1. EUROPEAN MEDICINES AGENCY (EMA)
(A) Tracto alimentario y metabolismo
Avalglucosidasa Alfa (Nexviadyme®) Genzyme
Indicación: Tratamiento enzimático de sustitución a largo plazo para el tratamiento de pacientes con enfermedad de Pompe (déficit de αlfaglucosidasa ácida).
Tipo: Autorizado el 24-6-2022, como medicamento huérfano; autorizado previamente en Estados Unidos (6-8-2021) como medicamento huérfano. Medicamento biológico, constituido por alfaglucosidasa ácida humana obtenida mediante tecnología de ADN recombinante.
Mecanismo: La avalglucosidasa alfa es una alfaglucosidasa ácida humana (AGA) recombinante que proporciona una fuente exógena de AGA. La avalglucosidasa alfa es una modificación de la alglucosidasa alfa en la que aproximadamente 7 estructuras de hexamanosa, cada una de las cuales incluye 2 mitades terminales de manosa-6-fosfato (bis-M6P), se conjugan con residuos de ácido siálico oxidado en la alglucosidasa alfa. La avalglucosidasa alfa presenta 15 veces más mitades de manosa-6-fosfato (M6P) en comparación con la alglucosidasa alfa natural. La unión a los receptores M6P en la superficie celular se produce a través de grupos de carbohidratos en la molécula de AGA, después de lo cual se internaliza y transporta a los lisosomas, donde sufre una división proteolítica que provoca un aumento de la actividad enzimática para degradar el glucógeno.
Eficacia clínica: Estudio multinacional, multicéntrico, aleatorizado y doble ciego para comparar la eficacia y la seguridad de Nexviadyme y alglucosidasa alfa una vez cada dos semanas durante 12 meses (49 semanas) en 100 pacientes de 16 a 78 años con enfermedad de Pompe sin tratamiento previo al inicio del tratamiento. El estudio incluyó una fase abierta, a largo plazo, de seguimiento de hasta 5 años para todos los pacientes, en la que los pacientes del grupo de alglucosidasa alfa cambiaron al tratamiento con Nexviadyme. La variable primaria del estudio fue el cambio en el % previsto de la capacidad vital forzada (CVF) en posición erguida desde el momento basal hasta los 12 meses (semana 49): 2,89% (Nexviadyme) vs 0,46% (alglucosidasa alfa). La variable secundaria fue el cambio en la distancia total caminada en 6 minutos (6-Minute Walk Test) desde el momento basal hasta los 12 meses (semana 49): 32,21 vs. 2,19 m.
Eventos adversos: Los más comunes son prurito (9,4%), erupción cutánea (8%), cefalea (7,2%), urticaria (6,5%), fatiga (6,5%), náuseas (5,8%) y escalofríos (5,1%).
Eladocagene Exuparvovec (Upstaza®) PTC
Indicación: Tratamiento de pacientes de 18 meses de edad y mayores con un diagnóstico clínico, molecular y genéticamente confirmado de deficiencia de L-aminoácido descarboxilasa aromática (AADC) con un fenotipo grave.
Tipo: Autorizado en condiciones excepcionales el 18-7-2022, como medicamento huérfano. Medicamento de terapia avanzada (terapia génica), constituido por 2,8 x 10(11) genomas vectorizados/0.5 mL, que expresa la enzima L-aminoácido aromático descarboxilasa humana (hAADC). Se trata de un vector recombinante no replicable basado en el virus adenoasociado serotipo 2 (AAV2) que contiene el ADNc del gen de la dopa descarboxilasa (DDC) humana bajo el control del promotor inmediato del citomegalovirus.
Mecanismo: La deficiencia de AADC es un defecto congénito de la biosíntesis de neurotransmisores con una herencia autosómica recesiva en el gen de la dopa descarboxilasa (DDC). El gen DDC codifica la enzima AADC, que convierte L-3,4 dihidroxifenilalanina (L-DOPA) en dopamina. Las mutaciones en el gen DDC conllevan una reducción o ausencia de la actividad de la enzima AADC, lo cual provoca una reducción en los niveles de dopamina y el fracaso de la mayoría de los pacientes con deficiencia de AADC para alcanzar los objetivos del desarrollo. Eladocagén exuparvovec es una terapia génica basada en vectores AAV2 recombinantes que contienen el cADN humano del gen DDC. Tras inyectarlo en el putamen, el medicamento da lugar a la expresión de la enzima AADC y a la posterior producción de dopamina y, en consecuencia, al desarrollo de la función motora en los pacientes con carencia de AADC tratados.
Eficacia clínica: Dos estudios clínicos que incluyeron a 20 pacientes con deficiencia grave de AADC, que fueron tratados con una dosis total de 1,8 × 10(11) o 2,4 × 10(11) genomas vectorizados en una única sesión quirúrgica. La variable principal de eficacia se determinó con la versión 2 de la Escala de Desarrollo Motor de Peabody (PDMS-2), que evalúa el desarrollo motor del niño hasta la edad de desarrollo de 5 años. La variable se evaluó a los 24 meses de la terapia génica, siendo la media de los mínimos cuadrados (LS) del cambio en la puntuación total de PDMS-2 con respecto al inicio de 104,4 puntos; la mejora con respecto al inicio en la puntuación total de PDMS-2 a los 12 meses de tratamiento fue de 76,1 puntos y se mantuvo hasta los 60 meses, alcanzando 108,2 puntos. Los pacientes que recibieron eladocagén exuparvovec a una edad más temprana mostraron una respuesta más rápida y un nivel final superior.
Eventos adversos: Los más comunes son insomnio, irritabilidad y discinesia. El más frecuente fue la discinesia (86%) y fue predominante durante los 2 primeros meses después del tratamiento.
Lonafarnib (Zokinvy®) EigerBio
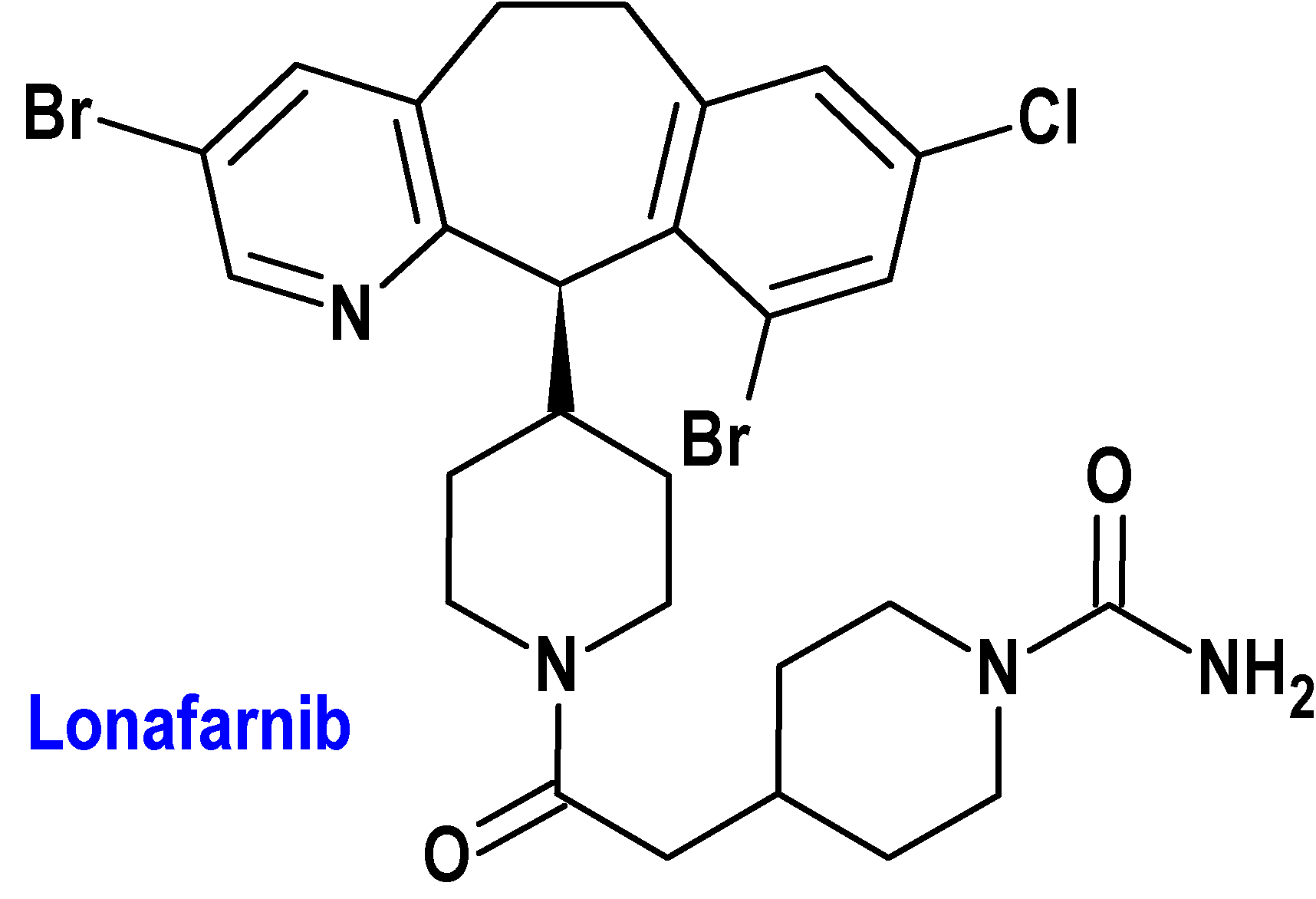
Indicación: Tratamiento de pacientes a partir de los 12 meses de edad con un diagnóstico confirmado genéticamente de síndrome de progeria de Hutchinson-Gilford, o una laminopatía progeroide con déficit de procesamiento asociada a una mutación heterocigótica de LMNA con acumulación de proteínas similares a la progerina o una mutación homocigótica o heterocigótica compuesta de ZMPSTE24.
Tipo: Autorizado el 18-7-2022, como medicamento huérfano y en circunstancias excepcionales; autorizado previamente en Estados Unidos (20-11-2020) como medicamento huérfano.
Mecanismo: La progreria es una anomalía genética progresiva que acelera el proceso de envejecimiento de los niños; forma parte del grupo de laminopatías, enfermedades genéticas debidas a mutaciones en el gen LMNA, que codifica para las láminas A y C, proteínas que componen la lámina que envuelve internamente el núcleo celular. La forma alterada de la proteína – conocida como progerina – lleva a la disrupción del ensamblaje normal de la envoltura nuclear, la función nuclear y la funcionalidad de la lamina A. La acumulación de progerina y proteínas similares a la progerina en las células dentro de las paredes de los grandes vasos sanguíneos provoca inflamación y fibrosis. Lonafarnib inhibe la farnesiltransferasa, impidiendo el proceso bioquímico de farnesilación y la posterior acumulación de progerina y proteínas similares a la progerina en la membrana nuclear interna, promoviendo así el mantenimiento de la integridad y la función celular.
Eficacia clínica: Dos estudios de fase 2, abiertos, de un solo centro y de un solo brazo. El primero se hizo sobre 28 pacientes que recibieron lonafarnib durante 24 a 30 meses con dosis iniciales de 115 mg/m2/12 h; tras 4 meses de tratamiento, a los que toleraron el tratamiento se les aumentó la dosis a 150 mg/m2/12 h a lo largo de 30 meses; tras completarlo, 26 fueron incorporados a un nuevo estudio en dos fases; en la primera recibieron tratamiento durante 5 años y en la segunda otros 3 adicionales. El segundo estudio contó con 35 pacientes, que recibieron 150 mg/m2/12 h de lonafarnib durante 12 a 36 meses. La variable primaria de eficacia fue la supervivencia sobre el conjunto de 62 pacientes tratados en ambos estudios, en una comparación histórica con pacientes no tratados. La esperanza de vida media de los pacientes tratados aumentó en un promedio de 0,44-0,47 años durante los primeros tres años de seguimiento y 4,3 años durante el último tiempo de seguimiento (11 años en total) en comparación con los pacientes no tratados.
Eventos adversos: Los más comunes son vómitos (86%), diarrea (78%), elevación de la aspartato aminotransferasa (64%), elevación de la alanina aminotransferasa (50%), disminución del apetito (41%), náuseas (38%), dolor abdominal (35%), fatiga (29%), pérdida de peso (27%), estreñimiento (18%) e infección del tracto respiratorio superior (11%). La mayoría se produjeron en las primeras 4 primeras semanas del tratamiento y, en general, disminuyeron de forma constante al aumentar la duración del tratamiento.
Olipudasa Alfa (Xenpozyme®) Genzyme
Indicación: Terapia enzimática de sustitución para el tratamiento de las manifestaciones no relacionadas con el sistema nervioso central (SNC) del déficit de esfingomielinasa ácida (Acid Sphingomyelinase Deficiency, ASMD) en pacientes pediátricos y adultos con tipo A/B o tipo B.
Tipo: Autorizado el 24-6-2022 como medicamento huérfano. Medicamento biológico; es una esfingomielinasa ácida humana obtenida mediante tecnología de ADN recombinante.
Mecanismo: Tratamiento de restauración exógena para paliar el déficit endógeno de esfingomielinasa ácida, con el fin de reducir la acumulación de esfingomielina en órganos de pacientes con déficit de esfingomielinasa ácida (ASMD).
Eficacia clínica: Tres estudios clínicos, uno en pacientes adultos (de fase 2/3, multicéntrico, aleatorizado, doble ciego y controlado con placebo), otro en pacientes pediátricos y un estudio de extensión en pacientes adultos y pediátricos, en los que participaron un total de 61 pacientes con ASMD. Las dos variables primarias de eficacia fueron las variaciones porcentuales de la capacidad de difusión pulmonar para el monóxido de carbono (22 vs. 3%, placebo) y del volumen del bazo (-39,4 vs +0,5%, placebo), desde el inicio hasta la semana 52.
Eventos adversos: Los más comunes son cefalea (32%), pirexia (25%), urticaria (22%), náuseas (20%), vómitos (17%), dolor abdominal (15%), mialgia (12%), prurito (10%) y aumento de la proteína C reactiva (10%).
(B) Sangre y sistema hematopoyético
Eptacog Beta activado (Cevenfacta®) Laboratoire français du Fractionnement et des Biotechnologies (LFB)
Indicación: Tratamiento en adultos y adolescentes (a partir de los 12 años) de episodios de sangrado y para la prevención de hemorragias en pacientes sometidos a cirugía o procedimientos invasivos en pacientes con hemofilia congénita con inhibidores de alta respuesta a los factores de coagulación VIII o IX (≥5 Unidades Bethesda, UB), así como en pacientes con hemofilia congénita con títulos bajos de inhibidores (UB <5), pero que se espera que tengan una respuesta anamnésica elevada a la administración de factor VIII (FVIII) o factor IX (FIX) o bien que sean refractarios a una mayor dosis de FVIII o FIX.
Tipo: Autorizado el 15-7-2022. Medicamento biológico, con estructura casi idéntica a la del Factor VII de coagulación humano producido a partir de leche de conejo mediante una tecnología de ADN recombinante.
Mecanismo: En condiciones normales, el FVIIa es el factor que inicia la coagulación tras su interacción con el factor tisular (FT) en la superficie celular. Eptacog Beta activado actúa como el factor de coagulación VIIa activando el factor X, que inicia el proceso de coagulación y, por lo tanto, controla el sangrado. Dado que el factor VII actúa directamente sobre el factor X, independientemente de los factores VIII y IX, el medicamento puede utilizarse para restablecer la hemostasia tanto en ausencia como en presencia de inhibidores.
Eficacia clínica: Tres estudios clínicos de fase 3 en un total de 60 pacientes de sexo masculino con hemofilia congénita A o B con inhibidores. Como variable principal de valoración de la eficacia clínica se utilizó la proporción de episodios de sangrado tratados con éxito con una respuesta «buena» o «excelente» (usando una escala de evaluación de cuatro puntos), independientemente de la gravedad, a 12 horas después de la administración inicial de este medicamento: 85% (81-90%) en uno de los estudios y 63% (61-66%) en otro; en el tercero la variable principal de valoración fue el porcentaje de cirugías u otros procedimientos invasivos con una respuesta «buena» o «excelente» al tratamiento 48 (±4) horas después de la última administración de este medicamento: 82%.
Eventos adversos: Los más comunes (todos ellos con una incidencia de 1,3%) son molestias en el lugar de la inyección, hematoma en el lugar de la infusión, hematoma posoperatorio, reacción relacionada con la infusión, aumento de la temperatura corporal, mareos y cefalea.
Valoctogene Roxaparvovoec (Roctavian®) Biomarin
Indicación: Tratamiento de la hemofilia A (deficiencia congénita de factor VIII) grave en pacientes adultos sin antecedentes de inhibidores del factor VIII y sin anticuerpos específicos contra el virus adenoasociado de serotipo 5 (AAV5) detectables.
Tipo: Autorizado condicionalmente el 24-8-2022, como medicamento huérfano. Medicamento de terapia avanzada (terapia génica) que expresa la variante SQ del factor de coagulación humano VIII (hFVIII-SQ) con el dominio B eliminado bajo el control de un promotor específico del hígado. Es un vector vírico adenoasociado de serotipo AAV5 recombinante y que no puede replicarse.
Mecanismo: Terapia génica basada en el vector vírico adenoasociado de serotipo 5 (AAV5) que, controlado por un promotor específico del hígado, causa la expresión de la variante SQ de un factor de coagulación humano VIII recombinante (hFVIII-SQ) con el dominio B eliminado. El hFVIII-SQ expresado reemplaza el factor de coagulación VIII ausente, necesario para la hemostasia eficaz. Después de la perfusión de valoctocogén roxaparvovec, el ADN del vector se procesa in vivo para formar transgenes episomales de longitud completa que persisten como las formas de ADN estables que respaldan la producción de hFVIII-SQ a largo plazo.
Eficacia clínica: Ensayo de fase 3 abierto y de un solo brazo, que incluyó a 123 pacientes adultos (mediana 30 años), con un seguimiento medio de 122 semanas. La variable principal de eficacia fue el cambio respecto al inicio en la actividad de factor VIII durante la semana 104 (2 años) después de la perfusión del medicamento: 22,7 UI/dl.
Eventos adversos: Los más comunes son aumento de los niveles de ALT (80%), AST (67%), LDH (54%), náuseas (37%) y cefalea (35%).
(D) Dermatología
Abedul, extracto (Filsuvez®) Amryt
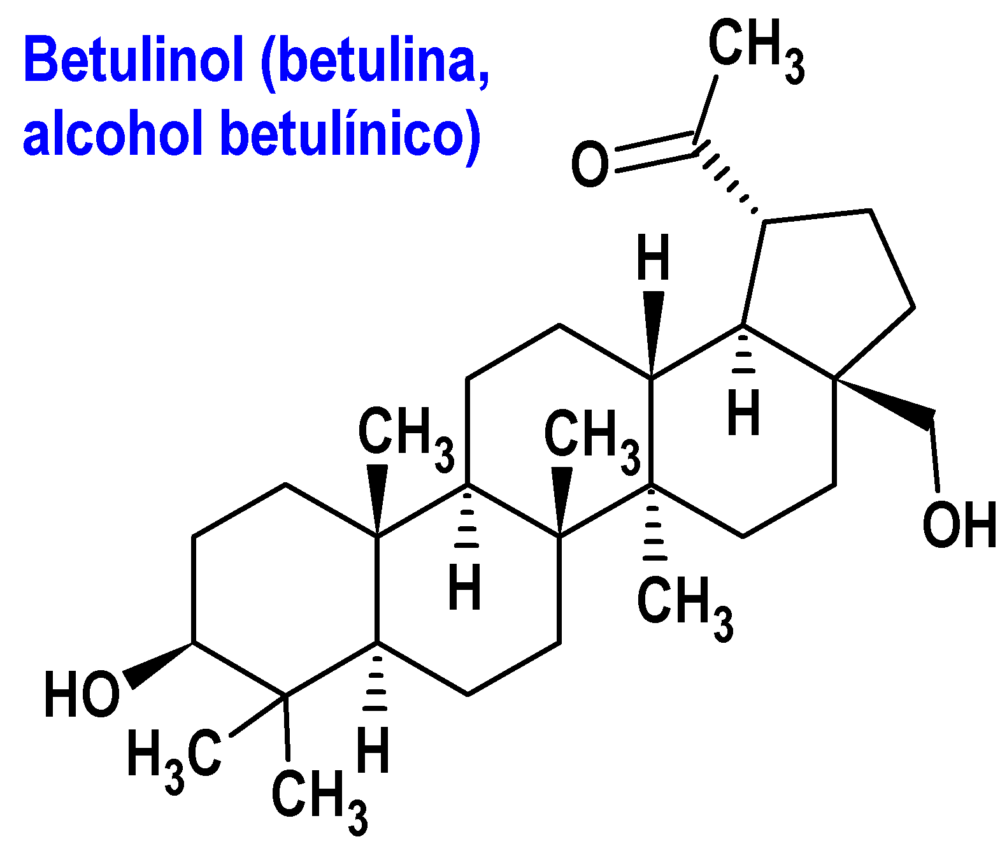
Indicación: Tratamiento de heridas de espesor parcial (HEP) asociadas a la epidermolisis bullosa (EB) distrófica y juntural en pacientes de 6 meses o mayores.
Tipo: Autorizado el 21-6-2022, como medicamento huérfano. Extracto seco y refinado de la corteza de dos especies de abedul (Betula pendula Roth y Betula pubescens Ehrh), equivalente a 0,5-1,0 g de corteza de abedul, conteniendo 84-95 mg de una mezcla de triterpenos compuesta por betulina, ácido betulínico, eritrodiol, lupeol y ácido oleanólico.
Mecanismo: Modula los mediadores inflamatorios y activan las vías intracelulares que intervienen en la diferenciación y la migración de los queratinocitos, así como en la curación y el cierre de las heridas, aunque se desconoce el mecanismo específico de acción en la cicatrización de heridas.
Eficacia clínica: Estudio de fase 3, aleatorizado, doble ciego y controlado con placebo en 223 adultos y niños, durante 90 días. La variable principal de eficacia fue la proporción de pacientes con primer cierre completo de la herida objetivo en 45 días: 41,3% vs 28,9% (placebo).
Eventos adversos: Los más comunes son complicaciones – aumento del tamaño, reapertura y dolor – en la herida (11,6% de los pacientes con EB y 2,9% con otras HEP), reacción en el lugar de aplicación (5,8% con EB), infecciones de la herida (4,0% con EB), prurito (3,1% con EB y 1,3% con otras HEP), dolor cutáneo (2,5% con otras HEP) y reacciones de hipersensibilidad (1,3% con EB).
(G) Sistema genitourinario y hormonal sexuales
Linzagolix (Yselty®) ObsEva
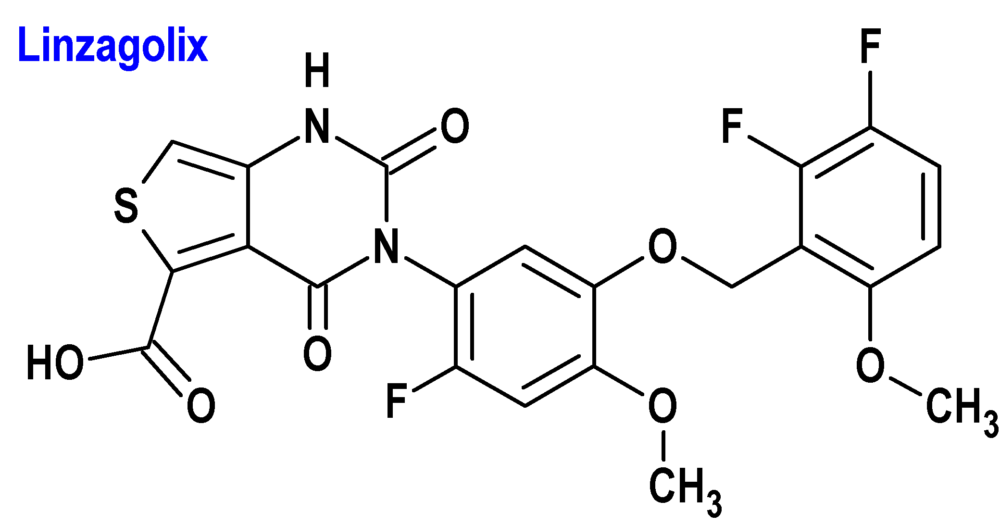
Indicación: Tratamiento de los síntomas moderados a graves de los miomas uterinos en mujeres adultas en edad fértil.
Tipo: Autorizado el 14-06-2022.
Mecanismo: Antagonista selectivo del receptor de la hormona liberadora de gonadotropinas (GnRH) que inhibe la señalización endógena de la GnRH al unirse de forma competitiva a los receptores de la GnRH en la hipófisis, modulando así el eje hipotálamo-hipófisogonadal. Provoca una supresión dependiente de la dosis de la hormona luteinizante (lutropina, LH) y la hormona folículo-estimulante (folitropina, FSH), lo que lleva a una disminución de las concentraciones sanguíneas de estradiol y progesterona. Dado que los fibromas uterinos se asocian con sangrado menstrual abundante, el principal beneficio del medicamento es una menor pérdida de sangre mensual.
Eficacia clínica: Dos estudios de fase 3, aleatorizados, doble ciego y controlados con placebo, en los que participaron 511 y 501 mujeres, respectivamente, de 52 semanas de tratamiento y 24 semanas de seguimiento posterior, con dosis de linzagolix de 100 mg y 200 mg, bien solo o con tratamiento hormonal complementario concomitante (THC: 1 mg de estradiol/0,5 mg de acetato de noretisterona). La variable principal de eficacia fue la respuesta positiva, definida como una pérdida – por ciclo – de sangre menstrual ≤80 ml y una reducción ≥50 % con respecto al momento basal durante los últimos 28 días previos a la semana 24: Placebo (29-35%); 100 mg (56%); 100 mg + THC (66-77%); 200 mg (71-78%); 200 mg + THC (76-94%).
Eventos adversos: Los más comunes son sofocos y dolores de cabeza.
(J) Antiinfecciosos sistémicos
Vacuna COVID-19 ARN bivalente Original/Omicron BA.1 (Comirnaty Original/Omicron BA.1®) Pfizer; (Spikevax Bivalent Original/Omicron BA.1®) Moderna
Vacunas adaptadas para ampliar la protección frente a la COVID-19, autorizadas el 1-9-2022 para su uso en adultos y adolescentes a partir de 12 años, al menos 3 meses después de la vacunación primaria o una dosis de refuerzo con una vacuna COVID-19, con el fin de atacar la subvariante Omicron BA.1 además de la cepa original de SARS-CoV-2. Los estudios demostraron que Comirnaty Original/Omicron BA.1 y Spikevax bivalente Original/Omicron BA.1 pueden desencadenar fuertes respuestas inmunitarias contra Omicron BA.1 y la cepa SARS-CoV-2 original en personas previamente vacunadas. En particular, fueron más eficaces para desencadenar respuestas inmunitarias contra la subvariante BA.1 que las vacunas originales. Asimismo, el Comité de Medicamentos Humanos de la EMA (CHMP) recomendó el día 12 de septiembre autorizar una vacuna bivalente adaptada dirigida a las subvariantes BA.4 y BA.5 de Omicron además de la cepa original de SARS-CoV-2, Comirnaty Original/Omicron BA.4-5, también para uso en personas mayores de 12 años que han recibido al menos un ciclo primario de vacunación contra COVID-19. El CHMP basó su opinión en particular en los datos clínicos disponibles con Comirnaty Original/Omicron BA.1. Los efectos secundarios observados con las vacunas adaptadas fueron comparables a los observados con las originales y, por lo general, fueron leves y de corta duración. Comirnaty Original/Omicron BA.1: Un estudio en adultos mayores de 55 años que habían recibido previamente 3 dosis de Comirnaty (vacunación primaria y un refuerzo) mostró que la respuesta inmune a la subvariante Omicron BA.1 fue mayor después de una segunda dosis de refuerzo de Comirnaty Original/Omicron BA.1 que después de una segunda dosis de la vacuna Comirnaty original (medida por el nivel de anticuerpos contra Omicron BA.1). Además, la respuesta inmunitaria a la cepa SARS-CoV-2 original fue comparable para ambas vacunas. En el estudio participaron más de 1.800 personas, de las cuales unas 300 recibieron Comirnaty Original/Omicron BA.1 en su composición final. Otro estudio en el que participaron más de 600 personas de entre 18 y 55 años que habían recibido previamente 3 dosis de Comirnaty mostraron que la respuesta inmunitaria a Omicron BA.1 fue mayor en las personas que recibieron un refuerzo con una vacuna que solo contenía Omicron BA. 1 que en los que recibieron un refuerzo con la vacuna Comirnaty original. Adicionalmente, el CHMP (Committee of Human Medicinal Products) de la EMA ha recomendado la autorización de una vacuna bivalente adaptada específicamente dirigida a las subvariantes BA.4 y BA.5 de Omicron (Comirnaty Original/Omicron BA.4-5). Spikevax bivalente Original/Omicron BA.1: Un estudio en el que participaron más de 800 adultos a partir de los 18 años, que encontró que una dosis de refuerzo de Spikevax bivalente Original/Omicron BA.1 indujo una respuesta inmunitaria más fuerte contra la cepa SARS-CoV-2 y la subvariante Omicron BA.1 en comparación con una dosis de refuerzo de la vacuna Spikevax original. El estudio comparó el nivel de anticuerpos en personas vacunadas previamente con una serie primaria y una dosis de refuerzo de Spikevax, y que recibieron una segunda dosis de refuerzo de Spikevax o Spikevax bivalente Original/Omicron BA.1. El Centro Europeo para la Prevención y el Control de Enfermedades (ECDC) y la Agencia Europea de Medicamentos (EMA) consideran que el uso de las vacunas COVID-19 adaptadas aportan una cantidad superior de anticuerpos capaces de neutralizar la variante Omicron BA.1, en comparación con las vacunas monovalentes actuales basadas en la cepa original. Además, los anticuerpos generados por estas vacunas adaptadas parecen ser capaces de neutralizar otros linajes y sublinajes de Omicron, incluidos BA.2, BA.2.75 y BA.5, de manera más eficiente que las vacunas actuales, aunque actualmente se desconoce en qué medida estas mejoras en la respuesta inmune hacia los linajes Omicron se traducirán en una mayor protección frente a la COVID-19. En cualquier caso, las vacunas adaptadas solo han sido aprobadas para su uso como dosis de refuerzo en personas que completaron al menos una serie primaria, sin importar qué vacunas se usaron para la serie primaria; por ello, el uso de estas vacunas adaptadas debe limitarse por el momento a la vacunación de refuerzo. El ECDC y la EMA indican que las vacunas monovalentes actuales que incorporan la cepa original siguen siendo útiles para una sensibilización eficaz y para inducir una protección inicial suficiente en individuos sin tratamiento previo.
Vacuna COVID-19 inactivada (Vacuna COVID-19 Valneva®) Valneva
Indicación: Inmunización activa para prevenir la COVID-19 causada por el virus SARS-CoV-2, en personas de entre 18 y 50 años.
Tipo: Autorizado el 24-6-2022. Medicamento biológico; vacuna purificada, adsorbida en hidróxido de aluminio y adyuvada con CpG 1018 (citosina-fosfato-guanina), conteniendo virus SARS-CoV-2 completo inactivado (cepa de Wuhan hCoV-19/Italy/INMI1-isl/2020), cultivado en células Vero.
Mecanismo: Genera respuestas tanto de anticuerpos neutralizantes de SARSCoV-2 como de inmunidad celular (Th1) contra la espícula (S) y otras proteínas presentes en la superficie, que pueden contribuir a la protección frente a la COVID-19.
Eficacia clínica: Estudio de fase 3 aún en curso, aleatorizado y comparativo con tratamiento activo y con enmascaramiento para el observador, en comparación con la vacuna frente a COVID-19 ChAdOx1-S recombinante (Vaxzevria®) en participantes adultos sanos (o con condiciones médicas estables) de cualquier género y de 18 años o más. Se aleatorizaron 2.975 (≥30 años) para recibir la pauta de vacunación de dos dosis de la VLA2001 (n = 1.978) o de ChAdOx1-S recombinante (n = 997), cada una con 28 días de separación. Además, 1.042 participantes de entre 18 y 29 años se incluyeron en un grupo de tratamiento sin aleatorizar para recibir VLA2001 sin enmascaramiento. Tanto la inmunogenicidad como la seguridad se evaluarán hasta el mes 12 tras la vacunación con la primera dosis. Las variables coprimarias de la inmunogenicidad se definieron como la superioridad de la media geométrica de los títulos (GMT) de la población de inmunogenicidad (804 vs. 577), así como la no inferioridad del índice de seroconversión (definida como el aumento en una proporción de 4 respecto del valor basal) de los anticuerpos neutralizantes específicos de SARS-CoV-2, dos semanas después de la administración de la segunda dosis (es decir, el día 43) en adultos de 30 años o más (población por protocolo): 97,4 vs. 98,9%.
Eventos adversos: Los más comunes son dolor a la palpación en el lugar de inyección (76%), fatiga (57%), dolor en el lugar de inyección (53%), cefalea (41%), mialgia (44%) y náuseas/vómitos (15%). La mayoría de las reacciones adversas fueron de intensidad leve y se resolvieron en un plazo de 2 días después de la vacunación.
Vacuna Hepatitis B recombinante (Prehevbri®) VBI Vaccines
Indicación: Inmunización activa frente a la infección causada por todos los subtipos conocidos del virus de la hepatitis B en adultos.
Tipo: Autorizado el 18-5-2022. Medicamento biológico; vacuna con la composición antigénica completa de los antígenos de superficie del virus de la hepatitis B (HBs), incluyendo el antígeno de superficie pequeño (S, 83%), el mediano (pre-S2, 6%) y el grande (pre-S1, 11%), obtenidos mediante tecnología de ADN recombinante, adsorbidos en hidróxido de aluminio y formulados en una estructura de partículas pseudovíricas.
Mecanismo: Confiere inmunidad frente a todos los subtipos conocidos de infección por el virus de la hepatitis B mediante la estimulación de una respuesta inmunitaria específica, determinada mediante la inducción de anticuerpos anti-HBs con un título ≥10 mUI/ml.
Eficacia clínica: Dos ensayos clínicos de fase 3, multicéntricos, aleatorizados, controlados y con doble enmascaramiento en adultos, en comparación con una vacuna autorizada frente a la hepatitis B (Engerix-B). Ambas vacunas se administraron en una pauta de 3 dosis a los 0, 1 y 6 meses. En el primer estudio (n= 1.441 adultos), la variable primaria fue la tasa de seroprotección, definida como el porcentaje de pacientes con unos títulos de anticuerpos anti-HBs ≥10 mUI/ml el día 196 del estudio: en sujetos ≥18 años en el grupo de PreHevbri fue no-inferior a la del grupo de Engerix-B el día 196 del estudio (91,4% vs. 76,5%) y en sujetos ≥45 años fue superior a la del grupo de Engerix-B (89,4% vs. 73,1 %). En el segundo estudio (n=2.345), el criterio de valoración principal del estudio fue comparar 3 lotes de PreHevbri y Engerix-B en cuanto a la respuesta inmunitaria evaluada midiendo la media geométrica de la concentración de anticuerpos anti-Hbs: 5.443 (Prehevbri) vs. 1.526 (Enferix-B) al día 196 y 2.093 vs. 473 al día 336.
Eventos adversos: Los más comunes son dolor en el lugar de la inyección (72%), sensibilidad en el lugar de la inyección (71%) y prurito/picor local (12%). Las reacciones sistémicas solicitadas más frecuentes fueron mialgia (42%), fatiga (38%) y cefalea (36%).
Vacuna Neumococo polisacárida conjugada 20-valente (Apexxnar®) Pfizer
Indicación: Inmunización activa para la prevención de la enfermedad invasiva y la neumonía causadas por Streptococcus pneumoniae en individuos de 18 años de edad y mayores.
Tipo: Autorizado el 14-2-2022. Medicamento biológico; vacuna constituida por 20 polisacáridos capsulares neumocócicos diferentes, todos adsorbidos en fosfato de aluminio y conjugados con la proteína transportadora CRM197, la cual modifica la respuesta inmune al polisacárido, pasando de una respuesta independiente a una dependiente de linfocitos T.
Mecanismo: La respuesta dependiente de linfocitos T conduce tanto a una producción de anticuerpos aumentada como a la generación de linfocitos B de memoria, lo que permite una respuesta de refuerzo en la reexposición a la bacteria. La vacunación induce la producción de anticuerpos séricos y la memoria inmunológica contra los serotipos contenidos en la vacuna. Eficacia clínica: Tres estudios clínicos de fase 3 en Estados Unidos y Suecia, en adultos que nunca habían recibido una vacuna antineumocócica o que habían sido vacunados previamente con Prevenar 13, PPSV23 o ambas. El estudio clínico pivotal fue aleatorizado, controlado con tratamiento activo, doble ciego y de no inferioridad, en sujetos de 18 años y mayores que previamente no habían recibido una vacuna antineumocócica (n= 3,889). Se midieron los títulos medios geométricos de la actividad opsonofagocítica específicos de serotipo antes de la primera vacunación y 1 mes después de cada vacunación. Las respuestas inmunes a todos los 13 serotipos comunes inducidas por Apexxnar fueron no-inferiores a las inducidas por Prevenar 13 a los mismos serotipos un mes después de la vacunación. Las respuestas inmunes a 6 de los 7 serotipos adicionales inducidas por Apexxnar fueron no-inferiores a las inducidas por PPSV23 a los mismos serotipos 1 mes después de la vacunación, pero la respuesta al serotipo 8 no cumplió con el criterio estadístico de no inferioridad definido.
Eventos adversos: Los más comunes (>10%) son cefalea, dolor articular y/o muscular, dolor/sensibilidad en el lugar de la inyección.
Lenacapavir (Sunlenca®) Gilead

Indicación: En combinación con otro(s) fármaco(s) antirretroviral(es), está indicado para el tratamiento de adultos con infección por el VIH-1 multirresistente a fármacos para los que, de otro modo, no es posible preparar una pauta de tratamiento antiviral supresor.
Tipo: Autorizado el 17-8-2022.
Mecanismo: Inhibidor selectivo multifase de la función de la cápside del VIH-1 que se une directamente a la interfaz entre las subunidades proteicas de la cápside (CA). Lenacapavir inhibe la replicación del VIH-1 al interferir en múltiples pasos fundamentales del ciclo de vida vírico, incluida la captación nuclear de ADN proviral del VIH-1 mediada por la cápside (al bloquear la unión de proteínas de importación nuclear a la cápside), el ensamblaje y la liberación del virus (al interferir en la función de Gag/Gag-Pol, reduciendo la producción de subunidades CA), y la formación del núcleo de la cápside (al alterar la velocidad de asociación de las subunidades de la cápside, lo que resulta en cápsides malformadas). Lenacapavir mostró actividad antiviral en cultivos celulares frente a todos los grupos del VIH-1 (M, N, O), incluidos los subtipos A, A1, AE, AG, B, BF, C, D, E, F, G, H; fue entre 15 y 25 veces menos activo frente a aislados del VIH-2 en comparación con el VIH-1.
Eficacia clínica: Ensayo multicéntrico, parcialmente aleatorizado, doble ciego y controlado con placebo, de 52 (un año) semanas de duración, que incluyó a 72 pacientes altamente tratados con tratamientos antirretrovirales previos con VIH-1 resistente a múltiples clases de fármacos. La variable principal de eficacia fue el porcentaje de pacientes que alcanzaron una reducción ≥0,5 log10 copias/ml desde el momento basal en el ARN del VIH-1 al final del período de monoterapia funcional: 87,5% vs. 16,7% (placebo).
Eventos adversos: Los más comunes son reacciones en la zona de inyección (63 %) y náuseas (4 %).
(L) Agentes antineoplásicos e inmunomoduladores
Capmatinib (Tabrecta®) Novartis
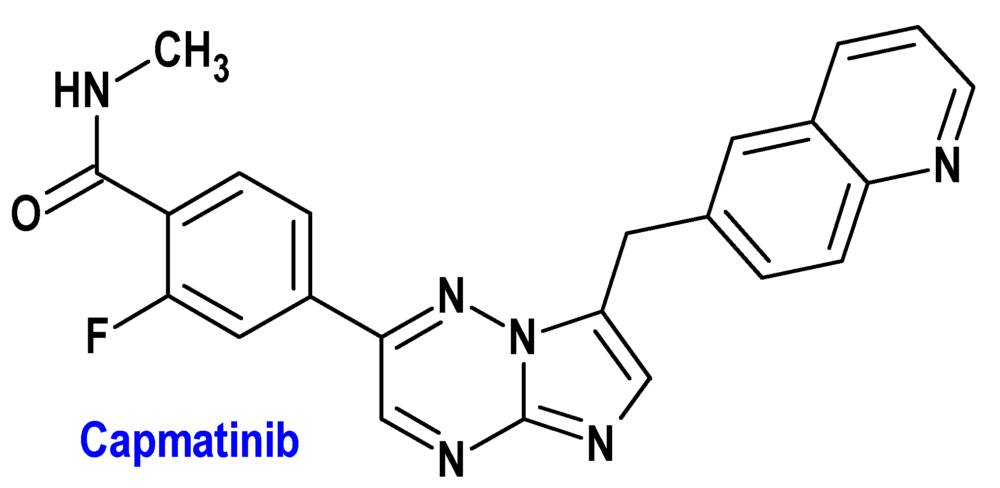
Indicación: Como monoterapia para el tratamiento de pacientes adultos con cáncer de pulmón de células no pequeñas (CPCNP) avanzado que albergan alteraciones que conducen a la omisión del exón 14 del gen del factor de transición epitelial mesenquimatoso (METex14), que requieren terapia sistémica después de un tratamiento previo con inmunoterapia y/o quimioterapia basada en platino.
Tipo: Autorizado el 20-6-2022; autorizado previamente en Estados Unidos (6-5-2020), como medicamento huérfano.
Mecanismo: Capmatinib es un inhibidor de la cinasa que se dirige al factor de transición epitelial mesenquimatoso (MET), incluida la variante mutante producida por la omisión del exón 14 (METex14). La omisión del exón 14 de MET da como resultado una proteína a la que le falta un dominio regulador que reduce su regulación negativa, lo que conduce a un aumento de la señalización de MET. Capmatinib inhibe la fosforilación de MET provocada por la unión del factor de crecimiento de hepatocitos o por la amplificación de MET, así como la fosforilación mediada por MET de proteínas de señalización y la proliferación y supervivencia de células cancerosas dependientes de MET.
Eficacia clínica: Estudio de fase 2 multicéntrico, no aleatorizado, abierto, multicohorte en 373 pacientes, que continuaron el tratamiento hasta progresión de la enfermedad documentada, falta de tolerancia al tratamiento o ausencia de beneficio clínico. La variable principal de eficacia fue la tasa de respuesta global en pacientes naïve (68%; 4% completa) y previamente tratados (41%; 0% completa). La mediana de la duración de la respuesta fue de 16,6 (naïve) y 9,7 meses (pretratados) y el porcentaje de pacientes con duración ≥12 meses fue del 47% y 32%, respectivamente.
Eventos adversos: Los más comunes son edema periférico (68%), náuseas (44%), fatiga (34%), aumento de creatinina en sangre (34%), vómitos (25%), disnea (23%), disminución del apetito (21%) y dolor de espalda (21%). Las más frecuentes de grado 3 o 4 son edema periférico (14%), aumento de lipasa (9,4%), aumento de ALT (8,1%), fatiga (8,1%), disnea (6,9%) y aumento de amilasa (5,6%). Las reacciones adversas graves que se presentaron en >2% de pacientes incluyeron disnea (5,6%), EPI/neumonitis (5,0%), celulitis (3,1%) y edema periférico (2,5%). Se notificaron reducciones de dosis en 31% e interrupciones permanentes del tratamiento en 12%.
Asciminib (Scemblix®) Novartis
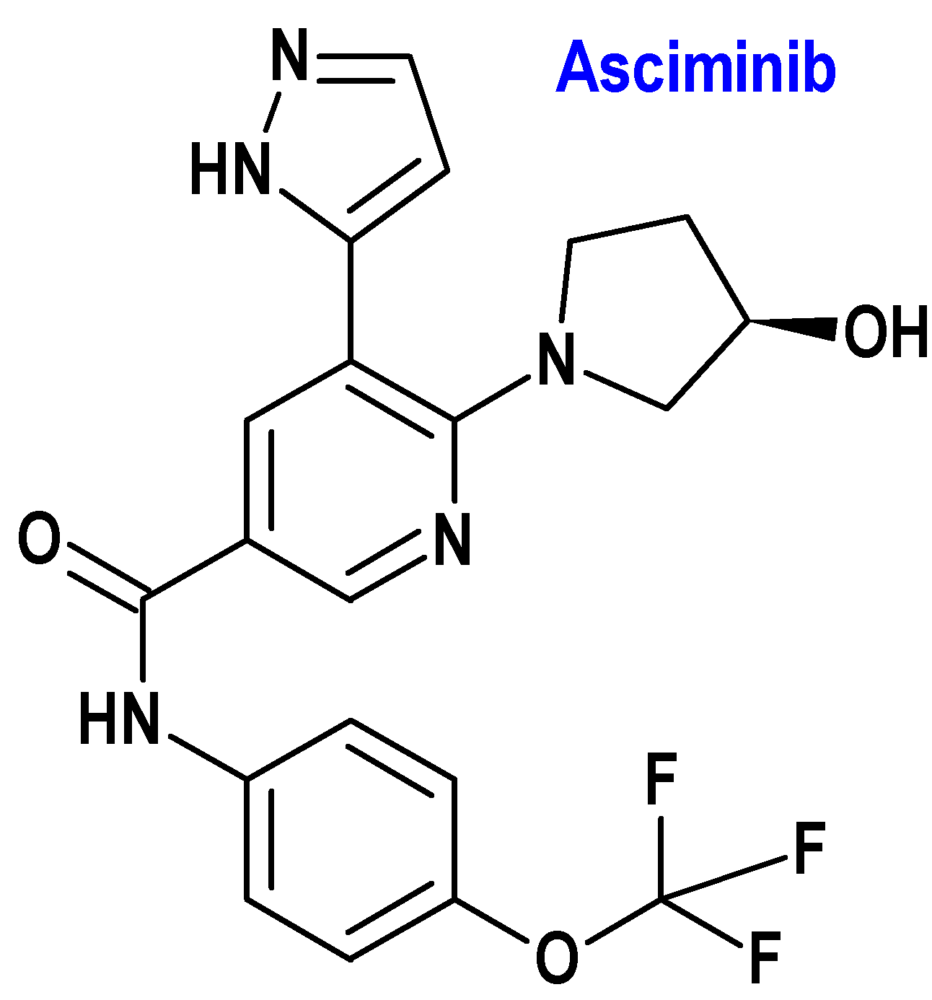
Indicación: Tratamiento de pacientes adultos con leucemia mieloide crónica en fase crónica con cromosoma Filadelfia positivo (LMC-FC Ph+) previamente tratado con dos o más inhibidores de la tirosina cinasa
Tipo: Autorizado el 25-8-2022, como medicamento huérfano; autorizado previamente en Estados Unidos (29-10-2021).
Mecanismo: Es un inhibidor de la tirosina cinasa ABL/BCR::ABL1. Asciminib inhibe la actividad cinasa ABL1 de la proteína de fusión BCR::ABL1 al dirigirse específicamente al bolsillo de miristoilo de ABL. El gen BCR está normalmente en el cromosoma número 22, mientras que el gen ABL lo está en el número 9; la mutación BCR-ABL ocurre cuando partes de los cromosomas 9 y 22 se desprenden y cambian de lugar, de tal manera que la parte del cromosoma 9 que se desprende incluye una parte del gen ABL. Cuando esta parte se traslada al cromosoma 22, parte del gen ABL se une al gen BCR, formando el gen BCR-ABL. Este cromosoma 22 mutado es conocido como “cromosoma Filadelfia” y la mutación no se hereda de los padres, sino que se trata de una mutación somática producida a lo largo de la vida.
Eficacia clínica: Estudio fase 3 multicéntrico, aleatorizado, abierto y controlado con bosutinib, que incluyó a 233 pacientes que continuaron el tratamiento hasta que se produjo una toxicidad inaceptable o el fracaso del tratamiento (la mediana de duración del tratamiento aleatorizado fue 103 semanas). La variable principal de eficacia fue la tasa de respuesta molecular mayor (relación BCR::ABL1 en la escala internacional ≤0,1%) a las 24 semanas: 25,5% (asciminb) vs. 13,2% (bosutinib).
Eventos adversos: Los más comunes (incidencia ≥20%) son dolor musculoesquelético (37%), infecciones respiratorias de vías altas (28%), trombocitopenia (28%), fatiga (27%), dolor de cabeza (24%), artralgia (22%), elevación de las enzimas pancreáticas (21%), dolor abdominal (21%), diarrea (21%) y náuseas (20%). Las reacciones adversas más frecuentes de grado≥3 (incidencia ≥5%) fueron trombocitopenia (19%), neutropenia (16%), elevación de las enzimas pancreáticas (12%), hipertensión (8,7%) y anemia (5,3%). Las reacciones adversas graves sucedieron en el 12,4% y las más frecuentes (incidencia ≥1%) fueron derrame pleural (2,5%), infecciones respiratorias de vías bajas (2,2%), trombocitopenia (1,7%), pirexia (1,4%), pancreatitis (1,1%), dolor torácico no-cardiaco (1,1%) y vómitos (1,1%).
Teclistamab (Tecvayli®) Janssen Cilag
Indicación: Monoterapia para el tratamiento de pacientes adultos con mieloma múltiple en recaída y refractario, que han recibido al menos tres tratamientos previos, incluidos un agente inmunomodulador, un inhibidor del proteasoma y un anticuerpo anti-CD38 y han presentado progresión de la enfermedad al último tratamiento.
Tipo: Autorizado condicionalmente el 23-8-2022, como medicamento huérfano. Medicamento biológico, constituido por un anticuerpo biespecífico humanizado de inmunoglobulina G4-prolina, alanina, alanina (IgG4-PAA) dirigido contra los receptores del antígeno de maduración de los linfocitos B (BCMA) y CD3, producido en una línea celular de mamífero (ovario de hámster chino) mediante tecnología de ADN recombinante.
Mecanismo: Teclistamab es un anticuerpo biespecífico IgG4-PAA de tamaño completo que se dirige al receptor de CD3 expresado en la superficie de los linfocitos T y al antígeno de maduración de linfocitos B (BCMA), que se expresa en la superficie de las células malignas de mieloma múltiple del linaje B , así como en los linfocitos B en fase avanzada y en las células plasmáticas. Gracias a sus puntos de unión dobles, teclistamab es capaz de atraer a los linfocitos T CD3+ a la proximidad de las células BCMA+, lo que provoca la activación de los linfocitos T y la subsiguiente lisis y muerte de las células BCMA+, que está mediada por la perforina secretada y varias granzimas almacenadas en las vesículas secretoras de los linfocitos T citotóxicos. Este efecto se produce sin tener en cuenta la especificidad de los receptores de los linfocitos T o sin dependencia de las moléculas del complejo principal de histocompatibilidad (CPH) de clase 1 en la superficie de las células que presentan el antígeno. Durante el primer mes de tratamiento, se observó la activación de los linfocitos T, la redistribución de los linfocitos T, la reducción de los linfocitos B y la inducción de las citocinas séricas.
Eficacia clínica: Ensayo de fase 1/2, de un solo grupo, abierto y multicéntrico, de XX semanas de duración, que incluyó a 165 pacientes con mieloma múltiple en recaída o refractario. La variable principal de eficacia fue la tasa de respuesta global, según lo determinado por la evaluación del Comité de Revisión Independiente utilizando los criterios del International Myeloma Working Group (IMWG) de 2016: respuesta global: 63%; respuesta completa: 6,7%; respuesta parcial 4,2%- La duración (mediana) de la respuesta fue de 18,4 meses.
Eventos adversos: Los más comunes son hipogammaglobulinemia (75%), síndrome de liberación de citocinas (72%), neutropenia (71%), anemia (55%), dolor musculoesquelético (52%), fatiga (41%), trombocitopenia (40%), reacción en la zona de inyección (38%), infección de las vías respiratorias altas (37%), linfopenia (35%), diarrea (28%), neumonía (28%), náuseas (27%), fiebre (27%), cefalea (24%), tos (24%), estreñimiento (21%) y dolor (21%). Se notificaron reacciones adversas graves en el 65% de los pacientes, incluyendo neumonía (16%), COVID-19 (15%), síndrome de liberación de citocinas (8%), sepsis (7%), fiebre (5%), dolor musculoesquelético (5%), lesión renal aguda (4,8%), diarrea (3,0%), celulitis (2,4%), hipoxia (2,4%), neutropenia febril (2,4%) y encefalopatía (2,4%).
Edgartigimod Alfa (Vyvgart®) Argenx
Indicación: Complemento de la terapia estándar para el tratamiento de pacientes adultos con miastenia gravis generalizada (MGG) con anticuerpos positivos frente a receptores de acetilcolina (AChR).
Tipo: Autorizado el 18-8-2022, como medicamento huérfano; autorizado previamente en Estados Unidos (17-12-2021). Medicamento biológico, constituido por un fragmento cristalizable (Fc) derivado de la inmunoglobulina humana recombinante G1 (IgG1).
Mecanismo: Es un fragmento de anticuerpo humano IgG1 diseñado para aumentar la afinidad con el receptor neonatal para el Fc (FcRn). Efgartigimod alfa se une al FcRn, lo que provoca una reducción de los niveles de IgG circulantes, incluidos los autoanticuerpos IgG patogénicos, pero sin afectar a los niveles de otras inmunoglobulinas (IgA, IgD, IgE o IgM), ni a los de la albúmina. Los autoanticuerpos IgG son la causa subyacente de la patogénesis de la miastenia gravis, ya que deterioran la transmisión neuromuscular al unirse a los receptores de la acetilcolina (AChR), al receptor de tirosina cinasa muscular (MuSK) o a la proteína 4 asociada con el receptor de lipoproteínas de baja densidad (LRP4).
Eficacia clínica: Ensayo multicéntrico, aleatorizado, con enmascaramiento doble y controlado con placebo de 26 semanas de duración, que incluyó a 167 pacientes. La eficacia clínica se midió mediante la escala de actividades de la vida diaria específica de la miastenia gravis (MG-AVD), con una puntuación total que oscila entre 0 y 24, siendo las puntuaciones más altas las que indican un mayor deterioro; en este estudio, se consideró que los pacientes con MGG respondían al tratamiento si había una reducción de ≥2 puntos en la puntuación total de MG-AVD en comparación con el estado inicial. La variable primaria de eficacia fue la comparación del porcentaje de pacientes respondedores durante el primer ciclo de tratamiento: 68% (edgartigimod alfa) vs. 30% (placebo).
Eventos adversos: Los más comunes son infecciones respiratorias de vías altas (10,7%) e infecciones urinarias (9,5%).
(N) Sistema nervioso
Lasmiditan (Rayvow®) Lilly
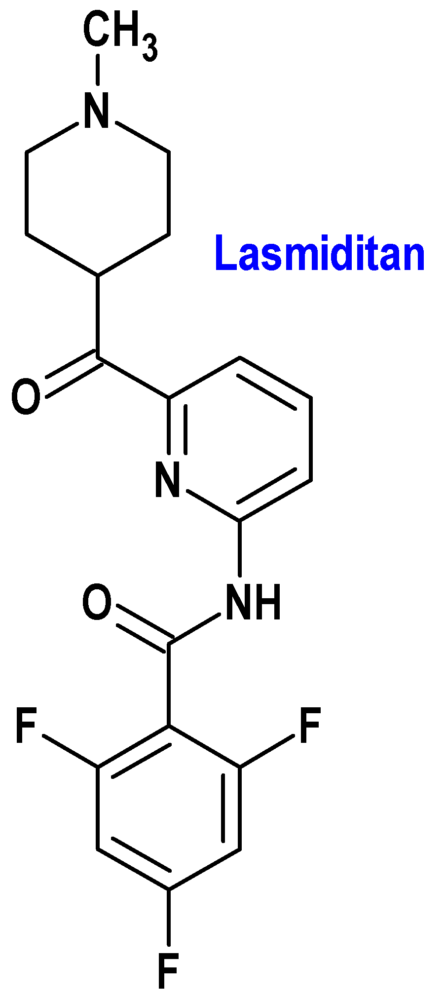
Indicación: Tratamiento agudo de la fase de cefalea de los ataques de migraña, con o sin aura en adultos.
Tipo: Autorizado el 17-8-2022; autorizado previamente en Estados Unidos (11-10-2019).
Mecanismo: Agonista del receptor de 5-hidroxitriptamina 1F (5-HT1F), de alta afinidad, que actúa a nivel central. Se desconoce el mecanismo de acción preciso, aunque los efectos terapéuticos de lasmiditán en el tratamiento de la migraña presumiblemente implican efectos agonistas en el receptor 5-HT1F, una disminución de la liberación de neuropéptidos y una inhibición de las vías del dolor, incluido el nervio trigémino. Lasmiditán mostró una selectividad >440 veces mayor para el receptor 5HT1F frente a los receptores 5-HT1B y 5-HT1D. No tiene efecto vasoconstrictor en las arterias coronarias humanas ex vivo, arterias mamarias internas humanas ex vivo o arterias meníngeas medias humanas ex vivo, probablemente debido a su baja afinidad por el receptor 5-HT1B, que posee efecto vasoconstrictor.
Eficacia clínica: Tres ensayos de fase 3, aleatorizados, controlados con placebo y doble ciego, que incluyeron a 5910 pacientes adultos, con 3-8 ataques de migraña al mes y migraña al menos moderadamente incapacitante. Las variables principales de eficacia fueron la proporción de pacientes con ausencia de dolor y la proporción de pacientes sin el síntoma más molesto (fotofobia, mayoritariamente), en comparación con el placebo, 2 horas después del tratamiento: 28-39% vs. 15-21% (placebo) y 41-49% vs. 30-34% (placebo), respectivamente.
Eventos adversos: Los más comunes son mareos (20%), somnolencia (7,8%), fatiga (7,7%), parestesia (6,8%), náuseas (4,9%), vértigo (2,6%), hipoestesia (2,5%) y debilidad muscular (2,3%).
2. FOOD & DRUG ADMINISTRATION (FDA)
(A) Tracto alimentario y metabolismo
Vutrisiran (Amvuttra®) Alnylam
Indicación: Tratamiento de la polineuropatía de la amiloidosis hereditaria mediada por transtiretina en adultos.
Tipo: Autorizado el 13-6-2022, como medicamento huérfano; no autorizado aún en la Unión Europea. Medicamento biológico constituido por un pequeño ácido ribonucleico de interferencia (ARNip) bicatenario químicamente modificado y dirigido al ARN mensajero (ARNm) de transtiretina (TTR), unido covalentemente a un ligando que contiene tres residuos de N-acetilgalactosamina (GalNAc) para facilitar la entrega del ARNip a los hepatocitos.
Mecanismo: El conjugado de ARNip-GalNAc de doble cadena causa la degradación del ARNm de la TTR tanto mutante como de tipo salvaje, a través de la interferencia de ARN, lo que da como resultado una reducción de la transtiretina sérica y de sus depósitos en los tejidos.
Eficacia clínica: Ensayo aleatorizado, abierto y controlado con placebo de 9 meses de duración, que incluyó a 199 pacientes. La variable principal de eficacia fue el cambio desde el inicio hasta el mes 9 en la puntuación modificada de deterioro de la neuropatía +7 (mNIS+7): -2,2 vs. +14,8.
Eventos adversos: Los más comunes son artralgia (11%), disnea (7%) y reducción de los niveles de vitamina A (7%).
(L) Agentes antineoplásicos e inmunomoduladores
Spesolimab (Spevigo®) Boehringer Ingelheim
Indicación: Tratamiento de los brotes de psoriasis pustulosa generalizada en adultos.
Tipo: Autorizado el 1-9-2022, como medicamento huérfano; no autorizado aún en la Unión Europea. Medicamento biológico constituido por un anticuerpo IgG1 monoclonal humanizado contra el IL-36R humano, producido en células de ovario de hámster chino mediante tecnología de ADN recombinante.
Mecanismo: Inhibe la señalización de la interleucina-36 (IL-36) al unirse específicamente al receptor IL-36R. La unión de spesolimab a IL-36R evita la activación posterior de IL-36R por ligandos afines (IL-36 α, β y γ) y la activación posterior de vías proinflamatorias y profibróticas.
Eficacia clínica: Ensayo aleatorizado, doble ciego y controlado con placebo, que incluyó a 53 pacientes con al menos una puntuación de 3 (moderado) en la escala GPPPGA (Generalized Pustular Psoriasis Physician Global Assessment), una supuntuación de pústulas de al menos 2 (leve) y, al menos, el 5% del área de la superficie corporal cubierta con eritema y presencia de pústulas. La variable principal de eficacia fue la proporción de sujetos con una subpuntuación de pústulas de GPPPGA de 0 (que indica que no hay pústulas visibles) en la semana 1 después del tratamiento: 54% vs 6% (placebo).
Eventos adversos: Los más comunes (>5%) son astenia, fatiga, cefalea, náusea y vómitos (9%); prurito, hematoma y sensación de quemazón en el punto de infusión IV e infección del tracto urinario (6%).
Deucravacitinib (Sotyktu®) Bristol Myers Squibb
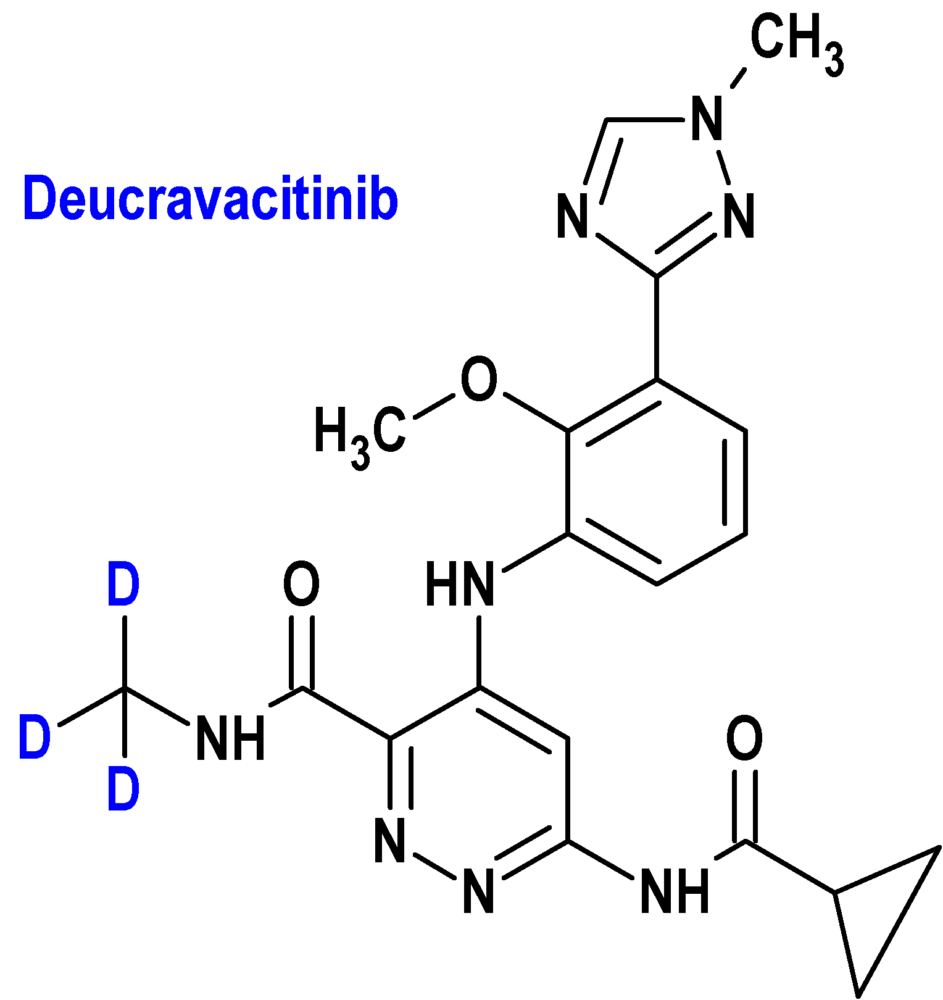
Indicación: Tratamiento de adultos con psoriasis en placas de moderada a grave que son candidatos para terapia sistémica o fototerapia.
Tipo: Autorizado el 9-9-2022; no autorizado aún en la Unión Europea. La molécula presenta la peculiaridad de contener tres átomos de deuterio (D), que sustituyen a otros tantos átomos de hidrógeno (H). El deuterio (hidrógeno-2) es un isótopo estable del hidrógeno (hidrógeno-1) que representa el 0,015% del total de átomos de hidrógeno existentes; su núcleo atómico está formado por un protón y un neutrón, mientras que el del hidrógeno solo contiene un protón.
Mecanismo: Inhibidor selectivo de la tirosina cinasa 2 (TYK2), un miembro de la familia de cinasas Janus (JAK). Deucravacitinib se une al dominio regulador de TYK2, estabilizando una interacción inhibidora entre los dominios regulador y catalítico de la enzima, dando como resultado la inhibición alostérica de la activación mediada por el receptor de TYK2 y de la activación de los transductores de señal y activadores de la transcripción (STAT). Las cinasas JAK, incluida la TYK2, funcionan como pares de homodímeros o heterodímeros en las vías JAK-STAT, donde TYK2 se empareja con JAK1 para mediar en múltiples vías de citocinas y con JAK2 para transmitir señales. El deucravacitinib redujo la expresión génica asociada a la psoriasis en la piel psoriásica de manera dependiente de la dosis, incluidas reducciones en los genes regulados por la vía de la IL-23 y la vía del IFN tipo I.
Eficacia clínica: Dos estudios de fase 3 de 52 semanas de duración, multinacionales, multicéntricos, aleatorizados, doble ciego, controlados con placebo y con un comparador activo (apremilast), incluyendo a un total de 1.684 pacientes con psoriasis en placas de moderada a grave, candidatos para fototerapia o terapia sistémica, con una afectación del área de superficie corporal ≥10%, una puntuación del índice de gravedad y área de psoriasis (PASI) ≥12 y una evaluación global del médico (sPGA) estática ≥3 (moderada o grave). Las variables principales de eficacia fueron el porcentaje de pacientes que alcanzaron el Índice de gravedad y área de psoriasis de 75 (PASI 75; reducción de al menos un 75% en la puntuación PASI basal) y el porcentaje de pacientes que alcanzaron una puntuación estática de 0 o 1 en la Evaluación global del médico (sPGA 0/1; claro o casi claro) en la semana 16 frente a placebo: (PASI 75) 53-58% (deucravacitinib) vs. 35-40% (apremilast) vs. 9-13% (placebo); (sPGA 0/1) 50-54% (deucravacitinib) vs. 32-34% (apremilast) vs. 7-9% (placebo). Los criterios de valoración secundarios clave incluyeron el porcentaje de pacientes que lograron PASI 75, PASI 90 y sPGA 0/1 en comparación con apremilast en semana 24: (PASI 75) 58-69% (deucravacitinib) vs. 38% (apremilast); (PASI 90) 32-42% (deucravacitinib) vs. 20-22% (apremilast); (sPGA 0/1) 49-59% (deucravacitinib) vs. 30-31% (apremilast).
Eventos adversos: Los más comunes son infecciones de las vías respiratorias superiores (19%), aumento de la creatina fosfocinasa en sangre (2,7%), herpes simple (2,0%), úlceras bucales (1,9%), foliculitis (1,7%) y acné (1,4%). El 2,4 % de los pacientes con deucravacitinib, el 3,8 % con placebo y el 5,2 % con apremilast experimentaron reacciones adversas que llevaron a la suspensión del tratamiento.
Eflapegrastim (Rolontis®) Spectrum
Indicación: Disminuir la incidencia de infección, manifestada por neutropenia febril, en pacientes adultos con neoplasias malignas no mieloides que reciben fármacos anticancerosos mielosupresores asociados con una incidencia clínicamente significativa de neutropenia febril.
Tipo: Autorizado el 9-9-2022, no autorizado aún en la Unión Europea. Medicamento biológico constituido por un factor estimulante de colonias de granulocitos (G-CSF) producido por acoplamiento covalente de un análogo de G-CSF humano y un fragmento Fc de inmunoglobulina humana G4 (IgG4), ambos de origen recombinante, a través de un único enlazador de polietilenglicol de 3,4 kDa. El dominio G-CSF recombinante en eflapegrastim es una variante del G-CSF humano con dos sustituciones de serina en las posiciones 17 y 65, y sin metionina N-terminal adicional.
Mecanismo: Es un factor de crecimiento de granulocitos humanos recombinante que se une a los receptores de G-CSF en las células progenitoras mieloides y los neutrófilos, desencadenando vías de señalización que controlan la diferenciación, proliferación, migración y supervivencia celular.
Eficacia clínica: Dos estudios de no inferioridad con control activo (pegfilgrastim), abiertos, aleatorizados y 1:1 de diseño similar, que incluyeron un total de 643 pacientes con cáncer de mama en etapa inicial, tratadas con docetaxel y ciclofosfamida. La variable principal de eficacia fue la duración (mediana) de la neutropenia en las pacientes tratadas: 0,20-0,31 días (eflapegrastim) vs. 0,35-0,39 días (pegfilgrastim).
Eventos adversos: Los más comunes (>20%) son fatiga (58%), náusea (52%), diarrea (40%), dolor óseo (38%), cefalea (29%), pirexia (28%), anemia (25%), erupción cutánea (25%), mialgia (22%), artralgia (21%) y dolor de espalda (20%). El 4 % de los pacientes interrumpieron el tratamiento debido a eventos adversos.
(M) Sistema músculo-esquelético
Toxina Daxibotulínica A (Daxxify®) Revance
Indicación: Mejora temporal en la apariencia de las líneas (arrugas) glabelares de moderadas a severas asociadas con la actividad del músculo corrugador y/o prócer en pacientes adultos.
Tipo: Autorizado el 7-9-2022; no autorizado aún en la Unión Europea. Medicamento biológico constituido por toxina botulínica purificada procedente de cultivos de Clostridium botulinum tipo A.
La toxina daxibotulínica A está formulada conjuntamente con un péptido excipiente estabilizador (RTP004), que mejora la unión de la neurotoxina a las superficies neuronales, lo que puede aumentar la probabilidad de internalización de la neurotoxina.
Mecanismo: Bloquea la transmisión colinérgica en la unión neuromuscular al inhibir la liberación de acetilcolina. Cuando se inyecta en el músculo esquelético, la toxina se interna en la terminal nerviosa, trasladándose al citosol neuronal donde escinde SNAP25, una proteína necesaria para el acoplamiento de la membrana de la vesícula sináptica y la posterior liberación de acetilcolina, lo que produce una disminución de la función muscular dependiente de la dosis. La recuperación de la actividad es gradual y resulta de la degradación de la cadena ligera de la neurotoxina en las neuronas con una contribución de la formación de brotes axonales. La reinervación muscular conduce a una reversión lenta de los efectos farmacológicos de la toxina.
Eficacia clínica: Dos ensayos clínicos aleatorizados, doble ciego, multicéntricos y controlados con placebo, que incluyó a 609 sujetos adultos con líneas glabelares de al menos una gravedad moderada en el ceño fruncido máximo. La variable principal de eficacia fue la tasa de sujetos con una puntuación de 0 o 1 (ninguna o leve) y una mejora de al menos 2 puntos desde el inicio para las evaluaciones del investigador y del sujeto en la semana 4, valorados por los investigadores y los sujetos utilizando una escala de 4 puntos (0=ninguno; 1=leve; 2=moderado; 3=grave) en comparación con el placebo: 92-86% vs. 1-2% (investigadores) y 76-77% vs. 0% (sujetos).
Eventos adversos: Los más comunes son reacciones en el punto de inyección (dolor, eritema, edema; 9%), cefalea (6%), ptosis palpebral (caída del párpado superior; 2%) y paresia facial (1%).
