1. INTRODUCCIÓN
El cáncer de pulmón es la causa principal de muerte por cáncer, en la mayoría de los países occidentales. Aunque es conocido que más del 80% de los tumores pulmonares son debidos al tabaco, el hábito de fumar es todavía muy frecuente en nuestra sociedad (1). Además, el cáncer de pulmón está aumentando en individuos que no han fumado nunca, lo que limita el diseño de medidas de prevención. Uno de los factores que contribuye a la elevada mortalidad de este tipo de cáncer es que la mayoría de los casos (>80%) son diagnosticados en fases tardías y, por lo tanto, son todavía muy escasos aquellos que son abordables quirúrgicamente. Otro factor que contribuye a la alta mortalidad del cáncer pulmonar es la poca eficacia de los tratamientos tradicionales (2). En los últimos años, como consecuencia de los esfuerzos en investigación, se ha progresado extraordinariamente en el desarrollo de estrategias que ofrezcan un mejor tratamiento. Además, se han desarrollado terapias individualizadas y de precisión, destinadas a inhibir la función de moléculas que se activan genéticamente en el cáncer o a fortalecer la respuesta inmunológica del organismo frente al cáncer. Estas nuevas estrategias terapéuticas han permitido mejorar la supervivencia de algunos grupos de pacientes. No obstante, aunque importantes, estos avances todavía requieren de estudios biológicos en profundidad porque en los diversos casos de cáncer, incluido el cáncer de pulmón, existe una gran heterogeneidad que depende de características moleculares específicas.
Estas características moleculares individuales van a determinar el comportamiento clínico del tumor (distinto pronóstico, distinta respuesta a tratamientos, distinto nivel de resistencia, distinta evolución, etc.). Si conseguimos entender mejor las peculiaridades biológicas que determinan el comportamiento diferente de cada tumor pulmonar podremos abordar cada caso mediante estrategias terapéuticas individualizadas.
La primera evidencia de que el sistema inmunológico es capaz de reconocer antígenos no propios presentados por células cancerosas y, eventualmente, de eliminarlos fue presentado por Paul Ehrlich a finales del siglo XIX (3). Posteriormente se propuso el concepto de inmunotolerancia el cual postula que, para impedir la acción del sistema inmunitario, los tumores eluden el reconocimiento y la función de los linfocitos T (3). Dicha inmunotolerancia es una característica de prácticamente todos los tumores, confiriéndoles la capacidad de escapar de la destrucción por el sistema inmune del huésped (4). Más recientemente, para referirse a este mismo concepto se ha acuñado el término “inmunoedición del cáncer” que postula tres fases secuenciales: eliminación, equilibrio y escape (5). Brevemente el concepto inmunoedición del cáncer propone que la presión selectiva sostenida ejercida por un sistema inmunitario competente para eliminar células cancerosas desencadena la aparición de clones tumorales que pueden evitar la destrucción por el sistema inmune, alterando el estado de equilibrio y, finalmente, escapar, permitiendo así el crecimiento tumoral. En esta revisión se hará referencia a los términos inmunotolerancia o inmunoescape, indistintamente, para describir la capacidad de las células tumorales de escapar de la acción del sistema inmunológico.
La elucidación de todas las estrategias moleculares que desarrollan las células tumorales para facilitar la inmunotolerancia durante el crecimiento y la progresión del cáncer es un tema de intensa investigación. A parte de permitirnos comprender las bases biológicas del desarrollo tumoral, los mecanismos que generan inmunotolerancia repercuten también en la respuesta del paciente a los tratamientos con inmunoterapia, lo cual puede servir para establecer nuevos biomarcadores y permitir el diseño de nuevos fármacos. Algunas de las estrategias de inmunotolerancia se basan en la represión fisiológica de la respuesta inmune. En una reacción inmunitaria, después de la presentación del antígeno a los linfocitos de células T se produce un incremento en los niveles de ciertos inhibidores de la respuesta inmune. Este es un mecanismo fisiológico conocido como resistencia inmune adaptativa y actúa como un bucle de retroalimentación negativa para atenuar la respuesta de las células T y mantenerlas dentro de un rango fisiológico deseado, previniendo así la sobreestimulación de la auto-reactividad (5). Entre estas moléculas que frenan la respuesta inmune se encuentran CTLA-4 (linfocitos T citotóxicos asociados a antígeno 4) y PD1 (proteína de muerte celular programada 1) y su ligando PD-L1 (6-7). Por lo tanto, el bloqueo de la actividad de estas moléculas restaurará e incrementará la actividad de las células T. La mayoría de los tratamientos de inmunoterapia disponibles actualmente en tumores sólidos están basados en anticuerpos monoclonales diseñados para inhibir la función de CTLA-4, PD-1 y PD-L1, aunque existen otros compuestos en investigación clínica. De forma genérica estos tratamientos se denominan inhibidores del punto de control inmunitario (ICI, del inglés, immune checkpoint inhibitors)
2. EL MICROAMBIENTE TUMORAL LOCAL Y LA INMUNOSUPRESIÓN EN CÁNCER DE PULMÓN
La capacidad de muchos tumores sólidos para suprimir la respuesta inmune del huésped se evidencia por las características inmunosupresoras del microambiente local del tumor. Así pues, se utiliza el término “tumor inflamado” (o caliente) cuando el entorno o microambiente tumoral es rico en infiltrados de varios tipos de células relacionadas con la respuesta inmunitaria, incluidos los linfocitos T CD8+ y CD4+, mientras el término “tumor no inflamado” (o frío) se refiere a aquellos tumores cuyo microambiente está empobrecido en este tipo de células (8). Por otro lado, para iniciar la activación de los linfocitos T CD8+ (CTL) un antígeno debe ser presentado por el complejo mayor de histocompatibilidad clase I (en humanos, HLA-I) de las células tumorales a los CTL (9). Así, los niveles del complejo HLA-I en las células tumorales también pueden ser un indicador del estado del microambiente tumoral (8-9). Finalmente, como se mencionó anteriormente, PD-L1 está directamente implicado en la represión de la actividad de los linfocitos T, por lo que la presencia de altos niveles de PD-L1 indican una respuesta inmune activa asociándose, por lo tanto, a tumores “inflamados” (8-9). Los resultados de algunos estudios atestiguan una relación entre niveles globalmente bajos en el microambiente tumoral de CTLs, del complejo HLA-I y de PD-L1 y un alto grado de inmunotolerancia (10). Algunos perfiles metabólicos y los niveles de ciertas citoquinas en tumores se han asociado también a inmunotolerancia en varios tipos de cáncer, incluyendo el cáncer de pulmón de célula no pequeña (CPCNP), el más frecuente. Ese es el caso de VEGF, que desencadena la elevación en los niveles de PD-1, CTLA-4, o de TIM3, y TGFβ, que mejoran la función de los linfocitos T- reguladores al mismo tiempo que atenúan la actividad de los T-citotóxicos y de las células natural killer (11-12). En un trabajo reciente, en el que se llevó a cabo un estudio de inmunohistoquimica con más de 400 piezas tumorales de CPCNP en estadios quirúrgicos, se observó que dos tercios eran completamente negativos o tenían bajos negativos niveles del complejo HLA-I (proteínas HLA-I y B2M). Esto ocurría tanto en los carcinomas de células escamoso como en los adenocarcinomas (13). En el mismo estudio se encontró una asociación entre inmunotinción positiva del complejo HLA-I y altos niveles de PD-L1 e infiltración intratumoral de CTLs. Los niveles altos de estas moléculas también se asociaron con una respuesta más favorable a inhibidores de PD-L1 y PD1 (13-14).
3. ALTERACIONES GENÉTICAS DE MOLÉCULAS QUE REGULAN LA RESPUESTA INMUNE ESTÁN GENÉTICAMENTE ALTERADAS EN EL CÁNCER DE PULMÓN
El proceso neoplásico promueve la inmunotolerancia, evitando así que el sistema inmune del huésped destruya la célula tumoral. En muchos casos se han identificado eventos genéticos, específicos del tumor, que están detrás de dichos mecanismos de inmunoescape. A continuación, se expone el estado actual del conocimiento sobre los eventos genéticos que ocurren en las células en el curso del desarrollo del tumor que facilitan la inmunotolerancia.
3.1. Alteraciones genéticas que dificultan el reconocimiento inmune y presentación de antígenos
La especificidad de la activación de CTLs contra las células tumorales se basa en el reconocimiento de neoantígenos tumorales, procesados y presentado adecuadamente por el complejo HLA-I en la superficie membrana de las células cancerosas (12,15). El complejo HLA- I consta de una molécula de cadena pesada (HLA-I) (ya sea HLA-A, HLA-B o HLA-C) y una cadena ligera invariable (B2M). La proteína B2M actúa como chaperona, manteniendo la estabilidad estructural del complejo HLA-I y su posición en la superficie celular. B2M es también esencial para el plegamiento de HLA-I y su transporte a la superficie celular para la presentación antigénica (15). La inactivación del complejo HLA-I por alteraciones de los genes HLA-I (principalmente HLA-A) o en B2M ha sido reportado en el cáncer humano. Aunque las alteraciones del gen HLA-A también han sido reportadas en varios tipos de cáncer, su naturaleza altamente polimórfica hace que las alteraciones en este gen sean difíciles de detectar. Es por ello por lo que su prevalencia puede haber sido subestimada (13, 16). Por otra parte, no es bien conocido hasta qué punto las alteraciones en uno sólo de los genes HLA-I afecta al reconocimiento antigénico ya que la pérdida de función de un gen HLA-I podría ser compensada por otro. El caso de las mutaciones en B2M es diferente porque la proteína B2M no es polimórfica ni existen genes de la misma familia con funciones similares. B2M también es esencial para asegurar la maduración y el anclaje a la superficie celular de otros complejos HLA. Las mutaciones en B2M asociadas a cáncer fueron detectadas por primera vez durante la secuenciación genomas y exomas de varios tipos de tumores, incluidas las neoplasias malignas hematopoyéticas (17). En los cánceres de pulmón, se detectó la inactivación bialélica de B2M en todos los tipos histopatológicos de este tipo de cáncer (13). Usando una gran cohorte de tumores primarios de pulmón y de líneas celulares de cáncer observamos que alrededor de 5% de todos los CPCNP y de cáncer de pulmón de célula pequeña (CPCP) primarios contenían mutaciones inactivadoras en este gen, lo que se correlacionó fuertemente con la pérdida de la complejo HLA-I. Asimismo, mutaciones de pérdida de función en B2M han sido detectadas en melanomas, asociadas a la adquisición de resistencia al tratamiento con ICI (18). En cáncer pulmonar la inactivación de genética de B2M también fue identificado en un tumor a la progresión al tratamiento con antiPD-1 (19).
Las moléculas del complejo HLA clase I se localizan en la superficie de células por un proceso de maduración que comienza en el retículo endoplasmático y requiere la participación de otras proteínas, incluyendo CANX, CALR, PDIA3, TAP1, TAP2 y TAPBP (16,20). Por lo tanto, el deterioro de la función de HLA-I en un tumor también puede surgir como consecuencia de alteraciones en cualquiera de estas otras proteínas que afectan a la producción, ensamblaje o transporte a la superficie celular del complejo. En este sentido, la presencia de mutaciones inactivadoras en algunas de las moléculas involucradas en estos procesos (por ejemplo, CALR, PDIA3 o TAP1) se han detectado en cáncer de pulmón, aunque con una frecuencia muy baja (13,20).
3.2. Alteraciones genéticas que afectan la respuesta a interferón gamma (IFNγ)
El IFNγ es una citoquina soluble producida predominantemente por el linfocito T y las células natural killer en respuesta a una variedad de estímulos inflamatorios o inmunitarios (20-23). El IFNγ se une a su receptor afín, que está compuesto por dos subunidades: IFNGR1 e IFNGR2, promoviendo el reclutamiento y activación de las enzimas Janus kinasas (JAK1 y JAK2) las cuales a su vez fosforilan al transductor de señal y activador de la transcripción 1 (STAT1) molécula que, una vez fosforilada y activada, se transloca al núcleo donde se une a elementos promotores específicos y modula transcripción de genes relacionados con la respuesta inmune, incluyendo PD-L1 (21-22). La señalización de IFNγ afecta el desarrollo tumoral porque inhibe directamente el crecimiento de células tumorales y promueve la apoptosis (22-23). Algunos estudios han mostrado la presencia de alteraciones genéticas en IFNGR1/2 o JAK1/2, las cuales convierten a las células tumorales en refractarias a IFNγ (24–27). La información acerca de anormalidades en señalización del IFNγ en el cáncer se ha acumulado desde estos primeros estudios, y su importancia es cada vez más reconocida, especialmente desde el advenimiento de la inmunoterapia. En el melanoma, mutaciones inactivadoras en moléculas que regulan la respuesta a IFNγ (p. ej., JAK1, JAK2 e IFNGR1) han sido identificadas y se ha comprobado que están asociados con la resistencia a tratamiento con ICI (25-26). Pacientes con melanoma cuyos tumores portaban pérdidas de número de copias en genes relacionados con la respuesta a IFNγ mostraron una peor respuesta al tratamiento con el anticuerpo anti-CTLA4, ipilimumab (26). En cáncer de pulmón, la investigación a este respecto es todavía muy limitada, aunque recientemente se publicó la presencia de mutaciones de pérdida de función de JAK1, JAK2 y IFNGR1 en este tipo de cáncer (27). Estas las mutaciones afectaron al 5-10% de los tumores, confirmándose una frecuencia similar en las bases de datos como, por ejemplo, el TCGA. En este trabajo también se demostró que las células que contenían mutaciones inactivadoras de JAK2 eran refractarias al tratamiento con IFNγ y que dichas mutaciones podían co-existir con mutaciones en otros oncogenes o genes supresores tumorales comunes en cáncer de pulmón, tales como como KRAS y STK11, pero no con la inactivación de genes implicados en el reconocimiento inmunológico (B2M, HLA-A, PDIA3 y TAP1) (27). Un resumen de las alteraciones genéticas en estas vías en cáncer de pulmón el cáncer se proporciona en la Fig. 1. Para que la respuesta inmune sea completa es necesario que la célula tumoral genere una respuesta a la estimulación con IFNγ. Así pues, la presencia de una firma genética con niveles altos de transcritos relacionados con la respuesta a esta citoquina está emergiendo como un biomarcador predictivo positivo para el tratamiento con ICI, con implicaciones pronósticas (28).
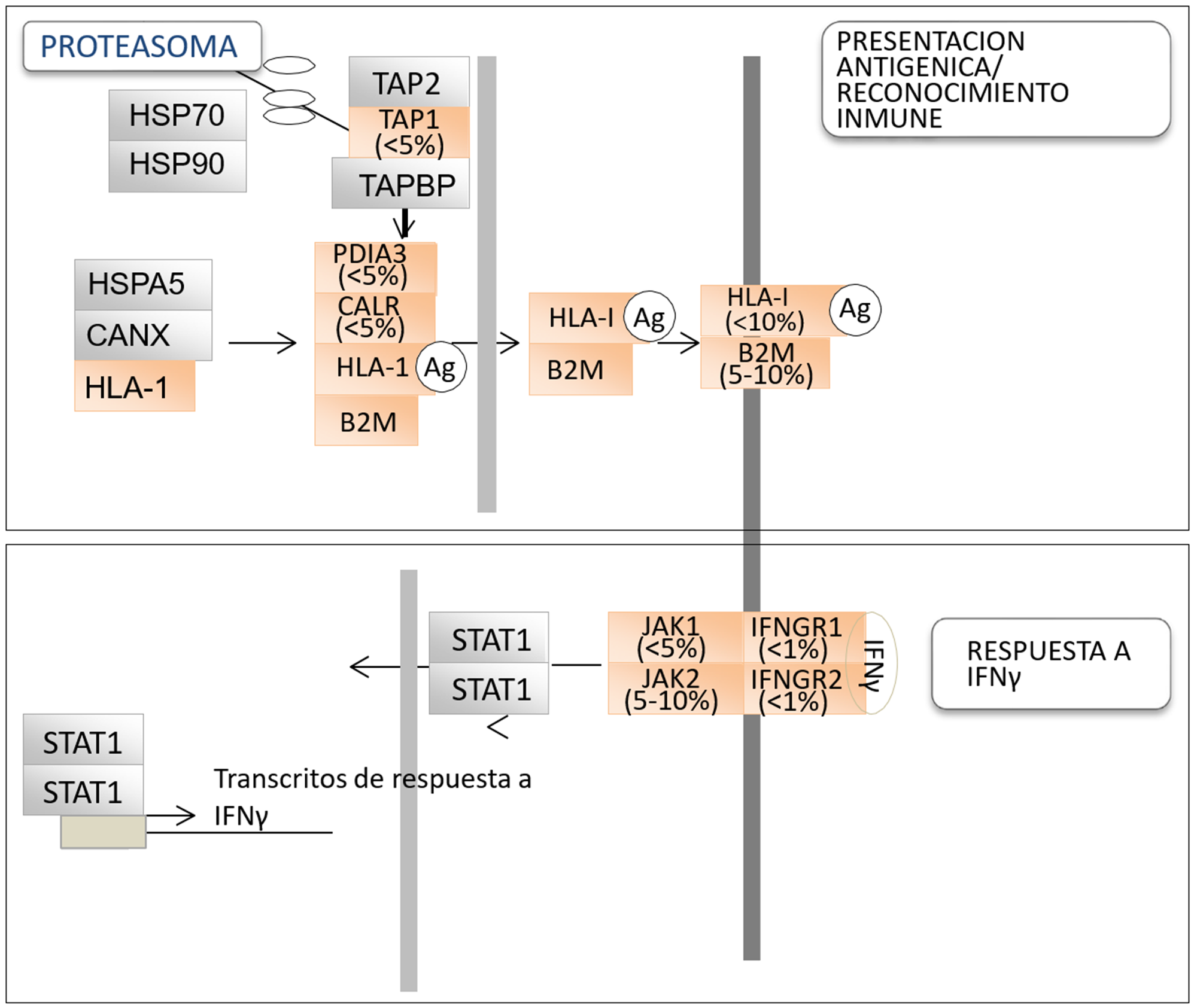
Figura 1. Representación esquemática de las distintas moléculas que intervienen en la presentación del antígeno y el inmunoreconocimiento (panel superior arriba) y en la respuesta a IFNγ (panel inferior). Ag, antígeno. El código de colores para las proteínas indica aquellas en las que se han identificado mutaciones inactivadoras bialélicas en el cáncer alguna vez (naranja) o nunca (gris). La frecuencia de inactivación de estas mutaciones en cáncer de pulmón, según la base de datos TCGA, también se indica.
3.3. Otras alteraciones genéticas que afectan a la respuesta inmune anti-tumoral
Como se mencionó anteriormente, niveles elevados de PD-L1 han sido descritos en distintos tipos de cáncer. Es interesante destacar que tanto JAK2, como CD274 (gen que codifica para PD-L1) y PDCD1LG2 (gen que codifica para PDL2) están todos situados en la misma región cromosómica (9p23- p24.2), región que puede estar amplificada en el cáncer, asociado con elevados niveles de PD-L1 y de infiltración de CTL y con tasas altas de respuesta a antiPD1/anti- PD-L1(29-30). En el CPCNP, la amplificación de CD274 ocurre en aproximadamente un 6% de los tumores (30).
Además, en muchos tipos de cáncer se ha observado una disrupción de la región 3′-UTR del gen CD274 asociado a niveles muy elevados de PD-L1 (31). En conjunto, estas observaciones sugieren que la activación genómica de CD274, por varios mecanismos que desencadenan su regulación positiva, podría facilitar la evasión inmunológica en distintos tipos de cáncer, incluyendo el cáncer de pulmón, promoviendo que tales tumores sean particularmente sensibles al tratamiento con ICI.
Otro factor importante que se ha asociado a una mayor inmunogenicidad y mejor respuesta a ICIs, es el número total de mutaciones somáticas, adquiridas por las células durante el desarrollo del tumor. Una mayor frecuencia de mutaciones en genes codificantes se ha asociado a una mayor carga de neoantígenos. Esto aumenta las posibilidades de que un antígeno capaz de estimular una reacción inmune se exprese en la superficie de la célula tumoral y pueda ser reconocido por el sistema inmunitario (32). Para expresar de forma cuantitativa la frecuencia mutacional en tumores en este contexto, se ha acuñado el término carga mutacional tumoral (TMB, del inglés Tumor Mutational Burden). Este valor se refiere al número total de mutaciones somáticas y no sinónimas que contiene un tumor, normalmente determinado mediante el uso de tecnologías de secuenciación de alto rendimiento. Los tipos de cáncer con una TMB alta incluyen carcinomas de pulmón, melanomas, carcinomas uroteliales y cánceres colorrectales con inestabilidad de microsatélites (33, 34). En el caso del cáncer de pulmón, la TMB elevada se asocia al hábito de fumar, dado que los carcinógenos asociados al tabaco generan alteraciones en el DNA y, por lo tanto, un elevado número de mutaciones y alteraciones cromosómicas (35). La proteína APOBEC (ARNm de apolipoproteína B enzima de edición similar a un polipéptido catalítico) ayuda a proteger de las infecciones virales. En el cáncer la desregulación de esta proteína se ha asociado a elevadas tasas de mutaciones y, por lo tanto, con un índice de TMB elevado (36, 37). Varios estudios han asociado el TMB alto con una mayor respuesta a tratamientos con ICI, independientemente de los niveles de PD-L1 (34, 38-39).
3.4. Efectos de las alteraciones en oncogenes y genes supresores tumorales en la inmunotolerancia
Las anormalidades en algunos de los denominados genes del cáncer, tanto oncogenes como genes supresores de tumores, que codifican para proteínas involucradas en las vías canónicas que promueven el crecimiento tumoral también se han asociado a inmunotolerancia. A continuación, se resumen algunos de los datos obtenidos hasta la fecha a este respecto.
3.4.1. Efectos de la inactivación de STK11 y de los genes del complejo remodelador de cromatina SWI/SNF
Casi el 30% de los adenocarcinomas de pulmón albergan mutaciones somáticas deletéreas en STK11 (40). Estas mutaciones predominan en los tumores de individuos fumadores y, a menudo, coexisten con mutaciones otros genes como TP53 o KRAS, que son esenciales para el desarrollo del cáncer (41, 41). El gen STK11 codifica para una proteína con actividad serina-treonina quinasa que tiene varios sustratos y funciones. De entre los sustratos mejor estudiados es la quinasa dependiente de AMP, AMPK, que participa en el control del metabolismo energético (42). STK11 también se ha asociado con la respuesta inmune. En modelos de ratón, la inactivación de Stk11 se ha asociado con la acumulación de neutrófilos con efectos supresor de células T, en el microambiente inmunitario tumoral, junto con cambios en los niveles de ciertas citoquinas, por ejemplo, aumento de Cxcl7, Cxcl3, Cxcl5, Csf3, GCsfIl33 y IL1a y una disminución de Ccl5 y Cxcl12 (43). Además, los neutrófilos asociados a los tumores de estos ratones demostraron producir niveles elevados de citoquinas asociadas al desarrollo tumoral, incluyendo IL6, en comparación con los neutrófilos de pulmones normales, y niveles más altos de fosfo-Stat3 (pStat3), de acuerdo con el papel de IL6 en la mediación de la activación de Stat3. Estas observaciones apoyan hallazgos previos en los cuales tumores de pulmón inducidos por carcinógenos de ratones que carecen Stat3 tiene un aumento en la producción de quimioquinas proinflamatorias y una disminución en la expresión de moléculas del complejo MHC clase I (44). Además, las células de CPCNP humano en las que STAT3 ha sido silenciado muestran las mismas características (44). De acuerdo con estas observaciones, el tratamiento con anti-PD1 en los modelos murinos con tumores deficientes en Stk11, resultó ineficaz mientras que el tratamiento con un anticuerpo neutralizante de IL6 o un anticuerpo que elimina los neutrófilos produjo beneficios terapéuticos (43). La inactivación de STK11 también se correlacionó con una inmunotinción negativa de PD-L1 y con una peor respuesta a ICI.. En concreto, se observó que los pacientes con CPCNP cuyos tumores portaban alteraciones concurrentes en KRAS y STK11 respondieron peor a ICI que los pacientes cuyos tumores sólo portaban las mutaciones en KRAS (45-46). De acuerdo con estas observaciones, se reportó una asociación significativa entre la presencia de mutaciones inactivadoras en STK11 con una tinción negativa o baja de PD-L1 y con niveles bajos de infiltración de CTL, en una cohorte de pacientes con CPNM sin tratamiento previo (27). Utilizando líneas celulares de cáncer de pulmón deficientes para STK11, se restituyó la expresión de STK11 sin observar cambios en los niveles de PD-L1 ni en moléculas involucradas en la presentación antigénica (complejo HLA-I), independientemente del estado de KRAS. Más recientemente, se ha demostrado que STK11 mejora la expresión de la proteína STING (STimulator of INterferon Genes) que media la producción de interferón tipo I (47).
En conjunto, estas observaciones apoyan el papel de STK11 en la inducción de un entorno inmunosupresor.
Por otra parte, las mutaciones inactivadoras en distintos miembros del complejo remodelador de la cromatina SWI/SNF son comunes en muchos tipos de cáncer. De entre los componentes genéticamente alterados más conocidos están SMARCB1, asociados a tumores rabdoides infantiles y SMARCA4. Ambos genes supresores están inactivados también en CPCNP (48).
Recientemente, en un estudio utilizando la tecnología de edición genética CRISPR/CAS9, se encontró que la eliminación de SMARCA4 y de otros miembros del complejo sensibilizaba a las células a ser eliminadas por las células T (49), lo que implica que la presencia de mutaciones en diferentes miembros del complejo SWI/SNF puede potenciar la respuesta inmunológica.
3.4.2. Efectos de la activación genética de receptores de factores de crecimiento: el caso de MET
Las alteraciones genéticas que conducen a la activación de ciertos receptores de factores de crecimiento con actividad de tirosina quinasa son comunes en determinados tipos de cáncer de pulmón.
Algunas de estas alteraciones se han relacionado con la expresión constitutiva de PD-L1 y con una respuesta pobre a ICIs (27, 50). Tal es el caso de las mutaciones de los genes que codifican para los receptores EGFR, RET y ALK, entre otros, las cuales predominan en adenocarcinomas de pulmón de individuos no fumadores, por lo que los tumores están caracterizados por tener un bajo TMB. Esto puede ayudar a explicar su baja capacidad de respuesta a las ICI a pesar de tener altos niveles de PD-L1. No obstante, los datos no son del todo concluyentes ya que los pacientes con cáncer de pulmón cuyos tumores tienen activación genética de EGFR o ALK fueron excluidos de la mayoría de los ensayos clínicos de fase II/III con ICIs o tuvieron que ser tratados previamente con inhibidores tirosina quinasa específicos (TKI, del inglés tyrosine kinase inhibitor) como criterios de inclusión. Por otra parte, la proteína MET es el receptor del factor de crecimiento de hepatocitos (HGF) que activa múltiples vías de señalización, incluyendo angiogénesis y proliferación celular (67). El oncogén MET se activa mediante mutaciones (METex14) o por amplificación génica. Estas alteraciones se detectan en el 3-4% de los CPCNP (51) y, a diferencia de otros receptores, las mutaciones no se detectan exclusivamente en tumores de individuos no fumadores. La activación aberrante de MET en cáncer de pulmón induce la expresión de moléculas que regulan negativamente la respuesta inmune (p.e. PD-L1, PDL-2 y SOCS1) y de factores de angiogénesis y vasculogénesis (p.e. VEGFA y NRP1), que son esenciales para establecer inmunosupresión (27). A pesar de los niveles altos de PD-L1 en células tumorales con activación de MET, los pacientes de CPCNP con estas alteraciones no presentaban un claro beneficio a ICI, mostrando tasas de respuesta inferiores a las esperadas (52).
3.4.3. Efectos de otras vías relacionadas con el cáncer
Además de lo anteriormente mencionado, anormalidades en otras vías moleculares relacionadas con el desarrollo tumoral se han asociado con un microambiente intratumoral inmunosupresor. Por ejemplo, la inactivación genética de PTEN, un regulador negativo de la vía de señalización PI3K-AKT, se ha asociado con una expresión constitutiva de PD-L1 y con otras características inmunosupresoras, efecto que se incrementa cuando ésta existe simultáneamente con la inactivación del gen supresor tumoral KEAP1 (53-54). Asimismo, se ha observado que la activación de la señalización por Wnt1 induce inmunotolerancia asociada a una menor abundancia de células T y al silenciamiento transcripcional de las quimioquinas CC/CXC en células dendríticas (55). Otro importante elemento inmunosupresor es TIM-3, también conocido como HAVCR2. Se ha demostrado que TIM-3 actúa como un regulador negativo de la respuesta inmune anti-tumoral al estimular la depleción de células T. El bloqueo de la actividad de TIM-3 indujo una mejora en la inmunidad del huésped contra el cáncer y aumentó la producción de interferón-gamma (IFNγ) en las células T. La expresión de TIM-3 se ha detectado en diferentes tipos de cáncer incluido el CPCNP. Un número creciente de estudios preclínicos muestran que el bloqueo de la actividad de TIM3 podría constituir un nuevo tratamiento de inmunoterapia en enfermos con cáncer (56).
3.5. El papel de la regulación epigenética en la inmunotolerancia y en la respuesta a ICIs
Nuestra comprensión del papel de anomalías epigenéticas durante el desarrollo del cáncer ha aumentado considerablemente en las últimas dos décadas. Derivado de este incremento en nuestro conocimiento se ha constatado que cambios en la metilación del ADN o en las modificaciones de las marcas de histonas también influyen en la respuesta del sistema inmune durante el desarrollo tumoral y en la respuesta a las ICI. En un estudio reciente utilizando un análisis masivo de todo el genoma, mediante microarrays de metilación que incluye cerca de
850.000 CpG, se identificó un perfil específico de sitios CpG metilados, la llamada firma EPIMMUNE capaces de predecir una respuesta favorable a terapia antiPD-1 en pacientes con CPCNP (57). Además, en algunos tumores se ha observado represión transcripcional por modificaciones epigenéticas en genes involucrados en los procesos de inmunidad innata y adaptativa. Estas alteraciones son reversibles mediante la utilización de agentes demetilantes del DNA, como la 5-azacitidina, o inhibidores de histona desacetilasas (HDACi), como el entinostat o el ricolinostat (58). Este último HDACi promueve cambios asociados con una mayor activación de las células T y de células presentadoras de antígenos en pacientes con CPCNP (59). De la misma manera, la inhibición del represor transcripcional EZH2 incrementaba la expresión de un subconjunto de genes proinmunogénicos, lo que convierte a EZH2 en una diana terapéutica potencial para su uso en combinación con ICI (60). Finalmente, la des-represión transcripcional de retrovirus endógenos (ERV) se asoció con una elevada abundancia de ARN de doble cadena, que promueve el incremento en la expresión de genes relacionados con la respuesta inmune, tales como los del complejo HLA-I, al mismo tiempo que se promueve la represión transcripcional de EZH2 (61-62). Estos resultados sugieren que fármacos que promuevan la des-represión de ERVs pueden tener un papel importante para potenciar la respuesta a terapias basadas en ICI. La figura 2 resume todas las alteraciones genéticas y epigenéticas en las células tumorales que pueden influir en la respuesta inmune del huésped en CPCNP.
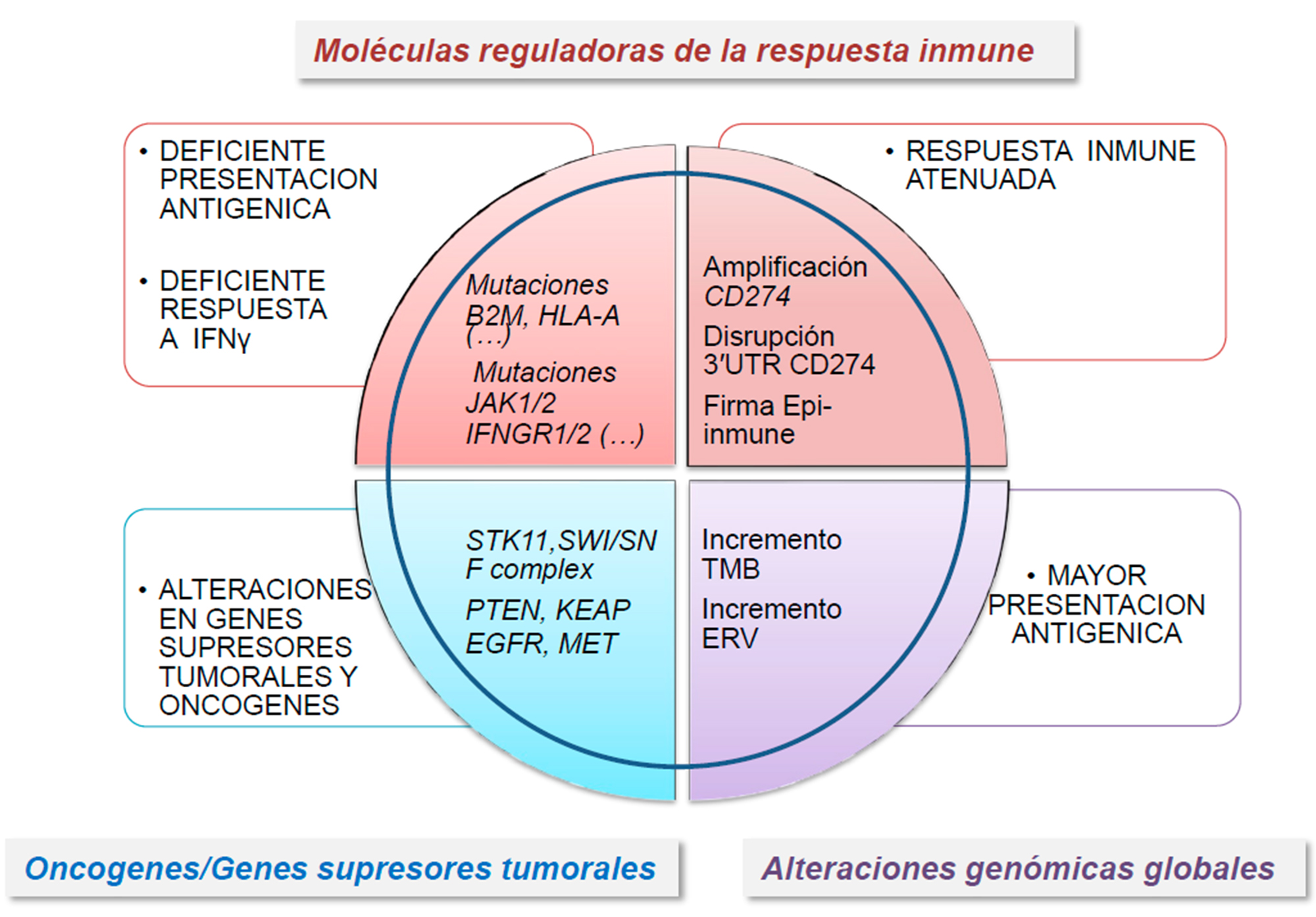
Figura 2. Resumen actual de las alteraciones genéticas que pueden afectar la capacidad del huésped de inducir una respuesta inmune antitumoral
4. INHIBIDORES DEL PUNTO DE CONTROL INMUNITARIO EN EL TRATAMIENTO DEL CÁNCER DE PULMÓN DE CÉLULAS NO PEQUEÑAS: ESTADO ACTUAL
Las opciones terapéuticas para CPCNP han mejorado dramáticamente en los últimos años. De forma similar al desarrollo de inhibidores de la tirosina quinasa (TKI) (6, 63), el advenimiento de los tratamientos con ICI ha dado lugar a respuestas clínicas, en algunos casos impresionantes y duraderas, con implicaciones pronósticas, incrementando la supervivencia global a 5 años. El éxito clínico con la primera generación de ICIs, que consisten en anticuerpos monoclonales cuya diana es CTLA-4 o la interacción PD-1/PD-L1, fue demostrado en ensayos clínicos prospectivos en varios tipos de cáncer (63-65). En la figura 3 se muestra una representación esquemática del funcionamiento de los ICIs que tienen como diana PD-1/PD-L1 y se indican aquellos que son habitualmente utilizados en la práctica clínica.
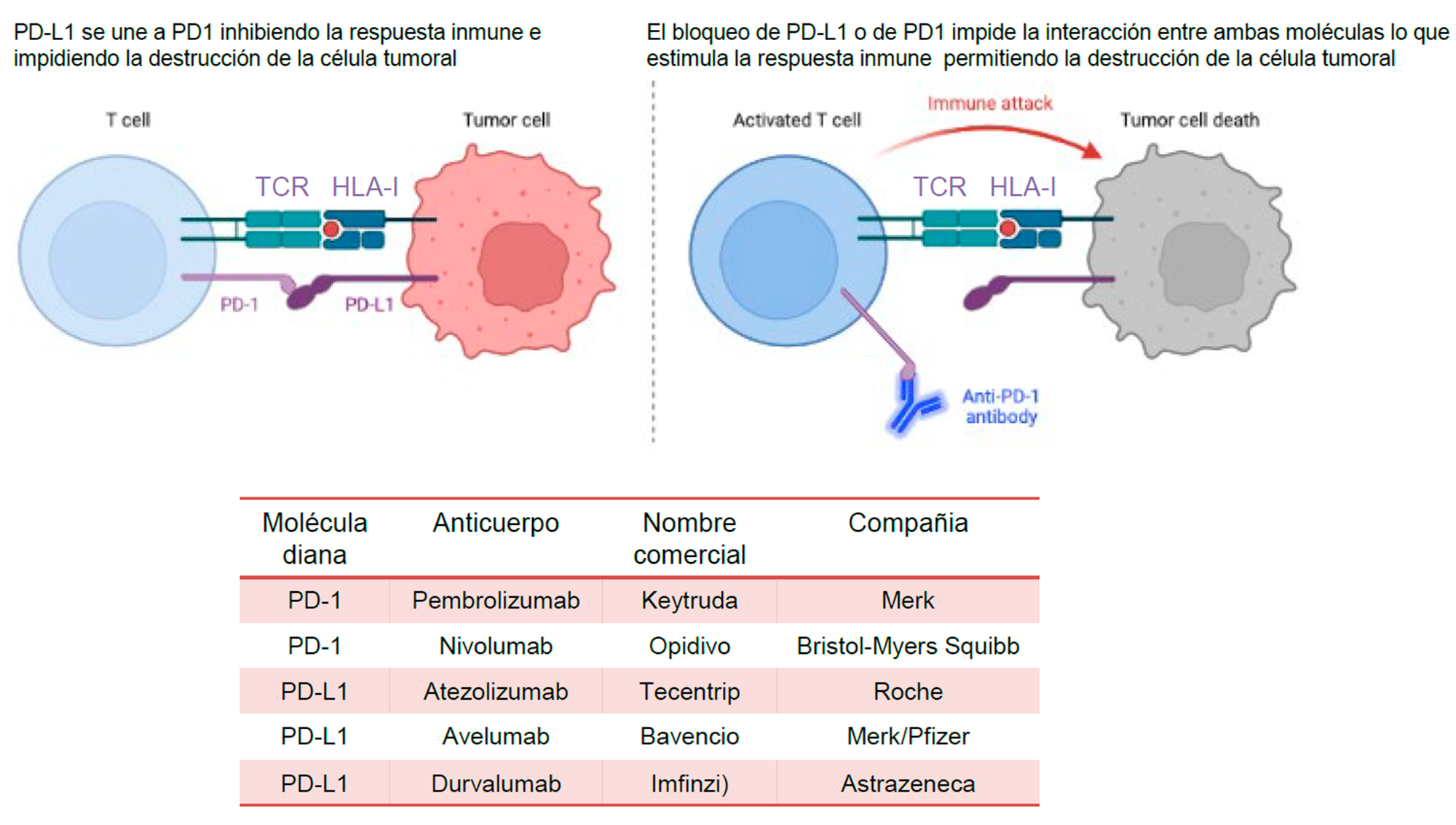
Figura 3. En la parte superior de la imagen se muestra una representación esquemática del reconocimiento de la célula tumoral por el linfocito T (izquierda). En este proceso interviene también el complejo HLA-I. A la derecha se muestra el modo de acción de los anticuerpos monoclonales antiPD-1 (o antiPD-L1). En la parte inferior de la figura se muestra una tabla con algunas de las características de los anticuerpos anti-PD1 y antiPD-L1 más comunes.
Nivolumab y pembrolizumab fueron los primeros anticuerpos antiPD-1 que mostraron beneficio clínico en pacientes con CPCNP avanzado, después de progresar a quimioterapia de primera línea basada en platino. Por otra parte, el atezolizumab fue el primer antiPD-L1 en obtener aprobación para la misma indicación clínica, lo que generó el desarrollo de numerosos ensayos con ICI en tratamientos de primera línea (66-68). Así pues, en el ensayo KEYNOTE 024, pacientes con CPCNP metastásico con elevados niveles de proteína PD-L1, determinados mediante inmunohistoquímica, fueron aleatorizados para recibir bien pembrolizumab o bien el doblete estándar de platino (69). Los resultados mostraron una mayor eficacia del pembrolizumab para todos los parámetros examinados, incluyendo la supervivencia global, indicando la superioridad de este tratamiento con respecto al tratamiento habitual. Estos datos promovieron su aprobación por los organismos reguladores de Estados Unidos (Administración de Alimentos y Medicamentos-FDA) y por la Agencia Europea del Medicamento (EMA) para su administración como agente único para el tratamiento de primera línea de pacientes con CPCNP cuyos tumores tuvieran altos niveles de PD-L1 (≥50% de células tumorales positivas). Sin embargo, en el ensayo clínico CHECKMATE 026 no se encontró beneficio en la supervivencia global cuando se trataron pacientes con CPCNP y niveles moderadamente positivos de PD-L1 (≥5% de células tumorales positivas) con nivolumab en primera línea, al compararse con pacientes que recibieron el tratamiento estándar, lo que sugería que el anti-PD1 en monoterapia y en primera línea solo representa un beneficio en pacientes cuyos tumores expresan PD-L1- alto (70). Por otro lado, los estudios KEYNOTE 189 y 407 demostraron que, para los CPCNP en estadios avanzados, el tratamiento con pembrolizumab en primera línea, en combinación con platino, mejora la supervivencia global, independientemente de los niveles de PD-L1 en el tumor (71-72). Esta combinación recibió la aprobación de la FDA. Finalmente, el estudio de fase III PACIFIC, pacientes con CPCNP en estadio III irresecable y sin progresión de la enfermedad después de dos ciclos de tratamiento basado en platino y radioterapia, fueron aleatorizados en una proporción de 2:1 para recibir durvalumab o placebo. El ensayo mostró un claro beneficio del durvalumab, que mejoró tanto la supervivencia libre de progresión como la supervivencia global. Estos resultados han sido validados en ensayos clínicos posteriores y han servido para cambiar el tratamiento estándar en esta población de pacientes (73). Otros anti-PD-L1 han sido y están siendo testado como terapia de consolidación en el CPCNP de estadio III.
En el CPCNP en estadios resecables (IB-IIIA) se ha comprobado que el uso de ICI más quimioterapia de forma neoadyuvante mejora la supervivencia libre de enfermedad, en comparación con el tratamiento de quimioterapia estándar, lo que llevó a la aprobación de esta estrategia terapéutica por la FDA en marzo de 2022 (74). Por otra parte, dentro del entorno de tratamiento de adyuvancia, el atezolizumab y el pembrolizumab, comparados frente a placebo después de la cirugía y quimioterapia adyuvante, mejoraron significativamente la supervivencia libre de enfermedad (75-76). Desde octubre de 2021, el atezolizumab adyuvante está aprobado por la FDA en pacientes con CPCNP en estadios II-IIIA completamente resecados con expresión de PD-L1⩾1% en células tumorales, mientras que la EMA aprobó el atezolizumab adyuvante en junio de 2022, pero solo para estadios II-IIIA con expresión de PD-L1⩾50 %.
5. RETOS PENDIENTES DE LA INMUNOTERAPIA EN EL CÁNCER DE PULMÓN
La experiencia acumulada de los fármacos dirigidos en el cáncer de pulmón ha demostrado la importancia de los contextos moleculares y genéticos para predecir respuestas favorables. Sin embargo, el escenario con ICI es mucho más complejo porque la diana terapéutica en este caso, ya sea PD1 o PD-L1, no está alterada genéticamente en la célula tumoral, al menos en la mayoría de los casos. En el cáncer de pulmón, la tinción de PD-L1 es actualmente el biomarcador ampliamente aceptado para predecir la respuesta a un tratamiento antiPD1 o antiPD-L1. A pesar de esto, algunos aspectos deben ser tenidos en cuenta. En primer lugar, menos de la mitad de los pacientes con tumores positivos para PD-L1 se benefician del tratamiento con ICI, mientras que algunos pacientes con tumores PD-L1 negativos responden positivamente a estas terapias (69). En segundo lugar, hay algunas limitaciones técnicas que conviene tener en cuenta a la hora de valorar las tinciones de PD-L1, porque los niveles de PD-L1 en las células tumorales son dinámicos y están reguladas por la actividad de los linfocitos T infiltrantes (6). Esto significa que la tinción de PD-L1 dentro de cada tumor puede generar un patrón heterogéneo que dificulta la estandarización de la inmunotinción (9-77). Así, por lo tanto, es necesario diseñar estudios que puedan identificar marcadores predictivos y establecer algoritmos que, al combinar perfiles genético-moleculares con información clínica y patológica, ayuden a seleccionar pacientes que responderán al tratamiento con ICI. En los apartados anteriores se ha descrito una serie de alteraciones genéticas y moleculares que, al influir en la capacidad de respuesta del sistema inmune del huésped, pueden también repercutir en la respuesta a ICIs.
Además, es necesario abordar otro importante reto clínico relacionado con el tratamiento de la ICI, el fenómeno de hiperprogresión, que se refiere al gran aumento de las tasas de crecimiento tumoral y deterioro clínico observado en algunos pacientes durante el tratamiento con ICI (78-79). A pesar de que infrecuente (<10% de los casos), su gran influencia sobre la calidad de vida y supervivencia del paciente exige que se dediquen esfuerzos a comprender sus implicaciones clínicas y causas moleculares.
En los últimos años, la llegada de la era de la inmunoterapia ha traído consigo nuevas ideas e innovaciones en el tratamiento del cáncer de pulmón de células pequeñas (CPCP). A diferencia del CPCNP, el CPCP tiene un origen neuroendocrino. Este tipo de cáncer exhibe un rápido crecimiento y tiene una elevada capacidad metástasis, lo que le confiere altas tasas de mortalidad. El CPCP representa alrededor del 10-15% de todos los casos de cáncer de pulmón y, aproximadamente el 70% de los casos diagnosticados se encuentran en estadios avanzados (80). Durante varias décadas hasta la actualidad, el etopósido combinado con el platino ha sido la terapia estándar de primera línea para el tratamiento del CPCP. No obstante, la tasa de supervivencia a 5 años en este tipo de cáncer sigue siendo muy baja, de alrededor del 2 % con supervivencias globales d unos 10 meses (81). En los últimos años se han llevado a cabo algunos ensayos clínicos para determinar el posible valor terapéuticos de los ICIs en este tipo de cáncer. Los resultados de los ensayos aleatorizados de fase III CASPIAN e IMpower-133 indicaron que la combinación de ICI con quimioterapia basada en platino y etopósido mejoraron la supervivencia global de los pacientes con CPCP, habiéndose convertido en el nuevo tratamiento estándar de primera línea (82-83). Aunque los ICI más quimioterapia mejoran la supervivencia global de los pacientes con CPCP, los beneficios observados hasta ahora no representan un gran avance. Por ello, es necesario identificar biomarcadores predictivos más adecuados y explorar nuevos tratamientos. El uso de más de un ICI simultáneamente, de ICI combinado con otros fármacos o los anticuerpos bi-específicos, representan nuevas estrategias que pueden llegar a ser prometedoras para el tratamiento del CPCP.
6. CONCLUSIONES
El desarrollo de las terapias basadas en ICI ha aportado enormes beneficios terapéuticos en el tratamiento del cáncer de pulmón, por lo que estos compuestos se han incorporado rápidamente en el arsenal terapéutico. Sin embargo, no todos los pacientes responden a estos tratamientos y, de los que lo hacen, más de la mitad progresarán. Las razones que explican la falta de respuesta inicial o la progresión tumoral en estos pacientes no están todavía del todo comprendidas. La compresión exhaustiva de las alteraciones genéticas, tanto las intrínsecas del tumor como las adquiridas durante el tratamiento, están proporcionando información muy valiosa. En particular aquellas alteraciones que permiten a los tumores evadir la respuesta inmunológica del huésped, como aquellas en componentes de sistema de presentación antigénica y de respuesta a IFNγ, merecen más atención. Las alteraciones en genes de estas vías, junto con los niveles de PD-L1 y la determinación de TMB, deberían ser incorporados en ensayos clínicos diseñados para medir la efectividad de los ICI en el tratamiento del CPCNP. Otros oncogenes o genes supresores de tumores bien establecidos, como MET y STK11, así como determinadas alteraciones epigenéticas, también juegan un papel en la inmunotolerancia, aunque los mecanismos todavía no se conocen bien. En definitiva, descifrar el conjunto completo de factores intrínsecos que permiten a los tumores escapar de la vigilancia y acción del sistema inmunitario del huésped sin duda tendrá implicaciones para la selección de pacientes en los tratamientos con ICI. Además, las investigaciones en esta dirección permitirán identificar nuevas dianas para el diseño de nuevas estrategias farmacológicas en el tratamiento del cáncer.
Conflicto de intereses
Se indica que, en el caso de ser premiados, se aceptarán las bases que rigen la convocatoria. Asimismo, se informa de la ausencia de conflicto de intereses del autor.
7. REFERENCIAS
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209-249.
- Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung cancer. Ann Glob Health. 2019;85(1):8.
- Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol.2004;22:329–60.
- Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer.12:252–264.
- Jordan EJ, Kim HR, Arcila ME, Barron D, Chakravarty D, Gao J, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. KEYNOTE-006 investigators. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372:2521–2532.
- Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18:197–218.
- Wei SC, Duffy CR, Alison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov.2018;8:1069–108.
- Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362.
- Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+T cells in tumors. J Exp Med. 2015;212:139–148.
- Johnston CJC, Smyth DJ, Dresser DW, Maizels RM. TGF-b in tolerance, development and regulation of immunity. Cell Immunol. 2016;299:14–22.
- Pereira C, Gimenez-Xavier P, Pros E, Pajares MJ, Moro M, Gomez A, et al. Genomic profiling of patient-derived xenografts for lung cancer identifies B2M inactivation impairing immunorecognition. Clin Cancer Res. 2017;23:3203–3213.
- Brambilla E, Le Teuff G, Marguet S, Lantuejoul S, Dunant A, Graziano S, et al. Prognostic effect of tumor lymphocytic infiltration in resectable non-small-cell lung cancer. J Clin Oncol.2016;34:1223–1230.
- Springer S. Transport and quality control of MHC class I molecules in the early secretory pathway. Curr Opin Immunol. 2015;34:83–90.
- Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152–8.
- Palomero T, Couronné L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA FYN kinase Peripher T cell lymphomas. Nat Genet. 2014;46:166–170.
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med.2016;375:819–829.
- Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420–1435.
- Saigi M, Alburquerque-Bejar JJ, Sanchez-Cespedes M. Determinants of immunological evasion and immunocheckpoint inhibition response in non-small cell lung cancer: the genetic front. Oncogene. 2019 Aug;38(31):5921-5932.
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–848.
- Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109.
- Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998;95:7556–7561.24.
- Ribas A. Releasing the brakes on cancer immunotherapy. N Engl J Med. 2015;373:1490–92.
- Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov.2017;7:188–201.
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167:397–404.
- Saigi M, Alburquerque-Bejar JJ, Mc Leer-Florin A, Pereira C, Pros E, Romero OA, et al. MET-oncogenic and JAK2-Inactivating alterations are independent factors that affect regulation of PD-L1 expression in lung cancer. Clin Cancer Res. 2018;24:4579–4587.
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–2940.
- Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 is upregulated by simultaneous amplification of the PD-L1 and JAK2 genes in non-small cell lung cancer. J Thorac Oncol. 2016;11:62–71.
- Goodman AM, Piccioni D, Kato S, Boichard A, Wang H-Y, Frampton G, et al. Prevalence of PDL1 amplification and preliminary response to immune checkpoint blockade in solid tumors. JAMA Oncol. 2018;4:1237–1244.
- Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant PD-L1 expression through 3’-UTR disruption in multiple cancers. Nature. 2016;534:402–406.
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218.
- Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–1.
- Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51:202–206.
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128.
- Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity.Cancer Discov. 2015;5:704–12.
- Wang S, Jia M, He Z. Liu X-S. APOBEC3B and APOBEC mutational signature as potential predictive markers for immunotherapy response in non-small cell lung cancer. Oncogene. 2018;37:3924–36.
- Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med.2018;378:2093–2104.
- Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell. 2018;33:843–852.
- Sánchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662.
- Sánchez-Cespedes M. A role for LKB1 gene in human cancer beyond the Peutz-Jeghers syndrome. Oncogene. 2007;26:7825–32.
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28.
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76:999–1008.
- Ihara S, Kida H, Arase H, Tripathi LP, Chen YA, Kimura T, et al. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis.Cancer Res. 2012;72:2990–2999.
- Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, et al. Co- occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;8:860–877.
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018;8:822–835.
- Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, Tange S, et al. Suppression of STING Associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019;9:34–45.
- Romero OA, Sanchez Cespedes M. The SWI/SNF genetic blockade: effects in cell differentiation, cancer and developmental diseases. Oncogene. 2014;33:2681–9.
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science. 2018;359:770–775.
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, et al. EGFR Mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res.2016;22:4585–4593.
- Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–859.
- Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, Chen R, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29:2085–2091.
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–216.
- Best SA, De Souza DP, Kersbergen A, Policheni AN, Dayalan S,Tull D, Rathi V, Gray DH, Ritchie ME, McConville MJ, Sutherland KD. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 2018;27:935–943.
- Kerdidani D, Chouvardas P, Arjo AR, Giopanou I, Ntaliarda G, Guo YA, Tsikitis M, Kazamias G, Potaris K, Stathopoulos GT, Zakynthinos S, Kalomenidis I, Soumelis V, Kollias G, Tsoumakidou M. Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma.Nat Commun. 2019;10:1405.
- He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther. 2018;11:7005–7009.
- Duruisseaux M, Martínez-Cardús A, Calleja-Cervantes ME, Moran S, Castro de Moura M, Davalos V, Piñeyro D, Sanchez-Cespedes M, et al. Epigenetic prediction of response to anti- PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med. 2018;6:771–781.
- Vendetti FP, Topper M, Huang P, Dobromilskaya I, Easwaran H, Wrangle J, Baylin SB, Poirier JT, Rudin CM. Evaluation of azacitidine and entinostat as sensitization agents to cytotoxic chemotherapy in preclinical models of non-small cell lung cancer. Oncotarget. 2014;6:56–70.
- Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, Dries R, Li Y, Liu S, et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 2017;7:852–867.
- Zingg D, Arenas-Ramirez N, Sahin D, Rosalia RA, Antunes AT, Haeusel J, Sommer L, Boyman O. The histone methyltransferase EZH2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017;20:854–867.
- Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi M, CP, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018;24:1143–1150.
- Rizvi H, Sanchez-Vega F, La K, ChatilaW, Jonsson P, Halpenny D, Plodkowski A, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti- programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–641.
- Lee CK, Man J, Lord S, Links M, Gebski V, Mok T, Yang JC. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J Thorac Oncol. 2017;12:403–407.
- Roach C, Zhang N, Corigliano E. Development of a companion diagnostic PD-L1 immunohistochemistry assay for pembrolizumab therapy in non-small-cell lung cancer. Appl Immunohistochem Mol Morphol. 2016;24:392–7.
- Fabrizio DA, Milbury C, Yip WK, Ramamurthy X, Bai V, Pattani P, et al. Analytic validation of tumor mutational burden as a companion diagnostic for combination immunotherapy in non small cell lung cancer. Ann Oncol. 2018;29 (Suppl 8):viii14–viii57.
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ,Srinivas S, Tykodi SS, et al. CheckMate 025 Investigators Nivolumab versus everolimus in advanced renalc ell carcinoma. N Engl J Med. 2015;373:1803–1813.
- Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med.2015;373:123–135.
- Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han JY, Molina JR, et al. Pembrolizumab versus docetaxel for previously treated, PDL1-positive, advanced non-small- cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550.
- Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously reated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265.
- Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, et al. KEYNOTE-024 InvestigatorsPembrolizumab versus chemotherapy for PD-L1-positive non- small-cell lung cancer. N Engl J Med. 2016;375:1823–1833.
- Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, Felip E, van den Heuvel MM, Ciuleanu TE, et al. First-line Nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376:2415–2426.
- Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, Hermes B, Çay Şenler F, Csőszi T, Fülöp A, et al. Pembrolizumab plus hemotherapy for squamous non-small cell lung cancer. N Engl J Med. 2018;379:2040–2051.
- Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med.2018;378:2078–2092.
- Spigel DR, Faivre-Finn C, Gray JE, Vicente D, Planchard D, Paz-Ares L, Vansteenkiste JF, Garassino MC, et al. Five year survival outcomes from the PACIFIC trial: durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. J Clin Oncol 2022; 40: 1301–1311.
- Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, Felip E, Broderick SR, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386:1973-1985.
- Felip E, Altorki N, Zhou C, Csőszi T, Vynnychenko I, Goloborodko O, Luft A, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, openlabel, phase 3 trial. Lancet 2021; 398: 1344–1357.
- Paz-Ares L, O’Brien MER, Mauer M, et al. VP3-2022: pembrolizumab (pembro) versus placebo for early-stage non-small cell lung cancer (NSCLC) following complete resection and adjuvant chemotherapy (chemo) when indicated: randomized, triple-blind, phase III EORTC-1416- LCG/ETOP 8-15 – PEARLS/KEYNOTE-091 study. Ann Oncol 2022; 33: 451–453.
- Tsao MS, Kerr KM, Dacic S, Yatabe Y, Hirsch FR. IASLC Atlas of PD-L1 testing in lung cancer. (2017) https://www.iaslc.org/ publications/iaslc-atlas-pd-l1-testing-lung-cancer.
- Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, Chaput N, Eggermont A, Marabelle A, Soria JC, Ferté C. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017;23:1920–1928.
- Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–4250.
- Saltos A, Shafique M and Chiappori A. Update on the biology, management, and treatment of small cell lung cancer (SCLC). Front Oncol 2020; 10: 1074.
- Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N,Hotta K, Trukhin D, Statsenko G, Hochmair MJ, et al. Durvalumabplus platinum-etoposide versus platinum-etoposide in first-line treatment of extensivestage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet 2019; 394: 1929-1939.
- Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, Huemer F, et al. first-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220-2229.
