unión con el objetivo de bloquear estas rutas angiogénicas.
Los derivados de la 2-fenilaminopirimidina han sido revolucionarios en la lucha contra el cáncer ya que actúan como inhibidores específicos de enzimas con actividad tirosina quinasa. Imatinib es un derivado de la molécula 2- fenilaminopirimidina que se desarrolló siguiendo un diseño racional de medicamentos. Para ello se inició la búsqueda de un posible inhibidor mediante un sondeo de muchos datos (en inglés «High-throughput screening») sobre compuestos químicos y sus efectos, hasta identificar la molécula 2-fenilaminopirimidina. Este fue el compuesto inicial que se probó y posteriormente se modificó introduciendo los grupos metil y benzamida. Más tarde se le fueron incorporando otros grupos químicos para garantizar una actividad antitumoral basada en la inhibición de la tirosin-kinasa que condujo al diseño del imatinib (5).
En el año 2001 se aprobó imatinib, primer miembro de un grupo de medicamentos dirigidos a inhibir la actividad de la enzima tirosina kinasa, actividad enzimática que se encuentra permanentemente activada en algunos tipos de células cancerosas. Según la IUPAC (International Union of Pure and Applied Chemistry o Unión Internacional de Química Pura y Aplicada) se nombra como 4-[(4-methylpiperazin-1-yl)methyl]-N-[4-methyl-3-[(4-pyridin-3-ylpyrimidin-2-yl)amino]phenyl]benzamide o (4-[4-metilpiperacin-1ilo)metil)]-N-[metil-3-[(4-piridin-3-ilpirimidin-2-ilo)amino]fenil]benzamida. Imatinib se comercializa en forma de mesilato de imatinib y se emplea en el tratamiento de leucemia mieloide crónica, leucemia linfobástica aguda, tumores del estroma gastrointestinal y otros tipos de enfermedades onocohematológicas. Este fármaco inhibe la proteína kinasa bcr-abl (Breakpoint Cluster Region-Abelson), codificada por el gen defectuoso bcr-abl o cromosoma Filadelfia, resultante de la fusión de la región bcr del cromosoma 22 con la región abl del cromosoma 9 [translocación t(9;22)].
Sin embargo, aunque imatinib inhibe la kinasa Bcr-abl con alta selectividad, no presenta especificidad absoluta ya que también inhibe al receptor PDGFR (factor de crecimiento derivado de plaquetas) y c-Kit. El desarrollo de resistencias a imatinib llevó al desarrollo de TKI de segunda generación que mostraron gran potencia y efectividad contra la mayoría de mutaciones excepto la mutación T315I (6). Esta última mutación consiste en el reemplazo de una treonina por una isoleucina en la posición 315 del gen bcr-abl.
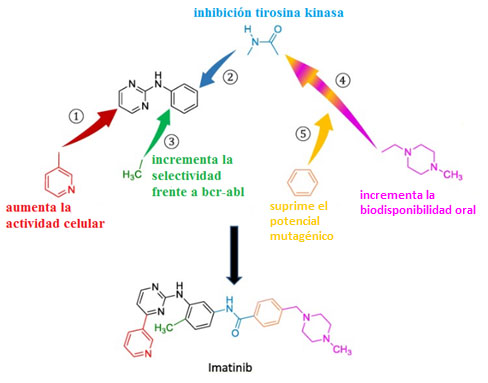
Figura 3. Fórmula química del imatinib y grupos químicos que contribuyen a mejorar su acción farmacológica. La fenilaminopirimidina, molécula central inhibidora de la tirosin-kinasa, se indica en negro. ① El grupo piridilo (grupo marcado en rojo) adicionado a la posición 3´ de la pirimidina incrementa la actividad celular, ② el sustituyente amídico (grupo marcado en azul) en el fenilo proporciona a la molécula la actividad inhibitoria de tirosina kinasas, ③ el grupo 6-metilo (grupo marcado en verde) adicionado al anillo central aminofenilo incrementa la selectividad del compuesto por bcr-abl. ④ la N-metilpiperazina (grupo marcado en violeta) incrementa la solubilidad acuosa y la biodisponibilidad oral del fármaco ⑤ se requiere la inserción en el grupo amida de un benceno (grupo marcado en amarillo) como espaciador para suprimir el potencial mutagénico de la anilina que se obtendría sin él. Bcr-abl: Breakpoint Cluster Region-Abelson. Modificada de Rossari et al. (6).
En las figuras 4 y 5 se presentan las fórmulas de algunos fármacos inhibidores del gen bcr-abl. Ponatinib (figura 4c) es el único TKI de tercera generación aprobado y el único activo contra la mutación Abl T315I gracias al grupo acetileno (en negro) que reemplaza el grupo pirimidinilamino (en rojo) de nilotinib favoreciendo una interacción con la isoleucina errónea de la posición 315 de la proteína bcr-abl. Estructuralmente es similar a nilotinib, pero además del grupo acetileno, contiene al igual que imatinib una metilpiperazina (en azul) en lugar del metilimidazol. Además, nilotinib posee un grupo piridina-pirimidina (en rojo) y ponatinib una porción imidazo[1,2-b]piridazina en la misma posición con el que harán puentes de hidrógeno en el bolsillo hidrofóbico de la proteína en el que son situados (6).
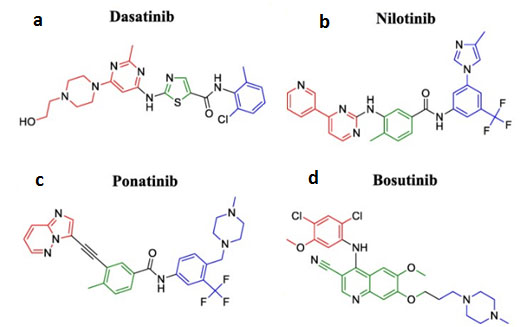
Figura 4. Estructura de fármacos inhibidores de bcr-abl. En verde: estructura central; en rojo y azul: sustituyentes. El círculo de puntos negros indica la parte de la molécula que funciona como conector con la región bisagra de la tirosin-kinasa en la proteína abl. Modificada de Rossari et al. (6).
La figura 5 esquematiza las fórmulas químicas de tres TKI diseñados para actuar sobre el dominio EGFR. Todos tienen en común el grupo quinazolina que actúa como conector a la región bisagra de la tirosin-kinasa (7). Gefitinib y afatinib incluyen radicales fluor y cloro en el grupo aminofenilo.
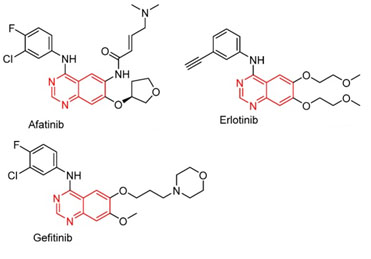
Figura 5. TKI dirigidos a EGFR (epidermal growth factor receptor – factor de crecimiento epidérmico). El grupo señalado en rojo funciona como conector a la región bisagra de la proteín kinasa Modificada de Cui et al. (7).
2. MATERIAL Y MÉTODOS
Se realizó una revisión bibliográfica de artículos científicos en la base de datos de PubMed-NCBI (National Center for Biotechnology Information), utilizando los descriptores en ciencias de la salud MeSH: “Antineoplastic Agents”, “Food-Drugs Interactions” desde 1 de enero de 2009 hasta 1 de marzo de 2019. Se excluyeron aquellos artículos que no estuvieran escritos en inglés o en español. Además, se consultaron guías editadas por la Sociedad Española de Farmacia Hospitalaria (SEFH) relacionados con interacciones farmacológicas, así como una guía publicada por la FDA (Food and Drug Administration), que recoge recomendaciones para el diseño de estudios sobre los efectos de los alimentos en la biodisponibilidad y bioequivalencia de fármacos.
3. RESULTADOS
Siguiendo los criterios de búsqueda mencionados en el apartado anterior, se localizaron 47 artículos, de los cuales se seleccionaron 55 para acotar la mini-revisión o por no cumplir con los objetivos de la misma. En estos artículos seleccionados se describen los distintos tipos de iAM y se examinan los factores implicados, desarrollando algunos ejemplos relacionados con TKI y otros antineoplásicos orales.
4. DISCUSIÓN
4.1. Definición
Se define por interacción alimento-medicamento (iAM) como aquella alteración en el comportamiento del medicamento debido a la ingestión conjunta con alimentos o a un estado de desnutrición o malnutrición por parte del paciente que altera la actividad terapéutica de un fármaco aumentándola, disminuyéndola, retardándola o incluso cualitativamente alterándola. Además, puede existir una interacción medicamento-alimento (iMA) consistente en la alteración de la utilización normal de los nutrientes debido al efecto de un fármaco (8,9,10). Esta mini-revisión se centrará fundamentalmente en las iAM.
Para poder describir los tipos de iAM es necesario recurrir a un concepto farmacocinético que adquiere gran importancia en el caso de la administración de fármacos de tipo extravasal (cualquier vía de administración con fase de absorción). Se trata del concepto de biodisponibilidad que detallaremos en el siguiente subapartado.
4.2. Biodisponibilidad
La biodisponibilidad es un parámetro biofarmacéutico y adimensional que cuantifica la disponibilidad fisiológica de un principio activo, es decir, cuantifica el porcentaje o proporción en que es capaz de acceder en forma inalterada a la circulación sistémica y a qué velocidad se produce dicho proceso (11).
Por tanto, para determinar la biodisponibilidad de un fármaco se recurre a dos conceptos diferentes: biodisponibilidad en magnitud y biodisponibilidad en velocidad. La biodisponibilidad en magnitud hace referencia a la fracción o cantidad de dosis aprovechada, se representa por F y se puede medir calculando el área bajo la curva (AUC).
La biodisponibilidad en velocidad tiene en cuenta la velocidad de absorción del fármaco y puede calcularse atendiendo a tres parámetros: a) la constante de absorción (ka), b) la concentración plasmática máxima alcanzada (Cmáx) y c) el tiempo en alcanzar la Cmáx (Tmáx). De manera que a mayor ka y/o Cmáx, mayor es la biodisponibilidad en velocidad, mientras que a mayor Tmáx, menor es la biodisponibilidad en velocidad.
4.3. Tipos de interacciones alimento-medicamento (iAM)
Dependiendo del tipo de iAM, éstas pueden clasificarse en: farmacéuticas, farmacocinéticas y farmacodinámicas (8, 12).
4.3.a. Interacciones farmacéuticas
Pueden producirse debido a una reacción directa entre la matriz del alimento y el fármaco o indirectamente por inducción de cambios en el medio gastrointestinal que conducen a una alteración en la absorción del mismo. Se refieren a los conceptos de compatibilidad, solubilidad y estabilidad por lo que cuando se estudian este tipo de iAM, es necesario recurrir al Sistema de Clasificación Biofarmacéutica (BCS), desarrollado por Amidon et al. en 1995 (13) que tiene en cuenta la solubilidad del fármaco y la permeabilidad intestinal al mismo.
El Sistema de Clasificación Biofarmacéutica basado en la Disposición del Fármaco (BDDCS) desarrollado por Wu y Benet surge en 2005 para complementar el antiguo BCS (14). Tanto el BCS como el BDDCS utilizan el concepto de solubilidad como uno de los dos criterios para su clasificación, pero en el segundo criterio, los dos sistemas difieren, ya que el BDDCS reemplaza el criterio de permeabilidad del antiguo BCS por la tasa de metabolismo. Al igual que el BCS, el BDDCS considera que un fármaco tiene alta solubilidad si la dosis terapéutica más alta de éste es soluble en una cantidad menor o igual a 250 mL de medio acuoso en un intervalo de pH de 1.2-7.5, según la FDA, y de 1.2-6.8 según la OMS a una temperatura de 37ºC. Además, el sistema BDDCS especifica que los fármacos con alto metabolismo poseen una tasa de metabolismo del 70 % de la dosis oral administrada, mientras que tras la administración de aquellos fármacos clasificados como de bajo metabolismo, más del 50 % de la dosis es excretada de manera inalterada. Sugiere además el sistema BDDCS que si la ruta mayoritaria de eliminación del fármaco fuese el metabolismo, entonces el fármaco tendría alta permeabilidad ya que para su metabolización es necesario que el fármaco penetre al interior de las células implicadas en dicho proceso. Sin embargo, si la ruta mayoritaria de eliminación fuese la excreción renal y biliar de fármaco inalterado (bajo metabolismo), entonces dicho fármaco debería ser clasificado como de baja permeabilidad. Por tanto, en base a los criterios de solubilidad y metabolismo, el BDDCS clasifica los fármacos en 4 clases (Figura 6a), mientras que desarrolla otra clasificación de los fármacos que dependen de la solubilidad y de la actividad de transportadores de absorción y/o eflujo gastrointestinal, que condicionan la permeabilidad del fármaco (Figura 6b).
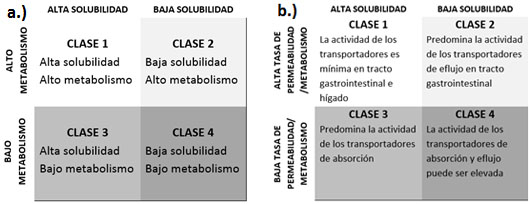
Figura 6. Criterios de clasificación de fármacos según el sistema BDDCS. La figura 6a clasifica atendiendo a solubilidad y metabolismo, mientras que la Figura 6b clasifica a los fármacos considerando solubilidad y permeabilidad. Modificada de Benet LZ. et al. (14).
De acuerdo a la figura 6b en los fármacos de clase 1 la ingesta de una comida rica en grasa no ejerce un efecto significativo en la biodisponibilidad del fármaco porque dado que la absorción de estos fármacos tiene lugar por difusión pasiva, la actividad de los transportadores en tracto gastrointestinal e hígado se considera clínicamente irrelevante y por tanto las interacciones entre el fármaco y el alimento por un transportador no se tienen en cuenta. Para los fármacos de clase 2, los efectos de los transportadores de eflujo (transportadores que expulsan las moléculas de fármaco al exterior de la célula) en el tracto gastrointestinal sí son relevantes (Figura 6b). Por ello, los fármacos de clase 2 pueden ver incrementada su biodisponibilidad en presencia de comidas ricas en grasa, bien debido a un incremento en su solubilidad o a la inhibición de los transportadores de eflujo por parte de los lípidos, limitando el regreso del fármaco al medio gastrointestinal y disminuyendo así el metabolismo enzimático. Por otro lado, para la absorción de los fármacos de clase 3 la actividad de los transportadores de absorción es importante ya que son fármacos con baja permeabilidad (Figura 6b). Por ello, es probable que cuando se administran fármacos de clase 3 con comidas ricas en grasa, se produzca una disminución de su biodisponibilidad debido al efecto inhibitorio de la grasa sobre los transportadores intestinales que median la absorción de estos fármacos. Para los fármacos de clase 4, parece que el efecto de la comida sobre la tasa de absorción es negativo o incluso nulo, aunque en algunos casos la tasa de absorción puede aumentar. Sin embargo, el número de estudios sobre el efecto de los alimentos en la biodisponibilidad de los fármacos de clase 4 es aún bastante limitado (11).
En algunos casos, las interacciones entre excipientes y alimentos pueden provocar cambios en la fisiología intestinal que influyan en la demostración de la bioequivalencia. Para los fármacos de clase 1 esperamos que los efectos de los alimentos en las medidas de la biodisponibilidad sean similares en los estudios de bioequivalencia. Sin embargo, para otras formas de liberación inmediata (clase 2, 3 y 4) y presentaciones de liberación modificada es más probable que una combinación de factores pueda influir en la disolución in vivo y/o en la absorción del fármaco. En estos casos, es difícil o muy difícil poder predecir la dirección y magnitud del efecto de los alimentos en la demostración de la bioequivalencia, sin estudios de bioequivalencia en situación de ingesta (16).
La figura 7 recoge de manera orientativa las características de solubilidad y permeabilidad, así como la tasa de metabolismo de algunos TKI.
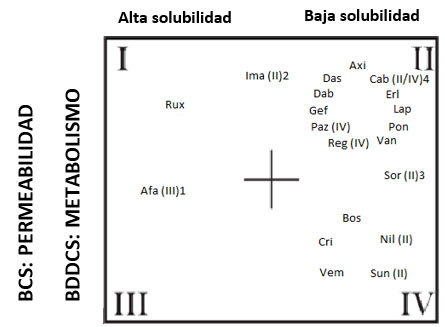
Figura 7. I/III: datos in vitro inconcluyentes. I/II: datos de solubilidad inconcluyentes. II/IV datos de permeabilidad inconcluyentes. Rux: ruxolitinib, Afa: afatinib, Ima: imatinib, Axi: axitinib, Das: dasatinib, Dab: dabrafenib, Gef: gefitinib, Paz: pazopanib, Reg: regorafenib, Van: vandetanib, Pon: ponatinib, Lap: lapatinib, Erl: erlotinib, Cab: cabozantinib, Sor: sorafenib, Bos: bosutinib, Cri: crizotinib, Vem: vemurafenib, Nil: nilotinib, Sun: sunitinib. Modificada de Herbrink et al. (17).
4.3.b. Interacciones farmacocinéticas
En estos casos el alimento altera la farmacocinética del medicamento afectando a los procesos de absorción, distribución, metabolismo y excreción (14). Este tipo de interacciones provocan una modificación de la concentración plasmática del fármaco y como consecuencia, de su concentración en el lugar de acción. En muchos fármacos la concentración en el lugar de acción o biofase, se relaciona directamente con su efecto terapéutico, y éste por tanto con la concentración plasmática. Por ello, la modificación de la concentración plasmática puede dar lugar a cambios en la actividad terapéutica, lo que hace imprescindible la monitorización de los niveles plasmáticos del fármaco (18). La biodisponibilidad depende de la absorción y del metabolismo del primer paso, por tanto, se considera que las interacciones entre medicamentos y alimentos más importantes son las que se producen en estos procesos (19). Se pueden dividir en presistémicas y postsistémicas.
Las primeras hacen referencia al efecto de primer paso, mientras que las segundas tienen lugar tras la entrada del fármaco en la circulación sistémica (12).
4.3.c.-Interacciones farmacodinámicas
Son debidas a la influencia que tiene un alimento sobre el efecto del fármaco en los receptores u órganos sobre los que actúa (18). Como ejemplos cabe destacar la actividad de ciertos componentes de los alimentos a nivel de proteínas implicadas en la transducción de señales (por ejemplo, el kaempferol, un flavonoide presente en la uva o el té verde, tiene alta afinidad por el factor de crecimiento epidérmico) o a nivel de proteínas con actividad enzimática (por ejemplo, la naringenina, un flavonoide presente en el pomelo o en la naranja amarga, inhibe la actividad de la aromatasa) (12).
4.4. Factores determinantes en las interacciones alimentomedicamento
Muchos son los factores implicados en la iAM, por lo que debe hablarse del carácter multifactorial de tales interacciones cuando el fármaco es administrado por vía oral. Por ello en este apartado se tratan las iAM desde un enfoque más específico, haciendo referencia a la vía oral y los antineoplásicos orales (11).
4.4.a.Tipo de alimento
Alimentos con alto contenido en grasa. La Guidance for Industry: Food-Effect Bioavalibility and Fed Bioequivalence Studies de la FDA describe como comida rica en grasa aquella compuesta por aproximadamente 150 kcal debidas a las proteínas, 250 kcal procedentes de los hidratos de carbono y 500-600 kcal atribuidas a las grasas, consumida en 30 min. Ingerir 900-1000 kcal en este tiempo es factible en el ámbito de un ensayo clínico, pero puede ser difícil en pacientes con cáncer que a menudo toman los antineoplásicos orales durante semanas, meses o años (16).
La ingesta de alimentos ricos en grasa puede provocar:
a) Retraso en la absorción del fármaco administrado por vía oral como consecuencia de un retraso en el vaciado gástrico. Los lípidos son los macronutrientes inhibidores más potentes del vaciado gástrico actuando por un mecanismo hormonal a través de la liberación de colecistoquinina, secretina y péptido inhibidor gástrico (11). Puesto que el lugar de absorción mayoritario de los fármacos es el intestino delgado, estos comenzarán a absorberse una vez abandonen el estómago. Si el fármaco es fácilmente absorbible, el vaciamiento gástrico será el factor limitante de la absorción, condicionando la biodisponibilidad en velocidad.
b) Incremento en la fracción de dosis absorbida de fármacos lipofílicos por un aumento en su solubilización. Por ejemplo, en el caso del lapatinib, un desayuno rico en grasas incrementa el AUC hasta 4,25 veces en comparación con la administración en ayunas (20).
c) Inducción de la secreción biliar, donde componentes de la misma, como las sales biliares, conducirían a la formación de micelas que ayudarían a la solubilización de fármacos lipofílicos (11).
d) Estimulación del transporte linfático intestinal que facilitaría la absorción de fármacos lipofílicos (11).
e) Inhibición de transportadores de eflujo dependientes de ATP, de los que son sustrato gran parte de antineoplásicos, presentes no solo en tumores y tejidos, sino también en la membrana de los enterocitos (21) (Tabla 1). La glicoproteína P (P-gp) por ejemplo, es una enzima presente en la membrana apical de los enterocitos capaz de captar moléculas de fármacos que están difundiendo a través de la membrana por absorción y devolverlas a la luz intestinal. Disminuye la captación de varios antineoplásicos orales con naturaleza anfifílica como son casi todos los TKI. Otros transportadores del epitelio intestinal son la proteína transportadora de aniones orgánicos (OATP), la proteína de farmacorresistencia múltiple (MDR) y la proteína de resistencia al cáncer de mama (MRP). Por ello, la inhibición de estos transportadores supondría un incremento de la biodisponibilidad en velocidad de dichos fármacos (17).
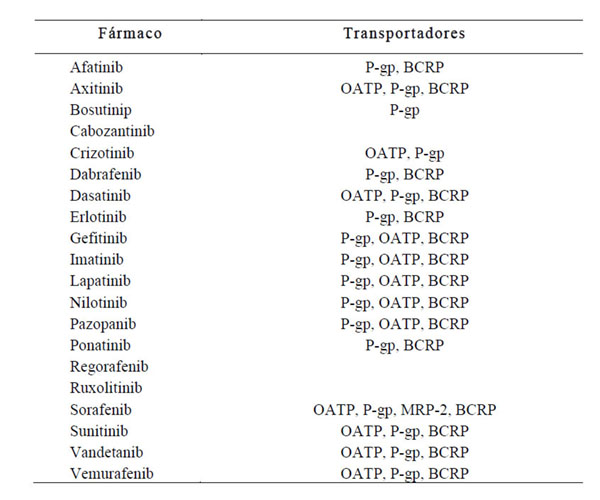
Tabla 1. Relación de transportadores intestinales y TKI.
BCRP, breast cancer resistance protein o proteína resistente del cáncer de mama; MRP-2 proteína de resistencia al cáncer de mama tipo 2; OATP, proteína transportadora de aniones orgánicos; P-gp, glicoproteína P.
f) Aumento del peristaltismo intestinal lo que podría conducir a una absorción incompleta, especialmente si el fármaco presenta una constante intrínseca de absorción baja, lo cual que reduciría la biodisponibilidad en magnitud (11).
Alimentos ricos en proteínas. De acuerdo a la revisión de Jianyuan et al. (11) la ingesta de una dieta hiperproteica puede producir:
a) Un incremento del flujo sanguíneo esplácnico lo cual suele favorecer la absorción del fármaco. En el caso de fármacos con bajo aclaramiento intrínseco, su eliminación es flujo independiente, mientras que para fármacos con alto aclaramiento intrínseco, un incremento del flujo sanguíneo hepático incrementa el metabolismo del fármaco de forma proporcional. Por tanto, la ingesta de alimentos disminuye la biodisponibilidad de este tipo de fármacos.
b) El aumento del catabolismo de las proteínas en aminoácidos que supone una competición entre éstos y el fármaco por los transportadores implicados en la absorción, como el PepT1.
c) Estimulación de la actividad metabólica de las enzimas, aumentando la velocidad de metabolización de los fármacos que son sustrato de las mismas.
Alimentos ricos en fibra. Asimismo Jianyuan et al. (11) consideran que debido a la ingesta de alimentos ricos en fibra:
a) Al igual que con los alimentos ricos en grasa, se induce un retraso en el vaciamiento gástrico.
b) Se produce adsorción de ácidos biliares postprandiales disminuyendo su efecto solubilizante sobre fármacos lipófilos y reduciéndose así su absorción.
c) La fermentación de ésta por la microbiota intestinal provoca una disminución del metabolismo intestinal del fármaco por un mecanismo competitivo.
Alimentos ricos en hidratos de carbono. También Jianyuan et al. (11) señalan:
a) Que el efecto de la ingesta de este tipo de alimentos sobre la absorción de los fármacos es poco predecible. No debemos olvidar que algunas interacciones fármacoalimento no tienen por qué ser sensibles al tipo de alimento, lo cual es ventajoso desde el punto de vista de suponer menor carga para el paciente por tener que elegir la categoría correcta de alimento en función del fármaco que esté tomando (10).
4.4.b. Reacciones físico-químicas entre fármaco y alimento
Cuando los fármacos son administrados vía oral, pueden producirse reacciones de tipo físico-químico en el tracto gastrointestinal (11). Una reacción física es la adsorción, que tiene lugar por ejemplo en el caso de la ingesta de fibra de manera concomitante con ciertos fármacos. Por reacciones químicas se entiende por ejemplo formación de precipitados o complejos. Los fármacos adsorbidos o precipitados no podrán ser absorbidos y la biodisponibilidad quedará reducida.
4.4.c.-Fisiología postprandial
La ingesta de comida provoca cambios fisiológicos que deben tenerse en cuenta pues pueden influir indirectamente en la absorción del fármaco. Entre ellos se encuentran:
a) Incremento en la viscosidad del lumen intestinal, que reduce la absorción del fármaco a través del epitelio de absorción. Por ejemplo, en el caso de la lenalidomida, se reduce un 20 % la fracción de dosis absorbida tras un desayuno rico en grasa, entre otras razones por la barrera física que supone el incremento de la viscosidad (11).
b) Retraso en el vaciado gástrico. Los fármacos que se absorben en el intestino sufrirán una disminución de la biodisponibilidad en velocidad (aumento de Tmáx) aunque la biodisponibilidad en magnitud no se verá influenciada.
c) Incremento del pH: En situación de ayuno, el pH gástrico es 1,7 mientras que en situación de postingesta, se incrementa hasta un valor de 5. El pH duodenal en cambio, pasa de un pH de 6,1 a 6,3. Puesto que muchos fármacos se absorben por difusión pasiva, la molécula debe estar en forma no ionizada para que tenga lugar la absorción. Por ello, fármacos de carácter ácido débil, se verán ionizados con el aumento de pH, mientras que las bases débiles estarán mayoritariamente en forma neutra y su absorción se verá favorecida. No obstante, también se debe tener en cuenta la estabilidad del fármaco tanto a pH ácido como básico. En el caso de erlotinib, el pH es uno de los factores más importantes para su absorción: A pH superior a 5 se produce un desplazamiento del equilibrio hacia la forma no ionizada disminuyéndose su solubilidad. Por ello, debe evitarse su coadministración con fármacos inhibidores de la bomba de protones, los cuáles como consecuencia de su mecanismo de acción aumentan el pH gástrico. En un estudio realizado por Van Leeuwen et al. (22) después de 14 días de administración concomitante de erlotinib con esomeprazol, con refresco tipo cola, el AUC0-12h y la Cmax aumentaban un 39 % y un 42 %, respectivamente en relación a los que habían bebido agua. También se observó que si los pacientes que tomaban únicamente erlotinib lo combinaban con refresco tipo cola, el AUC se elevaba ligeramente, aunque el resultado se consideró clínicamente irrelevante. Por tanto, la ingesta de un refresco tipo cola, al reducir el pH incrementa la solubilidad de erlotinib y por tanto su absorción, especialmente durante el uso concomitante con esomeprazol (22). Esto quizás podría ser extrapolado a otros TKI con solubilidad pH dependiente como cabozantinib, crizotinib, dabrafenib, gefitinib, ponatinib, sorafenib y vandetanib (17). Por lo que sería interesante evaluarlo en futuros ensayos con otras bebidas ácidas, como el zumo de naranja o diferentes bebidas carbonatadas.
d) Incremento del flujo de sangre esplácnico. Su efecto sobre la absorción del fármaco ya ha sido explicado en el apartado 4.4.a. de alimentos ricos en proteínas, por lo que no insistiremos.
e) Estimulación de la secreción biliar. Las micelas formadas gracias a las sales biliares provocan un aumento de la solubilidad de fármacos lipofílicos, pero también disminuyen la fracción fármaco libre susceptible de absorberse en la superficie de la membrana epitelial. Si la comida ingerida tiene un contenido elevado de grasa, se estimula la secreción de bilis, pero si es rica en fibra, esta última actúa como secuestrante biliar.
4.5. Ejemplos:
A continuación se exponen algunos ejemplos de iAM más estudiados y mejor conocidos:
Hypericum perforatum L. (hierba de San Juan): aunque se trata de una planta medicinal, la consideración como suplemento dietético hace conveniente que describamos las interacciones relacionadas con ella debido a su gran actividad inductora del citocromo P450 (CYP450). La hierba de San Juan está indicada en el tratamiento sintomático de estados de desánimo, pérdida de interés, cansancio y alteraciones del sueño. Las isoenzimas del CYP450 que más se afectan son la CYP3A4 y CYP2C9 (8). Numerosos estudios publicados analizan las posibles interacciones con imatinib. Frye et al. (23) analizaron el efecto de la hierba de San Juan en 12 pacientes que tomaban imatinib y se observó que el aclaramiento del fármaco aumentaba un 43% mientras que el AUC disminuía un 30 %. Además, la vida media y el Cmax descendían significativamente. Por ello debe evitarse el uso concomitante con este tipo de inductores debido al riesgo de fallo terapéutico que puede implicar (23).
Panax ginseng L. es una planta medicinal de la familia de las araliáceas, utilizada para estados de fatiga y agotamiento físico o intelectual, que contiene ginsenósidos capaces de inhibir las isoenzimas CYP3A4 y CYP2C9 (8) y también CYP2E1 (24). Se ha observado que algunos ginsenósidos son convertidos por la microbiota colónica en un derivado glucopiranosilico del protopanaxodiol, conocido como compuesto M1 o K, que interrumpe los procesos de angiogénesis (inhibición de VEF-A y FGF-2) e inhibe las actividades de las metaloproteinasas de matriz, necesaria para el crecimiento del cáncer (25). En el estudio de Bilgi et al. se observó que el uso concomitante con imatinib incrementa el riesgo de padecer hepatotoxicidad (26). Además, debido a los efectos estrogénicos de los ginsenósidos, el aporte conjunto del antineoplásico oral y el ginseng debe evitarse, particularmente en caso de pacientes afectados de cánceres hormono-dependientes. Otra posible interacción encontrada del ginseng está relacionada con la procarbazina, fármaco que se utiliza con otros medicamentos en determinados tipos de enfermedad de Hodgkin. Se ha registrado aumento de la probabilidad de padecer insomnio, dolor de cabeza, agitación, empeoramiento de la depresión y temblor (1). Esto se cree que puede deberse al incremento del metabolismo del ácido gamma-aminobutírico (GABA) y la afectación de la producción de corticoides.
Ajo (Allium sativum L .), su principal principio activo, la aliína, inhibe varias isoenzimas del CYP450, como la 2C9, 2C29 y 3A4, lo que afectaría a la vida media de los TKI. Además in vitro se ha visto que incrementa la actividad de transportadores como la P-gp, BCRP (breast cancer resistance protein) y MRP-2. Sin embargo, no hay evidencia suficiente para poder contraindicar su consumo en pacientes tratados con antineoplásicos orales (8, 27).
En relación a la vitamina C, destaca su uso en quimioterapia por su efecto antioxidante y su papel en la formación de colágeno. Tarumoto et al. (28) estudiaron en la línea celular KCL22/SR resistente a imatinib, el efecto del ácido ascórbico en la restauración de la sensibilidad a dicho fármaco. El Nrf2 es un factor de transcripción que regula la expresión de genes que codifican para la enzima glutation reductasa (enzima limitante de la síntesis de glutation reducido). Ante un estado de estrés oxidativo se produce la translocación de dicho factor de transcripción al núcleo para que se una al DNA y ejerza su función sobre la expresión génica. En el estudio se observó que la adición de ácido ascórbico a la línea celular con resistencia a imatinib suprime la translocación del Nrf2 al núcleo disminuyendo así los niveles de glutation y con ello restaurando la sensibilidad de la línea celular con resistencia a imatinib (28). Por otro lado, Lee et al.(29) realizaron un estudio en líneas celulares humanas de cáncer de pulmón no microcítico y observaron que el ácido ascórbico por su efecto antioxidante es capaz de disminuir las especies reactivas de oxígeno (ROS) tanto cuando se adiciona solo como cuando se asocia a gefitinib. En este estudio se concluye que puesto que el aumento intracelular de ROS se asocia a un incremento de mutaciones en el dominio kinasa del EGFR y por tanto a un aumento de resistencias de las células al TKI, la coadministración con ácido ascórbico podría disminuir la resistencia a este tipo de fármacos (29). Teniendo en cuenta estos datos bibliográficos, quizás la administración de TKI con alimentos ricos en vitamina C podría ser uno de los objetivos de futuros estudios.
Tras la ingesta de productos lácteos pueden formarse sales insolubles entre iones metálicos divalentes como el calcio, con fármacos como la estramustina alterándose la absorción del fármaco (8).
El zumo de pomelo (Citrus paradisi L .) contiene bergamotina y 6,7-dihidroxibergamotina (DHB), furanocumarinas que inhiben irreversiblemente el CYP3A4 de los enterocitos y ligeramente el CYP34A hepático, así como el CYP1A2 e isoenzimas de la subfamilia de CYP2C. También se ha visto que inhiben los transportadores ABCB1 (ATP-binding cassette sub-family B member 1) y OATP intestinales aunque no se ha demostrado que tenga efecto sobre la P-gp (30). En un estudio realizado en 21 individuos sanos se analizó la interacción entre el zumo de pomelo y nilotinib demostrando un aumento del 60 % en Cmax y un incremento del 29 % del AUC del fármaco (31). Sunitinib es otro TKI que es metabolizado principalmente por CYP3A4, y su biodisponibilidad también puede verse modificada como consecuencia de la ingesta de zumo de pomelo. Lo demuestra un estudio realizado por Van Erp et al. en 8 pacientes que tomaron 200 mL de zumo de pomelo tres veces al día durante un periodo de tiempo de tres días (30). Se observó que la biodisponibilidad de sunitinib aumentaba un 11 %, sin embargo se consideró clínicamente irrelevante.
Con respecto a otros zumos de frutas, en un estudio realizado in vitro por Fleisher et al. (30) se indica que dos componentes del zumo de naranja (tangerentina y nobiletina) incrementaban significativamente la concentración de dasatinib al provocar la inhibición de la P-gp. En relación al BCRP, cuatro ingredientes del zumo de pomelo (bergamotina, DHB, quercetina y kaempferol), dos del zumo de naranja (tangerentina y nobiletina) y uno del zumo de manzana (hesperetina) inhiben significativamente este transportador (32).
Por otro lado, el consumo continuo y elevado de vinagre podría reducir los niveles de potasio, por lo que en el caso de antineoplásicos orales que alteren la concentración plasmática de potasio en sangre podría ser necesario monitorizar este electrolito. Este es el caso de la abiraterona, medicamento antiandrógeno que se usa en el tratamiento del cáncer de próstata, donde la disminución del potasio en sangre es clasificada en la ficha técnica como una reacción adversa muy frecuente (8).
En relación a la ingesta de etanol las fichas técnicas de los TKI no recogen contraindicación con su consumo. Sin embargo, el alcohol puede agravar la toxicidad de múltiples fármacos debido a que su ingesta puede producir hipomagnesemia, hipoglucemia, hipertrigliceridemia, carencias vitamínicas o ataques de gota (33). Así por ejemplo en el caso de pacientes en tratamiento con sunitib y sorafenib donde las hipoglucemias son reacciones adversas frecuentes a estos fármacos, el consumo de alcohol podría considerarse desaconsejable. Lo mismo ocurre con ruxolitinib y las anormalidades lipídicas asociadas a éste. Además, se ha de tener en cuenta la relación de la ingesta de alcohol con el riesgo de aparición de varios cánceres como el de cabeza y cuello, esófago, hígado, mama y el colorrectal, por lo que cabría pensar que en pacientes con dichas patologías el consumo de alcohol debe restringirse.
Los mecanismos de la carcinogénesis alcohólica están basados en deficiencias nutricionales e hipovitaminosis, factores metabólicos, disminución de la actividad de enzimas reparadoras, producción de ROS o incremento de los niveles séricos de acetaldehído, el cual es producido a partir del etanol en el hígado y en el tracto gastrointestinal por las enzimas intestinales alcohol deshidrogenasa, el sistema microsomal de oxidación del etanol (MEOS) en el que participa el CYP450, específicamente el CYP2E1, y la catalasa (34).
Por otro lado, dado que el consumo crónico de alcohol puede originar esteatosis y/o hepatitis alcohólica y muchos de los antineoplásicos orales originan toxicidad hepática, no sería aconsejable su consumo durante el tratamiento con estos fármacos. Además, considerando que la ingesta crónica de alcohol induce la actividad del CYP2E1, la concentración plasmática de fármacos que se metabolizan por esta isoforma podría disminuir. Aunque los TKI no son metabolizados por CYP2E1, la ingesta de alcohol puede alterar la absorción de aquellos que son pH dependientes en tanto que el alcohol produce una estimulación de la secreción clorhidropéptica. Además la ingesta de alcohol retrasa el vaciado gástrico y aumenta la motilidad intestinal, factores que como ya se han descrito anteriormente, alteran la absorción de algunos fármacos (34).
A su vez el consumo crónico de alcohol causa una disminución de los niveles de ácido retinoico en el hígado ya que este es sintetizado a partir de retinol mediante varias vías enzimáticas que incluyen la alcohol deshidrogenasa y la acetaldehído deshidrogenasa, enzimas implicadas también en el metabolismo del alcohol (35). Por ello, fármacos derivados de retinoides como el bexaroteno, medicamento antineoplásico que se utiliza para el tratamiento de algunos tipos de cáncer como el linfoma cutáneo de células T, podrían ver disminuido su metabolismo cuando se co-administran con alcohol etílico debido a un mecanismo competitivo. Otro antineoplásico oral donde el consumo de etanol está contraindicado es la procarbazina ya que puede producirse depresión del sistema nervioso central y reacciones de tipo disulfiram debido a la inhibición de la enzima acetaldehído deshidrogenasa (1).
4.6. Consecuencias positivas en la coadministración de medicamentos y alimentos
La administración de un fármaco de manera concomitante con alimentos no siempre trae consecuencias negativas, siendo a veces beneficiosa y por tanto recomendable (3,17). Por ejemplo en el caso de los TKI existe una gran variabilidad en su absorción si se administran en ayuno o junto con alimentos. Así por ejemplo, es recomendable la administración de bosutinib con alimentos porque se ha estudiado que la coadministración con comida multiplica por 1,8 su Cmax y por 1,7 su AUC, en relación a la administración en ayunas (36). Por otro lado, es aconsejable administrar regorafenib con comidas ligeras con un contenido menor del 30 % de grasa, puesto que se ha encontrado que se alcanza el máximo nivel de concentración tanto de éste como de sus metabolitos farmacológicamente activos M-2 (N-óxido) y M-5 (N-óxido y N-desmetilo). En relación al lapatinib, su concentración plasmática se ve afectada por la hora de administración con respecto a la ingesta de alimentos. Cuando se administra 1 hora después de ingerir una comida se ha señalado que los valores medios del AUC son aproximadamente 2-3 veces mayores, en comparación a cuando es administrado 1 hora antes de la comida (37). En cuanto al imatinib, la administración con alimentos es aconsejable, puesto que se ha visto que las reacciones adversas gastrointestinales se minimizan (17).
La tabla 2 recoge algunas recomendaciones en relación a la administración de TKI con/sin alimentos (11). Sin embargo, en cuanto a las recomendaciones sobre la administración de estos fármacos con o sin comidas, existe controversia entre las emitidas por la FDA y por la Agencia Europea del Medicamento (EMA). Así por ejemplo, en el caso de olaparib la FDA no contraindica la toma del fármaco con comidas, mientras que la EMA recomienda su administración 1 hora antes de la comida o abstenerse de comer preferiblemente hasta dos horas más tarde. Asimismo en el caso de sorafenib, la EMA recomienda su administración con una comidas con bajo contenido en grasas mientras que la FDA aconseja la administración en ayunas. En el caso de afatinib, la FDA recomienda administrarlo 1 hora antes o 2 horas después de una comida, mientras que la EMA aconseja 3 horas antes de la comida o 1 hora después (3). Este es un aspecto muy importante que requiere investigación específica, ya que la concentración postprandial o en ayunas de transportadores y de sustratos metabólicos condicionan la absorción y metabolismo del fármaco (11).
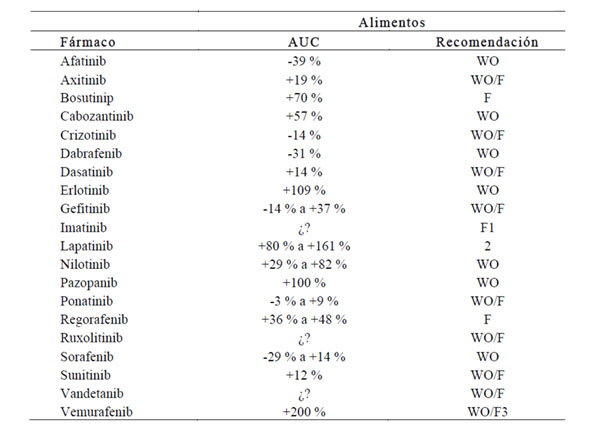
Tabla 2. Recomendaciones sobre la administración de diferentes TKI con o sin alimentos.
¿?, efecto desconocido; WO, sin comida; F, con comida; 1, Los alimentos sirven como protección gastrointestinal; 2, Administrar fármaco 1 hora antes o después de las comidas; 3, No existen aún recomendaciones. Modificada de Jianyuan et al. (11).
Como curiosidad, recientemente se está estudiando una interacción alimento-medicamento deseable y buscada. Se trata del efecto inhibitorio que tienen los flavonoides presentes en gran variedad de alimentos sobre la P-gp. Por lo que la administración del fármaco con alimentos ricos en flavonoides podría ser intencionada con el fin de aumentar la biodisponibilidad del primero (38).
5. CONSIDERACIONES Y ESTUDIOS A REALIZAR
Teniendo en cuenta todo lo comentado, creemos que para asegurar una terapia antioncológica oral eficaz y precisa es de enorme importancia tener en cuenta los aspectos que comentamos a continuación:
a) Considerar el estatus nutricional del paciente oncológico, ya que además de tener estado anímico bajo, apatía y anorexia, un estado nutricional deficiente condiciona una capacidad inmune deprimida y por tanto un estado defensivo del paciente bastante ineficiente para luchar contra los mecanismos celulares implicados en la promoción, progresión y fase terminal del cáncer (39). Es por ello que uno de los aspectos prioritarios en el tratamiento debe ser recuperar o al menos frenar la degradación del estatus nutricional que en el paciente oncológico pueda producirse.
b) Estudiar la existencia de pacientes metabolizadores rápidos y lentos, es decir con actividades enzimáticas elevadas o reducidas de la isoenzimas del citocromo P450 que condicionen la dinamia del fármaco y por tanto su biodisponibilidad. Por ello se realizarán previamente al aporte del fármaco estudios farmacogenéticos particularmente asociados a polimorfismos que afecten a la actividad de la isoenzimas más importantes del citocromo P450: CYP3A4 y CYP2C9 (40, 41).
c) Estudiar los beneficios del aporte de concentraciones más elevadas o más reducidas de los TKI a ciertas horas del día donde las concentraciones de los marcadores tumorales (proteínas de transporte como P-gp, OATP, BCRP, etc) sean más elevadas o estén más reducidas.
d) Analizar los efectos potenciadores del consumo de componentes dietéticos de reconocido papel en el control y prevención del cáncer a través de mecanismos epigenéticos. Como es sabido la epigenética incluye alteraciones en la expresión génica sin cambios en la secuencia de nucleótidos, mientras que la iniciación y progresión del cáncer incluye múltiples mecanismos epigenéticos. Debido a que algunos cambios metabólicos pueden revertirse con factores químicos o nutricionales, la epigenética puede formar parte de la estrategia y prevención de enfermedades neoplásicas. Así, componentes del ajo (dialildisulfuro, alilmercaptano), de la soja (genisteína, isofavonas) y el resveratrol, pueden inhibir la desacetilasa de histonas, mientras que la vitamina E o los folatos mediarían la metilación del DNA, conduciendo en ambos casos a una reducción de la expresión génica de la célula tumoral y de su proliferación (25). Hasta la fecha no se ha estudiado la posible interacción entre estos factores epigéneticos útiles en las primeras fases del cáncer con fármacos antitumorales que ya actúan en fases avanzados del mismo.
e) Estudiar la conveniencia de consumir diferentes tipos de dietas (p.ej. Dieta Mediterránea, dieta Atlántica) en pacientes tratados con antineoplásicos orales. Esta consideración implica multitud de aspectos comentados en esta mini-revisión y que atañen a diferentes tipos de alimentos presentes en este tipo de dieta y a su composición en hidratos de carbono, fibra dietética, ácidos grasos y componentes minoritarios (27). A este respecto el tipo de aceite culinario por el contenido en polifenoles (42, 43), el empleo de ajo y cebolla en cantidades importantes en la mayoría de los platos (p. ej. sofritos) podrían tener importancia, ya que consumidos en cantidades elevadas podrían reducir la actividad de las isoenzimas del citocromo P450, como ya hemos discutido (27).
Por último creemos que es importante destacar el papel de los relojes biológicos en el metabolismo de los xenobióticos y los mecanismos moleculares implicados (44). Es conocido que la concentración de muchas hormonas, enzimas y sustratos muestran ritmicidad circadiana (45, 46), habiéndose señalado a la cronodisrupción como antesala de probables patologías (45, 47). La cronoritmicidad está regulada por la existencia de genes CLOCK los cuales a su vez son capaces de modular la expresión de múltiples genes dando lugar a importantes variaciones rítmicas en la fisiología celular y de los tejidos (44, 46). Estos cambios a lo largo del día también afectan al metabolismo y acción terapéutica de los medicamentos y por ende a su farmacocinética y farmacodinamia según la hora del día, habiendo originado el concepto de cronofarmacología (44). Es por ello que parece obligado estudiar el papel de polimorfismos de los genes CLOCK en el metabolismo de los TKI y otros antitumorales. Además parece central conocer las posibles interacciones de estos genes CLOCK con los genes que expresan isoformas del Citocromo P450 implicadas en la metabolización de fármacos (p.ej. CYP3A4) y del alcohol (p.ej. CYP2E1) (10).
6. CONCLUSIONES
El conocimiento, cada vez mayor sobre las características fisiológicas, nutricionales y farmacológicas hace cada vez más factible predecir y prevenir los posibles efectos adversos derivados de las interacciones que se producen entre nutrientes y medicamentos. Para ello, deben analizarse los efectos de los alimentos sobre la biodisponibilidad del medicamento y su perfil farmacocinético, incluido el correspondiente a metabolitos (en particular con actividad farmacológica) para demostrar que son bioequivalentes la administración con alimentos y en condiciones de ayuno. Sin embargo, establecer un procedimiento normalizado para abordar las iAM no es sencillo puesto que las variaciones en los parámetros farmacocinéticos no parecen relacionarse de forma universal con cambios idénticos en la respuesta clínica en los pacientes. Asimismo, aún no existe consenso entre las recomendaciones ofrecidas por la FDA y la EMA sobre la administración de fármacos de forma conjunta o no con alimentos. Es por ello que se requiere un mayor número de estudios donde se analicen factores nutricionales y dietéticos que puedan originar cambios no esperados o indeseables en la acción terapéutica de los TKI. Además para garantizar el éxito terapéutico se debe exigir, estudio y conocimiento profesional amplio, y en particular de los factores nutricionales que además de inducir posibles iAM, originen cambios metabólicos y/o epigenéticos en la célula tumoral que impidan o al menos retrasen la progresión y desarrollo del cáncer.
Agradecimientos. Este trabajo se ha elaborado a partir de la ponencia “Interacciones de los nutrientes y alimentos que comprometen la acción terapéutica de fármacos antineoplásicos orales” impartida por D. Manuel Martínez Sesmero en la mesa redonda titulada Interacciones Fármaco-nutrientes que tuvo lugar en la Real Academia Nacional de Farmacia el 14 de marzo de 2019.
7. REFERENCIAS
- Collado Borrell R, Escudero Vilaplana V, Romero-Jiménez R. et al. Oral antineoplastic agent interactions with medicinal plants and food: an issue to take into account. J Cancer Res Clin Oncol. 2016; 142:2319–30.
- Romero Jiménez RM, Ortega Navarro C, Cuerda Compés C. La polifarmacia del paciente crónico complejo y la nutrición enteral. Nutr Hosp. 2017;34(1):58-69.
- Guo Yu, Dan-Na W, Yan Gong et al. Conflicting meal recommendations for oral oncology drugs: pose risksto patient care? Eur J Clin Pharmacol. 2018;74(6):833-42.
- Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13(1):1-14.
- Druker BJ, Lydon NB. Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J Clin Invest. 2000;105:3-7.
- Rossari F, Minutolo F, Orciuolo E. Past, present, and future of Bcr-Abl inhibitors: from chemical development to clinical efficacy. J Hematol Oncol. 2018;11(1):84.
- Cui JJ. A new challenging and promising era of tyrosine kinase inhibitors. ACS Med Chem Lett. 2014;5(4):272-4.
- San Miguel Samano MT, Sánchez Méndez JL. Interacciones alimento/medicamento. Inf Ter Sist Nac Salud. 2011;35(1):4-7.
- Quirós Ambel H, Martínez Sesmero JM. Drug-nutrient interactions in artificial nutritional support. An Real Acad Farm. 2018;84(2):226-37.
- Madurga Sanz M, Sánchez Muniz F. Food and drug adverse interactions: types, identification and update. An Real Acad Farm. 2018;84(2):216-25.
- Jianyuan D, Xiao Z, Zongmeng C, et al. A review of food–drug interactions on oral drug absorption. Drugs. 2017;77(17):1-16.
- Briguglio M, Hrelia S, Malaguti M. et al. Food bioactive compounds and their interference in drug pharmacokinetic/pharmacodynamic profiles. Pharmaceutics. 2018;10(4):277.
- Amidon GL, Lennernas H, Shah VP. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
- Benet LZ. The role of BCS (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classification system) in drug development. J Pharm Sci. 2013;102(1):1-3.
- Benet LZ, Broccatelli F, Oprea IT. BDDCS applied to over 900 drugs. The AAPSS journal. 2011;13:520-21.
- Guidance for industry: food-effect bioavailability and fed bioequivalence studies. U.S. Department of health and human services food and drug administration center for drug evaluation and research. 2002;2-4.
- Herbrink M, Nuijen B. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treatment Rev. 2015; 41:412–22.
- Aldaz Pastor A, Arocas Casañ V, Delgado Sánchez O. et al. Introducción a las interacciones farmacológicas. Sociedad Española de Farmacia Hospitalaria. 2012; 15-7.
- Santana Martínez S, Marcos Rodríguez JA. et al. Oral chemotherapy: food-drug interactions. Farm Hosp. 2015; 39:203-9.
- Koch KM, Reddy NJ, Cohen RB. et al. Effects of food on the relative bioavailability of lapatinib in cancer patients. J Clin Oncol. 2009;27:1191–6.
- Frederik E. Stuurman, Nuijen B, Beijnen JH. et al. Oral anticancer drugs: mechanism of low bioavailability and strategies for improvement. Clin Pharmacokinet. 2013; 52:399-414.
- Van Leeuwen RW, Peric R, Hussaarts KG. et al. Influence of the acidic beverage cola on the absorption of erlotinib in patients with non-small-cell lung cancer. J Clin Oncol. 2016; 34:1309-14.
- Frye RF, Fitzgerald SM, Lagattuta TF. et al. Effect of St John’s wort on imatinib mesylate pharmacokinetics. Clin Pharmacol. 2004; 24:1508-14.
- Fraga Elenes RA, Gordillo Bastidas D. Nutrición alternativa: efecto molecular de los suplementos. En: Nutrición molecular. Gordillo Bastidas D, Gordillo Bastidas E. (eds). McGraw Hill Education. Mexico. 2015, pp. 450-71.
- Flores Balcázar CH. Cáncer: regulación molecular con componentes dietéticos. En: Nutrición molecular. Gordillo Bastidas D, Gordillo Bastidas E. (eds). McGraw Hill Education. Mexico. 2015, pp. 307-28.
- Bilgi N, Bell K, Ananthakrishnan AN. et al. Imatinib and Panax ginseng: a potential interaction resulting in liver toxicity. Ann Pharmacother. 2010; 44(5):926-8.
- Sánchez Muniz F. Efectos nutracéuticos e interacciones con medicamentos del ajo. An Real Acad Farm. 2018. 30-4.
- Tarumoto T, Nagai T, Ohmine K. et al. Ascorbic acid restores sensitivity to imatinib via suppression of Nrf2-dependent gene expression in the imatinibresistant cell line. Exp Hematol. 2004; 32:375-81.
- Lee KE, Hahm E, Bae S. et al. The enhanced tumor inhibitory effects of gefitinib and L-ascorbic acid combination therapy in non-small cell lung cancer cells. Oncol Lett. 2017; 14:276-82.
- Van Erp NP, Baker SD, Zandvliet AS et al. Marginal increase of sunitinib exposure by grapefruit juice. Cancer Chemother Pharmacol. 2011; 67:695–703.
- Fleisher B, Unum J, Shao J. et al. Ingredients in fruit juices interact with dasatinib through inhibition of BCRP: a new mechanism of beverage-drug interaction. J Pharma Sci. 2015; 104:266-75.
- Yin OQ, Gallagher N, Li A. et al. Effect of grapefruit juice on the pharmacokinetics of nilotinib in healthy participants. J Clin Pharmacol. 2010; 50:188-94.
- Velasco Martín A. Farmacología y toxicología del alcohol etílico o etanol. An Real Acad Med Cir Vall. 2014; 51:242-8.
- Hernández Martín M. Relación entre el consumo de alcohol y el cáncer. Dialnet. 2018; 199-214.
- Clugston RD, Blaner WS. The adverse effects of alcohol on vitamin A metabolism. Nutrients. 2012; 4:356-71.
- Abbas R, Hug BA, Leister C. et al. A phase I ascending single-dose study for the safety, tolerability, and pharmacokinetics of bosutinib (SKI-606) in healthy adult subjects. Cancer Chemother Pharmacol. 2012; 69:221-7.
- Devriese LA, Koch KM, Mergui-Roelvink M. et al. Effects of low-fat and high-fat meals on steady-state pharmacokinetics of lapatinib in patients with advanced solid tumours. Invest New Drugs. 2014; 32:481-8.
- Bai J, Shengyu Z, Fan X. et al. Inhibitory effects of flavonoids on P-glycoprotein in vitro and in vivo: Food/herb-drug interactions and structure-activity relationships. Toxicol Appl Pharmacol. 2019;369:49-59.
- Ramírez Tortosa MC, Camblor Álvarez M, García Peris P. Nutricón y cáncer. En: Tratado de Nutrición. Tomo mIV. Nutrición cínica. Gil A. (ed.). Editorial Médica Panamericana, Buenos Aires, 2020, pp. 547-65.
- Nishiyama M, Eguchi H. Recent advances in cancer chemotherapy: current strategies, pharmacokinetics, and pharmacogenomics. Adv Drug Deliv Rev. 2009; 20;61:367-8.
- Lokiec F. (ed.) Drug interactions and pharmacogenetics. In Side effects of medical cancer therapy, Springer, Berlin, 2018.
- Sánchez-Muniz FJ. Aceite de oliva, clave de vida en la Cuenca Mediterránea. An Real Acad Nac Farm. 2007:73:653-92.
- Bastida S, Sánchez-Muniz FJ. Frying a cultural way of cooking in the Mediterranean diet. En: The Mediterranean Diet: An Evidence-based Approach. Preedy VR, Watson RR. (eds.). Elsevier. Amsterdam, 2015, pp. 217-34.
- Dallmann R, Okyar A, Lévi F. Dosing-time makes the poison: Circadian regulation and pharmacotherapy. Trends Mol Med. 2016; 22(5):430-445.
- Sánchez-Muniz FJ, Simón Martín C. Clock genes, chronodisruption, nutrition and obesity. Curr Res Diabetes Obes J 2017; 3(2):CRDJ. MS.ID. 555607.
- Simón Martín C, Sánchez-Muniz FJ. Cronodisrupción y desequilibrio entre cortisol y melatonina ¿Una antesala probable de las patologías crónicas degenerativas más prevalentes? JONNPR 2017; 2(11):619-633.
- Garaulet Aza M, Gómez Abellán P. Clock genes. Circadian rhythms and predisposition to obesity. An Real Acad. Farm. 2016; special Issue:44-54.