Fármacos de nueva aprobación – Newly approved drugs:
(B) SANGRE Y SISTEMA HEMATOPOYÉTICO: Hemofilia A y B: Marstacimab (Hympavzi®; EMA/FDA) y Concizumab (Alhemo®; EMA/FDA).(C) SISTEMA CARDIOVASCULAR: Taquicardia supraventricular: Landiolol (Rapiblyk®; FDA). Síndrome de quilomicronemia: Olezarsen (Tryngolza®; FDA). (J) ANTIINFECCIOSOS SISTÉMICOS: Infección urinaria: Sulopenem + Probenecid (Orlynvah®; FDA). Meningitis meningocócica (profilaxis): Vacuna Meningococo pentavalente (grupos A, B, C, W, Z) (Penbraya®) Pfizer (EMA/FDA). (L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES: Cáncer de pulmón: Ensartinib (Ensacove®; FDA). Adenocarcinoma gástrico: Zolbetuximab (Vyloy®; EMA/FDA). Cáncer de mama: Inavolisib (Itobeni®; FDA). Leucemia linfoblástica: Obecabtagene Autoleucel (Aucatzyl®; FDA). Leucemia aguda: Revumenib (Rebuforj®; FDA). Cáncer de vías biliares: Zanidatamab (Zihera®; FDA). Cáncer de pulmón, cáncer de páncreas: Zenocotuzumab (Bizengri®; FDA). Hiperplasia adrenal congénita: Crinecerfont (Crenessity®; FDA). Enfermedad aguda de injerto contra huésped: Remestemcel-L (Ryoncil®; FDA). Carcinoma cutáneo de células escamosas: Cosibelimab (Unloxcyt®; FDA). (N) SISTEMA NERVIOSO: Amiloidosis por transtiretina: Acoramidis (Attruby®; FDA). (R) SISTEMA RESPIRATORIO: Fibrosis quística (mucoviscidosis): Venzacaftor + Tezacaftor + Deutivacaftor (Alyftrek®; FDA).(S) ÓRGANOS SENSORIALES: (V) VARIOS: Revascularización de urgencia: Vaso de ingeniería tisular acelular (Symvess®; FDA).
(B) SANGRE Y SISTEMA HEMATOPOYÉTICO
Marstacimab (Hympavzi®) Pfizer (EMA, UE; FDA, USA)
Indicación: Profilaxis de rutina para prevenir o reducir la frecuencia de episodios de sangrado en pacientes adultos y pediátricos de 12 años de edad y mayores con hemofilia A (deficiencia congénita del factor VIII) sin inhibidores del factor VIII, o hemofilia B (deficiencia congénita del factor IX) sin inhibidores del factor IX.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal humano de inmunoglobulina G tipo 1 (IgG1). Autorizado en la Unión Europea el 18 de noviembre de 2024, como medicamento huérfano (Orphan drug); autorizado en Estados Unidos (FDA) el 10 de octubre de 2024.
Mecanismo: Anticuerpo monoclonal humano dirigido contra el dominio Kunitz 2 (K2) de TFPI (inhibidor de la vía del factor tisular) para neutralizar la la actividad de éste y mejorar la coagulación. TFPI es el principal inhibidor de la cascada de coagulación extrínseca y regula negativamente la generación de trombina dentro de la vía extrínseca de coagulación al inactivar las funciones de proteasa del complejo FXa/FVIIa/TF. El TFPI se une al sitio activo del factor Xa y lo inhibe a través de su segundo dominio inhibidor de Kunitz (K2).
Eficacia clínica: Estudio abierto, multicéntrico, de dos fases en 116 pacientes adultos y pediátricos (de 12 años o más y ≥35 kg) con hemofilia A grave sin inhibidores del Factor VIII o hemofilia B grave sin inhibidores del Factor IX. La variable principal de eficacia se basó en la tasa media anualizada de hemorragia de las hemorragias tratadas con HYMPAVZI y con las registradas durante la fase de observación: 38,00 (fase de observación) va. 3,18 (HYMPAVZI). Otros objetivos del estudio incluyeron la evaluación de la profilaxis con HYMPAVZI en las incidencias de hemorragias espontáneas (30,93 vs. 2,44), hemorragias articulares (32,86 vs. 2,83), hemorragias totales (47,76 vs. 7,39) y hemorragias en articulaciones diana (23,18 vs. 1,84).
Eventos adversos: Los más comunes son reacciones en el lugar de inyección (9%), cefalea (7%) y prurito (3%).
Concizumab (Alhemo®) Novo Nordisk (EMA, UE)
Indicación: Profilaxis rutinaria de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor VIII) con inhibidores del FVIII y de 12 años de edad o mayores; o con hemofilia B (deficiencia congénita del factor IX) con inhibidores del FIX y de 12 años de edad o mayores.
Tipo: Medicamento sintético biológico constituido por un anticuerpo monoclonal humanizado de IgG4. Autorizado en la Unión Europea el 16 de diciembre de 2024; autorizado en Estados Unidos (FDA) el 20 de diciembre de 2024.
Mecanismo: Anticuerpo contra el inhibidor de la vía del factor tisular (anti-TFPI). El TFPI es un inhibidor del factor Xa (FXa). La unión de concizumab al TFPI evita la inhibición del FXa por parte del TFPI. El aumento de la actividad del FXa prolonga la fase de inicio de la coagulación y permite que se genere suficiente trombina para una hemostasia eficaz. Concizumab actúa independientemente del FVIII y el FIX.
Eficacia clínica: Un ensayo en fase 3, multinacional, multicéntrico y abierto para investigar la eficacia y la seguridad de concizumab en la profilaxis de episodios hemorrágicos en pacientes varones, 91 adultos (58 con HAwl y 33 con HBwI) y 42 adolescentes (22 con HAwl y 20 con HBwI) con hemofilia A o B con inhibidores. La variable primaria del estudio fue comparar el efecto de la profilaxis con concizumab con el de la ausencia de profilaxis (tratamiento a demanda con agentes bypass) en la reducción del número de episodios hemorrágicos en pacientes adultos y adolescentes con hemofilia A o B con inhibidores; e el cociente de las tasas de sangrados anualizadas (ABR) fue de 0,14 (p < 0,001), lo que corresponde a una reducción de la ABR del 86% para los sujetos en profilaxis con concizumab en comparación con la ausencia de profilaxis.
Eventos adversos: Los más comunes (>10%) reacciones en la zona de inyección, especialmente eritema (5,9%), cardenales (4,4%) y hematoma (4,1%); mayoritariamente leves. Acontecimientos tromboembólicos (0,9%) e hipersensibilidad (0,3%).
(C) SISTEMA CARDIOVASCULAR
Landiolol (Rapiblyk®) AOP (FDA, USA)
Indicación: Reducción a corto plazo de la frecuencia ventricular en adultos con taquicardia supraventricular, incluida la fibrilación auricular y el aleteo auricular.
Tipo: Medicamento sintético estándar constituido por el 3-[4-[(2S)-2-hidroxi-3-[2-(morfolin-4-carbonilamino)etilamino]propoxi]fenil]propanoato de [(4S)-2,2-dimetil-1,3-dioxolan-4-il]metilo (figura 1). Autorizado en Estados Unidos (FDA) el 22 de noviembre de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Antagonista selectivo de los receptores beta-1 adrenérgicos; inhibe los efectos cronotrópicos positivos de las catecolaminas en el corazón, donde se encuentran predominantemente los receptores beta-1. El landiolol no muestra ninguna actividad estabilizadora de la membrana ni actividad simpaticomimética intrínseca en la dosis aprobada.
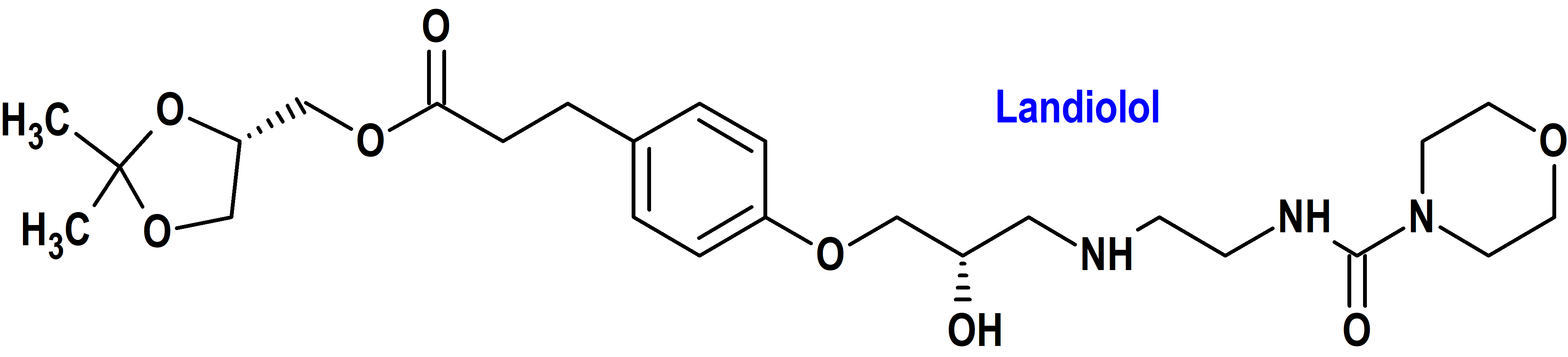
Eficacia clínica: 5 estudios aleatorizados, doble ciego y controlados con placebo, totalizando 317 adultos con taquicardia supraventricular , en los que landiolol disminuyó la frecuencia cardíaca en el 40-90% de los pacientes tratados en aproximadamente 10 minutos, en comparación con el 0-11% de los pacientes que recibieron placebo; la disminución de la frecuencia cardíaca se definió como una disminución >20% de la frecuencia cardíaca o una frecuencia cardíaca <100 latidos por minuto o al menos el cese intermitente de la arritmia.
Eventos adversos: El más común es hipotensión (10%).
Olezarsen (Tryngolza®) Ionis (FDA, USA)
Indicación: Tratamiento complementario a la dieta para reducir los triglicéridos en adultos con síndrome de quilomicronemia familiar (FCS).
Tipo: Medicamento sintético estándar constituido por un oligonucleótido antisentido conjugado con N-acetil-galactosamina (ASO-GalNAc3). Autorizado en Estados Unidos (FDA) el 19 de diciembre de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Oligonucleótido antisentido dirigido al ARNm de la apolipoproteína C-III (apoC-III), conjugado con un ligando que contiene tres residuos de N-acetilgalactosamina (GalNAc3) para permitir la distribución a los hepatocitos. El olezarsen se une al ARNm de apoC-III, lo que conduce a la degradación del ARNm y a una reducción de la proteína apoC-III sérica, lo que se traduce en un aumento de la depuración de triglicéridos (TG) y lipoporteínas de muy vaja densidad ( VLDL) en el plasma. El síndrome de quilomicronemia familiar (FCS) es un trastorno genético poco común que impide que el cuerpo descomponga las triglicéridos (TG) en el torrente sanguíneo, cuyos niveles normales son inferiores a 150 mg/dl y niveles superiores a 500 mg/dl se consideran extremadamente altos (hipertrigliceridemia grave). Las personas con FCS pueden tener niveles de TG de miles, lo cual puede derivar en dolor abdominal intenso, pancreatitis aguda y xantomas.
Eficacia clínica: Un ensayo clínico aleatorizado, controlado con placebo y doble ciego en 66 pacientes adultos con FCS y niveles de TG en ayunas de al menos 880 mg/dl (promedio de 2600 mg/dl). La variable principal de eficacia fue el cambio porcentual en los niveles de TG en ayunas desde el inicio hasta el mes 6: -42,5% en comparación con el grupo placebo. Los cambios porcentuales y absolutos medianos en los niveles de TG desde el inicio a lo largo del tiempo demostraron un efecto reductor constante durante el período de tratamiento de 12 meses.
Eventos adversos: Los más comunes (>5% de los pacientes tratados con olezarsen y con una frecuencia >3% mayor que con placebo) son reacciones en el punto de inyección (19 vs. 9%), trombocitopenia (12/4%) y artralgia (9/0%). Las reacciones adversas llevaron a la interrupción del tratamiento en el 7% de los pacientes tratados con olezarsen y en el 0% con placebo. Las razones más frecuentes para la interrupción del tratamiento fueron las reacciones de hipersensibilidad.
(J) ANTIINFECCIOSOS SISTÉMICOS
Sulopenem etzadroxilo + Probenecid (Orlynvah®) Iterum (FDA, USA)
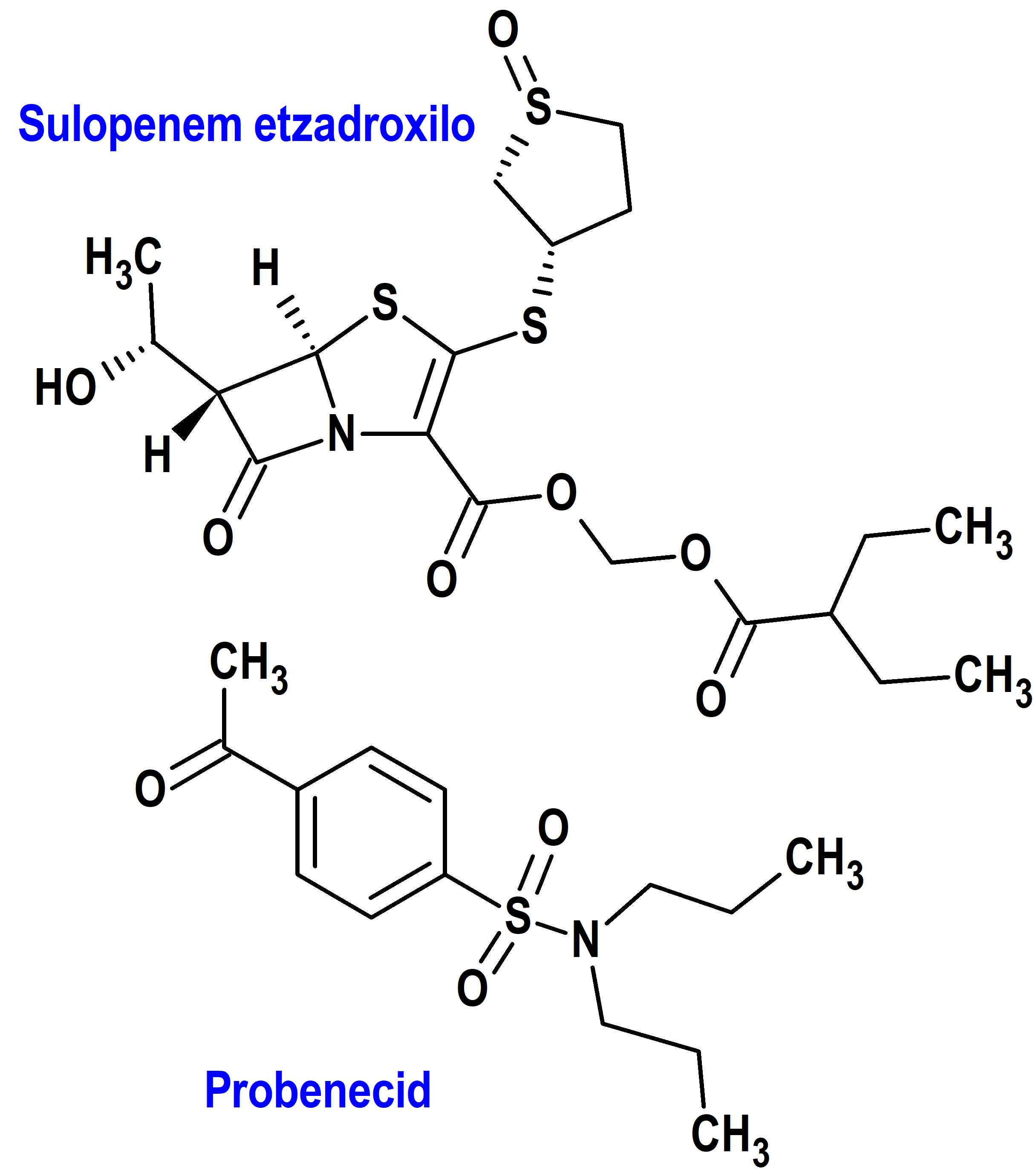
Indicación: Tratamiento de infecciones del tracto urinario no complicadas causadas por Escherichia coli, Klebsiella neumoniae o Proteus mirabilis en mujeres adultas que tienen opciones limitadas o nulas de tratamiento antibacteriano oral alternativo.
Tipo: Medicamento sintético estándar constituido por la combinación del éster (2-etil-1-oxobutoxi)metílico del ácido (5R,6S)-6-[(1R)-1-hidroxietil]-7-oxo-3-[(1R,3S)-1-oxotiolan-3-il]sulfanil-4-tia-1-azabiciclo[3.2.0]hept-2-eno-2-carboxílico (sulopenem etzadroxilo) y el ácido 4-(dipropilsulfamoil)benzoico (probenecid) (figura 2). Autorizado en Estados Unidos (FDA) el 25 de octubre de 2024 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como producto calificado para enfermedades infecciosas (Qualified Infectious Disease Product); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Combinación de un antibacteriano betalactámico carbapenémico (sulopenem etzadroxilo) y probenecid, un inhibidor tubular renal. El sulopenem etzadroxilo es un profármaco que se hidroliza al fármaco activo sulopenem después de la administración oral. El sulopenem tiene actividad contra bacterias aerobias y anaerobias grampositivas y gramnegativas resultante de la inhibición de la síntesis de la pared celular y está mediada por la unión del sulopenem a las proteínas de unión a la penicilina (PBP). En Escherichia coli, el sulopenem tiene afinidad de unión por las PBP en el siguiente orden: PBP2 > PBP1A > PBP1B > PBP4 > PBP3 > PBP5/6. El probenecid inhibe la eliminación renal de sulopenem mediada por OAT3, lo que produce un aumento de las concentraciones plasmáticas de sulopenem.
Eficacia clínica: Dos ensayos clínicos de fase 3, controlados, aleatorizados y doble ciego en los que participaron mujeres adultas con infecciones no complicadas del tracto urinario (UTI), administrándose en forma de un comprimido dos veces al día durante 5 días. En el ensayo 1 se aleatorizaron y trataron 2214 mujeres adultas, demostrando eficacia en pacientes con patógenos sensibles a amoxicilina/clavulanato con una tasa de respuesta compuesta (respuesta microbiológica y respuesta clínica combinadas) del 62% en comparación con una tasa de respuesta compuesta del 55% en el grupo de amoxicilina/clavulanato. En el ensayo 2 se aleatorizaron y trataron 1660 mujeres adultas con UTI, demostrando eficacia en pacientes con patógenos resistentes a la ciprofloxacina con una tasa de respuesta compuesta del 48% en comparación con una tasa de respuesta compuesta del 33% en el grupo de ciprofloxacina. Los ensayos clínicos que evaluaron el medicamento para el tratamiento de pacientes con UTI complicadas o infecciones intraabdominales complicadas no demostraron efectividad.
Eventos adversos: Los más comunes (>2%) son diarrea (10%), náuseas (4%), infección micótica vulvovaginal (2%), dolor de cabeza (2%) y vómitos (2%).
Vacuna Meningococo pentavalente (grupos A, B, C, W, Z) (Penbraya®) Pfizer (EMA, UE; FDA, USA)
Indicación: Inmunización activa de personas a partir de los 10 años de edad para prevenir la enfermedad invasiva causada por Neisseria meningitidis de los grupos A, B, C, W e Y, de acuerdo con las recomendaciones oficiales.
Tipo: Medicamento biológico constituido por polisacárido de Neisseria meningitidis de los grupos A1, C, W1 y Y1, junto con proteína de unión al factor H (fHbp) de las subfamilias A y B de Neisseria meningitidis del grupo B. Autorizado en la Unión Europea el 15 de noviembre de 2024; autorizado en Estados Unidos el 20 de octubre de 2024.
Mecanismo: Induce la producción de anticuerpos bactericidas específicos para los polisacáridos capsulares de Neisseria meningitidis de los grupos A, C, W e Y y para las variantes de fHbp de las subfamilias A y B de Neisseria meningitidis del grupo B. Los anticuerpos antimeningocócicos protegen frente a la enfermedad meningocócica invasiva a través de la actividad bactericida mediada por el complemento.
Eficacia clínica: La eficacia se infiere midiendo la inmunogenicidad con el ensayo de actividad bactericida en suero con complemento humano exógeno (hSBA) específico para cada grupo. Un título de hSBA ≥1:4 es el indicador de protección aceptado frente a la enfermedad meningocócica. La inmunogenicidad está documentada por datos procedentes de 3 estudios clínicos: en 2 estudios se evaluó la inmunogenicidad y la seguridad de dos dosis de Penbraya administradas a los 0 y 6 meses, o de MenB a los 0 y 6 meses y MenACWY-CRM a los 0 meses en participantes de 10 a 25 años de edad en EE. UU. y Europa. Ningún participante había recibido la vacuna MenB previamente. Tanto los participantes sin experiencia con ACWY como los participantes con experiencia con ACWY (que habían recibido 1 dosis de la vacuna conjugada MenACWY o monovalente MenC al menos 4 años antes de la inclusión) participaron en el estudio. En el tercer estudio se evaluó una pauta posológica ampliada de 2 dosis de Penbraya con un intervalo de 12 meses entre dosis en participantes de 10 a 14 años de edad que nunca habían recibido una vacuna antimeningocócica. Las tasas de serorrespuesta de Penbraya tras 2 dosis fueron no inferiores (margen de no inferioridad del 10%) a la vacuna de los grupos meningocócicos A, C, W-135 e Y conjugada con proteína CRM197 de Corynebacterium diphtheriae (MenACWY-CRM) tras 1 dosis tanto en la población sin experiencia con ACWY como en la población con experiencia con ACWY. Asimismo, las tasas de respuesta compuesta y serorrespuesta de Penbraya fueron no inferiores (margen de no inferioridad del 10%) a MenB tras 2 dosis. En otro estudio (estudio descriptivo de fase 2), tras 2 dosis de Penbraya administradas a los 0 y 12 meses, del 98,2 % al 99,1 % y del 92,9 % al 100 % de los participantes (n = 155 aleatorizados) lograron una serorrespuesta para los grupos A, C, W e Y y el grupo B, respectivamente. El 96,4 % de los participantes lograron una respuesta compuesta para el grupo B tras 2 dosis de Penbraya.
Eventos adversos: Los más comunes (≥15 %) tras la dosis 1 y la dosis 2, respectivamente, fueron dolor en el lugar de la inyección (89% y 84%), fatiga (52% y 48%), dolor de cabeza (47% y 40%), dolor muscular (26% y 23%), enrojecimiento en el lugar de la inyección (26% y 23%), hinchazón en el lugar de la inyección (25% y 24%), dolor en las articulaciones (20% y 18%) y escalofríos (20% y 16%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
Ensartinib (Ensacove®) Xcovery (FDA, USA)
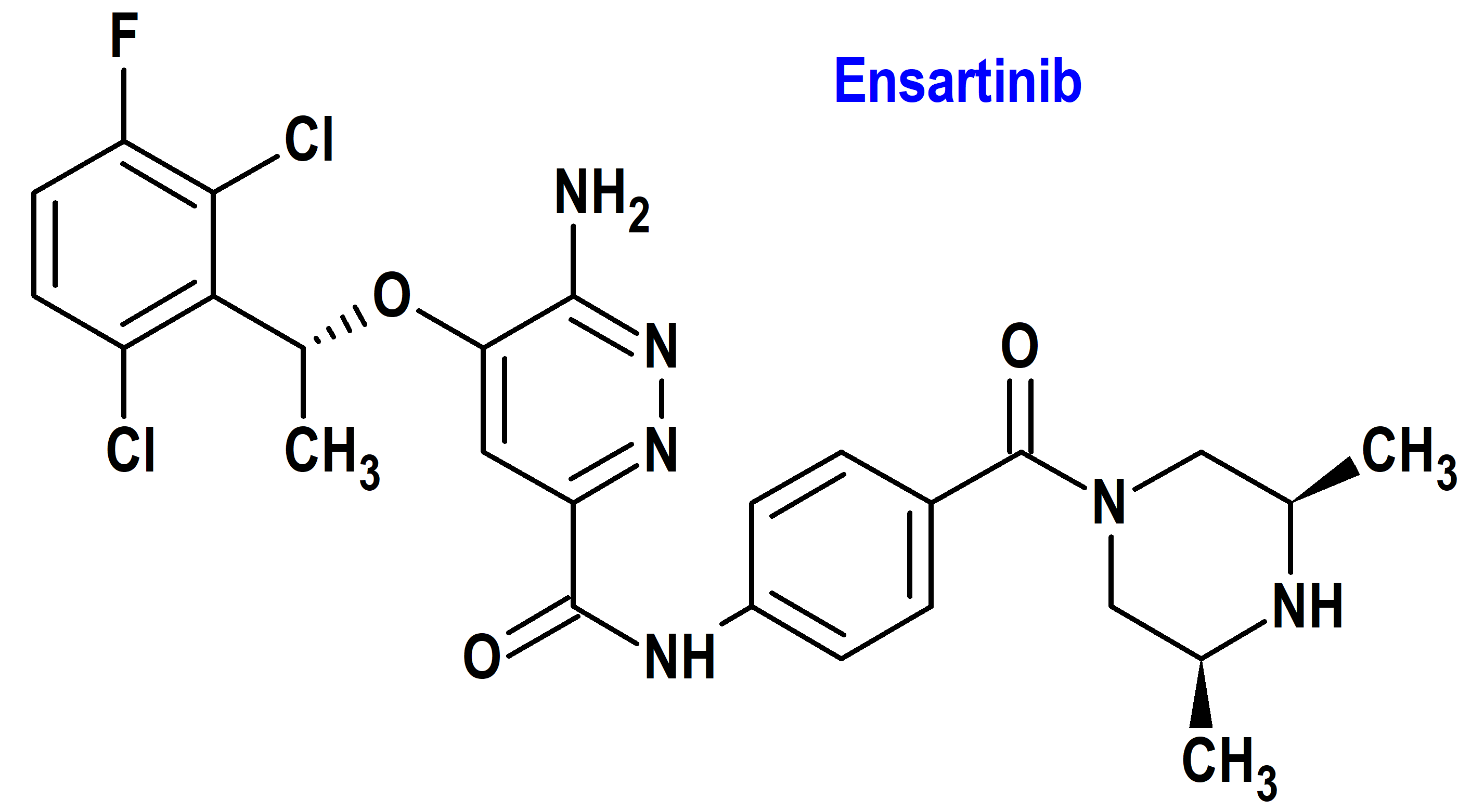
Indicación: Tratamiento de pacientes adultos con cáncer de pulmón de células no pequeñas (CPCNP) localmente avanzado o metastásico positivo para la quinasa del linfoma anaplásico (ALK) que no han recibido previamente un inhibidor de ALK.
Tipo: Medicamento sintético estándar constituido por la 6-amino-5-[(1R)-1-(2,6-dicloro-3-fluorofenil)etoxi]-N-[4-[(3R,5S)-3,5-dimetilpiperazina-1-carbonil]fenil]piridazina-3-carboxamida (figura 3). Autorizado en Estados Unidos (FDA) el 18 de diciembre de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la cinasa del linfoma anaplásico (ALK) y de otras cinasas, incluidas MET y ROS1. Ensartinib inhibe la fosforilación de ALK y sus proteínas de señalización descendentes AKT, ERK y S6, bloqueando así las vías de señalización mediadas por ALK e inhibiendo la proliferación en líneas celulares que albergan fusiones y mutaciones de ALK.
Eficacia clínica: Un ensayo abierto, aleatorizado, controlado con fármaco activo (crizotinib) y multicéntrico en 290 pacientes. La variable principal de eficacia fue la supervivencia libre de progresión (SLP) y la variable secundaria de eficacia clave fue la supervivencia general (SG). La mediana de la SLP fue de 25,8 meses con ensartinib vs. 12,7 meses con crizotinib. No hubo diferencia estadísticamente significativa en la SG.
Eventos adversos: Los más comunes (≥20%) son erupción cutánea, dolor musculoesquelético, estreñimiento, tos, prurito, náuseas, edema, pirexia y fatiga. Se produjeron reacciones adversas graves en el 23% de los pacientes, especialmente neumonía (4,9%), hemorragia (2,1%), exantema (2,1%) y celulitis (1,4%). La interrupción permanente debida a una reacción adversa se produjo en el 12% de los pacientes, principalmente por aumento de ALT (2,1%), aumento de AST (2,1%), neumonitis/EPI (2,1%), aumento de bilirrubina en sangre (1,4%) y aumento de bilirrubina conjugada (1,4%).
Zolbetuximab (Vyloy®) Astellas (EMA, UE)
Indicación: Tratamiento de primera línea, en combinación con quimioterapia basada en platino y fluoropirimidina, de pacientes adultos con adenocarcinoma gástrico o de la unión gastroesofágica (UGE) HER2 negativo localmente avanzado irresecable o metastásico cuyos tumores son positivos para Claudina (CLDN) 18.2.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal quimérico (ratón/humano IgG1) dirigido contra la molécula de unión estrecha CLDN18.2. Autorizado en la Unión Europea (EMA) el 19 de septiembre de 2024 como medicamento huérfano (Orphan drug); autorizado en Estados Unidos (FDA) el 18 de octubre de 2024.
Mecanismo: Se une de forma selectiva a líneas de células transfectadas con CLDN18.2 o las que expresan de forma endógena CLDN18.2, reduciendo las células CLDN18.2 positivas mediante citotoxicidad celular dependiente de anticuerpo (ADCC) y citotoxicidad dependiente del complemento (CDC). Los medicamentos citotóxicos aumentan la expresión de CLDN18.2 en células cancerígenas humanas y mejoraban las actividades ADCC y CDC inducidas por zolbetuximab.
Eficacia clínica: Dos estudios de fase 3, doble ciegos, aleatorizados y multicéntricos que incluyeron a 1.072 pacientes con adenocarcinoma gástrico o de la UGE localmente avanzado irresecable o metastásico, cuyos tumores eran positivos para CLDN18.2, negativos para HER2. La variable primaria de eficacia fue la supervivencia libre de progresión (SLP): medianas de 11,0 vs. 8,9 y de 8,2 vs. 6,8 meses, en los dos estudios; las variables secundarias fueron la supervivencia global (SG: medianas de 18,2/15,6 y 14,3/12,2 meses), la tasa de respuesta objetiva (TRO: 48,1/47,5 y 42,5/45,4%) y la duración de la respuesta (DDR: medianas de 9,0/8,1 y 6,3/6,1 meses).
Eventos adversos: Los más comunes son náuseas (77%), vómitos (67%), apetito disminuido (42%), neutropenia (31%), recuento de neutrófilos disminuido (28%), peso disminuido (22%), pirexia (17%), hipoalbuminemia (17%), edema periférico (14%), hipertensión (9%), dispepsia (8%) y escalofríos (5%); se produjeron reacciones adversas graves en el 45 % de los pacientes, especialmente vómitos y el 20% suspendió permanentemente zolbetuximab por reacciones adversas, principalmente por vómitos y náuseas. Las reacciones adversas llevaron a la interrupción de alguna dosis de zolbetuximab en el 61% de los pacientes.
Inavolisib (Itovebi®) Genentech (FDA, USA)
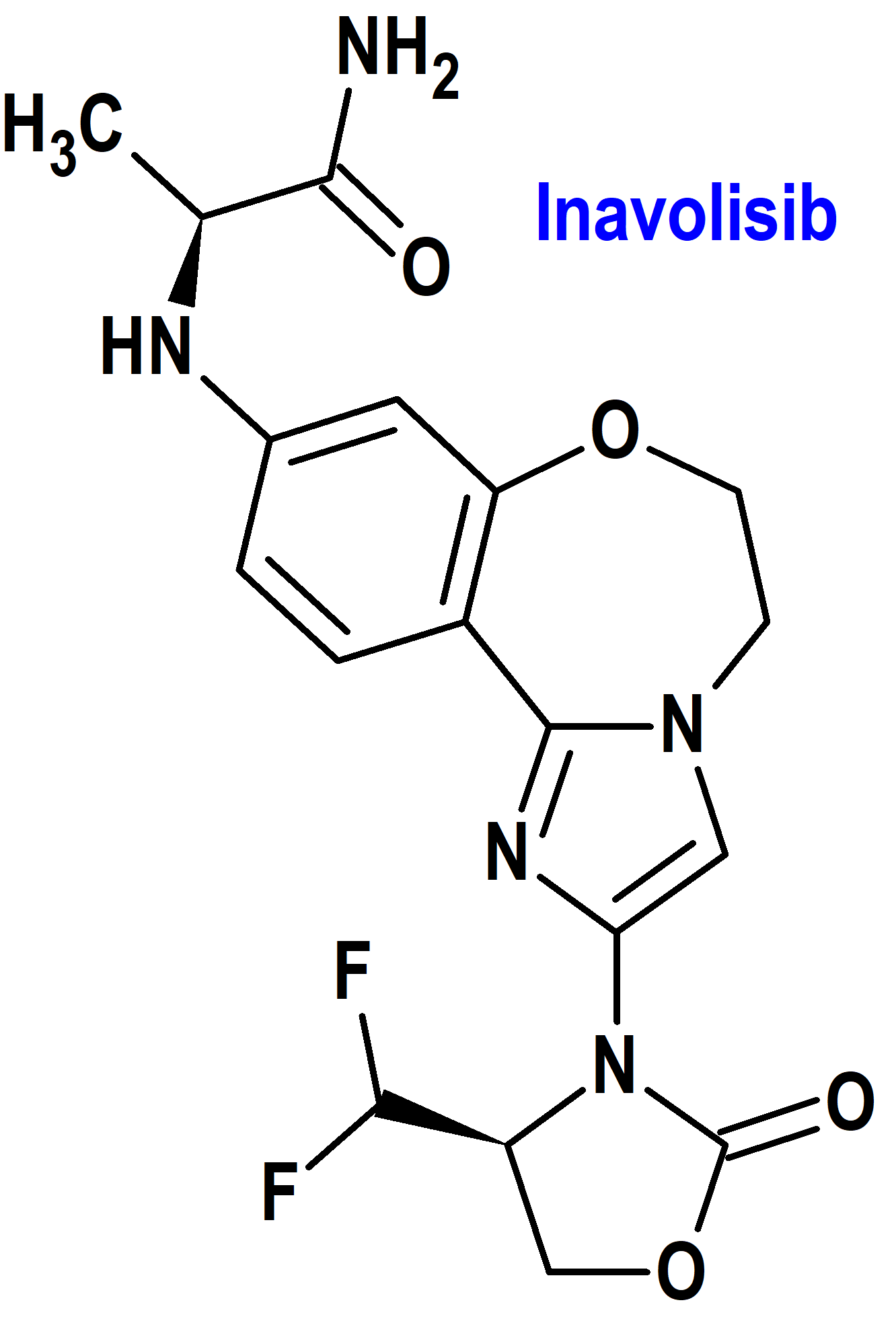
Indicación: Tratamiento, en combinación con palbociclib y fulvestrant, de adultos con cáncer de mama localmente avanzado o metastásico, resistente al sistema endocrino, con mutación de PIK3CA, positivo para el receptor hormonal (HR) y negativo para el receptor 2 del factor de crecimiento epidérmico humano (HER2).
Tipo: Medicamento sintético estándar constituido por la (2S)-2-[[2-[(4S)-4-(difluorometil)-2-oxo-oxazolidin-3-il]-5,6-dihidroimidazo[1,2-d][1,4]benzoxazepin-9-il]amino]propanamida (figura 4). Autorizado en Estados Unidos (FDA) el 11 de octubre de 2024 mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la fosfatidilinositol 3-cinasa (PI3K) con actividad inhibidora predominantemente contra PI3Kα. Induce la degradación de la subunidad alfa catalítica de PI3K mutada p110α (codificada por el gen PIK3CA), inhibiendo la fosforilación del objetivo descendente AKT, reduciendo la proliferación celular e induciendo la apoptosis en líneas celulares de cáncer de mama con mutación de PIK3CA. La combinación de inavolisib con palbociclib y fulvestrant aumenta la inhibición del crecimiento tumoral en comparación con cada tratamiento solo o con las combinaciones dobles.
Eficacia clínica: Un ensayo aleatorizado, doble ciego, controlado con placebo y multicéntrico en 325 pacientes con cáncer de mama metastásico o localmente avanzado, con resistencia endocrina, mutación de PIK3CA, HR-positivo, HER2-negativo, cuya enfermedad progresó durante o dentro de los 12 meses posteriores a la finalización de la terapia endocrina adyuvante y que no habían recibido terapia sistémica previa para la enfermedad metastásica o localmente avanzada. La variable principal de eficacia fue la supervivencia libre de progresión (SLP): mediana de 15,0 meses con inavolisib + palbociclib + fulvestrant y de 7,3 meses en el grupo de placebo + palbociclib + fulvestrant. Entre las variables secundarias de eficacia, la mediana de la duración de la respuesta (DOR) fue de 18,4 y 9,6 meses, respectivamente. El análisis provisional de la supervivencia global basado en la fracción de información del 63 % no alcanzó la significación estadística, pero respaldó la evaluación general de riesgo-beneficio con una reducción del riesgo del 36% (HR=0,64).
Eventos adversos: Los más comunes (≥20%) son disminución de neutrófilos, disminución de hemoglobina, aumento de glucosa en ayunas, disminución de plaquetas, disminución de linfocitos, estomatitis, diarrea, disminución de calcio, fatiga, disminución de potasio, aumento de creatinina, aumento de ALT, náuseas, disminución de sodio, disminución de magnesio, erupción cutánea, disminución del apetito, infección por COVID-19 y dolor de cabeza. Se produjeron reacciones adversas graves en el 24% de los casos y reacciones adversas mortales en el 3,7%. Se produjo una interrupción permanente del tratamiento debido a una reacción adversa en el 6% de los casos, interrupciones de la dosis en el 69% de los casos y reducciones de la dosis en el 14%.
Obecabtagene Autoleucel (Aucatzyl®) Autolus (FDA, USA)
Indicación: Tratamiento de adultos con leucemia linfoblástica aguda de precursores de células B en recaída o refractaria.
Tipo: Medicamento de terapia avanzada (génica), constituido por células T autólogas modificadas genéticamente dirigidas a CD19. Autorizado en Estados Unidos (FDA) el 8 de noviembre de 2024 y designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inmunoterapia de células T autólogas modificadas genéticamente dirigidas a CD19 que comprende las células T del propio paciente que se transducen con un vector lentiviral para expresar un receptor de antígeno quimérico anti-CD19 (CAR). El CAR está compuesto por un fragmento variable de cadena única (scFv) anti-CD19 murino unido a los dominios coestimuladores 4-1BB y CD3-zeta.
Eficacia clínica: Un ensayo clínico abierto, multicéntrico y de un solo brazo en el que participaron 112 adultos con leucemia linfoblástica aguda de células B CD19-positivas en recaída o refractaria, que debían haber sufrido una recaída tras una remisión de 12 meses o menos de duración, haber sufrido una recaída o haber sufrido una leucemia linfoblástica aguda refractaria tras dos o más líneas previas de terapia sistémica, o haber sufrido una recaída o una recaída refractaria tras 3 o más meses de un trasplante alogénico de células madre. Las variables principales de eficacia fueron la tasa y la duración de la remisión completa (RC) lograda dentro de los 3 meses posteriores a la infusión. Las medidas de resultado adicionales fueron la tasa y la duración de la remisión completa general, que incluye la remisión completa y la remisión completa con recuperación hematológica incompleta (RCi), en cualquier momento. De los 65 pacientes evaluables para la eficacia, 27 pacientes (42%) lograron la RC dentro de los 3 meses y la duración media de la RC lograda dentro de los 3 meses fue de 14,1 meses.
Eventos adversos: Los más comunes (≥20%) incluyeron el síndrome de liberación de citocinas (CRS), infecciones, dolor musculoesquelético, infecciones virales, fiebre, náuseas, trastornos infecciosos bacterianos, diarrea, neutropenia febril, ICANS, hipotensión, dolor, fatiga, dolor de cabeza, encefalopatía y hemorragia. El CRS se produjo en el 75% (3% de Grado 3) y las toxicidades neurológicas se produjeron en el 64% (12% de Grado ≥3), incluido el síndrome de neurotoxicidad asociada a células efectoras inmunitarias (ICANS) en el 24% (7% de Grado ≥3).
Revumenib (Rebuforj®) Sundax (FDA, USA)
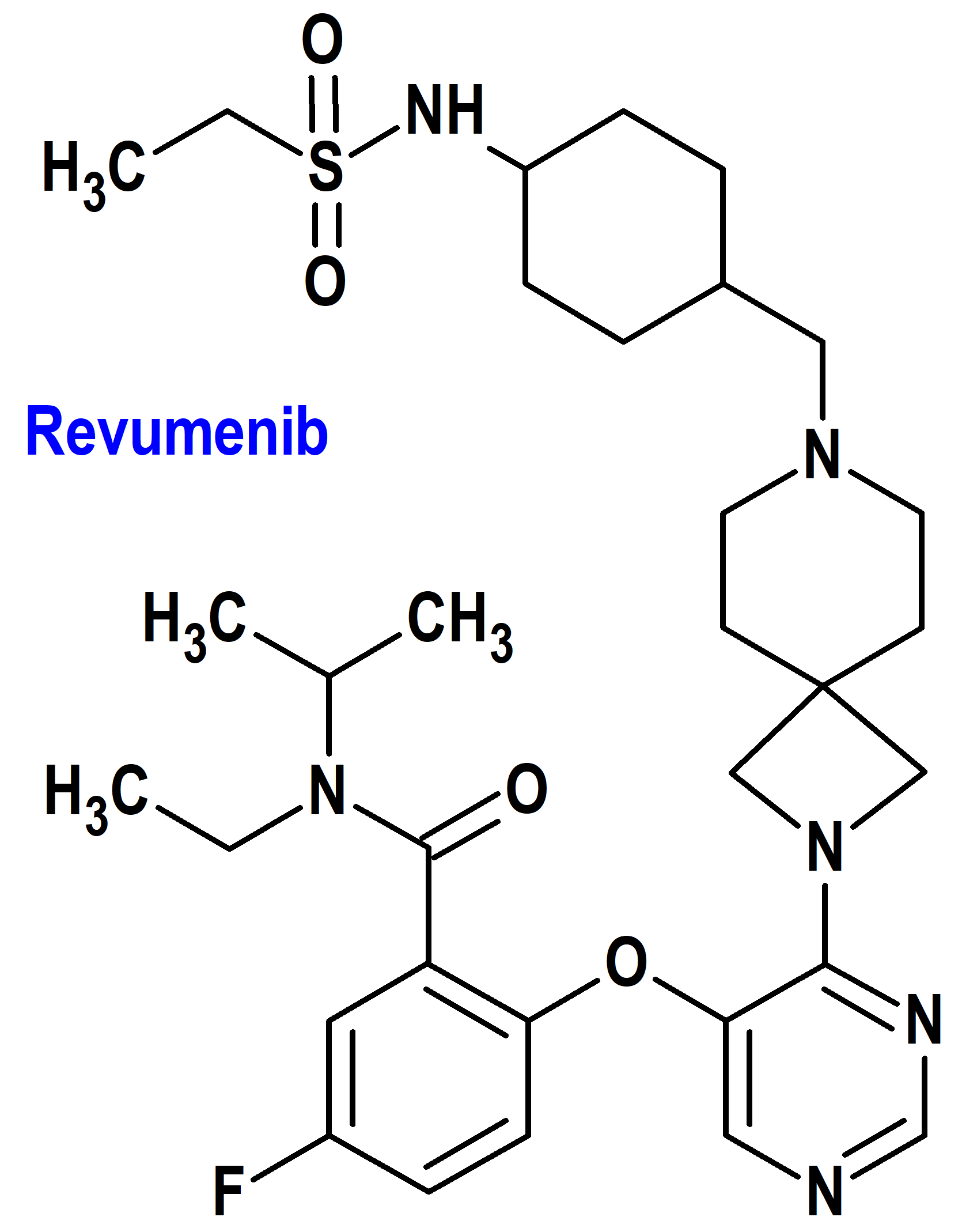
Indicación: Tratamiento de leucemia aguda con presencia de una translocación KMT2A en células de la médula ósea.
Tipo: Medicamento sintético estándar constituido por la N-etil-2-[4-[7-[[4-(etilsulfonilamino)ciclohexil]metil]-2,7-diazaspiro[3.5]nonan-2-il]pirimidin-5-il]oxi-5-fluoro-N-propan-2-ilbenzamida (figura 5). Autorizado en Estados Unidos (FDA) el 15 de noviembre de 2024 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la menina, bloquea la interacción de la lisina metiltransferasa 2A (KMT2A) de tipo salvaje y de las proteínas de fusión KMT2A con la menina. La unión de las proteínas de fusión KMT2A con la menina está implicada en las leucemias agudas con reordenamiento de KMT2A (KMT2Ar) a través de la activación de una vía transcripcional leucemogénica. La inhibición de la interacción menina-KMT2A con revumenib altera la transcripción de múltiples genes, incluidos los marcadores de diferenciación.
Eficacia clínica: Un ensayo multicéntrico abierto con una cohorte de un solo brazo en 104 pacientes adultos y pediátricos (de al menos 30 días de vida) con leucemia aguda en recaída o refractaria (R/R) con una translocación KMT2A; se excluyeron los pacientes con una duplicación parcial en tándem 11q23. Las variables principales de eficacia fueron la remisión completa (RC) más RC con recuperación hematológica parcial (RCh), la duración de RC+RCh y la conversión de dependencia de transfusiones a independencia. La tasa de RC+RCh fue del 21,2% y la duración media de RC+RCh fue de 6,4 meses. De los 22 pacientes que alcanzaron RC o RCh, la mediana de tiempo hasta la RC o la RCh fue de 1,9 meses. Entre los 83 pacientes dependientes de transfusiones de glóbulos rojos (RBC) y/o plaquetas al inicio, 12 (14%) se volvieron independientes de las transfusiones de glóbulos rojos y plaquetas durante cualquier período de 56 días posterior al inicio. De los 21 pacientes que no recibían transfusiones de glóbulos rojos ni de plaquetas al inicio del estudio, 10 (48%) permanecieron independientes de las transfusiones durante cualquier período de 56 días posterior al inicio.
Eventos adversos: Los más comunes (≥20%) son hemorragia, náuseas, aumento de fosfato, dolor musculoesquelético, infección, aumento de aspartato aminotransferasa, neutropenia febril, aumento de alanina aminotransferasa, aumento de la hormona paratiroidea intacta, infección bacteriana, diarrea, síndrome de diferenciación, prolongación del intervalo QT del electrocardiograma, disminución de fosfato, aumento de triglicéridos, disminución de potasio, disminución del apetito, estreñimiento, edema, infección viral, fatiga y aumento de la fosfatasa alcalina. Se produjeron reacciones adversas mortales en 4 (3%) pacientes. Se notificaron reacciones adversas graves en 99 (73%) pacientes, especialmente infección (24%), neutropenia febril (19%), infección bacteriana (17%), síndrome de diferenciación (12%), hemorragia (9%) y trombosis (5%). Las reacciones adversas que llevaron a la interrupción de la dosis se produjeron en el 42% de los pacientes, principalmente prolongación del intervalo QT del electrocardiograma, neutropenia febril, síndrome de diferenciación, infección, hipocalemia y náuseas. Las reacciones adversas que llevaron a la reducción de la dosis se produjeron en el 10% de los pacientes, más frecuentemente por prolongación del intervalo QT del electrocardiograma. Las reacciones adversas que llevaron a la interrupción permanente se produjeron en el 12% de los pacientes, debido mayoritariamente a infección e insuficiencia respiratoria.
Zanidatamab (Zihera®) Jazz (FDA, USA)
Indicación: Tratamiento de adultos con cáncer de vías biliares (CVB) previamente tratado, irresecable o metastásico HER2 positivo (IHC 3+).
Tipo: Medicamento biológico constituido por un anticuerpo humanizado, similar a IgG, biespecífico, dirigido a HER2. Autorizado en Estados Unidos (FDA) el 20 de noviembre de 2024 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo biespecífico dirigido a HER2 que se une a dos sitios extracelulares de HER2, dando como resultado la internalización que conduce a una reducción del receptor en la superficie de la célula tumoral. Induce citotoxicidad dependiente del complemento (CDC), citotoxicidad celular dependiente de anticuerpos (ADCC) y fagocitosis celular dependiente de anticuerpos (ADCP), dando como resultado la inhibición del crecimiento tumoral y la muerte celular.
Eficacia clínica: Un ensayo abierto, multicéntrico, de un solo brazo en 62 pacientes con cáncer de vías biliares HER2-positivo (IHC 3+) metastásico o irresecable que habían recibido al menos un régimen de quimioterapia sistémica con gemcitabina previo y con función cardíaca adecuada (definida como FEVI de al menos el 50%). Las principales medidas de resultados de eficacia fueron la tasa de respuesta objetiva, un 52% (3,2% completa y 48% parcial) y la duración de la respuesta, con una mediana de 14,9 meses (más de 6 meses en el 59% de los pacientes y más de 12 en el 44%).
Eventos adversos: Los más comunes (± 20%) son diarrea, reacción relacionada con la infusión, dolor abdominal y fatiga. Se produjeron reacciones adversas graves en el 53% de los pacientes, especialmente obstrucción biliar (15%), infección del tracto biliar (8%), sepsis (8%), neumonía (5%), diarrea (3,8%), obstrucción gástrica (3,8%) y fatiga (2,5%). La interrupción permanente debido a una reacción adversa ocurrió en el 2,5% de los pacientes, incluyendo fracción de eyección reducida y neumonitis. Las interrupciones de la dosis debido a una reacción adversa se produjeron en el 41%, debido a diarrea, aumento de la alanina aminotransferasa, aumento de la aspartato aminotransferasa, disminución de la fracción de eyección, neumonía, colangitis, fatiga, obstrucción biliar, dolor abdominal, aumento de la creatinina en sangre y disminución del potasio. Se produjeron reducciones de la dosis debido a una reacción adversa en el 4% de los pacientes, especialmente por diarrea, náuseas y pérdida de peso.
Zenocotuzumab (Bizengri®) Merus (FDA, USA)
Indicación: Tratamiento de adultos con cáncer de pulmón de células no pequeñas (CPCNP) avanzado irresecable o metastásico que alberga una fusión del gen neuregulina 1 (NRG1) con progresión de la enfermedad durante o después de una terapia sistémica previa; tratamiento de adultos con adenocarcinoma pancreático avanzado irresecable o metastásico que alberga una fusión del gen neuregulina 1 (NRG1) con progresión de la enfermedad durante o después de una terapia sistémica previa.
Tipo: Medicamento biológico, de terapia avanzada (génica, celular somática), constituido por un anticuerpo humanizado biespecífico dirigido a HER2 y HER3, de tipo inmunoglobulina G1 (IgG1) de longitud completa y bajo en fucosa. Autorizado en Estados Unidos (FDA) el 4 de diciembre de 2024 como medicamento huérfano (Orphan drug), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo biespecífico que se une a los dominios extracelulares de HER2 y HER3 expresados en la superficie de las células, incluidas las células tumorales, inhibiendo la dimerización de HER2:HER3 y previniendo la unión del gen neuregulina 1 (NRG1) a HER3. Zenocutuzumab disminuye la proliferación celular y la señalización a través de la vía de la fosfoinosítido 3-cinasa (PI3K)-AKT-diana de la rapamicina en mamíferos (mTOR). Además, zenocutuzumab media la citotoxicidad celular dependiente de anticuerpos (ADCC).
Eficacia clínica: Un estudio clínico multicéntrico, abierto y de múltiples cohortes que incluyeron a a 64 pacientes adultos con cáncer de pulmón de células no pequeñas (CPCNP) avanzado o metastásico con fusión NRG1 positiva que tuvieron progresión de la enfermedad después del tratamiento estándar para su enfermedad. Las variables principales de eficacia la tasa de respuesta global confirmada (33%; 1,6% completa y 31% parcial) y la duración de la respuesta (mediana de 7,4 meses; en un 43% fue de al menos 6 meses) determinada por una revisión central independiente ciega (BICR). En otro estudio clínico multicéntrico, abierto y de múltiples cohortes se incluyeron a 30 pacientes adultos con adenocarcinoma de páncreas avanzado o metastásico con fusión NRG1 positiva que tuvieron progresión de la enfermedad después del tratamiento estándar. Las variables principales de eficacia la tasa de respuesta global confirmada (40%; 3,3% completa y 37% parcial) y la duración de la respuesta (rango de 3,7 a 16,6 meses; en un 67% fue de al menos 6 meses) determinada por una revisión central independiente ciega (BICR).
Eventos adversos: Los más comunes (≥10%) fueron diarrea, dolor musculoesquelético, fatiga, náuseas, reacciones relacionadas con la infusión, disnea, erupción cutánea, estreñimiento, vómitos, dolor abdominal y edema; las anomalías de laboratorio de grado 3 o 4 más frecuentes (≥10 %) fueron aumento de la gamma-glutamil transferasa, disminución de la hemoglobina, disminución del sodio y disminución de las plaquetas. Se produjeron reacciones adversas graves en el 23 % de los pacientes, especialmente anemia, trombocitopenia, taquicardia, dolor abdominal, hemorragia hemorroidal, náuseas, ictericia colestásica, COVID-19, absceso hepático, fractura traumática, aumento de la creatinina en sangre, dolor de espalda, síndrome mielodisplásico y trastorno respiratorio. Se produjeron interrupciones de la dosis debido a una reacción adversa, excluidas las debidas a reacciones relacionadas con la infusión, en el 33 % de los pacientes.
Crinecerfont (Crenessity®) Neurocrine (FDA, USA)
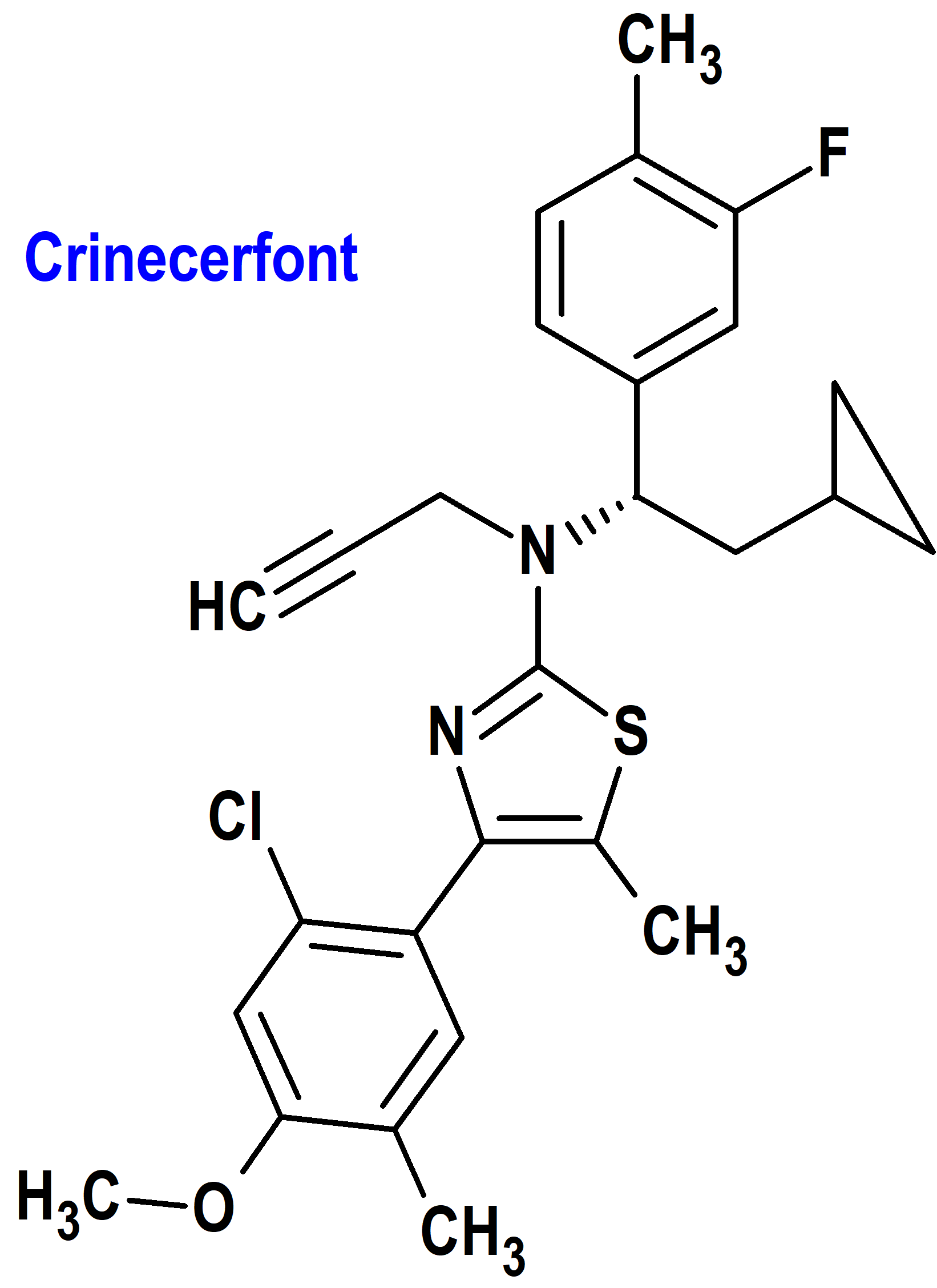
Indicación: Tratamiento complementario al reemplazo de glucocorticoides para controlar los andrógenos en pacientes adultos y pediátricos de 4 años de edad y mayores con hiperplasia suprarrenal congénita clásica.
Tipo: Medicamento sintético estándar constituido por la 4-(2-cloro-4-metoxi-5-metilfenil)-N-[(1S)-2-ciclopropil-1-(3-fluoro-4-metilfenil)etil]-5-metil-N-prop-2-inil-1,3-tiazol-2-amina (figura 6). Autorizado en Estados Unidos (FDA) el 13 de diciembre de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Antagonista selectivo del receptor del factor liberador de corticotropina (CRF) tipo 1. Crinecerfont bloquea la unión del CRF a los receptores CRF tipo 1 en la hipófisis, pero no a los receptores CRF tipo 2. La unión de Crinecerfont a los receptores CRF tipo 1 inhibe la secreción de la hormona adrenocorticotrópica (ACTH) de la hipófisis, reduciendo así la producción de andrógenos suprarrenales mediada por ACTH.
Eficacia clínica: Dos ensayos aleatorizados, doble ciego y controlados con placebo en 182 adultos y 103 niños con CAH clásica. En el primer ensayo, 122 adultos recibieron Crenessity dos veces al día y 60 recibieron placebo dos veces al día durante 24 semanas. Después de las primeras cuatro semanas del ensayo, la dosis de glucocorticoides se redujo a niveles de reemplazo y luego se ajustó en función de los niveles de androstenediona. La variable principal de eficacia fue el cambio desde el inicio en la dosis diaria total de glucocorticoides mientras se mantenía el control de la androstenediona al final del ensayo (-27% vs. -10%). En el segundo ensayo, 69 pacientes pediátricos recibieron Crenessity dos veces al día y 34 recibieron placebo dos veces al día durante 28 semanas. La variable principal de eficacia fue el cambio desde el inicio en el nivel sérico de androstenediona en la semana 4. El grupo que recibió Crenessity experimentó una reducción estadísticamente significativa desde el inicio en el nivel sérico de androstenediona, en comparación con un aumento promedio desde el inicio en el grupo placebo. Al final del ensayo, los pacientes asignados a Crenessity pudieron reducir su dosis diaria de glucocorticoides en un 18% mientras mantenían el control de los niveles de androstenediona en comparación con un aumento de la dosis diaria de glucocorticoides de casi el 6% en los pacientes asignados a placebo.
Eventos adversos: Los más comunes son fatiga (25% vs. 15% con placebo), vértigo (8/3%), artralgia (7/0%) y dolor de espalda (6/3%).
Remestemcel-L (Ryoncil®) Mesoblast (FDA, USA)
Indicación: Tratamiento de la enfermedad de injerto contra huésped aguda refractaria a esteroides (SR-aGVHD) en pacientes pediátricos de 2 meses de edad y mayores.
Tipo: Medicamento de terapia avanzada (celular somática), constituido por células estromales mesenquimales humanas (hMSC) derivadas de médula ósea alogénica. Autorizado en Estados Unidos (FDA) el 18 de diciembre de 2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track) y mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: La enfermedad de injerto contra huésped aguda refractaria a los esteroides es una afección grave y potencialmente mortal que puede presentarse como una complicación del trasplante alogénico de células madre hematopoyéticas (sanguíneas) (alo-HSCT). Las células madre mesenquimales humanas (hMSC) sirven para regular a la baja las respuestas inflamatorias y producir citocinas antiinflamatorias y factores de crecimiento para promover la reparación tisular.
Eficacia clínica: Un estudio multicéntrico de un solo brazo en 54 participantes pediátricos con SR-aGVHD después de someterse a un trasplante alogénico de células madre hematopoyéticas. Las variables principales de eficacia fueron en la tasa y la duración de la respuesta al tratamiento 28 días después de iniciar el tratamiento con Ryoncil. Los participantes del estudio que tuvieron una respuesta parcial o mixta al tratamiento (mejoría de la condición en un órgano sin cambios – parcial – o con un empeoramiento de la condición – mixta – en otro órgano) recibieron infusiones adicionales una vez por semana durante cuatro semanas adicionales. Dieciséis participantes del estudio (30%) tuvieron una respuesta completa al tratamiento, mientras que 22 participantes del estudio (41%) tuvieron una respuesta parcial.
Eventos adversos: Los más comunes son infecciones, fiebre, hemorragia, edema, dolor abdominal e hipertensión.
Cosibelimab (Unloxcyt®) Checkpoint (FDA, USA)
Indicación: Tratamiento de adultos con carcinoma escamocelular cutáneo metastásico (mCSCC) o CSCC localmente avanzado (laCSCC) que no son candidatos para cirugía curativa o radiación curativa.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal lambda IgG1 humano. Autorizado en Estados Unidos (FDA) el 13 de diciembre de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo bloqueador del ligando de muerte programada 1 (PD-L1) humano. El PD-L1 puede expresarse en células tumorales y células inmunitarias que se infiltran en tumores y puede contribuir a la inhibición de la respuesta inmunitaria antitumoral en el microambiente tumoral. La unión del PD-L1 a los receptores PD-1 y B7.1 que se encuentran en las células T y las células presentadoras de antígenos suprime la actividad citotóxica de las células T, la proliferación de células T y la producción de citocinas. El cosibelimab se une al PD-L1 y bloquea la interacción entre el PD-L1 y sus receptores PD-1 y B7.1. Esta interacción libera los efectos inhibidores del PD-L1 sobre la respuesta inmunitaria antitumoral. También se ha demostrado que el cosibelimab induce citotoxicidad mediada por células dependiente de anticuerpos.
Eficacia clínica: Un ensayo multicéntrico, multicohorte, abierto, en 109 pacientes con carcinoma espinocelular metastásico o carcinoma espinocelular laxo que no eran candidatos a cirugía curativa o radioterapia curativa. Las variables principales de eficacia fueron la tasa de respuesta objetiva (ORR) y la duración de la respuesta (DOR). La ORR fue del 47% para los pacientes con carcinoma espinocelular metastásico y del 48% para los pacientes con carcinoma espinocelular laminar. La DOR mediana no se alcanzó (rango: 1,4+, 34,1+) en los pacientes con carcinoma espinocelular metastásico y fue de 17,7 meses (rango: 3,7+, 17,7) en los pacientes con carcinoma espinocelular laminar.
Eventos adversos: Los más comunes (≥10%) son fatiga, dolor musculoesquelético, erupción cutánea, diarrea, hipotiroidismo, estreñimiento, náuseas, dolor de cabeza, prurito, edema, infección localizada e infección del tracto urinario.
(N) SISTEMA NERVIOSO
Acoramidis (Attruby®) Bridgebio (FDA, USA)
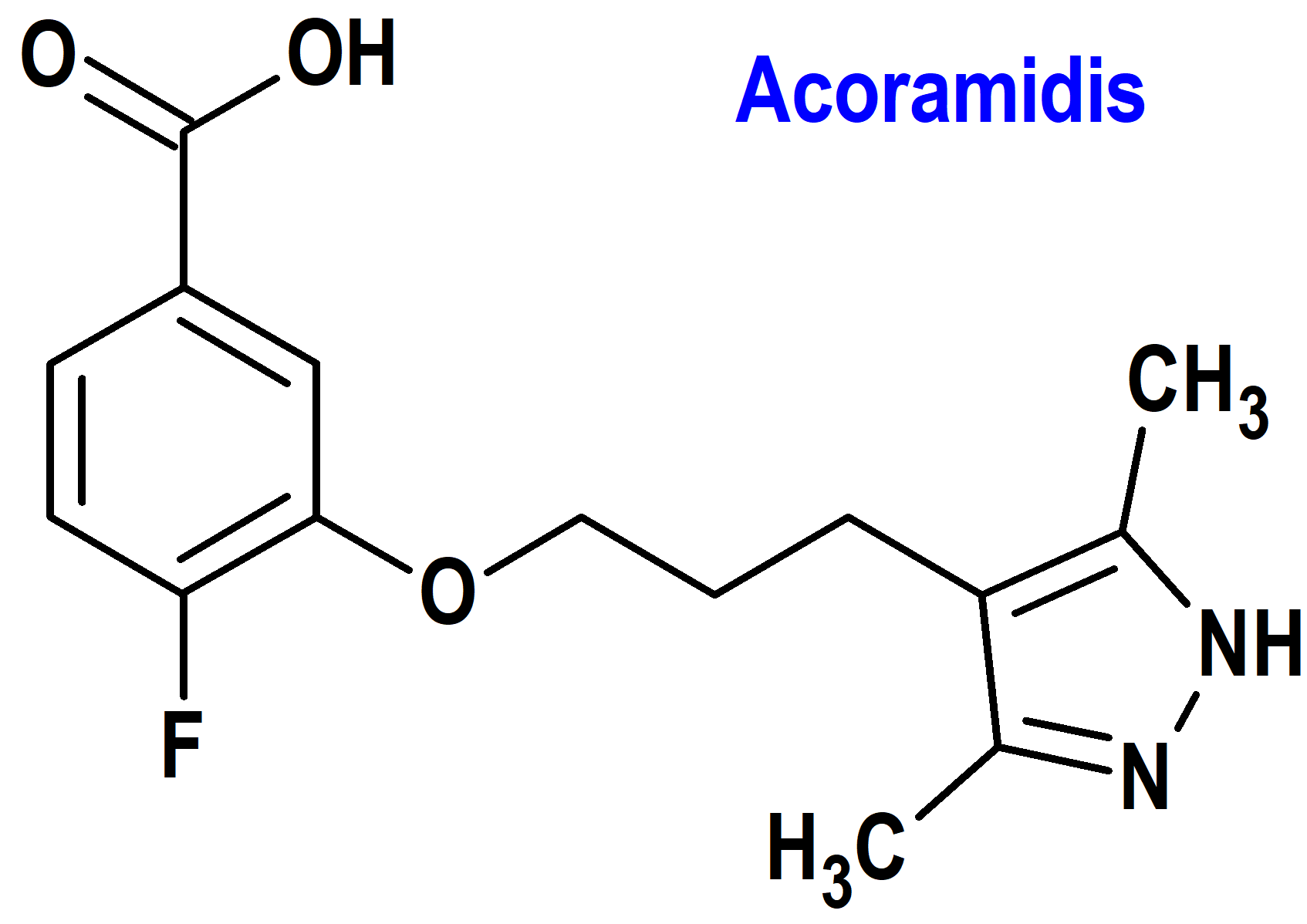
Indicación: Tratamiento de la adultos con miocardiopatía debida a amiloidosis mediada por transtiretina (ATTR-CM) de tipo salvaje (wATTR-CM) o hereditaria (hATTR-CM) para reducir la muerte y la hospitalización relacionadas con problemas cardíacos.
Tipo: Medicamento sintético estándar constituido por el ácido 3-[3-(3,5-dimetil-1H-pirazol-4-il)propoxi]-4-fluorobenzoico (figura 7). Autorizado en Estados Unidos (FDA) el 22 de noviembre de 2024 como medicamento huérfano (Orphan drug); evaluado favorablemente en la Unión Europea (CHMP, EMA) el 12 de diciembre de 2024.
Mecanismo: Estabilizador selectivo de la transtiretina (TTR), una proteína plasmática, tetramérica, cuya función es la de transportar tiroxina y proteína ligada al retinol. La TTR se sintetiza fundamentalmente en el hígado, aunque también en menor cuantía en otras localizaciones como los plexos coroideos o la retina. Acoramidis se une a la TTR en los sitios de unión de la tiroxina y retarda la disociación del tetrámero de TTR en sus monómeros constituyentes, el paso limitante de la velocidad en la amiloidogénesis.
Eficacia clínica: Un estudio multicéntrico, internacional, aleatorizado, doble ciego y controlado con placebo en 611 pacientes adultos con ATTR-CM de tipo salvaje o hereditario. Las variables principales de eficacia fueron la mortalidad por todas las causas y la frecuencia acumulada de hospitalizaciones relacionadas con enfermedades cardiovasculares (HCV) durante 30 meses. Al final de este periodos, más pacientes que tomaban acoramidis en comparación con placebo estaban vivos (81% frente a 74%) y hubo menos HVC ( media de 0,3 vs. 0,6 por año).
Eventos adversos: Los más comunes son diarrea y dolor abdominal superior. La mayoría son leves y se resuelven sin necesidad de suspender el tratamiento.
(R) SISTEMA RESPIRATORIO
Venzacaftor + Tezacaftor + Deutivacaftor (Alyftrek®) Vertex (FDA, USA)

Indicación: Tratamiento de la fibrosis quística (FQ, mucoviscidosis) en pacientes de 6 años de edad y mayores que tienen al menos una mutación F508del u otra mutación sensible en el gen regulador de la conductancia transmembrana de la fibrosis quística (CFTR).
Tipo: Medicamento sintético estándar constituido por la combinación a dosis fija de venzacaftor ((14S)-8-[3-(2-dispiro[2.0.24.13]heptan-7-iletoxi)pirazol-1-il]-12,12-dimetil-2,2-dioxo-2λ6-tia-3,9,11,18,23-pentazatetraciclo[17.3.1.111,14.05,10] tetracosa-1(22),5(10),6,8,19(23),20-hexaen-4-ona), tezacaftor (1-(2,2-difluoro-1,3-benzodioxol-5-il)-N-[1-[(2R)-2,3-dihidroxipropil]-6-fluoro-2-(1-hidroxi-2-metilpropan-2-il)indol-5-il]ciclopropano-carboxamida) y deutivacaftor (N-[2-terc-butil-4-[1,1,1,3,3,3-hexadeuterio-2-(trideuteriometil)propan-2-il]-5-hidroxifenil]-4-oxo-1H-quinolina-3-carboxamida) (figura 8). Autorizado en Estados Unidos (FDA) el 20 de diciembre de 2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: El vanzacaftor y el tezacaftor se unen a diferentes sitios de la proteína CFTR y tienen un efecto aditivo para facilitar el procesamiento celular y el tráfico de formas mutantes seleccionadas de CFTR (incluido F508del-CFTR) para aumentar la cantidad de proteína CFTR entregada a la superficie celular en comparación con cada molécula sola. El deutivacaftor potencia la probabilidad de apertura del canal (o activación) de la proteína CFTR en la superficie celular. El efecto combinado de vanzacaftor, tezacaftor y deutivacaftor es un aumento de la cantidad y la función de CFTR en la superficie celular, lo que resulta en un aumento de la actividad de CFTR medida tanto por el transporte de cloruro mediado por CFTR como por el cloruro en el sudor en pacientes con fibrosis quística.
Eficacia clínica: Dos ensayos aleatorizados, doble ciego y controlados con fármaco activo de 52 semanas de duración que compararon ALYFTREK y una combinación a dosis fija que contenía elexacaftor, tezacaftor e ivacaftor (ELX/TEZ/IVA). En los dos ensayos participaron un total de 971 pacientes de 12 años o más con fibrosis quística que tenían al menos una mutación F508del u otras mutaciones que respondían a ELX/TEZ/IVA en el gen CFTR. En ambos ensayos, el criterio de valoración principal evaluó la no inferioridad en el cambio absoluto medio en ppFEV1 (porcentaje previsto del volumen espiratorio forzado en 1 segundo) desde el inicio hasta la semana 24 y un criterio de valoración secundario clave evaluó el cambio absoluto medio desde el inicio en el cloruro en el sudor hasta la semana 24 en los grupos de tratamiento con ALYFTREK y ELX/TEZ/IVA. En el ensayo 1, el tratamiento con ALYFTREK dio como resultado una diferencia media de mínimos cuadrados de 0,2 puntos porcentuales en el cambio absoluto en el ppFEV1 desde el inicio hasta la semana 24 en comparación con ELX/TEZ/IVA, junto con una diferencia de -8,4 puntos en cloruro en el sudor. En el ensayo 2, la diferencia media de mínimos cuadrados fue de 0,2 puntos porcentuales en el cambio absoluto en el ppFEV1 y una diferencia de -2,8 puntos en cloruro en el sudor.
Eventos adversos: Los más comunes son tos (25%), nasofaringitis (21%), infección del tracto respiratorio superior (21%), dolor de cabeza (16%), dolor orofaríngeo (14%), gripe (11%), fatiga (11%), aumento de ALT (8%), erupción cutánea (8%), aumento de AST (7%) y congestión sinusal (7%). La proporción de pacientes que interrumpieron el tratamiento prematuramente debido a reacciones adversas fue del 3,8%.
(V) VARIOS
Vaso de ingeniería tisular acelular (Symvess®) Humacyte (FDA, USA)
Indicación: Uso en adultos como conducto vascular para lesiones arteriales de las extremidades cuando se necesita una revascularización urgente para evitar la pérdida inminente de la extremidad y el injerto de vena autóloga no es factible.
Tipo: Medicamento de terapia avanzada (ingeniería tisular) constituido por un conducto vascular diseñado con tejido acelular y compuesto de proteínas de la matriz extracelular (ECM) humana que se encuentran normalmente en los vasos sanguíneos humanos. Se fabrica mediante un proceso que utiliza células musculares lisas vasculares humanas derivadas de tejido aórtico humano. Autorizado en Estados Unidos (FDA) el 19 de diciembre de 2024 mediante revisión prioritaria (Priority Review) y designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Conducto vascular acelular que exhibe actividad mecánica y biológica capaz de soportar las fuerzas asociadas con el flujo sanguíneo arterial y la sutura durante la implantación, además puede favorecer la unión y proliferación celular. Tiene un diámetro interno de 6 mm y una longitud de 42 cm. Es fabricado mediante un proceso de ingeniería de tejidos en el que células musculares lisas vasculares humanas derivadas de tejido aórtico humano considerado adecuado para trasplante se almacenan, se expanden y se siembran en un andamio de malla tubular. El andamio sembrado de células se cultiva en un sistema de biorreactor biomimético para generar una construcción tubular intermedia que contiene células musculares lisas vasculares y la matriz extracelular que las células depositaron. Un proceso final de descelularización elimina el material genético y celular humano mientras se mantiene la estructura de la matriz extracelular y la actividad mecánica y biológica.
Eficacia clínica: Estudio prospectivo, multicéntrico y de un solo brazo en 54 pacientes con traumatismo vascular potencialmente mortal o que amenazaba la extremidad. La variable principal de eficacia fue la permeabilidad primaria (flujo sanguíneo presente sin ninguna intervención) y la permeabilidad secundaria (al menos una intervención necesaria para mantener el flujo sanguíneo) 30 días después de la implantación. De los 54 pacientes evaluados, 36 (67%) mantuvieron la permeabilidad primaria y 39 (72%) mantuvieron la permeabilidad secundaria el día 30. Cinco pacientes (9%) se sometieron a la amputación de la extremidad tratada dentro de los primeros 30 días y ocho (15%) pacientes se sometieron a la amputación de la extremidad tratada al final del estudio (mes 36).
Eventos adversos: Los más graves incluyen la rotura del injerto, la falla anastomótica y la trombosis. Dado que el producto utiliza células de un donante humano y reactivos de origen humano y bovino (vaca), no es descartable que pueda producirse la transmisión de enfermedades infecciosas o agentes infecciosos.
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE)
Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
Producto Calificado para Enfermedades Infecciosas. (Qualified Infectious Disease Product): un medicamento antibacteriano o antifúngico para uso humano destinado a tratar infecciones graves o potencialmente mortales, incluidas aquellas causadas por un patógeno resistente a antibacterianos o antifúngicos, incluidos patógenos infecciosos nuevos o emergentes.
