1. INTRODUCCIÓN
1.1. Statins: A brief but intense history
HMG-CoA reductase inhibitors, commonly known as statins, are widely prescribed as antihypercholesterolemic drugs and, nowadays, they are one of the most used worldwide (1). The mechanism of action of statins consists on blocking the active site of the HMG-CoA reductase, the first and the key rate-limiting enzyme in the mevalonate pathway, then reducing mevalonate availability to the cholesterol biosynthesis (2,3). Since their discovery in 1976, when mevastatin was isolated from Penicillium citrinum (4), a broad range of statins have been manufactured, both natural by fermentation products of some fungi or chemically modified (5). Apart from their synthesis origin, statins can be classified depending on their solubility in lipophilic (atorvastatin, simvastatin, lovastatin, fluvastatin, cerivastatin, and pitavastatin) or hydrophilic (pravastatin and rosuvastatin) statins (6). In this sense, lipophilic statins are more suitable to be metabolized by the cytochrome P(450) system than hydrophilic, making the former more easily to cross through membranes and, therefore, expectedly more potent (7).
In addition to this well-known lipid-lowering effect of statins, other pleotropic effects in different organs and levels have been attributed to these drugs during the last decades. Many authors have attempted to describe the possible mechanisms by which statins exert their pleiotropic effects, being the mechanism that involves the participation of small GTPase (Rho, Rac and Ras) proteins and the role of isoprenoids the most accepted one (8) (Figure 1). Small GTPase proteins play an essential role in nearly every aspect of cell biology, being crucial in each on/off processes by GDP phosphorylation and GTP hydrolyzation (9) and participating in cell signaling, cell differentiation and proliferation, myelination, cytoskeleton dynamics or endocytotic/exocytotic transport processes among others (6). Apart from its fundamental role in cholesterol synthesis, mevalonate is also the precursor of bile acids, lipoproteins, steroid hormones and provides metabolites for protein prenylation, for instance farnesylation and geranyl-geranylation. Therefore, decreasing mevalonate availability by HMG-CoA inhibition, statins also promote a reduction in isoprenylation, which is crucial in the small GTPase (Rho, Rac and Ras) family proteins activity and their membrane translocation (10).
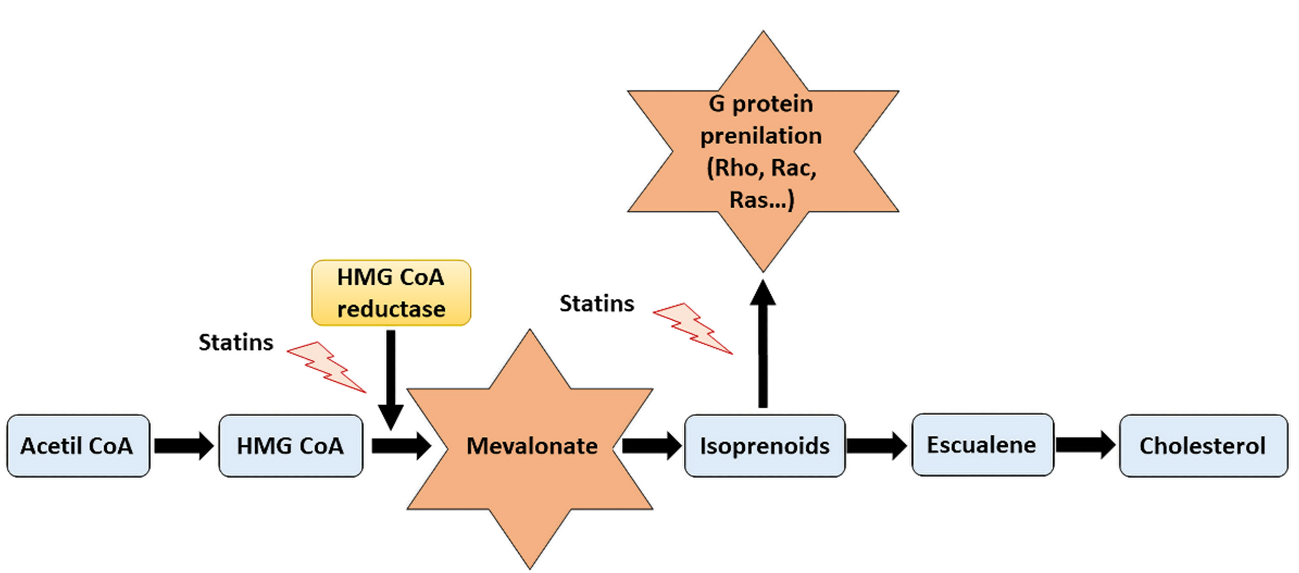
Figure 1. Representative scheme of cholesterol synthesis pathway and the actions of statins. By inhibiting the mevalonate synthesis, statins reduce cholesterol production as well as other relevant mechanisms implicated in essential cellular processes, including G proteins prenylation.
Diving in the bibliography, there are a vast number of articles and reviews discussing about the potential pleiotropic effects of statins, but they are mainly focused on some specific organ or pathology. The aim of the present review is to summarize these pleiotropic effects all along from the well-known cardiovascular studies to the potential use of statins in other pathologies such as kidney damage, cancer, autoimmune or infectious diseases.
1.2. An overview of the pleiotropic effects of statins
Among the pleiotropic effects attributed to statins, anti-inflammatory actions are the most studied in several tissues and diseases due to their relevance in a wide range of disorders. Statins modulate the inflammatory response by a complex lipid-independent mechanism. Statins reduce proinflammatory cytokines levels in vitro, such as TNFα, IL-6, MCP-1, IL-1 or IL-1β, mostly related to NF-kB pathway (11,12), as well as the inhibition of intracellular adhesion molecule-1 (ICAM-1), necessary for the immune cells recruitment (13). Although the mechanisms involved in their anti-inflammatory effects remain unclear, the critical role of the small GTPase proteins has been proposed. In this sense, Rho and Ras family proteins participate in the activation of NF-kB pathway via c-Jun NH2-terminal kinase, leading to an induction of cytokines and chemokines relevant for inflammation (14). Additionally, several human studies demonstrated that statins reduce C reactive protein (CRP) levels, contributing to reduce proinflammatory responses (12,15,16). Another mechanism described to participate in the beneficial effects of statins is their ability to modulate redox statement by impairing NAPDH oxidase (NOX) activation and function. Thus, NOX activation depends on an early anchorage of Rac proteins (p21 rac2) to the cell membrane which must be previously prenylated (17). This anchorage triggers the translocation of cytosolic subunits of NAPDH oxidase p47phox and p67phox, leading to NOX activation (18). The reduced isoprenylation induced by statins reduced Rac proteins translocation to the cell membrane, blocking NOX activation and the subsequent synthesis of reactive oxygen species (ROS)(19,20).
The participation of statins in the regulation of cell death has also been proposed by modulating programmed cell death apoptosis, the self-degradative system of autophagy or a programmed cell death characterized by inflammation known as pyroptosis (21–23). In this way, statins are able to modify levels of BCL-2, BCL-xL and BAX proteins, all of them involved in promoting apoptosis, as described using several statins both in vitro (24–26) and in vivo (27,28). However, while most of the studies attribute a pro-apoptotic property to statins, others indicate that statins did not modulate apoptosis or even they have anti-apoptotic effects (21,29). Regarding autophagy, several studies described the enhancement of autophagy promoted by statins as the upregulation not only of key proteins directly involved in this process, such as LC3-II, ERK1/2 or AKT (22,30–34), but also of indirectly related proteins, such as SKP2 (35,36). The role of statins inhibiting pyroptosis is closely related to their anti-inflammatory properties such as NLRP3 inflammasome, caspase-1, IL-18, IL-1b and GSDMD reduction (23). However, this mechanism presents several controversy since some other authors attributed anti-pyroprosis effects to statins (37). Other additional properties of statins involving the participation of several mechanisms include their participation in cellular phenotypic changes reducing proliferation and fibrosis, the downregulation platelet activation and coagulation, oxidative stress and angiogenesis (38) (Figure 2).
Unfortunately, although during the last decades, increasing evidences supporting the above-described effects of statins have been showed, it is mandatory to highlight the difficulty to separate cholesterol lowering beneficial effects of statins from their pleiotropic effects, especially in those human pathologies cursing with high cholesterol in which statins are used as a concomitant drug (39).
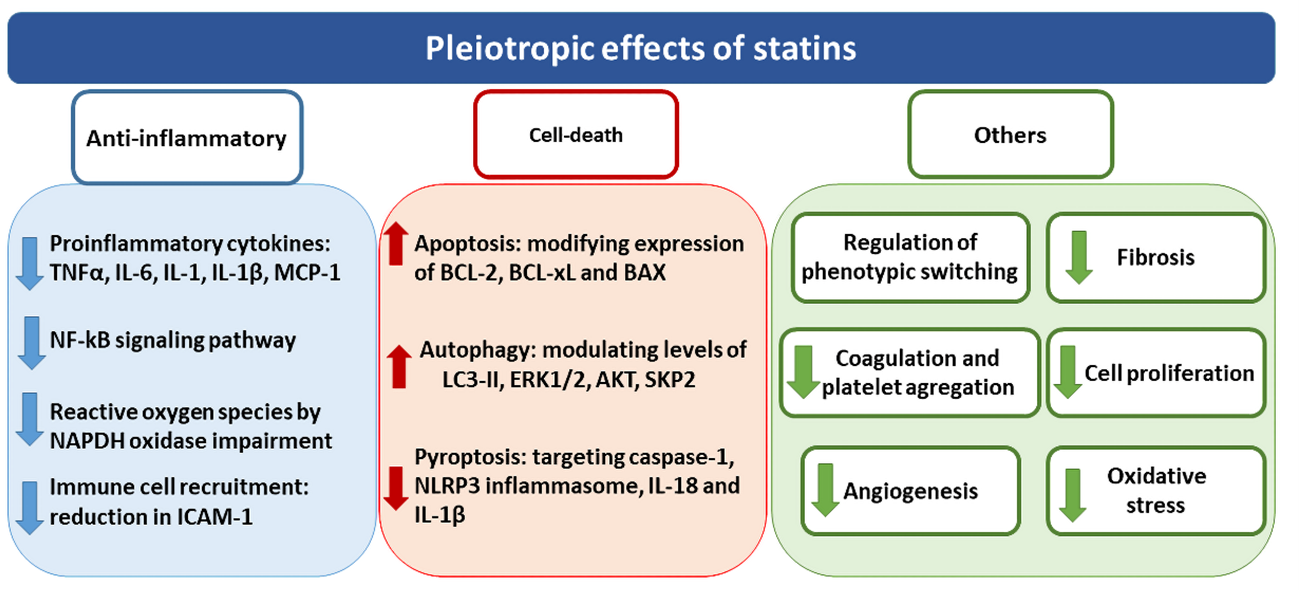
Figure 2. Overview of the pleiotropic effects attributed to statins. Pleiotropic effects of statins include the modulation of anti-inflammatory responses, cell-death mechanisms as well as other different cellular processes such as fibrosis, oxidative stress and proliferation.
2. STATINS, A WALK THROUGH PATHOLOGIES: FROM CARDIOVASCULAR TO COVID-19 DISEASES
2.1. Statins in cardiovascular pathologies
Nowadays, cardiovascular diseases (CVDs) are the leading cause of death and morbidity in developed countries and comprehends a huge range of pathologies, from atherosclerosis, aneurysms, acute coronary disease, or cardiac insufficiency, to some other complications related to other concomitant pathologies, such as chronic kidney disease (CKD)(40). Since most of the CVDs are also associated with high cholesterol levels, plenty number of these patients are under concomitant statins therapy. Apart from the obvious cholesterol lowering properties, some other pleiotropic benefits, including anti-inflammatory and pro-apoptotic responses, have also been attributed to benefits of these drugs in CVDs.
On the one hand, several in vitro studies have suggested a reduction in oxidative stress and inflammation mediated by statins as a potential mechanism of its beneficial effects in CVDs. More concretely, some authors demonstrated that statins reduce the proinflammatory and/or oxidative stress induced by TNF-α, angiotensin II (Ang II), aldosterone or C reactive protein (CRP) in cultured vascular smooth muscle cells (VSMCs)(41–44). Accordingly, statins inhibited the activation of NF-kB, activator protein 1 (AP-1)/C-jun, and hypoxia-inducible transcription factor (HIF-1) in human umbilical endothelial cells (HUVECs) in vitro (45). Statin treatment also decreased cellular proliferation and oxidative stress induced by advanced glycation end-products (AGEs) in cultured VSMCs of diabetic vasculopathy by blocking AGEs-receptor interaction and the subsequent activation of NF-kB, ERK, and p38 MAPK pathways (46). Another approach concluded that the withdrawal of in vitro statin treatment may cause a rebound increase of Ang II-mediated phosphorylation of p38 MAPK and ERK1/2, not only losing the previous exerted inhibitory effects, but further increasing the effects of Ang II (47). Apart from inflammation, other mechanisms have been described to be modulated by statins such as proliferation, migration or cell death. Thereby, stains reduced proliferation and migration responses in cultured VSMCs induced by AGEs or lysophosphatidic acid (LPA) by activating the ERK5-Nrf2 pathway or inhibiting Rac-1 activation respectively (48,49). Similarly, statins reduced migration and proliferation responses induced by lipopolysaccharide (LPS), thrombospondin-1 (TSP-1) and PDGF in cultured human aortic SMCs (HASMCs)(50–52). However, other authors established opposite effects depending on statin dose in human epidermal microvessel endothelial cells (HMVECs) in vitro. Here, while low doses of statins enhanced migration, proliferation and cell viability responses induced by fibroblast growth factor 2, higher doses resulted in the opposite effects, suggesting that statins regulate at least two different pathways, being one of them related to the mevalonate pathway (53). In cultured human atrial fibroblasts, another type of vascular cells, statins treatment decreased cell proliferation via Rho geranylgeranylation inhibition (54). More recently, it has been demonstrated that statins decreased TGF-β1 induced differentiation in human ventricular fibroblast by decreasing SMAD2/3 pathway activation (55).
Statins have been also described to modulate apoptosis-cell death by decreasing Rho prenylation in rat and human cultured VSMCs (56–58). Accordingly, some authors concluded that statin-induced apoptosis in VSMCs is mediated by the impairment of RhoA/Rac-1 protein activity, and the subsequent damage on cytoskeletal integrity (59). This proapoptotic effects induced by statins have been also corroborated in other vascular cell types such as HUVECs, cardiac fibroblasts, and pulmonary artery SMCs (60–62). Another cellular process implicated in the cell survival/death responses has showed to be regulated also by statins. Hence, in mouse coronary arterial myocytes, statins induced autophagy via Rac1 and mTOR inhibition (63). Likewise, statin-induced autophagy was demonstrated in HUVECs, and rat aortic VSMCs (60,64). Contrarily, a relatively recent described form of cellular homeostasis called pyroptosis, has been described to be downregulated by statins in HUVECs, associated to caspase-1, NLRP3 inflammasome, GSDMD, IL-1β, and IL-18 decreased levels (23).
Likewise, the in vivo approaches using statins in experimental CVDs showed similar results. Therefore, statins reduced the inflammatory component in the atherosclerotic lesion in experimental atherosclerosis in rabbit, by reducing NF-kB pathway activation and their related proinflammatory factors synthesis, cyclooxygenase (COX)-2 and matrix metalloproteinase (MMP)-3 expression as well as macrophage infiltration in the affected areas (65–67). The antioxidant ability was also demonstrated in another atherosclerosis model in rabbits in which statin administration reduced superoxide production and reverses the impairment of endothelium-dependent relaxation in aortas, leading to atherosclerosis progression reduction (68). In the same way, in an experimental atherosclerosis model in ApoE -/- mice, statins suppressed Mcp1 and Tnf-α gene expression levels, decreasing the size of atherosclerotic lesions (50). Another experimental model of atherosclerosis in ApoE -/- mice showed a novel insight in how statin treatment may affect this disease. Atorvastatin treatment potentiated SMAD pathway within aortic VSMCs, leading to elevated ECM protein deposition and amelioration of atherosclerosis, suggesting a new mechanism of statins involved in plaque stability (69).
Linked to the atherosclerotic lesion, intimal hyperplasia induced by the dysregulated proliferation of VSMCs in the vessels is a well-documented hypertrophic response that has been reported in all types of vascular reconstructive procedures including arterial bypass, stenting, angioplasty, vascular access grafting, etc. (70). In experimental jugular vein graft in rabbits, pravastatin administration reduced cell proliferation and increased cell apoptosis, suppressing intimal hyperplasia of the vein graft (71). Similarly, pitavastatin suppressed vein graft intimal hyperplasia in other experimental model in rabbits, inhibiting cell proliferative activity and decreasing Midkine expression, a heparin-binding growth factor involved in inflammation (72). In balloon-injury rabbit carotid arteries, pitavastatin prevented neointimal thickening as well as the accumulation of fibronectin/type I collagen in the intima (73).
Despite this, there is a controversy regarding statins effects in fibrotic CVDs. Although an increased fibrotic response seems to be beneficial in atherosclerotic lesions, it has been also reported the pathological role of the fibrotic process in other vascular disorders. Vascular remodeling plays an important role in the development and progression of some CVDs, such as hypertension and the previously mentioned atherosclerosis. One of the most related systems associated with this phenomenon is the renin-angiotensin system (74). In experimental hypertension induced by Ang II administration in rats, statin treatment suppressed Rho activation, resulting in the reduction of type I/type IV collagen deposition, aortic CCN2 protein and gene expression and MMP-9 gene levels overall reducing vascular fibrosis (75). Further research determined that atorvastatin treatment attenuates Ang II-induced oxidative stress and ECM alterations, such as collagen accumulation in the media layer, and increases endothelial NO synthase expression leading to a reduction in wall thickness and wall-to-lumen ratio (both vascular remodeling indicators), thus preventing from Ang II-induced vascular remodeling (76). These results were also demonstrated under simvastatin treatment, showing that this type of statin diminished SMAD activation triggered by Ang II in the aorta, as well as reduced overexpression of profibrotic factors and ECM components (77). Similarly, in a rat model of Ang II-induced cardiovascular hypertrophy, statin treatment inhibited Rac1 activation in the left ventricle (LV), decreasing heart hypertrophy (78). In another experimental model of hypertension using spontaneous hypertensive rats (SHRs), atorvastatin treatment resulted in a marked reduction of two hypertrophic indexes, the left-ventricle-weight-to-bodyweight ratio (LVW/BW) and the cardiomyocyte-transverse diameter (79). Moreover, p27 pro-apoptotic marker protein expression was increased by statin treatment, showing the beneficial effects of atorvastatin on left ventricular remodeling (80). The dysregulation of cell proliferation and apoptosis have been identified as the essential processes for the control of LV remodeling (81). Similar results were obtained in another study using SHRs, in which statin treatment decreased LVW/BW ratio (82). The latter went further, and determined heart-ultrastructure changes, showing that atorvastatin prevented Connexin43 (Cx43) gap-junctional disorganization in heart cardiomyocytes. Cx43 is the predominant gap junction protein in heart and is involved in the control of cell-to-cell communication to modulate contractility and the electrical coupling of cardiac myocytes (83). Apart from reducing cardiac remodeling, a different report using SHRs suggested that atorvastatin treatment may exert beneficial effects in calcium-regulating proteins, by restoring the reduced Sarco/endoplasmic reticulum CA²+ -ATPase activity (SERCA)(84), which is a key pump in charge of intracellular calcium homeostasis and dynamics (85).
Beneficial effects of statins have also been evaluated in myocardial infarction (MI), one of the most common manifestations of CVDs worldwide (86). In this way, plenty of in vivo studies have tried to describe the role that statins may exert in this pathology, independently of their lipid-lowering effects. In a mouse model of MI induced by myocardial ischemia-reperfusion (IR) injury, simvastatin treatment has shown to produce protective effects, reducing the area of injury (87). Similarly, in rat models of MI generated also by myocardial IR, statin pretreatment has demonstrated to confer significant cardio-protection, by decreasing creatine kinase levels and vascular permeability following reperfusion (88). In another IR-MI study, in which intravenous application of fluvastatin was carried 20 minutes before the onset of ischemia, infarcted size was reduced, as well as regional myocardial perfusion/function seemed to improved (89). In a similar approach, the application of atorvastatin before the onset of ischemia ameliorated reperfusion injury in rat hearts, improving vasodilatory capacity and the inhibition of neutrophil extravasation (90). The cardioprotective effects that statins exert against IR injury in rats has been associated to the inhibition of the RhoA signaling (91). Analyses with bigger mammals reported similar results. Thereby, IR-MI in pigs was significantly ameliorated in rosuvastatin-treated animals, showing lower infarct size without affecting cholesterol levels (92).
All these promising results obtained both in vitro and in vivo related to the non-lipid lowering use of statins made them to be considered pharmacological compounds with clinical relevance in the treatment of cardiovascular disorders. Therefore, many clinical trials and some other small-scale studies in human have been performed for years. Recent studies have showed encouraging and statistically significant results, even though they should be skeptically considered, due to small sample sizes, and taken as preliminary outcomes. For instance, in a study in which 20 people undergoing carotid endarterectomy without previous statin treatment were randomized to receive 80mg/d intensive atorvastatin treatment (n=11) or no statins (n=9) for one-month, intensive atorvastatin treatment diminished blood inflammatory markers, such as PGE2 plasma levels, NF-kB activity, and MCP-1 and COX-2 mRNA expression in patients with atherosclerosis (93). Interestingly, in normocholesterolemic healthy volunteer individuals, statins (atorvastatin and pravastatin, 20mg per day for 1 week) inhibited Rac1, but not RhoA/Rho-kinase activity, in circulating leukocytes (94). In another case the effects of statin therapy in the inflammatory profile of human epicardial adipose tissue (EAT) was analyzed, showing a strong association between statin therapy, EAT thickness and inflammatory profile in patients with aortic stenosis (95).
Although the pleiotropic effects of statins may seem quite convincing in vitro and in vivo, the clinical transcendence of those effects are yet being discussed. All the above mentioned promising preliminary studies led to the initiation of many large clinical trials. The interpretation of some of the obtained outcomes was focused on the evaluation of C-reactive protein (CRP), a marker of chronic inflammation that can serve also as an indicator of some CVDs (96). One of the first trials in this area was the A-Z trial enrolling patients with acute coronary syndrome treated with intensive and low dosage of simvastatin. The results showed similar CRP levels in all evaluated groups in the first month, but a reduction in CRP levels at the fourth month of treatment in those intensively treated (97) was found. The MIRACL study, another acute-coronary-syndrome trial using atorvastatin, reported a reduction in the recurrent ischemic events in the first 16 weeks in atorvastatin-treated patients (98). One of the most important statin-related clinical trials is JUPITER (NCT00239681). It consisted in a primary prevention trial using rosuvastatin and reported a reduction of cholesterol LDL levels, relative risk and CRP by 50%, 44% and 37%, respectively. The further analysis of the JUPITER results suggested greater benefits to those expected only from lipid-lowering effects (99). A more recent primary prevention trial, the HOPE-3 study (NCT00468923), proposed that the outcomes reduction observed in rosuvastatin patients was mainly due to lipid-lowering effects (100). Although the assessment of CRP plasma levels could help to differentiate between lipid-lowering and non-lipid-lowering anti-inflammatory effects of statins, the relationship of this marker and cardiovascular events such as stroke (commonly a primary endpoint) is controversial (101,102). At this point, it is necessary to highlight that the American Diabetes Association has now recommended the use of statins for all patients with diabetes over the age of 40 regardless of the LDL level, given the cardiovascular benefit of these drugs (103).
Altogether, it is difficult to distinguish between the lipid lowering benefits and the pleiotropic effects of statins in the CVDs clinical trials. It might be claimed that the contribution of the pleiotropic effects of statins in these patients remains unknown for two reasons: 1) these effects are correlated with the inhibition of cholesterol biosynthesis and, therefore, they are difficult to quantify; 2) the clinical trials using non-statin lipid-lowering agents are tested in statin-using patients (regulatory requirements) (39). Related to the latter, the clinical trials performed with another type of cholesterol-lowering drug, PCSK9i, such as GLAGOV (NCT01813422), SPIRE-1 and 2 (NCT01975376; NCT01975389) and FOURIER (NCT01764633) studies, have not been able to conclude whether statin pleiotropy has clinical effects, as these trials have been done on background statin therapy (104). Future studies comparing statin treatment to PCSK9i-only treatment could give further insight into the pleiotropy-related clinical effects of statins in CVDs.
2.2.Statins in renal pathologies
Nowadays, renal diseases are increasing their importance in terms of mortality worldwide. CKD is one of the fastest-growing global causes of death and it is expected to become the fifth common cause of death worldwide by 2040, being increased at the end of the century to the second position (105,106). The incidence of this disorder varies depending on the country, the age and ethnicity, and it is considered to be around 11% in high-income countries (107). One of the most prevalent complications related to renal diseases is cardiovascular disorders, especially hypertension (108).
Therefore, it is understandable that many patients with renal diseases are taking statins, since they are recommended in an impairment of cardiovascular health (109). This fact generated several studies trying to describe the effects that statins may have in kidney diseases per se. However, there is a quite controversy because, although some studies have reported beneficial effect of statins in renal disorders (110,111), some others showed a lack of effect (112).
At cellular level, several in vitro studies have shown an insight into how statins could exert their reno-protective effects, both decreasing inflammation and fibrosis processes. In Ang II treated human mesangial cells, simvastatin has shown to decrease protein expression levels of the proinflammatory cytokines TNF-α, IL-1β, IL-6 and MCP-1, as well as a reduction in the NF-κB pathway activation, and oxidative stress reduction by suppression of the NADPH oxidase (NOX2 and p47phox) pathway (113). Likewise, in HK2 proximal tubular epithelial cells, some statins (lovastatin, simvastatin and pravastatin) were reported to reduce the levels of activated cytoplasmatic and membrane associated RhoA and Rac1, leading to a diminish in several factors related to the epithelial-to-mesenchymal transition (EMT) phenomenon, such as E-cadherin, Cytokeratin-19 and Fn-EDA (114), a process which plays an important role in the initial steps of renal fibrosis (115).
In HEK-293 cells treated with advanced glycation end products (AGEs), decreased ROS generation and decreased NF-κB expression have been also observed after atorvastatin or rosuvastatin treatment (116). In cultured podocytes, pitavastatin treatment significantly inhibited hyperglycemia-induced podocyte injury by reducing apoptotic responses (117).
Moving to in vivo studies, HMG-CoA reductase inhibitors have been widely studied in many different renal disease models. Several experimental models of diabetic nephropathy in mice and rats have reported beneficial effects after statin treatment. In streptozotocin induced diabetic rats, pitavastatin showed to decrease transforming growth factor beta (TGF-β) and MCP-1 mRNA expression, and Rho protein expression, overall improving glomerular fibrosis (118). Another study using the same induced-diabetic nephropathy, has shown that simvastatin treated rats presented improved structures of glomeruli, renal tubules and less interstitial collagen deposition compared to the untreated diabetic rats. Oxidative stress was also ameliorated, malondialdehyde (MDA) and nitric oxide (NO) decreased and glutathione (GSH) increased (119). In db/db mice with early-stage diabetic nephropathy, a similar pattern appeared. Statins reduced glomerular RhoA protein activation and subsequent ROCK1 induction, therefore, reducing endothelial damage in the glomeruli and blocking the permeabilization (120). On the other hand, statins treatment also and reduced fibronectin and type IV collagen deposition, inhibiting thus fibrosis (121). In a CKD model induced by unilateral ureteral obstruction (UUO) in rat and mice, simvastatin seemed to attenuate renal interstitial inflammation and fibrosis (122,123), further supporting the anti-inflammatory and antifibrotic actions of statins at kidney level. In a 5/6 nephrectomy induced-renal failure rat CKD model, rosuvastatin has shown to improve fibrotic features, because of the preservation of glomerular structure, and the reduction of tubular atrophy, interstitial fibrosis, and endothelial disfunction by restoring endothelial nitric oxide synthase (eNOS) protein expression in renal arteries (124). Similar results have been obtained in high-salt-intake rat model (125) and aged rat model (126), backing up the positive result of statins in in vivo models of CKD. Accordingly, experiments performed in bigger mammals reported similar results. Hence, in a CKD model of hypertensive and hypercholesterolemic pigs, simvastatin has shown to improve apoptosis imbalance in endothelial progenitor cells (EPCs), without blocking the migration of EPCs, which would consequently ameliorate renal fibrosis (127). Statin effects in acute kidney injury (AKI) has also been extensively studied. Thus, in a model of contrast-agent induced AKI in rats, atorvastatin has demonstrated to decrease ROCK1 protein expression and followed kidney injury, such as tubular vacuolization, degeneration, necrosis and infiltration of mononuclear cells (126). Similar results have been reported in other rat nephrotoxicity studies, in which pretreatment with statins seemed to reduce kidney damage by decreasing inflammation and oxidative stress in radiation-induced nephrotoxicity (128).
Regarding apoptosis, simvastatin treatment decreased Caspase-3 protein expression, and restored BCL-2/BAX balance in streptozotocin induced diabetic rats, showing significant improvement (119). Similarly, during the evaluation of renal damaged induced by aging, some apoptosis-related proteins, such as Caspase-3 and BAX, decreased after statin treatment, as well as the BCL-2/BAX ratio imbalance (126). In this study, some fibrotic markers including MMP-9 and TGF-β protein expression decreased, and protein expression of tissue inhibitors of metalloproteinase (TIMP)-1 increased after statin treatment, suggesting an improvement of the fibrotic process as well (126). In cisplatin-induced nephrotoxicity and in gentamicin-induced nephrotoxicity in rats, statins therapy reduced apoptosis via MAPK pathway attenuation (129,130). In the same way, rosuvastatin has shown to reduce proinflammatory and apoptosis-related factors and to attenuate some fibrotic pathways, including the SMAD-dependent and SMAD-independent pathways in cyclosporine-induced nephropathy in rats (131).
At human level, several clinical trials have aimed to prove the potential benefits of statin treatment on renal diseases. Although statins have been demonstrated to be highly effective in early stages of CKD, the effects in patients with advanced CKD have not been appreciated, despite the LDL lowering effect (132). For instance, in the SHARP trial (NCT00125593), statin therapy was shown to modify atherosclerotic events in CKD patients (109). However, other studies were not able to evidence successful results. That is the case of chronic renal failure secondary to Autosomal Dominant Polycystic Kidney Disease (ADPKD) clinical trial (NCT02511418), in which simvastatin, apart from decreasing the lipid profile, did not modify renal parameters (133). Similarly, the ESPLANADE trial (NCT00199927) reported that fluvastatin treatment, in combination with an angiotensin-converting enzyme inhibitor and an angiotensin receptor blockade therapy, only reduced lipid concentrations in CKD patients with residual proteinuria, but did not modify renal function (134). Despite this, scientists are trying to search for new alternatives and improvements to tackle these diseases using statins. Therefore, more studies are currently being carried out on ADPKD (NCT03273413 on phase 4 and NCT04284657 on phase 2), with pravastatin, and also in CKD patients with proteinuria (NCT03550859 and NCT03543774) both of them on the recruiting phase, and using rosuvastatin and simvastatin, respectively. A systematic review and meta-analysis suggested that statins slightly reduce proteinuria and estimated glomerular filtration rate (eGFR) in CKD patients, but they did not reduce the risk of kidney failure events (135). In another systematic review and meta-analysis performed with diabetic kidney disease patients, authors suggested that statins might have beneficial effects on reducing albuminuria, but there was no strong evidence that statins had an effect on overt proteinuria or eGFR outcomes in these patients (136).
Considering all these results, further studies are required to demonstrate the potential use of statins in patients with kidney damage. Maybe, the above mentioned ongoing clinical trials are going to contribute to open new avenues in this regard.
2.3. Statins in cancer
Cancer is one of the most important and dreaded diseases along the final of the 20th Century and recent years. Its incidence is far from stopping and it keeps increasing each year, counting more than 17 million of affected people worldwide (137). The risk of suffering cancer exists during the whole lifetime, claiming for high awareness from the different healthcare systems. Ensuring tumoral prevention, as far as possible, and cancer patients’ care comprehend the key points to fulfill the control of this disease. Therefore, a huge range of therapies has been proved, both for preventing and treating the disease, as well as several adjunctive treatments.
In this way, statins have been proposed as potential anticarcinogenic drugs, both in primary and secondary cancer prevention (38,138,139), due to their properties in modulating cellular cycle and cell death. Some authors have attributed the effects of statins to reducing mesothelioma cells viability in vitro by inducing apoptosis (140), and their antiproliferative effects by stopping the transition between G1-S phases (141) related to the regulation of Ras protein (142). However, other studies showed that this effect was independent on Ras protein pathway (143) so, although several authors have tried to clarify these mechanisms, nowadays they still remain unclear.
BCL-2 family proteins are molecules with the ability of either promote or inhibit apoptosis. These proteins control the intrinsic apoptotic pathway by regulating mitochondrial membrane permeability (144). Different studies with statins demonstrated an increase in apoptosis induced by downregulation of BCL-2 expression levels (145). Although statin pro-apoptotic effects have been reported both in cancer and non-cancer cells, some tumoral cells types appear to be more suitable to HMG-CoA reductase inhibitors-induced apoptosis (146). Accordingly, statins seemed to augment apoptosis by reducing levels of BCL2 in different carcinogenic cells such as MCF-7 and MDA-MB-231 (breast cancer cells)(147,148), several osteosarcoma lines cells (149), COLO 205 and HCT 116 (human cancer colon cells)(150) and HepG2, SMMC-7721 and MHCC-97H (hepatocellular carcinoma cells)(151). Interestingly, several of these studies attributed the modulation of apoptosis to RhoA regulation, suggesting that the reduction of RhoA prenylation mediated by statins inhibits their translocation to the membrane, inducing cytosolic accumulation of RhoA where it is able to trigger the intrinsic signaling pathway of apoptosis (148,149). Curiously, another study using simvastatin in MG63 cells (osteosarcoma cell line) described the protective effect from oxidative stress-induced apoptosis by decreasing the caspase-3 and 9 expression and the Bcl2 downregulation induced by H2O2 (152). Other authors also demonstrated statins modulation of apoptosis by modulating some other complex mechanisms such as AKT/FOXO1 pathway in prostate cancer cells (153) or PTEN/AKT pathway in breast tumor cells (154). Among isoprenilation processes, geranylgeranylation has been described to be a key mechanism in apoptosis mediated by statins in tumors cell lines. In this way, the addition of geranylgeranyl pyrophosphate (GGPP) abrogated statins-induced apoptosis in human acute myeloid leukemia (155), hepatoma (156) and melanoma cell lines (157). However, another recent study carried out also in tumor cell lines, reported the participation of both geranylgeranylation and farnesyilation depletion in the intrinsic apoptosis activation induced by simvastatin (158). Apart from apoptosis, simvastatin have also showed to activate another cell death response, termed pyroptosis, in non-small cell lung cancer (NSCLC), suggesting an additional potential anti-tumor mechanism for statins by regulating another cell death mechanism (37).
Statins have also been described to exert anti-metastatic effects by modulating autophagy, a very relevant survival cellular process, at different levels (30). In this sense, the activation of the AMPK-TOR signaling pathway induced by cellular geranylgeranyl diphosphate depletion is showed to be the main mechanism of statins-induced autophagy, although some authors have described that statins can also activate this mechanism by inducing accumulation of nuclear p53 and by direct inhibition of histone deacetylases (30). In the same manner, many studies performed in tumoral tissues revealed that statins increased Beclin1 and LC3-II levels, two crucial proteins in the autophagic process (30), suggesting a novel Rho A GTPase-independent mechanism involved in the statin-induced anti-metastasis effects (31–33,159).
In addition to cell death and autophagy processes, another relevant phenomenon implicated in cancer diseases is the cell phenotype switching, since a shift from proliferative to invasive phenotype is one of the critical steps in metastasis (160). Thus, high doses of statins have been reported to influence cell migration and cell invasion in culture melanoma cells and to reduce local tumor growth and pulmonary metastases in nude mice injected with statin-treated melanoma cells (161,162). Interestingly, The Cancer Genome Atlas (TCGA) project, which profiled the mutational status and expression levels of all the genes in diverse cancers, revealed changes in genes implicated in cholesterol metabolism (163), suggesting a control of them during phenotype changes. Considering the latter and the antihypercholesterolemic effects of statins, this mechanism could also be promoting its anti-cancerogenic effects. Supporting this theory, cholesterol synthesis inhibitors, including statins, are reported to counteract CD271/Trk-A-dependent dissemination of melanoma cells associated to increased cholesterol synthesis (164). The neurotrophin receptor CD271 seems to confer a kind of therapy resistance and it is crucial in controlling melanoma growth vs. invasiveness (164,165) and, although it was identified as melanoma-initiating cell marker, high levels of CD271 have been also correlated to metastasis (166,167). The EMT is one of the most studied cellular-phenotype-switching processes, and it is considered pivotal for driving metastasis. In several in vitro studies performed in lung adenocarcinoma and breast cancer cells, statins have showed to suppress EMT process, diminish cell migration and decrease the metastasis process (168,169).
The potentially beneficial effects of statins in cancer have also been tested in experimental animal models. Consequently with in vitro studies, several researchers highlight the modulation of pathways related to cell survival and resistance to apoptosis, such as the AKT pathway, as the potential mechanism implicated in the benefits of statins in vivo, although the exact mechanisms involved still remain unclear (170,171). Supporting this theory, simvastatin treatment reduced breast tumor growth in a mouse model inoculated with MDA-MB-231 breast cancer cells by reducing AKT phosphorylation and Bcl-xL (an antiapoptotic factor) levels by NF-κB pathway inhibition (172). Similarly, in an experimental mice model of colon cancer, simvastatin administration promoted smaller cancer volume, larger necrotic areas and high apoptotic scores (150). In the same way, in a murine pulmonary metastasis model, statin treatment inhibited tumor growth and metastasis, improving the survival rate (173). Another study, performed in an experimental mice model of B16BL6 melanoma cells injection, demonstrated that statins administration reduced lung metastasis, cell migration, invasion, and adhesion by suppressing Rho/RhoA/ROCK pathway (174).
Despite all the aforementioned, the attempts to translate these beneficial results to humans have reported controversial results. Therefore, while some meta-analyses have concluded that statins have neutral or no effects in cancer incidence or progression (175,176) and the addition of statins to standard anti-cancer therapy did not appear to improve overall survival or progression-free survival in patients with advanced cancer (177), other authors indicated an association between statin use and reduced mortality in 13 cancer types (178). Interestingly, a report performed in lung adenocarcinoma patients after lung resection, showed an opposite effect of statins treatment depending on the presence of p53 mutation (168). Hence, while in patients with mutant p53, statin users showed significantly better survival than non-statin users, statins significantly worsened the prognosis of patients with wild type p53 (168). Several recently concluded phase 2 clinical trial have evaluated the potential benefits of statins in prostate cancer (NCT01992042) (NCT01759836), although no results have been reported yet. Other active phase 2 clinical trial aims to evaluate whether simvastatin treatment may stop tumor cells growth and reduce the aggressiveness of breast cancer cells (NCT03454529) or may prevent from liver cancer in patients with cirrhosis (NCT02968810). Similarly, there are two ongoing phase 3 clinical trial currently testing statin effects on cancer. One is evaluating atorvastatin use as adjuvant in breast cancer (NCT04601116) and the another one is assessing the potential benefits of statins treatment in breast cancer patients with dyslipidemia (NCT03971019). In addition, one of the most remarkable clinical trials called: “Statins for the Primary Prevention of Heart Failure in Patients Receiving Anthracycline Pilot Study” (SPARE-HF), is evaluating if statin pretreatment could prevent heart failure (HF) incidence induced by the anthracycline chemotherapy, which is used in several common cancers (NCT03186404).
In summary, although preclinical studies indicate that statins can modulate some cancer-related pathways, the current controversial in vivo results highlight the necessity of further research in this field to clearly demonstrate a beneficial effect of statins in human cancer. Maybe, future results obtained in all the above-mentioned ongoing clinical trials can throw some light on the potential use of statins in this disease.
2.4. Statins in brain pathologies
Central Nervous System (CNS) constitutes only 2% of total body, but it requires the 25% of total cholesterol, which demonstrates the relevance of cholesterol in this system. Furthermore, considering that cholesterol cannot cross the blood brain barrier (BBB), de novo cholesterol synthesis and transport by astrocytes to neurons is especially necessary (179–181). The main function of cholesterol in the CNS is attributable to the lipid rafts, which are necessary for neuronal bodies in the transmission of pre- and post-synaptic connections, and synaptic vesicles formation (179,182,183). Although only lipophilic statins are traditionally thought to cross the BBB, recent evidences showed that also hydrophilic statins may pass through BBB, as demonstrated by statin detection in cerebral spinal fluid (CSF) in humans (184). In this sense, some receptors such as organic anionic transporters or monocarboxylic acid transporters have been suggested to mediate hydrophilic statins transfer trough BBB (179,180,184). Due to the wide range of articles studying statins effects in different brain diseases, the studies are listed below in individual sections.
2.4.1. Alzheimer disease
Dementia is a broad term to describe a clinical syndrome of progressive cognitive decline, but there are several subtypes depending of their etiology (185). Among them, the most common neurodegenerative disease responsible for dementia is Alzheimer Disease (AD)(185), which is characterized by the presence of amyloid plaques, composed by Amiloid-β (Aβ) peptides generated from amyloid precursor protein, and neurofibrillary tangles (NFT), formed by hyperphosphorylated Tau (p-Tau) proteins on uncommon sites (186,187). Some studies have related AD with an altered cholesterol metabolism and, although the exactly molecular pathways linking cholesterol to AD phenotypes remains unclear(188), the presence of the Apolipoprotein (apo) E4 variant is considered the major genetic risk factor for AD. Supporting these data, some authors have concluded that statins may improve cognitive decline and risk in AD patients, being this protection greater in patients homozygous for ApoE4 (189,190).
At molecular level, statins have showed to reduced p-Tau protein aggregation by inhibiting the mevalonate pathway and decreasing Ab secretion in isogenic induced pluripotent stem cell (iPSC)-derived AD neurons in vitro (188). In the same way, statins promoted extracellular Aβ peptide degradation by the microglia, activating the Insulin-Decreasing Enzyme (IDE) in association with exosomes in vitro (191). Similar results were showed in culture rat neurons and CSF from guinea pig, in which simvastatin decreased Aβ40 and Aβ42 levels associated to reduced plasma and de novo brain cholesterol synthesis (192). On the other hand, statins administration in an experimental Alzheimer model induced by streptozotocin in rats demonstrated neuroprotective effects of statins without reducing plasma cholesterol levels (193). In addition, statins have showed to decreased NFT burden in a mouse model of tauopathy, which was associated to their anti-inflammatory but not to their cholesterol-lowering properties (194). Statins also exhibit a neurotrophic effect in brain cells independent of the mevalonate pathway, as showed by the upregulation of neurotrophins in neurons, microglia, and astrocytes via the PPARα-CREB pathway (195).
As showed for other drugs, the obtained results from clinical trials using statins in AD and dementia patients become controversial since the outcomes reached in clinical studies depend on multiple factors (196). Thus, a research performed in statin users of 65 years of age or older showed that different exposure to statins was associated to lower AD incidence in women and men, but this reduction varied depending on the race/ethnicity and the type of statin used (196). Another clinical trial showed an improvement in the proinflammatory state of AD associated to statin treatment but there were no differences in the cognitive decline among groups (197). In addition, as observed in vivo, simvastatin treatment reduced brain cholesterol levels in AD patients within 12 weeks, but there were no modifications in AD markers levels (198).
Studies evaluating statins safety in this pathology showed also controversial results, while some authors associated statins with dementia risk (199), different reports affirmed the cognitive safety of these drugs (200). Accordingly, there are more recent works about cognitive effects of statins showing opposite results. Thus, while some authors observed and proposed a mechanism by which statins induce cognitive decline (201), other studies evaluated statin use during six years and cognition in the elderly, and concluded that statins could not increase the risk of dementia (202). In a similar way that the former, a systematic review including the most relevant articles about statins and AD determined that there are not clear evidences showing an improvement in cognition and, therefore, there are not major clinical benefits from statins in the treatment of AD (203). Contrary, and supporting the latter article, an observational study re-analyzing previous studies in AD patients suggested that statins may benefit AD patients even more those homozygous for ApoE4 (189). Thus, although statins use in AD patients could be a promising option, further studies are necessary to confirm this hypothesis.
2.4.2. Parkinson disease
Parkinson Disease (PD) is another neurodegenerative affection in which the dopaminergic neurons from substantia nigra progressively lose their function because of the accumulation of the a-synuclein originating the Lewy bodies (204). Some studies have associated a-synuclein aggregations with cholesterol and lipid levels (205), as well as with oxidative stress and inflammation. Accordingly, several authors suggested that statins could be a potential therapeutic target for PD (204,206).
In addition to dopamine and substantia nigra, other neurotransmitters and brain areas are involved in the pathophysiology of PD, including the hippocampus and cerebral cortex (207). Atorvastatin treatment reduced 6-hydroxydopamine (6-OHDA)-induced toxicity and ROS production in cortical, hippocampal and striatal brain sections from rat ex vivo, suggesting that statins could contribute to the decrease of neuroinflammation and ROS production in PD in a cholesterol-independent manner (208). At molecular level, simvastatin reversed the downregulation of PI3K/Akt phosphorylation, and reduced neuroinflammatory mediators and caspase-3 activation induced by 6-OHDA in PC-12 cells (209). Accordingly, high doses of atorvastatin and simvastatin improved, at short and long term, the total locomotor activity in an experimental rat model of PD induced by 6-OHDA (210).
Another potential mechanism implicated in PD and other neurodegenerative diseases is autophagy downregulation (211). Human neuroblastoma cultured cells treated with rosuvastatin showed increased gene levels of Beclin-1 in a dose-dependent manner. In the same way, in an in vitro model of PD induced by rotenone, treatment with rosuvastatin increased autophagy and diminished the α-synuclein aggregations (212). Regarding the latter, in other in vitro PD models, statins administration showed to diminish the accumulation of α-synuclein by a cholesterol-dependent process (213). These effects were also studied in animal models of PD, in which lovastatin administration was associated to lower brain oxidized cholesterol levels, decreased α-synuclein aggregates and improved the neurological function in mice (205).
Contrary to these results, clinical trials and meta-analyses studies about the role of statins in PD remains controversial. Recent research highlighted the complexity of statins and PD showing that short-term use of statins were associated with an increased risk of PD, but the long-term use were not (214,215). However, in another recent meta-analysis, authors observed that statins can reduce the risk of PD, but concluded that additional randomized controlled trials and observational studies are necessary (216).
In conclusion, although there are clear in vitro and in vivo evidences suggesting beneficial effects of statins in PD patients, the controversial clinical results make necessary further studies. Maybe, the two ongoing clinical trials testing the potential benefit of statin therapy in PD patients (NCT04064294, NCT02787590) could clarify this concept.
2.4.3. Stroke
According to the World Health Organization, stoke is defined as a “rapidly developing clinical signs of focal (or global) disturbance of cerebral function, with symptoms lasting 24 hours or longer, or leading to death, with no apparent cause other than of vascular origin (217). However, the American Stroke Association for the 21st century has expanded this definition (proposed in 1970) including any objective evidence of permanent brain, spinal cord, or retinal cell death that could be attributed to a vascular etiology using pathological or imaging evidence with or without clinical symptoms (218). Interestingly, brain nitric oxide (NO) levels have been proposed to play a dual effect in stroke depending of their source. In this sense, while NO derived from inducible or neuronal nitric oxide synthases (iNOS and nNOS respectively) demonstrated to exert neuronal injury in the early and late stage of ischemic stroke, NO derived from the eNOS plays a neuroprotective effect (219). At this point, statins have showed to increase eNOS synthesis by both cholesterol-dependent and non-dependent regulation (220). Regarding this, elevated LDL and oxidized LDL downregulated eNOS expression in human and bovine endothelial cells in vitro, which was prevented by statins treatment (221,222). On the other hand, statins can also modulate eNOS expression by inhibiting RhoA geranylgeranylation and, consequently, the eNOS negative synthesis promoted by this factor. According to these results, experimental mice models of cerebral ischemia suggested an improvement in stroke outcome mediated by statins-induced eNOS upregulation and stabilization via FFPP and GPP inhibition (223–225).
The relevance of MMPs activity in the initial neuro-inflammatory cascade and in recovery phases of stroke has been also widely reported (226). Interestingly, statins decrease tissue plasminogen activator (tPA)-induced MMP-9 levels in culture astrocytes by inhibiting Rho activity (227), as well as MMP-2 and MMP-9 levels in a rat model of cerebral infarction induced by middle cerebral artery occlusion (MCAO)(228). Another important mechanism implicated in stroke severity is the exacerbated ROS production. In this point, statins have showed to play beneficial effects in the cerebrovascular function in a MCAO model and in obese Zucker rats, in both cases by inhibiting NADPH oxidase activity (229,230).
The potential effects of statins in stroke have been tested in several clinical trials during the last decades. The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial, performed in patients with recent stroke or transient ischemic attack, showed that statin use prevented strokes (231,232). At molecular level, despite articles suggesting that beneficial effects of statins in stroke are mediated by both lipid lowering and pleiotropic effects (233), other recent studies attribute these effects only to LDL cholesterol reduction, highlighting the difficulty in separating cholesterol levels reduction from pleiotropic effects (234,235). Therefore, even though the beneficial effects of statins in stroke seems to be clear, future studies evaluating the role of the pleiotropic effects of statins are necessary.
2.4.4. Depression
Although the molecular mechanisms underlying the physiological symptoms of depression remain unclear, some evidences suggest that serotonergic dysfunctions and cortisol dysregulation, loss of neuroplasticity and neuroprotection, or microglia and their associated cytokines, among others, could play a key role in this pathology. Major depressive disorder course with neuroinflammation and ROS exacerbation which causes the subsequent impairment of the neurotransmitters system (236–238). The ability of statins regulating the proinflammatory response has led to some studies testing the potential role of these drugs in depression treatment (184,239). Simvastatin treatment ameliorated depression and anxiety-like behaviors in experimental models of depression induced by chronic mild stress and LPS by decreasing neuroinflammation (240). Additionally, statins reduced oxidative stress and, as was previously demonstrated in vitro (195), prevented brain-derived neurotrophic factor (BD NF) reduction in an LPS-induced depression mice model (241). All together, these results suggest a potentially beneficial role of statins in depression-like animal models in an independent-cholesterol way.
Despite some controversial results (184), a vast number of studies claim the beneficial effects of statins in various psychiatric disorders including depression, in combination with conventional psychotropic medication (239). Although previous studies suggested that lipophilic statins could increase depression risk, a cohort study did not found differences between hydrophilic and lipophilic statins and the risk of incident depression (242). Accordingly, a recent systematic review revealed the effectivity of statins in the treatment of depression, as well as no higher risk of suffering depressive symptoms in statin-users (243). Although all these data support the beneficial effects of using statins in mood-related problems such as depression and anxiety, there are some recent completed or recruiting clinical trials (NCT03435744, NCT04301271) that could shed light on this topic in the future.
Finally, even though there are less data about the role of statins in other brain diseases, their potential role in many other neurological affections and pathologies have been suggested in epilepsy, schizophrenia, spinal cord injuries, CNS cancers, Autism, Huntington disease, Rett Syndrome and X Fragile syndrome (179,180,184).
2.5. Statins in autoimmune diseases
Autoimmune diseases include those disorders in which immune system fails to differentiate its own healthy tissues from foreign, inducing an immune response to self-tissue triggering an abnormal inflammatory response (244). Some of the most common autoimmune diseases are celiac disease, rhabdomyolysis, autoimmune hepatitis, systemic lupus erythematosus (SLE), psoriasis, rheumatoid arthritis (RA), Crohn’s disease (CD) or multiple sclerosis (MS). Taking into account the described potential anti-inflammatory properties of statins, some investigations have tested their use as adjunctive therapy in some autoimmune diseases.
In this sense, an in vitro study suggested that statins could improve thyroid function in patients with Hashimoto’s thyroiditis by lymphocyte apoptosis activation (245). Another study performed in cultured peripheral blood mononuclear cells (PBMCs) from RA patients showed that atorvastatin treatment decreased the expression of several proinflammatory cytokines such as IL-17A, TNFa and IL-6 (246). In agreement with these reports, statins diminished spleen TNFα, IL-6, IFN-gamma and MCP-1 expression in a murine collagen-induced arthritis (CIA) experimental model, as well as delayed CIA development (247,248). Similar results were found in other experimental models miming autoimmune disorders such as experimental autoimmune encephalomyelitis (EAE), MS or SLE in which statins showed to exert anti-inflammatory effects (249–251). However, other authors concluded that atorvastatin did not have protective effects in a SLE mice model (252), although these differences could be attributed to the different SLE model used and/or the kind of statin. Despite these preclinical evidences, statin effects in human autoimmune disorders remain unclear. Some statins have showed to promote an improvement in SLE patients by reducing TNF-α expression, improving endothelial function and decreasing plasma CRP concentration, which lead into better managing of the related cardiovascular complications associated to this pathology (253,254). Similar results were observed in RA patients, whose levels of CRP, Th1/Th2 and CD4/CD8 ratios, and cytokine production and NF-kB activation in cultured IL-1b-stimulated synoviocytes were reduced after statin treatment (255–257).
Several clinical trials have tested statins use in autoimmune diseases. In this sense, high-dose of simvastatin showed to reduce secondary progressive MS development without changes in immune system and was well tolerated and safe in a phase 2 clinical trial (NCT00647348)(258). Similarly, a recruiting phase 2 clinical trial aimed to evaluate the mechanisms implicated in these benefits (NCT03896217), but the current status of this study is unknown. In relapsing-remitting MS patients, another phase 2 clinical trial using high-dose of atorvastatin has also showed to exert beneficial effects alone or in combination with interferon (INF)-β, by increasing the expression of the anti-inflammatory cytokine IL-10 (NCT00616187)(259). In RA, there is a concluded phase 4 clinical trial in which the use of methotrexate in combination with statins in comparison to methotrexate alone has been evaluated, although not results have been published yet (NCT04177173). Similarly, in other autoimmune diseases, such as celiac disease (NCT03011931) or psoriasis (NCT02432040), several completed clinical trials have tried to test the potential beneficial effects of statins, but there are not results published.
To sum up, despite the experimental evidences suggesting statins as a potential adjunctive therapy in autoimmune disorders, mainly based in their anti-inflammatory and immune-modulatory effects, more clinical studies are necessary to confirm this principle.
2.6. Statins in infectious diseases
Apart from non-communicable diseases, statin beneficial effects have been also proposed to treat infectious diseases induced by bacteria, fungi, parasites or viruses, (260–262). A summary of studies evaluating the potential effects of statins in different types of pathogen infections are showed below.
2.6.1. Bacterial infections
The antibacterial properties of statins have been reported in a wide variety of studies. Several authors have attributed the statins-associated antibacterial properties to their basic activity as HMG-CoA reductase inhibitors, since this enzyme is essential in the isoprene biosynthesis in bacterial cells. However, other pleiotropic effects, such as cell growth regulation or apoptosis induction, have also been proposed (263).
The effect of statins have been directly demonstrated in Staphylococcus aureus by affecting bacterial viability and biofilm formation, by inhibiting multiple macromolecular synthesis pathways (264,265). In this line, recent in vitro studies postulated statins as potential topical antibacterial agents to treat skin infections induced by S. aureus, Escherichia coli, Pseudomonas aeruginosa or Serratia marcescens, proposing different mechanism of action such as teichoic acid structures disruption or decreasing cell surface alanine residues surfaces (266). Other in vitro studies, showed that statins may reduce Mycobacterium tuberculosis burden in human and mice macrophages by enhancing autophagy and phagosome maturation, being proposed as adjuvants agents in the treatment of M. tuberculosis infection (267,268). Similarly, the interference of statins with host nonsterol intermediates of cholesterol biosynthetic pathway in Salmonella Typhimurium have been proposed as a potential mechanism to reduce infection in host cells (269). In addition, the antibacterial properties of statins have also been described in other bacteria including Enterobacter cloacae, Streptococcus pneumoniae or Haemophilus influenzae (270).
Similar results were found for some bacteria in experimental mice models. Thus, treatment of experimental skin infection by methicillin-resistant S. aureus with statins significantly reduced the bacterial burden and inflammatory cytokines in the infected wounds (265). Statin treatment also reduced viable chlamydial counts as well as inflammatory cell infiltrates in the lung tissue in experimental Chlamydia pneumoniae infection in mice (271).
The association of reduced bacterial infection in patients with previous statin therapy was demonstrated in the early 2000s. Thereby, in 2005 a study demonstrated that prior therapy with statins may be associated with a reduced rate of severe sepsis and intensive care unit admission, suggesting the development of prospective controlled trials to further demonstrate whether statins may have a role in the primary prevention of sepsis (272). In 2006, in a retrospective cohort analysis, authors showed a significant survival benefit associated with continuing statin therapy in bacteraemic patients, supporting new studies using these drugs as adjuvant agents in sepsis (273). Similarly, another study performed ten years later also highlighted that prior therapy with statins was associated with reduced onset of acute bacterial infections and better outcome in adult patients (274). An additional research concluded that an appropriate antibiotic therapy in combination with statins were associated with lower risk of mortality from bloodstream infections in solid-organ transplant recipients (275). In the same way, and more recently, a meta-analysis approach concluded that there is a reduced risk of Clostridium difficile infection among patients using statins versus non-users (276). Similarly, another recent article based on the current antimicrobial resistance era, proposed the potential use of several non-antibiotic agents with antimicrobial activity, including statins, to help clinicians to combat problematic infections (263). Studies comparing anti-bacterial effects of different statins demonstrated that simvastatin generally exerted the greatest antibacterial activity against Gram-positive bacteria compared to atorvastatin, rosuvastatin, and fluvastatin. However, against Gram-negative bacteria, atorvastatin generally exhibited similar or slightly better activity compared to simvastatin, and both were more potent than rosuvastatin and fluvastatin (277).
Despite all these results suggesting the potential use of statins as a novel adjuvant antibiotic, ironically, some authors suggested that the current extensive use of statins for cardiovascular protection might result in selective pressures for antimicrobial resistance (277). Therefore, elucidating the exact mechanism implicated in the antibacterial activity of statins is essential prior to repurposing statins as a novel adjuvant antibiotic.
2.6.2. Fungal infections
HMG-CoA conversion to mevalonate plays a key role in ergosterol biosynthesis, the major sterol of the yeast cell membrane in fungi, being therefore this step considered a potential therapeutic target to treat fungal infections (278). The number of available antifungal agents is limited, which difficult the treatment of opportunistic fungal infections (279). Some authors have reported the antifungal activity of statins against a wide range of pathogenic yeasts and molds, especially Candida, Aspergillus and Zygomycetes (280–283). In some studies, statins have showed to potentiate the efficacy of available antifungal drugs. In this sense, rosuvastatin and fluvastatin acted synergistically and additively with amphotericin B in inhibiting the fungal growth of Zygomycetes species (281). Similarly, statins potentiated the efficacy of other antifungal drugs, azoles (miconazole, ketoconazole, itraconazole and fluconazole) in a synergistic fashion against a wide range of fungi organisms including some Candida and Aspergillus species (279). In addition, lovastatin synergized with itraconazole against planktonic cells and biofilms of Candida albicans through the regulation on ergosterol biosynthesis pathway (284), as well as simvastatin synergized with fluconazole against Candida species (285). Additionally, simvastatin inhibits Candida glabrata growth and affect the maintenance of mitochondrial DNA (286), possibly by mechanisms involving the interference with ergosterol biosynthesis, the major sterol of the yeast cell membrane (287).
In the clinic, several observational cohort studies have showed that patients using statins have a reduced risk of candidemia-related complications(283). In this sense, in patients with type 2 diabetes mellitus who underwent gastrointestinal surgery, statins administration correlated with lower incidence of cultures positive for Candida species (288). In a single-center matched-cohort study, the obtained results suggested that statins may provide a survival benefit in candidemia (289). Similarly, in a multicenter cohort study of hospitalized adults with candidemia, authors postulated that the use of statins might have a beneficial effect on outcomes of patients with candidemia (290). However, although all these studies describe the positive role of statins in fungal infections, there are some other studies reporting no effects of these drugs in fungal infections. In a retrospective case-control study, authors determined that statins did not decrease risk of invasive mold infections (291). In the same way, a retrospective study showed that statins use did not improve candidemia (292).
Altogether, despite all these promising results, additional prospective, randomized controlled clinical trials are necessary to investigate the real clinical benefit of statins on fungal infections.
2.6.3. Protozoan Parasite infections
Several reports claim the beneficial effects of the use of statins in the battle against parasite infections, in their majority, in combination with other drugs better than alone. In the case of Trypanosoma cruzi infection, known for provoking Chagas disease, an interesting study described that atorvastatin treatment reduced parasite infection in vitro, and showed that the combination of this statin with the current therapeutic option used in this infection (Benznidazole) gave synergistic effects against both blood (trypomastigotes) and intracellular (amastigotes) forms of the parasite (293). Beyond the in vitro studies, simvastatin treatment for one month diminished the infection-produced cardiac leukocyte infiltration and inflammatory mediators, as well as reduced the presence of blood trypomastigotes and heart amastigotes during the acute phase of Chagas disease of the Colombian strain of Trypanosoma cruzi in mice(294). However, authors stated that only simvastatin was not enough for decreasing mortality, pointing out the absence of a combination with a drug used to treat this disease as a limitation of the study (294). Markedly, a phase 2 clinical trial for Chagas disease using antichagasic therapy plus atorvastatin is now in recruiting phase (NCT04984616), and aims to evaluate whether this combined treatment is safe and more efficacious in reducing general inflammation than an antiparasitic therapy alone, by improving endothelial and cardiac functions.
Shifting to leishmaniasis, caused by the parasite Leishmania, different grades of the disease can be observed, from skin lesions to severe organ damage and failure. In several leishmaniasis experimental mouse models, the use of simvastatin was evaluated. Thus, topical simvastatin diminished the severity of the ear swelling, and pre-treatment with a low cholesterol diet and systemic simvastatin reduced footpad lesions in footpad infection (295). In both therapies, the parasite load was lessened, thus diminishing the severity of leishmaniasis. The mechanism proposed by the authors was through the ability of statins to inhibit the HMG-coA inside the macrophages, main objective of leishmania parasite (295). However, the authors did not explore a combination of simvastatin with other drugs, which could be interesting for future studies.
Another parasitic disease that could be treated with statins is toxoplasmosis, induced by Toxoplasma gondii. This parasite uses and accumulates isoprenoids from the host, and can produce their own isoprenoids. Atorvastatin treatment inhibit host isoprenoids formation in cultured fibroblast, thus increasing parasite production of isoprenoids, which could be blocked using zoledronic acid or fosmidomycin (296). The combination of these drugs showed a synergistic effect, ceasing parasite growth. Moreover, in the same study, the administration of atorvastatin in a mouse model infected with null Toxoplasma mutants for the enzyme responsible of isoprenoid synthesis, showed a markedly increase in mice survival when comparing to the infected mice with the not mutated Toxoplasma gondii (296). Considering these results, authors proposed a therapeutic approach with a combination of both treatments against host and parasite isoprenoids synthesis (296).
On the other hand, Malaria is an acute febrile illness caused by Plasmodium parasites, and one of the most severe complication of this infection is cerebral malaria, which has been associated with cerebral endothelial dysfunction and increase mortality of patients (297). Considering the previous described effects of statins, the use of these drugs and Ang II receptor blockers (ARB) to diminish hypertension and protect the cerebral endothelium has been assessed in mice (298). Thus, Plasmodium berghei-infected mice treated with an antimalarial drug plus combination of atorvastatin and ARB showed increase survival rates when compared to those only treated with the antimalarial drug (298). Moreover, endothelial activation markers in mice treated with the combining therapy were lower, as well as cerebral hemorrhages (298). To further study these promising results in patients, two clinical trials testing statins in malaria disease have been performed. Firstly, a phase 1 study evaluating the use of rosuvastatin combined with other drugs terminated due to toxicity problems with the main drug, a new antimalarial therapeutic option (NCT00811356). Secondly, another phase 1 study evaluating the combination of antimalarial and several drugs, including rosuvastatin, is currently in recruiting state (NCT05236530).
As a conclusion, all these studies bring to light that the novel combining therapy of a statin with an antiparasitic drug offers promising effects as a treatment of these diseases, although further studies are necessary to better clarify this idea.
2.6.4. Viral infections
Several evidences demonstrated the inhibitory effects of statins on viral infections by targeting specific mechanisms of the mevalonate pathway. In this sense, statins were able to interrupt the replication of hepatitis C virus (HCV) RNA in cultured hepatoma cells through the inhibition of a necessary geranylgeranylated host protein (299). Similarly, another study demonstrated that statins inhibited human immunodeficiency virus (HIV)-1 infection in cultured primary cells, in animal models and in chronically infected individuals, suggesting the decrease of Rho activity as the potential mechanism (300). HIV-1 replication was also reduced in culture CD4 T cells treated with statins by an increase in the levels of cyclin-dependent kinase inhibitor 1, also called p21cip1, related to mevalonate pathway and independent of p53 (301). Likewise, Dengue virus replication was decreased in human cultured PBMCs treated with statins, in this case thought their interference in the cholesterol biosynthesis (302). A posterior study performed in endothelial and epithelial cultured cells reported a higher effect of statins on dengue virus virion assembly than on replication, and suggested other mechanisms independent of isoprenoid levels (303). Experiments in cultured human endothelial cells demonstrated an antiviral activity of statins against human cytomegalovirus (CMV) also suggesting the involvement of the non-sterol isoprenoid arm of the mevalonate pathway as a potential mechanism (304). More recently, an in-silico prediction highlighted the potential beneficial effects of statins in several viral infections by interfering in the prenylation of viral proteins (305).
Apart from the traditional modulation of cholesterol levels, the anti-inflammatory effects of statins have also been postulated to be involved in their antiviral activity. Thus, statins impaired CMV replication in cultured endothelial cells by inhibiting viral antigen expression, DNA synthesis and viral particle production, all of them conceivably by reducing NF-κB pathway activation (306). Similar results were obtained in Epstein-Barr virus (EBV), in which statin administration inhibited NF-kB activation, induced apoptosis and interrupted alternative splicing genes, cell cycle and cell-cell interaction in cultured B lymphocytes (307,308). The translation of these results to experimental mice models, reported a delayed development of EBV-lymphomas in severe combined immunodeficiency mice treated with statins (307).
It is also worth noting the postulation of statins in other relevant and lethal diseases induced by virus that can result in epidemic or even in pandemic situations, such as Ebola virus disease (EVD), Influenza virus infections or Coronavirus infections (MERS, SARS-CoV1 and SARS-CoV2). In this sense, some authors have demonstrated that statin treatment suppress Ebola virus infectivity by interfering glycoprotein processing, which open new avenues for further studies in adequate animal models of EVD (309). Based on the anti-inflammatory and immunomodulatory effects of statins, some authors postulated the use of these drugs both in treatment and prophylaxis of the Influenza virus (310). In the same way, statins have been also postulated as vaccine adjuvant against influenza HA1 in mice and cynomolgus monkeys considering its ability to prolong antigen retention, enhance antigen presentation and T cell activation (311). Contrary, a recent study concluded that there was no significant difference in influenza vaccine efficiency by statin use in acute myocardial infarction patients (312). On the other hand, some authors postulated that treatment with statins could be a new approach to reduce mortality caused by seasonal and pandemic influenza (313). In a clinical trial performed in patients with acute respiratory distress syndrome (ARDS) (HARP-2, ISRCTN88244364), the use of statins did not show clinical benefits (314), although it is important to highlight that this disease can be induced by other causes different than virus infections. Despite these results, a later study of the HARP-2 trial, that classified ARDS patients into hyper-inflammatory and hypo-inflammatory sub-phenotypes, showed that simvastatin treatment was associated with improved survival in the hyper-inflammatory but not in the hypo-inflammatory sub-phenotype group (315). Regarding coronavirus infections, some authors postulated the potential use of statins in the treatment of the Middle East Respiratory Syndrome (MERS) infection based on the potential of statins modulating MYD88 levels and inhibiting NF-κB pathway activation (316), but there are no studies evaluating this hypothesis. Nevertheless, the potential benefices of statins treatment in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infection, that result in the recent COVID-19 pandemic, have been explored. Our group postulated, for the first time, that statins can help in the fight against COVID-19 by interfering in virus-receptors expression and function, lipid raft disruption, autophagy activation, and attenuation of both inflammatory response and coagulation (317). Subsequent studies have demonstrated some of our proposed mechanisms in vitro (318,319) and showed that the use of statins for primary prevention was associated with lower risks of hospitalization for COVID-19 and of in-hospital death from COVID-19 (320). There are also several completed and ongoing clinical trials evaluating the potential use of statins treatment in COVID-19 patients (NCT04952350; NCT05542095; NCT04348695), whose results could further define if statins are really a therapeutic option in COVID-19 disease.
Conflict of interest
None
Acknowledgements
RRRD, ATM and MRO are financially supported by grants from Instituto de Salud Carlos III (ISCIII). Sara Borrell program (grant number CD20/00042) to RRRD; grant number PI20/000140 to MRO and Red de Investigación Renal RICORS2040 KIDNEY DISEASE (ISCIII) RD21/0005/0002 to MRO; PFIS program (grant number FI18/00222) to A.T.M. LME is financially supported by Comunidad Autónoma de Madrid (CAM) FEDER-a way to build Europe (B2017/BMD-3751 NOVELREN-CM) granted to MRO.
4. REFERENCIAS
- Kantor ED, Rehm CD, Haas JS, Chan AT, Giovannucci EL. Trends in Prescription Drug Use Among Adults in the United States From 1999-2012. JAMA. 2015 Nov;314(17):1818–31.
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990 Feb;343(6257):425–30.
- Rodwell VW, Nordstrom JL, Mitschelen JJ. Regulation of HMG-CoA reductase. Adv Lipid Res. 1976;14:1–74.
- Endo A, Kuroda M, Tsujita Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogenesis produced by Penicillium citrinium. J Antibiot (Tokyo). 1976 Dec;29(12):1346–8.
- Illingworth DR, Tobert JA. HMG-CoA reductase inhibitors. Adv Protein Chem. 2001;56:77–114.
- Corsini A, Bellosta S, Baetta R, Fumagalli R, Paoletti R, Bernini F. New insights into the pharmacodynamic and pharmacokinetic properties of statins. Pharmacol Ther. 1999 Dec;84(3):413–28.
- Bytyçi I, Bajraktari G, Bhatt DL, Morgan CJ, Ahmed A, Aronow WS, et al. Hydrophilic vs lipophilic statins in coronary artery disease: A meta-analysis of randomized controlled trials. J Clin Lipidol. 2017;11(3):624–37.
- Takemoto M, Liao JK. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001 Nov;21(11):1712–9.
- Reiner DJ, Lundquist EA. Small GTPases. WormBook. 2018 Aug;2018:1–65.
- Iannelli F, Lombardi R, Milone MR, Pucci B, De Rienzo S, Budillon A, et al. Targeting Mevalonate Pathway in Cancer Treatment: Repurposing of Statins. Recent Pat Anticancer Drug Discov. 2018;13(2):184–200.
- Nakagomi A, Shibui T, Kohashi K, Kosugi M, Kusama Y, Atarashi H, et al. Differential Effects of Atorvastatin and Pitavastatin on Inflammation, Insulin Resistance, and the Carotid Intima-Media Thickness in Patients with Dyslipidemia. J Atheroscler Thromb. 2015;22(11):1158–71.
- Ascer E, Bertolami MC, Venturinelli ML, Buccheri V, Souza J, Nicolau JC, et al. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis. 2004 Nov;177(1):161–6.
- Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. 2001 Jun;7(6):687–92.
- Montaner S, Perona R, Saniger L, Lacal JC. Multiple signalling pathways lead to the activation of the nuclear factor kappaB by the Rho family of GTPases. J Biol Chem. 1998 May;273(21):12779–85.
- Khurana S, Gupta S, Bhalla H, Nandwani S, Gupta V. Comparison of anti-inflammatory effect of atorvastatin with rosuvastatin in patients of acute coronary syndrome. J Pharmacol Pharmacother. 2015;6(3):130–5.
- van de Ree MA, Huisman M V, Princen HMG, Meinders AE, Kluft C. Strong decrease of high sensitivity C-reactive protein with high-dose atorvastatin in patients with type 2 diabetes mellitus. Atherosclerosis. 2003 Jan;166(1):129–35.
- Freeman JL, Abo A, Lambeth JD. Rac “insert region” is a novel effector region that is implicated in the activation of NADPH oxidase, but not PAK65. J Biol Chem. 1996 Aug;271(33):19794–801.
- Ando S, Kaibuchi K, Sasaki T, Hiraoka K, Nishiyama T, Mizuno T, et al. Post-translational processing of rac p21s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J Biol Chem. 1992 Dec;267(36):25709–13.
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004 Mar;4(3):181–9.
- Delbosc S, Morena M, Djouad F, Ledoucen C, Descomps B, Cristol J-P. Statins, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are able to reduce superoxide anion production by NADPH oxidase in THP-1-derived monocytes. J Cardiovasc Pharmacol. 2002 Oct;40(4):611–7.
- Wood WG, Igbavboa U, Muller WE, Eckert GP. Statins, Bcl-2, and apoptosis: cell death or cell protection? Mol Neurobiol. 2013 Oct;48(2):308–14.
- Ashrafizadeh M, Ahmadi Z, Farkhondeh T, Samarghandian S. Modulatory effects of statins on the autophagy: A therapeutic perspective. J Cell Physiol. 2020 Apr;235(4):3157–68.
- Wu L-M, Wu S-G, Chen F, Wu Q, Wu C-M, Kang C-M, et al. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis. 2020 Jan;293:26–34.
- Dong Q, Yang Y, Song L, Qian H, Xu Z. Atorvastatin prevents mesenchymal stem cells from hypoxia and serum-free injury through activating AMP-activated protein kinase. Int J Cardiol. 2011 Dec;153(3):311–6.
- Xu SZ, Zhong W, Watson NM, Dickerson E, Wake JD, Lindow SW, et al. Fluvastatin reduces oxidative damage in human vascular endothelial cells by upregulating Bcl-2. J Thromb Haemost. 2008 Apr;6(4):692–700.
- Mück AO, Seeger H, Wallwiener D. Class-specific pro-apoptotic effect of statins on human vascular endothelial cells. Z Kardiol. 2004 May;93(5):398–402.
- Patassini S, Giampà C, Martorana A, Bernardi G, Fusco FR. Effects of simvastatin on neuroprotection and modulation of Bcl-2 and BAX in the rat quinolinic acid model of Huntington’s disease. Neurosci Lett. 2008 Dec;448(1):166–9.
- Rajtík T, Čarnická S, Szobi A, Mesárošová L, Máťuš M, Švec P, et al. Pleiotropic effects of simvastatin are associated with mitigation of apoptotic component of cell death upon lethal myocardial reperfusion-induced injury. Physiol Res. 2012;61(Suppl 2):S33-41.
- Doyon M, Hale TM, Huot-Marchand J-E, Wu R, de Champlain J, DeBlois D. Does atorvastatin induce aortic smooth muscle cell apoptosis in vivo? Vascul Pharmacol. 2011;54(1–2):5–12.
- Zhang J, Yang Z, Xie L, Xu L, Xu D, Liu X. Statins, autophagy and cancer metastasis. Int J Biochem Cell Biol. 2013 Mar;45(3):745–52.
- Shi Y, Felley-Bosco E, Marti TM, Stahel RA. Differential effects of lovastatin on cisplatin responses in normal human mesothelial cells versus cancer cells: implication for therapy. PLoS One. 2012;7(9):e45354.
- Hu M-B, Zhang J-W, Gao J-B, Qi Y-W, Gao Y, Xu L, et al. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct Pathol. 2018;42(5):409–15.
- Al-Qatati A, Aliwaini S. Combined pitavastatin and dacarbazine treatment activates apoptosis and autophagy resulting in synergistic cytotoxicity in melanoma cells. Oncol Lett. 2017 Dec;14(6):7993–9.
- Yang Z, Su Z, DeWitt JP, Xie L, Chen Y, Li X, et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine. 2017 May;19:49–59.
- Vosper J, Masuccio A, Kullmann M, Ploner C, Geley S, Hengst L. Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation. Oncotarget. 2015 Feb;6(5):2889–902.
- Wang S-T, Ho HJ, Lin J-T, Shieh J-J, Wu C-Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017 Feb;8(2):e2626.
- Wang F, Liu W, Ning J, Wang J, Lang Y, Jin X, et al. Simvastatin Suppresses Proliferation and Migration in Non-small Cell Lung Cancer via Pyroptosis. Int J Biol Sci. 2018;14(4):406–17.
- Ahmadi M, Amiri S, Pecic S, Machaj F, Rosik J, Łos MJ, et al. Pleiotropic effects of statins: A focus on cancer. Biochim Biophys acta Mol basis Dis. 2020 Dec;1866(12):165968.
- Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res. 2017 Jan;120(1):229–43.
- Mendis S, Davis S, Norrving B. Organizational update: the world health organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke. 2015 May;46(5):e121-2.
- Ortego M, Bustos C, Hernandez-Presa MA, Tunon J, Diaz C, Hernandez G, et al. Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis. 1999 Dec;147(2):253–61.
- Bruder-Nascimento T, Callera GE, Montezano AC, Belin de Chantemele EJ, Tostes RC, Touyz RM. Atorvastatin inhibits pro-inflammatory actions of aldosterone in vascular smooth muscle cells by reducing oxidative stress. Life Sci. 2019 Mar;221:29–34.
- Ortego M, Gomez-Hernandez A, Vidal C, Sanchez-Galan E, Blanco-Colio LM, Martin-Ventura JL, et al. HMG-CoA reductase inhibitors reduce I kappa B kinase activity induced by oxidative stress in monocytes and vascular smooth muscle cells. J Cardiovasc Pharmacol. 2005 May;45(5):468–75.
- Li J, Li J-J, He J-G, Nan J, Guo Y, Xiong C. Atorvastatin decreases C-reactive protein-induced inflammatory response in pulmonary artery smooth muscle cells by inhibiting nuclear factor-kappaB pathway. Cardiovasc Ther. 2010;28(1):8–14.
- Dichtl W, Dulak J, Frick M, Alber HF, Schwarzacher SP, Ares MPS, et al. HMG-CoA reductase inhibitors regulate inflammatory transcription factors in human endothelial and vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003 Jan;23(1):58–63.
- Yoon S-J, Yoon YW, Lee BK, Kwon HM, Hwang K-C, Kim M, et al. Potential role of HMG CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp Mol Med. 2009 Nov;41(11):802–11.
- Tristano AG, Castejon AM, Castro A, Cubeddu LX. Effects of statin treatment and withdrawal on angiotensin II-induced phosphorylation of p38 MAPK and ERK1/2 in cultured vascular smooth muscle cells. Biochem Biophys Res Commun. 2007 Feb;353(1):11–7.
- Hwang A-R, Han J-H, Lim JH, Kang YJ, Woo C-H. Fluvastatin inhibits AGE-induced cell proliferation and migration via an ERK5-dependent Nrf2 pathway in vascular smooth muscle cells. PLoS One. 2017;12(5):e0178278.
- Kaneyuki U, Ueda S, Yamagishi S, Kato S, Fujimura T, Shibata R, et al. Pitavastatin inhibits lysophosphatidic acid-induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth muscle cells by suppressing Rac-1-mediated reactive oxygen species generation. Vascul Pharmacol. 2007 Apr;46(4):286–92.
- Fukuda K, Matsumura T, Senokuchi T, Ishii N, Kinoshita H, Yamada S, et al. Statins meditate anti-atherosclerotic action in smooth muscle cells by peroxisome proliferator-activated receptor-γ activation. Biochem Biophys Res Commun. 2015 Jan;457(1):23–30.
- Seymour K, Stein J, Han X, Maier KG, Gahtan V. Statins and nitric oxide donors affect thrombospondin 1-induced chemotaxis. Vasc Endovascular Surg. 2014;48(7–8):470–5.
- Helkin A, Bruch D, Wilson DR, Gruessner AC, Bader RR, Maier KG, et al. Intraluminal Delivery of Simvastatin Attenuates Intimal Hyperplasia After Arterial Injury. Vasc Endovascular Surg. 2019 Jul;53(5):379–86.
- Katsumoto M, Shingu T, Kuwashima R, Nakata A, Nomura S, Chayama K. Biphasic effect of HMG-CoA reductase inhibitor, pitavastatin, on vascular endothelial cells and angiogenesis. Circ J. 2005 Dec;69(12):1547–55.
- Porter KE, Turner NA, O’Regan DJ, Balmforth AJ, Ball SG. Simvastatin reduces human atrial myofibroblast proliferation independently of cholesterol lowering via inhibition of RhoA. Cardiovasc Res. 2004 Mar;61(4):745–55.
- Rizvi F, Siddiqui R, DeFranco A, Homar P, Emelyanova L, Holmuhamedov E, et al. Simvastatin reduces TGF-β1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: Role of protein phosphatase activation. Int J Cardiol. 2018 Nov;270:228–36.
- Guijarro C, Blanco-Colio LM, Ortego M, Alonso C, Ortiz A, Plaza JJ, et al. 3-Hydroxy-3-methylglutaryl coenzyme a reductase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circ Res. 1998 Sep;83(5):490–500.
- Blanco-Colio LM, Villa A, Ortego M, Hernández-Presa MA, Pascual A, Plaza JJ, et al. 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis. 2002 Mar;161(1):17–26.
- Guijarro C, Blanco-Colio LM, Massy ZA, O’Donnell MP, Kasiske BL, Keane WF, et al. Lipophilic statins induce apoptosis of human vascular smooth muscle cells. Kidney Int Suppl. 1999 Jul;71:S88-91.
- Kang S, Kim K, Noh J-Y, Jung Y, Bae O-N, Lim K-M, et al. Simvastatin induces the apoptosis of normal vascular smooth muscle through the disruption of actin integrity via the impairment of RhoA/Rac-1 activity. Thromb Haemost. 2016 Aug;116(3):496–505.
- Zhao W-B, Fu H, Chang F, Liu J, Wang J, Li F, et al. Effects of various doses of atorvastatin on vascular endothelial cell apoptosis and autophagy in vitro. Mol Med Rep. 2019 Mar;19(3):1919–25.
- Copaja M, Venegas D, Aránguiz P, Canales J, Vivar R, Catalán M, et al. Simvastatin induces apoptosis by a Rho-dependent mechanism in cultured cardiac fibroblasts and myofibroblasts. Toxicol Appl Pharmacol. 2011 Aug;255(1):57–64.
- Fouty BW, Rodman DM. Mevastatin can cause G1 arrest and induce apoptosis in pulmonary artery smooth muscle cells through a p27Kip1-independent pathway. Circ Res. 2003 Mar;92(5):501–9.
- Wei Y-M, Li X, Xu M, Abais JM, Chen Y, Riebling CR, et al. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol Biochem. 2013;31(6):925–37.
- Liu D, Cui W, Liu B, Hu H, Liu J, Xie R, et al. Atorvastatin protects vascular smooth muscle cells from TGF-β1-stimulated calcification by inducing autophagy via suppression of the β-catenin pathway. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2014;33(1):129–41.
- Bustos C, Hernández-Presa MA, Ortego M, Tuñón J, Ortega L, Pérez F, et al. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J Am Coll Cardiol. 1998 Dec;32(7):2057–64.
- Hernández-Presa MA, Martín-Ventura JL, Ortego M, Gómez-Hernández A, Tuñón J, Hernández-Vargas P, et al. Atorvastatin reduces the expression of cyclooxygenase-2 in a rabbit model of atherosclerosis and in cultured vascular smooth muscle cells. Atherosclerosis. 2002 Jan;160(1):49–58.
- Hernández-Presa MA, Ortego M, Tuñón J, Martín-Ventura JL, Mas S, Blanco-Colio LM, et al. Simvastatin reduces NF-kappaB activity in peripheral mononuclear and in plaque cells of rabbit atheroma more markedly than lipid lowering diet. Cardiovasc Res. 2003 Jan;57(1):168–77.
- Rikitake Y, Kawashima S, Takeshita S, Yamashita T, Azumi H, Yasuhara M, et al. Anti-oxidative properties of fluvastatin, an HMG-CoA reductase inhibitor, contribute to prevention of atherosclerosis in cholesterol-fed rabbits. Atherosclerosis. 2001 Jan;154(1):87–96.
- Rodríguez-Vita J, Sánchez-Galán E, Santamaría B, Sánchez-López E, Rodrigues-Díez R, Blanco-Colio LM, et al. Essential role of TGF-beta/Smad pathway on statin dependent vascular smooth muscle cell regulation. PLoS One. 2008;3(12):e3959.
- Lemson MS, Tordoir JH, Daemen MJ, Kitslaar PJ. Intimal hyperplasia in vascular grafts. Eur J Vasc Endovasc Surg Off J Eur Soc Vasc Surg. 2000 Apr;19(4):336–50.
- Yamanouchi D, Banno H, Nakayama M, Sugimoto M, Fujita H, Kobayashi M, et al. Hydrophilic statin suppresses vein graft intimal hyperplasia via endothelial cell-tropic Rho-kinase inhibition. J Vasc Surg. 2005 Oct;42(4):757–64.
- Fujita H, Banno H, Yamanouchi D, Kobayashi M, Yamamoto K, Komori K. Pitavastatin inhibits intimal hyperplasia in rabbit vein graft. J Surg Res. 2008 Aug;148(2):238–43.
- Kitahara M, Kanaki T, Toyoda K, Miyakoshi C, Tanaka S, Tamaki T, et al. NK-104, a newly developed HMG-CoA reductase inhibitor, suppresses neointimal thickening by inhibiting smooth muscle cell growth and fibronectin production in balloon-injured rabbit carotid artery. Jpn J Pharmacol. 1998 Jun;77(2):117–28.
- Mulvany MJ. Small artery remodeling in hypertension. Curr Hypertens Rep. 2002 Feb;4(1):49–55.
- Ruperez M, Rodrigues-Diez R, Blanco-Colio LM, Sanchez-Lopez E, Rodriguez-Vita J, Esteban V, et al. HMG-CoA reductase inhibitors decrease angiotensin II-induced vascular fibrosis: role of RhoA/ROCK and MAPK pathways. Hypertens (Dallas, Tex 1979). 2007 Aug;50(2):377–83.
- Ricca RJ. Infected mesenteric lymphangioma. N Y State J Med. 1991 Aug;91(8):359–61.
- Rodrigues Diez R, Rodrigues-Diez R, Lavoz C, Rayego-Mateos S, Civantos E, Rodriguez-Vita J, et al. Statins inhibit angiotensin II/Smad pathway and related vascular fibrosis, by a TGF-beta-independent process. PLoS One. 2010 Nov;5(11):e14145.
- Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of Rac1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U S A. 2006 May;103(19):7432–7.
- Kang L, Fang Q, Hu S-J. Regulation of phospholamban and sarcoplasmic reticulum Ca2+-ATPase by atorvastatin: implication for cardiac hypertrophy. Arch Pharm Res. 2007 May;30(5):596–602.
- Kang L, Ge C-J, Hu S-J. Beneficial effect of atorvastatin on left ventricular remodeling in spontaneously hypertensive rats. Pharmacology. 2007;80(2–3):120–6.
- Tea BS, Dam T V, Moreau P, Hamet P, deBlois D. Apoptosis during regression of cardiac hypertrophy in spontaneously hypertensive rats. Temporal regulation and spatial heterogeneity. Hypertens (Dallas, Tex 1979). 1999 Aug;34(2):229–35.
- Chen H, Yao L, Chen T, Yu M, Wang L, Chen J. Atorvastatin prevents connexin43 remodeling in hypertrophied left ventricular myocardium of spontaneously hypertensive rats. Chin Med J (Engl). 2007 Nov;120(21):1902–7.
- Severs NJ. Pathophysiology of gap junctions in heart disease. J Cardiovasc Electrophysiol. 1994 May;5(5):462–75.
- Yao L, Chen G-P, Lu X, Zheng L-R, Mou Y, Hu S-J. Effects of atorvastatin on calcium-regulating proteins: a possible mechanism to repair cardiac dysfunction in spontaneously hypertensive rats. Basic Res Cardiol. 2009 May;104(3):258–68.
- Chemaly ER, Troncone L, Lebeche D. SERCA control of cell death and survival. Cell Calcium. 2018 Jan;69:46–61.
- Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019 Study. J Am Coll Cardiol. 2020 Dec;76(25):2982–3021.
- Jones SP, Trocha SD, Lefer DJ. Pretreatment with simvastatin attenuates myocardial dysfunction after ischemia and chronic reperfusion. Arterioscler Thromb Vasc Biol. 2001 Dec;21(12):2059–64.
- Di Napoli P, Taccardi AA, Grilli A, De Lutiis MA, Barsotti A, Felaco M, et al. Chronic treatment with rosuvastatin modulates nitric oxide synthase expression and reduces ischemia-reperfusion injury in rat hearts. Cardiovasc Res. 2005 Jun;66(3):462–71.
- Tiefenbacher CP, Kapitza J, Dietz V, Lee C-H, Niroomand F. Reduction of myocardial infarct size by fluvastatin. Am J Physiol Heart Circ Physiol. 2003 Jul;285(1):H59-64.
- Filusch A, Buss S, Hardt S, Katus HA, Kuecherer HF, Hansen A. Evaluation cardioprotective effects of atorvastatin in rats by real time myocardial contrast echocardiography. Echocardiography. 2008 Oct;25(9):974–81.
- Bulhak A, Roy J, Hedin U, Sjöquist P-O, Pernow J. Cardioprotective effect of rosuvastatin in vivo is dependent on inhibition of geranylgeranyl pyrophosphate and altered RhoA membrane translocation. Am J Physiol Heart Circ Physiol. 2007 Jun;292(6):H3158-63.
- Bulhak AA, Gourine A V, Gonon AT, Sjöquist P-O, Valen G, Pernow J. Oral pre-treatment with rosuvastatin protects porcine myocardium from ischaemia/reperfusion injury via a mechanism related to nitric oxide but not to serum cholesterol level. Acta Physiol Scand. 2005 Feb;183(2):151–9.
- Martín-Ventura JL, Blanco-Colio LM, Gómez-Hernández A, Muñoz-García B, Vega M, Serrano J, et al. Intensive treatment with atorvastatin reduces inflammation in mononuclear cells and human atherosclerotic lesions in one month. Stroke. 2005 Aug;36(8):1796–800.
- Rashid M, Tawara S, Fukumoto Y, Seto M, Yano K, Shimokawa H. Importance of Rac1 signaling pathway inhibition in the pleiotropic effects of HMG-CoA reductase inhibitors. Circ J. 2009 Feb;73(2):361–70.
- Parisi V, Petraglia L, D’Esposito V, Cabaro S, Rengo G, Caruso A, et al. Statin therapy modulates thickness and inflammatory profile of human epicardial adipose tissue. Int J Cardiol. 2019 Jan;274:326–30.
- McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, et al. Dissociation of C-Reactive Protein Localizes and Amplifies Inflammation: Evidence for a Direct Biological Role of C-Reactive Protein and Its Conformational Changes. Front Immunol. 2018;9:1351.
- de Lemos JA, Blazing MA, Wiviott SD, Lewis EF, Fox KAA, White HD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. JAMA. 2004 Sep;292(11):1307–16.
- Schwartz GG, Olsson AG, Ezekowitz MD, Ganz P, Oliver MF, Waters D, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. JAMA. 2001 Apr;285(13):1711–8.
- Ridker PM, Danielson E, Fonseca FAH, Genest J, Gotto AMJ, Kastelein JJP, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008 Nov;359(21):2195–207.
- Yusuf S, Bosch J, Dagenais G, Zhu J, Xavier D, Liu L, et al. Cholesterol Lowering in Intermediate-Risk Persons without Cardiovascular Disease. N Engl J Med. 2016 May;374(21):2021–31.
- Bos MJ, Schipper CMA, Koudstaal PJ, Witteman JCM, Hofman A, Breteler MMB. High serum C-reactive protein level is not an independent predictor for stroke: the Rotterdam Study. Circulation. 2006 Oct;114(15):1591–8.
- Ahmadi-Ahangar A. Predictive ability of C-reactive protein for stroke. Vol. 7, Caspian journal of internal medicine. 2016. p. 151–2.
- 10. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2019. Diabetes Care. 2019 Jan;42(Suppl 1):S103–23.
- Oesterle A, Liao JK. The Pleiotropic Effects of Statins – From Coronary Artery Disease and Stroke to Atrial Fibrillation and Ventricular Tachyarrhythmia. Curr Vasc Pharmacol. 2019;17(3):222–32.
- Foreman KJ, Marquez N, Dolgert A, Fukutaki K, Fullman N, McGaughey M, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet (London, England). 2018 Nov;392(10159):2052–90.
- Ortiz A, Sanchez-Niño MD, Crespo-Barrio M, De-Sequera-Ortiz P, Fernández-Giráldez E, García-Maset R, et al. The Spanish Society of Nephrology (SENEFRO) commentary to the Spain GBD 2016 report: Keeping chronic kidney disease out of sight of health authorities will only magnify the problem. Nefrologia [Internet]. 2019 Jan [cited 2021 Dec 21];39(1):29–34. Available from: https://pubmed.ncbi.nlm.nih.gov/30503082/
- Webster AC, Nagler E V, Morton RL, Masson P. Chronic Kidney Disease. Lancet (London, England). 2017 Mar;389(10075):1238–52.
- Duni A, Dounousi E, Pavlakou P, Eleftheriadis T, Liakopoulos V. Hypertension in Chronic Kidney Disease: Novel Insights. Curr Hypertens Rev [Internet]. 2020 Apr 16 [cited 2022 Nov 7];16(1):45–54. Available from: https://pubmed.ncbi.nlm.nih.gov/30987570/
- Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet (London, England). 2011 Jun;377(9784):2181–92.
- Fried LF. Effects of HMG-CoA reductase inhibitors (statins) on progression of kidney disease. Kidney Int. 2008 Sep;74(5):571–6.
- Navaneethan SD, Pansini F, Perkovic V, Manno C, Pellegrini F, Johnson DW, et al. HMG CoA reductase inhibitors (statins) for people with chronic kidney disease not requiring dialysis. Cochrane database Syst Rev. 2009 Apr;(2):CD007784.
- Haynes R, Lewis D, Emberson J, Reith C, Agodoa L, Cass A, et al. Effects of lowering LDL cholesterol on progression of kidney disease. J Am Soc Nephrol. 2014 Aug;25(8):1825–33.
- Zhang F, Sun D, Chen J, Guan N, Huo X, Xi H. Simvastatin attenuates angiotensin II‑induced inflammation and oxidative stress in human mesangial cells. Mol Med Rep. 2015 Feb;11(2):1246–51.
- Patel S, Mason RM, Suzuki J, Imaizumi A, Kamimura T, Zhang Z. Inhibitory effect of statins on renal epithelial-to-mesenchymal transition. Am J Nephrol. 2006;26(4):381–7.
- Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003 Dec;112(12):1776–84.
- Nabi R, Alvi SS, Shah A, Chaturvedi CP, Iqbal D, Ahmad S, et al. Modulatory role of HMG-CoA reductase inhibitors and ezetimibe on LDL-AGEs-induced ROS generation and RAGE-associated signalling in HEK-293 Cells. Life Sci. 2019 Oct;235:116823.
- Ohigashi M, Kobara M, Takahashi T, Toba H, Wada T, Nakata T. Pitavastatin suppresses hyperglycaemia-induced podocyte injury via bone morphogenetic protein-7 preservation. Clin Exp Pharmacol Physiol. 2017 Mar;44(3):378–85.
- Ohigashi M, Imai N, Toba H, Kobara M, Nakata T. Pitavastatin Exhibits Protective Effects on Podocytes Accompanied by BMP-7 Up-Regulation and Rho Suppression. Pharmacology. 2016;97(5–6):265–76.
- Al-Rasheed NM, Al-Rasheed NM, Bassiouni YA, Hasan IH, Al-Amin MA, Al-Ajmi HN, et al. Simvastatin ameliorates diabetic nephropathy by attenuating oxidative stress and apoptosis in a rat model of streptozotocin-induced type 1 diabetes. Biomed Pharmacother. 2018 Sep;105:290–8.
- Peng H, Luo P, Li Y, Wang C, Liu X, Ye Z, et al. Simvastatin alleviates hyperpermeability of glomerular endothelial cells in early-stage diabetic nephropathy by inhibition of RhoA/ROCK1. PLoS One. 2013;8(11):e80009.
- Kolavennu V, Zeng L, Peng H, Wang Y, Danesh FR. Targeting of RhoA/ROCK signaling ameliorates progression of diabetic nephropathy independent of glucose control. Diabetes. 2008 Mar;57(3):714–23.
- Vieira JMJ, Mantovani E, Rodrigues LT, Dellê H, Noronha IL, Fujihara CK, et al. Simvastatin attenuates renal inflammation, tubular transdifferentiation and interstitial fibrosis in rats with unilateral ureteral obstruction. Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc – Eur Ren Assoc. 2005 Aug;20(8):1582–91.
- Michli E, Gulmi FA, Chou S-Y, Mooppan UMM, Kim H. Atorvastatin preserves renal function in chronic complete unilateral ureteral obstruction. J Urol. 2007 Feb;177(2):781–5.
- Liu Y, Liu M, Zhang Y, Kang L, Chen P, Wang Z, et al. Protective effects of rosuvastatin in experimental renal failure rats via improved endothelial function. Biol Res Nurs. 2013 Jul;15(3):356–64.
- Fiore MC, Jimenez PM, Cremonezzi D, Juncos LI, García NH. Statins reverse renal inflammation and endothelial dysfunction induced by chronic high salt intake. Am J Physiol Renal Physiol. 2011 Aug;301(2):F263-70.
- Zhao J, Cheng Q, Ye P, Yang G, Liu S, Ao Q, et al. Atorvastatin improves pathological changes in the aged kidney by upregulating peroxisome proliferator-activated receptor expression and reducing matrix metalloproteinase-9 and transforming growth factor-β1 levels. Exp Gerontol. 2016 Feb;74:37–42.
- Lavi R, Zhu X-Y, Chade AR, Lin J, Lerman A, Lerman LO. Simvastatin decreases endothelial progenitor cell apoptosis in the kidney of hypertensive hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2010 May;30(5):976–83.
- Talebpour Amiri F, Hamzeh M, Naeimi RA, Ghasemi A, Hosseinimehr SJ. Radioprotective effect of atorvastatin against ionizing radiation-induced nephrotoxicity in mice. Int J Radiat Biol. 2018 Feb;94(2):106–13.
- Kaushik S, Tomar A, Puthanmadhom Narayanan S, Nag TC, Arya DS, Bhatia J. Pitavastatin attenuates cisplatin-induced renal injury by targeting MAPK and apoptotic pathways. J Pharm Pharmacol. 2019 Jul;71(7):1072–81.
- Jaikumkao K, Pongchaidecha A, Thongnak L-O, Wanchai K, Arjinajarn P, Chatsudthipong V, et al. Amelioration of Renal Inflammation, Endoplasmic Reticulum Stress and Apoptosis Underlies the Protective Effect of Low Dosage of Atorvastatin in Gentamicin-Induced Nephrotoxicity. PLoS One. 2016;11(10):e0164528.
- Nam HK, Lee SJ, Kim MH, Rho JH, Son YK, Lee SM, et al. Rosuvastatin attenuates inflammation, apoptosis and fibrosis in a rat model of cyclosporine-induced nephropathy. Am J Nephrol. 2013;37(1):7–15.
- Deshmukh A, Mehta JL. Statins and renal disease: friend or foe? Curr Atheroscler Rep. 2011 Feb;13(1):57–63.
- Zand L, Torres VE, Larson TS, King BF, Sethi S, Bergstralh EJ, et al. Renal hemodynamic effects of the HMG-CoA reductase inhibitors in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant [Internet]. 2016 Aug 1 [cited 2022 Nov 7];31(8):1290–5. Available from: https://pubmed.ncbi.nlm.nih.gov/26614268/
- Ruggenenti P, Perna A, Tonelli M, Loriga G, Motterlini N, Rubis N, et al. Effects of add-on fluvastatin therapy in patients with chronic proteinuric nephropathy on dual renin-angiotensin system blockade: the ESPLANADE trial. Clin J Am Soc Nephrol. 2010 Nov;5(11):1928–38.
- Su X, Zhang L, Lv J, Wang J, Hou W, Xie X, et al. Effect of Statins on Kidney Disease Outcomes: A Systematic Review and Meta-analysis. Am J kidney Dis Off J Natl Kidney Found. 2016 Jun;67(6):881–92.
- Qin X, Dong H, Fang K, Lu F. The effect of statins on renal outcomes in patients with diabetic kidney disease: A systematic review and meta-analysis. Diabetes Metab Res Rev. 2017 Sep;33(6).
- Fitzmaurice C, Akinyemiju TF, Al Lami FH, Alam T, Alizadeh-Navaei R, Allen C, et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018 Nov;4(11):1553–68.
- Chan KKW, Oza AM, Siu LL. The statins as anticancer agents. Clin cancer Res an Off J Am Assoc Cancer Res. 2003 Jan;9(1):10–9.
- Katz MS. Therapy insight: Potential of statins for cancer chemoprevention and therapy. Nat Clin Pract Oncol. 2005 Feb;2(2):82–9.
- Rubins JB, Greatens T, Kratzke RA, Tan AT, Polunovsky VA, Bitterman P. Lovastatin induces apoptosis in malignant mesothelioma cells. Am J Respir Crit Care Med. 1998 May;157(5 Pt 1):1616–22.
- Keyomarsi K, Sandoval L, Band V, Pardee AB. Synchronization of tumor and normal cells from G1 to multiple cell cycles by lovastatin. Cancer Res. 1991 Jul;51(13):3602–9.
- Bouterfa HL, Sattelmeyer V, Czub S, Vordermark D, Roosen K, Tonn JC. Inhibition of Ras farnesylation by lovastatin leads to downregulation of proliferation and migration in primary cultured human glioblastoma cells. Anticancer Res. 2000;20(4):2761–71.
- DeClue JE, Vass WC, Papageorge AG, Lowy DR, Willumsen BM. Inhibition of cell growth by lovastatin is independent of ras function. Cancer Res. 1991 Jan;51(2):712–7.
- Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ [Internet]. 2018 [cited 2022 Nov 7];25(1):65–80. Available from: https://pubmed.ncbi.nlm.nih.gov/29149100/
- Agarwal B, Bhendwal S, Halmos B, Moss SF, Ramey WG, Holt PR. Lovastatin augments apoptosis induced by chemotherapeutic agents in colon cancer cells. Clin cancer Res an Off J Am Assoc Cancer Res. 1999 Aug;5(8):2223–9.
- Wong WWL, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002 Apr;16(4):508–19.
- Mück AO, Seeger H, Wallwiener D. Inhibitory effect of statins on the proliferation of human breast cancer cells. Int J Clin Pharmacol Ther. 2004 Dec;42(12):695–700.
- Aberg M, Wickström M, Siegbahn A. Simvastatin induces apoptosis in human breast cancer cells in a NFkappaB-dependent manner and abolishes the anti-apoptotic signaling of TF/FVIIa and TF/FVIIa/FXa. Thromb Res. 2008;122(2):191–202.
- Fromigué O, Haÿ E, Modrowski D, Bouvet S, Jacquel A, Auberger P, et al. RhoA GTPase inactivation by statins induces osteosarcoma cell apoptosis by inhibiting p42/p44-MAPKs-Bcl-2 signaling independently of BMP-2 and cell differentiation. Cell Death Differ. 2006 Nov;13(11):1845–56.
- Cho S-J, Kim JS, Kim JM, Lee JY, Jung HC, Song IS. Simvastatin induces apoptosis in human colon cancer cells and in tumor xenografts, and attenuates colitis-associated colon cancer in mice. Int J cancer. 2008 Aug;123(4):951–7.
- Zhang W, Wu J, Zhou L, Xie H-Y, Zheng S-S. Fluvastatin, a lipophilic statin, induces apoptosis in human hepatocellular carcinoma cells through mitochondria-operated pathway. Indian J Exp Biol. 2010 Dec;48(12):1167–74.
- Zhao X-H, Xu Z-R, Zhang Q, Yang Y-M. Simvastatin protects human osteosarcoma cells from oxidative stress-induced apoptosis through mitochondrial-mediated signaling. Mol Med Rep. 2012 Feb;5(2):483–8.
- Deng J-L, Zhang R, Zeng Y, Zhu Y-S, Wang G. Statins induce cell apoptosis through a modulation of AKT/FOXO1 pathway in prostate cancer cells. Cancer Manag Res. 2019;11:7231–42.
- Ma Q, Gao Y, Xu P, Li K, Xu X, Gao J, et al. Atorvastatin Inhibits Breast Cancer Cells by Downregulating PTEN/AKT Pathway via Promoting Ras Homolog Family Member B (RhoB). Biomed Res Int. 2019;2019:3235021.
- Xia Z, Tan MM, Wong WW, Dimitroulakos J, Minden MD, Penn LZ. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia. 2001 Sep;15(9):1398–407.
- Kah J, Wüstenberg A, Keller AD, Sirma H, Montalbano R, Ocker M, et al. Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol Rep. 2012 Sep;28(3):1077–83.
- Saito A, Saito N, Mol W, Furukawa H, Tsutsumida A, Oyama A, et al. Simvastatin inhibits growth via apoptosis and the induction of cell cycle arrest in human melanoma cells. Melanoma Res. 2008 Apr;18(2):85–94.
- Alizadeh J, Zeki AA, Mirzaei N, Tewary S, Rezaei Moghadam A, Glogowska A, et al. Mevalonate Cascade Inhibition by Simvastatin Induces the Intrinsic Apoptosis Pathway via Depletion of Isoprenoids in Tumor Cells. Sci Rep. 2017 Mar;7:44841.
- Semenzato G, Amadori G, Tosato F, Sarasin P, Cazzaro G, Gasparotto G. Letter: Lymphocytes in chronic lymphatic leukaemia. Lancet (London, England). 1976 Jan;1(7952):207.
- Kumar N, Cramer GM, Dahaj SAZ, Sundaram B, Celli JP, Kulkarni R V. Stochastic modeling of phenotypic switching and chemoresistance in cancer cell populations. Sci Rep. 2019 Jul;9(1):10845.
- Glynn SA, O’Sullivan D, Eustace AJ, Clynes M, O’Donovan N. The 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, simvastatin, lovastatin and mevastatin inhibit proliferation and invasion of melanoma cells. BMC Cancer. 2008 Jan;8:9.
- Pich C, Teiti I, Rochaix P, Mariamé B, Couderc B, Favre G, et al. Statins Reduce Melanoma Development and Metastasis through MICA Overexpression. Front Immunol. 2013;4:62.
- Kuzu OF, Noory MA, Robertson GP. The Role of Cholesterol in Cancer. Cancer Res [Internet]. 2016 Apr 15 [cited 2022 Nov 7];76(8):2063–70. Available from: https://pubmed.ncbi.nlm.nih.gov/27197250/
- Restivo G, Diener J, Cheng PF, Kiowski G, Bonalli M, Biedermann T, et al. low neurotrophin receptor CD271 regulates phenotype switching in melanoma. Nat Commun. 2017 Dec;8(1):1988.
- Lehraiki A, Cerezo M, Rouaud F, Abbe P, Allegra M, Kluza J, et al. Increased CD271 expression by the NF-kB pathway promotes melanoma cell survival and drives acquired resistance to BRAF inhibitor vemurafenib. Cell Discov. 2015;1:15030.
- Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010 Nov;18(5):510–23.
- Civenni G, Walter A, Kobert N, Mihic-Probst D, Zipser M, Belloni B, et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011 Apr;71(8):3098–109.
- Nishikawa S, Menju T, Takahashi K, Miyata R, Chen-Yoshikawa TF, Sonobe M, et al. Statins may have double-edged effects in patients with lung adenocarcinoma after lung resection. Cancer Manag Res. 2019;11:3419–32.
- Koohestanimobarhan S, Salami S, Imeni V, Mohammadi Z, Bayat O. Lipophilic statins antagonistically alter the major epithelial-to-mesenchymal transition signaling pathways in breast cancer stem-like cells via inhibition of the mevalonate pathway. J Cell Biochem. 2018 Sep;
- Testa JR, Tsichlis PN. AKT signaling in normal and malignant cells. Oncogene. 2005 Nov;24(50):7391–3.
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007 Jun;129(7):1261–74.
- Ghosh-Choudhury N, Mandal CC, Ghosh-Choudhury N, Ghosh Choudhury G. Simvastatin induces derepression of PTEN expression via NFkappaB to inhibit breast cancer cell growth. Cell Signal. 2010 May;22(5):749–58.
- Tsubaki M, Takeda T, Kino T, Obata N, Itoh T, Imano M, et al. Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am J Cancer Res. 2015;5(10):3186–97.
- Kidera Y, Tsubaki M, Yamazoe Y, Shoji K, Nakamura H, Ogaki M, et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rho-associated coiled-coil-containing protein kinase pathway. J Exp Clin Cancer Res. 2010 Sep;29(1):127.
- Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statins and cancer risk: a meta-analysis. JAMA. 2006 Jan;295(1):74–80.
- He Y, Li X, Gasevic D, Brunt E, McLachlan F, Millenson M, et al. Statins and Multiple Noncardiovascular Outcomes: Umbrella Review of Meta-analyses of Observational Studies and Randomized Controlled Trials. Ann Intern Med. 2018 Oct;169(8):543–53.
- Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, et al. Statin therapy in the treatment of active cancer: A systematic review and meta-analysis of randomized controlled trials. PLoS One. 2018;13(12):e0209486.
- Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2012 Nov;367(19):1792–802.
- Fracassi A, Marangoni M, Rosso P, Pallottini V, Fioramonti M, Siteni S, et al. Statins and the Brain: More than Lipid Lowering Agents? Curr Neuropharmacol. 2019;17(1):59–83.
- Ling Q, Tejada-Simon M V. Statins and the brain: New perspective for old drugs. Prog Neuropsychopharmacol Biol Psychiatry. 2016 Apr;66:80–6.
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001 Apr;12(2):105–12.
- Hering H, Lin C-C, Sheng M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci Off J Soc Neurosci. 2003 Apr;23(8):3262–71.
- Sooksawate T, Simmonds MA. Effects of membrane cholesterol on the sensitivity of the GABA(A) receptor to GABA in acutely dissociated rat hippocampal neurones. Neuropharmacology. 2001;40(2):178–84.
- McFarland AJ, Anoopkumar-Dukie S, Arora DS, Grant GD, McDermott CM, Perkins A V, et al. Molecular mechanisms underlying the effects of statins in the central nervous system. Int J Mol Sci. 2014 Nov;15(11):20607–37.
- Duong S, Patel T, Chang F. Dementia: What pharmacists need to know. Can Pharm J (Ott). 2017;150(2):118–29.
- Di Paolo G, Kim T-W. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011 May;12(5):284–96.
- Vanden Dries V, Stygelbout V, Pierrot N, Yilmaz Z, Suain V, De Decker R, et al. Amyloid precursor protein reduction enhances the formation of neurofibrillary tangles in a mutant tau transgenic mouse model. Neurobiol Aging. 2017 Jul;55:202–12.
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, et al. Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-β in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell. 2019 Mar;24(3):363-375.e9.
- Geifman N, Brinton RD, Kennedy RE, Schneider LS, Butte AJ. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer’s disease. Alzheimers Res Ther. 2017 Feb;9(1):10.
- Wang C, Najm R, Xu Q, Jeong D-E, Walker D, Balestra ME, et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med. 2018 May;24(5):647–57.
- Tamboli IY, Barth E, Christian L, Siepmann M, Kumar S, Singh S, et al. Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J Biol Chem. 2010 Nov;285(48):37405–14.
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001 May;98(10):5856–61.
- Tramontina AC, Wartchow KM, Rodrigues L, Biasibetti R, Quincozes-Santos A, Bobermin L, et al. The neuroprotective effect of two statins: simvastatin and pravastatin on a streptozotocin-induced model of Alzheimer’s disease in rats. J Neural Transm. 2011 Nov;118(11):1641–9.
- Boimel M, Grigoriadis N, Lourbopoulos A, Touloumi O, Rosenmann D, Abramsky O, et al. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J Neuropathol Exp Neurol. 2009 Mar;68(3):314–25.
- Roy A, Jana M, Kundu M, Corbett GT, Rangaswamy SB, Mishra RK, et al. HMG-CoA Reductase Inhibitors Bind to PPARalpha to Upregulate Neurotrophin Expression in the Brain and Improve Memory in Mice. Cell Metab. 2015 Aug;22(2):253–65.
- Zissimopoulos JM, Barthold D, Brinton RD, Joyce G. Sex and Race Differences in the Association Between Statin Use and the Incidence of Alzheimer Disease. JAMA Neurol. 2017 Feb;74(2):225–32.
- Pressman P, Gottfried JA. Journal Club: a randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2012 Jul;79(4):e33-6.
- Serrano-Pozo A, Vega GL, Lutjohann D, Locascio JJ, Tennis MK, Deng A, et al. Effects of simvastatin on cholesterol metabolism and Alzheimer disease biomarkers. Alzheimer Dis Assoc Disord. 2010;24(3):220–6.
- Orsi A, Sherman O, Woldeselassie Z. Simvastatin-associated memory loss. Pharmacotherapy. 2001 Jun;21(6):767–9.
- Rojas-Fernandez CH, Goldstein LB, Levey AI, Taylor BA, Bittner V. An assessment by the Statin cognitive safety task force: 2014 update. J Clin Lipidol. 2014;8(3 SUPPL):S5–16.
- Tan B, Rosenfeldt F, Ou R, Stough C. Evidence and mechanisms for statin-induced cognitive decline. Expert Rev Clin Pharmacol. 2019 Apr;1–10.
- Samaras K, Makkar SR, Crawford JD, Kochan NA, Slavin MJ, Wen W, et al. Effects of Statins on Memory, Cognition, and Brain Volume in the Elderly. J Am Coll Cardiol. 2019 Nov;74(21):2554–68.
- Mejías-Trueba M, Pérez-Moreno MA, Fernández-Arche MÁ. Systematic review of the efficacy of statins for the treatment of Alzheimer’s disease. Clin Med. 2018 Feb;18(1):54–61.
- Gelders G, Baekelandt V, Van der Perren A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J Immunol Res. 2018;2018:4784268.
- Koob AO, Ubhi K, Paulsson JF, Kelly J, Rockenstein E, Mante M, et al. Lovastatin ameliorates alpha-synuclein accumulation and oxidation in transgenic mouse models of alpha-synucleinopathies. Exp Neurol. 2010 Feb;221(2):267–74.
- Saeedi Saravi SS, Saeedi Saravi SS, Khoshbin K, Dehpour AR. Current insights into pathogenesis of Parkinson’s disease: Approach to mevalonate pathway and protective role of statins. Biomed Pharmacother. 2017 Jun;90:724–30.
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004 Oct;318(1):121–34.
- Massari CM, Castro AA, Dal-Cim T, Lanznaster D, Tasca CI. In vitro 6-hydroxydopamine-induced toxicity in striatal, cerebrocortical and hippocampal slices is attenuated by atorvastatin and MK-801. Toxicol Vitr an Int J Publ Assoc with BIBRA. 2016 Dec;37:162–8.
- Xu Y-Q, Long L, Yan J-Q, Wei L, Pan M-Q, Gao H-M, et al. Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci Ther. 2013 Mar;19(3):170–7.
- Kumar A, Sharma N, Gupta A, Kalonia H, Mishra J. Neuroprotective potential of atorvastatin and simvastatin (HMG-CoA reductase inhibitors) against 6-hydroxydopamine (6-OHDA) induced Parkinson-like symptoms. Brain Res. 2012 Aug;1471:13–22.
- Tan C-C, Yu J-T, Tan M-S, Jiang T, Zhu X-C, Tan L. Autophagy in aging and neurodegenerative diseases: implications for pathogenesis and therapy. Neurobiol Aging. 2014 May;35(5):941–57.
- Kang SY, Lee S-B, Kim HJ, Kim H-T, Yang HO, Jang W. Autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity in an in vitro model of Parkinson’s disease. Neurosci Lett. 2017 Mar;642:20–6.
- Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, et al. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J Neurochem. 2008 Jun;105(5):1656–67.
- Carroll CB, Wyse RKH, Grosset DG. Statins and Parkinson’s: A complex interaction. Vol. 34, Movement disorders : official journal of the Movement Disorder Society. United States; 2019. p. 934–5.
- Jeong S-M, Jang W, Shin DW. Association of statin use with Parkinson’s disease: Dose-response relationship. Mov Disord. 2019 Jul;34(7):1014–21.
- Yan J, Qiao L, Tian J, Liu A, Wu J, Huang J, et al. Effect of statins on Parkinson’s disease: A systematic review and meta-analysis. Medicine (Baltimore). 2019 Mar;98(12):e14852.
- Donkor ES. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res Treat. 2018;2018:3238165.
- Sacco RL, Kasner SE, Broderick JP, Caplan LR, Connors JJB, Culebras A, et al. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013 Jul;44(7):2064–89.
- Chen Z-Q, Mou R-T, Feng D-X, Wang Z, Chen G. The role of nitric oxide in stroke. Med Gas Res. 2017;7(3):194–203.
- Laufs U. Beyond lipid-lowering: effects of statins on endothelial nitric oxide. Eur J Clin Pharmacol. 2003 Mar;58(11):719–31.
- Hernández-Perera O, Pérez-Sala D, Navarro-Antolín J, Sánchez-Pascuala R, Hernández G, Díaz C, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998 Jun;101(12):2711–9.
- Martínez-González J, Raposo B, Rodríguez C, Badimon L. 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition prevents endothelial NO synthase downregulation by atherogenic levels of native LDLs: balance between transcriptional and posttranscriptional regulation. Arterioscler Thromb Vasc Biol. 2001 May;21(5):804–9.
- Endres M, Laufs U, Liao JK, Moskowitz MA. Targeting eNOS for stroke protection. Trends Neurosci. 2004 May;27(5):283–9.
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, et al. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1998 Jul;95(15):8880–5.
- Sawada N, Liao JK. Targeting eNOS and beyond: emerging heterogeneity of the role of endothelial Rho proteins in stroke protection. Expert Rev Neurother. 2009 Aug;9(8):1171–86.
- Montaner J, Ramiro L, Simats A, Hernández-Guillamon M, Delgado P, Bustamante A, et al. Matrix metalloproteinases and ADAMs in stroke. Cell Mol Life Sci. 2019 Aug;76(16):3117–40.
- Wang S, Lee S-R, Guo S-Z, Kim WJ, Montaner J, Wang X, et al. Reduction of tissue plasminogen activator-induced matrix metalloproteinase-9 by simvastatin in astrocytes. Stroke. 2006 Jul;37(7):1910–2.
- Liu XS, Zhang ZG, Zhang L, Morris DC, Kapke A, Lu M, et al. Atorvastatin downregulates tissue plasminogen activator-aggravated genes mediating coagulation and vascular permeability in single cerebral endothelial cells captured by laser microdissection. J Cereb blood flow Metab Off J Int Soc Cereb Blood Flow Metab. 2006 Jun;26(6):787–96.
- Hong H, Zeng J-S, Kreulen DL, Kaufman DI, Chen AF. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol. 2006 Nov;291(5):H2210-5.
- Erdös B, Snipes JA, Tulbert CD, Katakam P, Miller AW, Busija DW. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol Heart Circ Physiol. 2006 Mar;290(3):H1264-70.
- Amarenco P, Benavente O, Goldstein LB, Callahan A 3rd, Sillesen H, Hennerici MG, et al. Results of the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial by stroke subtypes. Stroke. 2009 Apr;40(4):1405–9.
- Tramacere I, Boncoraglio GB, Banzi R, Del Giovane C, Kwag KH, Squizzato A, et al. Comparison of statins for secondary prevention in patients with ischemic stroke or transient ischemic attack: a systematic review and network meta-analysis. BMC Med. 2019 Mar;17(1):67.
- Aznaouridis K, Masoura C, Vlachopoulos C, Tousoulis D. Statins in Stroke. Curr Med Chem. 2019;26(33):6174–85.
- Amarenco P, Kim JS, Labreuche J, Charles H, Abtan J, Béjot Y, et al. A Comparison of Two LDL Cholesterol Targets after Ischemic Stroke. N Engl J Med. 2020 Jan;382(1):9.
- Wechsler LR. Statins and Stroke – It’s Complicated. Vol. 382, The New England journal of medicine. United States; 2020. p. 81–2.
- Brites D, Fernandes A. Neuroinflammation and Depression: Microglia Activation, Extracellular Microvesicles and microRNA Dysregulation. Front Cell Neurosci. 2015;9:476.
- Horowitz MA, Zunszain PA. Neuroimmune and neuroendocrine abnormalities in depression: two sides of the same coin. Ann N Y Acad Sci. 2015 Sep;1351:68–79.
- Bakunina N, Pariante CM, Zunszain PA. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology. 2015 Mar;144(3):365–73.
- Kim S-W, Kang H-J, Jhon M, Kim J-W, Lee J-Y, Walker AJ, et al. Statins and Inflammation: New Therapeutic Opportunities in Psychiatry. Front psychiatry. 2019;10:103.
- Yu X-B, Zhang H-N, Dai Y, Zhou Z-Y, Xu R-A, Hu L-F, et al. Simvastatin prevents and ameliorates depressive behaviors via neuroinflammatory regulation in mice. J Affect Disord. 2019 Feb;245:939–49.
- Taniguti EH, Ferreira YS, Stupp IJ V, Fraga-Junior EB, Doneda DL, Lopes L, et al. Atorvastatin prevents lipopolysaccharide-induced depressive-like behaviour in mice. Brain Res Bull. 2019 Mar;146:279–86.
- Dave C V, Winterstein AG, Park H, Cook RL, Hartzema AG. Comparative risk of lipophilic and hydrophilic statins on incident depression: A retrospective cohort study. J Affect Disord. 2018 Oct;238:542–6.
- Yatham MS, Yatham KS, Ravindran A V., Sullivan F. Do statins have an effect on depressive symptoms? A systematic review and meta-analysis. J Affect Disord. 2019;257(June):55–63.
- Duan L, Rao X, Sigdel KR. Regulation of Inflammation in Autoimmune Disease. Vol. 2019, Journal of immunology research. 2019. p. 7403796.
- Gullu S, Emral R, Bastemir M, Parkes AB, Lazarus JH. In vivo and in vitro effects of statins on lymphocytes in patients with Hashimoto’s thyroiditis. Eur J Endocrinol. 2005 Jul;153(1):41–8.
- de Oliveira PSS, da Paixão ABF, da Rocha Junior LF, Branco Pinto Duarte AL, Pereira MC, Barreto de Melo Rêgo MJ, et al. Atorvastatin inhibits IL-17A, TNF, IL-6, and IL-10 in PBMC cultures from patients with severe rheumatoid arthritis. Immunobiology. 2020 May;225(3):151908.
- Leung BP, Sattar N, Crilly A, Prach M, McCarey DW, Payne H, et al. A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J Immunol. 2003 Feb;170(3):1524–30.
- Yamagata T, Kinoshita K, Nozaki Y, Sugiyama M, Ikoma S, Funauchi M. Effects of pravastatin in murine collagen-induced arthritis. Rheumatol Int. 2007 May;27(7):631–9.
- de Oliveira DM, de Oliveira EML, Ferrari M de FR, Semedo P, Hiyane MI, Cenedeze MA, et al. Simvastatin ameliorates experimental autoimmune encephalomyelitis by inhibiting Th1/Th17 response and cellular infiltration. Inflammopharmacology. 2015 Dec;23(6):343–54.
- Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol. 2004 Jan;172(2):1273–86.
- Aprahamian T, Bonegio R, Rizzo J, Perlman H, Lefer DJ, Rifkin IR, et al. Simvastatin treatment ameliorates autoimmune disease associated with accelerated atherosclerosis in a murine lupus model. J Immunol. 2006 Sep;177(5):3028–34.
- Graham KL, Lee LY, Higgins JP, Steinman L, Utz PJ, Ho PP. Failure of oral atorvastatin to modulate a murine model of systemic lupus erythematosus. Arthritis Rheum. 2008 Jul;58(7):2098–104.
- Ferreira GA, Navarro TP, Telles RW, Andrade LEC, Sato EI. Atorvastatin therapy improves endothelial-dependent vasodilation in patients with systemic lupus erythematosus: an 8 weeks controlled trial. Rheumatology (Oxford). 2007 Oct;46(10):1560–5.
- Sahebkar A, Rathouska J, Derosa G, Maffioli P, Nachtigal P. Statin impact on disease activity and C-reactive protein concentrations in systemic lupus erythematosus patients: A systematic review and meta-analysis of controlled trials. Autoimmun Rev. 2016 Apr;15(4):344–53.
- An J, Alemao E, Reynolds K, Kawabata H, Solomon DH, Liao KP, et al. Cardiovascular Outcomes Associated with Lowering Low-density Lipoprotein Cholesterol in Rheumatoid Arthritis and Matched Nonrheumatoid Arthritis. J Rheumatol. 2016 Nov;43(11):1989–96.
- Kanda H, Yokota K, Kohno C, Sawada T, Sato K, Yamaguchi M, et al. Effects of low-dosage simvastatin on rheumatoid arthritis through reduction of Th1/Th2 and CD4/CD8 ratios. Mod Rheumatol. 2007;17(5):364–8.
- Lazzerini PE, Lorenzini S, Selvi E, Capecchi PL, Chindamo D, Bisogno S, et al. Simvastatin inhibits cytokine production and nuclear factor-kB activation in interleukin 1beta-stimulated synoviocytes from rheumatoid arthritis patients. Clin Exp Rheumatol. 2007;25(5):696–700.
- Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet (London, England). 2014 Jun;383(9936):2213–21.
- Paul F, Waiczies S, Wuerfel J, Bellmann-Strobl J, Dörr J, Waiczies H, et al. Oral high-dose atorvastatin treatment in relapsing-remitting multiple sclerosis. PLoS One. 2008 Apr;3(4):e1928.
- Parihar SP, Guler R, Brombacher F. Statins: a viable candidate for host-directed therapy against infectious diseases. Nat Rev Immunol [Internet]. 2019 Feb 1 [cited 2022 Nov 7];19(2):104–17. Available from: https://pubmed.ncbi.nlm.nih.gov/30487528/
- Miró-Canturri A, Ayerbe-Algaba R, Smani Y. Drug Repurposing for the Treatment of Bacterial and Fungal Infections. Front Microbiol [Internet]. 2019 [cited 2022 Nov 7];10(JAN). Available from: https://pubmed.ncbi.nlm.nih.gov/30745898/
- Hui DS, Lee N, Chan PK, Beigel JH. The role of adjuvant immunomodulatory agents for treatment of severe influenza. Antiviral Res [Internet]. 2018 Feb 1 [cited 2022 Nov 7];150:202–16. Available from: https://pubmed.ncbi.nlm.nih.gov/29325970/
- Lagadinou M, Onisor MO, Rigas A, Musetescu D-V, Gkentzi D, Assimakopoulos SF, et al. Antimicrobial Properties on Non-Antibiotic Drugs in the Era of Increased Bacterial Resistance. Antibiot (Basel, Switzerland). 2020 Mar;9(3).
- Graziano TS, Cuzzullin MC, Franco GC, Schwartz-Filho HO, de Andrade ED, Groppo FC, et al. Statins and Antimicrobial Effects: Simvastatin as a Potential Drug against Staphylococcus aureus Biofilm. PLoS One. 2015;10(5):e0128098.
- Thangamani S, Mohammad H, Abushahba MFN, Hamed MI, Sobreira TJP, Hedrick VE, et al. Exploring simvastatin, an antihyperlipidemic drug, as a potential topical antibacterial agent. Sci Rep. 2015 Nov;5:16407.
- Ko HHT, Lareu RR, Dix BR, Hughes JD. In vitro antibacterial effects of statins against bacterial pathogens causing skin infections. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2018 Jun;37(6):1125–35.
- Parihar SP, Guler R, Khutlang R, Lang DM, Hurdayal R, Mhlanga MM, et al. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J Infect Dis. 2014 Mar;209(5):754–63.
- Guerra-De-Blas PDC, Torres-González P, Bobadilla-Del-Valle M, Sada-Ovalle I, Ponce-De-León-Garduño A, Sifuentes-Osornio J. Potential Effect of Statins on Mycobacterium tuberculosis Infection. J Immunol Res. 2018;2018:7617023.
- Catron DM, Lange Y, Borensztajn J, Sylvester MD, Jones BD, Haldar K. Salmonella enterica serovar Typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation. Infect Immun. 2004 Feb;72(2):1036–42.
- Masadeh M, Mhaidat N, Alzoubi K, Al-Azzam S, Alnasser Z. Antibacterial activity of statins: a comparative study of atorvastatin, simvastatin, and rosuvastatin. Ann Clin Microbiol Antimicrob. 2012 May;11:13.
- Erkkilä L, Jauhiainen M, Laitinen K, Haasio K, Tiirola T, Saikku P, et al. Effect of simvastatin, an established lipid-lowering drug, on pulmonary Chlamydia pneumoniae infection in mice. Antimicrob Agents Chemother. 2005 Sep;49(9):3959–62.
- Almog Y, Shefer A, Novack V, Maimon N, Barski L, Eizinger M, et al. Prior statin therapy is associated with a decreased rate of severe sepsis. Circulation. 2004 Aug;110(7):880–5.
- Kruger P, Fitzsimmons K, Cook D, Jones M, Nimmo G. Statin therapy is associated with fewer deaths in patients with bacteraemia. Intensive Care Med. 2006 Jan;32(1):75–9.
- Nassaji M, Ghorbani R, Afshar RK. The Effect of Statins Use on the Risk and Outcome of Acute Bacterial Infections in Adult Patients. J Clin Diagn Res. 2015 Nov;9(11):OC09-12.
- Hsu J, Andes DR, Knasinski V, Pirsch J, Safdar N. Statins are associated with improved outcomes of bloodstream infection in solid-organ transplant recipients. Eur J Clin Microbiol Infect Dis Off Publ Eur Soc Clin Microbiol. 2009 Nov;28(11):1343–51.
- Wijarnpreecha K, Panjawatanan P, Thongprayoon C, Ungprasert P. Statins & risk of Clostridium difficile infection: A meta-analysis. Indian J Med Res. 2019 Oct;150(4):359–64.
- Ko HHT, Lareu RR, Dix BR, Hughes JD. Statins: antimicrobial resistance breakers or makers? PeerJ. 2017;5:e3952.
- Andrade-Pavón D, Sánchez-Sandoval E, Rosales-Acosta B, Ibarra JA, Tamariz J, Hernández-Rodríguez C, et al. The 3-hydroxy-3-methylglutaryl coenzyme-A reductases from fungi: a proposal as a therapeutic target and as a study model. Rev Iberoam Micol. 2014;31(1):81–5.
- Nyilasi I, Kocsubé S, Krizsán K, Galgóczy L, Pesti M, Papp T, et al. In vitro synergistic interactions of the effects of various statins and azoles against some clinically important fungi. FEMS Microbiol Lett. 2010 Jun;307(2):175–84.
- Qiao J, Kontoyiannis DP, Wan Z, Li R, Liu W. Antifungal activity of statins against Aspergillus species. Med Mycol. 2007 Nov;45(7):589–93.
- Galgóczy L, Lukács G, Nyilasi I, Papp T, Vágvölgyi C. Antifungal activity of statins and their interaction with amphotericin B against clinically important Zygomycetes. Acta Biol Hung. 2010 Sep;61(3):356–65.
- Macreadie IG, Johnson G, Schlosser T, Macreadie PI. Growth inhibition of Candida species and Aspergillus fumigatus by statins. FEMS Microbiol Lett. 2006 Sep;262(1):9–13.
- Tavakkoli A, Johnston TP, Sahebkar A. Antifungal effects of statins. Pharmacol Ther. 2020;208:107483.
- Zhou Y, Yang H, Zhou X, Luo H, Tang F, Yang J, et al. Lovastatin synergizes with itraconazole against planktonic cells and biofilms of Candida albicans through the regulation on ergosterol biosynthesis pathway. Appl Microbiol Biotechnol. 2018 Jun;102(12):5255–64.
- Menezes EA, Vasconcelos Júnior AA de, Silva CLF, Plutarco FX, Cunha M da C dos SO, Cunha FA. In vitro synergism of simvastatin and fluconazole against Candida species. Rev Inst Med Trop Sao Paulo. 2012 Aug;54(4):197–9.
- Westermeyer C, Macreadie IG. Simvastatin reduces ergosterol levels, inhibits growth and causes loss of mtDNA in Candida glabrata. FEMS Yeast Res. 2007 May;7(3):436–41.
- Liu G, Vellucci VF, Kyc S, Hostetter MK. Simvastatin inhibits Candida albicans biofilm in vitro. Pediatr Res. 2009 Dec;66(6):600–4.
- Spanakis EK, Kourkoumpetis TK, Livanis G, Peleg AY, Mylonakis E. Statin therapy and decreased incidence of positive Candida cultures among patients with type 2 diabetes mellitus undergoing gastrointestinal surgery. Mayo Clin Proc. 2010 Dec;85(12):1073–9.
- Forrest GN, Kopack AM, Perencevich EN. Statins in candidemia: clinical outcomes from a matched cohort study. BMC Infect Dis. 2010 Jun;10:152.
- Cuervo G, Garcia-Vidal C, Nucci M, Puchades F, Fernández-Ruiz M, Mykietiuk A, et al. Effect of statin use on outcomes of adults with candidemia. PLoS One. 2013;8(10):e77317.
- Thompson JN, Huycke MM, Greenfield RA, Kurdgelashvili G, Gentry CA. Case-control study of statin prevention of mould infections. Mycoses. 2011 Sep;54(5):e481-5.
- Welch ML, Liappis AP, Kan VL. Candidemia outcomes not improved with statin use. Med Mycol. 2013 Feb;51(2):219–22.
- Araujo-Lima CF, Peres RB, Silva PB, Batista MM, Aiub CAF, Felzenszwalb I, et al. Repurposing Strategy of Atorvastatin against Trypanosoma cruzi: In Vitro Monotherapy and Combined Therapy with Benznidazole Exhibit Synergistic Trypanocidal Activity. Antimicrob Agents Chemother. 2018 Sep;62(9).
- Silva RR, Shrestha-Bajracharya D, Almeida-Leite CM, Leite R, Bahia MT, Talvani A. Short-term therapy with simvastatin reduces inflammatory mediators and heart inflammation during the acute phase of experimental Chagas disease. Mem Inst Oswaldo Cruz. 2012 Jun;107(4):513–21.
- Parihar SP, Hartley M-A, Hurdayal R, Guler R, Brombacher F. Topical Simvastatin as Host-Directed Therapy against Severity of Cutaneous Leishmaniasis in Mice. Sci Rep. 2016 Sep;6:33458.
- Li Z-H, Ramakrishnan S, Striepen B, Moreno SNJ. Toxoplasma gondii relies on both host and parasite isoprenoids and can be rendered sensitive to atorvastatin. PLoS
