1. INTRODUCCIÓN
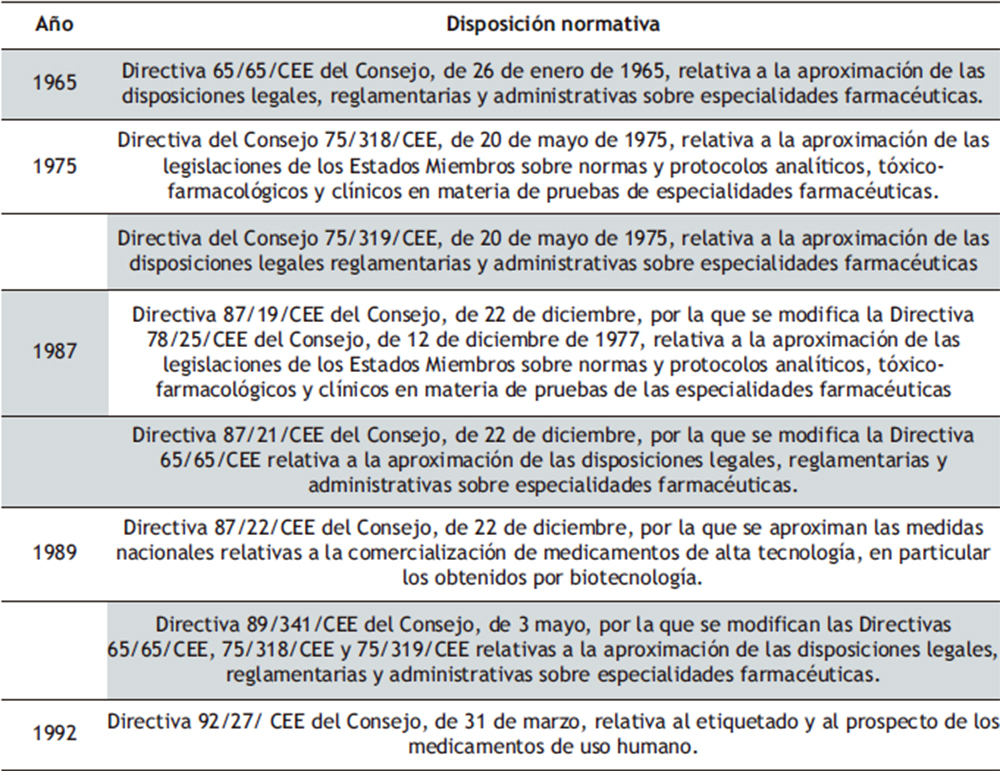
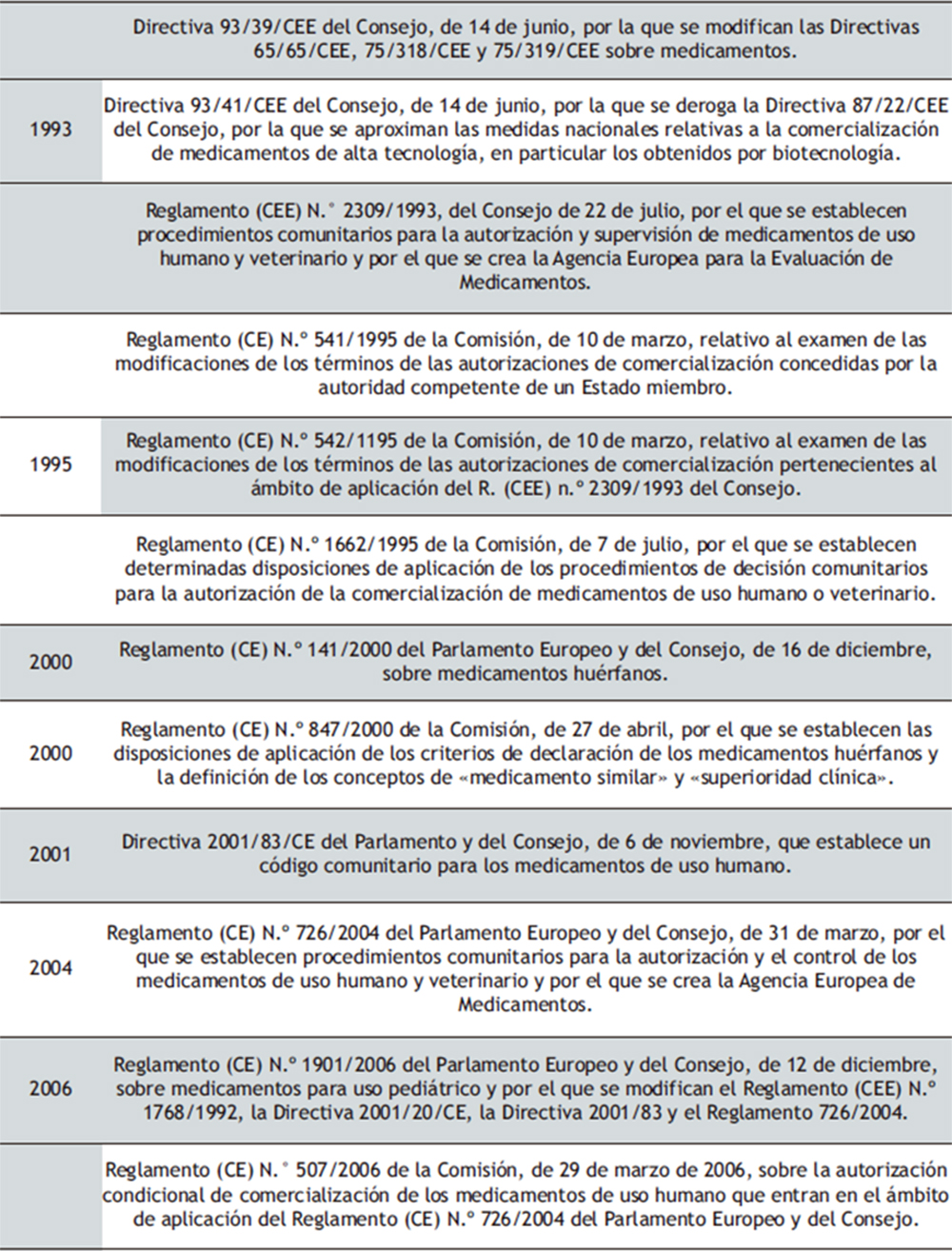
La regulación del sector farmacéutico en la UE ha sido muy extensa, especialmente en lo que se refiere al marco regulatorio de la otorgación de AC de medicamentos de uso humano (tabla 1). Uno de los hitos más relevantes en la regulación comunitaria del sector farmacéutico fue la promulgación de la Directiva 65/65/CEE (1). Los siguientes avances acontecidos en el siglo XX respecto a la producción normativa y sistematización de esta por parte del legislador de la UE han sido objeto de pormenorizado estudio por ALBA ROMERO (2,3).
Hay que tener en cuenta que en las últimas décadas se han promulgado las dos principales normas jurídicas reguladoras de la AC de los medicamentos de uso humano en el territorio regulatorio de la UE, a saber, la Directiva 2001/83/CE y el Reglamento (CE) N.º 726/2004(4). Sin embargo, en los próximos años tales normas jurídicas serán sustituidas con motivo del desarrollo legislativo previsto en la Estrategia Farmacéutica para Europa, adoptada por la CE adoptó el 25/11/2020(5,6). La citada propuesta persigue la generación de un entorno farmacéutico con visión de futuro y centrado en los pacientes en el que la industria farmacéutica de la UE pudiese innovar, progresar y mantener su liderazgo mundial.
Tabla 1. Cronología de principales normas jurídicas reguladoras de la AC de medicamentos de uso humano en la UE (1965-2023).

Con objeto de proporcionar respuesta global a los retos actuales a los que se enfrenta el sector, se había previsto la propuesta de revisión de la legislación farmacéutica que aborda todo el ciclo de vida de los medicamentos. Los objetivos que se persiguen en el marco de la referida Estrategia son los siguientes(7): (i) respaldar la innovación, impulsar la competitividad y atractivo de la industria farmacéutica; (ii) facilitar la accesibilidad y asequibilidad de los medicamentos –para ello, se reducirá la carga administrativa, se acelerarán los procedimientos y se reducirán los plazos de AC de los medicamentos–; (iii) optimizar la eficiencia regulatoria –es decir, la regulación gozará de mayor agilidad y flexibilidad–; (iv) mejorar la disponibilidad de los medicamentos de uso humano en los EM –para ello, se optimizará la seguridad en las cadenas legales de suministro–; (v) reforzar la calidad y la sostenibilidad ambiental; y, (vi) combatir la resistencia a los agentes antimicrobianos.
Con respecto al primer objetivo de la futura legislación farmacéutica, esto es, brindar apoyo a la innovación a los operadores del sector, conviene destacar algunos problemas actuales a los que el nuevo marco regulatorio debe hacer frente, y que han sido reflejados tanto en la literatura como en los análisis ex post de la normativa promulgada por las instituciones de la UE. Estos retos son los siguientes: (i) el creciente coste en materia de I+D de nuevos fármacos(8); (ii) el relevante gasto farmacéutico por los sistemas nacionales de salud(9,10); (iii) las barreras en la financiación de determinados medicamentos –esto es, la asequibilidad se ve comprometida, especialmente en el caso de los medicamentos huérfanos, debido a su elevado precio–(11,12); (iv) los retrasos en el acceso a las terapias innovadoras por parte de los pacientes (13); (v) el insuficiente fomento de la innovación(9), especialmente en las áreas de necesidades médicas insatisfechas, como es el caso de los antimicrobianos(14); (vi) la necesidad del fortalecimiento de la salud pública tras la pandemia de la COVID-19(15) –en concreto, se ha evidencia la necesidad de disponer de procedimientos simplificados y ágiles para conceder AC de medicamentos de uso humano que, estando en las últimas etapas de desarrollo, permitan abordar una emergencia de salud pública–; y (vii) el establecimiento de un marco normativo que contemple adecuadamente la innovación, que aborde convenientemente las terapias vanguardistas, disruptivas y emergentes (16–21), y que suprima las cargas burocráticas innecesarias.
En consecuencia, en fecha 26/04/2023 tuvo lugar la publicación del paquete farmacéutico, constituido tres propuestas normativas (tabla 2), si bien es cierto que este hito se ejecutó con un ligero retraso con respecto al cronograma propuesto habida cuenta de la evidente envergadura de las medidas propuestas.
Directiva del Parlamento Europeo y del Consejo sobre el Código comunitario relativo a los medicamentos de uso humano y por el que se deroga la Directiva 2001/83/CE y la Directiva 2009/35/CE.
Reglamento del Parlamento Europeo y del Consejo sobre por el que se establecen los procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y por el que se regula la EMA, por el que se modifican el Reglamento (CE) N.º 1934/2007 y el Reglamento (UE) N.º 536/2014 y se deroga el Reglamento (CE) N.º 726/2004, el Reglamento (CE) N.º 141/2000 y el Reglamento (CE) N.º 1901/2006.
Reglamento (UE) N.º 2020/1043 del Parlamento Europeo y del Consejo de 15 de julio de 2020 relativo a la realización de ensayos clínicos y al suministro de medicamentos para uso humano que contengan organismos modificados genéticamente o estén compuestos por estos organismos, destinados a tratar o prevenir la enfermedad coronavírica (COVID-19).
Recomendación del Consejo sobre la intensificación de las medidas de la UE para luchar contra la resistencia a los antimicrobianos de acuerdo con el concepto «Una sola salud». Tabla 2. Propuestas de disposiciones normativas contenidas en el nuevo paquete farmacéutico (2023).
Tabla 2. Propuestas de disposiciones normativas contenidas en el nuevo paquete farmacéutico (2023).

El presente trabajo tiene por objeto analizar la situación regulatoria de la innovación farmacéutica en el futuro corpus normativo de la UE (figura 1). En particular, se analizan las novedades más relevantes de las dos disposiciones de hard law previstas en el nuevo paquete farmacéutico.
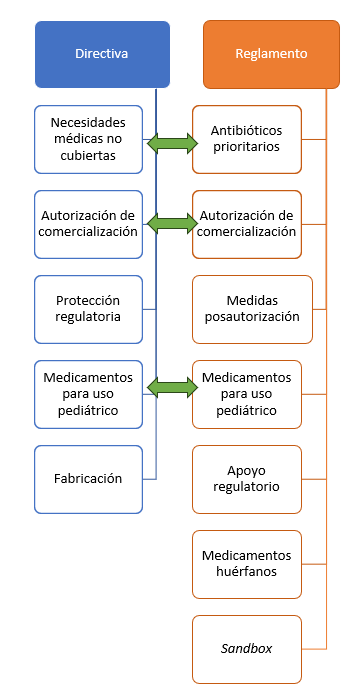
Figura 1. Principales ejes de la innovación farmacéutica en el nuevo paquete normativo de la UE.
2. PERÍODO DE PROTECCIÓN REGULATORIA
El acceso de los pacientes a los medicamentos en los EM se ha logrado de forma limitada en los últimos años, sin llegar a constituirse claramente un mercado interior único de los medicamentos en la UE. Se ha constatado que los pacientes tienen un acceso desigual a los medicamentos en toda la UE, toda vez que en los Estados miembros más pobres los medicamentos innovadores se comercializan del orden de varios años tras su introducción en el mercado de los EM más ricos (22).
Se regulan los plazos de protección de los datos regulatorios obrantes en el expediente de registro para los medicamentos de uso humano autorizados por cualquier procedimiento regulatorio y el período de exclusividad comercial.
La PD establece un sistema equilibrado de incentivos modulados o variables para los laboratorios farmacéuticos que recompense a los operadores que cumplen importantes objetivos de salud pública. La CE confía en que este sistema «generará ingresos para las empresas que desarrollen con éxito estos medicamentos revolucionarios, sin contribuciones financieras directas de los Estados miembros»(23).
2.1. Protección de datos regulatorios. Exclusividad comercial (art. 80 de la PD)
Los laboratorios farmacéuticos que comercialicen medicamentos de referencia tendrán un período mínimo de protección regulatoria de ocho años, conforme al siguiente esquema: (i) seis años de protección de datos, que se aplicará en todos los EM, con independencia de que el medicamento no esté autorizado o se haya suspendido o revocado su AC, y (ii) dos años de exclusividad comercial.
Las empresas podrán beneficiarse de periodos adicionales de protección, aumentando el periodo hasta un máximo de doce años para los medicamentos innovadores (en la actualidad es de once años). De esta manera, se recompensa tanto la innovación como el reposicionamiento de medicamentos de uso humano.
El período de protección de datos de seis años podría verse reducido voluntariamente por parte del laboratorio TAC. Se habilita la concesión a un tercero el empleo de sus datos con el fin de obtener una AC basada en el consentimiento.
Los períodos de protección de datos y de exclusividad comercial de los medicamentos de uso humano no se aplicarán en situaciones excepcionales. Así, si se concede una licencia obligatoria para hacer frente a una emergencia de salud pública, estos plazos se mantendrán suspendidos mientras se mantenga vigente la referida licencia obligatorio. La PR incorpora un procedimiento específico para obtener una AC temporal en casos de emergencia de salud pública.
Por tanto, los períodos de protección de datos y de exclusividad comercial deben suspenderse cuando se haya emitido una licencia obligatoria para hacer frente a una emergencia de salud pública. Dicha suspensión se limitará exclusivamente respecto a la licencia obligatoria otorgada y su beneficiario; y deberá cumplir con el objeto, el ámbito territorial, la duración y el objeto de la licencia obligatoria otorgada.
2.2. Plazos regulatorios de protección de datos (art. 81 de la PD)
El inicio de la protección de datos regulatoria se computará desde la fecha de concesión de la AC del medicamento, bien en el primer Estado miembro, o bien, en la UE.
Se establece la posibilidad de prorrogar el período de protección de datos de medicamentos de uso humano inicialmente establecido en seis años, hasta un total de diez años. Las prórrogas aplicables son las siguientes:
(1) De dos años –ampliable a tres años– si se inicia y se mantiene un suministro continuado y en cantidad suficiente para cubrir las necesidades de los pacientes en todos los EM en los dos años siguientes a la concesión de la AC.
Cn el objetivo de que un mayor número de operadores materialicen su investigación en medicamentos autorizados, se establece un incentivo especial en los siguientes supuestos: (i) PYMEs; (ii) organizaciones sin fines de lucro; y (iii) entidades que, en el momento de la concesión de la AC, no hayan obtenido más de cinco AC.
(2) De seis meses si el medicamento responde a una necesidad médica no cubierta. Esta prórroga sólo podrá aplicarse a los medicamentos que han sido objeto de una AC condicional cuando hayan obtenido una AC en un plazo máximo de cuatro años tras la obtención de la AC condicional.
(3) De seis meses para medicamentos que contengan un nuevo principio activo, y para el que se hayan realizado ensayos clínicos comparativos. Se prevé un desarrollo reglamentario por parte de la EMA, organismo que establecerá las directrices científicas sobre los criterios para proponer un comparador pertinente para un ensayo clínico. La utilización de los datos obtenidos a través de ensayos comparativos ayudará a las autoridades nacionales a evaluar mejor la rentabilidad de un nuevo medicamento, y también agilizará el acceso a nuevas terapias por parte de los pacientes.
(4) De un año si el medicamento de uso humano obtiene una indicación terapéutica adicional en los que se demuestra un beneficio clínico significativo en comparación con los tratamientos existentes. Dicha indicación terapéutica adicional podrá obtenerse durante la vigencia del período de protección de datos de dicho medicamento, no siendo necesario incorporar esa indicación en el momento de la solicitud de AC. Esta prórroga de un año sólo podrá concederse una única vez, no pudiendo acumularse al incorporar nuevas indicaciones terapéuticas para un medicamento concreto.
2.3. Prórroga del plazo de protección de datos de los medicamentos suministrados en los Estados miembros (art. 82 de la PD)
Para la aplicación de la prórroga del plazo de protección de datos de los medicamentos suministrados en los EM, el TAC deberá solicitar una modificación de la AC correspondiente en unos plazos determinados. Tales solicitudes se presentarán a los EM pertinentes.
Los EM tienen la posibilidad de renunciar a la condición de comercialización del medicamento en el territorio de su competencia a efectos de la prórroga de la protección de datos.
La renuncia a la prórroga explicada se podrá realizar a través de una declaración de no objeción para prolongar el período de protección de datos.
2.4. Protección de datos para reposicionamiento de medicamentos (art. 84 de la PD)
El reposicionamiento de los medicamentos existentes se vería incentivado con una única ampliación del plazo de protección de datos en cuatro años. Para ello, se exige: (i) la ejecución de estudios clínicos o preclínicos o no clínicos en relación con una indicación terapéutica, en la que se demuestre un beneficio clínico significativo; y (ii) que el medicamento haya obtenido una AC a través de un expediente abreviado y que no se haya beneficiado previamente de protección de datos, o bien, hayan transcurrido veinticinco años desde la concesión de la AC del medicamento.
2.5. Exención a la protección de los derechos de propiedad intelectual (art. 85 de la PD)
Se ha evidenciado la aplicación heterogénea de la cláusula Bolar(24–26) en la UE, y la necesidad de facilitar la introducción de medicamentos genéricos y de medicamentos biosimilares, asegurando la disponibilidad de estos el primer día tras la expiración de la patente del medicamento de referencia. Por ello, se ampliará la regulación previa en torno a la exención a la protección de los derechos de propiedad intelectual.
Se asegurará así la aplicación armonizada de la disposición Bolar en todos los EM. Se ampliará su alcance, de modo que será aplicable a: (i) los medicamentos híbridos, biohíbridos y a las variaciones posteriores; (ii) la evaluación de las tecnologías sanitarias; (iii) la fijación de precios y decisiones de reembolso.
3. NECESIDADES MÉDICAS NO CUBIERTAS
Pese a la evolución frenética de la ciencia y de la tecnología, actualmente hay enfermedades que carecen de tratamiento o estos son subóptimos(27–29), pues la I+D ejecutada por parte de la industria farmacéutica no siempre se centra en las necesidades médicas no cubiertas, quedando el mercado desabastecido en determinadas áreas terapéuticas, entre las que pueden citarse las enfermedades infecciosas, toda vez que la AC de medicamentos antimicrobianos redunda en un interés de la salud pública.
Como hemos anticipado, el desarrollo de nuevos agentes antimicrobianos no constituye una línea de investigación prioritaria para la industria farmacéutica. Dada la rápida aparición de resistencias a los antibióticos disponibles(30), se prevé que en el futuro el arsenal terapéutico sea muy reducido o inexistente para el tratamiento de las enfermedades infecciosas producidas por patógenos multirresistentes. Se ha considerado oportuno la creación de un sistema de incentivos en forma de vales o cupones para fomentar el desarrollo de nuevos antimicrobianos de carácter prioritario.
3.1. Medicamentos que abordan una necesidad médica no cubierta (art. 83 de la PD)
Los criterios que definen los medicamentos que responden una necesidad médica no cubierta son: (i) que una de sus indicaciones terapéuticas se relaciona con una enfermedad gravemente debilitante o potencialmente mortal; (ii) que no exista ningún medicamento autorizado en la UE para tratar dicha enfermedad, o bien, en el caso de que existan, la enfermedad se asocia a una alta mortalidad o morbilidad se prevé que los diversos elementos de esas definiciones, basadas en criterios de necesidades médicas no cubiertas se especifiquen con mayor grado de detalle por la CE, teniendo en cuenta los informes publicados por la EMA; y (iii) que el empleo del medicamento traiga consigo una reducción significativa de la mortalidad o morbilidad en la población diana.
3.2. Incentivos para el desarrollo de antibióticos prioritarios
3.2.a. Otorgación del derecho de un cupón de exclusividad de datos transferibles (art. 40 de la PR)
Se considera antimicrobiano prioritario a aquel antimicrobiano cuyos datos preclínicos o no clínicos y clínicos demuestran un beneficio clínico significativo respecto a la resistencia a los antimicrobianos existentes. Tal medicamento deberá cumplir con, al menos, una de las siguientes características: (i) se agrupa en una nueva familia terapéutica; (ii) tiene un mecanismo de acción diferente con respecto al resto de antimicrobianos previamente autorizados en la UE; (iii) contiene un principio activo no autorizado previamente en la UE y que está indicado para el tratamiento de un organismo multirresistente y una infección grave o potencialmente mortal.
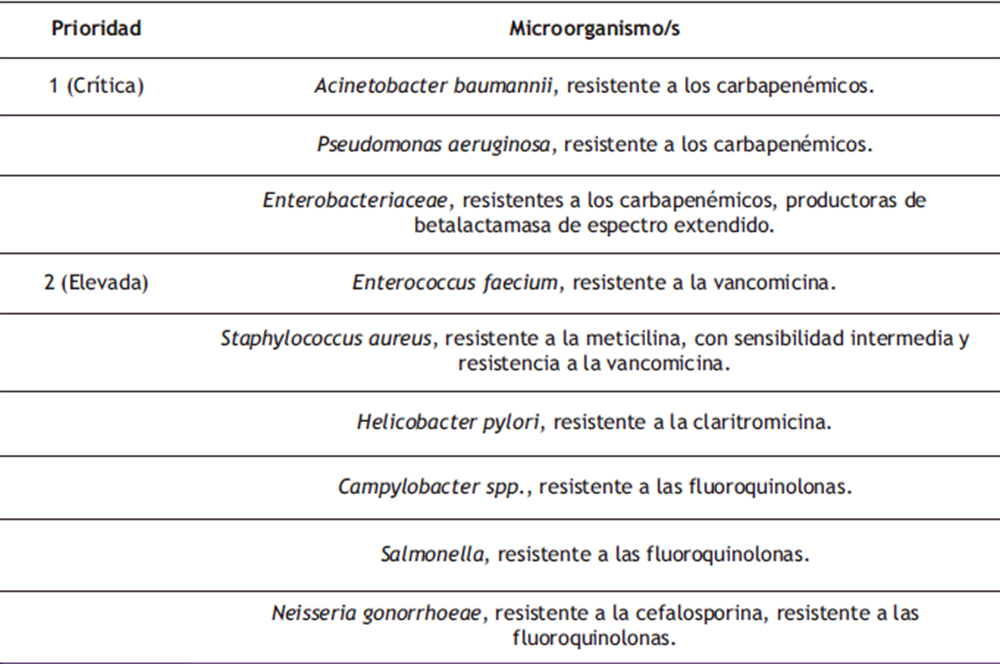
De cara a la calificación de un antimicrobiano como prioritario, se tendrá en cuenta la Lista de patógenos prioritarios para la I+D de nuevos antibióticos publicada por la Organización Mundial de la Salud (tabla 3), así como las listas equivalentes establecidas en la UE.
La CE podrá conceder un cupón de exclusividad de datos transferibles a los laboratorios desarrolladores de nuevos antimicrobianos. Este cupón beneficiará a su titular con el aumento de la protección de datos de un medicamento autorizado por un período de doce meses.
Se imponen dos obligaciones al solicitante para la otorgación del cupón: (i) demostrar la capacidad para suministrar el antimicrobiano prioritario en cantidad suficiente para cubrir las necesidades de los pacientes de la UE; e (ii) informar sobre el apoyo financiero público directo recibido para la I+D del antimicrobiano prioritario.
El TAC deberá informar del apoyo financiero directo recibido de cualquier fuente en todo el mundo en un plazo de treinta días tras la concesión de una AC. Esta información será de fácil acceso para el público en un portal web específico del laboratorio.
Tabla 3. Fragmento de la lista de patógenos prioritarios para la I+D de nuevos antibióticos de la Organización Mundial de la Salud(31).
3.2.b. Uso y la transferencia del cupón (art. 41 de la PR)
El empleo del cupón conllevará la ampliación del período de doce meses para el antimicrobiano prioritario, o bien, para cualquier otro medicamento autorizado a elección del titular del cupón.
El cupón podrá ser utilizado por el TAC del antibiótico prioritario, o bien, este podrá transferirlo a un tercero. En cualquier caso, para el empleo del cupón, el medicamento deberá haberse autorizado por el procedimiento centralizado de registro en los últimos cuatro años, a contar desde la solicitud de uso del cupón.
Los cupones podrán ser utilizados en una única ocasión, y en un plazo máximo de cinco años tras su concesión, siempre se mantenga vigente la AC del antimicrobiano prioritario.
Con relación a las transferencias de los cupones, estas sólo podrán realizarse en única ocasión, y deberán ser comunicadas por parte del TAC del antimicrobiano prioritario a la EMA en un plazo de treinta días. El valor de la transacción podrá ser monetario, o cualquier otro acordado entre las partes. El TAC deberá informar a la EMA del valor de la transacción realizada entre las partes; la EMA, facilitará al público esa información.
3.2.c. Validez del cupón (art. 42 de la PR)
La validez del cupón expirará cuando: (i) se haya usado, esto es, la CE haya conferido una ampliación del plazo de protección de datos por un año, tras la correspondiente solicitud; (ii) tras cinco años tras la fecha de su concesión; (iii) cuando no se haya satisfecho una solicitud de suministro, contratación o compra del antimicrobiano prioritario en la UE, de forma previa a la transferencia del cupón.
Se habilita a terceros el registro de un medicamento genérico o híbrido empleando como medicamento de referencia aquel que tiene el antimicrobiano prioritario cuando se retire la AC del antimicrobiano prioritario antes de que expiren los períodos de protección de datos y de exclusividad comercial.
3.2.d. Duración de la aplicación de los incentivos (art. 43 de la PR)
Se limita la duración de los cupones al acontecimiento del primero de los siguientes hitos: (i) quince años tras la entrada en vigor de la PR, o bien, (ii) la otorgación de diez cupones de exclusividad de datos transferibles.
4. AUTORIZACIONES DE COMERCIALIZACIÓN
La Directiva seguirá regulando el procedimiento descentralizado, el procedimiento nacional y el procedimiento de reconocimiento mutuo. El Reglamento regularizaría el procedimiento centralizado.
En la PR se establecen tres nuevos tipos específicos de AC que tienen carácter temporal y están sometidas a unas reglas especiales: (i) AC concedida en circunstancias excepcionales; (ii) AC de carácter condicional; y, (iii) AC temporales de emergencia.
4.1. Procedimientos de registro tradicionales
4.1.a. Procedimientos descentralizado, de reconocimiento mutuo y nacional (arts. 5 a 28 de la PD)
Como principales novedades deben destacarse la digitalización en los expedientes de registro(32) –Electronic Common Technical Document–; y la reducción de los plazos para la otorgación de la AC.
Con relación a la segunda de estas medidas, se prevé la reducción del período total de los procedimientos descentralizado, de reconocimiento mutuo y nacional de AC de medicamentos de uso humano del orden de treinta días, concretamente de doscientos diez días –período en vigor–(33,34) a ciento ochenta días. Una vez el CHMP haya emitido un dictamen, la CE deberá emitir una decisión relativa a la AC nacional en un plazo de cuarenta y seis días (en lugar de sesenta y siete, como sucede actualmente). Así se reducirá la media actual del orden de cuatrocientos días que median entre la presentación y la resolución de la AC.
4.1.b. Procedimiento centralizado (arts. 3 a 29 de la PR)
Los medicamentos de uso humano que deben ser objeto de una autorización por el procedimiento centralizado de registro son los referidos en el anexo I. Del futuro anexo I se eliminan los medicamentos biotecnológicos obtenidos por los métodos del hibridoma y del anticuerpo monoclonal y, se incorporan los siguientes grupos de medicamentos: (i) medicamentos de uso humano con una sustancia activa no autorizada en la UE antes del 20 de mayo de 2004 –con algunas exclusiones–; (ii) medicamentos autorizados con una AC para uso pediátrico; y (ii) antimicrobianos prioritarios.
Los medicamentos de alta tecnología, los medicamentos para uso pediátrico y los medicamentos con nuevos principios activos se han incorporado en el referido listado para mantener un elevado nivel de evaluación científica de los nuevos medicamentos.
Se establece un procedimiento específico para la concesión de AC de medicamentos genéricos por el procedimiento centralizado.
4.1.c. Agencia Europea del Medicamento (arts. 135 a 170 de la PR)
La EMA continuará siendo el organismo encargado de coordinar los recursos científicos que los EM ponen a su disposición para la evaluación, control y farmacovigilancia de los medicamentos(35,36).
Se reestructura la EMA. Se suprime, o bien, se sustituye un buen número de comités científicos –comités de medicamentos huérfanos, pediátrico, de medicamentos de terapias avanzada y de medicamentos tradicionales a base de plantas– por grupos de trabajo. Tan solo se mantendrán los comités principales: CHMP y Pharmacovigilance Risk Assessment Committee o Comité Europeo de Farmacovigilancia (PRAC). De este modo, se proporciona mayor apoyo a los comités principales a través de grupos de trabajo y grupos de expertos, evitando las duplicidades y el uso no optimizado de la experiencia y de los recursos de la EMA.
4.2. Autorización concedida en circunstancias excepcionales (art. 18 de la PR)
Antes de abordar las particularidades del novedoso procedimiento regulador de la AC en circunstancias excepcionales, debe advertirse de la necesidad de diferenciar adecuadamente las actuales AC condicionales, actualmente reguladas en el Reglamento (CE) N.° 507/2006 (tabla 1), que tendrían una regulación adaptada en la regulación de la AC concedida en circunstancias excepcionales(37).
Estas AC se otorgarán en aquellos casos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y seguridad del medicamento de uso humano en las condiciones normales de uso. En tales casos, la CE podrá conceder una AC sujeta a unas condiciones específicas, bajo el cumplimiento de tres requisitos concretos.
La validez de estas AC será de dos años desde la otorgación de la autorización para la nueva indicación terapéutica, y posteriormente, según la frecuencia determinada por la EMA.
4.3. Autorización de carácter condicional (art. 19 de la PR)
El marco regulatorio actual ha permitido la concesión de AC a terapias innovadoras y prometedoras, pero este debe ser objeto de reforma habida cuenta las desigualdades en el acceso a los medicamentos en los diferentes EM.
Los medicamentos prometedores que tienen el potencial de abordar significativamente las necesidades médicas insatisfechas deben beneficiarse de un respaldo científico mejorado y precoz. De esta manera, se facilitará un acceso temprano a las nuevas terapias.
Para responder a las expectativas legítimas de los pacientes, y tener en cuenta el progreso imparable de la ciencia y las terapias, se requieren procedimientos ágiles de evaluación de medicamentos con gran interés terapéutico y, por tanto, es preciso regular unas AC condicionales, sometidas a unas condiciones particulares y revisables periódicamente.
Estas AC podrán ser concedidas, bien para una solicitud nueva, o bien, para ampliar la AC de una indicación terapéutica existente. Se requiere, en sendos casos: (i) una motivación suficiente, basada en el beneficio en una necesidad médica no cubierta; (ii) que se requieran datos clínicos adicionales; y (iii) que el beneficio de la disponibilidad inmediata en el mercado supere los riesgos derivados de la incertidumbre por la ausencia de datos clínicos.
Se habilitará la concesión de AC condicionales para medicamentos de uso humano en situaciones de emergencia cuando no se hayan facilitado datos preclínicos o farmacéuticos completos.
La AC condicional estará sometidas a unas obligaciones específicas, que deberán ejecutarse en los plazos habilitados para su cumplimiento. Tales obligaciones serán revisadas por la EMA periódicamente.
La validez de la AC condicional será de un año, renovable durante los tres primeros años tras la concesión de la AC condicional, y posteriormente, cada dos años.
4.4. Autorización temporal de emergencia (arts. 30-39 de la PR)
En cualquier situación de emergencia de salud pública es de gran interés que puedan desarrollarse y estar disponibles dentro de la UE medicamentos seguros y eficaces tan pronto como sea posible. En el caso de las vacunas destinadas a controlar la pandemia provocada en el marco de la COVD-19, las instituciones de la UE hicieron uso del procedimiento de rolling review con objeto de agilizar la AC de dichos medicamentos(38).
En casos de emergencia sanitaria, la CE podrá conceder una AC para medicamentos de uso humano destinados al tratamiento, la prevención o el diagnóstico de una enfermedad o afección grave o potencialmente mortal que esté directamente relacionada con una emergencia de salud pública, sin necesidad de aportar un expediente completo.
4.3.a. Criterios de concesión (art. 31 de la PR)
Los criterios para la concesión de una AC temporal de emergencia son los siguientes: (i) que no exista otro método satisfactorio de tratamiento, prevención o diagnóstico autorizado o suficientemente disponible en la UE; o, si existiese, que la otorgación de la AC contribuya a abordar la emergencia de salud pública; (ii) sobre la evidencia científica disponible, la EMA concluye que el medicamento podría ser eficaz para tratar, prevenir o diagnosticar la enfermedad o afección directamente relacionada con la emergencia de salud pública, y que la relación beneficio/riesgo estimada sea favorable. En otras palabras, se debe valorar que el beneficio derivado de la disponibilidad inmediata en el medicamento supera el riesgo derivado de la incertidumbre de la ausencia de determinados datos clínicos.
4.3.b. Opinión científica. Decisión de la Comisión Europea (arts. 32 y 33 de la PR)
Se establece una regulación específica relativa a la opinión científica por parte del CHMP para la concesión de estas AC temporales de emergencia, la cual deberá emitirse sin demoras indebidas Basándose en el dictamen del CHMP, la CE deberá adoptar una decisión sobre la AC, sin retrasos indebidos.
La CE podrá establecer las condiciones específicas con respecto a la AC temporal de emergencia y, en concreto, las condiciones de fabricación, uso, suministro, control de la seguridad y el cumplimiento de las Normas de Correcta Fabricación (NCF) y las Buenas Prácticas de Farmacovigilancia (BPFV).
Podrán establecerse un plazo y unas condiciones específicas para: (i) exigir la finalización de los estudios en curso; o bien, (ii) la realización de estudios adicionales para garantizar la eficacia y seguridad del medicamento, así como minimizar su impacto medio ambiental.
4.3.c. Validez (art. 34 de la PR)
La vigencia de la AC temporal de emergencia expirará cuando la CE ponga fin al reconocimiento de la emergencia de salud pública.
4.3.d. Modificación, suspensión y revocación (art. 35 de la PR)
Las condiciones de modificación, suspensión y revocación de la AC temporal de emergencia por parte de la CE s podrán ser acordadas en tres supuestos: (i) se dejan de cumplir los requisitos por los cuales se otorgó la AC; (ii) con motivo de la protección de la salud pública; (iii) el TAC no ha cumplido las obligaciones específicas, o bien, con las condiciones y obligaciones impuestas en la AC.
4.3.e. Concesión de AC o AC condicional tras la AC temporal de emergencia (art. 36 de la PR)
El TAC podrá presentar: una solicitud de AC de carácter: ordinario; condicional y temporal de emergencia.
4.3.f. Período de transición (art. 37 de la PR)
Tras la suspensión o revocación de una AC temporal de emergencia, los EM, en circunstancias excepcionales, podrán permitir un período de transición con el fin de que los pacientes continúen con los tratamientos con medicamentos de uso humano autorizados a través del procedimiento de emergencia que venían recibiendo.
4.3.g. Anulación de la AC concedida en virtud del art. 3.2 de la Propuesta de la Directiva (art. 39 de la PR)
Se habilita la retirada de las autorizaciones temporales de uso de medicamentos no autorizados con motivo de la defensa nuclear, biológica, química y radiológica (NBQr). Si la CE concede AC temporales de emergencia, los EM deberán revocar las autorizaciones nacionales concedidas para medicamentos que contengan el mismo principio activo que los medicamentos que han recibido una AC temporal de emergencia por la CE.
5. MEDIDAS POST-AUTORIZACIÓN
5.1. Actualización de la AC relacionadas con el desarrollo científico y tecnológico (art. 45 de la PR)
En relación con la actualización de una AC debido al desarrollo científico y tecnológico, se establece una regulación muy similar con respecto al art. 90 de la PD. Se imponen unos requisitos determinados para los laboratorios TAC.
5.2. Dictamen científico sobre los datos presentados por entidades sin ánimo de lucro para el reposicionamiento de medicamentos (art. 48 de la PR)
Las entidades sin ánimo de lucro podrán presentar a la EMA o a las agencias nacionales de los EM las evidencias preclínicas o clínicas significativas para una nueva indicación terapéutica que tenga una utilidad probable en una necesidad médica no cubierta.
La EMA podrá realizar una evaluación de la relación beneficio/riesgo del medicamento en la nueva indicación terapéutica, basándose en la evidencia científica disponible.
El dictamen de la EMA respecto a la evaluación científica del medicamento de uso humano se pondrá a disposición del público y de las autoridades competentes de los EM. Si el dictamen de la EMA es favorable, el TAC deberá presentar una modificación de la AC para incorporar la nueva indicación terapéutica, relacionada con una necesidad médica no cubierta.
6. APOYO REGULATORIO PREVIO A LA AUTORIZACIÓN
6.1. Asesoramiento científico a empresas y entidades sin ánimo de lucro (art. 58 de la PR)
Las empresas podrán solicitar asesoramiento científico y regulatorio a la EMA para: (i) el desarrollo de medicamentos de gran interés para la salud pública o con elevado grado de innovación; (ii) el desarrollo de medicamentos que cubren una necesidad médica no cubierta; (iii) el reposicionamiento de medicamentos ya autorizados.
Se proporcionará un asesoramiento científico adaptado a las PYMEs. Estas entidades se beneficiarán de apoyo reglamentario, administrativo y procedimental, con una reducción, aplazamiento o exención de tasas.
La EMA podrá solicitar asesoramiento a expertos y a otros organismos de la UE en casos debidamente justificados.
6.2. Asesoramiento científico paralelo (art. 59 de la PR)
Las organizaciones sin ánimo de lucro recibirán apoyo administrativo, así como una reducción, aplazamiento o exención de tasas, y podrán recibir un asesoramiento científico paralelo.
6.3. Refuerzo del apoyo científico y regulatorio para los medicamentos prioritarios (art. 60 de la PR)
La EMA ofrecerá apoyo científico y regulatorio, así como mecanismos de evaluación simplificados para determinados medicamentos, en los que el solicitante de la AC evidencie que: (i) es probable que aborde una necesidad médica no cubierta; (ii) sea un medicamento huérfano y es probable que aborde una necesidad médica insatisfecha; y (iii) se espera que sea de gran interés por su relevancia para la salud pública, por su elevado grado de innovación, o bien, por ser antimicrobianos prioritarios.
También ofrecerá apoyo científico y regulatorio a los laboratorios que desarrollen medicamentos para diagnosticar, prevenir o tratar una enfermedad resultante de amenazas sanitarias transfronterizas de carácter grave.
6.4. Recomendaciones científicas sobre el estado regulatorio (art. 61 de la PR)
La EMA podrá prestar asesoramiento científico a los laboratorios farmacéuticos o a las autoridades competentes de los EM para las solicitudes de AC que deben seguir obligatoriamente el procedimiento centralizado. En otras palabras, la EMA informará, en un plazo de sesenta días –prorrogable a noventa días– desde la recepción de la solicitud, si tales solicitudes entran dentro del ámbito de aplicación del anexo I de la PR por considerarse que el producto responde a la definición de medicamento de terapias avanzadas.
6.5. Decisiones sobre el status regulatorio (art. 62 de la PR)
La CE podrá decidir si el producto entra dentro del ámbito de aplicación del anexo I de la PR por iniciativa propia, o bien, por petición de los EM por su de desacuerdo motivado con la recomendación de la EMA.
7. MEDICAMENTOS HUÉRFANOS
A los medicamentos huérfanos –o, como indica la profesora CABEZAS LÓPEZ, medicamentos sin interés comercial(39)– se les aplican unos requisitos específicos(11,40–42), diferentes al de los medicamentos para uso pediátrico(43–45).
7.1. Criterios para su designación (art. 63 de la PR)
Se mantienen los dos criterios de prevalencia y de ausencia de ningún método satisfactorio autorizado en la UE, destinado al diagnóstico, prevención o tratamiento de dicha afección o que, de existir, el medicamento aporte un beneficio considerable a quienes padecen dicha afección. La supresión del criterio basado en el rendimiento de la inversión puede justificarse en que, hasta la fecha, no habría sido aplicado.
7.2. Concesión de la designación (art. 64 de la PR)
La designación puede solicitarse en cualquier etapa del desarrollo del medicamento de uso humano, siempre que se presente antes de la AC.
La EMA resolverá, autorizando o denegando la designación de medicamento huérfano en un plazo de noventa días tras la recepción de la solicitud; tal decisión será puesta a disposición del público.
7.3. Transferencia de la designación (art. 65 de la PR)
Se habilita la transferencia de la designación huérfana del solicitante de tal designación a un tercero, previa autorización por parte de la EMA. La EMA resolverá, autorizando o denegando la transferencia de la designación de medicamento huérfano en un plazo de treinta días tras la recepción de la solicitud.
7.4. Validez de la designación (art. 66 de la PR)
Se establece un plazo de validez de las designaciones de medicamentos huérfanos por un período del orden de siete años, que podrá ser prorrogado cuando el promotor del medicamento huérfano pueda aportar pruebas de que los estudios pertinentes que avalan el uso del medicamento designado como huérfano en las condiciones de uso son prometedoras con respecto a la presentación de una solicitud futura. Una vez obtenida una AC, la designación huérfana quedará anulada. El solicitante de la designación huérfana podrá solicitar su anulación en cualquier momento.
7.5. Registro de designaciones de medicamentos huérfanos (art. 67 de la PR)
El registro público de medicamentos designados como huérfanos será creado y gestionado por la EMA. Tal registro será de acceso público e incluirá, al menos, la información estipulada.
7.6. Colaboración en el desarrollo de medicamentos huérfanos (arts. 68 y 73 de la PR)
Se regula la asistencia de la EMA en la elaboración del protocolo y el apoyo a la investigación de medicamentos huérfanos. También la contribución financiera de la UE relacionada con los medicamentos huérfanos. Así, se establecen tasas reducidas en relación con los medicamentos huérfanos.
7.7. Autorización de comercialización de medicamentos huérfanos (art. 69 de la PR)
Para un mismo medicamento de uso humano podrán solicitarse AC independientes, en función de si las indicaciones terapéuticas solicitadas son de utilidad para el diagnóstico, prevención o tratamiento de una enfermedad rara, u otro tipo de enfermedades.
Para la concesión de una AC de un medicamento huérfano, el solicitante deberá demostrar que se ha concedió previamente la designación de medicamento huérfano por parte de la EMA.
7.8. Medicamentos huérfanos que abordan una gran necesidad médica no cubierta (art. 70 de la PR)
Se regulan las condiciones en las que se considera que los medicamentos huérfanos abordan una gran necesidad médica no cubierta: (i) no existe ningún medicamento autorizado en la UE para tratar dicha enfermedad, o bien, en el caso de que existan, el solicitante demuestra que el medicamento huérfano tiene un beneficio significativo y traerá un avance terapéutico excepcional; y (ii) el empleo del medicamento trae consigo una reducción significativa de la mortalidad o morbilidad en la población diana.
Una AC basada en datos bibliográficos no podrá avalar que el medicamento de uso humano responde a una necesidad médica no cubierta.
7.9. Exclusividad comercial (art. 71 de la PR)
Tras la concesión de una AC a un medicamento huérfano, deberá respetarse un período de exclusividad del mercado, durante el cual, los EM no podrán conceder una AC ni una prórroga de una AC existente para la misma indicación terapéutica para medicamentos similares a ese medicamento huérfano.
La AC podrá concederse para la misma indicación terapéutica a un medicamento similar si el TAC del medicamento huérfano original: (i) ha dado su consentimiento a un solicitante dado; (ii) no puede mantener abastecido el mercado de la UE; o bien, (iii) si el segundo solicitante puede establecer en la solicitud que el medicamento similar al previamente autorizado es más seguro, más efectivo o clínicamente superior.
Se propone una modulación en la exclusividad en el mercado, que oscilará entre nueve y trece años, en función del tipo de medicamento huérfano, toda vez que los medicamentos huérfanos que abordan una gran necesidad médica no cubierta supondrían un avance terapéutico excepcional.
La duración estándar de la exclusividad comercial se contará desde la fecha en que se concedió la primera AC del medicamento huérfano en la UE. Tendrá una duración de nueve años con carácter general, y se reducirá a cinco años para los medicamentos cuya solicitud de AC esté basada en datos bibliográficos al considerar que la innovación generada por ese medicamento es menos significativa.
Todo laboratorio TAC de un medicamento huérfano podrá beneficiarse de períodos adicionales de exclusividad en el mercado si: (i) abordan una gran necesidad médica no cubierta: un año adicional; (ii) los medicamentos huérfanos autorizados reciben indicaciones terapéuticas autorizadas: dos años adicionales o cuatro años adicionales, si reciben dos autorizaciones para una o dos nuevas indicaciones terapéuticas para diferentes condiciones huérfanas, respectivamente; (ii) comercializan el medicamento en todos los EM de la UE: un año adicional.
Una vez expirado el período de exclusividad comercial del medicamento huérfano de referencia, podrá iniciarse la comercialización de los medicamentos biosimilares. En los últimos dos años de exclusividad comercial del medicamento huérfano de referencia, podrá presentarse, validarse y evaluarse la AC de los medicamentos biosimilares.
8. MEDICAMENTOS PARA USO PEDIÁTRICO
8.1. Incentivos
8.1.a. Art. 86 de la PD
Se mantiene la posibilidad de prorrogar los efectos de la patente o del CCP por un período de seis meses si la solicitud de AC del medicamento de referencia incluye los resultados de los estudios realizados conforme a un PIP. La prórroga se concederá con independencia de que el medicamento de uso humano reciba una indicación terapéutica aprobada en la población pediátrica, exigiendo que los resultados de los estudios realizados consten en la ficha técnica y en el prospecto del medicamento.
Para los medicamentos de uso pediátrico autorizados por el procedimiento descentralizado o de reconocimiento mutuo, tal prórroga sólo se aplicará cuando el medicamento se comercialice en todos los EM.
En el caso de solicitud de modificación de la AC por nuevas: (i) indicaciones terapéuticas, (ii) dosis, (iii) vías de administración, si el solicitante obtiene la prórroga de un año de exclusividad comercial, no podrá disfrutar de esta prórroga de seis meses.
Se establecen incentivos para los medicamentos de uso humano que obtengan nuevas indicaciones terapéuticas pediátricas cuando aporten un beneficio clínico significativo en comparación con las terapias existentes en la UE.
Si el TAC tiene intención de retirar un medicamento de ese grupo del mercado, deberá informar a la EMA en un plazo determinado.
8.1.b. Recompensas e incentivos (arts. 93 y 96 de la PR)
Se siguen aplicando requisitos específicos para apoyar el desarrollo de medicamentos para uso pediátrico. Si se concede una AC para uso pediátrico, tal medicamento podrá beneficiarse de los incentivos establecidos en la PD. En definitiva, teniendo en consideración los objetivos específicos y los impactos económicos y sociales de las medidas propuestas, se mantiene la extensión del CCP del orden de seis meses por completar un PIP.
Los medicamentos para uso pediátrico podrán ser beneficiados por los incentivos ofrecidos por los EM o por al UE para fomentar la I+D y la disponibilidad de este tipo de medicamentos.
8.2. Plan de investigación pediátrica
8.2.a. Introducción (art. 74 de la PR)
En determinados casos se requiere la realización de estudios de medicamentos de uso humano en la población pediátrica para: (i) garantizar que los medicamentos estén debidamente autorizados para su uso en la población pediátrica; (ii) mejorar la información disponible sobre el uso de medicamentos en la población pediátrica; y (iii) que los medicamentos se presenten en dosis y en formulaciones adecuadas para su uso en la población pediátrica.
El PIP debe presentarse durante el desarrollo del medicamento y antes de la presentación de la solicitud de AC.
Se podrá presentar un PIP inicial abreviado en dos supuestos: (i) que el principio activo sea novedoso en la UE y esté destinado a tratar una nueva condición pediátrica; y (ii) que la EMA acepte una solicitud motivada por parte del solicitante. En estos casos, que el plan inicial contendrá: (i) el calendario de las medidas propuestas; (ii) los detalles de las medidas que se conozcan en el momento de la presentación de la solicitud; (iii) la frecuencia de presentación de las versiones actualizadas del PIP; y, (iv) la fecha prevista de presentación del PIP final.
Se exime, pues, de la presentación de todas las medidas propuestas para la evaluación de las garantías sanitarias de medicamento, así como de las medidas tecnológicas de adaptación del medicamento a las necesidades de la población pediátrica.
Cuando el laboratorio solicitante de una AC prevea no poder completar el PIP, deberá emitir informe motivado a la EMA, organismo que deberá emitir resolución motivada de aceptación o denegación de la solicitud formulada en un plazo de veinte días.
8.2.b. Validación (art. 76 de la PR)
El PIP se debe presentar a la EMA antes del inicio de los ensayos clínicos. La EMA deberá validar la solicitud en un plazo de treinta días tras la recepción de la solicitud, prorrogable a sesenta días si realice un requerimiento documental al solicitante.
8.2.c. Autorización (art. 77 de la PR)
Una vez validado el PIP, la EMA deberá aprobar el plan inicial en un plazo de noventa días con carácter general, o de setenta días para planes abreviados. Las actualizaciones de los planes serán autorizados en un plazo máximo de treinta días; si en ese plazo no se ha emitido resolución expresa, la actualización se dará por autorizada. El PIP final derivado de un PIP abreviado deberá ser aprobado en un plazo máximo de sesenta días.
8.2.d. Prórrogas (arts. 81 y 82 de la PR)
Con el fin de garantizar que la investigación se lleve a cabo sólo cuando sea segura y ética, el solicitante puede presentar, mediante informe motivado y basado en motivos científicos, técnicos, o en la protección de la salud pública, la solicitud de prórroga al inicio o la finalización de las medidas previstas en el PIP en el momento de la presentación de dicho plan. Las prórrogas se acordarán cuando sea necesario realizar estudios en adultos en primer lugar, o cuando los estudios en la población pediátrica se prolonguen más en el tiempo. La duración de la prórroga de las medidas previstas en el PIP se determinará, caso por caso, por la EMA y tendrán una duración máxima de cinco años. Si la EMA acuerda una prórroga inicial de cinco años, el solicitante no podrá acogerse a una extensión.
El solicitante podrá presentar la solicitud de extensión de prórroga del PIP, mediante informe motivado, seis meses antes de la expiración de la prórroga y la EMA resolverá en un plazo máximo de sesenta días o de ciento veinte días si realiza un requerimiento documental al solicitante.
8.2.e. Modificación (arts. 84 y 85 de la PR)
Se permitirá modificar el PIP cuando el solicitante encuentre severas dificultades en su implementación. Así, si el solicitante considera que el plan resulta inviable o inapropiado, este podrá presentar las siguientes solicitudes: (i) modificación; (ii) prórroga; y (iii) dispensa. La EMA deberá resolver cualquiera de tales solicitudes en un plazo máximo de noventa días.
Se habilita a la EMA instar al solicitante a que proponga cambios en el PIP, que deberán ser ejecutados en un plazo de sesenta días. La EMA revisará dichos cambios y adoptará una decisión final de aceptación o denegación en un plazo de 30 días, o bien, podrá solicitar modificaciones adicionales a los cambios presentados. En estos casos, la EMA podrá adoptar una decisión definitiva en un plazo no superior a sesenta días.
La EMA elaborará disposiciones detalladas sobre el formato y el contenido que deberán seguir las solicitudes de acuerdo o modificación de un PIP, las solicitudes de dispensa y de aplazamiento.
La EMA previa consulta a la CE, a los EM y a las partes interesadas, elaborará los detalles del contenido de una solicitud, modificación, dispensa y prórroga del PIP.
8.2.f. Cumplimiento (art. 86 de la PR)
Para asegurar que los datos que respaldan la AC relativa a un medicamento para uso pediátrico se han desarrollado correctamente, el CHMP deberá comprobar el cumplimiento del PIP, así como cualquier exención y prórroga de este.
8.2.g. Adopción de decisiones regulatorias (art. 87 de la PR)
La EMA podrá consultar al CHMP o a sus grupos de trabajo para emitir su dictamen, el cual será puesto a disposición del público.
8.2.h. Suspensión (art. 88 de la PR)
Los solicitantes se ven obligados a notificar a la EMA su intención de suspender la realización del PIP, mediante informe motivado en un plazo no inferior de seis meses antes de tal suspensión. El informe será puesto a disposición del público.
8.2.i. Datos obtenidos (art. 90 de la PR)
Una vez concedida la AC del medicamento de uso humano, los datos derivados del PIP deberán reflejarse en la ficha técnica y en el prospecto: (i) los resultados de los estudios clínicos realizados de conformidad con el PIP, y (ii) las dispensas.
La CE incluirá en la AC una declaración que indique el debido cumplimiento de la solicitud con el PIP autorizado cuando corresponda.
8.3. Dispensas
8.3.a. Introducción (art. 75 de la PR)
Se mantienen las dispensas relativas a la presentación de los estudios y la información recogida de acuerdo con el PIP aprobado en las solicitudes de AC en las que se aprecie: (i) que es probable que el medicamento pueda ser nocivo, inseguro o ineficaz para una parte o la totalidad de la población pediátrica; (ii) que es probable que el medicamento no represente una ventaja terapéutica significativa respecto a otros tratamientos existentes para pacientes pediátricos; y, (iii) que la enfermedad o condición para la cual está destinado el medicamento sólo afecta a la población adulta.
8.3.b. Validación de la solicitud (art. 78 de la PR)
Las dispensas, se deben presentar a la EMA. La EMA deberá validar la solicitud en un plazo de noventa días tras la recepción de la solicitud, prorrogable a un máximo de ciento ochenta días si realice un requerimiento documental al solicitante. También podrá revisar, de oficio, en cualquier momento, las decisiones de las dispensas concedidas.
8.3.c. Registro de dispensas concedidas (art. 79 de la PR)
Para garantizar que la investigación en la población pediátrica sólo se lleve a cabo para satisfacer sus necesidades terapéuticas, la EMA mantendrá un listado público de las dispensas otorgadas, que será actualizado periódicamente.
8.3.d. Concesión de dispensas (arts. 80 y 83 de la PR)
Se regula, con carácter general, el procedimiento de concesión de dispensas tras una decisión negativa sobre un PIP. Y, en particular, las dispensas durante las situaciones de emergencia sanitaria. Durante las emergencias sanitarias se considera oportuno no retrasar la autorización precoz de un medicamento destinado al tratamiento o la prevención de una afección relacionada con la emergencia de salud pública. Por ello, se permitirá excluir temporalmente el cumplimiento de los requisitos relativos a los estudios pediátricos para otorgar una AC.
Para los medicamentos no autorizados previamente en la UE, se requiere que en la AC se incorpore una decisión de la EMA. Pues bien, esa decisión sólo se aplicará a los medicamentos destinados al diagnóstico, prevención o tratamiento de una enfermedad grave o potencialmente mortal o condición que estén directamente relacionados con la salud pública.
La decisión de la EMA incluirá los motivos para conceder dicha dispensa, así como la duración de esta. Antes de la expiración de la dispensa, el solicitante deberá presentar a la EMA un PIP o una solicitud de dispensa conforme al procedimiento habilitado.
8.4. Asesoramiento científico. Apoyo financiero (arts. 89 y 97 de la PR)
Aquellas personas físicas o jurídicas (empresas) que desarrollen un medicamento de uso humano destinado a uso pediátrico o al uso intrauterino podrá solicitar asesoramiento preliminar a la EMA con relación al diseño y la realización de los estudios y ensayos necesarios para demostrar la calidad, seguridad y eficacia del medicamento en la población pediátrica.
8.5. Autorizaciones de comercialización
8.5.a. Modificación de autorizaciones de comercialización previamente otorgadas (art. 91 de la PR)
Todos los ensayos clínicos que impliquen el uso de un medicamento autorizado en la población pediátrica serán presentados a la EMA o a los EM que hayan autorizado el medicamento en un plazo máximo de seis meses tras la finalización de los estudios. Esta obligación deberá cumplirse con independencia de que el TAC tenga la intención de solicitar una AC de una indicación pediátrica. Según la información obtenida, la CE podrá actualizar la ficha técnica y en el prospecto, y en su caso, modificar la AC.
8.5.b. Concesión (art. 92 de la PR)
Las solicitudes de AC de medicamentos para uso pediátrico contendrán los datos y documentos necesarios para establecer la calidad, eficacia y seguridad del medicamento en la población pediátrica, incluyendo los datos para respaldar la formulación adecuada, forma farmacéutica, concentración, vía de administración y el dispositivo de administración, de acuerdo con el PIP aprobado, el cual también deberá incluirse en la solicitud.
Las solicitudes de AC de medicamentos para uso pediátrico deberán incluir la presentación de datos relativos al uso del medicamento en esa población, que serán recopilados de acuerdo con un PIP aprobado.
El medicamento para uso pediátrico podrá tener la misma denominación que el medicamento que contenga el mismo principio activo y sea comercializado por el mismo TAC del medicamento autorizado en adultos. Es por ello, por lo que también se habilita a que los medicamentos para uso pediátrico también puedan tener indicaciones terapéuticas en adultos.
8.6. Ensayos clínicos pediátricos (art. 94 de la PR)
Para aumentar la transparencia, la base de datos de la UE sobre ensayos clínicos incluirá ensayos clínicos realizados en terceros países.
El resumen de los resultados de todos los ensayos clínicos pediátricos incluidos en la base de datos europea de datos clínico deberá estar disponible en un plazo de seis meses tras la finalización de los ensayos clínicos, salvo que no sea posible por razones debidamente justificadas.
8.7. Red europea (art. 95 de la PR)
Se establecerá una regulación de la red europea de representantes de pacientes, académicos, desarrolladores de medicamentos, investigadores y centros con experiencia en la realización de ensayos en la población pediátrica. Tal red será desarrollada por la EMA, cuyos objetivos se explicitan.
8.8. Informe anual (art. 98 de la PR)
La EMA deberá publicar un informe anual sobre las actuaciones relacionadas con el Capítulo VII de la Propuesta del Reglamento, regulador de los medicamentos para uso pediátrico.
9. ESPACIOS CONTROLADOS DE PRUEBAS
El ‘sandbox’ regulatorio es espacio controlado de pruebas en el cual es posible desarrollar, validar y probar en un entorno controlado soluciones normativas innovadoras o adaptadas que facilitan el desarrollo y la AC de medicamentos innovadores que probablemente entren en el ámbito de aplicación de la PR, de conformidad con un plan específico y por un tiempo limitado bajo supervisión de las autoridades regulatorias.
Los espacios controlados de pruebas permiten: (i) avanzar la regulación a través del aprendizaje normativo proactivo; (ii) mejorar el conocimiento normativo y encontrar los mejores medios para regular las innovaciones basadas en datos del mundo real(46), especialmente en una etapa muy temprana en el desarrollo de un medicamento de uso humano, lo cual puede ser relevante en un contexto de digitalización, incertidumbre y tecnologías disruptivas.
La CE, por recomendación de la EMA, podrá establecer un espacio controlado de pruebas de conformidad con un plan de espacio controlado de pruebas específico.
El espacio controlado de pruebas establecerá un marco normativo, incluyendo los requisitos científicos para el desarrollo, y en su caso, la ejecución de ensayos clínicos y la comercialización del medicamento. Estos ‘sandbox’ regulatorios estarán bajo el control de las autoridades competentes de los EM afectados.
La EMA se responsabilizará de supervisar a los medicamentos emergentes y de vanguardia. Podrá solicitar información y datos, y establecer debates preliminares con las partes interesadas.
La EMA podrá recomendar a la CE la creación de un espacio controlado de pruebas para medicamentos innovadores, para los que se prevé que entren dentro del ámbito de aplicación del presente Reglamento. Se establece una única excepción a lo anteriormente indicado, concretamente que el programa de desarrollo del medicamento se encuentre en fases avanzadas.
La decisión de la creación del espacio controlado de pruebas corresponde a la CE, por recomendación de la EMA. El espacio controlado de pruebas tendrá una duración limitada en el tiempo. En la decisión de su creación, la CE deberá indicar, entre otros datos, una fecha de vencimiento y las condiciones detalladas para su aplicación.
La CE podrá suspender o revocar un espacio controlado de pruebas en cualquier momento en determinados supuestos. También podrá: (i) prorrogar la duración de un ‘sandbox’ regulatorio; (ii) modificar las condiciones de la autorización de un espacio controlado de pruebas, por recomendación de la EMA, cuando se identifiquen riesgos para la salud pública que puedan ser mitigados.
9.1. Productos desarrollados bajo un espacio controlado de pruebas (art. 114 de la PR)
Los EM deben tener en cuenta el plan de un espacio controlado de pruebas antes de autorizar la realización de un ensayo clínico con productos candidatos a medicamento de uso humano afectados por el mismo.
Un medicamento de uso humano desarrollado bajo un espacio controlado de pruebas sólo podrá autorizarse por el procedimiento centralizado de registro. La AC mantendrá la misma vigencia que el ‘sandbox’ regulatorio, aunque podrá solicitarse prórroga de la AC por parte del TAC.
Los medicamentos desarrollados bajo un espacio controlado de pruebas indicarán claramente esta circunstancia en la ficha técnica y en el prospecto.
La AC de un medicamento desarrollado bajo un espacio controlado de pruebas podrá incluir excepciones a la regulación contenida en la legislación farmacéutica. Las excepciones serán excepcionales, proporcionales y justificadas y tendrán por objeto alcanzar los objetivos perseguidos en las condiciones de la AC.
La CE podrá suspender y/o revocar la AC del medicamento cuando el espacio controlado de pruebas regulatorio haya sido suspendido o revocado.
La CE deberá modificar urgentemente la AC del medicamento de uso humano con motivo de las medidas de mitigación de riesgos.
9.2. Disposiciones generales (art. 115 de la PR)
Los espacios controlados de pruebas no afectarán a las facultades de supervisión y control de las autoridades competentes. Por tanto, si se identificasen riesgos asociados con estos productos, las autoridades competentes: (i) adoptarán, con carácter inmediato, medidas cautelares de mitigación de riesgos, al tiempo que informan a la CE; (ii) se podrá suspender cautelarmente el desarrollo del medicamento en casos excepcionales.
Se mantiene el régimen general de responsabilidad civil en el desarrollo del producto(47).
Se deberán establecer las modalidades y las condiciones de funcionamiento de los espacios controlados de pruebas. Entre otros aspectos, se incorporarán los criterios de admisión y el procedimiento de la solicitud, selección, participación y retirara del ‘sandbox’ regulatorio, y los derechos y las obligaciones de las partes implicadas.
La EMA recopilará información de los EM, y presentará informes a la CE sobre los resultados de la implementación de los espacios controlados de pruebas.
10. FABRICACIÓN
El art. 148 de la PD detalla el procedimiento de registro del listado de todos los centros descentralizados de fabricación del titular de la autorización de fabricación. Con el fin de dar un marco legal a los nuevos avances científicos y la transformación tecnológica, se habilita así un marco legal flexible que permite la fabricación descentralizada o personalizada de determinados medicamentos(48), basándose en un sistema de gestión de riesgos y bajo la responsabilidad de una persona cualificada de un local central autorizado.
Los sitios descentralizados, por tanto, tan solo requieren ser registrados por la autoridad regulatoria competente del Estado miembro. A diferencia de los locales centralizados, estos no requieren una autorización de fabricación.
11. DISCUSIÓN
El legislador ha optado acertadamente por mantener la regulación de los medicamentos de uso humano autorizados con mayor grado de innovación a través de una disposición normativa de rango de Reglamento, el cual será directamente aplicable en todos los EM y será obligatorio en todos sus elementos, de conformidad con el art. 288 del Tratado de Funcionamiento de la UE.
En cuanto a los nuevos períodos de protección regulatoria, parece oportuno reseñar que la reforma no afecta al sistema de protección de la propiedad intelectual de la UE (patentes, marcas, derechos de autor, CCP). En la UE se mantiene el clásico sistema de incentivos de propiedad intelectual(49), sin perjuicio de la recentísima entrada en vigor de la regulación de la patente unitaria en numerosos EM, aunque no en España(50–52). Sin embargo, en nuestra opinión es necesaria una reforma de la regulación del CCP contenida en el Reglamento (UE) N.º 2019/933, del Parlamento Europeo y del Consejo, de 20 de mayo(53), habida cuenta de la evidente aplicación fragmentada de las medidas contenidas en el mismo.
Los incentivos para la reutilización y el reposicionamiento de medicamentos existentes facilitarán a los pacientes un acceso más rápido a las terapias que satisfagan sus necesidades sanitarias en nuevas indicaciones terapéuticas, posiblemente en enfermedades olvidadas o en patologías que cuentan con un reducido arsenal terapéutico.
La reforma podrá ampliar, con respecto al modelo actual, en determinadas circunstancias los plazos de protección de datos de los medicamentos de referencia. Hasta la fecha, se habilita un modelo fijo en el que la protección de datos de medicamentos de uso humano tiene una duración de ocho años y la exclusividad comercial, en dos años adicionales (ampliable a tres). La PD establece un período de protección de datos general de seis años, lo cual ha sido objeto de crítica por parte de ciertas patronales, al suponer una merma de dos años con respecto a la regulación actual(54). Este plazo podrá ser prorrogado bajo diferentes circunstancias y variables, hasta cuatro años adicionales, y un período de exclusividad comercial de dos años. El futuro periodo total de protección oscilará entre los ocho y los doce años, frente al período máximo de once años establecido actualmente.
De esta manera, el marco regulatorio fomenta la comercialización de medicamentos innovadores, y facilitaría el acceso a los mismos. Se prevé que la protección reglamentaria de doce años aumente el acceso a los medicamentos en un quince por ciento o, dicho con otras palabras, que sesenta y siete millones de personas más en la UE podrían beneficiarse potencialmente de un nuevo medicamento(23).
Con respecto a las necesidades médicas no cubiertas, se establecen medidas para fomentar el desarrollo de medicamentos para ampliar el arsenal terapéutico, y muy concretamente en el ámbito de los antimicrobianos prioritarios. Los medicamentos podrán ver ampliada la exclusividad de mercado de uno de sus por un período de un año, ya sean por el laboratorio que desarrolle el antibiótico, o bien, por parte de un tercero –el laboratorio tendría libertad de elección para escoger el medicamento que se beneficiaría del incentivo, o bien, puede transferir el cupón a un tercero–. Estos efectos impactarían a un máximo de diez medicamentos, o bien, durante un plazo de quince años tras la entrada en vigor del nuevo paquete farmacéutico.
En cuanto a las AC, la digitalización de los expedientes de registro de medicamentos de uso humano facilitará la integración de plataformas y sistemas regulatorios en toda la UE, y facilitará la reutilización de datos.
La presentación de las solicitudes de AC por medios electrónicos por parte de la industria farmacéutica generará un ahorro de los costes para todos los operadores del sector.
Se simplifican los procedimientos de registro de medicamentos de uso humano, reduciéndose los plazos para la obtención de una AC. En consecuencia, se aumenta la eficiencia de las evaluaciones de AC por parte de las autoridades regulatorias, disminuyendo el time to market, disminución de los costes para todos los laboratorios farmacéuticos (por ejemplo, al no tener que devengarse la tasa para la renovación de las AC), y facilitando la accesibilidad de los medicamentos.
Ya en 2002 el profesor VALVERDE LÓPEZ criticaba la inexistencia de un mercado común o único de los medicamentos en la UE, dado su impacto negativo sobre la protección de la salud de los pacientes(55). Conviene poner de manifiesto que las diferencias en el acceso a los medicamentos innovadores entre los diferentes EM no se deben a la evaluación previa a la concesión de una AC, al menos no en el procedimiento centralizado de registro.
Siguiendo a ALBA ROMERO, el procedimiento centralizado de registro de medicamentos presenta, fundamentalmente, tres ventajas: (i) la puesta a disposición del medicamento para todos los ciudadanos de la UE; (ii) la optimización en el uso empleo de las competencias especializadas de los evaluadores, y (iii) la garantía de que las industrias, debiendo adecuarse a normas más severas clínicas y preclínicas, presentarán AC de medicamentos con carácter innovador, real y ampliamente probado(3). La agilidad se verá incrementada con la nueva reforma normativa. Por tanto, son las decisiones de fijación del precio de venta y a la financiación por parte de los sistemas sanitarios públicos de los medicamentos autorizados –competencias reservadas a los EM(56)– las que realmente limitan el acceso a medicamentos novedosos, y muy especialmente a los medicamentos huérfanos en España, como ha puesto en relieve un reciente análisis ejecutado por CUÉLLAR RODRÍGUEZ(57).
Asimismo, la nueva estructura de la EMA, en la que se reducen el número de comités científicos, y se amplían las funciones de estos: (i) aumentará la capacidad científica de la red europea de regulación, y (ii) liberará recursos científicos para reforzar el apoyo previo a la AC destinado a los desarrolladores de medicamentos de uso humano.
Se habilitan nuevos procedimientos regulatorios para hacer frente a las emergencias sanitarias, y el acceso a terapias prometedoras por parte de los pacientes. De esta manera, se mejora el acceso a terapias innovadoras por parte de todos los pacientes de la UE.
Se proporcionará mayor asesoramiento científico a los futuros solicitantes de una AC de medicamentos prioritarios –por ejemplo, antibióticos y para el reposicionamiento de medicamentos–. El apoyo regulatorio alcanzará tanto a PYMEs como a entidades sin ánimo de lucro.
El asesoramiento científico de la EMA mejorará la calidad de las solicitudes de AC de los medicamentos innovadores, puesto que podrá prestarse años antes de solicitar una AC. De esta manera, se facilitará la toma de decisiones por parte de los laboratorios farmacéuticos. Se fomentará la comercialización de nuevos medicamentos prioritarios y de gran relevancia para la salud pública o que aborden necesidades médicas insatisfechas, así como nuevos medicamentos de terapias avanzadas.
En relación con los medicamentos huérfanos, actualmente estos gozan de un periodo de exclusividad comercial de diez años de duración. Aunque un sector doctrinal ha sugerido el mantenimiento de estos incentivos(58), la futura norma establece un plazo general del orden de nueve años, pero estos podrán ascender a un máximo de trece años. Por tanto, se podría aumentar hasta en tres años el período de exclusividad comercial de los medicamentos huérfanos. Salvo para los medicamentos de referencia que hayan sido AC por un procedimiento simplificado basado en datos bibliográficos, los medicamentos huérfanos disfrutarán de un aumento en período de exclusividad comercial de hasta tres años con respecto a la regulación vigente actualmente.
Gracias a la nueva regulación de los medicamentos para uso pediátrico, se prevé el desarrollo de un mayor número de medicamentos –al facilitarse un retorno de la inversión derivada de la investigación del medicamento en niños–, en particular en áreas de necesidades infantiles no cubiertas, y que su acceso al mercado sea más ágil.
El concepto de experimento jurídico no resulta novedoso desde el punto de vista doctrinal(59). Incluso algunas normas jurídicas del ordenamiento jurídico español ya han previsto la incorporación de ‘sandbox’ regulatorios en relación con los sistemas financieros(60). No obstante, la incorporación de los espacios controlados de pruebas en la legislación farmacéutica de la UE constituye un hito importante, toda vez que: (i) facilitará el uso de datos sanitarios del mundo real; (ii) incorporará un procedimiento regulatorio ágil para dar cabida a los avances científicos, la digitalización, la inteligencia artificial y los medicamentos de vanguardia; e (iii) impulsará la capacidad científica de la red reguladora de la UE.
Las disposiciones regulatorias analizadas podrán ser objeto de modificación a lo largo del complejo y dilatado proceso de tramitación normativa, pues se prevé un buen número de enmiendas en la discusión tripartida entre la CE, el Consejo de la UE y el Parlamento Europeo(61,62).
12. CONCLUSIÓN
El abismal y ágil desarrollo científico-tecnológico acontecido en los últimos años, unido a las actuales preocupaciones políticas, sociales y económicas justifica plenamente la actualización de la legislación farmacéutica de la UE.
El paquete normativo propuesto recientemente por la CE plantea reformas de gran calado en la regulación de los medicamentos de uso humano, aunque mantiene la tradicional ratio legis de las normas precedentes: garantizar la calidad, eficacia y seguridad de los medicamentos disponibles comercialmente.
En relación con la innovación, la futura regulación: (i) respaldará la innovación, (ii) fomentará la innovación, la I+D, haciendo especial énfasis en las necesidades médicas no cubiertas y ofrecerá apoyo regulatorio a PYMEs y entidades sin ánimo de lucro, (iii) promoverá la producción de medicamentos in house, (iv) agilizará y simplificará los procedimientos regulatorios para la obtención de una AC de medicamentos de uso humano, (v) introducirá incentivos eficaces para la innovación, reforzando los períodos de protección de exclusividad de datos y de exclusividad comercial, (vi) mejorará el acceso a medicamentos innovadores por parte de los pacientes de todos los EM.
Lista de abreviaturas
AC: autorización de comercialización.
ADN: Ácido desoxirribonucleico.
art./arts.: artículo/artículos.
CCP: Certificado complementario de protección.
CE: Comisión Europea.
CHMP: Committee for Medicinal Products for Human Use (Comité de Medicamentos de Uso Humano).
EM: Estados miembros.
EMA: European Medicines Agency (Agencia Europea del Medicamento).
I+D: Investigación y desarrollo.
PD: Propuesta de Directiva.
PIP: Plan de investigación pediátrica.
PR: Propuesta de Reglamento.
PYMEs: Pequeñas y medianas empresas.
TAC: Titular de la autorización de comercialización.
UE: Unión Europea.
13. REFERENCIAS
- Cabezas López MD, Martín Martín C, López Andújar G. Evolución de la regulación del medicamento en la Unión Europea. En: Gomis Blanco A, Rodríguez Nozal R, editores. De la botica de El Escorial a la industria farmacéutica: en torno al medicamento. Alcalá de Henares: Servicio de Publicaciones de la Universidad de Alcalá de Henares; 2015. p. 255-77.
- Alba Romero S, Gutiérrez Pérez MV. Evolución del Estatuto Jurídico del medicamento en la Unión Europea hasta la actual codificación. Not Unión Eur. 2001;197:73-86.
- Alba Romero S. Farmacia y Unión Europea. Madrid; 1994. 307 p.
- Noguera Peña A, del Castillo Rodríguez C. Derecho Farmacéutico y Legislación Farmacéutica en España y en la Unión Europea: concepto, evolución y fuentes. An Real Acad Nac Farm. 2021;87(3):273-320.
- Fernández-Rañada [López-Doriga] J. La estrategia farmacéutica para Europa. Cuad Derecho Farm. 2021;79:17-24.
- Carrión García de Parada FJ, González Díaz M. Incentivos a fármacos pediátricos y huérfanos en la nueva Estrategia Farmacéutica Europea. Comun En Prop Ind Derecho Competencia. 2022;95:103-19.
- Comisión Europea. Unión Europea de la Salud: la Comisión propone una reforma de la legislación farmacéutica para lograr unos medicamentos más accesibles, asequibles e innovadores [Internet]. Comisión Europea. 2023 [citado 1 de mayo de 2023]. Disponible en: https://ec.europa.eu/commission/presscorner/detail/es/ip_23_1843
- Martín Quero A. Innovación en salud: back to basics. Cuad Derecho Farm. 2020;72:36-41.
- Valverde [López] JL. The pharmaceuticals industry in trouble. Pharm Policy Law. 2013;15:51-69.
- Noguera Peña A, Del Castillo Rodríguez C. Equilibrio entre la innovación y el gasto público sanitario. El caso particular de los medicamentos biosimilares. Rev Derecho Estado. 2021;48:273-96.
- Blanco Delgado VM, Noguera Peña A. Medicamentos huérfanos: reflexiones jurídicas sobre precios y financiación pública. En: Ribón Seisdedos E, editor. Anuario Jurídico Secciones del ICAM 2022. Madrid: Sepin; Ilustre Colegio de la Abogacía de Madrid; 2022. p. 211-21.
- Moreno-Tapia Rivas I. El acceso a terapias innovadoras: una visión global. Cuad Derecho Farm. 2017;62:6-11.
- Calvente Cestafe N. La mejora en el acceso de la innovación terapéutica. Cuad Derecho Farm. 2020;74:29-42.
- del Llano Núñez-Cortés A. La resistencia antimicrobiana y por qué debería preocuparnos. Una respuesta desde la Unión Europea. Derecho Salud. 2022;32(Extra 1):124-9.
- del Llano Núñez-Cortés A. El Derecho de la Salud Pública: una propuesta de mejora normativa a propósito de la Covid-19. Derecho Salud. 2021;31(2):102-31.
- Faus Santasusana J. Medicina de precisión y medicamentos de terapia avanzada. En: Recuerda Girela MÁ, editor. Tecnologías disruptivas: regulando el futuro. Cizur Menor: Aranzadi; 2019. p. 77-98.
- Martínez-Máñez R. La revolución de la nanomedicina. En: Valdés Castrillón B, editor. Nanotecnología: promesas, realidades y retos. Málaga: Instituto de Academias de Andalucía; 2021. p. 91-130.
- Ruiz Antúnez [S.]. Regulación de medicamentos de terapia avanzada en la Unión Europea. Pharmatech. 2019;41:44-50.
- Puig Hermida M, Losada Garijo C, Nveñe García M. Innovación en medicamentos biosimilares y su regulación. En: Recuerda Girela MÁ, editor. Tecnologías disruptivas: regulando el futuro. Cizur Menor: Aranzadi; 2019. p. 157-77.
- Antich Isern P, Aparicio Blanco J. La frontera con los productos sanitarios en el actual marco legislativo español. An Real Acad Nac Farm. 2022;88(2):209-34.
- Amarilla Mateu N, Lozano Arjona M. Productos frontera, productos milagro y la protección de la salud pública. Derecho Salud. 2016;26(Extra 1):237-46.
- Gail M. Nueva legislación farmacéutica de la UE: un «equilibrio» entre los intereses de los pacientes y los intereses de la industria [Internet]. El Global. 2023 [citado 3 de mayo de 2023].
- Disponible en: https://elglobal.es/politica/nueva-legislacion-farmaceutica-de-la-ue-equilibrio-entre-los-intereses-de-los-pacientes-y-los-intereses-de-la-industria/
[Editorial]. ¿En qué consiste la reforma farmacéutica europea? [Internet]. Diariofarma. 2023 [citado 2 de mayo de 2023]. Disponible en: https://diariofarma.com/2023/04/26/en-que-consiste-la-reforma-farmaceutica-europea - Tudor EC. La cláusula Bolar como excepción a los derechos conferidos por una patente farmacéutica en Europa. Rev Estud Eur. 2018;71:300-8.
- Montañá Mora M. La «cláusula Bolar» de la Directiva 2004/27 como nueva excepción a los derechos de patente a cambio de la armonización del período de exclusividad de datos a 10 años. Not Unión Eur. 2009;288:5-22.
- Golmayo Sebastián E, López Alzaga R. Uso experimental y «cláusula bolar»: Dos límites a los derechos de patente parcialmente inexplorados. Un análisis de los artículos 61.b) y c) de la Ley de Patentes. En: Anuario Jurídico Secciones del ICAM 2021. Madrid: Sepin; Ilustre Colegio de Abogados de Madrid; 2021. p. 225-38.
- Ridley DB, Calles Sánchez A. Introduction of European priority review vouchers to encourage development of new medicines for neglected diseases. The Lancet. 2010;376(9744):922-7.
- Bombillar Sáenz FM. Aspectos éticos y jurídicos de la investigación y comercialización de medicamentos para enfermedades olvidadas. En: Farmamundi, editor. Una reflexión sobre el comercio internacional, la propiedad intelectual y el derecho a la salud. Farmamundi. Huesca; 2015. p. 47-66.
- Serrano M. La sociedad civil y las enfermedades raras. Arbor Cienc Pensam Cult. 2018;194(789):a459.
- [Editorial]. La lucha contra la resistencia bacteriana. Farm Rev Cons Gen Col Of Farm. 2013;390:50-8.
- [Editorial]. La OMS publica la lista de las bacterias para las que se necesitan urgentemente nuevos antibióticos [Internet]. Organización Mundial de la Salud. 2017 [citado 13 de junio de 2023]. Disponible en: https://www.who.int/es/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed
- Estruch M. Nuevo portal europeo para el envío de dossiers de registro (CESP). Pharmatech. 2013;1:62-4.
- Hernández Vázquez V. Presente y futuro de los procedimientos de registro de medicamentos en la Unión Europea. Cuad Derecho Farm. 2002;3:14-23.
- Montpart Costa E, Martín Barea MP. Procedimiento de registro de mutuo reconocimiento de medicamentos de uso humano. Offarm Farm Soc. 2001;20(7):93-100.
- Valverde López JL. Autorización y registro de medicamentos en Europa. En: Manuel García J, editor. Curso básico de derecho farmacéutico: 100 cuestiones esenciales. 3a ed. Madrid: Instituto de Legislación Farmacéutica; 2014. p. 212-22.
- Gardner JS. The European Agency for the Evaluation of Medicines and European Regulation of Pharmaceuticals. Eur Law J. 1996;2(1):48-82.
- Moreno Tapia [Rivas] I, Sabater J. La autorización condicional de medicamentos en la Unión Europea. Cuad Derecho Farm. 2006;17:21-4.
- Ruiz Sayritupac de Nué ME, González Leonor M del C. Procedimiento de autorización de las vacunas contra la COVID-19: Estados Unidos de América, Unión Europea y América Latina. Ars Pharm. 2022;64(1):28-52.
- Cabezas López MD, García López JA. Lecciones de legislación farmacéutica española. s.l.: Avicam; 2021. 352 p.
- Bombillar Sáenz FM. Acceso al medicamento y derecho a la protección de la salud: régimen jurídico de los medicamentos huérfanos en la Unión Europea. Cad Ibero-Am Direito Sanitário. 2014;3(3):123-48.
- Llopart Baker M. Medicamentos huérfanos: medidas de fomento de la investigación y desarrollo. Situación actual en la Unión Europea. Cuad Derecho Farm. 2018;66:58-63.
- Barranco Vela R. El estatuto jurídico de los medicamentos huérfanos en la Unión Europea: El derecho a la salud de los pacientes con enfermedades raras. En: Barranco Vela R, Bombillar
- Sáenz FM, editores. El acceso al medicamento: retos jurídicos actuales, intervención pública y su vinculación al derecho a la salud. Granada: Comares; 2010. p. 107-36.
- Bouchard C. Medicamentos pediátricos, una necesidad todavía no resuelta en Europa. Cuad Derecho Farm. 2004;11:10-3.
- Díaz de Escauriaza B. Novedades introducidas en materia de medicamentos pediátricos en virtud del Reglamento 1901/2006, de 12 de diciembre de 2006, sobre medicamentos de uso pediátrico. Cuad Derecho Farm. 2007;20:6-9.
- Valverde [López] JL, Ceci A. The Regulation of Paediatric Medicines in the EU. Pharm Policy Law. 2009;11:1-2.
- Martínez de Villarreal J. Bioinformática: El reto de los datos. Pharmatech. 2019;46:68-71.
- Vázquez Bulla C, Moretón Sanzarmiento MF. Responsabilidad civil y productos farmacéuticos. Madrid: Reus; 2020. 268 p.
- Noguera Peña A, del Castillo Rodríguez C. Medicamentos de elaboración o preparación no industrial: una nueva propuesta de clasificación. Ars Pharm. 2021;62(4):379-88.
- Carrión [García de Parada] FJ, Buitrón Patuel E. El Convenio de la Patente Europea: 40 años desde la Conferencia Diplomática de Munich. Comun En Prop Ind Derecho Competencia. 2017;80:65-77.
- Carbonell Vallès E, Manzanares Sanz R. Patente unitaria: qué cambia, qué permanece y cómo afecta. Cuad Derecho Farm. 2022;82:4-15.
- Balañá Vicente S. Patente unitaria en el marco de la cooperación reforzada. Evolución del sistema europeo de patentes a propósito de la reciente creación de la patente europea con efecto unitario. Comun En Prop Ind Derecho Competencia. 2013;68:51-81.
- Desaunettes-Barbero L, de Visscher F, Strowel A, Cassiers V, editores. The Unitary Patent Package & Unified Patent Court: Problems, Possible Improvements and Alternatives. s.l.: Ledizioni; 2023. 639 p.
- Carrión García de Parada FJ, Buitrón Patuel E. La reforma de Reglamento CCP y la excepción de fabricación para la exportación. Estado de la cuestión. Cuad Derecho Farm. 2019;68:6-17.
- [Editorial]. Farmaindustria se une a la Efpia en sus críticas al paquete farmacéutico [Internet]. Diariofarma. 2023 [citado 2 de mayo de 2023]. Disponible en: https://diariofarma.com/2023/04/27/farmaindustria-se-une-a-la-efpia-en-sus-criticas-a-la-legislacion-farmaceutica
- Valverde López JL. El estatuto jurídico del medicamento en la Unión Europea. En: Villanueva Cañadas E, editor. España y Europa, hacia un ideal sanitario común: recopilación comentada de textos comunitarios y nacionales en materia de sanidad y salud pública. Madrid: Ministerio de Sanidad y Consumo; 2002. p. 85-176.
- Llopart Vidal M. Acceso al mercado. Análisis de las sucesivas evaluaciones de medicamentos en el marco del procedimiento de financiación y precio. Cuad Derecho Farm. 2023;4-17.
- Cuéllar Rodríguez S. Retraso en la comercialización en España de los medicamentos novedosos autorizados por la Unión Europea. Panor Actual Medicam. 2023;47(463):477-83.
- Faus Santasusana J. Los incentivos en favor de los medicamentos huérfanos. Cuad Derecho Farm. 2020;72:42-52.
- Doménech Pascual G. Los experimentos jurídicos. Rev Adm Pública. 2004;164:145-87.
- Jarne Muñoz P. El espacio controlado de pruebas (regulatory sandbox) en el Anteproyecto de Ley de medidas para la transformación digital del sistema financiero. Actual Civ. 2018;11.
Gonzalo C. A propósito de la propuesta de revisión de la legislación farmacéutica europea [Internet]. El Global. 2023 [citado 8 de mayo de 2023]. Disponible en: https://elglobal.es/opinion/tribunas/a-proposito-de-la-propuesta-de-revision-de-la-legislacion-farmaceutica-europea/ - Faus [Santasusana] J. El futuro del medicamento en Europa, primeras reflexiones. Cápsulas Bol Inf Juríd. 2023;238:1-4.
