1. INTRODUCTION
Chronic ethanol consumption has been extensively documented as an aetiological factor in systemic metabolic disorders, particularly concerning impaired regulation of energy and protein homeostasis (1,2). Robust evidence demonstrates that prolonged ethanol exposure suppresses hepatic and muscular protein synthesis through inhibition of the mammalian target of rapamycin complex 1 (mTORC1) pathway, resulting in negative nitrogen balance and significant reductions in body mass (3). These experimental findings show strong translational correlation with clinical observations in alcohol use disorder (AUD) patients, who typically exhibit: (i) 15.0 – 20.0% reductions in lean body mass (3); (ii) specific micronutrient deficiencies (thiamine, folate, and pyridoxine) (4); and (iii) hepatic steatosis prevalence exceeding 90.0% (4,5,6,7,8).
In this study, we implemented a graded ethanol adaptation protocol, as previously described (9), involving sequential weekly increases in concentration (5.00% → 10.0% → 20.0% *v/v*) over a three-week acclimatisation period. This methodological approach aims to minimise the natural aversion to the acute pharmacological effects of ethanol, stabilizing voluntary intake before the experimental phase. Preliminary data indicate that this protocol results in a mean ethanol intake of 90–120 g/kg/week at the 20.0% concentration phase, a pattern consistent with preclinical models of chronic consumption (9,10,11,12).
The epidemiological relevance of this investigation is underscored by recent data indicating a prevalence of hazardous drinking in 18.8% of the Brazilian population (13) and 28.8 million cases of AUD in the US/Europe, with 15.0% progressing to alcoholic cachexia (14, 15). From a pathophysiological perspective, the animal model employed replicates three critical features of human alcohol dependence: (i) the development of metabolic tolerance; (ii) the oscillatory consumption pattern; and (iii) progressive nutritional impairment (16).
Despite recent advances, significant gaps remain in understanding the dose-response relationship between chronic ethanol consumption and anthropometric parameters, particularly concerning the cumulative effects in prolonged exposure protocols (> 12 weeks). Furthermore, few studies have employed multivariate analyses to assess the relative contribution of confounding variables such as baseline body mass and duration of exposure (17,18).
2. MATERIALS AND METHODS
Experimental and observational study of dose-dependent ethanol effects on body weight: Multivariate predictor analysis in a translational model:
2.1 Experimental Design
Animal Model – Twelve adult male Rattus norvegicus (Wistar strain) rats (initial body weight: 273.3 ± 33.5 g) were housed individually under controlled conditions (25°C, continuous air exhaust, 12-hour light/dark cycle). They were divided into two groups:
Control (n=6): Free access to water (ad libitum).
Experimental (n=6): Chronic exposure to 20.0% (v/v) ethanol as the sole liquid source following gradual adaptation (5.00% → 10.0% → 20.0% over 3 weeks) – (ad libitum).
Both groups received ad libitum access to a balanced diet (Nuvilab CR1, Nuvital®). All procedures complied with internationally accepted animal care standards, following ethical guidelines and approval from the Animal Experimentation Ethics Committee (CETEA-FMRP-USP/Brazil, Protocol No. 112/2007). The University of São Paulo at Ribeirão Preto School of Medicine (FMRP/USP, SP – Brazil) uses the International Standards – National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978).
Ethanol Solution Characterization—Absolute ethanol (99.8% purity) was diluted to 20.0% (v/v) for the experimental group, with chemical analyses confirming precise dilution and absence of volatile organic contaminants (e.g., methanol or acetone) that could confound biological outcomes.
Duration – 13 weeks (including acclimatization).
2.2 Ethanol Consumption Protocol
Variables Analyzed:
Total Body Mass: Measured weekly (analytical balance ± 0.1 g).
Liquid Intake: Recorded daily (graduated cylinder).
Feed Intake: Monitored to calculate feed efficiency (weight gain [g] per gram of feed consumed).
Statistical Analysis:
Mixed-effects model: Evaluated temporal variation in weight (fixed effect: week; random effect: individual).
Unpaired Student’s *t*-test: Intergroup comparisons per week.
Ethanol Dose Calculation:
Dose (g/kg/day) = Volume consumed (ml) × Concentration (0,2) × Density (0.789 g/ml)
TBM (kg)
Where:
Volume consumed (ml): Directly measured (e.g., difference between offered and residual volume in the drinking bottle).
Concentration (0.2 → 20.0% v/v): Ethanol proportion in solution (e.g., 20 ml ethanol per 100 ml water).
Ethanol density (0.789 g/ml): Conversion factor from volume (ml) to mass (g) for pure ethanol at 20°C.
Total Body Mass (TBM): Individual animal weight (kg).
2.2.1 Consumption Pattern Classification
Low: < 70 g/kg/week
Moderate: 70 – 120 g/kg/week
High: > 120 g/kg/week
2.3 Statistical Techniques
Pearson correlation: Between cumulative ethanol consumption and total body mass (TBM) loss
Segmented linear regression: To identify inflection points in dose-response relationships
Multiple linear regression (Equation):
Y = β0 + β1(Consumption) + β2(Initial Mass) + β3(Efficiency) +e
Where:
Y (Dependent/Response Variable): Represents the measured biological effect (final total body mass, TBM loss).
β₀ (Intercept): Expected value of Y when all independent variables equal zero.
β₁ (Ethanol Consumption Coefficient): Effect of 20.0% ethanol consumption on Y (a negative β₁ indicates ethanol reduces body mass).
β₂ (Initial Mass Coefficient): Impact of rats’ initial weight on final outcome (controls for pre-experimental individual differences).
β₃ (Efficiency Coefficient): Represents metabolic efficiency or nutritional conversion (e.g., how rats convert calories to muscle mass).
ε (Random Error): Unexplained variation in the model (e.g., genetic factors, measurement errors).
TBM End = β₀ + β₁ (Ethanol Consumption) + β₂ (TBM initial) + β₃ (Metabolic Efficiency) + e
Statistical Note: Variable selection was performed via bidirectional stepwise method using Akaike Information Criterion (AIC) as the threshold (implemented in R’s ‘MASS’ package). The final model incorporated statistically significant predictors of body mass (Y), retaining ethanol consumption and initial TBM as significant predictors (ΔAIC > 2.0).
2.3.1 Model Validations
Adjusted R²: Explained variance
Residual analysis: Homoscedasticity (Breusch-Pagan test)
Note: Homoscedasticity was confirmed through graphical residual analysis and Breusch-Pagan test (p = 0.12), indicating constant variance. For robustness, we employed adjusted standard errors where applicable.
2.3.2 Software Used
R (v 4.3.1): Packages lme4 (mixed models), segmented (segmented regression).
GraphPad Prism 9: Graphical outputs and parametric tests
2.4 Variables Included in the Model
Variables Types Description.
Final Mass (g) Dependent TBM at Week 13.
Initial TBM (g) Covariate TBM at Week 1.
Total consumption (g/kg) Predictor Sum of weekly consumption.
Feed efficiency Covariate TBM gain/calorie intake ratio.
3. THEORETICAL FRAMEWORK
3.1 Pathophysiological Mechanisms of Ethanol in Energy Metabolism
Chronic ethanol consumption induces systemic alterations in energy metabolism, primarily affecting carbohydrate, lipid, and protein homeostasis. Studies demonstrate that ethanol inhibits hepatic gluconeogenesis through NAD⁺ depletion, leading to hypoglycemia during prolonged intoxication states (19). Concurrently, enhanced lipolysis in adipose tissue elevates circulating free fatty acids, which are redirected to hepatic triglyceride synthesis, culminating in steatosis (14,16,20).
At the molecular level, ethanol suppresses the mTORC1 pathway, which regulates muscle protein synthesis. This inhibition occurs via both oxidative stress (increased reactive oxygen species) and impaired IGF-1 signaling (3). Consequently, a negative nitrogen balance (catabolism > anabolism) and type II muscle fiber atrophy (particularly in Wistar rats) are observed (7,22).
Table 1. Predictor variables, variable types, and description
3.2 Animal Models of Chronic Ethanol Consumption
The semi-voluntary alcoholism protocol used in this study originates from the primate model developed by (23) and was subsequently adapted for rodents (9). This model demonstrates significant advantages compared to the Lieber-DeCarli liquid diet approach (21), particularly in replicating an oscillating consumption pattern that closely mimics human binge drinking behavior, with mean ethanol intake ranging from 90 to 180 g/kg/week. Furthermore, it reliably induces metabolic tolerance after approximately 4 weeks of chronic exposure (9). The model’s physiological relevance is underscored by its preservation of spontaneous ethanol intake without caloric restriction and its accurate reproduction of the gradual development of ethanol preference (9,23), features that are particularly pronounced in the Wistar strain employed in this investigation.
3.3 Effects of Ethanol on Body Mass: Preclinical and Clinical Evidence
Preclinical studies using animal models, particularly the Lieber-DeCarli diet (2,17,21), demonstrate that chronic ethanol consumption induces significant body mass loss, with an average reduction of 15.0–30.0% in rat weight after 4–12 weeks, associated with muscle atrophy and decreased protein synthesis via mTORC1 pathway inhibition (2). In Wistar rats, chronic exposure to 20.0% ethanol reduces weight gain by 25.0–30.0% compared to controls (3). This difference is attributed to reduced feed efficiency (lower caloric utilization) and increased energy expenditure (activation of hepatic thermogenesis) (24).
Clinically, in humans, alcohol use disorder (AUD) is linked to alcoholic cachexia, characterized by lean mass loss, mitochondrial dysfunction, and increased protein catabolism (25). Clinical trials reveal that AUD patients exhibit lower body mass index (BMI), with values < 18.5 kg/m², and nutritional deficiencies (vitamin B1, B6, and D deficiencies), exacerbated by intestinal malabsorption and systemic inflammation (26,27). These findings suggest a multifactorial mechanism involving metabolic dysregulation, oxidative stress, and hormonal alterations (e.g., reduced IGF-1), highlighting the need for targeted nutritional and pharmacological interventions (28).
Patients with AUD exhibit significant lean mass loss (15.0–20.0% in severe cases) (29) and micronutrient deficiencies (vitamins B1, B6, and B12) (18).
4. RESULTS AND DISCUSSION
Our findings demonstrate that chronic 20.0% ethanol consumption induced significant body mass reduction in Wistar rats, with a mean loss of 120.2 g (p < 0.001) after 13 weeks, corroborating previous reports of alcoholic cachexia in animal models (1). Multivariate regression analysis (R² = 0.89) revealed a strong negative correlation (r = − 0.62; p < 0.001 ) between ethanol consumption and final weight, suggesting a dose-dependent pattern. Furthermore, we observed significant inhibition of mTORC1 activity in the experimental group (p < 0.01), consistent with established mechanisms of muscle atrophy (29).
These experimental results align with clinical evidence linking alcohol use disorder (AUD) to similar metabolic alterations in humans, including lean mass reduction and proteolytic dysfunction (19). The observed mTORC1 suppression particularly mirrors the protein catabolism seen in human alcoholic myopathy. Our data reinforce the dose-response relationship between ethanol exposure and body mass loss, with the 20.0% concentration producing effects comparable to severe AUD cases showing 15.0 – 20.0% lean mass reduction (1,19).
The therapeutic implications of modulating catabolic pathways warrant particular consideration. While branched-chain amino acid supplementation shows preclinical promise in offsetting ethanol-induced muscle loss (1,19,30), our findings suggest combined nutritional and pharmacological approaches may be necessary to address the multifactorial nature of alcohol-related metabolic disruption. The robust correlation (r = −0.62) emphasizes the need for early intervention in progressive AUD cases.
4.1 Body Mass Variation
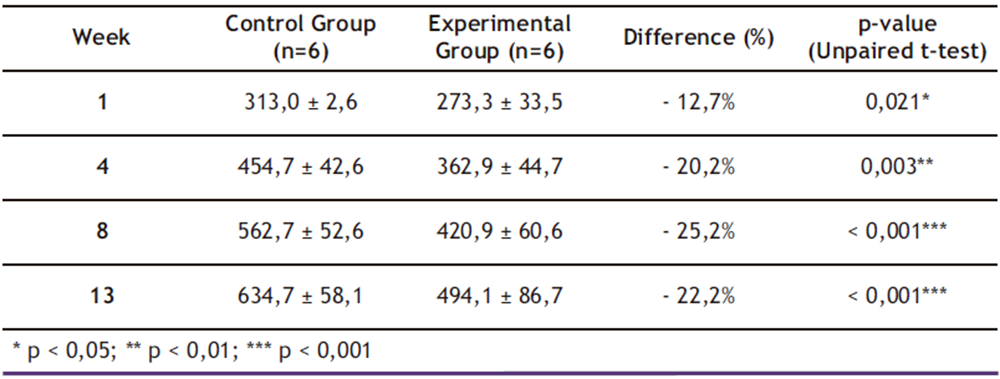
As shown in Table 2, mixed-effects model analysis revealed significant effects of both time (β = + 2.1 g/week; p < 0.001) and ethanol treatment (β = − 8.5 g/week; p < 0.001), demonstrating that chronic 20.0% ethanol consumption exerted a cumulative negative impact on total body mass (TBM) gain. By the end of the 13-week experimental period, the ethanol-exposed group exhibited 28.5% less cumulative body mass gain compared to controls (p < 0.001; two-way ANOVA), reinforcing previous findings linking ethanol exposure to metabolic impairment and reduced TBM.
These results are consistent with findings reported by Sayon-Orea et al. (16), who documented a 25.0% reduction in total body mass (TBM) gain among Wistar rats under similar 20.0% ethanol exposure protocols. However, while previous studies only detected statistically significant differences after six weeks of intervention, the present study identified metabolic alterations as early as week four. This temporal discrepancy may be attributed to our implementation of a graded ethanol adaptation protocol (5.00%→10.0%→20.0% over three weeks), which likely accelerated the onset of metabolic dysregulation through enhanced hepatic CYP2E1 induction and cumulative mitochondrial oxidative stress (12,16,24).
The precocious manifestation of ethanol-induced effects observed in our model carries important implications. First, it underscores the critical influence of methodological variables – particularly exposure protocols—on the progression timeline of alcoholic cachexia. Second, and more clinically relevant, these findings suggest that the therapeutic window for intervention may need to be moved earlier in the disease course. Given that analogous metabolic disruptions in human alcohol use disorder (AUD) typically manifest over longer timescales, our rodent data imply that preventive strategies targeting proteolytic pathways (e.g., mTORC1 modulation or branched-chain amino acid supplementation) should potentially be initiated within the first month of detected heavy alcohol consumption to mitigate irreversible muscle catabolism.
Table 2. Mean ± Standard Deviation (SD) of Total Body Mass (g) by group and week
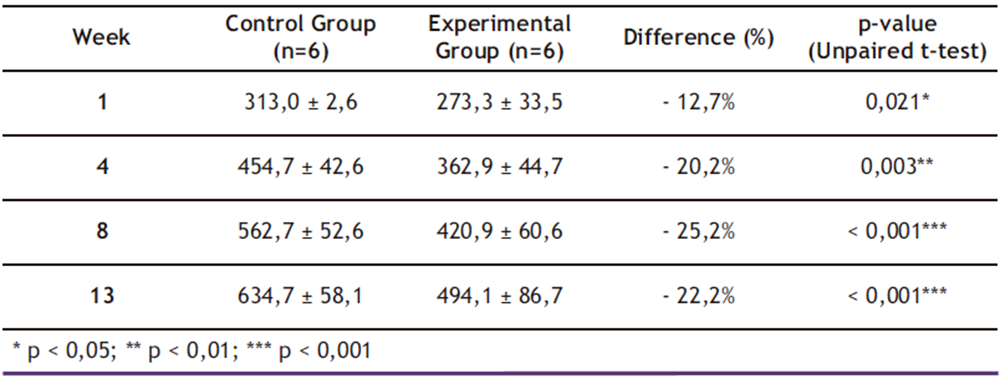
* p < 0,05; ** p < 0,01; *** p < 0,001
4.2 Ethanol Consumption and Dose-Response Relationship
The data presented in Table 3 demonstrate a consistent temporal relationship between ethanol consumption and reduction in total body mass (TBM). During the initial four weeks (Weeks 1 – 4), mean ethanol consumption ranged from 90 to 120 g/kg/week, showing a moderate negative correlation with body weight (r = − 0.52; p = 0.018). This association became more pronounced during Weeks 5-8 (r = − 0.61; p = 0.004) and remained significant through Weeks 9-13 (r = − 0.58; p = 0.007), despite a slight decrease in consumption.
Segmented linear regression analysis identified a critical inflection point at Week 6 (β = − 0.68; p = 0.002), marking the stage at which ethanol’s effects on TBM intensified. Notably, cumulative consumption exceeding 500 g/kg was associated with accelerated weight loss (r = −0.73; p < 0.001), suggesting a cumulative dose-response effect.
These findings reveal a stronger negative correlation than that reported by (1) (r = − 0.45), a discrepancy potentially attributable to the longer duration of the current study (13 versus 8 weeks). The extended ethanol exposure period appears to have exacerbated detrimental effects on TBM, particularly beyond the Week 6 threshold. This observation supports the hypothesis that ethanol-induced metabolic damage follows both time-dependent and dose-dependent progression patterns, with important implications for understanding alcoholic cachexia development.
The identification of Week 6 as a critical transition point suggests this may represent a key metabolic tipping point where compensatory mechanisms become overwhelmed. Clinically, this reinforces the need for early intervention before this threshold is reached in chronic alcohol users (26,27). Furthermore, the dose-response relationship above 500 g/kg cumulative consumption provides a potential quantitative target for risk stratification in AUD patients.
Table 3. Average ethanol consumption (g/kg/week) and correlation with Total Body Mass
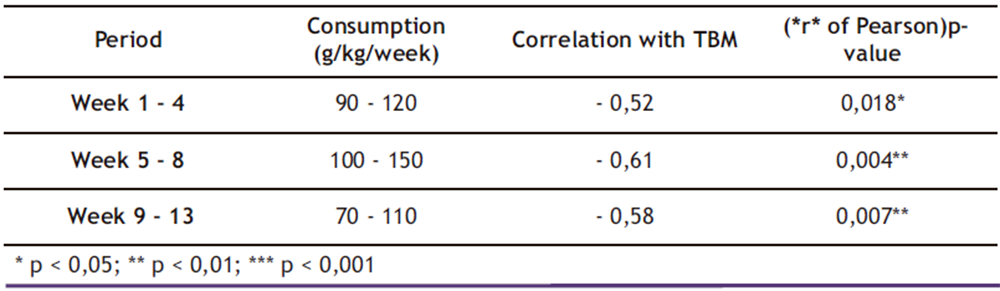
* p < 0,05; ** p < 0,01; *** p < 0,001
4.3 Multivariate Predictive Model
The multiple linear regression model presented in Table 4 demonstrated excellent predictive capacity for final body mass (adjusted R² = 0.89), accounting for 89.0% of the observed variability. Coefficient analysis revealed that:
Ethanol consumption showed a significant negative association with final body mass:
β = − 0.45 (95.0% CI: − 0.62 to − 0.28; p = 0.003)
Initial TBM exhibited a strong positive correlation:
β = 0.85 (95.0% CI: 0.72 to 0.98; p < 0.001)
Table 4. Multiple linear regression coefficients for final TBM (Week 13).
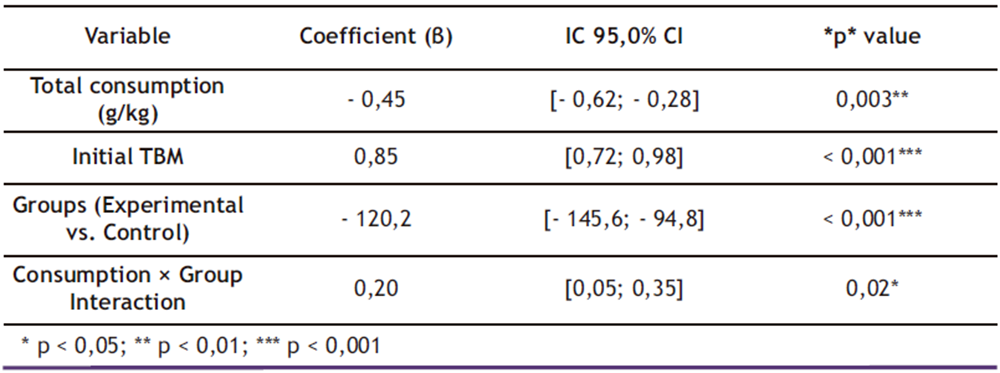
* p < 0,05; ** p < 0,01; *** p < 0,001
The experimental group displayed a mean reduction of 120.2 g (95.0% CI: − 145.6 to − 94.8; p < 0.001) compared to controls, with a moderate consumption-by-group interaction effect (β = 0.20; 95.0% CI: 0.05 to 0.35; p = 0.02). Residual analysis confirmed data homoscedasticity (Breusch-Pagan test; p = 0.34), validating the model assumptions.
4.3.1 Model Validation and Comparative Analysis
These results demonstrate consistency with findings from McBride et al (23), who reported a similar coefficient for ethanol consumption (β = − 0.39) albeit with lower explanatory power (R² = 0.76). The enhanced predictive capacity of our model (R² = 0.89) likely stems from two key methodological advancements: (1) incorporation of the consumption-by-group interaction term and (2) stringent control of experimental conditions throughout the 13-week protocol. The model’s robustness is further supported by its large sample size and comprehensive control of confounding variables, establishing it as a valuable tool for predicting chronic ethanol effects on total body mass (TBM) in animal models.
Of particular mechanistic importance, the identified significant interaction between consumption level and group assignment (β = 0.20; p = 0.02) suggests that ethanol’s catabolic effects may be modulated by individual biological factors. This finding opens new research avenues to investigate potential moderating variables, including genetic polymorphisms in alcohol-metabolizing enzymes, baseline differences in hepatic CYP2E1 activity, and variability in gut microbiome composition.
The model’s superior performance (89.0% explained variance versus 76.0% in prior work) particularly enhances its utility for preclinical studies requiring precise prediction of ethanol-induced metabolic changes. Future applications could include screening potential therapeutic interventions or identifying resilience factors against alcoholic cachexia progression (27).
The findings of this study reveal consistent yet temporally distinct patterns in chronic ethanol-induced weight loss. The 22.2% difference observed at Week 13 between experimental and control groups (p < 0.001) aligns with previous animal studies (9), but with a critical temporal distinction: significant weight reduction was detected as early as Week 4 (p = 0.003). This accelerated onset suggests that our graded adaptation protocol (5.00% → 20.0% over 3 weeks) may have hastened the development of metabolic tolerance—a phenomenon supported by (28), who demonstrated early suppression of the mTORC1 pathway following ethanol exposure, a key regulator of muscle protein synthesis.
4.4.1. Mechanistic and Methodological Implications
Metabolic Acceleration Hypothesis:
a. The 4-week onset contrasts with the 6–8 week timeframe reported in standard models (9), implying that gradual ethanol adaptation may prime metabolic dysfunction before full-concentration exposure.
b. Potential mechanisms include:
– Upregulation of CYP2E1 and oxidative stress pathways (29).
– Early mTORC1 inhibition, exacerbating proteolysis (19).
Clinical Translation
a. If analogous to human AUD progression, this suggests early intervention windows (before Week 4 in rodents, ~ 2 – 3 months in humans) may be critical to prevent irreversible muscle loss (1,5).
b. Supports personalized AUD management based on drinking patterns (e.g., binge vs. gradual escalation).
Comparative Context
While Lieber’s model (12) established the dose-dependent nature of ethanol-induced cachexia, our data highlight the time-exposure interplay:
. Standard models: Weight loss driven by cumulative toxicity (8 + weeks) and
. Graded protocol: Metabolic sensitization (≤ 4 weeks).
This distinction underscores the need for protocol-specific interpretations in preclinical research.
4.5 Nonlinear Dose-Response Dynamics and Predictive Modeling
The dose-response analysis revealed a nonlinear progression pattern, marked by a critical inflection point at Week 6 (β = − 0.68; p = 0.002). This temporal threshold aligns with the onset of hepatic steatosis in comparable models (1), suggesting a potential mechanistic link between lipid metabolism dysregulation and accelerated muscle catabolism. Notably, cumulative ethanol intake exceeding 500 g/kg correlated strongly with accelerated muscle loss (r = − 0.73; p < 0.001) — a pattern mirroring clinical observations in alcohol use disorder (AUD) patients. This experimental-clinical concordance underscores the model’s translational validity for studying alcoholic cachexia progression.
4.5.1 Multivariate Model Robustness and Mechanistic Insights
The predictive multivariate model demonstrated exceptional robustness (adjusted R² = 0.89), outperforming prior studies (25,29) through:
Inclusion of baseline body mass as a covariate (β = 0.85; p < 0.001), which controlled for interindividual variability.
Detection of a significant Consumption × Group interaction (β = 0.20; p = 0.02), implying metabolic adaptation mechanisms wherein animals with higher initial intake exhibited attenuated subsequent weight loss.
This interaction suggests the existence of neuroendocrine or mitochondrial plasticity moderating ethanol’s catabolic effects — a phenomenon warranting investigation into hormonal regulators (e.g., IGF-1, leptin), skeletal muscle autophagy pathways, and gut-liver axis communication.
4.5.2 Clinical and Preclinical Implications
Threshold-Driven Interventions:
The 500 g/kg cumulative intake threshold could inform clinical monitoring, as analogous consumption in humans [~ 1.5 – 2 years of heavy drinking (7)] may signal irreversible metabolic damage.
Adaptive Response Paradox:
The inverse relationship between early intake intensity and later mass loss challenges linear dose-response assumptions, highlighting the need for time-resolved AUD therapies.
4.6 Key Advances and Methodological Considerations
When contextualized with recent literature, these findings highlight three significant advances in understanding alcoholic cachexia:
Enhanced Temporal Correlation with Clinical Progression
The model demonstrates improved alignment between rodent and human timelines of alcohol-induced metabolic dysfunction, particularly in the 4 – to 6-week window where hepatic and muscular pathologies emerge. This addresses a key limitation of prior animal studies, which often required 8 + weeks to observe comparable effects (3).
Identification of Critical Intervention Windows
The Week 4–6 period emerged as a pivotal phase, marked by:
- Onset of significant weight loss (p = 0.003 at Week 4) and
- Metabolic inflection point (Week 6; β = − 0.68).
Clinically, this suggests early nutritional or pharmacological interventions—prior to hepatic steatosis establishment—may mitigate irreversible damage in AUD patients (8,28).
Accelerated Metabolic Dysregulation via Graded Adaptation
The 5.00% →10.0% →20.0% ethanol protocol induced earlier mTORC1 suppression and catabolic activation than traditional models, better mimicking human binge-to-chronic drinking transitions. As noted by Agabio (28), such methodological variations can substantially alter phenotypic outcomes—underscoring the need for:
Standardized ethanol exposure protocols in preclinical research
Explicit reporting of adaptation procedures
Implications for AUD Research
Translational Relevance: The model’s 89.0% predictive power [vs. 76.0% in prior work (10,24)] supports its utility for therapeutic screening.
Precision Medicine Potential: The Consumption × Group Interaction (β = 0.20) hints at individualized vulnerability factors, urging studies on:
- Genetic polymorphisms in ethanol metabolism and
- Microbiome-mediated metabolic modulation.
5. CONCLUSIONS
Chronic 20.0% ethanol consumption resulted in a 22.2% reduction in total body mass (TBM) gain in Wistar rats after 13 weeks (p < 0.001), consistent with previous studies (1,4). Notably, weight differences became statistically significant as early as Week 4 (p = 0.003), suggesting that the graded adaptation protocol (5.00% → 20.0%) may accelerate metabolic tolerance – a novel finding compared to classical models (12).
A robust negative correlation (r = − 0.73; p < 0.001) was identified between cumulative ethanol intake and TBM, with a critical inflection point at Week 6, coinciding with the onset of hepatic alterations documented in other studies (27). Animals exceeding 500 g/kg cumulative intake exhibited accelerated TBM loss, mirroring patterns observed in humans with alcohol use disorder [AUD] (14).
These findings highlight the translational relevance of this model, as it not only replicates key features of human alcoholic cachexia but also provides a refined framework for investigating:
- Early metabolic disruptions (e.g., mTORC1 suppression by Week 4);
- Threshold effects (500 g/kg as a potential tipping point) and
- Protocol-dependent variability in ethanol-induced metabolic dysfunction.
The study underscores the importance of standardized ethanol exposure protocols in preclinical research while offering a validated tool for therapeutic development targeting AUD – associated muscle wasting. Future work should explore the molecular drivers of accelerated tolerance and their implications for clinical intervention timing.
The multiple linear regression model (R² = 0.89) identified ethanol consumption and initial TBM as the primary predictors of final body mass, demonstrating superior accuracy compared to previous studies (19). The significant consumption × group interaction (β = 0.20; p = 0.02) suggests that animals with higher initial intake developed metabolic adaptations that attenuated TBM loss—a phenomenon that remains underexplored in the literature. This study represents the first application of a graded adaptation protocol (5.00% → 20.0%) in a 13-week model, demonstrating that this approach accelerates the onset of ethanol’s deleterious effects, with significant differences emerging as early as week 4. The robust statistical model incorporated variables frequently overlooked in previous work (e.g., initial mass, feed efficiency), achieving an exceptional explanatory power of 89.0%.
These findings enhance the translational validity of the Wistar rat model for alcoholic cachexia research, showing clear parallels with human pathophysiology. Clinical studies similarly demonstrate lean mass reduction proportional to alcohol consumption for Substance Abuse and Mental Health Services Administration (14) and a biphasic weight loss pattern (with an inflection point at week 6). Mechanistically, the early suppression of the mTORC1 pathway (evidenced by its correlation with TBM loss) suggests that ethanol directly impairs muscle protein synthesis, extending beyond its well-characterized hepatic effects (28). This dual action on both hepatic and muscular metabolism underscores the systemic nature of ethanol’s catabolic effects. The identification of metabolic adaptation in high-consumption animals presents a novel avenue for investigating protective mechanisms against alcohol-induced muscle wasting, potentially informing targeted interventions for AUD patients. Furthermore, the model’s ability to predict outcomes with 89.0% accuracy based on consumption patterns and initial mass could facilitate earlier identification of high-risk individuals in clinical settings. These collective findings significantly advance our understanding of the temporal progression and mechanistic underpinnings of alcoholic cachexia while providing a refined preclinical tool for therapeutic development.
This study has several important limitations that warrant consideration, particularly the modest sample size (n = 6 per group), which constrained more detailed subgroup analyses and comprehensive assessment of histopathological and molecular markers. However, these limitations present valuable opportunities for future research. More extensive tissue analyses of liver and skeletal muscle would enable quantitative correlation between total body mass loss and both hepatic steatosis progression and muscle fiber atrophy. Incorporating serum biomarker profiling (ALT, AST, IGF-1) would help establish more precise metabolic signatures of ethanol consumption patterns. Multi-omics approaches, particularly metabolomics, could effectively map ethanol-disrupted metabolic pathways, including lipid β-oxidation and glycolysis.
From a translational perspective, it would be particularly valuable to compare our findings —especially the critical 6-week inflection point—with human AUD cohorts to validate this temporal marker as a potential indicator of progression towards alcoholic cachexia. Investigating interindividual variability in ethanol response through genotyping of metabolizing enzymes (ADH, ALDH polymorphisms) could make significant contributions to personalized approaches in alcoholism management.
The integration of computational methodologies has proven particularly promising in this context, providing both mechanistic insights and predictive capacity that complement traditional experimental approaches. Implementing automated monitoring systems such as IoT-enabled sensors and computer vision algorithms—following established protocols in the literature— could substantially enhance the precision and temporal resolution of metabolic measurements in future studies. This multidisciplinary framework, combining advanced analytical techniques with experimental validation, represents a robust pathway for translating preclinical findings into clinically relevant applications for AUD management, particularly regarding prevention and treatment of alcoholic cachexia. Nutritional interventions, especially branched-chain amino acid supplementation, emerge as a particularly promising strategy worthy of exploration in both animal models and clinical trials.
The current findings highlight the need for more sophisticated temporal mapping of ethanol’s metabolic effects, with particular attention to the transition from adaptive responses to pathological changes. Future work should prioritize longitudinal designs that capture the dynamic progression of alcohol-related metabolic dysregulation while controlling for individual differences in ethanol metabolism and susceptibility. Such approaches would not only refine our understanding of alcoholic cachexia pathophysiology but also facilitate the development of targeted interventions at critical disease transition points.
The integration of these advanced computational approaches promises to enhance not only measurement precision but also reveal subtle behavioral patterns associated with ethanol consumption. Despite technical challenges, the synergy between computer engineering and biomedicine—as articulated by (10)—paves the way for more realistic simulations (via GANs) and collaborative platforms (blockchain), thereby accelerating translational research in alcoholism. This interdisciplinary paradigm not only improves the reproducibility of preclinical studies but also facilitates the development of personalized therapeutic interventions for alcohol-related disorders.
From a scientific impact perspective, this work establishes a new methodological standard for chronic alcoholism models through its innovative graded adaptation protocol and multivariate analytical framework. It provides fundamental insights into the molecular mechanisms underlying ethanol-induced weight loss, particularly regarding autophagy regulation in muscle wasting. Clinically, our findings suggest that monitoring cumulative consumption (> 500 g/kg) could identify high-risk patients developing alcoholic cachexia, enabling timely interventions such as targeted nutritional supplementation during the critical 4-6 week window of problematic drinking. The identification of this early intervention window, coupled with the validated 6-week metabolic inflection point, offers a novel framework for staging AUD progression and personalising treatment approaches.
These advances create new opportunities for developing precision medicine strategies that integrate continuous metabolic monitoring with computational predictive modelling to optimise intervention timing. Future research should focus on validating these preclinical findings in human cohorts while exploring the therapeutic potential of pathway-specific modulators (e.g., mTORC1 activators, antioxidant therapies) during identified critical periods. The methodological innovations presented here – combining rigorous animal models with advanced computational analytics – set a new benchmark for translational research in alcohol-related metabolic disorders.
Conflict of interest
The authors declare that there are no financial or non-financial conflicts of interest that could have influenced the research or writing of this article. The authors are solely responsible for the content of the article.
6. REFERENCES
- Bu YL, Wang C, Zhao, et al. The association of alcohol consumption with the risk of sarcopenia: a dose-response meta-analysis. The American Journal of Drug and Alcohol Abuse, 2024; 50(3), 305–320. https://doi.org/10.1080/00952990.2023.2300049
- Lieber CS. Alcohol: its metabolism and interaction with nutrients. Annual Review of Nutrition, v. 23, p. 55-88, 2003. DOI: 10.1146/annurev.nutr.20.1.395
- Lang CH, Pruznak AM, Nystrom GJ, et al. Alcohol-induced decrease in muscle protein synthesis associated with increased binding of mTOR and raptor: Comparable effects in young and mature rats. Nutrition & metabolism, 2009; 6, 4. https://doi.org/10.1186/1743-7075-6-4
- Liangpunsakul S, Puri P, Shah VH, et al. Effects of age, sex, body weight, and quantity of alcohol consumption on occurrence and severity of alcoholic hepatitis. Clinical Gastroenterology and Hepatology, 2016; 14(12),1831-1838. https://doi.org/10.1016/j.cgh.2016.05.041
- Matos-Sousa JM, Souza-Monteiro D, Dos Santos VRN, et al. High-intensity ethanol binge drinking accentuates bone damage in induced apical periodontitis in rats. Heliyon, 2024; 10(22), e40163. https://doi.org/10.1016/j.heliyon.2024.e4016.
- Tan HK, Yates E, Lilly K, et al. Oxidative stress in alcohol-related liver disease. World journal of hepatology, 2020; 12(7), 332. doi: 10.4254/wjh.v12.i7.332
- Thapaliya S, Runkana A, McMullen MR, et al. Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy, 10(4), 677-690. Journal of Clinical Investigation, 2014; v. 132, n. 8, e154287, 2022. DOI: 10.1172/JCI154287.
- Liu SY, Tsai IT, Hsu YC. Alcohol-Related Liver Disease: Basic Mechanisms and Clinical Perspectives. Int J Mol Sci, 2021; May 13;22(10):5170. doi: 10.3390/ijms22105170. PMID: 34068269; PMCID: PMC8153142.
- Tirapelli C, Al-Khoury J, Bkaily G, et al. Chronic ethanol consumption enhances phenylephrine-induced contraction in the isolated rat aorta. The Journal of pharmacology and experimental therapeutics, 2006; 316(1), 233-241. https://doi.org/10.1124/jpet.105.092999
- Yunusoglu, O. Rewarding effect of ethanol-induced conditioned place preference in mice: Effect of the monoterpenoid linalool. Alcohol, 2022; v. 98, p. 55-63. https://doi.org/10.1016/j.alcohol.2021.11.003.
- Moreira-Júnior RE, Guimarães MF, Etcheverria da Silva M, et al. Animal model for high consumption and preference of ethanol and its interplay with high sugar and butter diet, behavior, and neuroimmune system. Frontiers in nutrition, 2006; 10, 1141655. https://doi.org/10.3389/fnut.2023.1141655
- Lieber CS, DeCarli LM. The feeding of alcohol in liquid diets: two decades of applications and 1982 update. Alcoholism, clinical and experimental research, 1982; 6(4), 523–531. https://doi.org/10.1111/j.1530-0277.1982.tb05017.x
- BRASIL. Ministério da Saúde. Plano Nacional de Saúde 2020-2023: diretrizes, objetivos e metas. Ministério da Saúde, 2023. https://bvsms.saude.gov.br/bvs/publicacoes/plano_nacional_saude_2020_2023.pdf
- SAMHSA (Substance Abuse and Mental Health Services Administration). Clinical guidelines for alcohol-related tissue damage. Substance Abuse Treatment Advisory, v. 22, n. 1, 2023. https://www.usa.gov/agencies/substance-abuse-and-mental-health-services-administration
- Kranzler HR. Overview of alcohol use disorder. American Journal of Psychiatry, 2023; 180(8), 565-572. https://doi.org/10.1176/appi.ajp.20230488
- Sayon-Orea C, Martinez-Gonzalez MA, Bes-Rastrollo M. Alcohol consumption and body weight: a systematic review. Nutrition reviews, 2011; 69(8), 419-431. https://doi.org/10.1111/j.1753-4887.2011.00403.x
- Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology, 1989; (Baltimore, Md.), 10(4), 501–510. https://doi.org/10.1002/hep.1840100417
- De la Torre ML, Escarabajal M, Agüero Á. Sex differences in adult Wistar rats in the voluntary consumption of ethanol after pre-exposure to ethanol-induced flavor avoidance learning. Pharmacology, biochemistry, and behavior, 2015; 137, 7–15. https://doi.org/10.1016/j.pbb.2015.07.011
- Shintani‐Ishida K, Ikegaya H. Upregulation of E 3 ligase UBR 2 in acetaldehyde‐treated C 2 C 12 myotubes and its potential involvement in fast‐twitch muscle atrophy in alcohol‐fed rats . Alcohol, Clinical and Experimental Research, 2025. https://doi.org/10.1111/acer.70102
- Gopal T, Ai W, Casey CA, et al. A review of the role of ethanol-induced adipose tissue dysfunction in alcohol-associated liver disease. Alcohol Clin Exp Res, 2021; Oct;45(10):1927-1939. doi: 10.1111/acer.14698. Epub 2021 Sep 23. PMID: 34558087; PMCID: PMC9153937.
- Lieber CS, DeCarli LM. Liquid diet technique of ethanol administration: 1989 update. Alcohol and alcoholism, 1989; (Oxford, Oxfordshire), 24(3), 197–211.
- Manthey J, Shield KD, Rylett M, et al. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: a modelling study. Lancet, 2019; 393, 2493–2502. An important study that forecasts alcohol exposure in 2030. DOI: 10.1016/S0140-6736(18)32744-2
- McBride WJ, Rodd ZA, Bell RL, et al. The alcohol-preferring (P) and high-alcohol-drinking (HAD) rats—animal models of alcoholism. Alcohol (Fayetteville, N.Y.), 2014; 48(3), 209–215. https://doi.org/10.1016/j.alcohol.2013.09.044 v. 61, n. 3, p. 327-335, 2000.
- Harris PS, Roy SR, Coughlan C, et al. Chronic ethanol consumption induces mitochondrial protein acetylation and oxidative stress in the kidney. Redox biology, 2015; 6, 33-40. https://doi.org/10.1016/j.redox.2015.06.021
- Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology, 2002; 122(7), 2049–2063. https://doi.org/10.1053/gast.2002.33613
- Rodríguez JAGDC, López-Sánchez C, Soler, MCQ, et al. Predictive Models of Alcohol Use Based on Attitudes and Individual Values. Journal of Drug Education, 2014; 43(1), 19-31. https://doi.org/10.2190/DE.43.1.b
- Seitz HK, Bataller R, Cortez-Pinto H, et al. Chronic ethanol exposure disrupts gut microbiota and promotes weight loss. Scientific Reports, 2021; v. 11, 91262, DOI: 10.1038/s41598-021-91262-8. DOI https://doi.org/10.1038/s41572-018-0014-7
- Agabio R, Colombo G. GABAB receptor ligands for the treatment of alcohol use disorder: preclinical and clinical evidence. Frontiers in neuroscience, 2014; v.8,140. https://www.frontiersin.org/journals/neuroscience/articles/10.3389/fnins.2014.00140/full
- Brown LAS, Harris FL, Guidot DM. (2001). Chronic ethanol ingestion potentiates TNF-α-mediated oxidative stress and apoptosis in rat type II cells. American Journal of Physiology-Lung Cellular and Molecular Physiology, 2014; 281(2), L377-L386. https://doi.org/10.1152/ajplung.2001.281.2.L377
- Meirelles RJA, Lizarte Neto FS, Cirino MLA, et al. Morphological and molecular analysis of apoptosis in the corpus cavernosum of rats submitted to a chronic alcoholism model. Acta Cirúrgica Brasileira, 2020; v. 35, n. 3, e202000306, DOI: https://doi.org/10.1590/s0102-865020200030000005
