1. INTRODUCCIÓN: BREVE DESCRIPCIÓN DE LOS LAUREADOS
El premio Nobel de Química de 2022 ha sido concedido a Carolyn Ruth Bertozzi, Morten Peter Meldal y Karl Barry Sharpless “por el desarrollo de la química click y bioortogonal” (1,2).

Figura 1. Los tres galardonados con el premio Nobel de Química de 2022
Karl Barry Sharpless, nacido en Filadelfia en 1941, consiguió una licenciatura en Química por el Darthmouth College (1963) y un doctorado en Química por la Universidad de Stanford (1968) por un trabajo sobre la química del escualeno dirigido por Eugene Van Tamelen. Continuó su formación con estudios postdoctorales en la Universidad de Stanford (1968), uniéndose al laboratorio de James P. Collman, un especialista en química organometálica. Posteriormente, y dentro todavía de su etapa postdoctoral, trabajó en la Universidad de Harvard (1969) en el laboratorio de Konrad E. Bloch, un especialista en enzimología que había sido galardonado con el premio Nobel en 1964 por su trabajo sobre el metabolismo del colesterol y los ácidos grasos. Sharpless ha sido profesor en el Massachussetts Institute of Technology (1970-77), en la Universidad de Stanford (1977-80), de nuevo en el MIT (1980-90) y finalmente en el Scripps Research Institute (1990-actualidad). Cabe destacar que Sharpless es el segundo investigador en ser galardonado dos veces con el Premio Nobel de Química, habiéndolo recibido por primera vez en 2001 por el desarrollo de reacciones de oxidación enantioselectivas (figura 2).
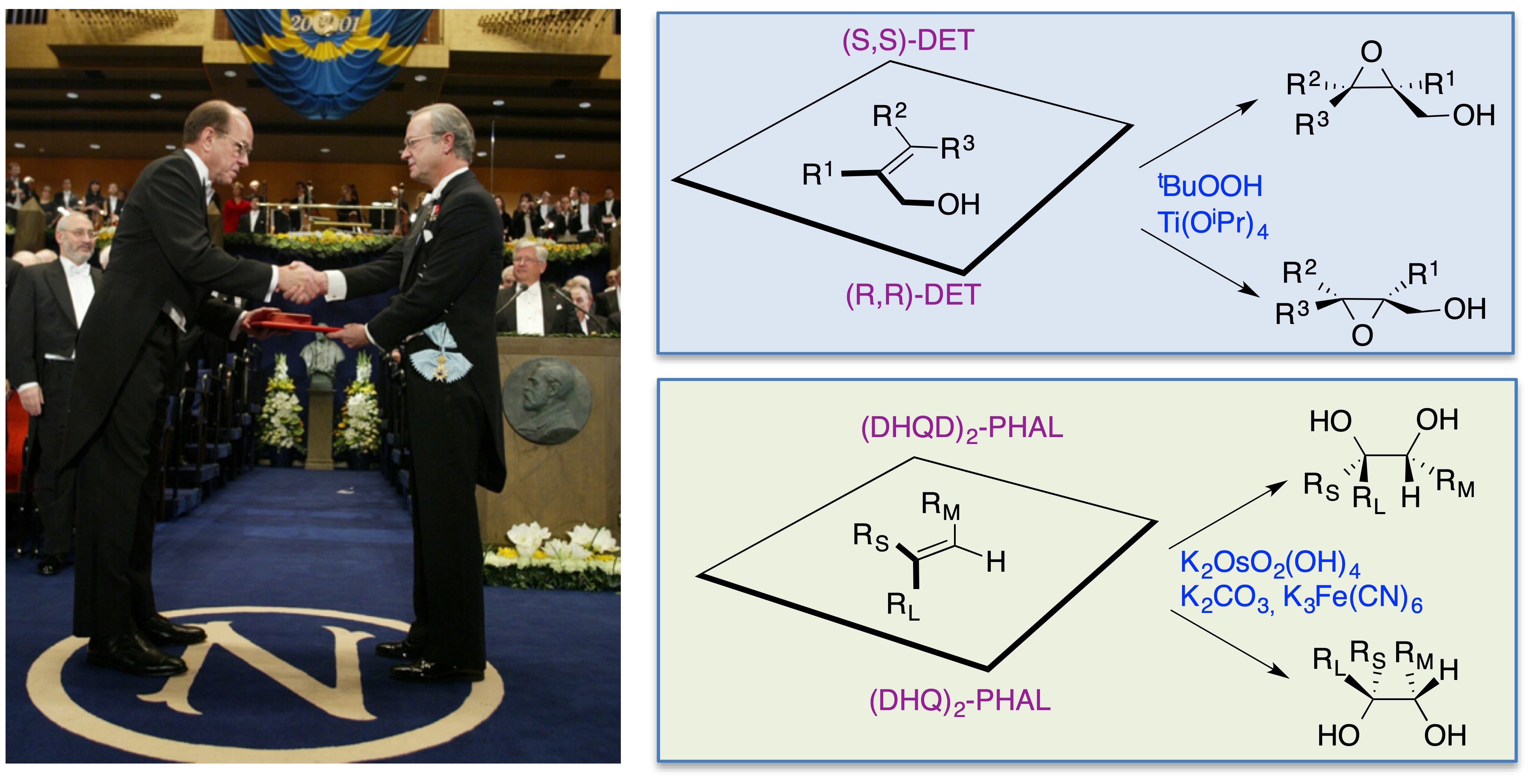
Figura 2. El primer Premio Nobel de Química de Sharpless, concedido en 2001
Morten Peter Meldal nació en Copenhague en 1954 y estudió Ingeniería Química en la Universidad Tecnológica de Copenhague, completando su licenciatura en 1980. Obtuvo el Doctorado en Química por la Universidad Tecnológica de Copenhague en 1983, trabajando bajo la dirección de Klaus Bock en el campo de la síntesis de carbohidratos. Se trasladó entonces a la Universidad de Cambridge para llevar a cabo estudios postdoctorales sobre química de péptidos en el grupo de Robert C. Sheppard del MRC Laboratory of Molecular Biology. Tras esta estancia, consiguió una plaza de profesor en la Universidad Tecnológica de Copenhague, donde estuvo entre 1986 y 1988. Se trasladó entonces al Centro de Investigación de Carlsberg de Copenhague, donde trabajó entre 1988 y 2011 y desarrolló el trabajo por el que ha recibido el Premio Nobel. Finalmente, en 2011 se trasladó a la Universidad de Copenhague, donde sigue trabajando en la actualidad.
Carolyn Ruth Bertozzi nació en Boston en 1966 y obtuvo su Licenciatura en Química por la Universidad Harvard en 1988, así como un doctorado en Química sobre síntesis de oligosacáridos por la Universidad de Berkeley en 1993, trabajando bajo la dirección de Mark D. Bednarski. Para sus estudios postdoctorales seleccionó el grupo del Dr. Steven D. Rosen en la Universidad de California en San Francisco), trabajando en las implicaciones de la química de oligosacáridos en inmnunología. Ha ocupado puestos docentes e investigadores en la Universidad de Berkeley (1996-2000), en el Howard Hughes Medical Institute (2000-2015) y en la Universidad de Stanford (2015-actualidad).
2. LA INTRODUCCIÓN DEL CONCEPTO DE QUÍMICA CLICK POR SHARPLESS
Pocos meses antes de la concesión de su primer premio Nobel, Sharpless, junto con un estudiante postdoctoral de su grupo (Harmuth C. Kolb) y otro antiguo doctorando que estaba trabajando en una empresa spin-off creada por el propio Sharpless (M. G. Finn), publicó un artículo que supuso la creación del concepto de química click y el desarrollo de sus aspectos teóricos (3). Es interesante destacar que Sharpless dedicó a este tema la parte final de su conferencia de aceptación de su primer premio Nobel, a pesar de haberle sido concedido por sus logros en un campo muy diferente, el de la oxidación enantioselectiva (4).
Esta contribución seminal comienza con algunas consideraciones generales sobre la síntesis orgánica, que pueden resumirse en los siguientes puntos:
- La finalidad última de la síntesis orgánica no es la preparación de compuestos, sino la obtención de determinadas propiedades asociadas a éstos.
- La síntesis orgánica convencional pone énfasis en la construcción de enlaces carbono-carbono y se basa principalmente en la química del grupo carbonilo.
- Sharpless sugiere que, por su dificultad, esta no es la mejor estrategia para obtener de forma ágil compuestos bioactivos.
- Propone, en su lugar, el empleo de reacciones modulares en las que se generan enlaces carbono-heteroátomo entre sustratos simples y asequibles.
- Indica, finalmente, que esta estrategia requiere el uso de reacciones simples y robustas para unir los fragmentos estructurales, proponiendo el nombre de química click para designar este tipo de reacciones.
Por tanto, estrictamente la expresión “química click“ no hace referencia a una reacción concreta, sino a un concepto ideal de reacción. Sharpless propone los siguientes como requisitos de una reacción click ideal:
- Se trata de un proceso modular, es decir, implica la unión de fragmentos preformados.
- Es robusta y presenta un amplio ámbito de aplicación.
- Transcurre con elevado rendimiento.
- Implica el uso de compuestos de partida sencillos.
- Puede llevarse a cabo en ausencia de disolvente, o empleando disolventes no tóxicos (idealmente agua).
- Genera subproductos no tóxicos.
- Es regioselectiva.
- Es enantioespecífica, es decir, genera productos estereoquímicamente diferentes cuando se parte de isómeros espaciales diferentes. En cambio, Sharpless no impone el requisito de que la reacción sea enantioselectiva.
- Es experimentalmente sencilla de realizar.
- Permite un fácil aislamiento del producto de interés, sin necesidad de cromatografía.
- Idealmente, es insensible al aire y al oxígeno.
Finalmente, Sharpless y sus colaboradores sugirieron algunas reacciones ya conocidas que pueden considerarse buenas candidatas al estatus de reacción click y que se esquematizan en la figura 3. De ellas, las primeras en investigarse con tal finalidad fueron las cicloadiciones [3 + 2].
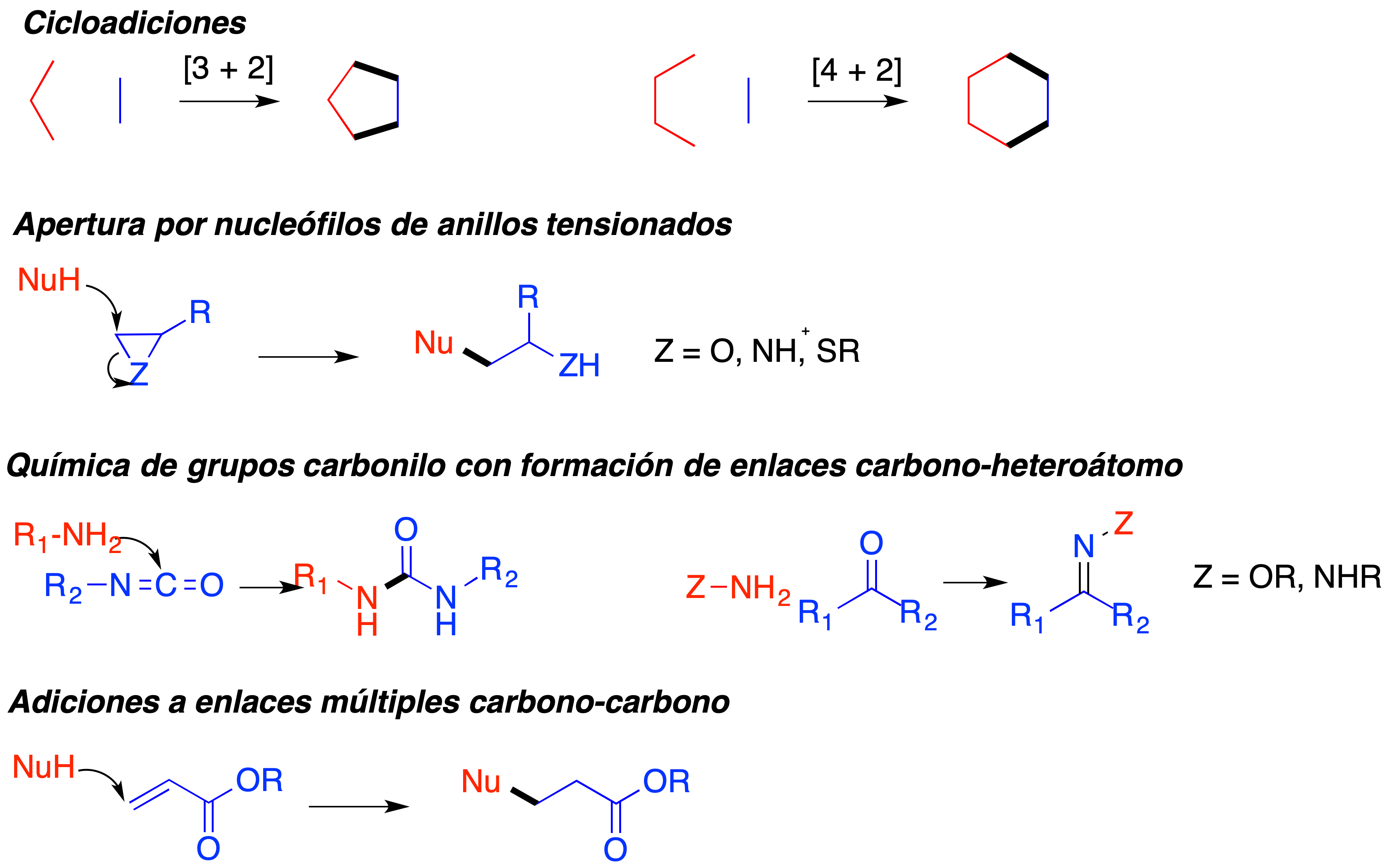
Figura 3. Algunas transformaciones candidatas a reacciones click propuestas en la publicación de Sharpless de 2001
3. LAS CICLOADICIONES [3+2] COMO REACCIONES CLICK
Las cicloadiciones [3 + 2] se basan generalmente en la reacción entre un reactivo que se describe como un 1,3-dipolo, por presentar separación de carga y deslocalización electrónica sobre un fragmento estructural de tres carbonos, y otro, llamado dipolarófilo, que reacciona con el primero para generar un ciclo de cinco miembros. Por este motivo, las reacciones [3 + 2] se describen habitualmente como cicloadiciones 1,3-dipolares (Figura 4).

Figura 4. Esquema general de una cicloadición 3 + 2 dipolar
La primera reacción de este tipo, en la que se obtuvo un derivado de pirazol por tratamiento del diazoacetato de metilo con fumarato de metilo fue descrita por Eduard Buchner en 1888 (5). Otros precursores del estudio de las cicloadiciones 1,3-dipolares fueron Erst Otto Beckmann, que investigó la química de las nitronas, incluyendo una cicloadición 1,3-dipolar con isocianato de fenilo (6), Arthur Michael, que utilizó por primera vez una azida como 1,3-dipolo (7) y Otto Dimroth, que estudió la reacción entre el ácido hidrazoico y el acetileno para dar triazol (8) (Figura 5).
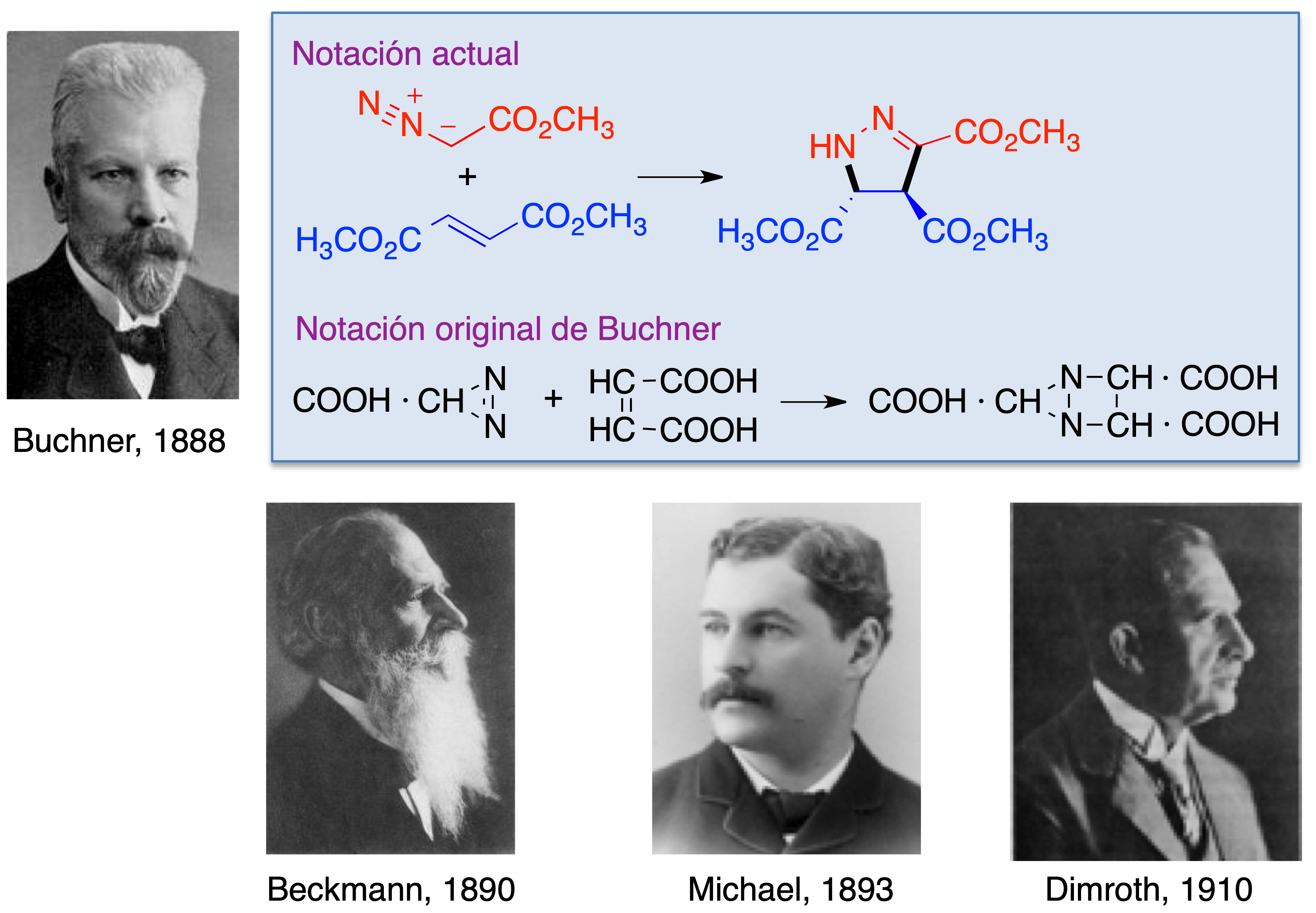
Figura 5. Los pioneros de las reacciones de cicloadición 1,3-dipolares
A partir de estos antecedentes fue Rolf Huisgen quien, a partir de 1957 y trabajando en la Universidad de Munich, estableció las cicloadiciones 1,3-dipolares como un nuevo campo de la síntesis orgánica. En concreto, los logros iniciales de Huisgen en esta área pueden resumirse como:
- Sistematización de los 1,3-dipolos existentes y diseño y estudio de otros (por ejemplo, las münchnonas).
- Empleo de nuevos dipolarófilos (por ejemplo, los arinos).
- Propuesta de un mecanismo de reacción concertado.
- Sistematización de la reacción en un artículo de revisión (9).
Debido a estos logros, la reacción entre azidas y alquinos para dar derivados de triazol se conoce como cicloadición de Huisgen. A pesar de su gran relevancia, tiene algunas limitaciones. Así, en su versión original requiere elevadas temperaturas y carece de regioselectividad, proporcionando mezclas de triazoles 1,4- y 1,5-disustituidos (Figura 6).
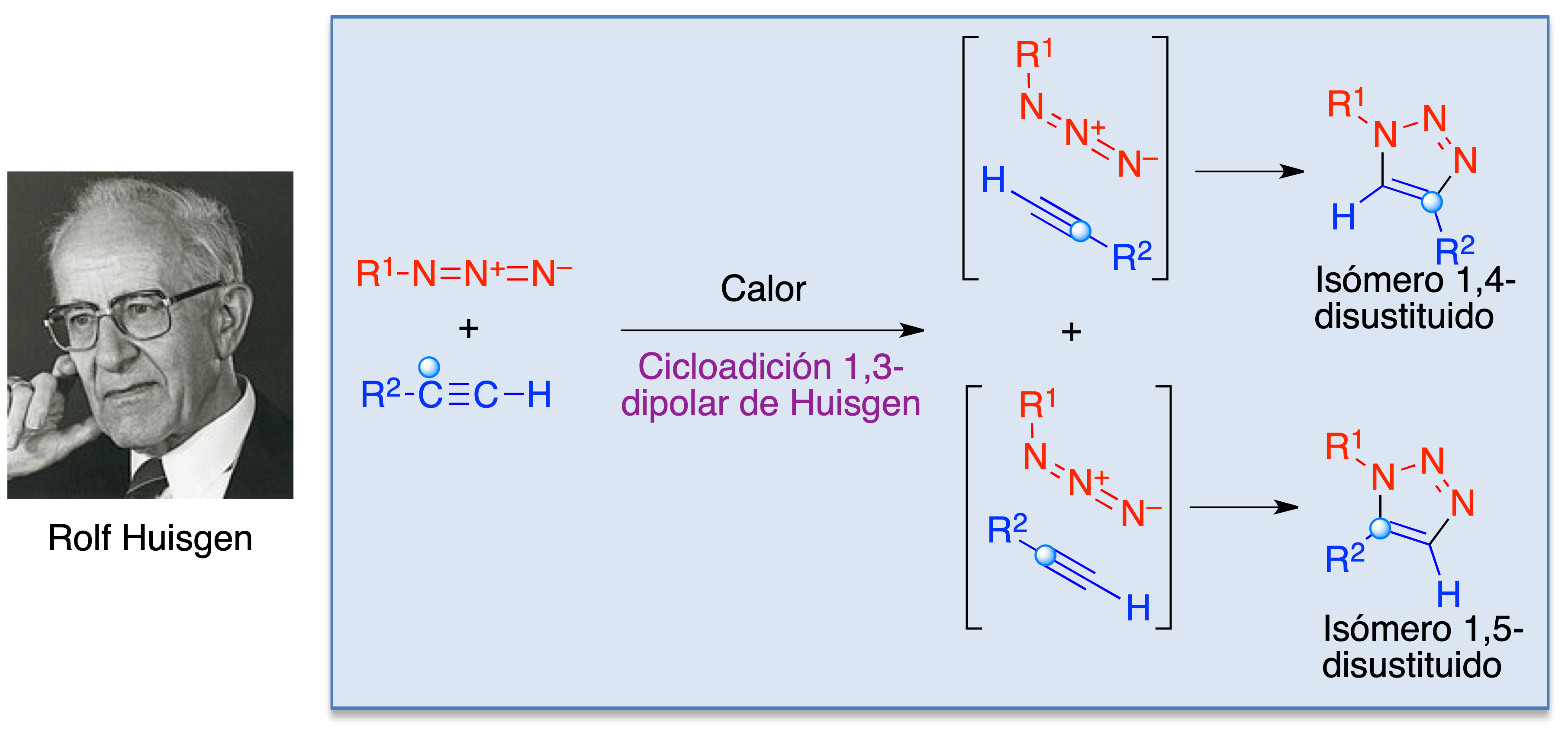
Figura 6. La cicloadición 1,3-dipolar de Huisgen
La falta de regioselectividad y la necesidad de altas temperaturas hacen que la versión original de la reacción de Huisgen sea en muchos casos poco práctica sintéticamente. Sin embargo, en 2002 se describió que, en presencia de sales de Cu(I), la reacción se convierte en regioselectiva, a favor del isómero 1,4-disustituido, y puede llevarse a cabo en condiciones suaves (Figura 7). Esta variante de la reacción de Huisgen se describe habitualmente como CuAAC (copper-catalyzed azide alkyne cycloaddition), y fue descrita, casi simultáneamente, por Meldal, en el Centro de Investigación de Carlsberg en Copenhague (10), y Fokin y Sharpless, en el Scripps Institute (11).

Figura 7. La reacción de Huisgen catalizada por cobre (CuAAC)
La catálisis por cobre facilitó el empleo generalizado de la reacción de Huisgen en en todos los campos de la síntesis orgánica, cuando se necesita unir dos fragmentos estructurales en una sola molécula (12,13,14,15). Fue tanto su impacto que es habitual considerarla la reacción click por antonomasia y designarla como tal, a pesar de que, como ya se ha comentado, “click“ hace referencia a un concepto de reacción, no a una reacción concreta. Es difícil consultar un número reciente de una revista dedicada a la Química Farmacéutica sin encontrar varios ejemplos de profármacos o fármacos multidiana obtenidos por conjugación de dos fragmentos estructurales mediante una reacción CuAAC. Como ejemplo, en la Figura 8 se muestran dos estructuras multidiana recientes, diseñadas, respectivamente, como antiinflamatorio en hipoxia cardíaca (16) y contra el cáncer (17).
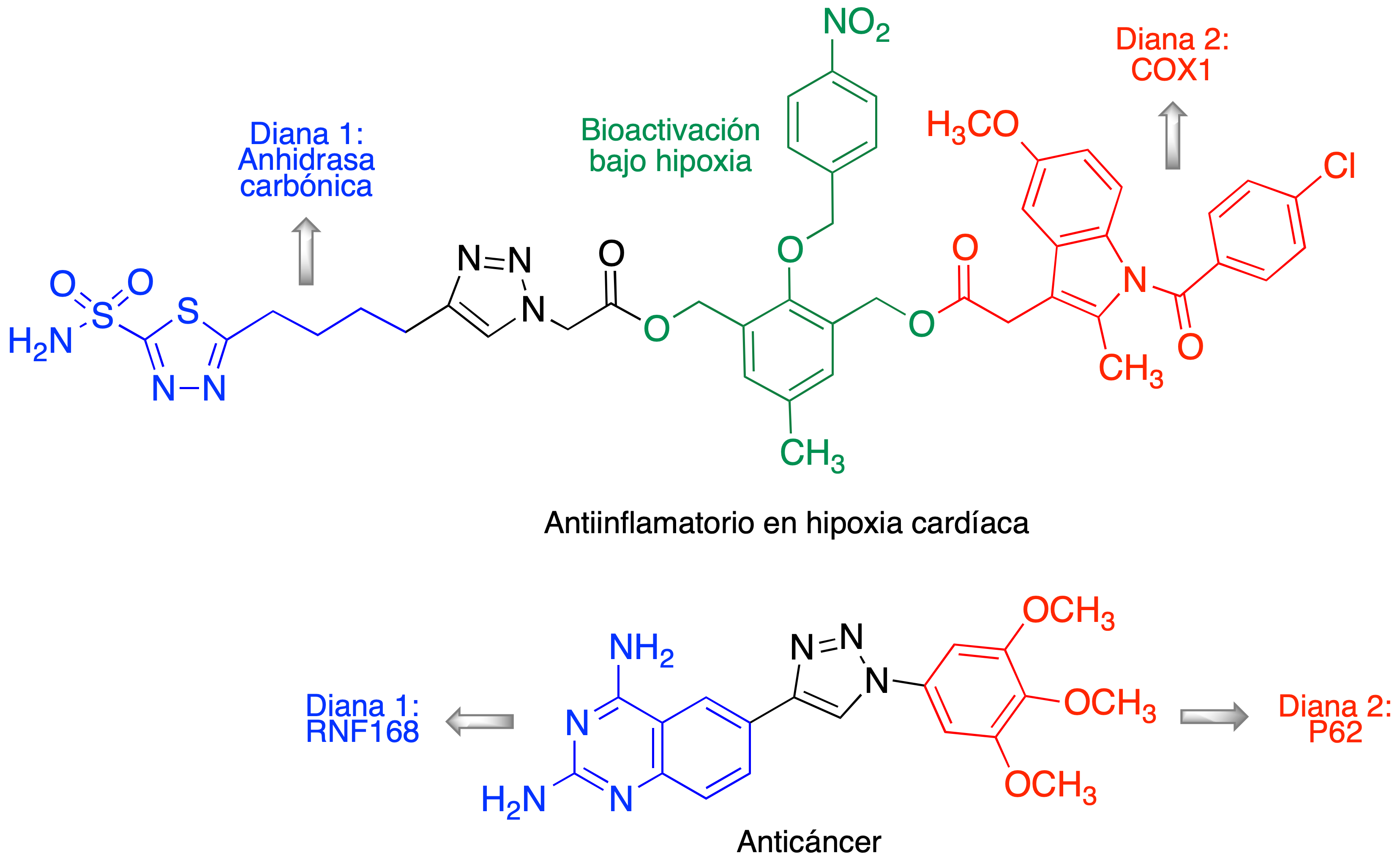
Figura 8. Dos ejemplos recientes de fármacos multidiana construidos mediante la reacción CuAAC
Similarmente, en la figura 9 se esquematiza el funcionamiento de una sonda fluorescente basada en rodamina y que es capaz de marcar selectivamente células de hepatocarcinoma por reconocimiento de receptores de asialoglicoproteínas (ASGPR). Tras la internalización del fármaco por endocitosis, el cambio de pH asociado a su entrada en el lisosoma conduce a la apertura de la rodamina para generar la correspondiente especie fluorescente (18).
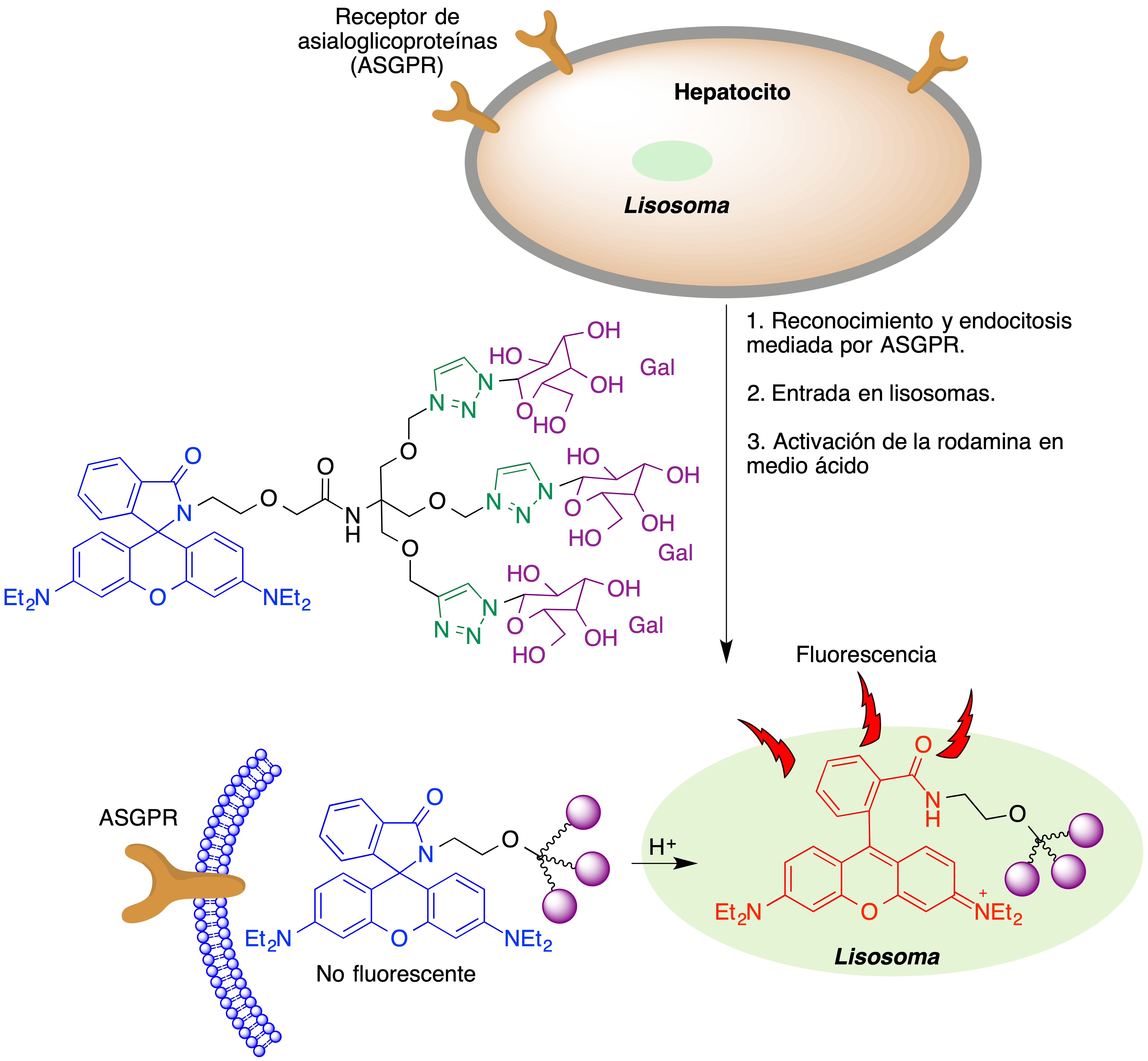
Figura 9. Marcaje selectivo de células de hepatocarcinoma basada en el reconocimiento de receptores de asialoglicoproteínas (ASGPR)
La reacción CuAAC ha encontrado también aplicación en la síntesis de fármacos derivados de triazol. Un ejemplo es la rufinamida, un fármaco antiepiléptico de introducción relativamente reciente para el tratamiento del síndrome de Lennox-Gastaut. Este fármaco ha sido sintetizado recientemente por nuestro grupo en condiciones mecanoquímicas, utilizando un molino de bolas vibratorio para llevar a cabo las tres etapas de la ruta (Figura 10).
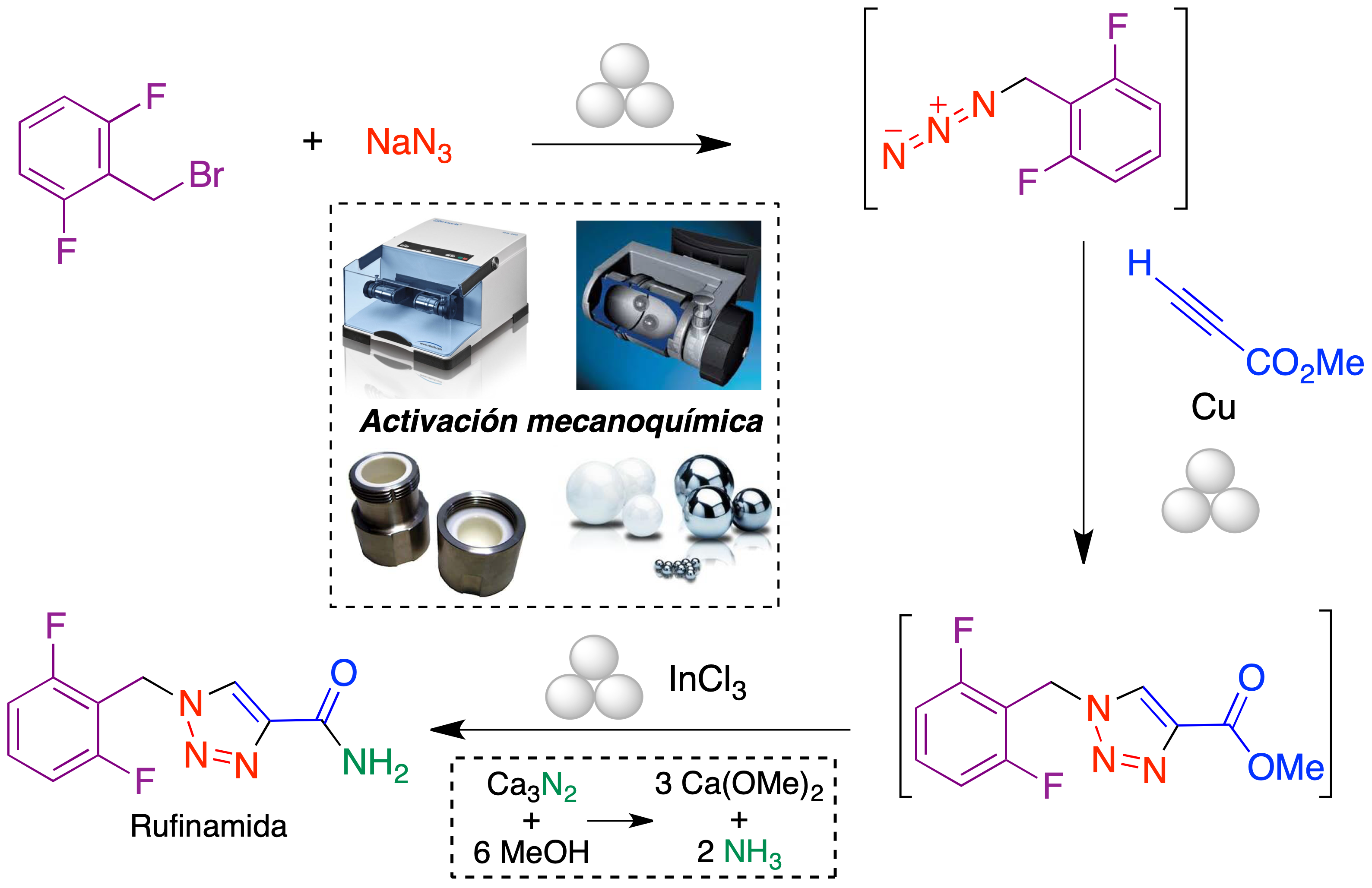
Figura 10. Síntesis mecanoquímica del fármaco antiepiléptico rufinamida
A partir del bromuro de 2,6-difluorobencilo, se llevaron a cabo una reacción de sustitución nucleofílica en la que se introduce el grupo azida, una cicloadición de Huisgen con propiolato de metilo catalizada por Cu0 y una reacción final de amidación (19). Una característica de este método es el empleo de nitruro de calcio como un precursor sólido de amoníaco, que resultó particularmente adecuado para las condiciones mecanoquímicas (20).
A pesar del éxito de la reacción CuAAc como método de síntesis, la toxicidad del cobre impide utilizar la reacción en la mayor parte de los medios biológicos, además de que estos catalizadores pueden desactivarse por coordinación con diversos grupos funcionales de las biomoléculas. En otras palabras, la reacción CuAAc no es, en principio, bioortogonal.
4. Química bioortogonal. Concepto y desarrollo inicial
Una reacción se considera bioortogonal si tiene lugar en condiciones fisiológicas y además no interfiere en las reacciones bioquímicas y por tanto se puede llevar a cabo en medios biológicos (21,22). Este término parece haber sido introducido por primera vez en la tesis doctoral de George Lemieux, dirigida por Bertozzi (23), y se inspira en el uso del término “ortogonal” en química de péptidos y en otros ámbitos de la química para describir grupos protectores que pueden considerarse independientes por poderse eliminar de forma selectiva, sin afectarse unos a otros (24).
Se plantea, por tanto, el problema de cómo lograr que la cicloadición de Huisgen tenga lugar a temperatura ambiente y en ausencia de catalizadores basados en metales de transición para lograr biocompatibilidad. Una posibilidad es utilizar sustratos que sean inestables y por tanto altamente reactivos, ya que una mayor energía del sustrato de una reacción se traduce en una menor energía de activación del proceso. (Figura 11.A). En el caso de la reacción de Huisgen, puede aprovecharse la elevada reactividad asociada a la tensión angular de alquinos cuyo triple enlace pertenece a un carbociclo de ocho miembros, estrategia que ha sido designada como strain-promoted azide–alkyne 1,3-dipolar cycloaddition (SpAAC) (Figura 11.B). La reactividad de estos compuestos se incrementa mediante la presencia de dos átomos de flúor, fuertemente aceptores, en la posición vecina al triple enlace. Empleando estos sustratos altamente reactivos la reacción de cicloadición dipolar tiene lugar en condiciones suaves, aunque, al faltar el efecto director del catalizador de cobre, proporciona los dos regioisómeros posibles (25) (Figura 11C). Otros derivados de ciclooctino utilizados habitualmente en química bioortogonal están resumidos en la figura 11D.

Figura 11. A, B. Incremento de reactividad en alquinos tensionados. C, D. Algunos ciclooctinos habituales en química bioortogonal y su empleo en la cicloadición de Huisgen.
Aunque se conocían ejemplos aislados, debidos a Blomquist (26) y Wittig (27), de reacciones de Huisgen llevadas a cabo a partir de ciclooctino sin necesidad de catalizadores metálicos, fue Bertozzi quien sistematizó esta transformación y publicó el primer ejemplo de una reacción 1,3-dipolar de un cicloalquino para el marcaje de una proteína de membrana en un entorno celular. Sus experimentos iniciales se basaron en un descubrimiento previo (28) que indicaba que la ruta biosintética habitual de los ácidos siálicos puede manipularse para lograr la presencia de ácidos siálicos modificados en la membrana celular mediante la administración de análogos de la N-acetilmanosamina, entre ellos la N-azidoacetilmanosamina peracetilada (Ac4ManNAz), como se indica en la Figura 12. El intermedio 1, procedente de la desacetilación del compuesto de partida seguida de fosforilación, es un hemiacetal que está en equilibrio con el hidroxialdehído 2. La reacción de éste con el fosfoenol piruvato proporciona 3, cuya ciclación seguida de hidrólisis genera el ácido azidosiálico.

Figura 12. Incorporación de ácido azidosiálico a las membranas celulares tras la administración de N-azidoacetilmanosamina peracetilada
De esta forma se consiguen células portadoras de grupos azida en los glicanos presentes en su superficie y que pueden por tanto reaccionar mediante el proceso SpAAC con un compuesto que contiene un fragmento de ciclooctino y otro de biotina unidos mediante un espaciador. Esta reacción conduce, por tanto, a la incorporación de moléculas de biotina a la membrana celular. Por otra parte, la proteína estreptavidina contiene un profundo bolsillo que presenta una gran afinidad por la biotina, de modo que la adición de estreptavidina marcada con isotiocianato de fluoresceína (FITC) conduce, finalmente, al marcaje de la membrana celular con moléculas de fluoresceína, altamente fluorescentes (29) (Figura 13). Investigaciones posteriores del grupo de Bertozzi permitieron la adaptación de esta metodología al marcaje de membranas celulares en organismos vivos (30,31).
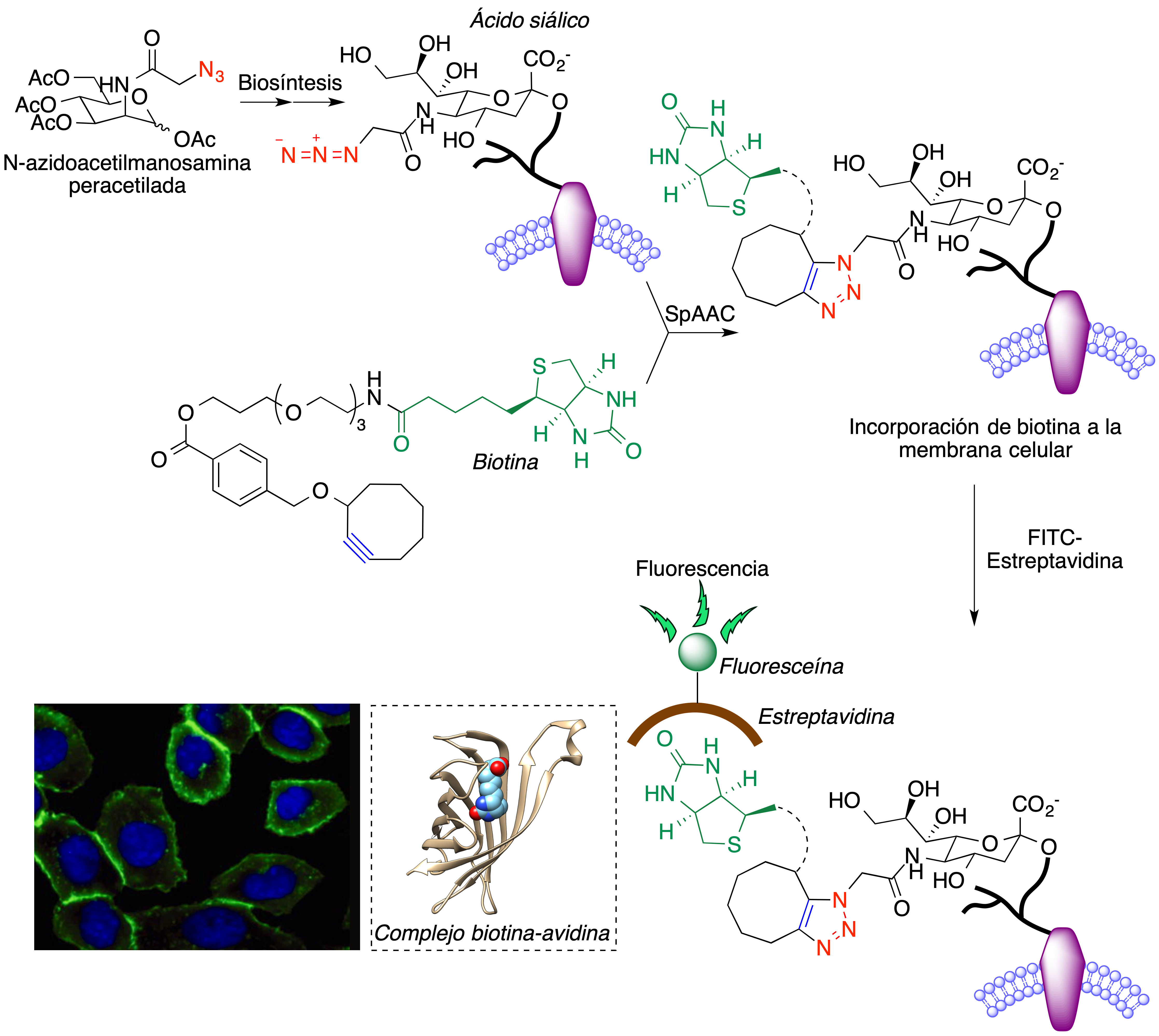
Figura 13. Marcaje fluorescente de células por el método de Bertozzi. La imagen del experimento de tinción celular se ha reproducido, con permiso, de la referencia 30; copyright (2007) National Academy of Sciences. El complejo estreptavidina-biotina se ha generado a partir de pdb 1mk5.
5. Reacciones bioortogonales diferentes a la cicloadición 1,3-dipolar
Además de la de Huisgen, se han utilizado otras reacciones de cicloadición como base para procesos de bioconjugación. La cicloadición [4 + 2] es la clásica reacción de Diels-Alder, en la que se forma un ciclo de seis eslabones a partir de un compuesto de partida que aporta cuatro átomos al nuevo anillo y que contiene dos dobles enlaces conjugados (dieno) y de un segundo compuesto, del que proceden los dos átomos restantes del nuevo anillo y que contiene un doble enlace aislado (denominado dienófilo). Aunque existen ejemplos de su uso en reacciones de bioconjugación, tiene el inconveniente de que el aducto puede revertir a los compuestos de partida (Figura 14.A). Una manera de superar este problema es emplear como dieno un sistema de 1,2,4,5-tetrazina, ya que en ese caso el aducto inicialmente generado evoluciona rápidamente por extrusión de una molécula de nitrógeno y no tiene la opción de regenerar los compuestos de partida mediante un mecanismo de tipo retro Diels-Alder (Figura 14.B). La reacción de la tetrazina con alquenos se clasifica como un proceso hetero Diels-Alder de demanda electrónica inversa (iEDDA) porque en ella el dienófilo es de baja densidad electrónica.
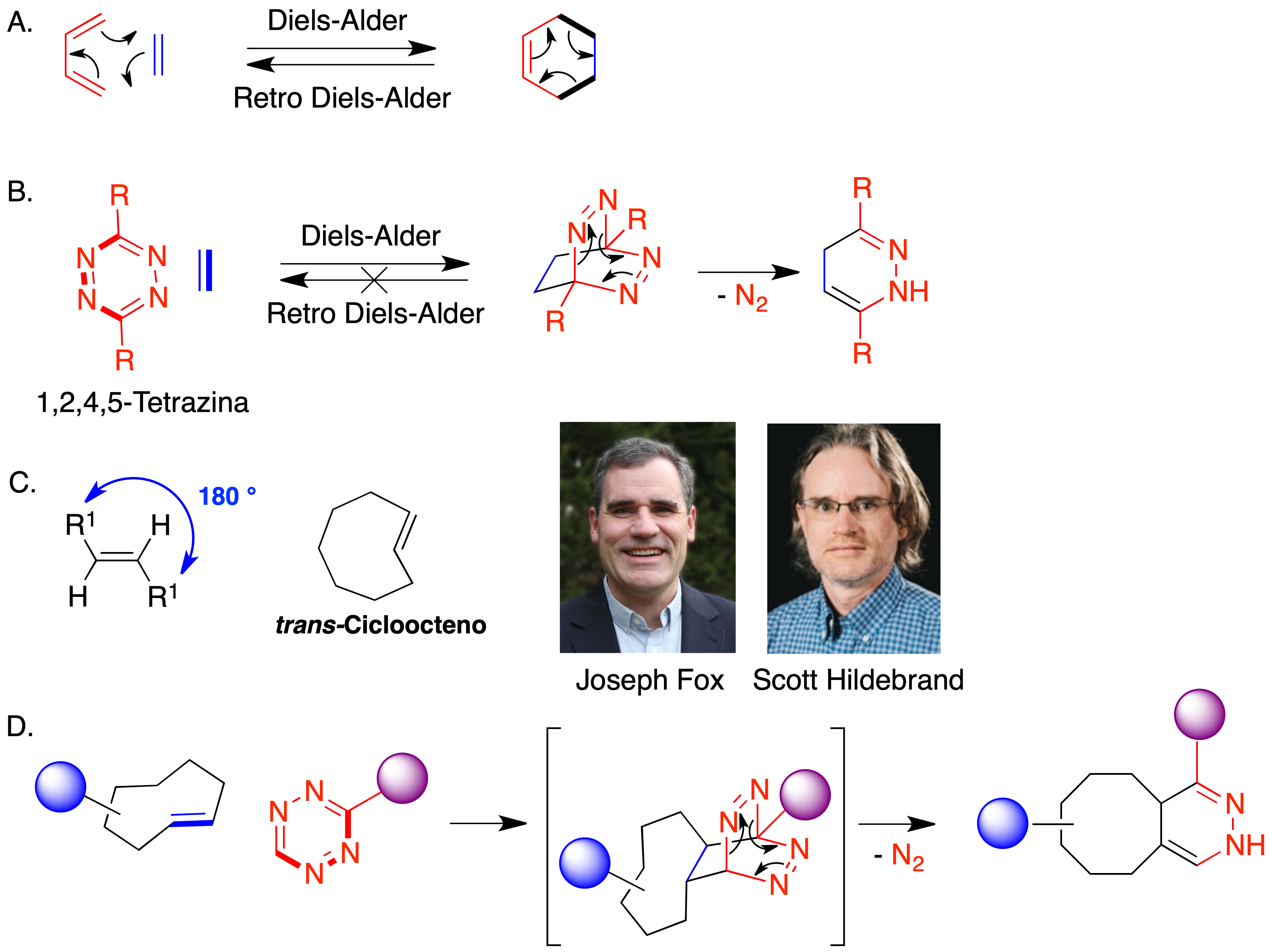
Figura 14. Química bioortogonal basada en reacciones hetero Diels-Alder
Los grupos de Fox (32) y Hilderbrand (33) desarrollaron de forma independiente la aplicación de esta reacción hetero Diels-Alder a la química bioortogonal. Para incrementar la velocidad de reacción, utilizaron como dienófilos derivados del trans-cicloocteno, altamente tensionados (Figura 14.C) Esto permite conseguir reacciones muy rápidas en condiciones fisiológicas, que han encontrado una aplicación muy extensa en el campo de la Química Biológica gracias a que su constante de velocidad (k ~ 105 M–1 s–1) es del mismo orden de magnitud que los procesos de asociación entre biomoléculas, lo que permite aplicaciones en las que la química sustituye o complementa a la biología (Figura 14.D).
6. Algunas aplicaciones de la química bioortogonal
Existe un número inmenso de aplicaciones de la química bioortogonal (34,35,36,37,38), que no es posible tratar con profundidad aquí. Se resumen a continuación algunos ejemplos seleccionados.
6.1 Distribución selectiva de fármacos
Una estrategia habitual para lograr la distribución selectiva de fármacos hacia tumores se basa en el empleo de anticuerpos dirigidos a antígenos específicos del tumor. De esta manera se puede lograr la fijación al tumor de anticuerpos portadores de sustituyentes de trans-ciclooctenilo. Unos días después, para asegurar la distribución completa del anticuerpo, se administra una cantidad equimolecular de un derivado radiomarcado de tetrazina de eliminación rápida, consiguiéndose un eficaz y rápido marcaje de las células tumorales con el radioisótopo (39) (Figura 15).

Figura 15. Fijación de radioisótopos en células tumorales por medio de una reacción click. La imagen del tumor en el ratón se ha reproducido de la referencia 39, con permiso de John Wiley and Sons.
La diferenciación entre las células normales y las tumorales es esencial para el diagnóstico y el tratamiento del cáncer, por lo que es de gran interés la posibilidad que ofrece la química bioortogonal de llevar a cabo reacciones específicas en la superficie de las células tumorales. Como se comentó previamente, es posible la incorporación de glúcidos sustituidos con grupos azida en los ácidos siálicos presentes en la cubierta glucoproteica de las células, lo que se ha aprovechado para el marcaje selectivo de células tumorales tanto in vitro como in vivo. Para ello, se razonó que un análogo de la N-azidoacetilmanosamina peracetilada que tuviera un grupo éter en la posición anomérica (4) no debería ser capaz de incorporarse directamente a la ruta biosintética que conduce al ácido azidosiálico, ya que, al no contener un fragmento de hemiacetal, no sería posible la formación de un grupo aldehído (compuesto 1) a través de su apertura. El objetivo era diseñar un grupo protector de este tipo cuya desprotección dependiera de una enzima sobreexpresada en tumores, de modo que fuera posible la generación del intermedio 1 y, a partir de él, de ácido azidosiálico, que marcaría selectivamente las células tumorales con grupos azida (Figura 16). Su presencia debería permitir la fijación posterior de compuestos con relevancia terapéutica o diagnóstica mediante reacciones bioortogonales, lo que se describe como la estrategia ATTACK (active tissue targeting via anchored click chemistry).

Figura 16. Fundamento bioquímico de la estrategia ATTACK.
Para esta finalidad, se diseñó un compuesto que contiene en la posición anomérica de la manosa un grupo de benzhidril éter, conectado a su vez a un resto de N-acetil lisina (5). Este compuesto se activa mediante una secuencia de reacciones que se inicia con la desacetilación del fragmento de lisina por una histona desacetilasa (HDAC), seguida por la hidrólisis del enlace amida por la catepsina L (CTSL) para dar el compuesto 6. Ambas enzimas están sobreexpresadas en muchos tumores y el hecho de que la activación requiera dos etapas enzimáticas ayuda a la selectividad hacia el tejido tumoral. Una tercera etapa, no enzimática, implica la descomposición espontánea de 6 que libera el compuesto 1, citado anteriormente como un intermedio de la biosíntesis del ácido azidosiálico (Figura 17) (40).
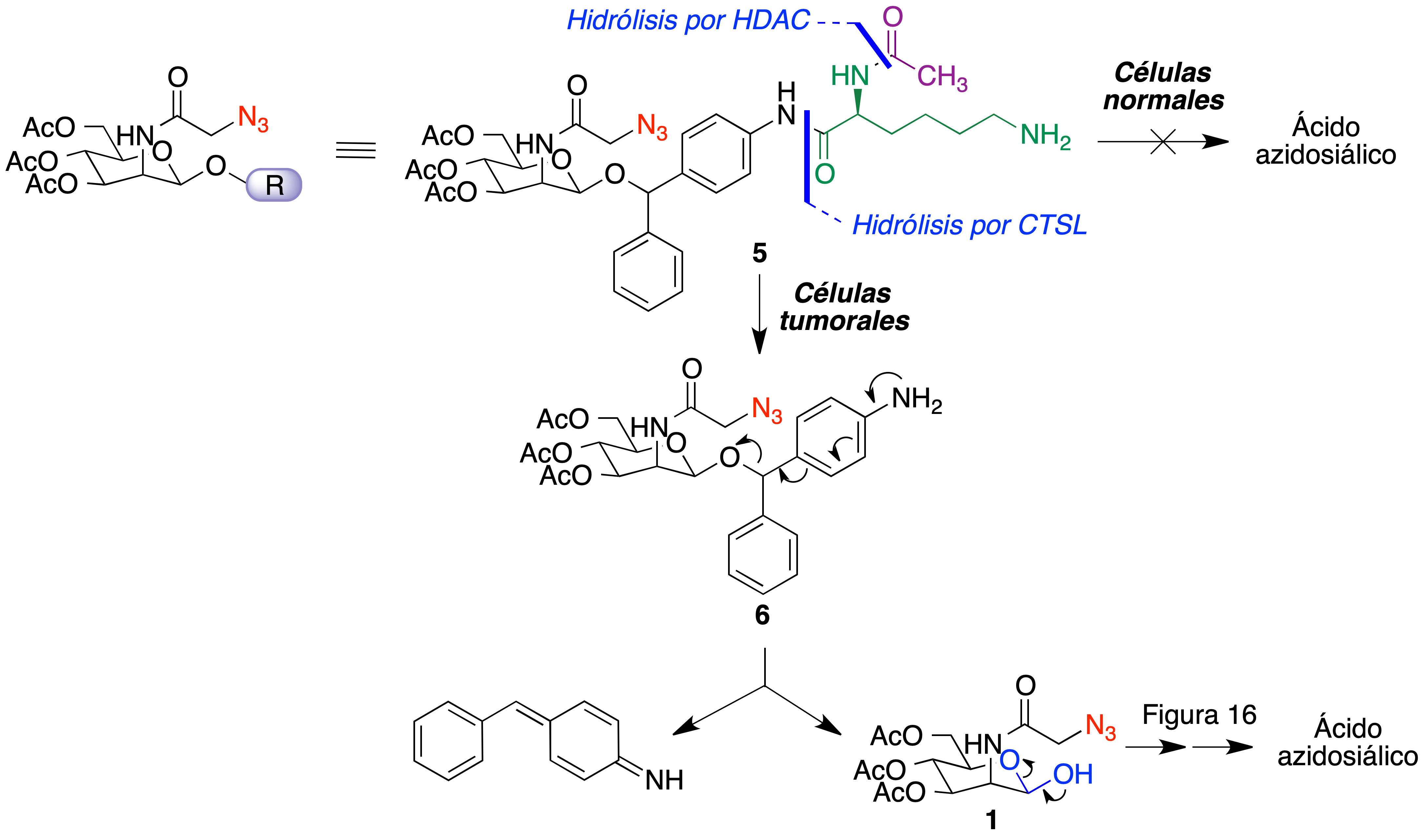
Figura 17. Un precursor de ácido azidosiálico para su activación selectiva en células tumorales
Esta técnica puede emplearse para lograr la distribución selectiva de fármacos antitumorales hacia el tejido tumoral, con algunas ventajas frente a la aproximación habitual basada en el empleo de anticuerpos monoclonales:
No es necesario identificar un antígeno específico en el tumor.
Implica el uso de moléculas pequeñas, mucho más baratas que los anticuerpos y susceptibles de administración oral.
Se esquematiza a continuación el proceso utilizado para conseguir el transporte selectivo de doxorubicina a tumores. La administración del compuesto 5 a ratones en los que se ha implantado un tumor permite la acumulación de grupos azida en las membranas de las células tumorales. Si se administra a continuación el compuesto 7, que contiene un alquino altamente tensionado y está unido al fármaco que se desea transportar mediante un espaciador, logra a la fijación del fármaco al tumor (Figura 18).
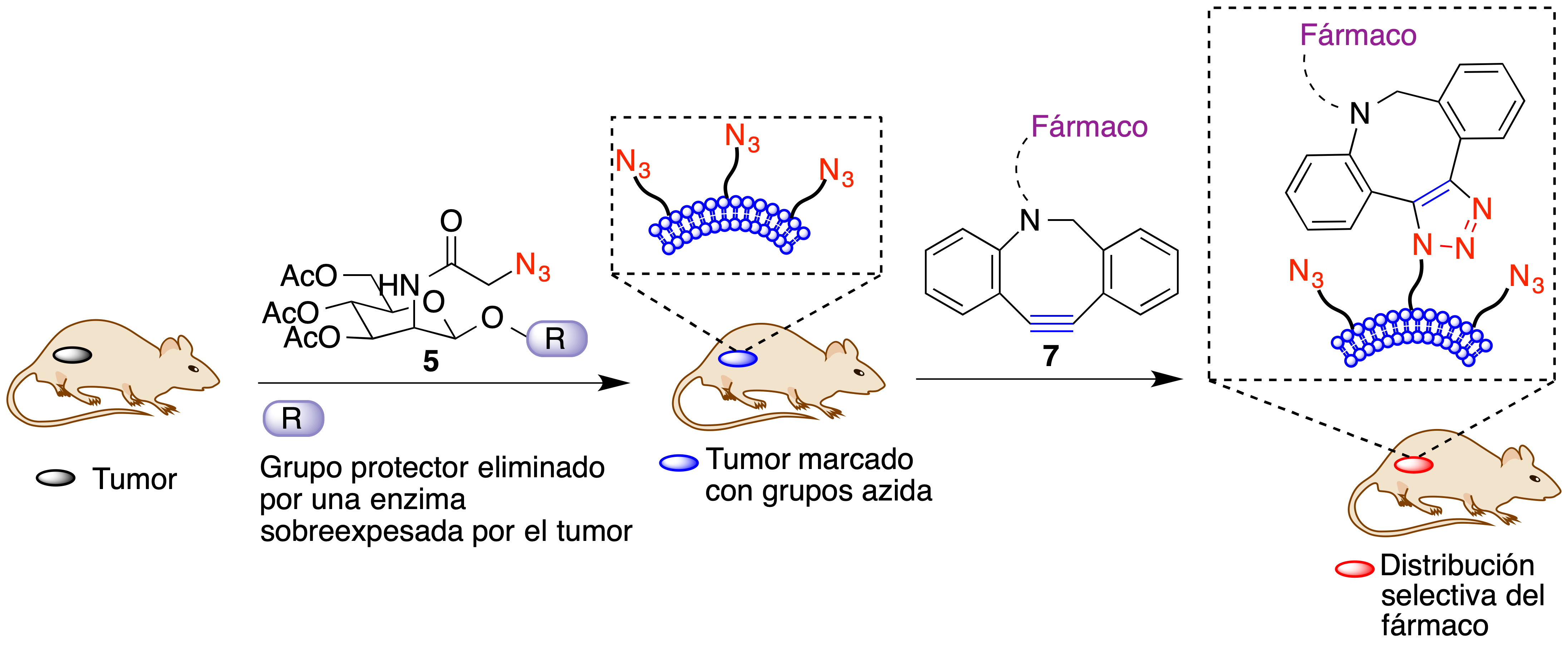
Figura 18. Fijación de fármacos a las membranas de células tumorales mediante la estrategia ATTACK.
Una vez lograda la fijación selectiva del fármaco a las células tumorales es necesaria su liberación. La proteasa catepsina B demostró la capacidad de hidrolizar el enlace amida correspondiente al residuo de arginina del espaciador, dando lugar al intermedio 8. Este, finalmente, se descompone espontáneamente y libera la doxorubicina (Figura 19).
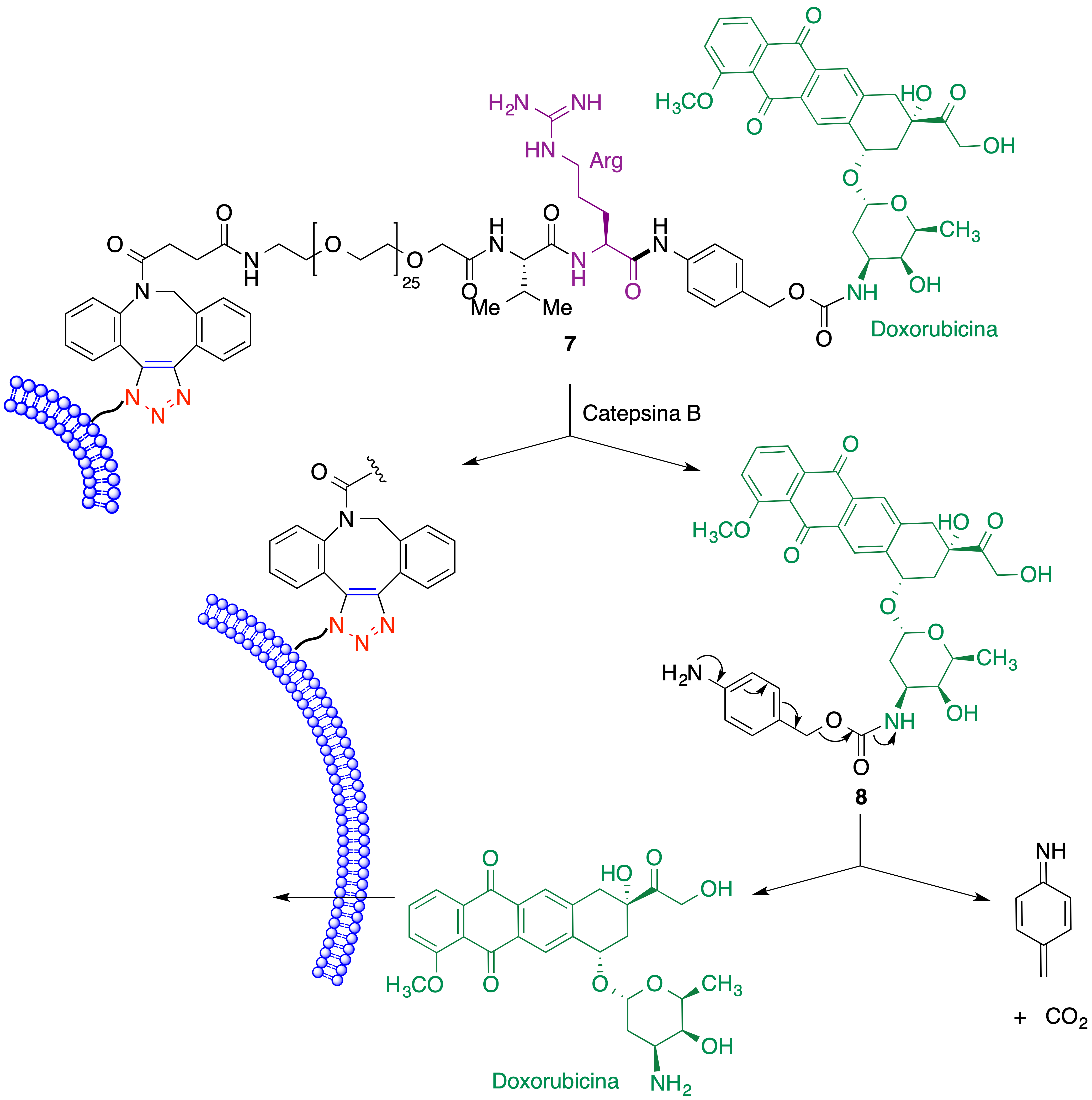
Figura 19. Liberación de doxorubicina junto a una célula tumoral como parte de la estrategia ATTACK
6.2 Profármacos basados en reacciones click
La inestabilidad química de los productos de reacciones click de carbamatos de trans-3-ciclooctenilo (41) proporciona una forma sencilla de liberar compuestos tales como fármacos, agentes de diagnóstico, etc. junto a una diana de interés (Figura 20).

Figura 20. Carbamatos de trans-3-ciclooctenilo para la liberación de fármacos mediada por una reacción click
Como un primer ejemplo de la activación selectiva de este tipo de profármacos en el tejido tumoral, se comentará a continuación una estrategia basada en el aprovechamiento del efecto efecto de permeacón y retención aumentada (enhanced permeability and retention, EPR). Este efecto se debe a las alteraciones del tejido vascular en el entorno de los tumores, ya que la neovasculatura generada por el fenómeno de angiogénesis es diferente de la de un tejido sano y se caracteriza por células endoteliales desorganizadas y con grandes fenestraciones, que permiten una mayor entrada y acumulación de nanopartículas en comparación con un tejido sano. En el ejemplo que se va a comentar, estas nanopartículas actúan como activadoras de un profármaco, que puede entrar tanto en el tejido sano como en el tumoral pero solo se activa en este último ya que en el sano no están presentes las nanopartículas (Figura 21).
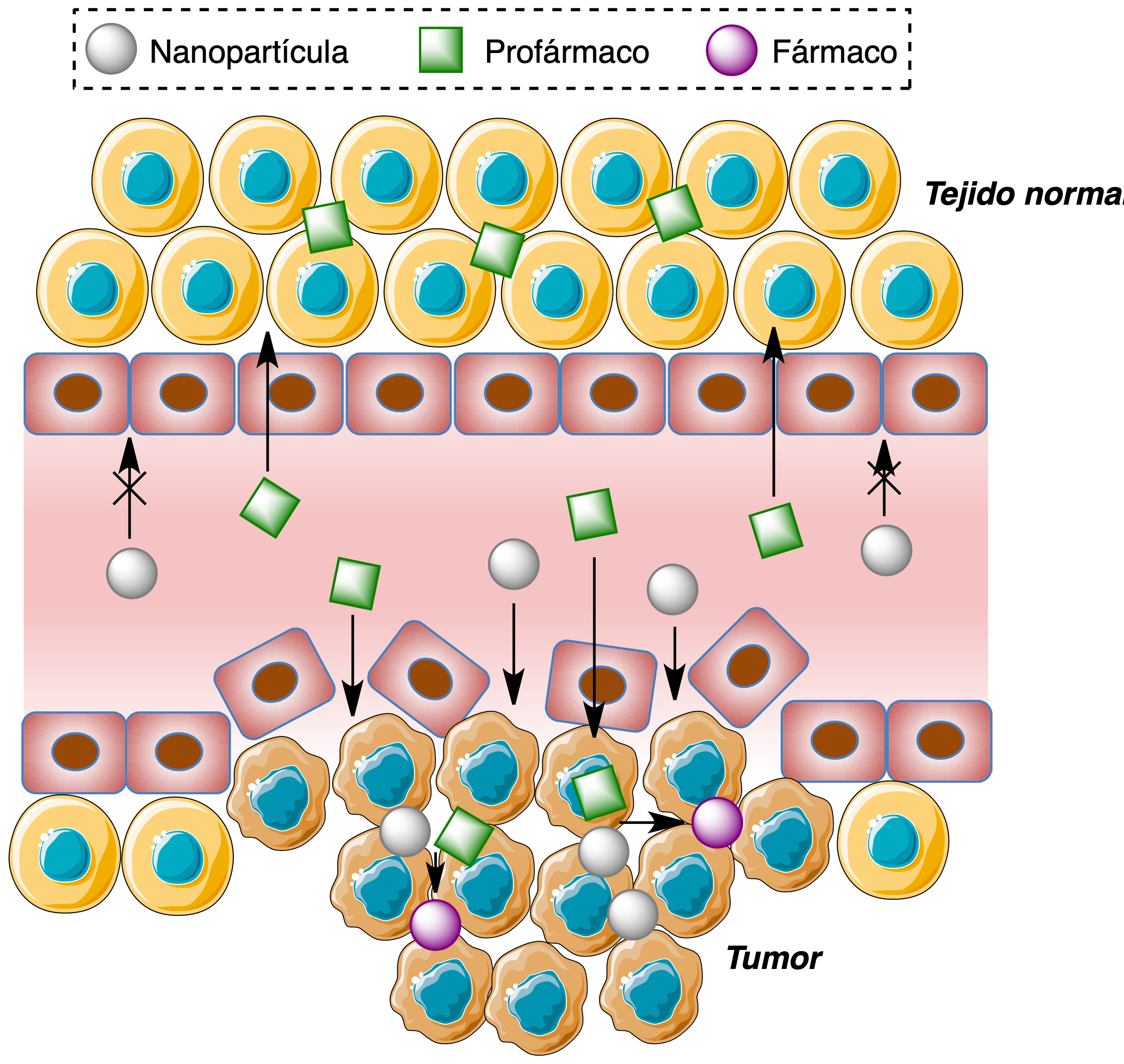
Figura 21. Distribución selectiva de nanopartículas mediada por el efecto EPR y su aplicación a la activación selectiva de profármacos
Las nanopartículas utilizadas son nanotubos de carbono que contienen como sustituyentes anillos de tetrazina, por lo que cuando entran en contacto con los fragmentos de ciclooctenilo de una molécula del profármaco tiene lugar la reacción hetero Diels-Alder para dar el compuesto 9, que se descompone según el mecanismo que se ha indicado previamente para liberar la doxorubicina (Figura 22).
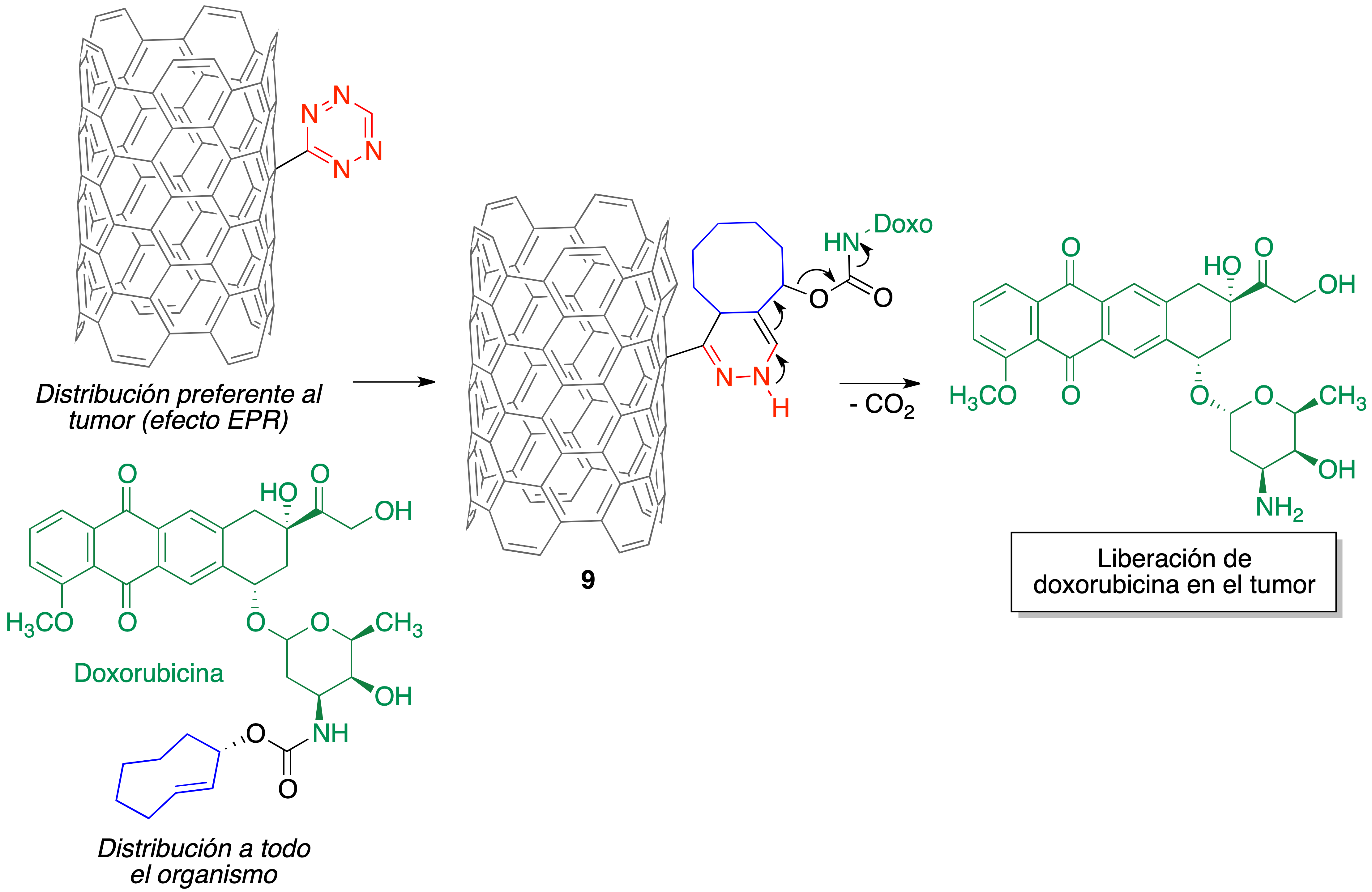
Figura 22. Liberación selectiva de doxorubicina en tumores a partir de un profármaco generado in situ mediante una reacción hetero Diels-Alder de nanotubos de carbono portadores de anillos de tetrazina.
6.3 Síntesis de fármacos en su lugar de acción
La posibilidad de llevar a cabo reacciones click en entornos biológicos, y más concretamente en el interior de cavidades de las proteínas, abre las puertas a un nuevo paradigma en el descubrimiento de fármacos. Se trata de una aproximación basada en fragmentos en la que cada uno de ellos se une a un bolsillo de la proteína y después se conectan mediante una reacción de Huisgen dentro de la propia proteína diana, permitiendo potencialmente un ajuste perfecto a ésta (Figura 23). Cabe esperar que, al utilizar como recipiente de reacción la propia proteína diana, esta estrategia conduzca a compuestos activos de forma más rápida y eficaz que el método alternativo que implica la síntesis convencional en el laboratorio de familias de compuestos para proceder posteriormente a su ensayo frente a la diana.
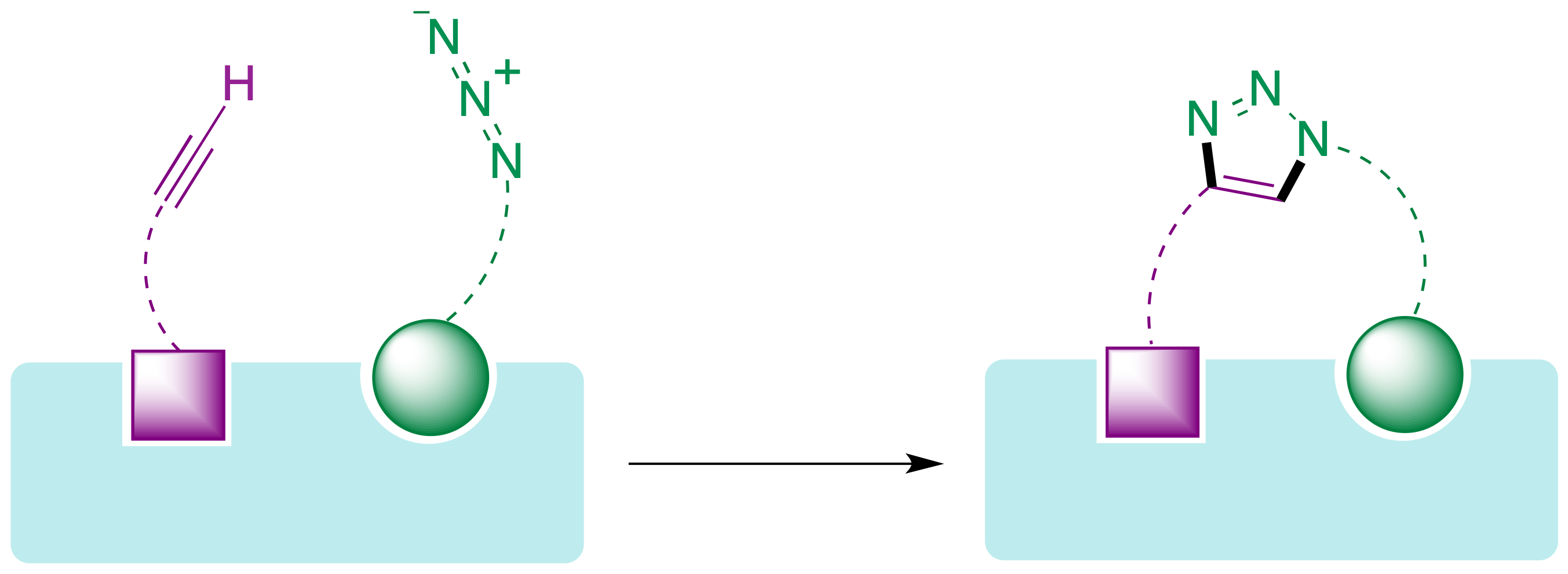
Figura 23. Diseño de fármacos basado en fragmentos que se conectan mediante una reacción click dentro de la proteína diana.
El trabajo pionero en esta área se debe una vez más a Sharpless, que propuso la técnica de diseño previamente descrita y la aplicó a la búsqueda de inhibidores de la enzima acetilcolinesterasa, una diana muy importante en el tratamiento de la enfermedad de Alzheimer (42,43). Teniendo en cuenta que esta enzima tiene dos sitios principales de unión de inhibidores, se buscó un compuesto que se uniera a ambos. Para ello, se sintetizaron diversos derivados de la tacrina, un inhibidor que se une a una región vecina a la tríada catalítica, y por otra parte una serie de derivados de fenantridinio, que se esperaba tuvieran afinidad por el sitio aniónico periférico de la enzima. En lugar de sintetizar en el laboratorio toda la colección de análogos, se pusieron en contacto con la enzima, en placas de pocillos, todas las combinaciones posibles tacrina-fenantridinio. Se esperaba que para algunas de ellas la disposición espacial y la distancia entre los grupos azida y etinilo, una vez que los sistemas heterocíclicos principales ocuparan sus lugares de unión, permitiera una reacción de Huisgen que generara un inhibidor divalente en el seno de la propia enzima inhibida.
De acuerdo con estas expectativas, una de las 83 combinaciones tacrina-fenantridinio estudiadas dio lugar a la formación del compuesto 10, que resultó ser el inhibidor más potente conocido de la enzima, con un valor de CI50 de 100 femtomolar. Cabe destacar que la reacción de Huisgen llevada a cabo en el seno de la enzima tuvo lugar rápidamente a temperatura ambiente, a pesar de la ausencia de catalizadores de cobre, y dio lugar a un solo regioisómero a causa de las restricciones espaciales impuestas por la enzima. En la figura 24 se incluye también el complejo que forma el compuesto 10 con la acetilcolinesterasa, que muestra la ocupación tanto del sitio de fijación de tacrina como del sitio aniónico periférico (44).
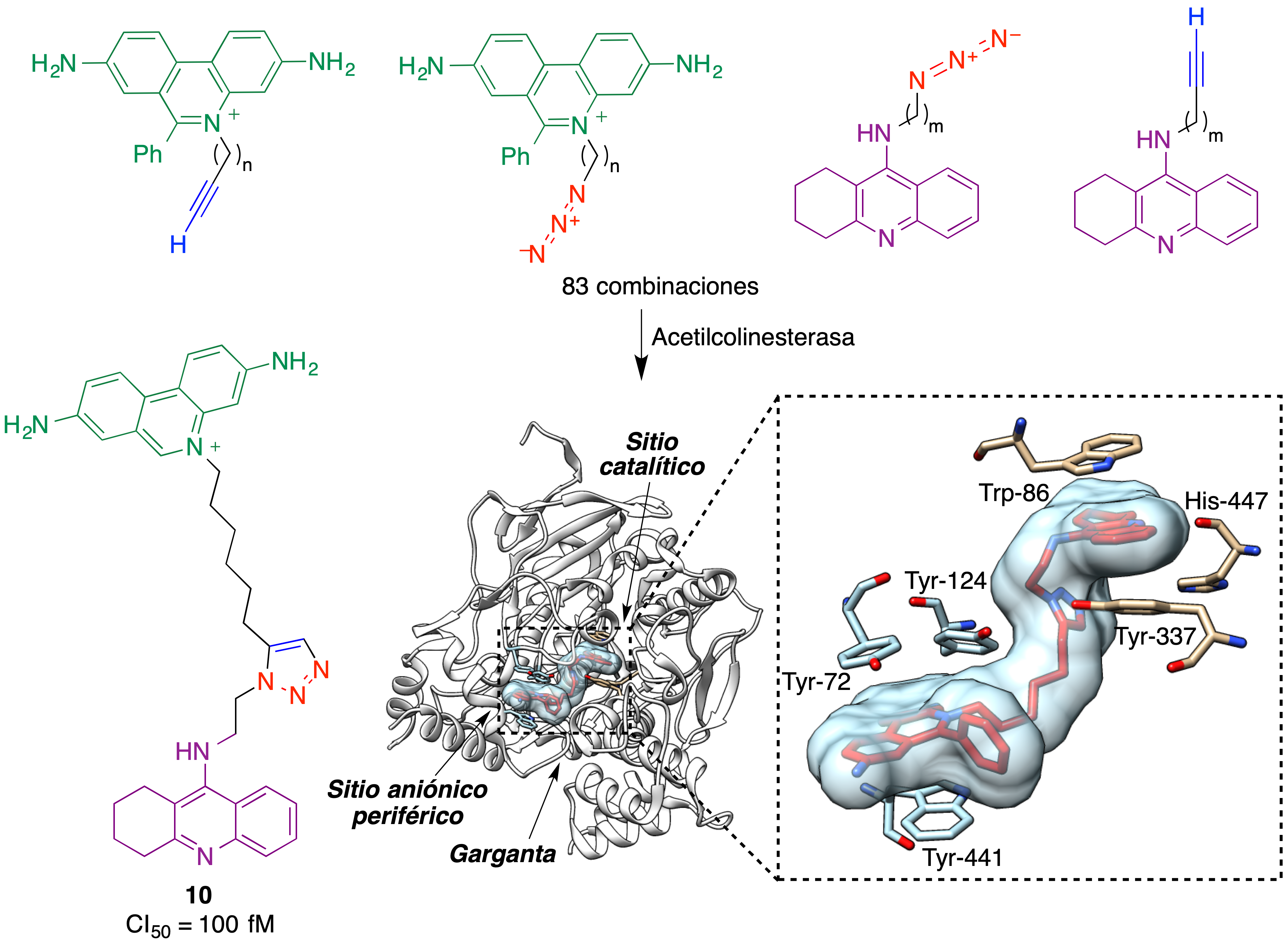
Figura 24. Síntesis de un inhibidor femtomolar de la acetilcolinesterasa (compuesto 10), llevada a cabo en el seno de la propia enzima. El complejo formado por 10 y la acetilcolinesterasa se ha generado a partir de PDB 1Q83.
La estrategia de síntesis in situ introducida por Sharpless ha sido generalizada por otros investigadores y ha encontrado múltiples aplicaciones en Química Farmacéutica y Química Biológica (45).
Una segunda situación en la que puede ser conveniente llevar a cabo la síntesis de un fármaco dentro del organismo se da cuando la especie con actividad terapéutica es una molécula de gran tamaño, lo que puede traer consigo una baja biodisponibilidad y otros problemas farmacocinéticos. Este es el caso de los fármacos de tipo PROTAC (proteolysis-targeting chimera), una estrategia emergente cuya finalidad es la de promover la hidrólisis de una proteína diana marcándola para su degradación por el proteasoma. Los PROTAC son agentes multidiana, diseñados para que presenten afinidad con la diana que se desea degradar y también con una proteína perteneciente a la familia de las ligasas E3, que tienen la capacidad de transferir unidades de ubiquitina a la proteína diana y conseguir así que sea sustrato del proteasoma.
Desde el punto de vista del diseño de fármacos, uno de los defectos de los PROTAC es que son moléculas excesivamente grandes, dada la necesidad de que contengan fragmentos capaces de unirse a dos dianas y además un espaciador. Esto puede traer consigo una baja biodisponibilidad y otros problemas farmacocinéticos. Como alternativa basada en la química bioortogonal, se ha propuesto la estrategia CLIPTAC (click-formed proteolysis-targeting chimeras), consistente en la administración por separado de un ligando de la proteína diana y otro de una ligasa E3, de forma que ambos fragmentos tienen tamaños moleculares más reducidos que el PROTAC completo y atraviesan fácilmente las membranas celulares. Una de estas moléculas contiene un fragmento de trans-cicloocteno y la otra uno de tetrazina para que tenga lugar una reacción hetero Diels-Alder que genere el PROTAC de forma intracelular, evitándose los problemas de distribución asociados a su administración como tal (Figura 25).
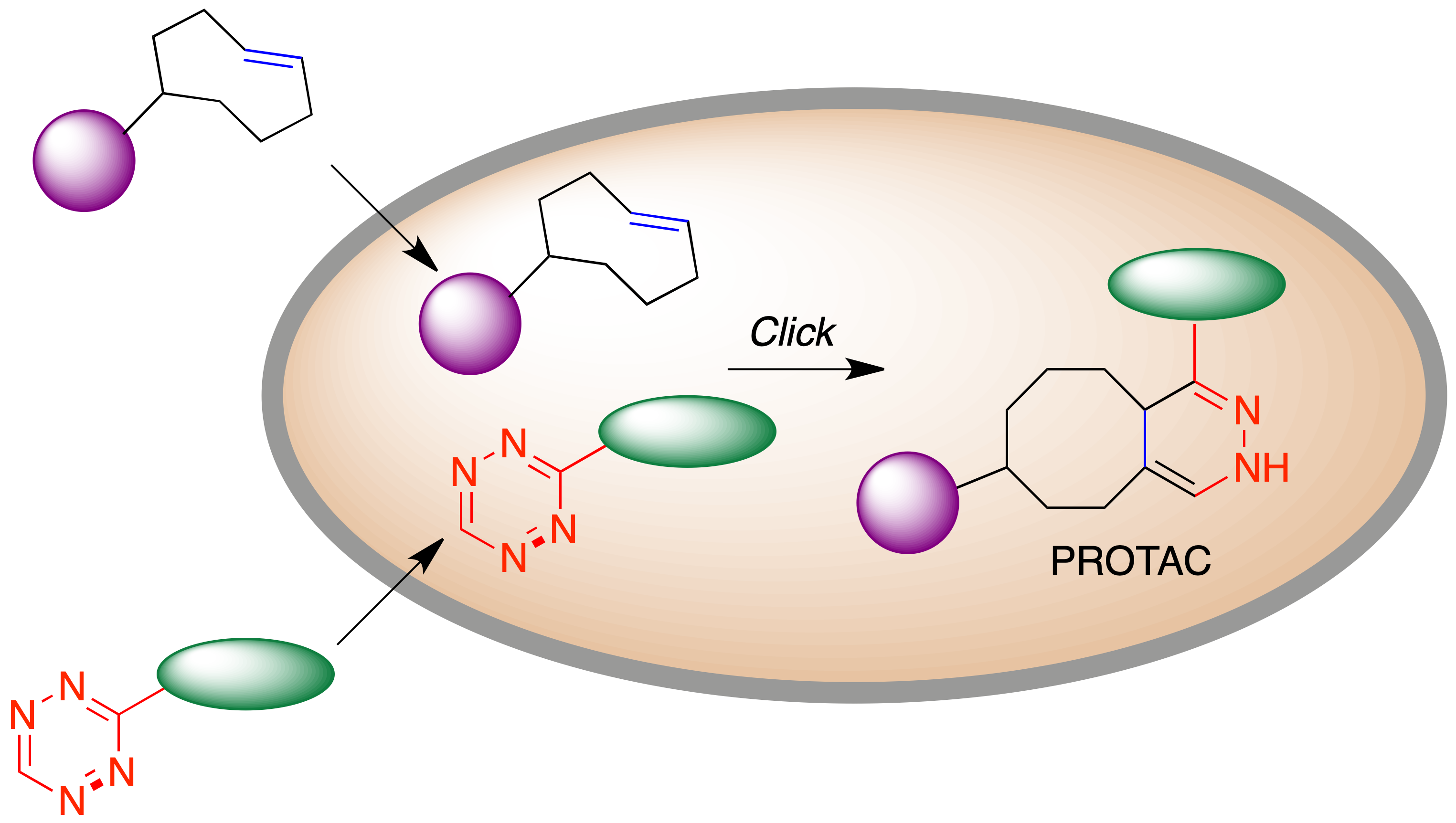
Figura 25. Fundamento de la estrategia CLIPTAC. Generación intracelular de PROTACs
Esta idea se ha aplicado a la degradación de oncoproteínas como posible base de tratamientos antitumorales. Como ejemplo, se representa en la Figura 26 la estructura de un PROTAC generado in situ y capaz de inducir la degradación de la proteína BDR4 (bromodomain-containing protein 4). Este PROTAC sirve de enlace entre el complejo E3-E2-ubiquitina y la proteína diana, que de esta forma puede ser ubiquitinilada y por tanto actuar como sustrato del proteasoma, como se esquematiza en la figura 26 (46).
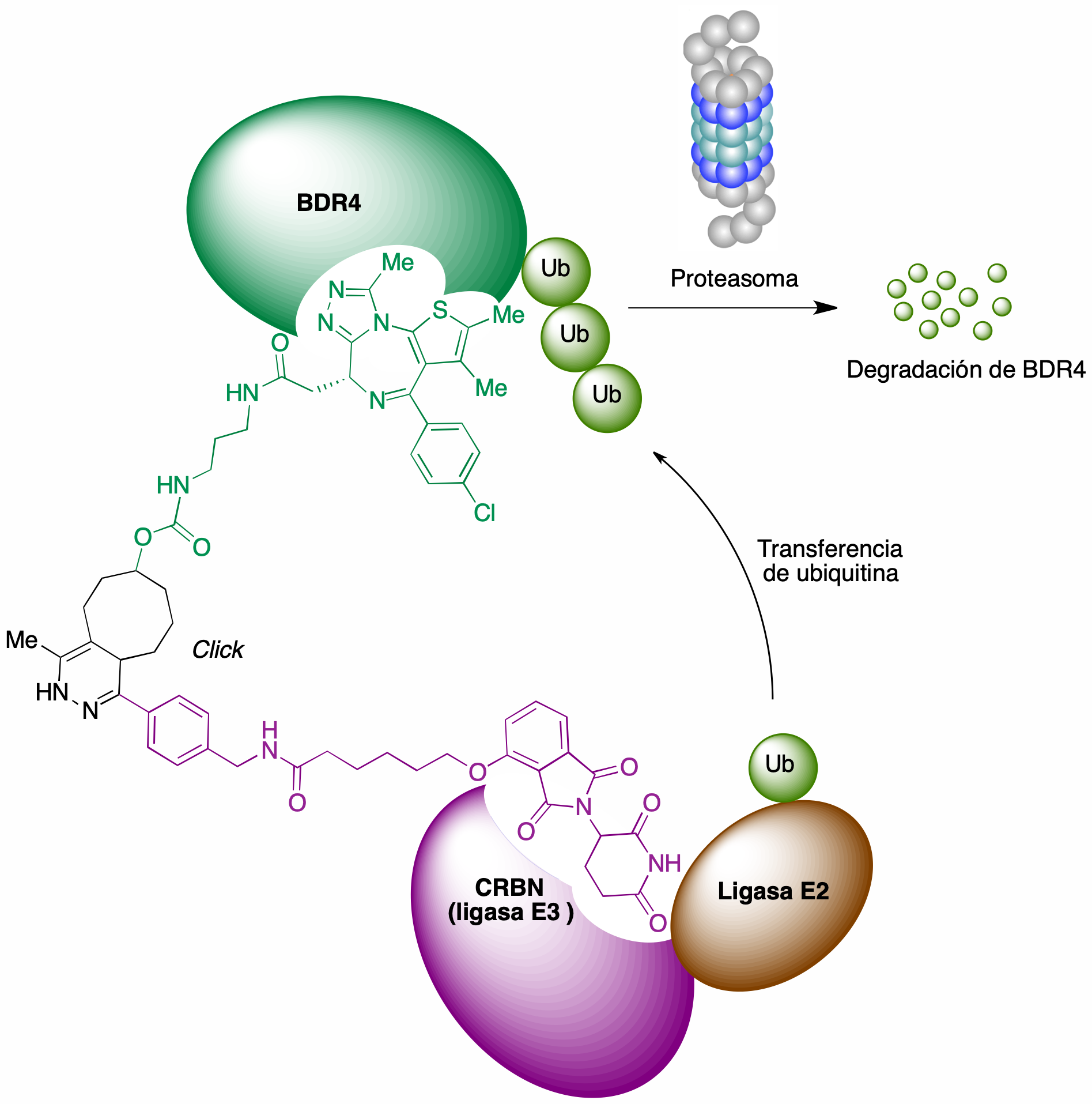
Figura 26. Un ejemplo de CLIPTAC, destinado a lograr la degradación de la oncoproteína BDR4
6.4 LYTACs: Degradación lisosomal de proteínas
Debido al gran interés actual en la degradación controlada de proteínas como estrategia terapéutica, se están investigando activamente metodologías alternativas al uso del proteasoma para degradar proteínas que se consideren dianas terapéuticas (47). En este contexto, Bertozzi publicó en 2020 sus primeros resultados sobre una técnica a la que ha llamado LYTAC (Lysosome Targeting Chimeras) y que se basa en la explotación de los mecanismos lisosomales de degradación de proteínas (48). A diferencia de los PROTACs, que normalmente afectan a proteínas citoplasmáticas, los LYTACs son capaces de degradar proteínas extracelulares y de membrana, que se estima que constituyen alrededor del 40% del proteoma, y por tanto tienen potencialmente una mayor generalidad como entidades terapéuticas. El proceso de degradación de una proteína extracelular por los lisosomas se inicia con su reconocimiento por receptores de membrana de la familia LTR (Lysosome-Targeting Receptors), de los cuales el más extendido es el receptor CI-M6PR (también llamado IGF2R), que reconoce glicoproteínas que terminan en residuos de manosa-6-fosfato y los transfiere a los lisosomas. La estrategia LYTAC consiste en unir covalentemente, por medio de una reacción click azida-alquino, un glucolipopéptido que contiene restos de manosa-6-fosfato a un anticuerpo capaz de reconocer la proteína que se desea degradar (Figura 27.A), siendo posible también el uso para este fin de ligandos con naturaleza de moléculas pequeñas.
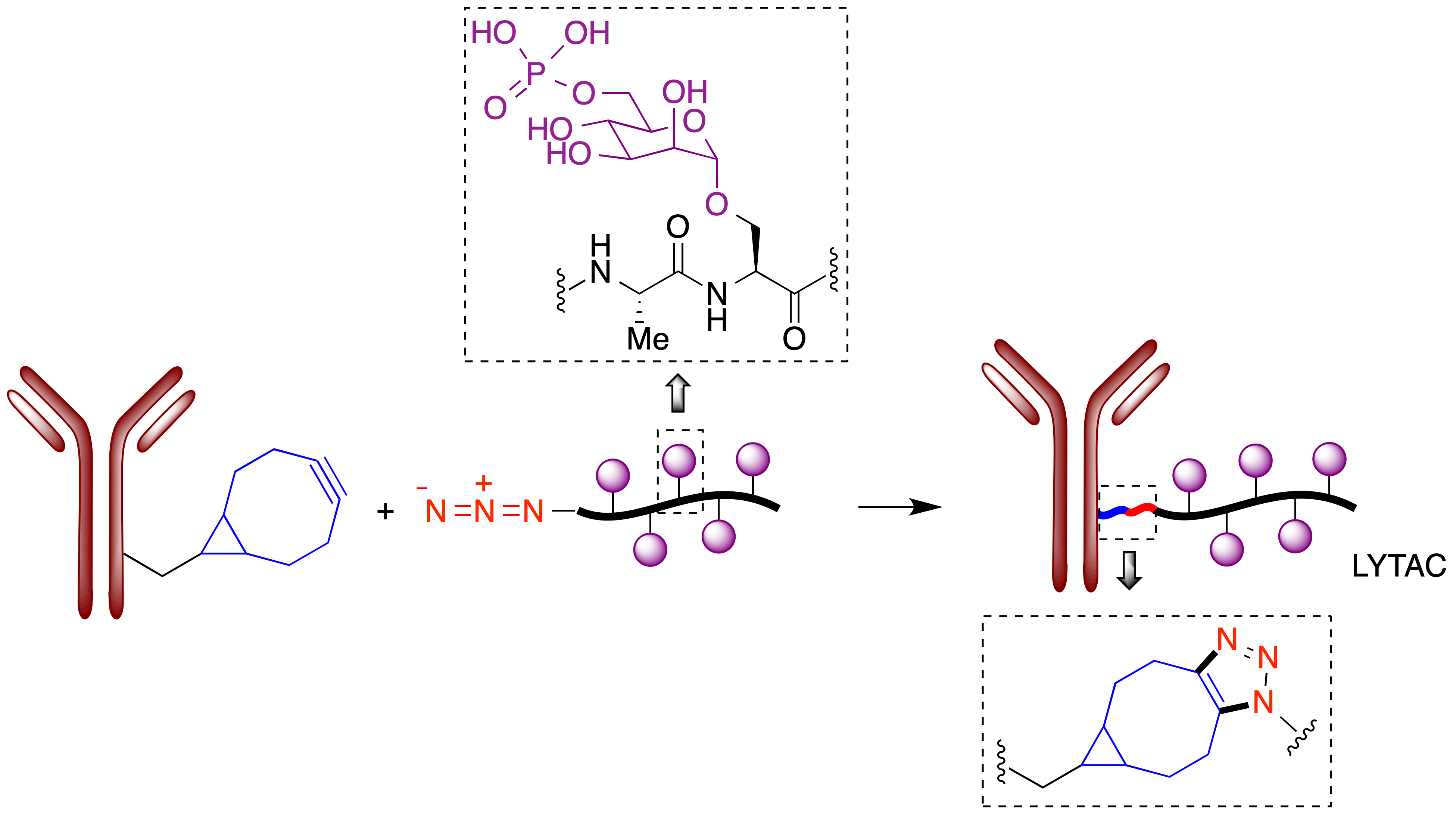
Figura 27. Unión covalente de un glucolipopéptido portador de residuos de manosa-6-fosfato a un anticuerpo por medio de una reacción click
Una vez que el anticuerpo ha reconocido a la proteína diana y que los fragmentos de manosa-6-fosfato son reconocidos por CI-M6PR, se desencadena un proceso de endocitosis que transfiere todo el conjunto al interior de un lisosoma donde tiene lugar la degradación hidrolítica de la proteína, reciclándose el receptor CI-M6PR (Figura 28).
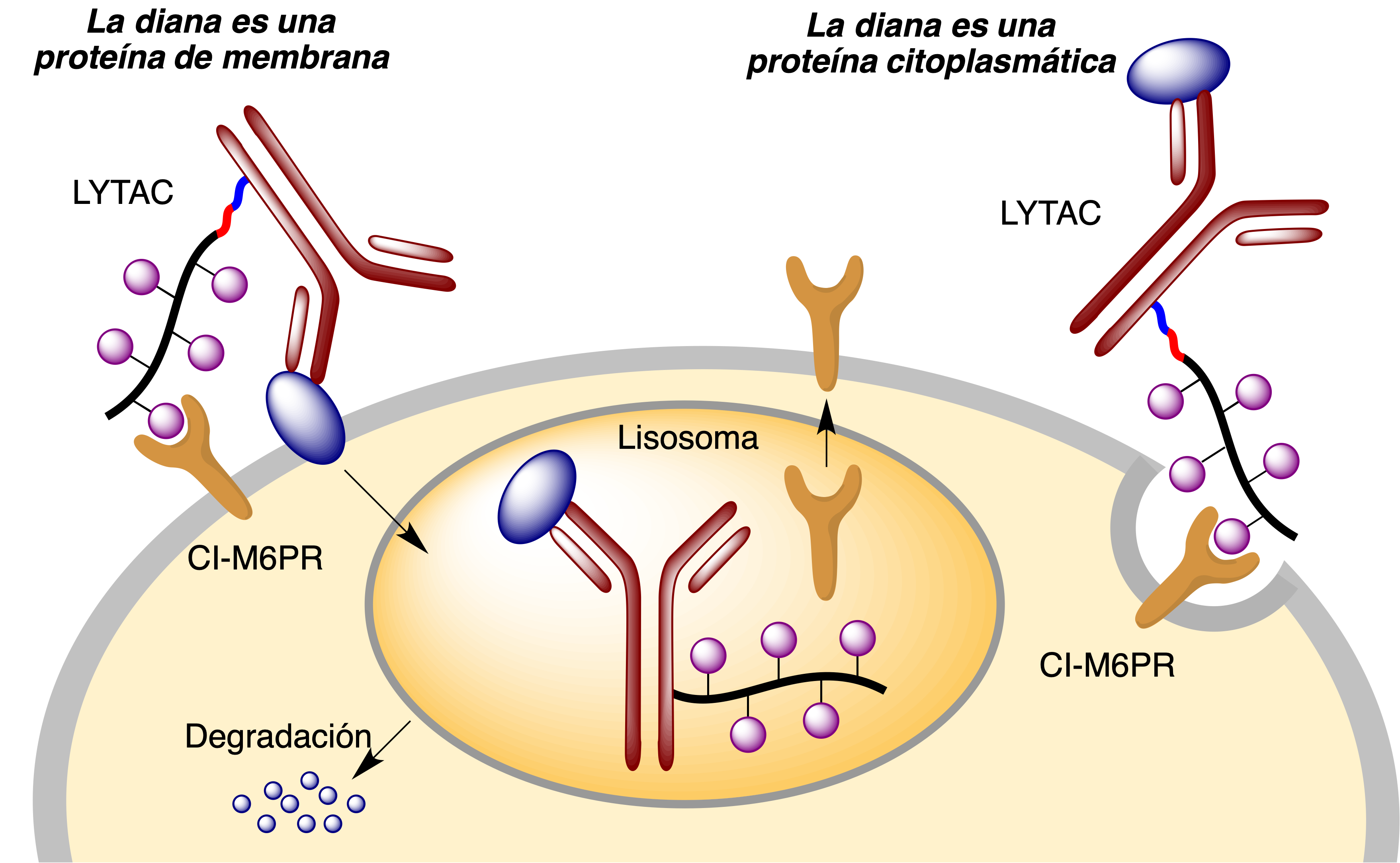
Figura 28. La estrategia LYTAC para la degradación lisosómica de proteínas
Aunque los receptores de la familia LTR están ampliamente distribuidos en el organismo, algunas de estas proteínas se expresan selectivamente en determinados tejidos, lo que hace posible diseñar LYTACs dirigidos selectivamente a un tejido determinado. Por ejemplo, Bertozzi ha utilizado ASGPR, una glicoproteína transmembrana expresada específicamente en el hígado, como receptor de LYTACs (49).
6.5 Proteómica química in situ
La proteómica química se basa en el uso de moléculas orgánicas como sondas para investigar la distribución y función de las proteínas, proporcionar información sobre las interacciones entre éstas y validar nuevas dianas farmacológicas. Esta nueva ciencia es actualmente esencial en el descubrimiento y desarrollo de fármacos (50,51,52,53).
La formación de imágenes basada en fluorescencia permite la visualización no invasiva de las biomoléculas para investigar sus funciones. Por este motivo, el descubrimiento de la proteína verde fluorescente, premiado con el premio Nobel de Química en 2008 (54) supuso una revolución en muchas áreas de la biología. El empleo de la química bioortogonal ha permitido llevar a cabo estudios celulares de la interacción entre moléculas pequeñas y proteínas (55). En una aproximación a la localización de la diana intracelular de un fármaco, se puede introducir en un cultivo celular un análogo de dicho fármaco que se haya modificado mediante la introducción de un fragmento de trans-cicloocteno en una parte de la molécula que, de acuerdo con estudios de relación estructura-actividad previos, no sea esencial para la interacción con su diana. De entre los miles de proteínas presentes en la superficie o el interior de la célula y que forman el proteoma de ésta, este compuesto debe unirse selectivamente a su diana. Si a continuación se adiciona un derivado de tetrazina, dotado de fluorescencia para facilitar la monitorización del proceso, debe tener lugar una reacción hetero Diels-Alder en la que se genera una unión covalente entre el fármaco y el fragmento de tetrazina. Puesto que el fármaco, a su vez, se encuentra unido a su diana, el proceso induce la emisión fluorescente por parte de ésta (Figura 29). Este método ha permitido la localización intracelular de la diana de un elevado número de fármacos, incluyendo el taxol, olaparib, foretinib y dasatinib, entre otros muchos (56).
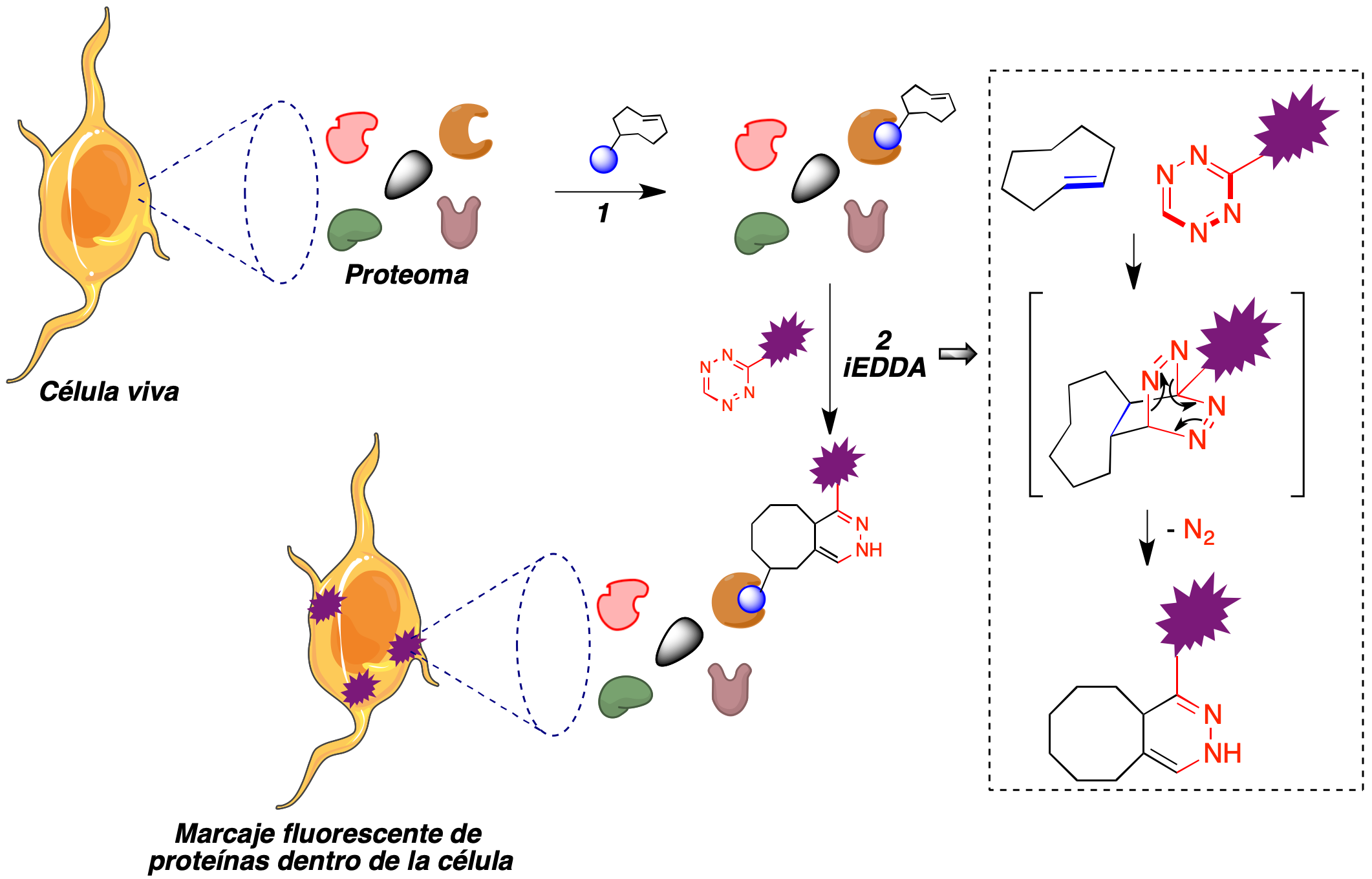
Figura 29. Localización de la diana intracelular de un fármaco basada en su marcaje covalente con derivados fluorescentes de tetrazina. Etapa 1: Unión a su diana de un fármaco, al que se ha incorporado un resto de trans-ciclooctenilo. Etapa 2: Marcaje fluorescente de la proteína unida al fármaco por medio de una reacción hetero Diels-Alder del fragmento de trans-ciclohexeno con un derivado fluorescente de 1,2,4,5-tetrazina.
Esta metodología permite también el marcaje fluorescente de proteínas para las que no se dispone de ligandos conocidos. En una primera etapa, se procede a la modificación de la proteína que se desea estudiar mediante la incorporación de un aminoácido no natural portador de un resto de trans-ciclooctenilo, utilizando técnicas de genética molecular. En una segunda etapa, la reacción del fragmento de trans-cicloocteno con un derivado fluorescente de 1,2,4,5-tetrazina conduce al marcaje covalente de la proteína (Figura 30).

Figura 30. Marcaje fluorescente de una proteína para la que no se conocen ligandos
7. CONCLUSIONES
Los avances de la ciencia no se deben solo a la realización de nuevos descubrimientos, sino también a la aparición de nuevos conceptos y marcos de referencia mentales que estimulen nuevos planteamientos y permitan abrir caminos previamente inexplorados. En este contexto, el Nobel de Química de 2022 ha premiado la invención y desarrollo de los conceptos de química click y química bioortogonal. La relevancia de los logros de los galardonados se pone de manifiesto en los grandes avances a los que han conducido en todos los campos de la química y áreas relacionadas.
Agradecimientos
El autor agradece al Ministerio de Ciencia, Innovación y Universidades (proyecto TED2021-129408B-I00) la financiación de la investigación de su grupo en el área de la organocatálisis sostenible.
8. REFERENCIAS
- Fernholm A. Their functional chemistry works wonders. Popular science background on the Nobel Prize in Chemistry 2022. https://www.nobelprize.org/prizes/chemistry/2022/popular-information.
- Ramström O. Click chemistry and bioorthogonal chemistry. Scientific background on the Nobel Prize in Chemistry 2022. https://www.nobelprize.org/prizes/chemistry/2022/advanced-information.
- Kolb HC, Finn MG, Sharpless KB. Click chemistry: Diverse chemical function from a few good reactions. Angew Chem Int Ed 2001; 40: 2004-2021.
- https://www.nobelprize.org/prizes/chemistry/2001/sharpless/lecture/
- Breugst M, Reissig H-U. The Huisgen reaction: Milestones of the 1,3-dipolar cycloaddition. Angew Chem Int Ed 2020; 59: 12293-12307.
- Beckmann E. Zur kenntniss der aldoxime. VII. Ber Dtsch Chem Ges 1890; 23: 3331-3341.
- Michael A. Ueber die einwirkung von diazobenzolimid auf acetylendicarbonsäuremethylester. J Prakt Chem 1893; 48: 94–95.
- Dimroth O, Fester G. Triazol und tetrazol aus stickstoffwasserstoffsäure. Ber Dtsch Chem Ges 1910; 43: 2219-2223.
- Huisgen R. 1,3-Dipolar cycloadditions. Past and future. Angew Chem Int Ed Engl 1963; 2: 565–598.
- Tomøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 2002; 67: 3057-3064.
- Rostovtsev W, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I) catalyzed regioselective ligation of azides and terminal alkynes. Angew Chem Int Ed 2002; 41: 2596-2599.
- Meldal M, Tornoe CW. Cu-catalyzed azide-alkyne cycloaddition. Chem Rev 2008; 108: 2952−3015.
- Hein JE, Fokin VV. Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem Soc Rev 2010; 39: 1302−1315.
- Liang LY, Astruc D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord Chem Rev 2011; 255: 2933−2945.
- Haldon E, Nicasio MC, Pérez PJ. Copper-catalysed azide-alkyne cycloadditions (CuAAC): An update. Org Biomol Chem 2015; 13: 9528−9550.
- Zhou W, Wang C, Liu Z, Gou S. Hypoxia-activated prodrugs with dual COX-2/CA Inhibitory effects on attenuating cardiac inflammation under hypoxia. J Med Chem 2022; 65: 13436-13451.
- Wang FC, Peng D, Ren T-T, Liu SP, Du J-R, Chen Z-H, Zhang T-T, Gu X, Li M, Cao S-L, Xu X. A 1,2,3-triazole derivative of quinazoline exhibits antitumor activity by tethering RNF168 to SQSTM1/P62. J Med Chem 2022; 65: 15028–15047.
- Hu W, Chen Z, Wang M, Luo T, Wang J. Design, synthesis and evaluation of HepDirect fluorescence probes mediated by asialoglycoprotein receptor. Dyes and Pigments 2018; 159: 471-478.
- Gómez-Carpintero C, Cabrero C, Sánchez JD, González JF, Menéndez JC. A sequential multistep process for the fully mechanochemical, one-pot synthesis of the antiepileptic drug rufinamide. Green Chem Lett Rev 2022; 15: 639-646
- Gómez-Carpintero C, Sánchez JD, González JF, Menéndez JC. Mechanochemical synthesis of primary amides. J Org Chem 2021; 86: 14232-14237
- Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed Engl. 2009; 48: 6974-6998.
- Avendaño C. Química bioortogonal. Real Academia Nacional de Farmacia, Monografía 34. Madrid, 2012.
- Lemieux GA. Modulating cell surface immunoreactivity and cellular targeting of diagnostic probes using bio-orthogonal chemistry. PhD Thesis, University of California, Berkeley, 2002.
- Kocienski PJ. Protecting groups, 3ª Ed. Thieme Verlag, 2005.
- Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. A comparative study of bioorthogonal reactions with azides. ACS Chem Biol 2006, 1, 644–648.
- Blomquist AT, Liu LH, Many-membered carbon rings. VII. Cycloöctyne. J Am Chem Soc 1953; 75: 2153-2154.
- Wittig G, Krebs A. Zur existenz niedergliedriger cycloalkine, I. Chem Ber 1961; 94: 3260-3275.
- Mahal LK, Yarema KJ, Bertozzi CR. Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science 1997; 276: 1125–1128.
- Agard NJ, Prescher JA, Bertozzi CR. A strain-promoted [3 + 2] azide−alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc 2004; 126: 15046-15047.
- Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA 2007; 104: 16793-16797.
- Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. In vivo imaging of membrane-associated glycans in developing zebrafish. Science 2008; 320: 664-667.
- Blackman ML, Royzen M, Fox JM. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J Am Chem Soc 2008; 130: 13518-13519.
- Devaraj NK, Weissleder R, Hilderbrand SA. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjug Chem 2008; 19: 2297-2299.
- Devaraj NK. The future of bioorthogonal chemistry ACS Cent Sci 2018; 4: 952−959.
- Scinto SL, Bilodeau DA, Hincapie R, Lee W, Nguyen SS, Xu M, am Ende CW, Finn MG, Lang K, Lin Q, Pezacki JP, Prescher JA, Robillard MS, Fox JM. Bioorthogonal chemistry. Nat Rev Primers 2021; 1: 30.
- Bird RE, Lemmel SA, Yu X, Zhou QA. Bioorthogonal chemistry and its applications. Bioconjug Chem 2021; 32: 2457−2479.
- Taiariol L, Chaix C, Farre C, Moreau E. Click and bioorthogonal chemistry: the future of active targeting of nanoparticles for nanomedicines? Chem Rev 2022; 122: 340–384.
- Wu D, Yang K, Zhang Z, Feng Y, Rao L, Chen X, Yu G. Metal-free bioorthogonal click chemistry in cancer theranostic. Chem Soc Rev 2022; 51: 1336-1376.
- Rossin R, Verkerk PR, van den Bosch SM, Vulders RCM, Verel I, Lub J, Robillard MS. In vivo chemistry for pretargeted tumor imaging in live mice. Angew Chem Int Ed 2010; 49: 3375–3378.
- Wang H, Wang R, Cai K, He H, Liu Y, Yen J, Wang Z, Xu M, Sun Y, Zhou X, Yin Q, Tang L, Dobrucki IT, 5, Dobrucki LW, Chaney EJ, Boppart SA, Fan TM, Lezmi S, ChenX, Yin L, Cheng J. Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat Chem Biol 2017; 13: 415–424.
- Versteegen RM, Rossin R, ten HoeveW, Janssen HM, Robillard RS. Click to release: instantaneous doxorubicin elimination upon tetrazine ligation. Angew Chem Int Ed 2013; 52: 14112 –14116
- Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Click chemistry in situ: Acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew Chem Int Ed 2002; 41: 1053-1057.
- Bourne Y, Sharpless KB, Taylor P, Marchot P. Steric and dynamic parameters influencing in situ cycloadditions to form triazole inhibitors with crystalline acetylcholinesterase. J Am Chem Soc 2016; 138: 1611−1621.
- Bourne Y, Kolb HC, Radic Z, Sharpless KB, Taylor P, Marchot P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc Natl Acad Sci USA 2004; 101: 1449-1454.
- Agnew HD, Coppock MB, Idso MN, Lai BT, Liang JX, McCarthy-Torrens AM, Warren CM, Heath JR. Protein-catalyzed capture agents. Chem Rev 2019; 119: 9950-9970.
- Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Cent Sci 2016; 2: 927−934.
- Cores A, Villacampa M. PROTAC: Redirigiendo los sistemas de degradación de proteínas a nuevos sustratos. An Real Acad Farm 2022, 88: 45-59.
- Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, Bertozzi CR. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020; 584:291-297.
- Ahn G, Banik SM, Miller CL, Riley NM, Cochran JR, Bertozzi CR. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat Chem Biol 2021: 17: 937–946.
- Doyle SK, Pop MS, Evans HL, Koehler AN. Advances in discovering small molecules to probe protein function in a systems context. Curr Opin Chem Biol 2016; 30: 28–36.
- Garbaccio MR, Parmee ER. The impact of chemical probes in drug discovery: A pharmaceutical industry perspective. Cell Chem Biol 2016; 23: 11-17.
- Jones LH, Neubert H. Clinical chemoproteomics – Opportunities and obstacles. Sci Transl Med 2017; 9: eaaf7951.
- Chen X, Wang Y, Ma N, Tian J, Shao Y, Zhu B, Wong YK, Liang Z, Zou C, Wang J. Target identification of natural medicine with chemical proteomics approach: probe synthesis, target fishing and protein identification. Sig Transduct Target Ther 2020; 5: 72.
- Tsien RY. Constructing and exploiting the fluorescent protein paintbox (Nobel lecture). Angew Chem Int Ed 2009; 48: 5612–5626.
- Cañeque T, Müller S, Rodríguez R. Visualizing biologically active small molecules in cells using click chemistry. Nat Rev Chem 2018; 1: 201-215.
- Kim YR, Kim YH, Kim SW, Lee YJ, Chae DE, Kim KA, Lee ZW, Kim ND, Choi JS, Choi IS, Lee KB. A bioorthogonal approach for imaging the binding between dasatinib and its target proteins inside living cells. Chem Commun 2016; 52: 11764–11767.
