Fármacos de nueva aprobación – Newly approved drugs:
(A) TRACTO ALIMENTARIO Y METABOLISMO: Esteatosis hepática no alcohólica: Resmetirom (Rezdiffra®; FDA). (C) SISTEMA CARDIOVASCULAR: Hipertensión arterial: Aprocitentan (Tryvio®; FDA). Hipertensión arterial pulmonar: Sotatercept (Winrevair®; FDA). (D) DERMATOLOGÍA: Molusco contagioso: Berdazimero (Zelsuvmi®; FDA). (J) ANTIINFECCIOSOS SISTÉMICOS: Infección urinaria complicada: Cefepima/Enmetazobactam (Exblifep®; EMA/FDA). COVID-19: Pemivibart (Pemgarda®; FDA). (L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES: Carcinoma urotelial: Erdafitinib (Barlversa®; FDA). Melanoma: Lifileucel (Amtagvi®; FDA). (M) SISTEMA MÚSCULO-ESQUELÉTICO: Distrofia muscular de Duchenne: Givinostat (Duvyzat®; FDA).
(A) TRACTO ALIMENTARIO Y METABOLISMO
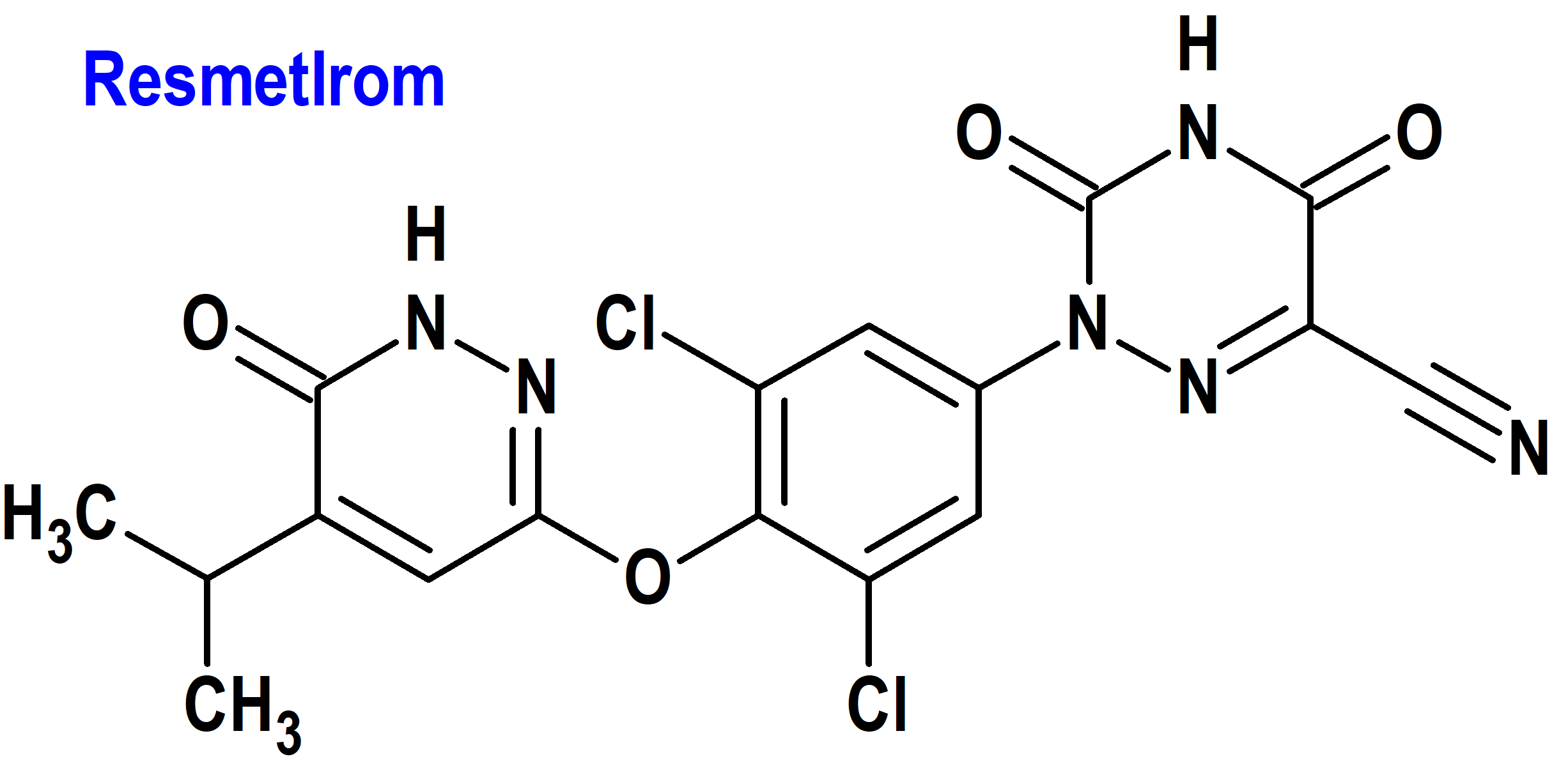
Resmetirom (Rezdiffra®) Madrigal (FDA, USA)
Indicación: Tratamiento, junto con dieta y ejercicio, de adultos con esteatohepatitis no alcohólica no cirrótica (NASH) con fibrosis hepática de moderada a avanzada (consistente con los estadios de fibrosis F2 a F3).
Tipo: Medicamento sintético estándar, constituido por 2-[3,5-dicloro-4-((6-oxo-5-(propan-2-il)-1,6-dihidropiridazin-3-il)oxi)fenil]-3,5-dioxo-2,3,4,5-tetrahidro-1,2,4-triazina-6-carbonitrilo. Autorizado en Estados Unidos (FDA) el 14-3-2024 por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Agonista parcial del receptor beta de la hormona tiroidea (THR-β). Resmetirom produce un 89% de la respuesta máxima en comparación con la triyodotironina (T3) en un ensayo funcional in vitro para la activación de THR-β. El mismo ensayo funcional para el agonismo del receptor alfa de la hormona tiroidea (THR-a) mostró una eficacia del 49% para el resmetirom en relación con T3. THR-β es la forma principal de THR en el hígado, y la estimulación de THR-β en el hígado reduce los triglicéridos intrahepáticos, mientras que las acciones de la hormona tiroidea fuera del hígado, incluso en el corazón y hueso, están mediados en gran medida a través de THR-α.
Eficacia clínica: Un ensayo aleatorizado, doble ciego y controlado con placebo de 54 meses de duración. En el ensayo, 888 sujetos fueron asignados aleatoriamente para recibir uno de los siguientes: placebo (294), 80 mg de resmetirom (298) o 100 mg (296) una vez al día, además de la atención estándar para NASH, que incluye asesoramiento sobre una dieta saludable y ejercicio. El criterio de valoración midió el grado de inflamación y cicatrización del hígado. A los 12 meses, las biopsias de hígado mostraron que una mayor proporción de sujetos que fueron tratados con resmetirom lograron una resolución de NASH o una mejora en la cicatrización del hígado en comparación con aquellos que recibieron el placebo. Un total del 26-27 % de los que recibieron 80 mg de resmetirom y el 24-36 % de los que recibieron 100 mg experimentaron una resolución de la esteatosis y ningún empeoramiento de las cicatrices hepáticas, en comparación con el 9-13 % de los que recibieron recibieron placebo (la variedad de respuestas refleja las lecturas de diferentes patólogos).
Eventos adversos: Los más comunes (>10%) son diarrea (23-33%) y náuseas (15-18%).
(C) SISTEMA CARDIOVASCULAR
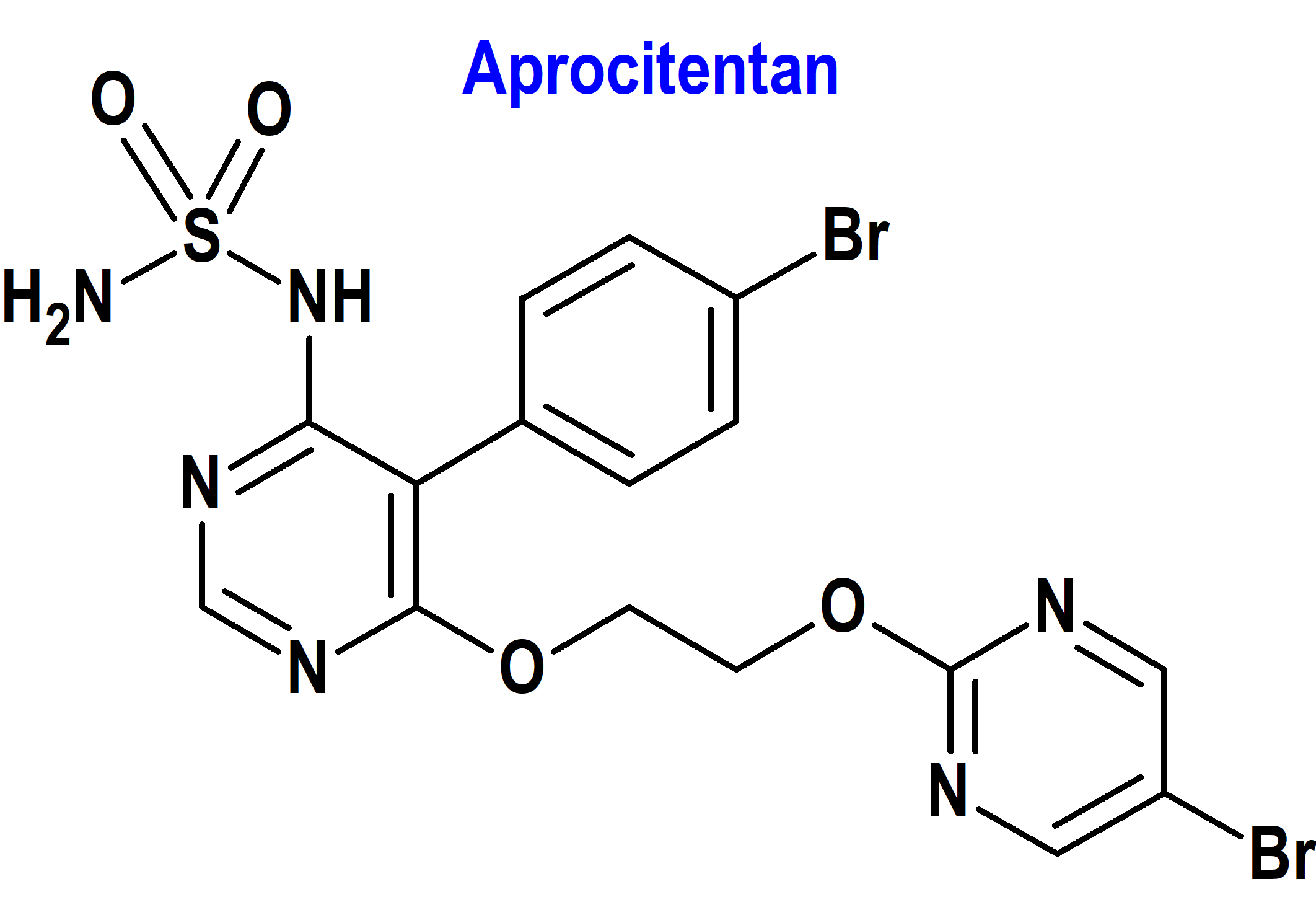
Aprocitentan (Tryvio®) Idorsia (FDA, USA)
Indicación: Tratamiento de la hipertensión en combinación con otros fármacos antihipertensivos, para reducir la presión arterial en pacientes adultos que no están adecuadamente controlados con otros fármacos.
Tipo: Medicamento sintético estándar constituido por N-[5-(4-bromofenil)-6-[2-[(5-bromo-2-pirimidinil)oxi]etoxi]-4-pirimidinil]-sulfamida. Autorizado en Estados Unidos (FDA) el 19-3-2024; no autorizado aún en la Unión Europea(EMA).
Mecanismo: Antagonista de los receptores de endotelina-1 (ET-1), inhibiendo la unión de ésta a los receptores ETA y ETB. ET-1, a través de sus receptores (ETA y ETB), media una variedad de efectos nocivos como vasoconstricción, fibrosis, proliferación celular e inflamación. En la hipertensión, la ET-1 puede causar disfunción endotelial, hipertrofia y remodelación vascular, activación simpática y aumento de la síntesis de aldosterona.
Eficacia clínica: Estudio multicéntrico de fase 3 de tres partes en adultos con presión arterial sistólica (PAS) ≥140 mmHg tratados con al menos tres medicamentos antihipertensivos. Después del período de preinclusión con placebo de 4 semanas, 730 pacientes fueron aleatorizados por igual para recibir 12,5 mg, 25 mg de aprocitentan o placebo una vez al día durante el período inicial de tratamiento doble ciego (DB) de 4 semanas (parte 1). Al final de las 4 semanas, todos los pacientes ingresaron al período de tratamiento simple ciego (parte 2), donde recibieron 25 mg de aprocitentan una vez al día durante 32 semanas. Al final de las 32 semanas, los pacientes fueron reasignados al azar para recibir 25 mg de aprocitentan o placebo, una vez al día, durante un período de retirada de DB de 12 semanas (parte 3). El criterio de valoración principal de eficacia fue el cambio en la PAS sentado (SiSBP) desde el inicio hasta la semana 4 durante la parte 1: -15,4 vs -11,6 mmHg en la presión sistólica y -10,4 vs. -6,4 en la diastólica. La persistencia del efecto antihipertensivo se demostró en la parte 3 del ensayo, en la que los pacientes que tomaban aprocitentan fueron realeatorizados para recibir placebo o 25 mg de aprocitentan después de un período durante el cual todos los pacientes fueron tratados con 25 mg. En los pacientes realeatorizados a placebo, la presión arterial sistólica media aumentó, mientras que en los pacientes realeatorizados a 25 mg de aprocitentan el efecto antihipertensivo se mantuvo y fue estadísticamente superior al placebo en la semana 40.
Eventos adversos: Los más comunes son edema/retención de líquidos (9%) y anemia (4%).
Sotatercept (Winrevair®) Merck Sharp Dohme (FDA, USA)
Indicación: Tratamiento de adultos con hipertensión arterial pulmonar (HAP, Grupo 1 de la Organización Mundial de la Salud, OMS) para aumentar la capacidad de ejercicio, mejorar la clase funcional (FC) de la OMS y reducir el riesgo de eventos de empeoramiento clínico.
Tipo: Medicamento biológico constituido por una proteína de fusión recombinante homodimérica que consta del dominio extracelular del receptor humano de activina tipo IIA (ActRIIA) unido al dominio Fc de IgG1 humana. Autorizado en Estados Unidos (FDA) el 26-3-2024 como medicamento huérfano (Orphan drug) y designado como terapia innovadora (Breakthrough Therapy); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Es un inhibidor de la señalización de la activina que se une a la activina A y otros ligandos de la superfamilia del TGF-b. Como resultado, sotatercept mejora el equilibrio entre la señalización proliferativa (mediada por ActRIIA/Smad2/3) y antiproliferativa (mediada por BMPRII/Smad1/5/8) para modular la proliferación vascular. En modelos animales redujo la inflamación e inhibió la proliferación de células endoteliales y del músculo liso en la vasculatura enferma. Estos cambios celulares se asocian con paredes vasculares más delgadas, reversión parcial de la remodelación del ventrículo derecho y mejora de la hemodinámica.
Eficacia clínica: Ensayo clínico doble ciego, controlado con placebo, multicéntrico y de grupos paralelos en el que 323 pacientes con HAP (Grupo 1 FC II o III de la OMS) que fueron aleatorizados 1:1 para recibir sotatercept o placebo, administrados por vía subcutánea una vez cada 3 semanas. El criterio de valoración principal de eficacia fue el cambio desde el valor inicial en la semana 24 en la distancia de caminata de 6 minutos (6 MWD). En el grupo sotatercept, la mediana de aumento ajustada con placebo en 6 MWD fue de 41 metros, produciendo una mejora con respecto al valor inicial de al menos 1 FC de la OMS en la semana 24. El tratamiento con sotatercept resultó en una reducción del 84 % en la aparición de muerte por cualquier causa o eventos de empeoramiento clínico de la HAP en comparación con el placebo, hasta que el último paciente completó la visita de la semana 24 (datos hasta el límite de datos; duración media de la exposición 33,6 semanas).
Eventos adversos: Los más comunes (≥10% en pacientes que recibieron sotatercept y 5% más que placebo) son dolor de cabeza, epistaxis, erupción cutánea, telangiectasia, diarrea, mareos y eritema.
(D) DERMATOLOGÍA
Berdazimero (Zelsuvmi®) LNHC (FDA, USA)
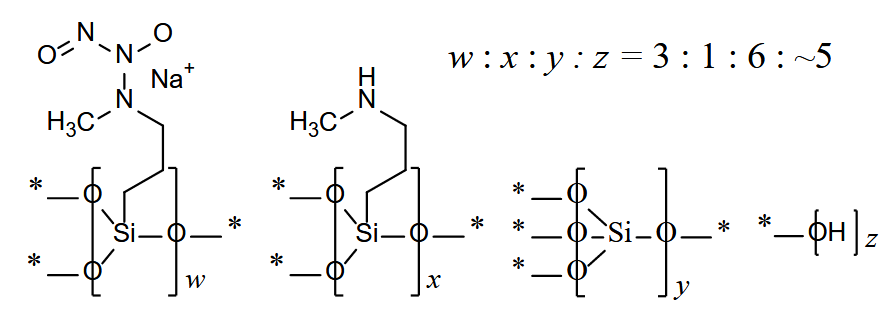
Indicación: Tratamiento tópico del molusco contagioso en adultos y pacientes pediátricos de 1 año de edad y mayores.
Tipo: Medicamento sintético estándar constituido por un polímero formado a partir de 1-hidroxi-3-metil-3-(3-(trimetoxisilil)propil)-1-triazen-2-óxido y silicato de tetraetilo, correspondiendo químicamente a poli[{[3-(metilamino)propil]silasesquioxano}-co-{[3-(1-metil-2-nitroso-2-oxidohidrazin-1-il)propil]silasesquioxano}-co-silicato (1:3:6 x)], parcialmente hidrolizado (Si: OH ~ 10: 5). Autorizado en Estados Unidos (FDA) el 5-1-2024; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Es un agente liberador de óxido nítrico. Se desconoce el mecanismo de acción para el tratamiento del molusco contagioso.
Eficacia clínica: 3 ensayos multicéntricos, aleatorizados, doble ciego, de grupos paralelos y controlados con placebo (vehículo) en un total de 1.598 sujetos con molusco contagioso. La variable primaria de eficacia fue la proporción de sujetos que lograron una eliminación completa las lesiones en la 12ª semana: 32,4 vs 19,7%; 30,0 vs 20,3% y 26 vs 22%.
Eventos adversos: Los más comunes son reacciones en el lugar de aplicación, que incluyen dolor (como sensación de ardor o escozor, 18,7%), eritema (11,7%), prurito (5,7%), exfoliación (5,0%), dermatitis (4,9%), hinchazón (3,5%), erosión ( 1,6%), decoloración (1,5%), vesículas (1,5%), irritación (1,2%) e infección (1,1%).
(J) ANTIINFECCIOSOS SISTÉMICOS
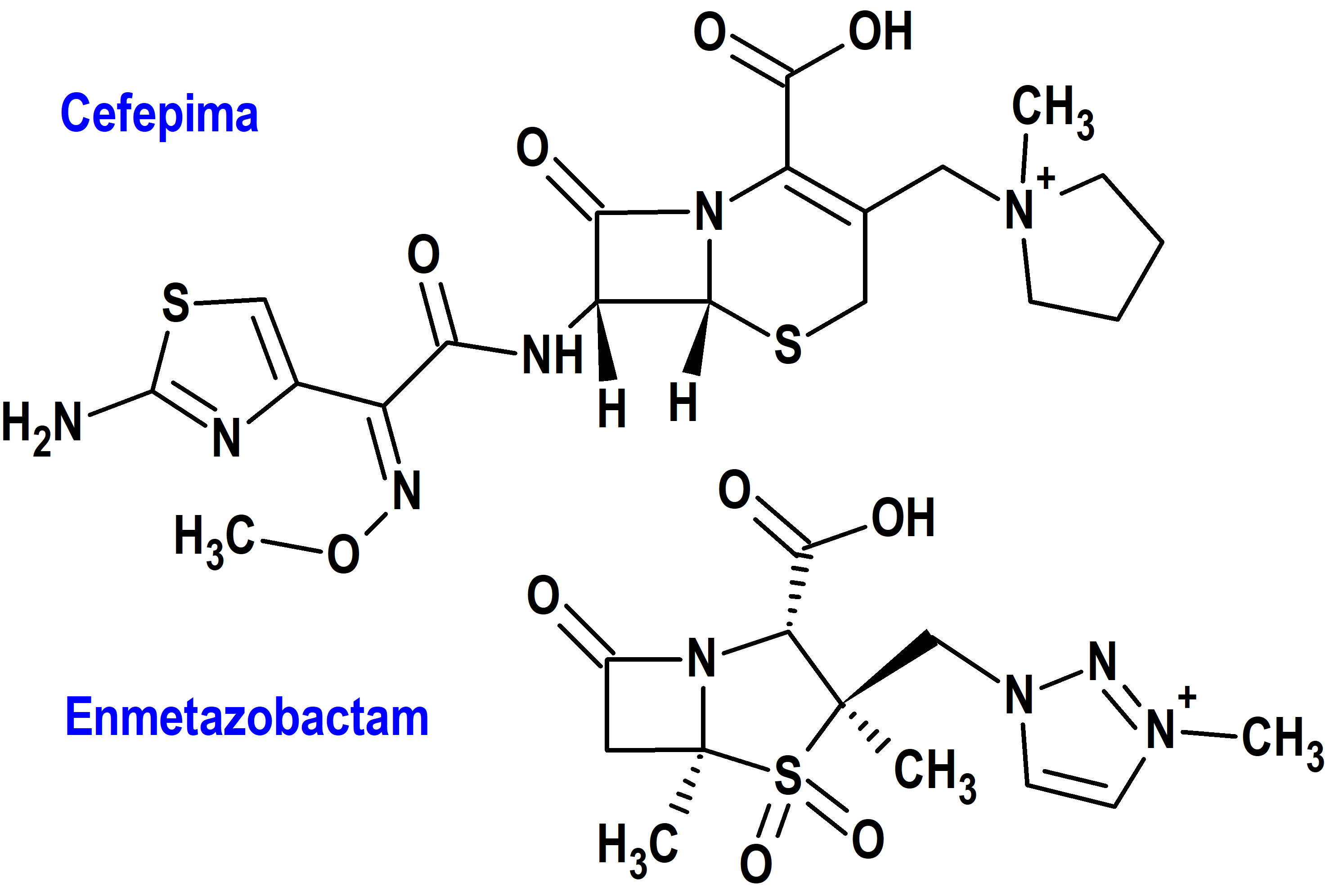
Cefepima/Enmetazobactam (Exblifep®) Allecra (FDA, USA) Advanz (EMA, UE)
Indicación: Tratamiento de pacientes de 18 años de edad y mayores con infecciones complicadas del tracto urinario, incluida pielonefritis, causadas por los siguientes microorganismos susceptibles: Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, Proteus mirabilis y complejo Enterobacter cloacae.
Tipo: Medicamento sintético estándar, constituido por una combinación de un antibiótico cefelosporínico previamente autorizado[cefepima: (6R,7R,Z)-7-(2-(2-aminotiazol-4-il)-2-(metoxiimino)acetamido)-3-((1-metilpirrolidinio-1-il)metil)8-oxo-5-tia-1-aza-biciclo[4.2.0]oct-2-eno-2-carboxilato] y un inhibidor de betalactamasasa [enmetazobactam: (2S,3S,5R)-3-metil-3-((3-metil-1H-1,2,3-triazol-3-io-1-il)metil)-7-oxo-4-tia-1-azabiciclo[3.2.0]heptano-2-carboxilato 4,4-dióxido]. Autorizado en Estados Unidos (FDA) el 22-2-2024 mediante revisión prioritaria (Priority Review); autorizado en la Unión Europea (CHMP/EMA) el 21-3-2024.
Mecanismo: Combinación de un antibacteriano betsalactámico del grupo de las cefalosporinas (cefepima) y un inhibidor de betalactamasas (enmetazobactam). La acción bactericida de la cefepima resulta de la inhibición de la síntesis de la pared celular. La cefepima penetra la pared celular de la mayoría de las bacterias grampositivas y gramnegativas para unirse a la proteína fijadora de penicilina (PBP). La cefepima es estable a la hidrólisis de algunas betalactamasas, incluidas las penicilinasas y cefalosporinasas producidas por bacterias gramnegativas y grampositivas, con la excepción de las betalactamasas de espectro extendido (ESBL), algunas oxacilinasas y las betalactamasas hidrolizantes de carbapenémicos. El enmetazobactam es un inhibidor de betalactamasas que protege a la cefepima de la degradación por ciertas serina betalactamasas como las SBLE.
Eficacia clínica: Un total de 1041 adultos con infecciones complicadas del tracto urinario, incluida pielonefritis, fueron aleatorizados en una proporción de 1:1 en un ensayo multinacional, doble ciego y de no inferioridad, que comparó cefepima/enmetazobactam (2/0,5 g, IV) con piperacilina/ tazobactam (4/0,5 g, IV), ambos cada 8 horas (IV durante 2 horas) durante 7 días, o hasta 14 días para pacientes con bacteriemia concurrente. La población microbiológicamente modificada por intención de tratar (mMITT) fue la población principal del análisis de eficacia e incluyó a todos los pacientes aleatorizados que recibieron cualquier fármaco del estudio y tenían al menos 1 patógeno gramnegativo inicial ≥105 unidades formadoras de colonias (UFC)/ml. en urocultivo o el mismo patógeno en sangre y urocultivo que no fuese resistente a cefepima/enmetazobactam o piperacilina/tazobactam (CIM≤8/8 mcg/ml o MIC≤64/4 mcg/ml, respectivamente). Se incluyeron un total de 345 y 333 pacientes en la población mMITT en los grupos de tratamiento, respectivamente. Las tasas compuestas de respuesta (curación clínica y respuesta microbiológica) fue del 79,1% (cefepima/enmetazobactam) vs. 58,9% (piperacilina/tazobactam), las de curación clínica fueron del 92,5 vs. 88,9% y las de respuesta microbiológica del 82,9 vs. 64,9%.
Eventos adversos: Los más comunes son (5% o más) aumento de los valores des transaminasas y de bilirrubina, dolor de cabeza y flebitis y otras reacciones en el lugar de la infusión.
Pemivibart (Pemgarda®) Inviviyd (FDA, USA)
Indicación: Profilaxis previa a la exposición de la enfermedad por coronavirus 2019 (COVID-19) en adultos y adolescentes (de 12 años o más que pesen al menos 40 kg) que actualmente no estén infectados con SARS-CoV-2 y que no hayan tenido una exposición reciente conocida a un individuo infectado con SARS-CoV-2 y que tengan un compromiso inmunológico de moderado a severo debido a una condición médica o a la recepción de medicamentos o tratamientos inmunosupresores y sea poco probable que genere una respuesta adecuada a la vacuna COVID-19.
Tipo: Medicamento biológico constituido por un anticuerpo monoclonal de tipo IgG1 frente a la proteína S (spike) del SARS-COV-2. Autorizado en Estados Unidos (FDA) el 22-3-2024 para el uso de emergencia (EUA), pero sin que el producto esté definitivamente aprobado; no autorizado aún en la Unión Europea (EMA).
Mecanismo: Anticuerpo monoclonal neutralizante de la proteína S (spike) del SARS-COV-2.
Eficacia clínica: Ensayo clínico de fase 3 en curso en adultos de al menos 18 años de edad en dos cohortes; la cohorte A es un ensayo abierto de un solo grupo en adultos con inmunodepresión de moderada a grave (n=306), mientras que la cohorte B es un ensayo aleatorizado 2:1 y controlado con placebo en el que adultos que no están inmunodeprimidos de moderados a graves recibieron pemivibart (n=317) o placebo (n=162). La FDA utilizó un enfoque de inmunopuente (immunobridging), basado en la relación entre los títulos de anticuerpos neutralizantes y la eficacia clínica identificada con otros mAb humanos contra el SARS-CoV-2, incluyendo adintrevimab y algunos otros mAb que fueron autorizados previamente para la prevención de COVID-19. Los títulos de anticuerpos neutralizantes séricos de pemivibart fueron consistentes con los niveles asociados con la eficacia en ensayos clínicos anteriores de adintrevimab y algunos otros mAb. La relación media geométrica entre el título calculado para pemivibart contra la variante relevante JN.1 (basado en un valor de EC50 del ensayo de neutralización de virus auténtico de 63,6 ng/mL) y el título calculado para adintrevimab contra Delta (basado en un valor de EC50 del ensayo de neutralización de virus auténtico similar de 7 ng/mL) fue 0,82 (IC90%: 0,80-0,85).
Eventos adversos: Los más comunes son infección del tracto respiratorio superior (6%), infección viral (4%), enfermedad similar a la influenza (3%), fatiga (3%), dolor de cabeza (2%), náuseas (2%), reacciones locales en el lugar de la perfusión (2%) y reacciones sistémicas relacionadas con la perfusión y reacciones de hipersensibilidad (4%) y anafilaxia (0,6%).
(L) AGENTES ANTINEOPLÁSICOS E INMUNOMODULADORES
Erdafitinib (Balversa®) Janssen (FDA, USA)
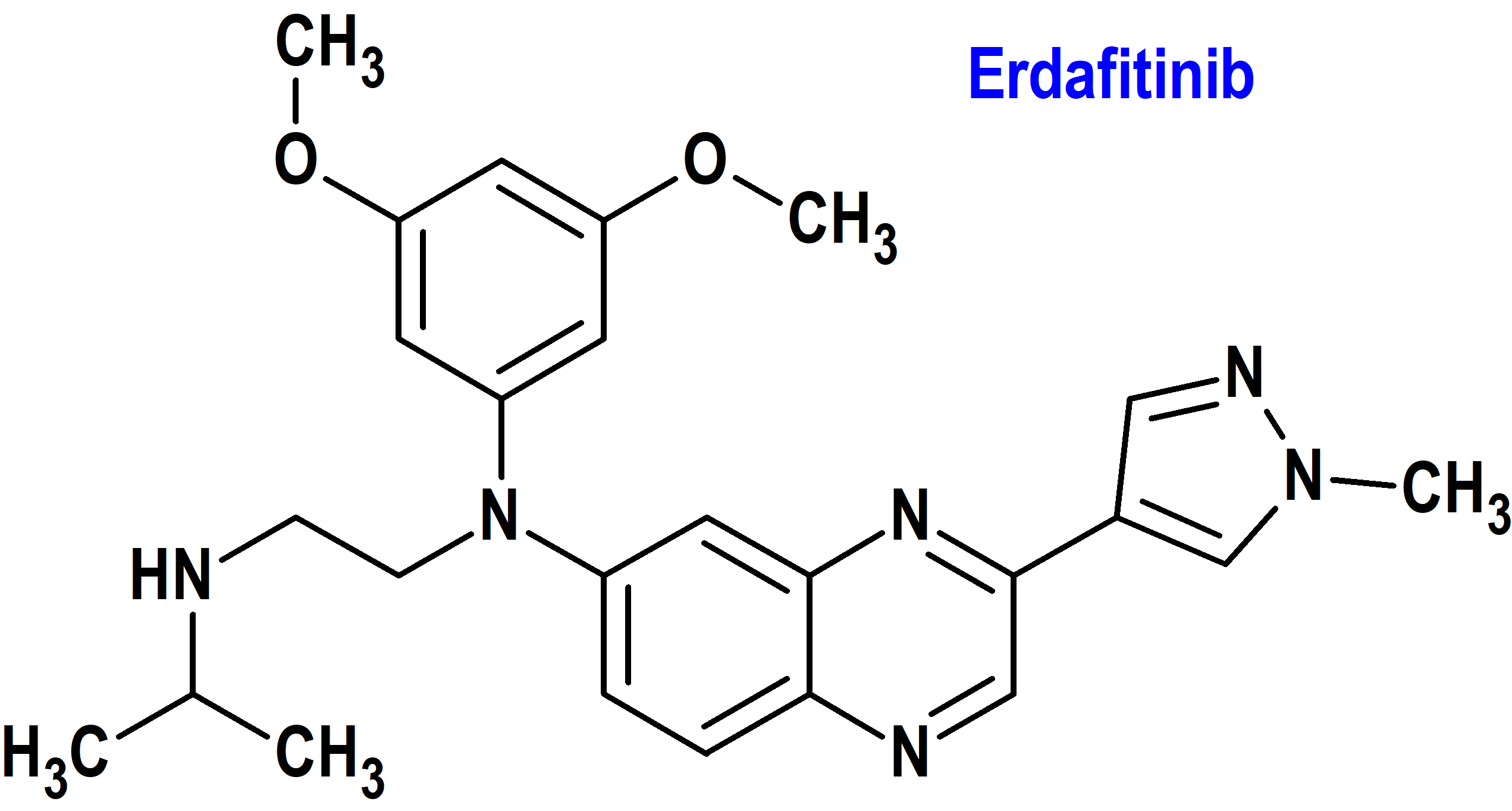
Indicación: Tratamiento de pacientes adultos con carcinoma urotelial localmente avanzado o metastásico (mUC) con alteraciones genéticas susceptibles de FGFR3 cuya enfermedad ha progresado durante o después de al menos una línea de terapia sistémica previa. Esta indicación modifica la previamente autorizada por la FDA para pacientes con mUC con alteraciones susceptibles de FGFR3 o FGFR2 después de una quimioterapia previa que contiene platino.
Tipo: Medicamento sintético estándar constituido por la N-(3,5-dimetoxifenil)-N’-(1-metiletil)-N-[3-(1-metil-1H-pirazol-4-il)-quinoxalin-6-il]etano-1,2-diamina. Autorizado en Estados Unidos (FDA) el 19-1-2024 de forma acelerada (Accelerated Approval), mediante revisión prioritaria (Priority Review); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la cinasa que se une e inhibe la actividad enzimática de FGFR1, FGFR2, FGFR3 y FGFR4. Erdafitinib inhibe la fosforilación y señalización del FGFR y disminuye la viabilidad celular en líneas celulares que expresan alteraciones genéticas del FGFR, incluidas mutaciones puntuales, amplificaciones y fusiones.
Eficacia clínica: Un ensayo aleatorizado y abierto de 266 pacientes con mUC que albergaban alteraciones seleccionadas del FGFR3 que habían recibido 1 o 2 tratamientos sistémicos previos, incluido un inhibidor de PD-1 o PD-L1. Los pacientes fueron aleatorizados para recibir erdafitinib o la quimioterapia elegida por el investigador (docetaxel o vinflunina). La variable principal de eficacia fue la supervivencia general (12,1 vs. 7,8 meses). Las variables secundarias fueron la supervivencia libre de progresión (5,6 vs. 2,7 meses) evaluada por el investigador y la tasa de respuesta objetiva (35,5 vs. 8,5%).
Eventos adversos: Los más comunes (>20%) son trastornos de las uñas, diarrea, estomatitis, sequedad de boca, síndrome de eritrodisestesia palmo-plantar, disgeusia, fatiga, piel seca, estreñimiento, disminución del apetito, alopecia, ojo seco, disminución de peso y anomalías de laboratorio (aumento de fosfato, potasio, calcio, fosfatasa alcalina, alanina aminotransferasa, aspartato aminotransferasa y creatinina; y disminución de hemoglobina, sodio y fosfato).
Lifileucel (Amtagvi®) Iovance (FDA, USA)
Indicación: Tratamiento de pacientes adultos con melanoma irresecable o metastásico previamente tratados con un anticuerpo bloqueador de PD-1, y si la mutación BRAF V600 es positiva, un inhibidor de BRAF con o sin un inhibidor de MEK.
Tipo: Medicamento de terapia avanzada (celular somática), constituido por células T autólogas derivadas de tejido tumoral del paciente, procedentes de una o más lesiones tumorales. Autorizado en Estados Unidos (FDA) el 16-2-2024 como medicamento huérfano (Orphan drug), de forma acelerada (Accelerated Approval), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), y designado como terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy, RMAT); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Es una inmunoterapia con linfocitos T cultivados procedentes de una o varias localizaciones tumorales del propio paciente, capaz de potenciar la respuesta inmune frente al melanoma.
Eficacia clínica: Un estudio clínico global, multicéntrico y de múltiples cohortes que incluyó a pacientes adultos con melanoma irresecable o metastásico que habían sido tratados previamente con al menos una terapia sistémica, incluido un anticuerpo bloqueador de PD-1, y si eran positivos para el Mutación BRAF V600, un inhibidor de BRAF o un inhibidor de BRAF con un inhibidor de MEK. La variable principal de eficacia consistió en la tasa de respuesta objetiva al tratamiento y la duración de la respuesta. Entre los 73 pacientes tratados, la tasa de respuesta objetiva fue del 31,5% (completa en el 4,1%) y del 27,4 % con una respuesta parcial. Entre los pacientes que respondieron al tratamiento, el 57%, el 48% y el 44% continuaron manteniendo respuestas sin progresión del tumor o muerte a los 6, 9 y 12 meses, respectivamente.
Eventos adversos: Los más comunes son escalofríos, fiebre, fatiga, taquicardia, diarrea, neutropenia febril, edema, erupción cutánea, hipotensión, caída del cabello, infección, hipoxia y sensación de dificultad para respirar.
(M) SISTEMA MÚSCULO-ESQUELÉTICO
Givinostat (Duvyzat®) Italfarmaco (FDA, USA)
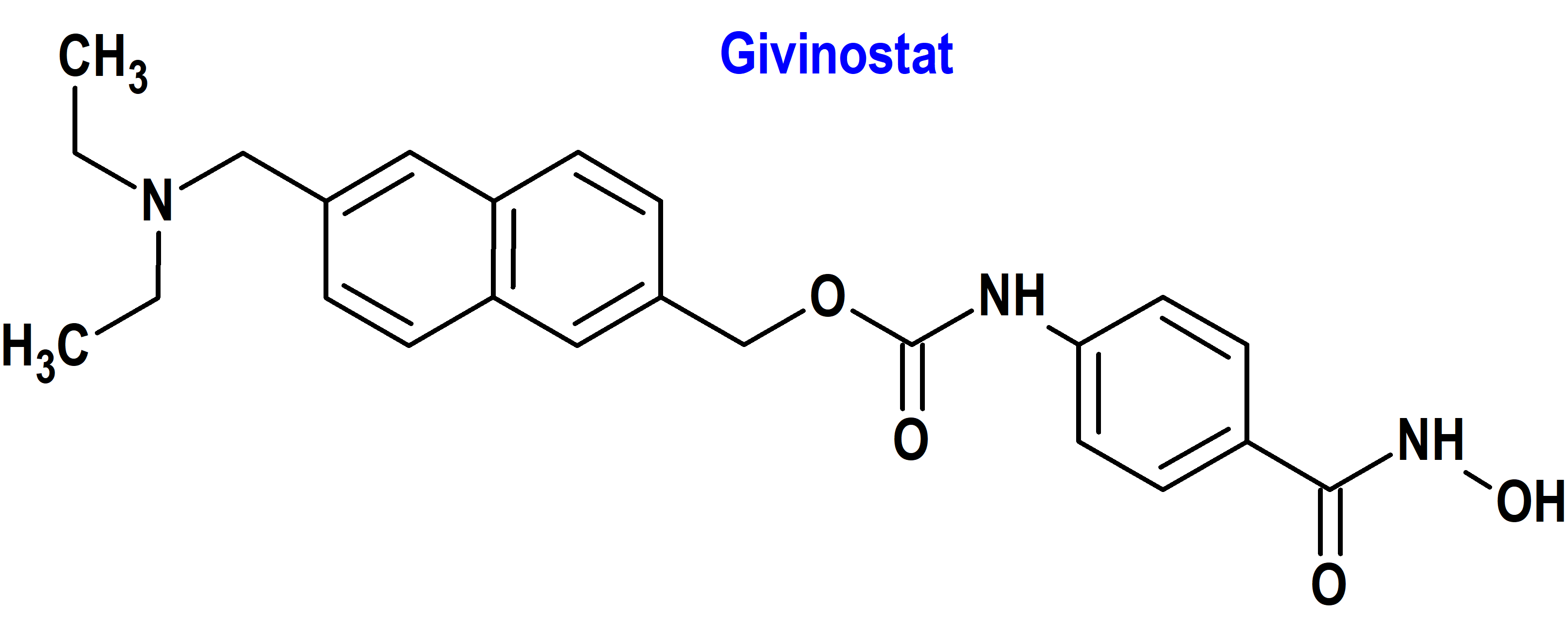
Indicación: Tratamiento de la distrofia muscular de Duchenne (DMD) en pacientes de 6 años de edad y mayores.
Tipo: Medicamento sintético estándar constituido por [6-(dietilaminometil)naftalen-2-il]metil [4(hidroxi- carbamoil)fenil]carbamato. Autorizado en Estados Unidos (FDA) el 21-3-2024 como medicamento huérfano (Orphan drug), por vía rápida (Fast Track), mediante revisión prioritaria (Priority Review), con bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD); no autorizado aún en la Unión Europea (EMA).
Mecanismo: Inhibidor de la histona desacetilasa de clase I y clase II (HDAC) y varias citocinas proinflamatorias. Esto reduce la expresión del factor de necrosis tumoral (TNF), de la interleucina 1a y b y de la interleucina 6
Eficacia clínica: Estudio aleatorizado, doble ciego y controlado con placebo de 18 meses de duración sobre 179 pacientes masculinos de 6 años en adelante. La variable principal clínica fue el cambio desde el inicio hasta el mes 18 en el tiempo de subida de 4 escaleras (4SC) en comparación con placebo, una medida de la función muscular. Los pacientes tratados con givinostat mostraron una menor disminución, en comparación con el placebo (1,25 vs. 3,03).
Eventos adversos: Los más comunes (>10%) son diarrea (37%), dolor abdominal (34%), trombocitopenia (33%), náuseas/vómitos (32%), hipertrigliceridemia (23%) y pirexia (13%).
PROCEDIMIENTOS ESPECIALES DE EVALUACIÓN Y AUTORIZACIÓN
Tanto la Agencia Europea de Medicamentos (European Medicines Agency, EMA), de la Unión Europea, como la Administración de Alimentos y Medicamentos (Food & Drug Administration, FDA), de Estados Unidos, disponen de diversos procedimientos de evaluación y autorización de medicamentos para incentivar el desarrollo de nuevos tratamientos para enfermedades que de otra manera no atraerían el interés de las empresas debido al elevado coste del desarrollo y la imposibilidad de retorno económico comercial, así como para facilitar la mejor y más rápida disponibilidad posible de medicamentos designados como especialmente relevantes atendiendo a las particulares características patológicas de algunos pacientes, así como a la gravedad de las patologías para los que son destinados y a su potencial repercusión social y epidemiológica, valorando si constituyen el primer tratamiento disponible o si presentan ventajas significativas sobre los tratamientos existentes. Estas designaciones y procedimientos son referenciados, en su caso, en las monografías de los medicamentos previamente descritas.
EMA (European Medicines Agency, UE)
- Medicamentos Prioritarios (Priority Medicines; PRIME): es un esquema de evaluación de la EMA para apoyar el desarrollo de medicamentos que se dirigen a una necesidad médica no cubierta, basándose en una interacción mejorada y un diálogo temprano con los desarrolladores de medicamentos prometedores, para optimizar los planes de desarrollo y acelerar la evaluación para que estos medicamentos puedan llegar antes a los pacientes, empleando para ello el asesoramiento científico y la evaluación acelerada.
- Evaluación acelerada (Accelerated assessment): reduce el plazo máximo para que el Comité de Medicamentos de Uso Humano (CHMP) revise una solicitud de autorización de comercialización de medicamentos, pasando de 210 a 150 días. Las solicitudes pueden ser elegibles para una evaluación acelerada si el CHMP decide que el producto es de gran interés para la salud pública y la innovación terapéutica.
- Autorización de comercialización condicional (Conditional marketing authorisation) para solicitudes de medicamentos que presenten datos clínicos menos completos que los normalmente requeridos, siempre que el beneficio de la disponibilidad inmediata del medicamento supere el riesgo inherente al hecho de que todavía se requieren datos adicionales, tal como aquellos destinados a tratar, prevenir o diagnosticar enfermedades gravemente debilitantes o potencialmente mortales, incluyendo a los medicamentos huérfanos.
- Autorización de comercialización en condiciones excepcionales (Exceptional circumstances) para medicamentos en los que el solicitante no puede proporcionar datos completos sobre la eficacia y la seguridad en condiciones normales de uso, porque la condición a tratar es rara o porque la recopilación de información completa no es posible o no es ético.
- Medicamento huérfano (Orphan drug): son designados como tales aquellos destinados a tratar enfermedades raras (en la Unión Europea son aquellas que afectan a menos de 5 de cada 10.000 habitantes), no resultan atractivos a los patrocinadores por su escasa rentabilidad y precisan por ello apoyo adicional para su desarrollo.
FDA (Food & Drug Administration, USA)
- Revisión prioritaria (Priority Review): evaluación de solicitudes de medicamentos que, de aprobarse, serían mejoras significativas en la seguridad o eficacia del tratamiento, diagnóstico o prevención de afecciones graves en comparación con las solicitudes estándar, considerando mejora significativa a la evidencia de mayor efectividad en el tratamiento, prevención o diagnóstico de la condición; eliminación o reducción sustancial de una reacción farmacológica limitante del tratamiento; mejora documentada del cumplimiento del paciente que se espera que conduzca a una mejora en los resultados graves; o evidencia de seguridad y eficacia en una nueva subpoblación.
- Bono para la revisión prioritaria de enfermedades pediátricas raras (Rare Pediatric Disease Priority Review Voucher, RPD): la FDA puede otorgar bonos o cupones de revisión prioritaria a los patrocinadores de aplicaciones de productos destinados para enfermedades pediátricas raras que cumplan con ciertos criterios. Este bono es un incentivo que el patrocinador recibe en forma de “cupón especial”, el cual puede ser empleado de dos maneras: para aplicar el sistema de revisión prioritaria de la FDA en cualquier otro de sus productos o venderlo a otra compañía interesada en que su propio medicamento sea revisado de forma prioritaria.
- Terapia innovadora (Breakthrough Therapy): medicamentos destinados a tratar una afección grave y cuya evidencia clínica preliminar indica que puede demostrar una mejora sustancial sobre la terapia disponible en una o varias variables clínicamente significativas, como la duración del efecto, la relevancia del resultado clínico observado mostrando una clara ventaja sobre la terapia disponible.
- Autorización acelerada (Accelerated Approval): medicamentos indicados en afecciones graves que cubran una necesidad médica no satisfecha, que puedan ser autorizados precozmente basándose en una a más variables subrogadas (una medida de laboratorio o signo físico que se usa como sustituto de una variable clínicamente significativa que es una medida directa sobre lo que siente un paciente, sus funciones o su supervivencia y que se espera que prediga el efecto de la terapia).
- Vía rápida (Fast Track): medicamentos que aborden enfermedades graves en las que puedan tener un impacto significativo sobre la supervivencia, el funcionamiento diario o la probabilidad de que la afección, si no se trata, progrese de una condición menos severa a una más severa, tales como el SIDA, la enfermedad de Alzheimer, la insuficiencia cardíaca y o cáncer.
- Medicamento huérfano (Orphan drug): designación de un medicamento potencialmente útil para prevenir, diagnosticar o tratar una enfermedad rara; es decir, con menos de 200.000 pacientes/año (los que supone una prevalencia aproximada de 7,5/10.000 habitantes, en la actualidad).
- Terapia avanzada de medicina regenerativa (Regenerative Medicine Advanced Therapy): cualquier medicamento de terapia celular, de ingeniería tisular, de células y tejidos humanos, o cualquier combinación de dichas terapias o productos, que esté destinado a tratar, modificar, revertir o curar una enfermedad o afección grave o potencialmente mortal; y que la evidencia clínica preliminar indica que el medicamento tiene el potencial de abordar necesidades médicas no cubiertas para dicha enfermedad o afección.
