1. INTRODUCCIÓN
1.1. Evolución de los patrones de consumo de fructosa
La fructosa es un monosacárido presente en frutas, verduras y miel, tanto en su forma libre como asociado a glucosa para formar la sacarosa. Por tanto, siempre ha sido un componente básico de nuestra dieta, pero sus patrones y frecuencia de consumo han cambiado notablemente en los últimos años (1). El consumo de fructosa ha aumentado exponencialmente por su uso como edulcorante en forma de jarabe de maíz rico en fructosa (del inglés, High-fructose corn syrup, HFCS), destacando su presencia en bebidas azucaradas y alimentos procesados (2). Debido a su elevado poder edulcorante, propiedades organolépticas y bajo coste, el uso industrial del HFCS se ha disparado, incrementándose el consumo anual por persona en Estados Unidos de 0,23 kg en los 1970s a 28,4 kg en los 2000s. Dicho consumo se ha relacionado con mayor riesgo de desarrollar síndrome metabólico (SM), enfermedades cardiovasculares (ECV) y obesidad (3,4).
1.2. Dietas: fructosa, tagatosa y dieta occidental
De forma similar al consumo de fructosa, existen otros tipos de dietas que favorecen el riesgo de desarrollar enfermedades no transmisibles relacionadas con la nutrición. En este trabajo determinamos los efectos que estas dietas pueden ejercer sobre la fisiología del organismo, incluyendo la modulación de la expresión de proteína y la metilación del material genético (5,6).
En primer lugar, se compararon los efectos del consumo de fructosa con los de su epímero estructural: la tagatosa, un edulcorante considerado como seguro. Su menor contenido calórico, menor índice glucémico y elevado poder edulcorante hacen que su popularidad se haya incrementado en los últimos años. La tagatosa no presenta afinidad por el transportador de carbohidratos del intestino delgado implicado en la absorción de azúcares, de manera que solo un 20-25% de la tagatosa es absorbida y el resto es utilizado por la microbiota como sustrato fermentable, razón por la que se considera un prebiótico (7,8). Asimismo, este monosacárido se asocia con un potencial efecto antidiabético, pues su consumo se ha relacionado con una disminución de los niveles postpandriales de glucosa (9).
No obstante, también se consideró el consumo de fructosa asociado a la ingesta de colesterol, representando la dieta occidental (del inglés, Western Diet, WD) un patrón de dieta actual propio de los países occidentales. La WD proporciona un elevado aporte calórico de baja calidad nutricional, caracterizándose por una ingestión reducida de frutas y verduras, frente a un consumo elevado de colesterol, grasas saturadas y azúcares añadidos (10). Esta dieta se ha asociado a una amplia variedad de enfermedades crónicas no transmisibles como la obesidad, diabetes y ECV, dado que altera el metabolismo lipídico y promueve un estado de inflamación en el organismo (11).
1.3. Metabolismo de la fructosa y enfermedad cardiovascular
Las generaciones más jóvenes están cada vez más expuestas a altas concentraciones de azúcares, en particular fructosa, lo cual es alarmante dada su relación con el desarrollo de ECV y SM. Estas enfermedades no transmisibles aparecen como resultado de la compleja interacción entre factores genéticos y ambientales, destacando el papel del metabolismo lipídico en su desarrollo (12). Las ECV, un conjunto de patologías crónicas y heterogéneas que afectan al corazón y al sistema circulatorio, son la principal causa de mortalidad global, representando en 2021 en torno a 1/3 de las muertes en la Unión Europea (13,14). El SM es un trastorno caracterizado por la presencia de varios factores de riesgo cardiovascular que aumentan la probabilidad de padecer patologías potencialmente mortales como infarto de miocardio, accidente cerebrovascular y diabetes tipo II (15,16). Según el Adult Treatment Panel III (ATP III), se diagnostica SM si se presentan al menos tres de las siguientes alteraciones: obesidad abdominal, hipertensión, hiperglucemia en ayunas, hipertrigliceridemia y bajos niveles de colesterol asociado a lipoproteínas de alta densidad (HDL) (17).
Según estudios observacionales, el consumo de fructosa es un factor de riesgo para la aparición de enfermedades crónicas en humanos. Asimismo, numerosos modelos animales han relacionado las dietas enriquecidas con fructosa con la manifestación de ECV y SM (18-20). Esto se debe al carácter altamente lipogénico de dicho azúcar, implicado en el desarrollo de dislipidemia, obesidad, resistencia a insulina y a leptina, hipertensión e hiperuricemia (2,21). La diferente estructura química de la glucosa (aldohexosa) y la fructosa (cetohexosa) explica las diferencias en su absorción y metabolismo. Mientras que el metabolismo de la glucosa está altamente regulado, la fructólisis o metabolismo de la fructosa presenta una menor regulación, afectando negativamente al metabolismo lipídico (1,22).
En torno a un 70-80% de la fructosa se metaboliza en el hígado, donde se transforma en fructosa-1-fosfato y es escindida en gliceraldehído y dihidroxiacetona (DHAP). Estas dos moléculas pueden ser convertidas en glucosa a través de la gluconeogénesis, que puede utilizarse para la síntesis de glucógeno (2). El gliceraldehído puede fosforilarse a gliceraldehído-3-fosfato (G3P) y convertirse en piruvato, que se descarboxila para rendir acetil-CoA. Esta molécula puede entrar en el ciclo del ácido cítrico (CAC) o ser precursor para la síntesis de ácidos grasos (AG). Por su parte, la DHAP puede transformarse en glicerol-3-fosfato que, junto con los AG, forma triglicéridos (TG) (23). Por el contrario, en el metabolismo de la glucosa intervienen la glucoquinasa y la fosfofructoquinasa-1, implicadas en la regulación de la ruta y que limitan la cantidad de acetil-CoA destinado a la síntesis de AG (24).
Por tanto, la fructosa constituye una fuente de triosas – fosfato involucradas en la síntesis de lípidos de novo, gluconeogénesis y glucogenogénesis (25). Como su metabolismo no está regulado, el CAC no es capaz de metabolizar el exceso de acetil-CoA generado tras la fructólisis, lo que promueve la síntesis de AG y TG en mucha mayor medida que la glucosa (26). Como resultado, el consumo de fructosa se relaciona con hipertrigliceridemia y un aumento de los niveles de TG en el hígado, favoreciendo el desarrollo de esteatosis y la secreción de lipoproteínas de muy baja densidad (VLDL) (27). La alteración del metabolismo lipídico afecta también a los niveles de colesterol total y colesterol asociado a lipoproteínas de baja densidad (LDL), dando lugar a una situación de dislipemia (28).
1.4. Consumo prenatal de fructosa y programación fetal
La teoría del desarrollo temprano de la salud y la enfermedad (del inglés, developmental origins of health and disease, DOHAD) sugiere que las exposiciones nutricionales inadecuadas durante tres periodos críticos (preconcepción, embarazo y lactancia) juegan un papel fundamental en el desarrollo de obesidad y otras enfermedades metabólicas en la descendencia. Esto añade una dimensión adicional de complejidad al estudio de la etiología de las enfermedades cardiovasculares y el síndrome metabólico, explicando por qué el estudio de los factores de riesgo biológicos junto con los hábitos de vida adulta es insuficiente para predecir la aparición de ECV (29,30). Originalmente, la DOHAD se centraba en la desnutrición materna, pero cada vez son más numerosos los estudios que ponen el foco en determinar cómo patrones inadecuados de consumo de macro- y micronutrientes durante la etapa prenatal son capaces de promover el desarrollo de obesidad y otros factores de riesgo para la aparición de ECV en la descendencia (29,31).
Recientemente, varios estudios realizados en ratas han relacionado la exposición prenatal a fructosa con el desarrollo de enfermedades crónicas no transmisibles en la descendencia durante su vida adulta, como obesidad, diabetes tipo II y resistencia a insulina (32). La literatura muestra diferentes resultados en función del género, con efectos más pronunciados en los machos, razón por la que el presente estudio se ha centrado en este género (33,34). Los efectos perjudiciales asociados al consumo de fructosa durante el embarazo son resultado de un fenómeno conocido como programación fetal, que es el conjunto de adaptaciones que experimenta el embrión cuando está sometido a un entorno intrauterino estresante y que favorecen el riesgo de desarrollar enfermedades crónicas durante la vida adulta (35).
1.5. Epigenética: metilación de promotores relacionados con el metabolismo lipídico
La epigenética es el mecanismo subyacente que explica las adaptaciones que tienen lugar como resultado de la exposición prenatal a fructosa, y se define como los cambios heredables en la expresión génica que tienen lugar sobre la estructura del ácido desoxirribonucleico (ADN) y cromatina sin afectar a la secuencia de ADN (36). Dentro de estas modificaciones epigenéticas se encuentra la metilación, que consiste en la incorporación de un grupo metilo, generalmente a la citosina, cuando esta se encuentra formando un dinucleótido con la guanina (CpG) (37).
Las regiones del genoma con un elevado número de repeticiones CpG reciben el nombre de islas CpG, y son especialmente susceptibles de ser metiladas. Estas son abundantes en los promotores génicos, teniendo un papel fundamental en la regulación de su expresión (38). Un mayor estado de metilación se suele relacionar con una menor expresión del gen afectado, dado que los grupos metilo ejercen un impedimento estérico que dificulta el acceso de la maquinaria de transcripción. Por tanto, la metilación se trata de un fenómeno fundamental para controlar múltiples procesos fisiológicos como la diferenciación celular, la impronta genómica o la inactivación del cromosoma X (39).
Las DNA-metiltransferasas (DNMTs) son las enzimas responsables de la metilación de la citosina. Por un lado, la DNA-metiltransferasa 3a (DNMT3a) y la DNA-metiltransferasa 3b (DNMT3b) establecen patrones de metilación de novo durante la embriogénesis temprana y gametogénesis; mientras que la DNA-metiltransferasa 1 (DNMT1) mantiene los patrones de metilación existentes durante la mitosis (40). La expresión y actividad de las DNMTs se encuentra finamente regulada a muchos niveles, desde la transcripción hasta el nivel postraduccional. En concreto, la regulación metabólica de la metilación del DNA comprende la regulación locus-dependiente, mediada por factores de transcripción; y aquella mediada por los niveles de metabolitos centrales, como es el caso de SAM (S-adenosilmetionina), sustrato de estas enzimas (41).
Factores que alteran la expresión o actividad de las DNMTs favorecen el establecimiento de patrones de metilación aberrantes, los cuales se relacionan con patologías como el cáncer o ECV. Aunque estos cambios en la metilación pueden ocurrir a lo largo de la vida adulta, la mayor parte del epigenoma se establece durante el desarrollo temprano del feto, tratándose de un periodo crítico durante el cual factores nutricionales pueden afectar notablemente a los patrones de metilación (42,43). De hecho, el metabolismo lipídico hepático es especialmente sensible a la regulación epigenética, existiendo numerosos estudios que relacionan la exposición prenatal a fructosa con la alteración de la metilación de promotores lipídicos en la descendencia (29). En concreto, la ingesta materna de fructosa durante el embarazo se ha relacionado con la hipermetilación de los promotores del receptor nuclear activado por proliferadores peroxisomales (PPARα y del receptor X hepático alfa (LXRα (32,33,44), así como con un mayor nivel de metilación general del ADN mitocondrial (45).
1.6. Otras enzimas implicadas en la metilación
Al igual que las DNMTs están implicadas en la metilación del ADN, existen otras enzimas que llevan a cabo procesos de metilación de componentes celulares diferentes al material genético. Este es el caso de la fosfatidiletanolamina N-metiltransferasa (Pemt), localizada en los hepatocitos e implicada en el metabolismo de los fosfolípidos. En concreto, cataliza la conversión de fosfatidiletanolamina (PE) a fosfatidilcolina (PC) mediante la transferencia de tres grupos metilos aportados por SAM, siendo responsable de la producción de un 30% de la PC del organismo (46,47). Además de su función estructural en las membranas celulares, la PC favorece la solubilización de los ácidos biliares en la bilis y la secreción de quilomicrones y VLDL (48). Como resultado, la alteración de los niveles de PC por disfunción de Pemt se asocia con el desarrollo de patologías. Una menor actividad de Pemt se ha relacionado con menores niveles de PC, menor secreción de lípidos, acumulación de TG en el hígado y, como resultado, desarrollo de hígado graso no alcohólico (6,49).
Por otro lado, la glicina N-metiltransferasa (GNMT) es una enzima implicada en la metilación de la glicina para formar sarcosina, utilizando SAM como donante de metilos. Esta enzima desempeña un papel fundamental en la regulación de la relación SAM/SAH (S-adenosilhomocisteína) en mamíferos, por lo que está implicada en la disponibilidad de metilos y su utilización por otras metiltransferasas, evitando fenómenos de metilación aberrante (50,51). Mutaciones en humanos afectando al gen de la GNMT y modelos animales knock-out para GNMT se han relacionado con un incremento en los niveles séricos de metionina y SAM, y con el desarrollo de patologías hepáticas como esteatosis y fibrosis (52).
1.7. Metabolismo de un carbono
SAM es la molécula utilizada por las DNMT, Pemt y GNMT como donante universal de grupos metilos y forma parte del ciclo de la metionina, englobado dentro de la ruta del metabolismo de un carbono. Esta ruta se divide en el ciclo del folato y el ciclo de la metionina, ligados a la transulfuración y conectados para facilitar la transferencia de un residuo de un carbono para la metilación del ADN y síntesis de macromoléculas (53). Tal y como se muestra en la Figura 1, el ciclo del folato comienza con el ácido fólico, que experimenta una serie de transformaciones bioquímicas para convertirse en 5-metil-tetrahidrofolato (5-metil-THF). Dicha molécula sirve como donante de metilos para la conversión de la homocisteína a metionina gracias a la acción de las enzimas metionina sintasa (MTR) y metionina sintasa reductasa (MTRR). En mamíferos, la remetilación de la metionina también se puede llevar a cabo por la enzima betaína-homocisteína metiltransferasa (BHMT) (53).
A continuación, la metionina es adenilada por la enzima metionina adenosiltransferasa (MAT1A) para generar SAM. Este proceso tiene lugar mayoritariamente en el hígado, donde se metaboliza el 50% de la metionina (6,53). Una vez es utilizado por las metiltransferasas como sustrato, SAM pasa a convertirse en SAH. La SAH hidrolasa (AHCY) transforma SAH en homocisteína, que se puede convertir en metionina, tal y como se ha mencionado anteriormente. La relación SAM/SAH debe estar estrechamente regulada para evitar fluctuaciones que puedan derivar en alteraciones de diversos procesos fisiológicos llevados a cabo por las metiltransferasas, así como en patologías hepáticas como hígado graso no alcohólico o cirrosis (54). Asimismo, la homocisteína puede ser eliminada a través de la vía de la transulfuración, generando sulfuro de hidrógeno (H2S) y cisteína como productos por acción de las enzimas cistationina gamma liasa (CSE) y cistationina beta sintasa (CBS) (53,55).
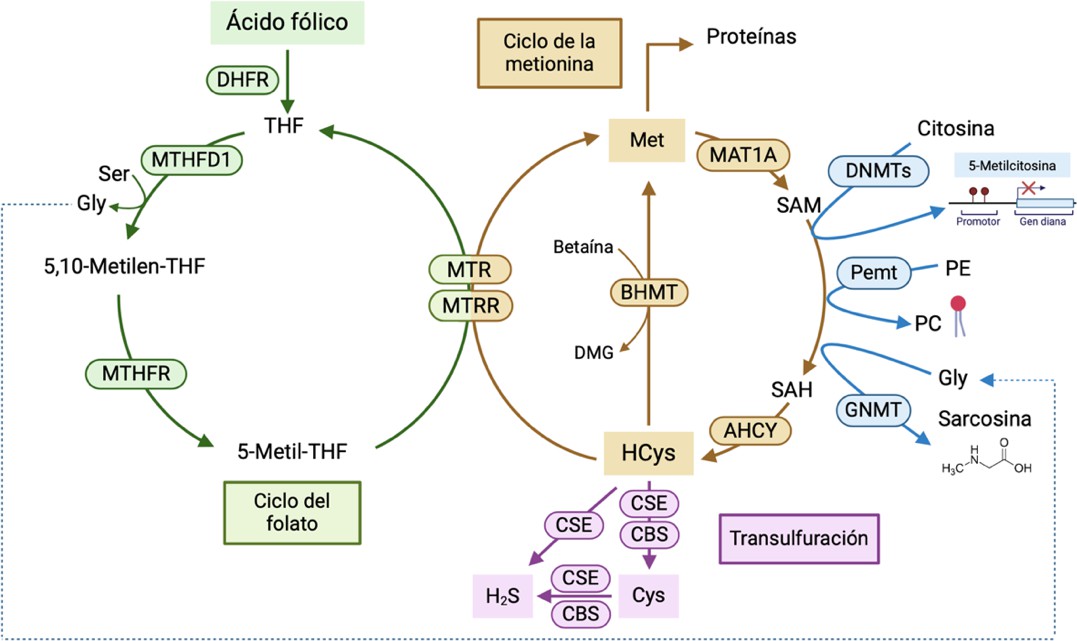
Figura 1. Metabolismo de un carbono (ciclo del folato + ciclo de la metionina), vía de la transulfuración y metilación de componentes celulares. AHCY (SAH hidrolasa); BHMT (betaína-homocisteína metiltransferasa); CBS (cistationina beta sintasa); CSE (cistationina gamma liasa); Cys (cisteína); DHF (dihidrofolato); DHFR (dihidrofolato reductasa); DMG (dimetilglicina); DNMTs (DNA-metiltransferasas); GNMT (glicina N-metiltransferasa); H2S (sulfuro de hidrógeno); MAT1A (metionina adenosiltransferasa); MTHFD1 (metilen-THF deshidrogenasa-1); MTHFR (metilen-THF-reductasa); MTR (metionina sintasa); MTRR (metionina sintasa reductasa); Pemt (fosfatidiletanolamina N-metiltransferasa); SAH (S-adenosilhomocisteína); SAM (S-adenosilmetionina); THF (tetrahidrofolato). Figura realizada con BioRender.
Considerando que el metabolismo de un carbono está estrechamente ligado a otras rutas metabólicas, la nutrición de la gestante puede afectar a la expresión y/o actividad de sus enzimas y alterar el epigenoma de la descendencia. Por tanto, el metabolismo de un carbono podría tratarse del vínculo entre la dieta, el metabolismo y la epigenética (54).
2. HIPÓTESIS
El consumo de fructosa durante el embarazo se asocia con el desarrollo de enfermedades crónicas no transmisibles en la descendencia a lo largo de la edad adulta, como enfermedades cardiovasculares, diabetes y obesidad. Estas alteraciones se deben al fenómeno de la programación fetal, producido por el establecimiento de marcas epigenéticas en el material genético de la descendencia. Dentro de estas marcas epigenéticas se encuentra la metilación, muy relacionada con el metabolismo de un carbono. En estudios anteriores hemos comprobado cómo el consumo pre- y postnatal de fructosa altera la expresión de algunos genes involucrados en dicha ruta metabólica. Esto podría derivar en cambios en los niveles de metabolitos que actúan como donantes o receptores de metilos, afectando a la actividad de las enzimas metiltransferasas (DNMTs, Pemt y GNMT). Consecuentemente, alteraciones de la actividad de las DNMTs se relacionarían con cambios en la metilación de promotores génicos y, en última instancia, con cambios en la expresión de las enzimas reguladas por dichos patrones epigenéticos. En el presente estudio se consideró el efecto del consumo prenatal de fructosa sobre la ruta del metabolismo de un carbono, así como las consecuencias del consumo directo de diversos tipos de dieta, que incluyen fructosa, tagatosa o dieta occidental.
3. OBJETIVOS
Teniendo en cuenta los antecedentes descritos, los objetivos de este estudio fueron:
Determinar si el consumo de fructosa en animales jóvenes genera alteraciones en la expresión de enzimas implicadas en la metilación del ADN (DNMTs) y otras metiltransferasas.
Comprobar si dichas modificaciones son específicas del consumo de fructosa, utilizando la tagatosa como epímero de la fructosa.
Estudiar si los efectos producidos por la fructosa se ven aumentados, disminuidos o invariables al añadir colesterol a la dieta, como un modelo de dieta occidental.
Estudiar si el consumo de fructosa durante el embarazo se asocia con alteraciones en la metilación de promotores génicos en la descendencia.
4. MATERIALES Y MÉTODOS
4.1. Diseño experimental
El diseño experimental (Figura 2) fue aprobado por el Comité Ético. Se utilizaron hembras de rata Sprague-Dawley alimentadas ad libitum con una dieta estándar para ratas (B&K Universal, Barcelona, España) que se cruzaron con machos de la misma cepa y se dividieron en dos grupos en función de la bebida recibida durante la gestación: madres control (agua) y madres fructosa (agua con fructosa al 10% (p/v)). Tras el parto, las camadas se ajustaron a 9 crías y, durante la lactancia, tanto las madres como su descendencia bebieron agua sin aditivos. Al finalizar este período, se seleccionaron las crías macho y se dejaron crecer hasta los 3 meses, edad a la que se inició el tratamiento dietético. Para ello, se dividieron en cuatro grupos distintos según la bebida y/o ingesta que recibirían durante los siguientes 21 días.
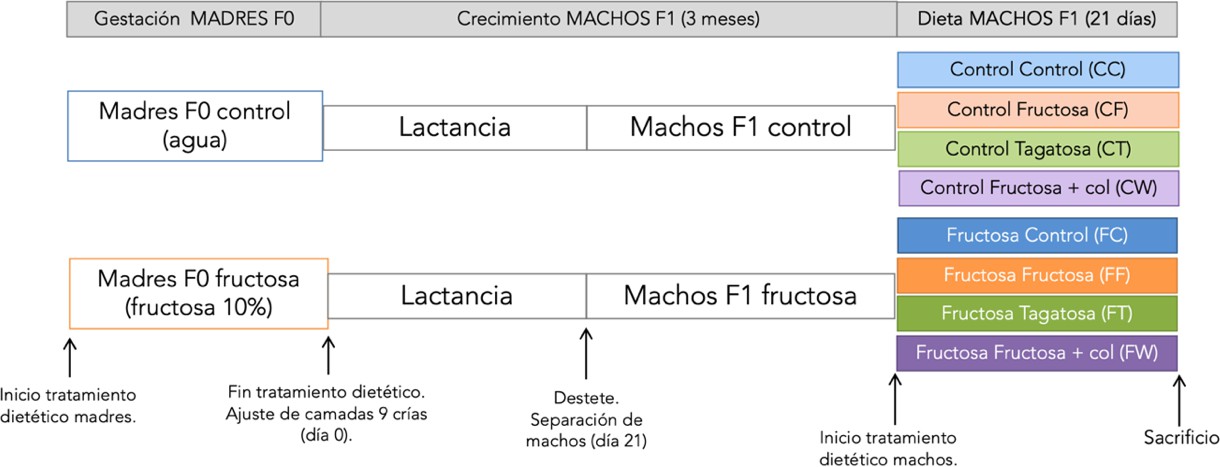
Figura 2. Diseño experimental
Se obtuvieron así 8 grupos experimentales, donde la primera letra hace referencia a la dieta de la madre durante la gestación y la segunda a la dieta consumida por la descendencia joven macho:
Grupo Control (CC y FC): dieta sólida estándar y agua sin aditivos.
Grupo Fructosa (CF y FF): dieta sólida estándar y agua con fructosa al 10% (p/v).
Grupo Tagatosa (CT y FT): dieta sólida estándar y agua con tagatosa al 10% (p/v).
Grupo Fructosa + Colesterol o Dieta Occidental (CF+Col y FF+Col): dieta sólida estándar con un 2% de colesterol añadido y agua con fructosa al 10% (p/v).
A los 21 días de tratamiento dietético, las ratas fueron sacrificadas. Se tomaron muestras de tejidos y sangre para determinar parámetros tisulares y plasmáticos.
4.2. Determinación de la expresión génica
Para la determinación de la expresión génica fue necesario extraer el ARN, tratarlo con DNAsas y transformarlo posteriormente en ADN complementario antes de proceder a realizar la PCR a tiempo real. Para la extracción del ácido ribonucleico (ARN), se realizó un homogenado del tejido, el cual se trató con BCP (bromocloropropano) y se sometió a centrifugación. Para purificar el ARN se utilizó el kit RiboPure™ (Ambion Inc., EE. UU.). La concentración y pureza del ARN purificado se determinaron utilizando el NanoDropTM, comprobando que las relaciones 260/280 y 260/230 se encontraban en el margen adecuado (1,8-2,2). Asimismo, mediante electroforesis en gel de agarosa al 2% se comprobó la integridad del ARN (visualizando las bandas correspondientes al ARN ribosómico).
El ARN purificado se trató con DNasas para eliminar posibles contaminaciones, utilizando el kit Turbo DNA-freeTM (Invitrogen, ThermoFisher, EE. UU.), y la pureza, concentración e integridad del ARN tratado se determinaron nuevamente. A continuación, se llevó a cabo una PCR convencional para comprobar la ausencia de ADN contaminante, utilizando primers del gen constitutivo de la proteína ribosómica S-29 (RPS29) (Tabla I). La ausencia de ADN se comprobó a través de una electroforesis en gel de agarosa al 2%, observándose únicamente la banda del control positivo. El ARN tratado con DNasas fue sometido a la acción de la transcriptasa inversa (RT) para obtener ADN complementario (ADNc). Se utilizó la transcriptasa inversa SuperScript II (Invitrogen, EE. UU.). Para comprobar la obtención de ADNc, se amplificó mediante PCR convencional una secuencia del gen RPS29 (Tabla I), y el producto de amplificación se visualizó a través de electroforesis en gel de agarosa al 2%.
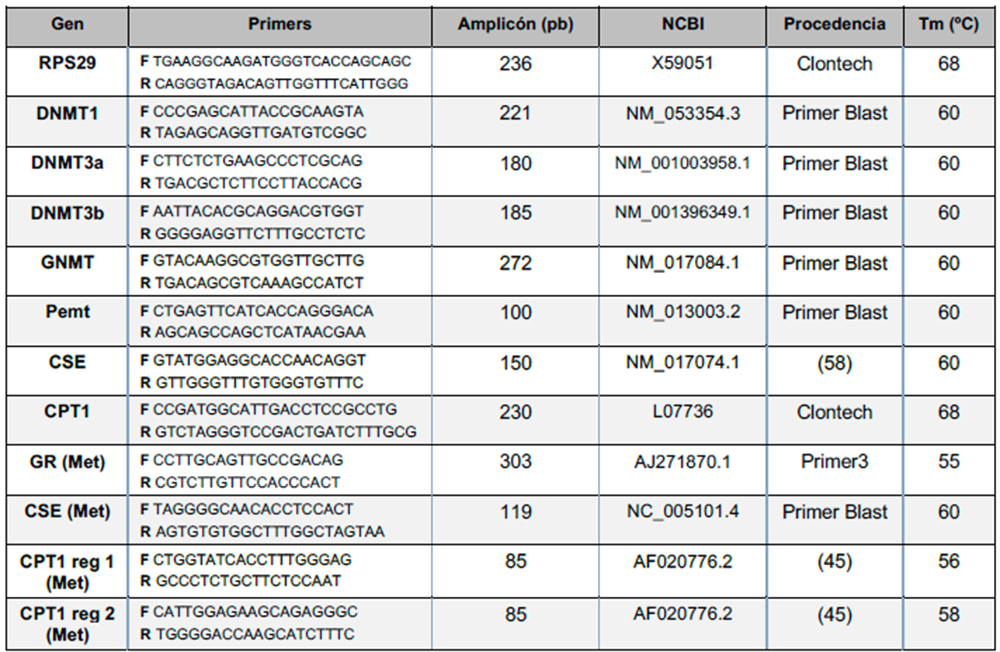
La expresión de los genes de interés (RPS29, DNMT1a, DNMT3a, DNMT3b, GNMT, Pemt, CSE, CPT1) se determinó a través de PCR a tiempo real (Q-PCR), empleando el ADNc obtenido y los primers recogidos en la Tabla I y utilizando el termociclador CFX96 Touch Real time PCR System (Biorad, EE. UU.). La cuantificación de la expresión se realizó mediante el método 2–∆∆Cp, por el que el valor de Cp (número de ciclos necesarios para superar el umbral de detección del equipo) obtenido es ajustado para el gen de interés utilizando como referencia el Cp del gen constitutivo RPS29 para esa misma muestra (55).
Previamente, fue necesario realizar rectas de calibrado de los genes estudiados mediante Q-PCR, para lo que se utilizaron diluciones seriadas de ADNc. El objetivo fue comprobar la eficiencia de los primers (E), cuyo valor numérico se encontró dentro del margen (1,8-2,2) y fue utilizado en los cálculos de expresión.
4.3. Determinación de la metilación de promotores
La determinación del nivel de metilación de promotores génicos se llevó a cabo según el protocolo descrito por Oakes et al. (56). Para la lisis, extracción y purificación del ADN se empleó el kit DNeasy Blood and Tissue Kit (Qiagen, EEUU). Muestras de, aproximadamente, 25 mg de hígado se lisaron mediante incubación con buffer ALT y proteinasa K. A continuación, se eliminó el ARN contaminante con RNasa A y se adicionó un buffer de lisis AL con tiocianato de guanidinio (inactiva las DNasa y RNasa A y elimina las proteínas). Por último, se obtuvo el ADN libre de contaminantes gracias a una columna de purificación.
El ADN purificado se dividió en dos alícuotas para cada una de las muestras estudiadas, una de las cuales se trató con la enzima de restricción McrBC (Takara Bio Inc., Japón). McrBC es una enzima de restricción que reconoce como secuencia diana 5´AC(Nn)GC3´ y que únicamente es capaz de cortar cuando las citosinas de dicha secuencia están metiladas. Para ello, 1 µg de ADN se trató con 1 µL de McrBC, 2 µL de buffer 10X, 2 µL de seroalbúmina bovina (BSA) 0,1%, 0,2 µL de GTP 100 mM y agua ultrapura estéril hasta completar el volumen a 20 µL. La mezcla se incubó a 37 ºC (temperatura óptima de la enzima) durante 16 h y posteriormente se inactivó la enzima a 56 ºC durante 20 minutos. La otra alícuota permaneció sin tratar, empleándose como control no tratado, de manera que se añadió 1 µL extra de buffer 10X en lugar de 1 µL de McrBC.
La determinación de la concentración y pureza del ADN purificado se llevó a cabo empleando NanoDrop® One (ThermoFisher, EEUU). Para comprobar que el tratamiento con la enzima fue satisfactorio, se realizó una electroforesis en gel de agarosa al 2%. En la Figura 3 se aprecia cómo las muestras no tratadas dan lugar a una única banda diferenciada, mientras que las muestras tratadas se corresponden con una estela que representa el ADN cortado en múltiples sitios por la enzima.

Figura 3. Gel de electroforesis del ADN sin tratar y tratado con McrBC. Las muestras control no tratadas se ven como bandas nítidas sin degradar, mientras que las tratadas presentan una estela a lo largo del carril debido a los múltiples cortes del ADN.
A continuación, para determinar el nivel de metilación de los promotores, se realizó una Q-PCR empleando tanto la alícuota de ADN tratado con McrBC como la alícuota sin tratar. Para ello, se diseñaron primers (Tabla I) que flanquean regiones con una alta proporción de CG, susceptibles de formar parte de islas CpG y ser metiladas. El diseño de los primers requirió, en primer lugar, buscar la secuencia del gen de interés en la base de datos GenBank, perteneciente al National Center for Biotechnology Information (NCBI, EEUU). Dicha secuencia, junto con aproximadamente unos 1000 pares de bases upstream de la misma, se introdujo en el programa informático MethPrimer, que identifica las regiones con una alta proporción de CG, con mayor posibilidad de formar parte de una isla CpG del promotor (57).
A continuación, la secuencia sugerida por MethPrimer fue introducida en el programa Primer Blast (NCBI), especificando los parámetros óptimos para la obtención de los primers. Una vez obtenidas varias posibles parejas, fue necesario analizar la secuencia amplificada por cada una de ellas, comprobando, en primer lugar, que la secuencia diana estaba presente (en la hebra codificante o en la no codificante); y, además, que la secuencia diana no apareciese en la secuencia de los cebadores. Teniendo en cuenta dichos criterios, se escogió una pareja de primers para cada promotor (Tabla I).
Tabla 1. Primeros empleados para amplificar el ARNm y para determinar el nivel de metilación de promotores en Rattus norvegicus.
Por tanto, un alto nivel de metilación en dichas regiones del promotor permite que la enzima McrBC reconozca la diana y corte el ADN en dicha localización, impidiendo la acción de la polimerasa y la amplificación por PCR de dicha región. Como resultado, las muestras tratadas con McrBC en las que dichas regiones estén metiladas se relacionarían con mayores valores de Cp obtenidos tras la Q-PCR en comparación con las muestras no tratadas. El termociclador utilizado para la amplificación fue CFX96 Touch Real-time PCR System (Biorad, EE. UU.). El porcentaje de metilación de la región promotora se determinó utilizando la diferencia de Cp obtenidos entre las muestras tratadas y sin tratar (ΛCp) utilizando la ecuación descrita por Oakes et al.: % Metilación = 100 · (1 – e-0,7ΛCp) (56) (Figura 4).
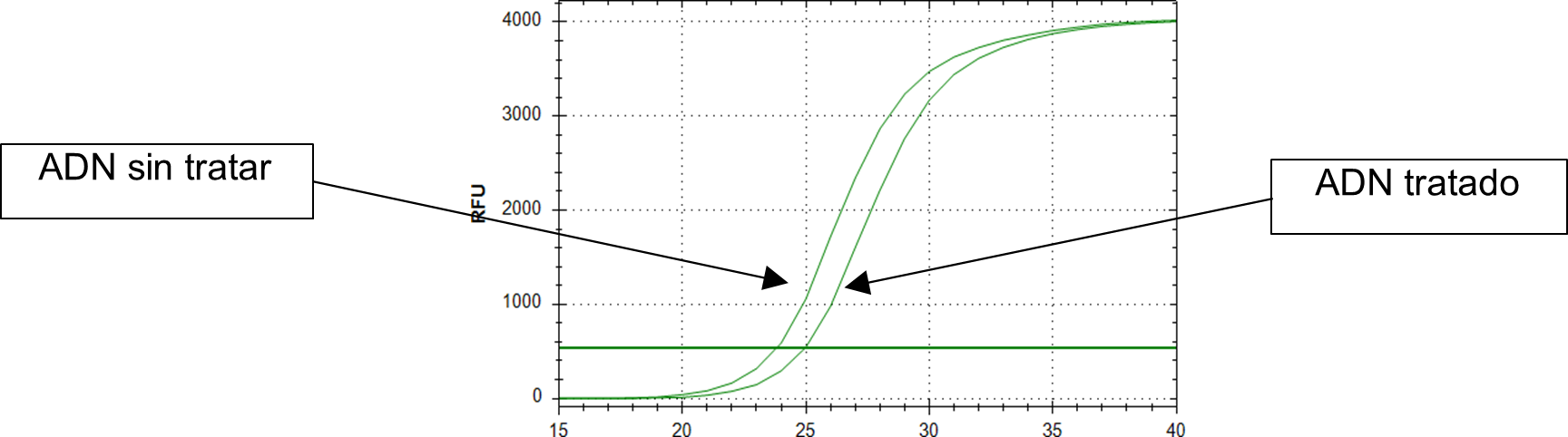
Figura 4. Curva de amplificación por PCR a tiempo real de una muestra de ADN tratado (derecha) y sin tratar (izquierda).
Previamente a la medida de la metilación de los genes diana ((CSE, CPT1 Región 1 (Reg 1), CPT1 Región 2 (Reg 2)), se midió la metilación de un gen control (receptor de glucocorticoides, GR) utilizando primers que amplificaban zonas del genoma ausentes de secuencias diana para la enzima (Tabla I). El objetivo fue comprobar que la concentración del ADN tratado con McrBC y no tratado era, aproximadamente, la misma; y que no se habían producido roturas inespecíficas en las muestras. Se aceptaron como correctas las muestras cuya diferencia de Cp del ADN tratado y del ADN control fuese igual o inferior a ± 0.3 ciclos (56).
4.4. Análisis estadístico de los datos
Los resultados se expresaron como media ± error estándar (ES) correspondiente a un número de 7 u 8 animales por grupo. Las diferencias estadísticas se analizaron mediante ANOVA de doble vía. Cuando las diferencias resultaron estadísticamente significativas (p<0,05), se analizaron mediante el test a posteriori de Bonferroni para determinar la significatividad de la varianza. Por un lado, los factores analizados fueron el efecto de consumo de fructosa por la madre durante la gestación (M) en la descendencia macho joven. Estas diferencias entre madres control vs. madres fructosa se indicaron con una almohadilla (#). Por otro lado, se analizó el efecto de los tratamientos que se administraron a la descendencia macho joven (D). Las diferencias entre estos, dentro del mismo grupo nutricional de las madres, se indicarán mediante un asterisco. Toda la determinación estadística se realizó utilizando el programa informático SPSS (versión 28).
5. RESULTADOS Y DISCUSIÓN
5.1. Consumo de fructosa vs. tagatosa
En base a estudios previos que mostraban que el consumo de fructosa alteraba la expresión de algunas enzimas del metabolismo de un carbono, se hipotetizó que esto podría modificar la expresión de las DNMTs y otras metiltransferasas, explicando así las alteraciones observadas en el epigenoma (33,56). Para determinar si dicho efecto era específico de este azúcar, se estudió comparativamente con la tagatosa, que, como se ha mencionado en la introducción, es un epímero de la fructosa.
5.1.1. Expresión génica de las DNMTs en hígado y corazón
Los resultados del análisis de varianza (ANOVA) de doble vía para la expresión de las DNMTs (DNMT1, DNMT3a, DNMT3b) tanto en hígado como en corazón (Tabla 2) muestran que la dieta afecta a la expresión de las DNMTs solo en el corazón, poniendo de manifiesto un efecto tisular dependiente.
Tabla 2. ANOVA de doble vía de los resultados obtenidos en la expresión DNMT1, DNMT3a y DNMT3b en hígado, así como DNMT1 y DNMT3b en corazón, en la descendencia macho. p: valor de probabilidad. Eta cuadrada (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
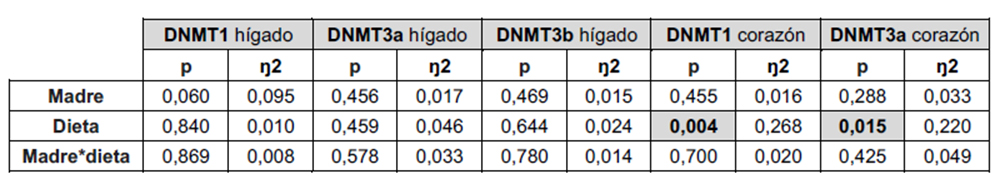
De acuerdo con los resultados de la ANOVA, no se observaron diferencias significativas en los niveles de expresión hepática de estos genes como resultado de la dieta (Figura 5). Sin embargo, es importante destacar que la regulación de las DNA-metiltransferasas no se limita únicamente a la transcripción y traducción de los genes que las codifican, sino que también involucra una regulación post-traduccional mediada por diferentes mecanismos, entre los que se encuentra la regulación metabólica, mediada por metabolitos centrales del metabolismo de un carbono (59). Por tanto, según los resultados de expresión obtenidos, aunque la dieta no genere diferencias en la expresión hepática de estas enzimas, no es posible concluir que no haya variaciones en su actividad mediadas por otros mecanismos.
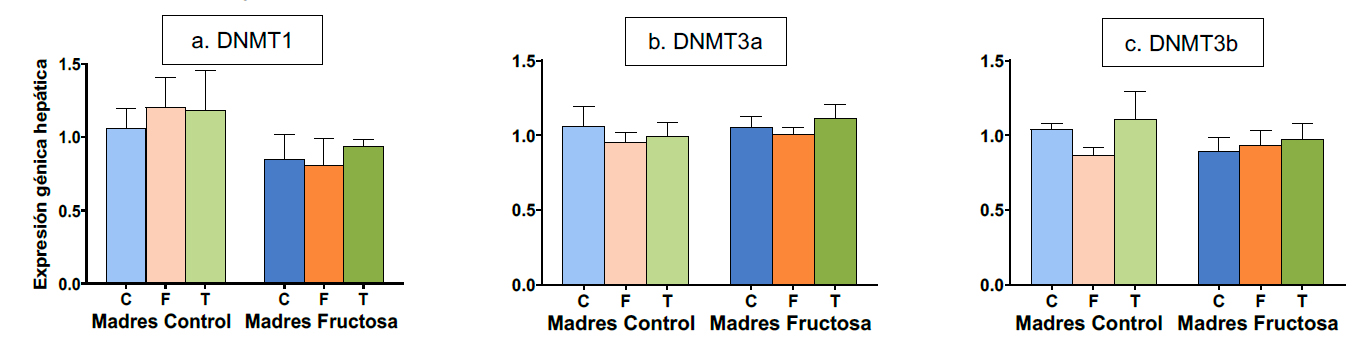
Figura 5. Expresión de los genes (u.a.) DNMT1 (a), DNMT3a (b), DNMT3b (c) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o tagatosa líquida (10%): barras verdes). Los datos representan la media ± E.S. de 7-8 ratas. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
No obstante, cuando se midió la expresión de DNMT1 y DNMT3a en corazón, sí que se encontraron diferencias significativas en función del carbohidrato consumido durante la vida postnatal. Como se muestra en la Figura 6, la expresión de DNMT1 y DNMT3a disminuyó significativamente en la descendencia de las madres control tras el consumo de tagatosa. Por tanto, la menor expresión cardiaca de estas enzimas sería un efecto específico de la tagatosa no observado tras la ingesta de fructosa.
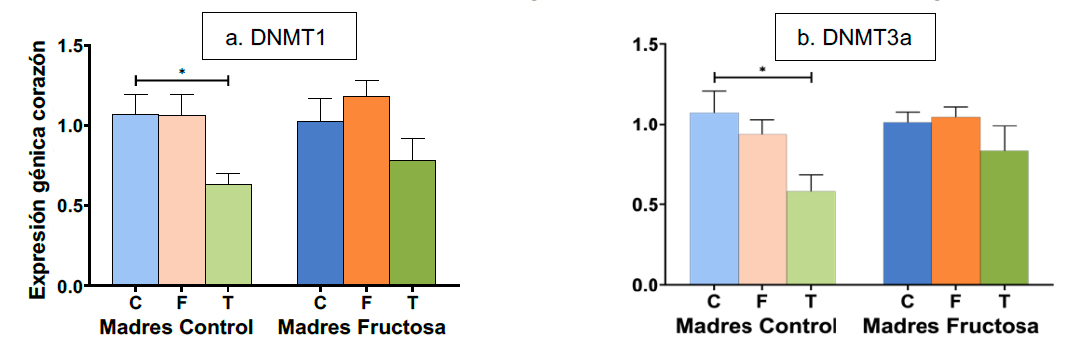
Figura 6. Expresión de los genes (u.a.) DNMT1 (a) y DNMT3a (b) en corazón en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o tagatosa líquida (10%): barras verdes). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.1.2. Expresión génica hepática de Pemt y GNMT
Como se ha mencionado previamente, otras enzimas que forman parte del ciclo de la metionina y que utilizan SAM como donante de metilos son Pemt, implicada en la síntesis de fosfatidilcolina, y GNMT, en la síntesis de sarcosina. Tal y como se muestra en la Tabla 3, el análisis de la ANOVA de doble vía muestra que la expresión hepática de GNMT presenta diferencias estadísticamente significativas debidas al factor DIETA, sin encontrarse diferencias significativas en la expresión de Pemt (Figura 7a).
Al analizar las diferencias encontradas en la expresión de GNMT, observamos que el consumo de carbohidratos por la descendencia de madres control y madres fructosa provocó menores niveles en la expresión de esta enzima, aunque sin alcanzar diferencias estadísticamente significativas con el grupo control (Figura 7b).
Tabla 3. ANOVA de doble vía de los resultados obtenidos en la expresión hepática de Pemt y GNMT en la descendencia macho. p: valor de probabilidad. Eta cuadrado (η2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
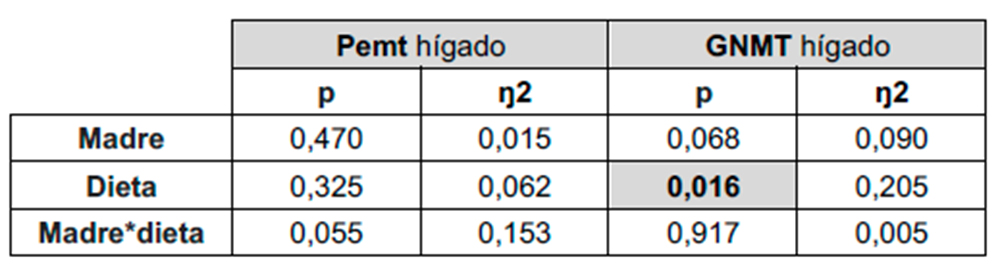
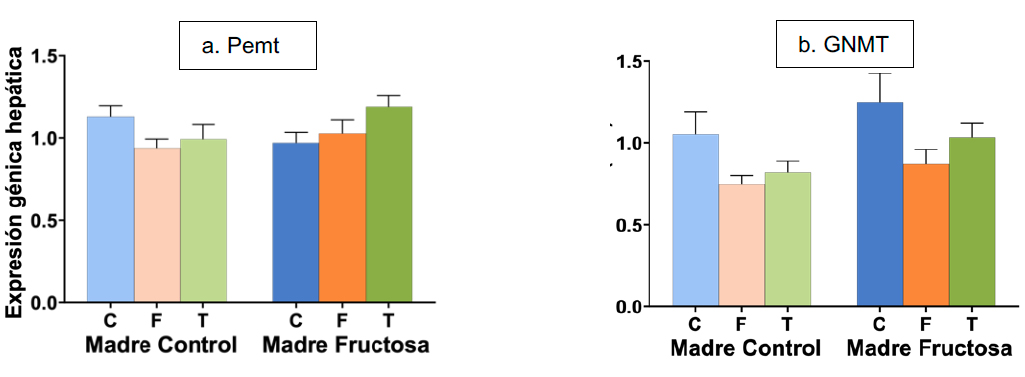
Figura 7. Expresión de los genes (u.a.) Pemt (a) y GNMT (b) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o tagatosa líquida (10%): barras verdes). Los datos representan la media ± E.S. de 7-8 ratas. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.1.3. Nivel de metilación del promotor de la CSE en hígado
Aunque no se observaron diferencias significativas en la expresión de las DNA-metiltransferasas como consecuencia del consumo de fructosa o tagatosa, teniendo en cuenta que la actividad de dichas enzimas no depende únicamente de su expresión, se evaluó la metilación del promotor de la enzima CSE, implicada en la ruta de la transulfuración y, por tanto, en el metabolismo de un carbono. Dado que la ingesta de fructosa pre- y postnatal se asocia con un mayor riesgo de ECV y SM, los cambios en la metilación de este promotor podrían estar potencialmente vinculados con alteraciones en el metabolismo y promover factores de riesgo para el desarrollo de estas patologías.
El estudio ANOVA de doble vía (Tabla 4) muestra que la metilación del promotor de CSE está afectada significativamente por el efecto MADRE. El análisis posterior mediante el test de Bonferroni permitió identificar que el consumo de fructosa por la descendencia de madres control dio lugar a niveles de metilación significativamente superiores a los observados como resultado del consumo de agua o tagatosa. Así, como se muestra en la Figura 8, el nivel de metilación del promotor de CSE se vio aumentado como consecuencia del consumo postnatal de fructosa.
Sorprendentemente, el aumento en el nivel de metilación observado en los machos descendientes de madres control que consumieron fructosa no se observó cuando éstos consumieron tagatosa, lo que pone de manifiesto el efecto específico del carbohidrato ingerido. Asimismo, el consumo materno de fructosa anuló drásticamente dicho aumento en la metilación del promotor de CSE en la descendencia cuando esta consumió dicho monosacárido (Figura 8). Este resultado parece indicar que el consumo prenatal de fructosa evitaría la hipermetilación observada en el promotor de CSE como resultado de la ingesta de fructosa durante la vida adulta a través del fenómeno de la programación fetal.
Tabla 4. ANOVA de doble vía de los resultados obtenidos en la metilación de la CSE hepática, en la descendencia macho. p: valor de probabilidad. Eta cuadrado (η2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
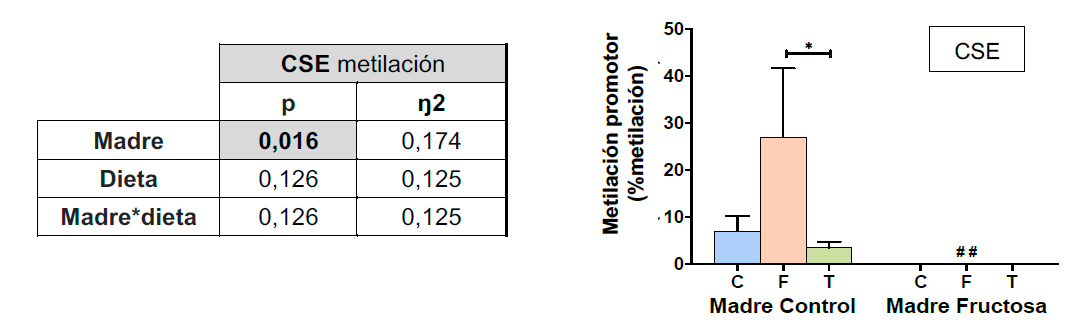
Figura 8. Metilación del promotor de la CSE en hígado de machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o tagatosa líquida (10%): barras verdes). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las almohadillas (#) indican diferencias estadísticamente significativas entre machos tratados con la misma dieta, pero procedentes de madres que recibieron diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.1.3. Nivel de metilación del promotor de la CSE en hígado
Expresión génica hepática de la CSE
Con el fin de determinar si los cambios observados en la metilación del promotor de CSE como resultado del consumo de fructosa podrían tener algún efecto sobre la expresión de la enzima codificada por este gen, se midió la expresión hepática de dicha enzima limitante de la transulfuración. En la Tabla 5 se muestran los resultados de la ANOVA de doble vía para la expresión de CSE, y se puede observar que el factor DIETA mostró diferencias estadísticamente significativas.
Tabla 5. ANOVA de doble vía de los resultados obtenidos en la expresión hepática de CSE en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
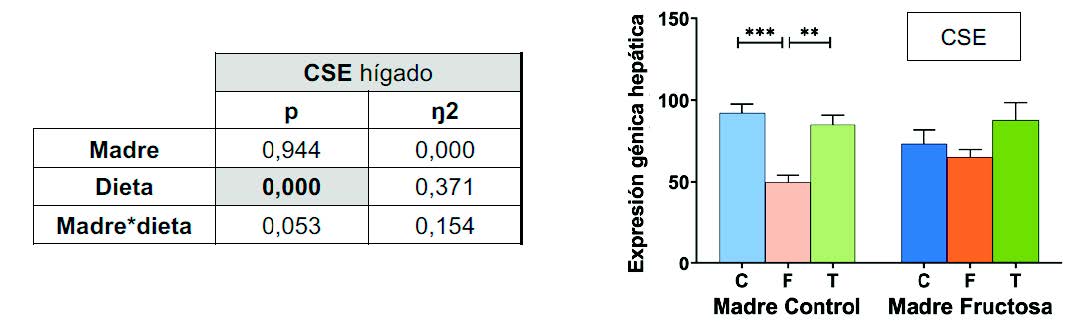
Figura 9. Expresión del gen CSE en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o tagatosa líquida (10%): barras verdes). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.1.3. Nivel de metilación del promotor de la CSE en hígado
Como se muestra en la Figura 9, la hipermetilación del promotor CSE se tradujo, como era de esperar, en una menor expresión de la enzima en los machos procedentes de madres control que consumieron fructosa. De nuevo, de acuerdo con lo observado en la metilación de dicho promotor (Figura 8), no se observaron diferencias en la expresión de la CSE entre los tres grupos experimentales en los machos descendientes de madres fructosa. Como se muestra en la Figura 1, la CSE está involucrada en la conversión de la homocisteína a través de la vía de la transulfuración, siendo la principal enzima responsable en la producción hepática de H2S (60).
Se ha descrito que niveles reducidos de H2S están asociados con el desarrollo de patologías como la hipertensión, arritmia o fallo cardiaco, pues este gasotransmisor desempeña un papel crucial en diversos procesos fisiológicos como la regulación de la función cardiaca, la inflamación y la vasodilatación (61,62). Esta relación refuerza el carácter perjudicial del consumo de fructosa, ya que una menor expresión de la CSE inducida por este monosacárido podría conducir a niveles más bajos de H2S, lo que, a su vez, podría favorecer el desarrollo de patologías.
5.1.5. Relación entre niveles de SAH y metilación
Como se ha mencionado en el apartado 5.1.1., aunque las dietas administradas no alteraron la expresión de las metiltransferasas en el hígado, los resultados obtenidos muestran cambios en los patrones de metilación del promotor de la CSE (Figura 8). Esto sugiere la existencia de cambios en la actividad de las DNA-metiltransferasas mediados por otro mecanismo diferente a la expresión génica. Por tanto, para intentar descifrar la causa de la alteración del metiloma, se evaluaron los niveles hepáticos de SAH, un producto de las metiltransferasas que afecta su actividad enzimática.
Los resultados muestran una tendencia hacia menores niveles hepáticos de SAH en la descendencia de madres control tras el consumo de fructosa, en comparación con el grupo que consumió tagatosa o agua. Esta tendencia no se observó en los hijos de madres fructosas (Tabla VI, Figura 10). Dicha disminución se relaciona inversamente con los niveles de metilación observados en el promotor de CSE (Figura 8), es decir, menores niveles de SAH se podrían asociar con un mayor nivel de metilación en la descendencia de madres control que consumieron fructosa. Dado que SAH regula negativamente a las DNMTs, podríamos estar ante la causa de las alteraciones observadas en el metiloma en este grupo experimental (63). Menores niveles de SAH debido al consumo de fructosa implicarían una mayor actividad enzimática de las DNMTs y una mayor metilación del promotor de CSE, reduciendo la expresión de la enzima. Por tanto, aunque no se observan cambios en la expresión de las DNMTs debido a la dieta, sí se vieron alteraciones en la metilación por posibles cambios en su actividad.
Tabla VI. ANOVA de doble vía de los resultados obtenidos en los niveles hepáticos de SAH en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
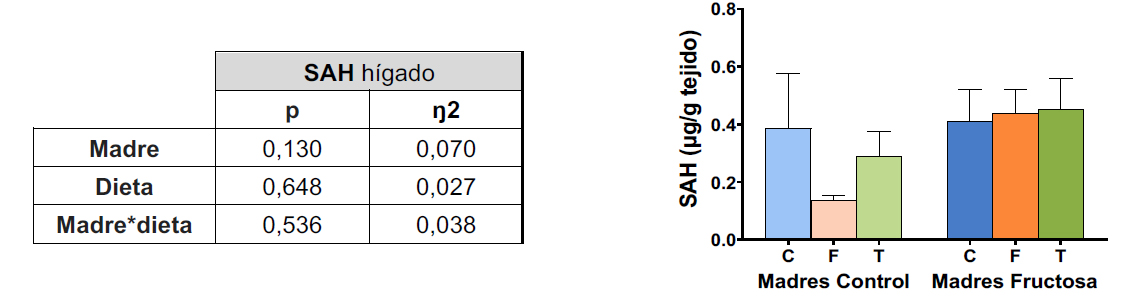
Figura 10. Niveles de SAH (μg/g tejido) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o tagatosa líquida (10%): barras verdes). Los datos representan la media ± E.S. de 7-8 ratas. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos. Resultados cedidos correspondientes a la tesis doctoral en preparación de Cristina Donis Rodríguez.
5. 2. Consumo de fructosa vs. fructosa + colesterol (dieta occidental)
Una vez analizados los efectos del consumo de fructosa, se determinaron los efectos del consumo de la dieta occidental, para lo que se empleó como modelo el consumo de fructosa asociada a colesterol.
5.2.1. Expresión génica de DNMTs en hígado y corazón
Tras el análisis estadístico de la ANOVA de doble vía, solo se observaron diferencias estadísticamente significativas en la expresión hepática de DNMT1 con el factor MADRE. Por otro lado, cuando se estudió la interacción MADRE*DIETA, la expresión de DNMT1 en corazón mostró diferencias cuasi significativas con un p=0,053. A su vez, al analizar la expresión de DNMT3a en este mismo tejido, se observó que el factor DIETA mostraba un valor de p=0,074, y esta diferencia fue incluso mayor cuando se analizó la interacción MADRE*DIETA con un p de 0,06 (Tabla VII).
Tabla VII. ANOVA de doble vía de los resultados obtenidos en la expresión DNMT1, DNMT3a y DNMT3b en hígado, así como DNMT1 y DNMT3b en corazón, en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
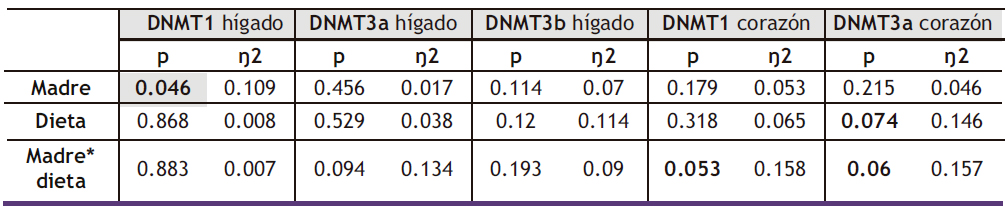
Al analizar las diferencias estadísticas mediante el test de Bonferroni, se observó que el consumo de dieta occidental mostró una menor expresión de DNMT3a en los descendientes de madres control que consumieron esta dieta cuando se comparaba con el grupo control, aunque sin alcanzar diferencias estadísticamente significativas (Figura 11 b). Sin embargo, la expresión de DNMT3b sí que presentó diferencias significativas, tanto por la dieta consumida por la descendencia como por efecto de la programación fetal (Figura 11c). Así, el consumo de la dieta occidental por la descendencia de madres control provocó niveles significativamente menores cuando se comparó con el grupo control. Sin embargo, dicho efecto inducido por la WD no fue encontrado en los descendientes de madres fructosa.
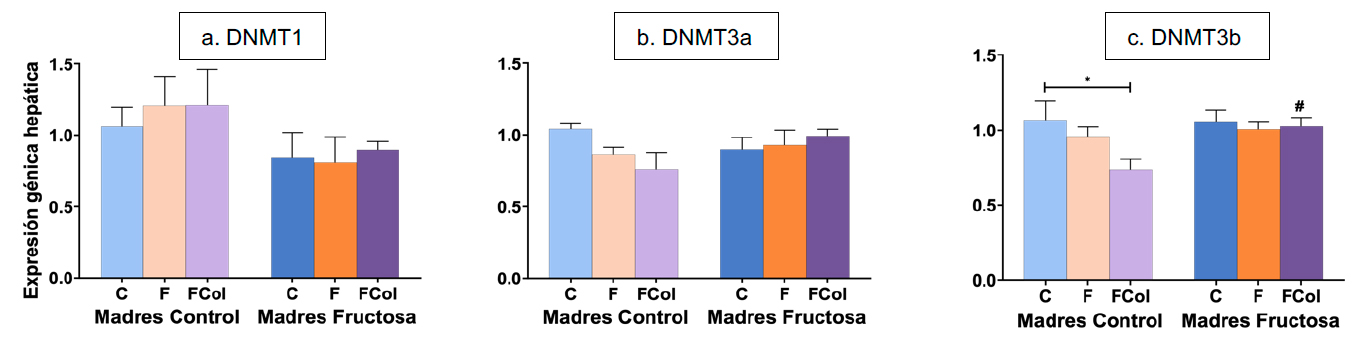
Figura 11. Expresión de los genes DNMT1 (a), DNMT3a (b), DNMT3b (c) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o fructosa + col (2%): barras moradas). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las almohadillas (#) indican diferencias estadísticamente significativas entre machos tratados con la misma dieta, pero procedentes de madres que recibieron diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.1.3. Nivel de metilación del promotor de la CSE en hígado
Por tanto, teniendo en cuenta su implicación en el establecimiento de los patrones de metilación de novo, una menor expresión de DNMT3a y DNMT3b promovida por la dieta occidental podría potencialmente resultar en un metiloma aberrante (40). Por otro lado, el efecto de la programación fetal debido al consumo materno de fructosa atenuó los efectos observados por la dieta occidental (WD) en la descendencia de madres control. Este resultado reafirma que la fructosa consumida por la madre durante la gestación podría actuar como un estresor intrauterino que altera la expresión de las enzimas del ciclo de la metionina y evita la disminución en la expresión de DNMT3a y DNMT3b que aparece tras la exposición a una dieta occidental en la juventud a través de la programación fetal.
Por otro lado, al analizar la expresión de las DNA metiltransferasas en el corazón, tras el test de Bonferroni se observaron niveles significativamente inferiores en la expresión de DNMT1 en los hijos de madres fructosa que consumieron fructosa asociada a colesterol (Figura 12a) en comparación con los que consumieron fructosa. Un perfil similar se observó tras analizar la expresión de DMNT3a (Figura 12b). Cabe destacar que los menores niveles observados en la descendencia de madres fructosa que consumieron dieta occidental mostraron diferencias estadísticamente significativas al compararlos con los de la descendencia de madres control que ingirieron esa misma dieta. Nuevamente, la nutrigenómica que estaba afectando a la expresión de las DNMTs tras el consumo de la dieta occidental resultó ser dependiente del tejido estudiado.
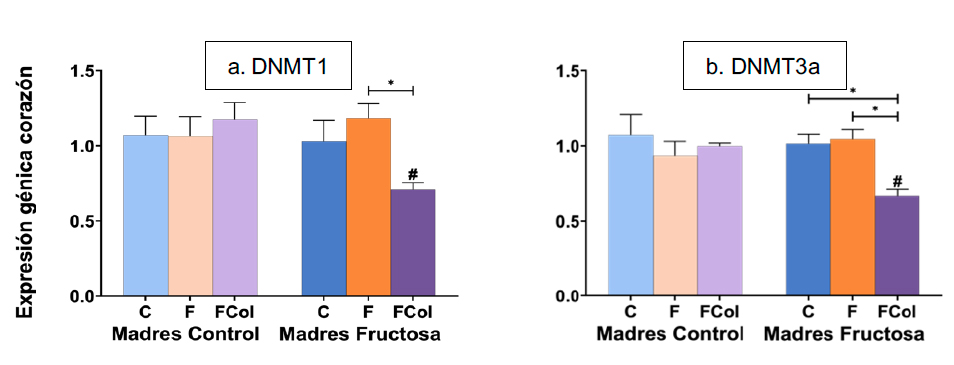
Figura 12. Expresión de los genes DNMT1 (a) y DNMT3a (b) (u.a.) en corazón en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o fructosa + col (2%): barras moradas). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las almohadillas (#) indican diferencias estadísticamente significativas entre machos tratados con la misma dieta, pero procedentes de madres que recibieron diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.2.2. Expresión génica hepática de Pemt y GNMT
El análisis de la ANOVA de doble vía mostró diferencias estadísticamente significativas en ambos genes debidas al factor DIETA (Tabla VIII). Posteriormente, cuando se analizaron dichas diferencias mediante el test de Bonferroni, y tal y como se muestra en la Figura 13, se observó que el consumo de la dieta occidental provocó una disminución en la expresión de ambas enzimas, independientemente de la dieta consumida por la madre durante la gestación. Por tanto, sería interesante indagar en las consecuencias de esta disminución en la expresión, ya que podría traducirse en una menor síntesis de sarcosina y fosfatidilcolina, con potenciales repercusiones en el desarrollo de diversas patologías (50,53).
Tabla VIII. ANOVA de doble vía de los resultados obtenidos en la expresión hepática de Pemt y GNMT en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
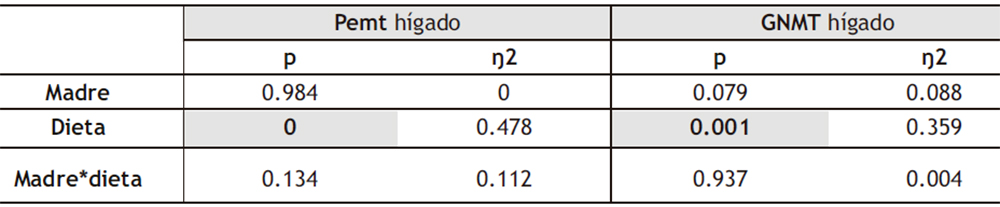
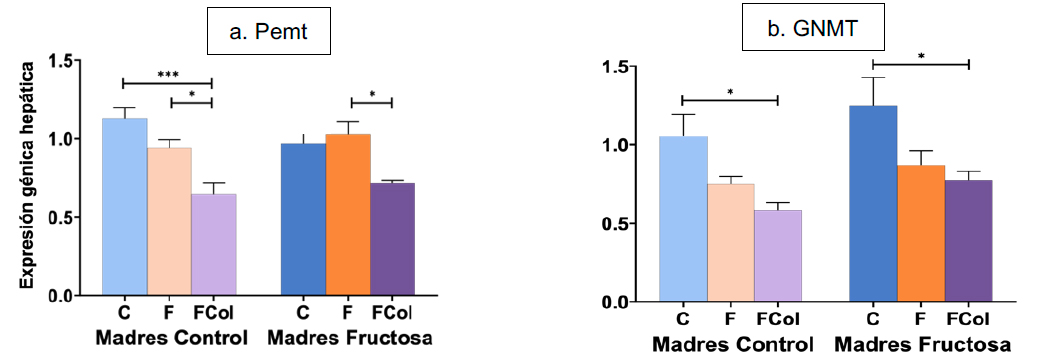
Figura 13. Expresión de los genes GNMT (a) y Pemt (b) (u.a.) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o fructosa + col (2%): barras moradas). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
5.2.3. Nivel de metilación del promotor de CPT1 en hígado
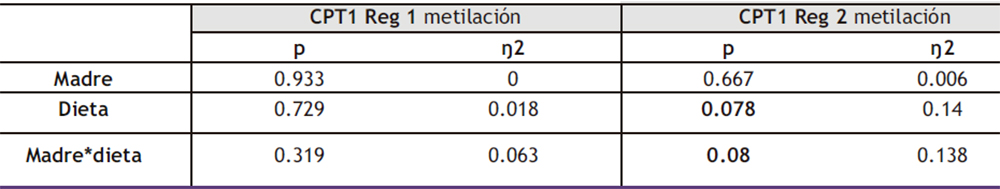
Dado que la expresión de DNMT3a y DNMT3b disminuyó como resultado del consumo de la dieta occidental (fructosa + colesterol) en los hijos de madres control sin afectar a la descendencia de madres fructosa, se midió el nivel de metilación del promotor de la enzima carnitina palmitoiltransferasa 1 (CPT1) para determinar cómo estos cambios en la expresión podían afectar al metiloma. En este caso y basándonos en la bibliografía, analizamos dos regiones del promotor. Como se observa en la Tabla IX, el estudio de ANOVA de doble vía muestra que la metilación de la región 2 del promotor de CPT1 está modulada por el factor DIETA y por la interacción MADRE*DIETA, aunque sin llegar a ser significativo en ningún caso.
El análisis posterior mediante el test de Bonferroni permitió identificar que el nivel de metilación del promotor del gen para CPT1 en la descendencia de madres control que consumió dieta occidental era menor cuando se comparó con la descendencia que consumió fructosa, tanto en la región 1 como en la región 2 del promotor (Figura 14). Además, se observó una tendencia hacia una menor metilación en la región 1 del promotor de CPT1 en comparación con los machos que consumieron agua descendientes de madres control (Figura 14a). Esto es coherente con los resultados de expresión de DNMT3a y DNMT3b obtenidos para dicho grupo experimental, pues una menor actividad de estas enzimas como resultado de su menor expresión supondría una menor metilación de los promotores génicos (Figura 11b,c). Por otro lado, cuando los individuos procedentes de madres que habían estado expuestas a fructosa durante la gestación consumieron la dieta occidental, se observó una tendencia hacia el aumento de la metilación de las dos regiones estudiadas (Figura 14). Por tanto, el fenómeno de la programación fetal inducido por la exposición a fructosa durante la gestación se volvía a observar en este caso.
5.2.4. Expresión génica hepática de la CPT1
Para determinar los efectos observados en la metilación del promotor del gen para CPT1, se midió la expresión génica de la enzima. La CPT1 es una enzima involucrada en la β-oxidación de los AG en las mitocondrias, catalizando la entrada de los AG de cadena larga desde el citosol hasta el interior del orgánulo, por lo que juega un papel fundamental en la homeostasis energética del organismo (64).
Tabla IX. ANOVA de doble vía de los resultados obtenidos en la metilación de CPT1 Reg 1 y CPT1 Reg 2 en hígado en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
A pesar de que el estudio ANOVA de doble vía no mostró diferencias estadísticamente significativas, el posterior test de Bonferroni permitió corroborar que el perfil de expresión coincidía con lo esperado en base a la metilación del promotor en la descendencia, tanto de madres control como de madres fructosa. Los mayores niveles de metilación observados tras consumo de fructosa por la descendencia de madres control en ambas regiones del promotor (Figura 14) se correlacionaban con los menores niveles de expresión observados por este grupo (Figura 15). Curiosamente, esa hipermetilación que se observaba en los descendientes de madres control tras la ingesta de fructosa desaparecía al combinar dicho azúcar con el consumo de colesterol, y coincidía con la normalización de la expresión de CPT1 a los niveles observados en la progenie que tomó agua sin aditivos.
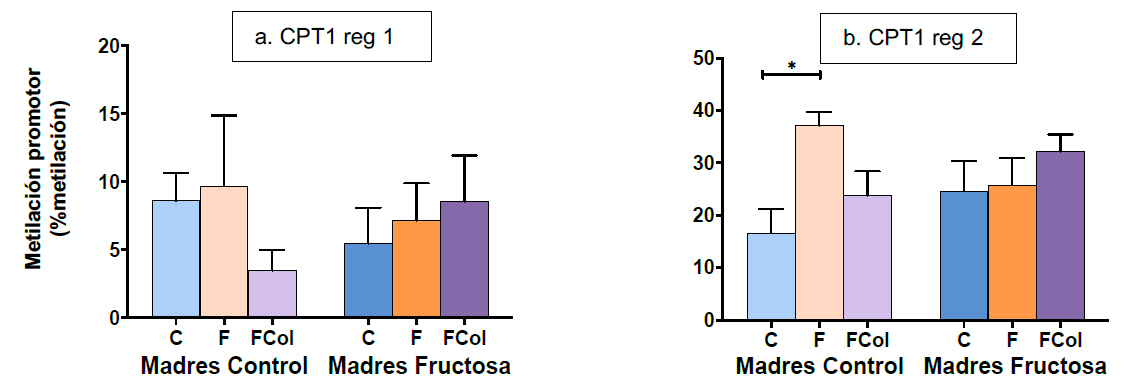
Figura 14. Metilación de los promotores CPT1 Región 1 (a) y CPT1 Región 2 (b) en hígado de machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o fructosa + col (2%): barras moradas). Los datos representan la media ± E.S. de 7-8 ratas. Los asteriscos (*) indican diferencias estadísticamente significativas entre machos tratados con diferente dieta. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
Tabla X. ANOVA de doble vía de los resultados obtenidos en la expresión hepática de CPT1 en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
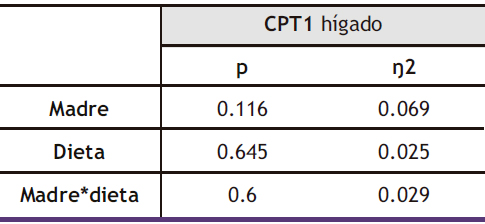
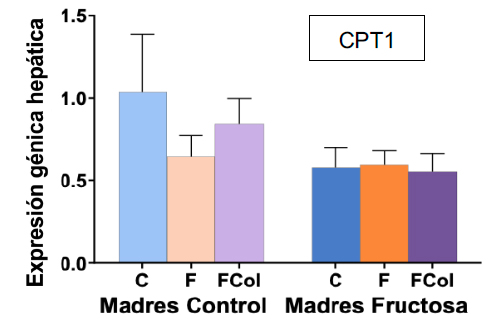
Figura 15. Expresión del gen CPT1 (u.a.) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o fructosa + col (2%): barras moradas). Los datos representan la media ± E.S. de 7-8 ratas. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
Por otro lado, los individuos descendientes de madres fructosa presentaron niveles prácticamente idénticos de expresión de CPT1, independientemente de la dieta que recibieron durante su juventud. Esto resulta llamativo teniendo en cuenta las diferentes tendencias de expresión de la enzima observadas en la descendencia de madres control como resultado de la dieta que consumieron en su vida postnatal. Este resultado podría indicar que, de nuevo, la ingesta materna de fructosa promueve la programación fetal insensibilizando a la descendencia ante los cambios en la expresión de la enzima CPT1 promovidos por factores nutrigenómicos, en este caso las dietas estudiadas. Por tanto, esta falta de respuesta a dietas potencialmente lipogénicas en la expresión de una enzima limitante de una vía tan importante del metabolismo lipídico como es la CPT1 podría promover la aparición de enfermedades metabólicas que cursen con esteatosis y/o dislipemia (65).
5.2.5. Relación entre niveles de SAH y metilación
Al medir los niveles de SAH en los machos jóvenes que consumieron fructosa + colesterol, no se obtuvieron cambios significativos entre grupos (Tabla XI). No obstante, a diferencia de lo observado en los hijos de madres control que consumieron fructosa, en los que se observó una tendencia hacia menores niveles de SAH, cuando la ingesta de dicho monosacárido se asoció a colesterol, se encontró la tendencia opuesta (Figura 16) y niveles similares a los encontrados en los machos control.
Tabla XI. ANOVA de doble vía de los resultados obtenidos en los niveles hepáticos de SAH en la descendencia macho. p: valor de probabilidad. Eta cuadrado (ŋ2): proporción de la varianza atribuida a la madre, a la dieta o a la interacción madre*dieta.
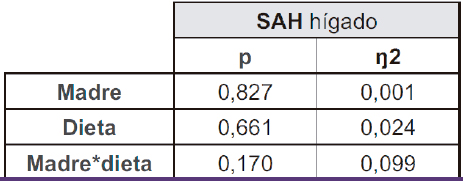
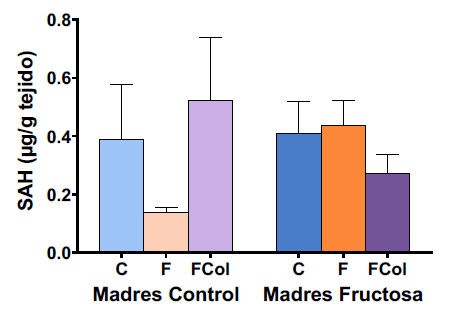
Figura 16. Niveles de SAH (μg/g) en hígado en machos F1 descendientes de madres control y madres fructosa tras consumir diferentes dietas durante 21 días (control: barras azules; fructosa líquida (10%): barras naranjas; o fructosa + col (2%): barras moradas). Los datos representan la media ± E.S. de 7-8 ratas. Las diferencias entre los factores y su interacción se analizaron utilizando ANOVA de doble vía con un test a posteriori de Bonferroni para detectar las diferencias significativas entre grupos.
Considerando la retroalimentación negativa que ejerce dicho metabolito contra las DNMTs, la existencia de elevados niveles de SAH es coherente con la menor tasa de metilación encontrada en la región 1 del promotor de CPT1 en los hijos de madres control que consumieron una dieta occidental. Por tanto, la actividad de DNMT3a y DNMT3b estaría disminuida de manera importante por la acción combinada de los mecanismos transcripcional, definido por una menor expresión génica, y de retroalimentación negativa de la actividad enzimática en este grupo. De la misma manera, la tendencia a disminuir los niveles de SAH en los descendientes de madres fructosa que tomaron la dieta occidental (Figura 16) podría ser la causante de la tendencia a una mayor metilación del promotor de CPT1 observada en ese mismo grupo, principalmente en la región 2 del mismo (Figura 14).
No obstante, considerando que SAM también es sustrato de Pemt y GNMT, es probable que los elevados niveles de SAH detectados en los descendientes de madres control que consumieron una dieta occidental también estuvieran ejerciendo una acción inhibitoria sobre la actividad de dichas enzimas. De hecho, la literatura contempla la relación SAM/SAH como uno de los principales reguladores de su actividad (46,66). Por tanto, la existencia de este potencial efecto inhibitorio sobre la actividad enzimática, junto a la disminución de su expresión génica observada, podría favorecer el desarrollo de patologías de diversa naturaleza, resultado de la disminución de los niveles de PC y sarcosina.
6. CONCLUSIONES
El consumo de fructosa por individuos jóvenes aumentó drásticamente la metilación del promotor de la CSE hepática en la descendencia de madres control, probablemente a través del aumento de la actividad de las DNMTs, como consecuencia de la disminución de los niveles de SAH. Este aumento no se observó tras el consumo de tagatosa, lo que implica que la alteración del metiloma y la consecuente disminución de la expresión de la CSE son dependientes del carbohidrato ingerido.
Sin embargo, la hipermetilación del promotor de la CSE no se observó en la descendencia de madres que consumieron fructosa durante la gestación, lo que indicaría que dicho monosacárido es capaz de anular la alteración del metiloma promovida por el consumo postnatal de fructosa a través del fenómeno de la programación fetal.
La ingesta postnatal de fructosa, cuando se asoció a colesterol, disminuyó la expresión hepática de DNMT3a y DNMT3b en los descendientes de madres control. Esta menor expresión, junto con una posible menor actividad de las DNMTs ejercida por los elevados niveles de SAH encontrados en este grupo experimental, estaría promoviendo una disminución de los niveles de metilación del promotor de CPT1 y, como resultado, un aumento de la expresión de esta enzima.
En los individuos descendientes de madres fructosa que consumieron una dieta occidental, la expresión de CPT1 resultó ser prácticamente idéntica entre grupos. Esto reafirma la hipótesis de que el consumo materno de fructosa promueve la programación fetal, insensibilizando a la descendencia ante los cambios en la expresión de la enzima CPT1 promovidos por factores nutrigenómicos. Esta falta de adaptación a cambios dietéticos en esta enzima del catabolismo de ácidos grasos podría potenciar el desarrollo de esteatosis y dislipemia en dichos individuos.
El consumo de una dieta occidental disminuyó significativamente la expresión de Pemt y GNMT, independientemente de la dieta recibida por la madre. Esto pone de manifiesto que la alteración de la metilación inducida por la dieta no es exclusiva de los promotores génicos, sino que también afecta a otros componentes celulares, en este caso el metabolismo de fosfolípidos y de glicina.
Listado de abreviaturas
#5-metil-THF: 5-metil-tetrahidrofolato
ADN: ácido desoxirribonucleico
ADNc: ADN complementario
AG: ácidos grasos
AHCY: SAH hidrolasa
ANOVA: análisis de Varianza
ARN: ácido ribonucleico
ATP III: Adult Treatment Panel III
BCP: bromocloropropano
BHMT: betaína-homocisteína metiltrasnsferasa
CAC: ciclo del ácido cítrico
CBS: cistationina beta sintasa
CC: machos de madres control-control
CF: machos de madres control-fructosa
CF+col: machos de madres control-dieta occidental
Cp: crossing point
CpG / CG: dinucleótido citosina – guanina
CT: machos de madres control-tagatosa
CPT1: carnitina palmitoiltransferasa 1
CSE: cistationina gamma liasa
DHAP: dihidroxiacetona
DNMTs: DNA-metiltransferasas
DNMT1: DNA-metiltransferasa 1
DNMT3a: DNA-metiltransferasa 3a
DNMT3b: DNA-metiltransferasa 3b
DOHAD: developmental origins of health and disease (teoría del desarrollo temprano de la salud y la enfermedad)
ECV: enfermedad cardiovascular
ES: error estándar
FC: machos de madres fructosa-control
FF: machos de madres fructosa-fructosa
FF+col: machos de madres fructosa-dieta occidental
FT: machos de madres fructosa-tagatosa
G3P: gliceraldehído-3-fosfato
GNMT: glicina-N-metiltransferasa
GR: glucocorticoides
GTP: guanosina trifosfato
H2S: sulfuro de hidrógeno
HDL: lipoproteínas de alta densidad
HFCS: High Fructose Corn Syrup (Jarabe de Maíz Rico en Fructosa)
LDL: lipoproteínas de baja densidad
LXRα receptor X hepático alfa
MAT1A: metionina adenosiltransferasa MTHFD1: metilen-THFdeshidrogenasa-1 MTHFR: metilen-THF-reductasa
MTR: metionina sintasa
MTRR: metionina sintasa reductasa
NCBI: National Center for Biotechnology Information
PC: fosfatidilcolina
PCR: reacción en cadena de la polimerasa
PE: fosfatidiletanolamina
Pemt: fosfatidiletanolamina N-metiltransferasa
PPARα: receptor nuclear activado por proliferadores peroxisómicos
Q-PCR: PCR a tiempo real
RT: transcriptasa inversa
SAH: S-adenosilhomocisteína
SAM: S-adenosilmetionina
SM: Síndrome Metabólico
THF: tetrahidrofolato TG: triglicéridos
VLDL: lipoproteínas de muy baja densidad
WD: Western Diet (Dieta Occidental)
7. REFERENCIAS
- Febbraio MA, Karin M. “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 2021;33(12):2316-2328.
- Zhang DM, Jiao RQ, Kong LD. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients. 2017;9(4):335.
- Tappy L, Lê KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90(1):23-46.
- Jung S, Bae H, Song WS, et al. Dietary Fructose and Fructose-Induced Pathologies. Annu Rev Nutr. 2022;42:45-66.
- Mirisola MG. The Nutriepigenome. Genes (Basel). 2023;14(11):1997.
- Dahlhoff C, Desmarchelier C, Sailer M, et al. Hepatic methionine homeostasis is conserved in C57BL/6N mice on high-fat diet despite major changes in hepatic one-carbon metabolism. PLoS One. 2013;8(3):e57387.
- Roy S, Chikkerur J, Roy SC, et al. Tagatose as a Potential Nutraceutical: Production, Properties, Biological Roles, and Applications. J Food Sci. 2018;83(11):2699-2709.
- Mayumi S, Kuboniwa M, Sakanaka A, et al. Potential of Prebiotic D-Tagatose for Prevention of Oral Disease. Front Cell Infect Microbiol. 2021;11:767944.
- Espinosa I, Fogelfeld L. Tagatose: from a sweetener to a new diabetic medication?. Expert Opin Investig Drugs. 2010;19(2):285-294.
- Malesza IJ, Malesza M, Walkowiak J, et al. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells. 2021;10(11):3164.
- Clemente-Suárez VJ, Beltrán-Velasco AI, Redondo-Flórez L, et al. Global Impacts of Western Diet and Its Effects on Metabolism and Health: A Narrative Review. Nutrients. 2023;15(12):2749.
- Giussani M, Lieti G, Orlando A, et al. Fructose Intake, Hypertension and Cardiometabolic Risk Factors in Children and Adolescents: From Pathophysiology to Clinical Aspects. A Narrative Review. Front Med. 2022;9:792949.
- Francula-Zaninovic S, Nola IA. Management of Measurable Variable Cardiovascular Disease’ Risk Factors. Curr Cardiol Rev. 2018;14(3):153-163.
- Causes of death statistics. [Internet]. Eurostat 2021. [Actualizado mar 2024; citado 24 may 2024]. Disponible en: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Causes_of_death_statistics#Major_causes_of_death_in_the_EU_in_2021
- Cho Y, Lee SY. Useful Biomarkers of Metabolic Syndrome. Int J Environ Res Public Health. 2022;19(22):15003.
- Bovolini A, Garcia J, Andrade MA, et al. Metabolic Syndrome Pathophysiology and Predisposing Factors. Int J Sports Med. 2021;42(3):199-214.
- Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2(5-6):231-237.
- Tran LT, Yuen VG, McNeill JH. The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol Cell Biochem. 2009;332(1-2):145-159.
- Hsieh CC, Liao CC, Liao YC, et al. Proteomic changes associated with metabolic syndrome in a fructose-fed rat model. J Food Drug Anal. 2016;24(4):754-761.
- Moreno-Fernández S, Garcés-Rimón M, Vera G, et al. High Fat/High Glucose Diet Induces Metabolic Syndrome in an Experimental Rat Model. Nutrients. 2018;10(10):1502.
- Moeller SM, Fryhofer SA, Osbahr AJ 3rd, et al. The effects of high fructose syrup. J Am Coll Nutr. 2009;28(6):619-626.
- Geidl-Flueck B, Hochuli M, Németh Á, et al. Fructose- and sucrose- but not glucose-sweetened beverages promote hepatic de novo lipogenesis: A randomized controlled trial. J Hepatol. 2021;75(1):46-54.
- Ferraris RP, Choe JY, Patel CR. Intestinal Absorption of Fructose. Annu Rev Nutr. 2018;38:41-67.
- Li L, Wang M, Ma Q, et al. Role of glycolysis in the development of atherosclerosis. Am J Physiol Cell Physiol. 2022;323(2):C617-C629.
- Merino B, Fernández-Díaz CM, Cózar-Castellano I, et al. Intestinal Fructose and Glucose Metabolism in Health and Disease. Nutrients. 2019;12(1):94.
- Tappy L. Fructose metabolism and noncommunicable diseases: recent findings and new research perspectives. Curr Opin Clin Nutr Metab Care. 2018;21(3):214-222.
- Mortera RR, Bains Y, Gugliucci A. Fructose at the crossroads of the metabolic syndrome and obesity epidemics. Front Biosci. 2019;24(2):186-211.
- Stanhope KL, Medici V, Bremer AA, et al. A dose-response study of consuming high-fructose corn syrup-sweetened beverages on lipid/lipoprotein risk factors for cardiovascular disease in young adults. Am J Clin Nutr. 2015;101(6):1144-1154.
- Bokor S, Csölle I, Felső R, et al. Dietary nutrients during gestation cause obesity and related metabolic changes by altering DNA methylation in the offspring. Front Endocrinol. 2024;15:1287255.
- Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261(5):412-417.
- Koletzko B, Godfrey KM, Poston L, et al. Nutrition During Pregnancy, Lactation and Early Childhood and its Implications for Maternal and Long-Term Child Health: The Early Nutrition Project Recommendations. Ann Nutr Metab. 2019;74(2):93-106.
- Ando Y, Yamada H, Munetsuna E, et al. Maternal high-fructose corn syrup consumption causes insulin resistance and hyperlipidemia in offspring via DNA methylation of the Pparα promoter region. J Nutr Biochem. 2022;103:108951.
- Rodrigo S, Fauste E, de la Cuesta M, et al. Maternal fructose induces gender-dependent changes in both LXRα promoter methylation and cholesterol metabolism in progeny. J Nutr Biochem. 2018;61:163-172.
- Kendig MD, Ekayanti W, Stewart H, et al. Metabolic Effects of Access to Sucrose Drink in Female Rats and Transmission of Some Effects to Their Offspring. PLoS One. 2015;10(7):e0131107.
- Munetsuna E, Yamazaki M, Ohashi K, et al. Epigenetics and fructose metabolism: A new mechanism of fructose effects (Chapter 21). In: Patel BV. Molecular Nutrition: Carbohydrates; 2019. 353-368.
- Moreno-Fernandez J, Ochoa JJ, Lopez-Frias M, et al. Impact of Early Nutrition, Physical Activity and Sleep on the Fetal Programming of Disease in the Pregnancy: A Narrative Review. Nutrients. 2020;12(12):3900.
- Meng H, Cao Y, Qin J, et al. DNA methylation, its mediators and genome integrity. Int J Biol Sci. 2015;11(5):604-617.
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010-1022.
- Sergeeva A, Davydova K, Perenkov A, Vedunova M. Mechanisms of human DNA methylation, alteration of methylation patterns in physiological processes and oncology. Gene.2023;875:147487.
- Sharma U, Rando OJ. Metabolic Inputs into the Epigenome. Cell Metab. 2017;25(3):544-558.
- Janke R, Dodson AE, Rine J. Metabolism and epigenetics. Annu Rev Cell Dev Biol. 2015;31:473-496.
- Chen C, Wang Z, Ding Y, et al. DNA Methylation: From Cancer Biology to Clinical Perspectives. Front Biosci. 2022;27(12):326.
- Li Y, Pollock CA, Saad S. Aberrant DNA Methylation Mediates the Transgenerational Risk of Metabolic and Chronic Disease Due to Maternal Obesity and Overnutrition. Genes. 2021;12(11):1653.
- Ohashi K, Munetsuna E, Yamada H, et al. High fructose consumption induces DNA methylation at PPARα and CPT1A promoter regions in the rat liver. Biochem Biophys Res Commun. 2015;468(1-2):185-189.
- Mizuno G, Yamada H, Munetsuna E, et al. High-fructose corn syrup intake increases hepatic mitochondrial DNA copy number and methylation in adolescent rats. Nutr Res. 2023;110:57-65.
- Vance DE. Physiological roles of phosphatidylethanolamine N-methyltransferase. Biochim Biophys Acta. 2013;1831(3):626-632.
- Li J, Xin Y, Li J, Chen H, Li H. Phosphatidylethanolamine N-methyltransferase: from Functions to Diseases. Aging Dis. 2023;14(3):879-891
- van der Veen JN, Kennelly JP, Wan S, et al. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim Biophys Acta Biomembr. 2017;1859(9 Pt B):1558-1572.
- Kim YC, Seok S, Byun S, et al. AhR and SHP regulate phosphatidylcholine and S-adenosylmethionine levels in the one-carbon cycle. Nat Commun. 2018;9(1):540.
- Johnson AA, Cuellar TL. Glycine and aging: Evidence and mechanisms. Ageing Res Rev. 2023;87:101922.
- Wang YC, Wu MT, Lin YJ, et al. Regulation of Folate-Mediated One-Carbon Metabolism by Glycine N-Methyltransferase (GNMT) and Methylenetetrahydrofolate Reductase (MTHFR). J Nutr Sci Vitaminol (Tokyo). 2015;61 Suppl:S148-S150. doi:10.3177/jnsv.61.S148
- Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47(4):1191-1199. doi:10.1002/hep.22159
- Clare CE, Brassington AH, Kwong WY, et al. One-Carbon Metabolism: Linking Nutritional Biochemistry to Epigenetic Programming of Long-Term Development. Annu Rev Anim Biosci. 2019;7:263-287. doi:10.1146/annurev-animal-020518-115206
- Mato JM, Martínez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol. 2013;12(2):183-189.
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res.2001;29(9):e45.
- Oakes CC, La Salle S, Robaire B, et al. Evaluation of a quantitative DNA methylation analysis technique using methylation-sensitive/dependent restriction enzymes and real-time PCR. Epigenetics. 2006;1(3):146-152.
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427-1431.
- Shang Y, Siow YL, Isaak CK, O K. Downregulation of Glutathione Biosynthesis Contributes to Oxidative Stress and Liver Dysfunction in Acute Kidney Injury. Oxid Med Cell Longev. 2016;2016:9707292.
- Loaeza-Loaeza J, Beltran AS, Hernández-Sotelo D. DNMTs and Impact of CpG Content, Transcription Factors, Consensus Motifs, lncRNAs, and Histone Marks on DNA Methylation. Genes (Basel). 2020;11(11):1336.
- Kabil O, Motl N, Banerjee R. H2S and its role in redox signaling. Biochim Biophys Acta. 2014;1844(8):1355-1366.
- Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587-590.
- Kolluru GK, Shackelford RE, Shen X, et al. Sulfide regulation of cardiovascular function in health and disease. Nat Rev Cardiol. 2023;20(2):109-125.
- Soda K. Polyamine Metabolism and Gene Methylation in Conjunction with One-Carbon Metabolism. Int J Mol Sci. 2018;19(10):3106.
- Schlaepfer IR, Joshi M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology. 2020;161(2):bqz046.
- Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C(2):77-85.
- Vryer R, Saffery R. Metabolic regulation of DNA methylation in mammalian cells (Chapter 19). En: Tollefsbol TG (edit). Handbook of epigenetics. The new molecular and medical genetics; 2017. 293-305.
