1. INTRODUCCIÓN
1.1. La fructosa
La fructosa es un monosacárido de seis carbonos, clasificado estructuralmente como una cetohexosa, que se encuentra de forma natural en las frutas, la miel y ciertas verduras. Junto a la glucosa, ha sido comúnmente utilizada como fuente de azúcares añadidos, principalmente en bebidas azucaradas. En el contexto de la dieta occidental, también conocida como Western diet, su presencia se ha visto notablemente incrementada por el consumo de azúcar de mesa (sacarosa), un disacárido compuesto por glucosa y fructosa, y de jarabe de maíz rico en fructosa (HFCS, del inglés: High-Fructose Corn Syrup). Dicho jarabe se obtiene por procesado industrial del almidón de maíz a través de una hidrólisis enzimática del almidón y posterior isomerización de parte de la glucosa obtenida a fructosa. Se comercializa en diferentes formatos que contienen proporciones variables de glucosa y fructosa, siendo el más comúnmente usado el HFCS-55, el cual contiene 55% de fructosa y 45% de glucosa en forma libre. Debido a su bajo coste, mayor solubilidad en agua y alto poder edulcorante, el HFCS ha sido ampliamente utilizado por la industria alimentaria en la elaboración de alimentos procesados (1).
En consecuencia, la ingesta de fructosa en la población en forma de HFCS ha experimentado un aumento acusado desde finales de los años 70, alcanzando un valor máximo a finales de los 90, comparado con otros tipos de azúcares (Figura 1) (2). Esta tendencia ha suscitado una creciente preocupación en la comunidad científica, ya que múltiples trabajos de investigación han encontrado evidencias sólidas, tanto en estudios realizados en humanos como en animales de experimentación, que relacionan la ingesta de fructosa con el desarrollo del síndrome metabólico. El síndrome metabólico se ha convertido en uno de los principales problemas de salud pública en el siglo XXI y es un cuadro clínico que se diagnostica cuando un paciente presenta tres de las siguientes cinco alteraciones: hipertensión, obesidad abdominal, hiperglucemia en ayunas, hipertrigliceridemia y bajos niveles plasmáticos de colesterol en lipoproteínas de alta densidad (HDL). El síndrome metabólico aumenta el riesgo de padecer enfermedades cardiovasculares, obesidad, diabetes tipo 2 y enfermedad del hígado graso no alcohólico (NAFLD) (3,4). Estas enfermedades están englobadas dentro de lo que se conoce como enfermedades no transmisibles (ENT), una de las principales causas de muerte a nivel global, tanto en países desarrollados como en vías de desarrollo. Aunque estas enfermedades tienen cierto componente genético, una dieta pobre en alimentos de origen vegetal y marcada por los patrones de dieta occidental es un nexo causal en el aumento en la incidencia de ENT crónicas (5).
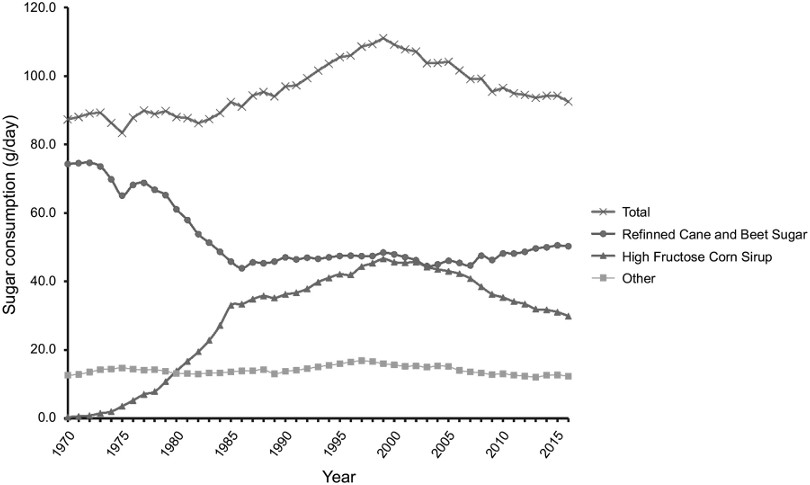
Figura 1. Evolución del consumo per cápita (g/día) de jarabe de maíz de alta fructosa en comparación con otros tipos de azúcares utilizados en la industria en EE. UU. desde 1970 a 2016 (2).
Aunque la fructosa y la glucosa son isómeros estructurales, tienen idéntico peso molecular, fórmula química y aportan el mismo valor energético (4 kcal/g); ambas moléculas presentan diferencias significativas a nivel fisiológico y metabólico (Figura 2) (1). La fructosa tiene un poder edulcorante mayor que la glucosa, 1.7 frente a 0.75, y tiene un índice glucémico menor, 23 frente a 100, respectivamente. Además, tanto su absorción intestinal como su metabolismo hepático son completamente diferentes. A nivel intestinal, el transporte de glucosa es un proceso que requiere energía y está mediado por el cotransportador de sodio-glucosa 1 (SLGT1, del inglés: sodium-glucose linked transporter), mientras que la entrada de la fructosa en el enterocito se realiza a través de un transporte pasivo facilitado mediado por el transportador de glucosa GLUT5 (6). En lo que respecta a su metabolismo, la glucosa es metabolizada a través de la glucólisis por la mayoría de los tejidos del organismo y esta ruta está controlada, entre otros factores, por la insulina. En cambio, la fructosa se metaboliza predominantemente en el hígado, intestino y riñón en un proceso conocido como fructólisis. Sorprendentemente, el metabolismo de la fructosa no estimula la secreción de insulina o leptina, dos de las hormonas más importantes que regulan el metabolismo energético y el apetito, siendo este uno de los factores clave en el desarrollo del síndrome metabólico (1).
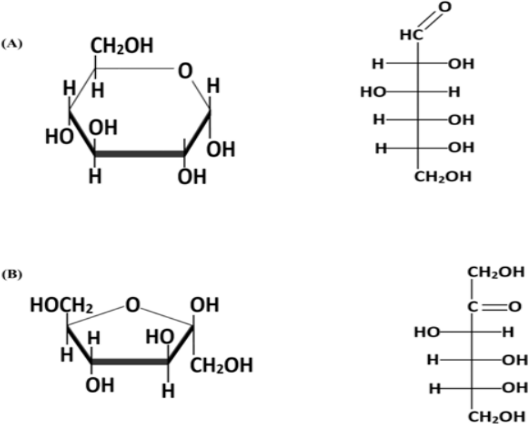
Figura 2. Estructuras de la glucosa (A) y la fructosa (B) (1)
1.2. Metabolismo y absorción de la fructosa
Tras su ingesta, la fructosa es absorbida en el enterocito mediante un sistema de transporte facilitado mediado por GLUT5. De aquí, pasa a la circulación sistémica a través del transportador GLUT2 (figura 3) (6). Estudios experimentales llevados a cabo en ratones evidencian cómo la completa eliminación del transportador GLUT5 produce una malabsorción de la fructosa, dando lugar a la producción de gases y otros trastornos intestinales. A pesar de que el transportador GLUT2 presenta menos afinidad por la fructosa (Km = 11 mM) que GLUT5 (Km = 6 mM), es probable que tenga mayor relevancia en su captación a nivel hepático, ya que GLUT5 apenas se expresa en dicho tejido (7). A diferencia de la glucosa, que se distribuye ampliamente por los tejidos periféricos bajo control de la insulina, la fructosa es captada casi en su totalidad por el hígado, donde se metaboliza el 70% de la fructosa de forma rápida y sin necesidad de respuesta de la insulina (1,5). La expresión del transportador GLUT5 está altamente regulada por la proteína de unión al elemento respuesta de carbohidratos (ChREBP, del inglés: Carbohydrate Responsive-Element Binding Protein), que a su vez se activa por medio de metabolitos secundarios de la fructólisis. Además, ChREBP también induce la expresión de genes involucrados en la lipogénesis, favoreciendo la síntesis y acumulación de ácidos grasos en el hígado y en otros órganos como el páncreas o el intestino delgado (3,4). Por tanto, se ha comprobado que un mayor consumo de fructosa potenciará sus efectos perjudiciales en el organismo al inducir la expresión de GLUT5, lo que favorece su propia absorción intestinal.
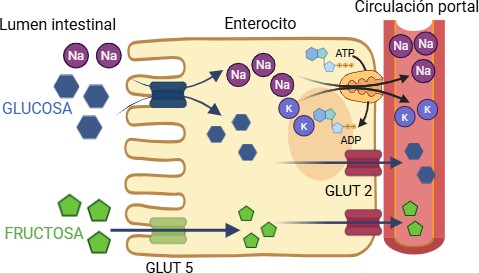
Figura 3. Transporte intestinal de glucosa y fructosa. Paso hacia el interior del enterocito a través de SGLT1 (sodium-glucose linked transporter 1) y GLUT5, respectivamente, y su posterior paso al torrente sanguíneo a través de GLUT2. Imagen realizada con Biorender (www.biorender.com).
Es a nivel hepático donde existen las principales diferencias entre ambos monosacáridos. La glucosa en el hígado entra en la glucólisis, donde es fosforilada a glucosa-6-fosfato por la enzima glucoquinasa y, posteriormente, la fosfofructoquinasa-1 la transforma en fructosa 1,6-bisfosfato (figura 4). Ambas reacciones están estrictamente reguladas, de tal manera que la glucoquinasa es activada por la insulina, mientras que la fosfofructoquinasa-1 presenta una retroalimentación negativa por producto (ATP y citrato). En cambio, en el hígado la fructosa es metabolizada a través de una ruta conocida como fructólisis (figura 4). A través de esta ruta, la fructosa es fosforilada a fructosa-1-fosfato por la enzima cetohexoquinasa (KHK), para posteriormente ser escindida en dos productos: la dihidroxiacetona fosfato y el gliceraldehído, a través de una reacción catalizada por la aldolasa B.
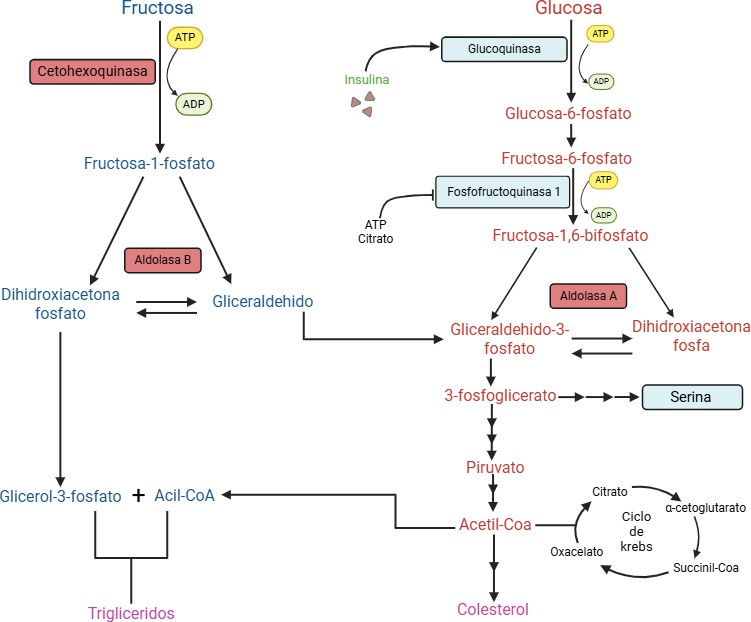
Figura 4. Metabolismo hepático de la glucosa y la fructosa. Imagen realizada con Biorender (www.biorender.com).
Finalmente, ambos compuestos se pueden transformar en gliceraldehído 3-fosfato, que se incorpora a la glucólisis, en una etapa posterior a las catalizadas por la glucoquinasa y la fosfofructoquinasa 1, saltándose así dos de los puntos claves de regulación de la glucólisis. A diferencia de estas dos enzimas claves, la KHK no presenta ningún mecanismo de control y, de hecho, su expresión y actividad está estimulada por la presencia de fructosa. Como resultado, el consumo excesivo de fructosa conduce a una producción descontrolada de triosas fosfato que conduce a una mayor formación de acetil coenzima A (CoA) y glicerol-3-fosfato, precursores esenciales para la síntesis de ácidos grasos y triglicéridos (3).
Este proceso no está exento de consecuencias metabólicas. De esta manera, el gasto excesivo de adenosín trifosfato (ATP) para la fosforilación de la fructosa por la KHK conduce a una depleción de ATP y a un aumento de los niveles de adenosín difosfato (ADP). Este ADP es transformado en adenosín monofosfato (AMP) por la AMP desaminasa (AMPD) y, posteriormente, a ácido úrico por la vía de las purinas. La hiperuricemia resultante tiene graves consecuencias metabólicas; entre ellas, promueve el estrés oxidativo, disminuye la cantidad de óxido nítrico y provoca disfunción endotelial. Estas alteraciones están implicadas en el mecanismo patogénico de la hipertensión arterial y la lesión renal (3,5).
Diversos autores, entre ellos Herman y Birnbaum, han demostrado cómo el metabolismo de la fructosa promueve una mayor secreción hepática de lipoproteínas de muy baja densidad (VLDL) ricas en triglicéridos, incrementando así el riesgo de dislipidemia y de acumulación de grasa visceral (3). Por otro lado, la fructosa, al alterar la señal hormonal de la leptina e insulina, provoca un desequilibrio energético, ya que no estimula adecuadamente las respuestas homeostáticas que controlan la saciedad (3,4).
Por tanto, el metabolismo específico de la fructosa, caracterizado por su rápida conversión hepática en lípidos, elevada producción de ácido úrico y su deficiente regulación hormonal, constituye un factor determinante en el desarrollo del síndrome metabólico y sus patologías asociadas. Esta singularidad explica su mayor impacto en comparación con otros azúcares y justifica la creciente preocupación por su papel en la fisiopatología de enfermedades metabólicas y cardiovasculares.
1.3. Patologías asociadas al consumo de fructosa
El consumo excesivo de fructosa se ha relacionado de forma directa con múltiples alteraciones características del síndrome metabólico, tales como la obesidad, la hipertensión arterial, la resistencia a la insulina, la hipertrigliceridemia, las enfermedades cardiovasculares y el estrés oxidativo, siendo el hígado el órgano más afectado debido a su papel central en el metabolismo de este azúcar (1,3).
Numerosos estudios han demostrado que una elevada ingesta de fructosa induce la activación de la lipogénesis de novo, lo que conlleva una acumulación de triglicéridos en el hígado. Esta acumulación promueve el desarrollo de dislipidemia, caracterizada por un aumento de VLDL, un descenso del colesterol en HDL y un aumento de las lipoproteínas de baja densidad (LDL) pequeñas y densas, altamente aterogénicas. Estos cambios se asocian con un mayor riesgo de enfermedad cardiovascular y aterosclerosis en sujetos con alto consumo de azúcares añadidos, particularmente fructosa (1,4).
Además del impacto lipídico, el metabolismo hepático de la fructosa promueve la producción de ácido úrico. Tal y como se ha detallado en el apartado de metabolismo de la fructosa, la conversión de fructosa en fructosa-1-fosfato genera como subproducto el ADP, que a su vez se transforma en AMP. El AMP puede sufrir una desaminación dando lugar a inosina monofosfato (IMP), o bien una fosforilación, transformándose en inosina. Ambas moléculas son catabolizadas a hipoxantina y posteriormente a xantina, que por medio de la xantina oxidasa se convierte en ácido úrico (3). La hiperuricemia provocada por la fructosa se ha relacionado con la aparición de resistencia a la insulina, inflamación sistémica y estrés oxidativo (6). Estos efectos son especialmente preocupantes en individuos con predisposición a enfermedades metabólicas como obesidad o diabetes tipo 2.
Por otra parte, se ha demostrado que el consumo de fructosa favorece la acumulación de grasa visceral, ya que, como hemos comentado anteriormente, su metabolismo se salta dos de los puntos clave de regulación de la glucólisis, fomentando la producción descontrolada de triosas fosfato y su posterior conversión en ácidos grasos y triglicéridos (3,7). Además, se ha descrito que la fructosa activa los factores de transcripción ChREBP y la proteína de unión al elemento respuesta de esteroles tipo 1c (SREBP-1c, del inglés: Sterol response element binding protein 1c); ambos dos promueven la expresión de genes involucrados en la lipogénesis. A su vez, también se ha observado cómo dietas ricas en fructosa disminuyen la expresión del receptor activado por proliferadores de peroxisomas alfa (PPARα, del inglés Peroxisome proliferator-activated receptor alpha), que a su vez favorece la oxidación de ácidos grasos. Por tanto, una mayor síntesis de ácidos grasos y triglicéridos, sumada a su reducido catabolismo, favorece una mayor liberación de VLDL al torrente sanguíneo, generando así hipertrigliceridemia (8).
En conclusión, las investigaciones llevadas a cabo hasta el momento respaldan que el consumo elevado de fructosa provoca cambios desfavorables en el metabolismo lipídico y energético, con graves consecuencias sobre la función hepática y la salud cardiovascular. Estos efectos metabólicos adversos son especialmente relevantes en situaciones de una alta ingesta de forma prolongada, como ocurre en poblaciones expuestas donde abunda el consumo de bebidas azucaradas y productos ultraprocesados (3).
1.4. Epigenética y programación fetal
Según Bird, la epigenética es “la adaptación estructural de regiones cromosómicas con el fin de registrar, señalar o perpetuar estados de actividad alterados” (9). Otros autores la han descrito como la ciencia que estudia los cambios producidos en la expresión de los genes que son hereditarios, y que no se deben a alteraciones en la secuencia genética del ácido desoxirribonucleico (ADN) (10). Muchas enfermedades metabólicas, como la diabetes tipo 2, la obesidad o el hígado graso no alcohólico, están sujetas a cambios epigenéticos, de los cuales se pueden distinguir tres principales mecanismos reguladores, que son: la metilación del ADN, la modificación de las histonas y los microácidos ribonucleicos (miARN) (11). Todos ellos juegan un papel clave en la regulación de la diferenciación celular, la expresión génica específica de cada célula, la impronta parental, la inactivación del cromosoma X, así como la estabilidad y estructura genómicas (12).
El embarazo representa una etapa de alta vulnerabilidad metabólica, tanto para la madre como para el feto. Durante la gestación, se producen numerosos cambios fisiológicos y metabólicos destinados a asegurar el adecuado desarrollo fetal, entre ellos una mayor sensibilidad a estímulos nutricionales. La salud física y mental, el estilo de dieta, así como el ambiente al que se vea expuesta la madre durante el embarazo, tendrán un papel crucial que marcará la salud metabólica del feto, tanto en sus primeros años de vida como en la edad adulta. Alteraciones endocrinas, agentes infecciosos o la biodisponibilidad de nutrientes son algunos de los factores claves más importantes involucrados en la programación fetal. Según Marciniak y colaboradores, “la programación fetal tiene lugar cuando el entorno óptimo en el que crece el feto se ve alterado por factores hostiles, especialmente durante los períodos críticos del desarrollo de los órganos esenciales” (13).
Además, varios estudios experimentales con modelos animales han demostrado que cambios metabólicos desencadenados durante los periodos prenatal y postnatal están estrechamente relacionados con el desarrollo de alteraciones metabólicas durante la edad adulta (14). Siguiendo este modelo, se cree que el consumo de dietas que contienen fructosa por parte de la madre ejerce un efecto adverso sobre la programación metabólica del feto que podría desencadenar enfermedades metabólicas a largo plazo (15).
Todas estas modificaciones se transmiten de generación en generación a través de mecanismos epigenéticos, definidos como la serie de alteraciones en la expresión génica, sin afectar a la secuencia de ADN. Algunos de los mecanismos mejor descritos son la metilación del ADN, la acetilación de histonas y los ARN no codificantes. Estas modificaciones son hereditarias y reversibles, y alteran la estructura y condensación de la cromatina, afectando de esta manera a la expresión del fenotipo (16).
Las modificaciones epigenéticas pueden alterar la morfología de los órganos del feto y comprometer el funcionamiento normal de las células (16). Se ha comprobado que la malnutrición y la sobrealimentación son perjudiciales para el desarrollo normal del feto. Por un lado, un déficit nutricional durante la gestación puede derivar en malformaciones en el feto que están directamente relacionadas con el desarrollo del síndrome metabólico en la edad adulta. Por otro lado, la descendencia de madres con problemas de obesidad o diabetes tipo 2 es más propensa a padecer obesidad en los primeros años de vida o en la edad adulta (17).
Así pues, el estudio de los efectos de la ingesta de fructosa en el embarazo es un campo de creciente interés, no solo por su importancia clínica inmediata en la propia gestación, sino también por su trascendencia para la salud de la descendencia.
Los principales mecanismos epigenéticos incluyen la metilación del ADN, que consiste en la adición de grupos metilo a residuos de citosina (a menudo en islas CpG en regiones promotoras), regulando la expresión génica (18). Este proceso es catalizado por ADN metiltransferasas (DNMT) que utilizan S-adenosilmetionina (SAM) como donante de metilos, y su alteración es clave en la embriogénesis (19). Otro mecanismo es la modificación de histonas, las proteínas que empaquetan el ADN en nucleosomas. La acetilación de histonas por enzimas HAT relaja la cromatina, favoreciendo la transcripción, mientras que las HDAC la compactan, reprimiendo la expresión génica (20). Finalmente, los ARN no codificantes (ARNnc) son moléculas de ARN que no se traducen a proteínas, pero regulan la expresión génica (21). Estos incluyen los microARN (miARN), que inhiben la traducción o degradan los ARNm diana, y los ARN no codificantes largos (ARNlnc) y circulares (ARNcirc), que modulan la transcripción y la estabilidad de otros ARN (22).
1.4.1 Fructosa durante el embarazo
Múltiples trabajos científicos han demostrado que el consumo excesivo de fructosa en madres gestantes, tanto en forma de sacarosa o HFCS, puede tener efectos perjudiciales en la descendencia adulta, tales como el desarrollo de obesidad, resistencia a la insulina e hipertensión (1,5). En este proceso, la placenta desempeña una función crítica, ya que actúa como órgano regulador para el transporte de nutrientes de la madre al feto, por lo que su correcto funcionamiento es primordial para el desarrollo óptimo del feto (23). Estudios realizados en modelos animales han puesto de manifiesto la importancia de la placenta en la salud de la descendencia. Estos trabajos han demostrado que las placentas de madres expuestas a un consumo elevado de fructosa mostraban un mayor estrés oxidativo, además de una mayor producción de ácido úrico y alteraciones en los transportadores de glucosa (24,25,26).
Pese a que se ha demostrado la correlación entre el consumo de fructosa y el desarrollo de enfermedades cardiovasculares o la obesidad, su consumo no está contraindicado en mujeres embarazadas (15). Datos experimentales han evidenciado que esta ingesta podría producir modificaciones en el desarrollo perinatal del feto, afectando a la glucemia, insulinemia, lipidemia y a las vías de señalización de la leptina. Así, se ha observado cómo la descendencia de madres alimentadas con dietas ricas en fructosa presenta dislipidemia y una mayor síntesis hepática de ácidos grasos (27).
Otros autores han demostrado que un consumo elevado de fructosa durante la gestación puede provocar modificaciones epigenéticas persistentes en la descendencia. Un ejemplo bien documentado es la alteración en la metilación del promotor del gen LXRα (receptor X hepático alfa), un receptor nuclear clave del metabolismo lipídico y del colesterol (15). En estudios realizados con ratas, se observó que los machos descendientes de madres alimentadas con dietas ricas en fructosa presentaban una hipermetilación en la región promotora del gen LXRα, lo que se asoció con una menor expresión del receptor y sus genes diana, como el receptor SR-B1 (del inglés: Scavenger receptor class B type 1), el receptor ABCG5 (del inglés: ATP Binding Cassette Subfamily G Member 5) y el colesterol 7-alfa hidroxilasa (citocromo p450 tipo 7 A1, CYP7A1). En el grupo de las descendientes hembra sucedió lo contrario, tanto en la metilación del promotor de LXRa como en la expresión del gen y sus genes diana. Todo esto sugiere la existencia de diferencias dependientes del sexo en la programación epigenética inducida por la fructosa. También se observó que en la descendencia macho los niveles de ácido fólico, implicado en el metabolismo de un carbono, aumentaron y lo opuesto sucedió en las hembras. Estos datos ponen de manifiesto que el estado de metilación de la región promotora del gen de LXRα puede estar relacionado con la disponibilidad de folato, clave para la síntesis de nucleótidos, la regeneración de la metionina y la metilación del ADN (15).
1.5. Metabolismo de la serina
La L-serina es un aminoácido de vital importancia en la homeostasis celular. La principal fuente de este aminoácido son las proteínas, donde su contenido puede variar entre un 2-5%. A nivel endógeno, se puede sintetizar a partir del 3-fosfoglicerato, un intermediario de la glucólisis, y de la glicina.
Aunque no es un aminoácido esencial en el sentido estricto de la palabra, se ha descrito como “condicionalmente esencial” durante situaciones de gran necesidad metabólica o estrés fisiológico, es decir, en una situación de enfermedad inflamatoria, cáncer o enfermedad infecciosa (24). Funcionalmente, la L-serina participa en la biosíntesis de nucleótidos y lípidos de membrana, así como en la de los aminoácidos glicina y cisteína, y en la síntesis de glutatión, molécula que actúa como regulador del estado redox de la célula (28,29).
El primer paso en la síntesis de serina consiste en la oxidación del 3-fosfoglicerato (3PG), un intermediario de la glucólisis, a 3-fosfohidroxipiruvato (3-PHP) utilizando como coenzima NAD+, que se reduce a su vez a NADH+H+, a través de una reacción catalizada por la 3-fosfoglicerato deshidrogenasa (PHGDH), enzima limitante y reguladora de la ruta. El NADH formado podría utilizarse como dador de electrones en la cadena respiratoria para generar energía, conectando así esta ruta con el metabolismo energético (28). En segundo lugar, el 3-fosfohidroxipiruvato reacciona con glutamato y experimentan una transaminación que da lugar a 3-fosfoserina y alfa-cetoglutarato (aKG), en un proceso catalizado por la 3-fosfoserina aminotransferasa (PSAT1). Esta etapa constituye una reacción anaplerótica del ácido cítrico, dado que genera alfa-cetoglutarato, que es un intermediario de dicho ciclo. Finalmente, se produce la hidrólisis irreversible del grupo fosfato por medio de la fosfoserina fosfatasa (PSPH), dando lugar a L-serina y la liberación de un grupo fosfato inorgánico.
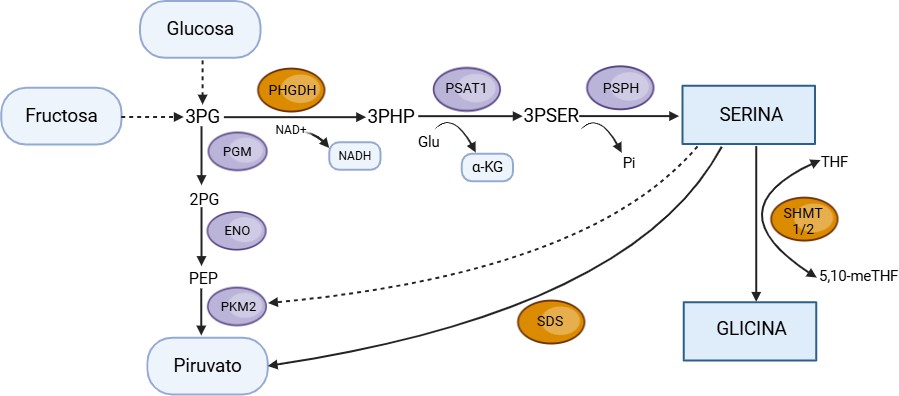
Figura 5. Representación esquemática del metabolismo de la serina a partir de 3-fosfoglicerato en el hígado. 2-PG: 2-fosfoglicerato; 3PG: 3-fosfoglicerato; 3PHP: 3-fosfohidroxipiruvato; 3PSER: 3-fosfoserina; α-KG: α-cetoglutarato; NADH: dinucleótido de nicotinamida adenina; PEP: fosfoenolpiruvato; PHGDH: 3-fosfoglicerato deshidrogenasa; PKM2: piruvato quinasa M2; ENO: enolasa; PSAT1: 3-fosfoserina aminotransferasa 1; PSPH: fosfoserina fosfatasa; SDS: serina deshidrogenasa; SHMT1: serina hidroximetiltransferasa 1; SHMT2: serina hidroximetiltransferasa 2; THF: tetrahidrofolato; 5,10-meTHF: 5,10-metilentetrahidrofolato. Imagen realizada con Biorender (www.biorender.com).
La L-serina es también un intermediario crítico con una influencia clara en la regulación epigenética. De hecho, está directamente implicada en el metabolismo de un carbono a través de la vía del folato/metionina. La conversión de serina en glicina mediante la acción de la serina hidroximetiltransferasa (SHMT) produce el 5,10-metilentetrahidrofolato (5,10-CH₂-THF) a partir del tetrahidrofolato (THF), reponiendo la reserva de un carbono (28). El 5,10-CH₂-THF es crucial para la síntesis de purinas, pirimidinas y la formación de 5-metiltetrahidrofolato (5-CH₃-THF), que posteriormente cede su grupo metilo a la homocisteína, dando lugar a metionina, en una reacción catalizada por la metionina sintasa. A continuación, la metionina producida se metila para formar S-adenosilmetionina (SAM), el principal donante de grupos metilo en la metilación del ADN, el ARN y las proteínas, integrando así el metabolismo de la serina con la regulación epigenética (28,29). Por otro lado, la homocisteína también puede servir de precursora para la síntesis de glutatión.
La serina ejerce un papel importante en el desarrollo de las células tumorales, dado que su metabolismo genera multitud de precursores necesarios para mantener el elevado grado de división celular que caracteriza a este tipo de células.
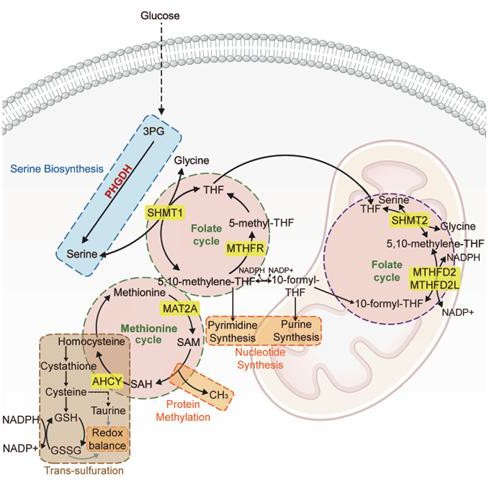
Figura 6. Interconexión de la biosíntesis de serina con el ciclo del folato-metionina y el metabolismo de un carbono. Imagen extraída de (28).
Desde el punto de vista fisiológico, la serina también regula procesos inmuno-metabólicos implicados en la homeostasis pulmonar, especialmente en células epiteliales y macrófagos. Se ha descrito que, en presencia de agentes infecciosos, la serina promueve la biosíntesis de lípidos y la producción mitocondrial de ATP, mecanismos esenciales para preservar la integridad de la barrera epitelial y la respuesta inmune innata frente a patógenos respiratorios (30).
1.5.1 Transporte de la serina
Durante el periodo perinatal, la placenta ejerce una función esencial en el desarrollo del feto, ya que actúa como intermediario en el transporte de nutrientes entre la madre y el feto. Entre las moléculas que son transportadas de la madre al feto a través de la placenta se encuentran los aminoácidos. Este proceso de transferencia está altamente regulado e incluye 2 etapas: en primer lugar, los nutrientes son captados por las microvellosidades (MVM) de la membrana plasmática del sincitiotrofoblasto a partir de la sangre materna, y posteriormente, son liberados al sistema circulatorio del feto a través de la membrana basal (31).
El sincitiotrofoblasto es la principal barrera para la transferencia de nutrientes entre la madre y el feto. En primer lugar, los aminoácidos deben atravesar la membrana plasmática de las microvellosidades (MVM) orientada a la madre y, posteriormente, la membrana basal orientada al feto. Además, en el sincitiotrofoblasto los aminoácidos pueden sufrir transformaciones, pudiendo ser convertidos en nuevos aminoácidos (Figura 7).
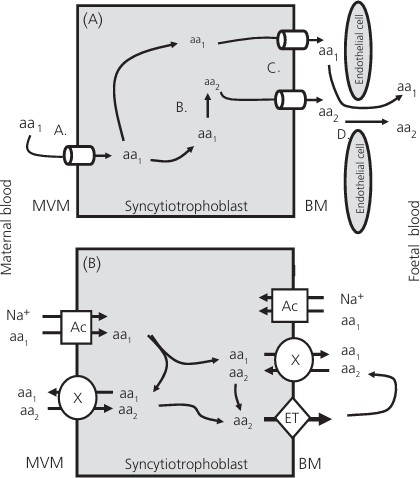
Figura 7. Mecanismos de transporte de aminoácidos. A: Los aminoácidos (aa) de la sangre materna son captados por transportadores de la membrana apical del sincitiotrofoblasto placentario. B: Dentro del sincitiotrofoblasto, los aminoácidos se metabolizan para producir nuevos aminoácidos. C: Los aminoácidos son transportados a través de la membrana basal por proteínas transportadoras. D: Los aminoácidos se difunden en la sangre fetal a través de las fenestraciones del endotelio capilar. (B) Los aminoácidos son captados por transportadores acumulativos (Ac) o intercambiadores (X) (31).
Se ha visto que la concentración de aminoácidos es mayor en el plasma de los fetos que en el de la madre, lo que indica que el transporte de aminoácidos a través de la placenta se da de manera activa (31).
Existen tres sistemas para el transporte de aminoácidos a través de la placenta, clasificados en función de su especificidad y mecanismo de acción. Estos incluyen el sistema A, dentro del cual se incluyen los transportadores de aminoácidos neutros acoplados a sodio (SNAT, del inglés: sodium-coupled neutral amino acid transporters) 1 (SNAT1), 2 (SNAT2) y 4 (SNAT4). Se trata de un sistema activo dependiente de sodio, específico para aminoácidos pequeños y neutros, como la serina. Está estrechamente relacionado con el suministro de precursores metabólicos al feto, tanto es así que se ha observado que una disminución de su actividad provoca una restricción del crecimiento intrauterino (RCIU) (32). Por otro lado, se encuentra el sistema L, el cual es un intercambiador independiente de sodio involucrado en el transporte de aminoácidos neutros de cadena larga ramificada, como la leucina o la isoleucina. Dichos aminoácidos son esenciales en la síntesis proteica y señalización celular, activando la vía mTOR (del inglés: mammalian target of Rapamycin), responsable de regular la proliferación celular. Por último, el sistema ASC incluye los transportadores alanina-serina-cisteína (ASC) 1 (ASCT1) y 2 (ASCT2). También transporta serina y otros aminoácidos neutros de cadena corta, como la cisteína y la alanina. Actúa como un transportador facilitador, promoviendo el equilibrio homeostático intracelular de estos aminoácidos (32,33). Como ya hemos comentado anteriormente, aunque la serina es considerada un aminoácido no esencial, es clave en el desarrollo del feto, ya que actúa en la síntesis de fosfolípidos, nucleótidos, glutatión y componentes del sistema nervioso central (33). Además de la capacidad de la placenta de sintetizar serina de novo a partir del intermediario glucolítico 3-fosfoglicerato, hay evidencias de que también puede ser transportada de manera activa desde la circulación materna al feto, particularmente en situaciones en las que la demanda fetal sobrepase la capacidad biosintética placentaria (31).
Algunos estudios han demostrado que, en situaciones adversas exhibidas durante el embarazo, como, por ejemplo, la restricción del crecimiento intrauterino (RCIU), el funcionamiento normal del sistema A se ve alterado, lo que provoca una disminución del transporte de serina y otros aminoácidos esenciales de la placenta al feto (32). Asimismo, hábitos poco saludables de la madre como fumar, la inactividad física, la depresión, el estrés o desórdenes neurológicos pueden modular negativamente la programación fetal del neonato, además de afectar a la función placentaria y, por consiguiente, a la biodisponibilidad de nutrientes para el adecuado desarrollo del feto (13).
2. HIPÓTESIS
Con la evidencia científica expuesta anteriormente, en el presente trabajo nos planteamos la hipótesis de que el consumo materno de fructosa durante la gestación puede afectar al metabolismo de la serina y glicina, lo que podría alterar sus niveles plasmáticos, tanto en las madres gestantes como en sus fetos.
Para corroborar esta hipótesis, planteamos los siguientes objetivos.
3. OBJETIVOS
3.1. Objetivo general
El principal objetivo de este trabajo experimental es estudiar el impacto del consumo materno de fructosa durante la gestación sobre la expresión de los genes involucrados en la biosíntesis de serina, su transformación en glicina y su conversión a piruvato en ratas gestantes y evaluar si este consumo materno pudiera afectar a la descendencia hembra cuando está gestante y a sus fetos.
3.2. Objetivos específicos
Para alcanzar el objetivo general, proponemos los siguientes objetivos específicos:
Estudiar el efecto de la ingesta de fructosa durante la gestación en el metabolismo de la serina en el hígado de ratas gestantes.
Estudiar el metabolismo de la serina en el hígado de gestantes descendientes de madres-fructosa expuestas o no a este carbohidrato durante su propia gestación.
Evaluar el metabolismo y transporte de la serina en la placenta de las ratas descendientes de madres con fructosa, como principal órgano encargado del transporte de nutrientes de la madre al feto, a través del estudio de la expresión génica de los transportadores implicados.
Estudiar el metabolismo de la serina en el hígado de los fetos con el fin de evaluar si existe un efecto de transmisión intergeneracional de las alteraciones metabólicas asociadas al consumo de fructosa durante la gestación.
4. Materiales y métodos
4.1. Diseño experimental
Se utilizaron muestras de hígado y placenta de rata Sprague-Dawley. Los protocolos de manejo animal fueron revisados y aprobados por un Comité de Ética de Investigación institucional, siguiendo las directrices para el cuidado de animales de experimentación.
Se llevaron a cabo dos diseños experimentales:
Diseño experimental 1:
Ratas hembra Sprague-Dawley se cruzaron con machos y, a lo largo de toda la gestación, se les permitió el acceso libre a agua sin aditivo (grupo Control) o bien agua suplementada con un 10% de fructosa (p/v) (grupo Fructosa) o 10% de glucosa (grupo Glucosa). Los tres grupos experimentales fueron los siguientes:
Gestantes F0 que consumieron agua durante la gestación (Control, C).
Gestantes F0 que consumieron fructosa 10% (p/v) durante la gestación (Fructosa, F)
Gestantes F0 que consumieron glucosa 10% (p/v) durante la gestación (Glucosa, G)
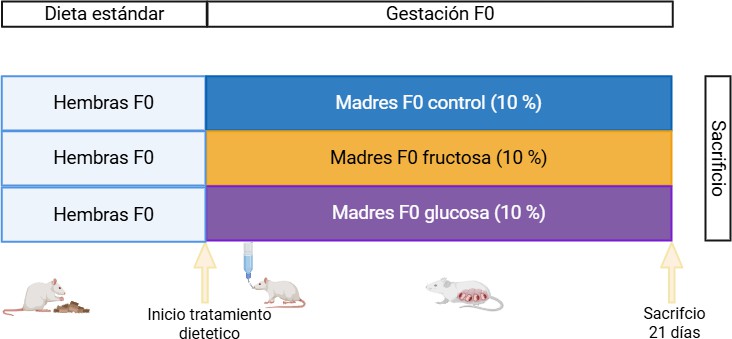
Figura 8. Diseño experimental del procedimiento 1. Control: Ratas gestantes que consumieron agua durante la gestación. Fructosa: Ratas gestantes cuyas madres consumieron fructosa (10 %) durante la gestación. Glucosa: Ratas gestantes cuyas madres consumieron glucosa (10 %) durante la gestación. Imagen realizada con Biorender (www.biorender.com).
Diseño experimental 2:
Ratas hembra Sprague-Dawley se cruzaron con machos y a lo largo de toda la gestación, se les permitió el acceso libre a agua sin aditivo o bien agua suplementada con un 10% de fructosa (p/v) (generación F0). Tras parir, se realizó un ajuste de las camadas a 9 crías y se les administró agua sin aditivo durante la lactancia. Cuando las crías alcanzaron los 20 días de edad, se destetaron y se les dejó crecer, permitiéndoles el acceso a agua sin aditivo (generación F1). Las hembras F1 a las 8 semanas de edad se cruzaron con machos controles y se dividieron, tal y como se describe en la figura 9, creándose así tres grupos experimentales:
Gestantes F1 cuyas madres consumieron agua durante la gestación y ellas consumieron agua durante su propia gestación (Control-Control, CC).
Gestantes F1 cuyas madres consumieron fructosa 10% (p/v) durante la gestación y ellas consumieron agua sin aditivo durante su gestación (Fructosa-Control, FC).
Gestantes F1 cuyas madres consumieron fructosa 10% (p/v) durante la gestación y ellas consumieron fructosa 10% (p/v) durante su gestación (Fructosa-Fructosa, FF).
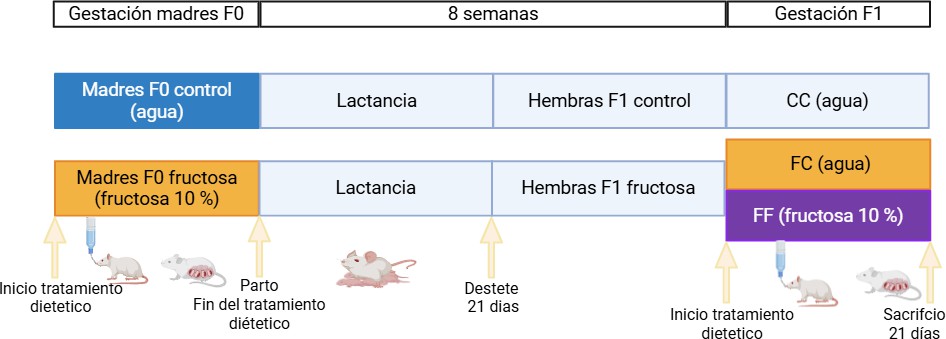
Figura 9. Diseño experimental del procedimiento 2. CC: Las ratas gestantes y sus madres consumieron agua durante la gestación. FC: Ratas gestantes cuyas madres consumieron fructosa (10 %) durante la gestación y ellas tomaron agua sin aditivo durante la gestación. FF: Ratas gestantes cuyas madres consumieron fructosa (10 %) durante la gestación y ellas tomaron fructosa. Imagen realizada con Biorender (www.biorender.com).
Las gestantes (5 hembras por grupo experimental) fueron sacrificadas en el día 21 de gestación, y se recogió el hígado y las placentas. Los fetos se pesaron y fueron sacrificados para obtener el hígado. En este caso, se llevó a cabo un pool de los plasmas e hígados de fetos procedentes de la misma madre, que fueron almacenados a -80 ºC.
4.2. Estudio de la expresión génica del ARNm
4.2.1. Homogenización de los tejidos y extracción del ARN total
4.2.1. Homogenización de los tejidos y extracción del ARN total
Para la extracción del ácido ribonucleico total (ARN) se utilizó el kit RibopureTM (Invitrogen, ThermoFisher, EE. UU.). Para ello, se homogeneizaron 100 mg de hígado materno o fetal en un TissueLyser LT (Qiagen, EE. UU.) con TRI Reagent® (Invitrogen, ThermoFisher, EE. UU.) que, gracias al fenol y al tiocianato de guanidina, inactiva las nucleasas y lisa las células. A continuación, las muestras se dejaron reposar durante 5 minutos a temperatura ambiente para descomponer los complejos nucleoproteicos. Posteriormente, el homogeneizado se centrifugó a 16.000 xg durante 1.º minuto a 4 ºC. Seguido, se añadió bromocloropropano (Invitrogen, ThermoFisher, EE. UU.) al sobrenadante, lo que provocó la separación de ARN en la fase acuosa superior, quedando las proteínas y el ADN en la interfase y en la fase orgánica inferior. Para ello, las muestras se centrifugaron de nuevo a 16.000 xg y 4 °C durante 10 minutos. La fase superior, donde se encuentra el ARN, se recogió para proceder a la purificación final de este último.
4.2.2. Purificación del ARN
La purificación final del ARN se realizó por unión de este a unos filtros de fibra de vidrio y, tras repetidos lavados y su posterior elución con un tampón bajo en sales. Brevemente, 600 µL procedentes de la extracción del ARN del apartado anterior, se transfirieron a un cartucho con filtro de kit RibopureTM (Invitrogen, ThermoFisher, EE. UU.) previamente insertado en un tubo Eppendorf limpio. A continuación, se centrifugaron los tubos a 13.500 xg y temperatura ambiente durante 30 segundos y se desechó el líquido filtrado, ya que el ARN permanece unido al filtro del cartucho. Después, se añadieron 500 µl de solución de lavado (Invitrogen, ThermoFisher, EE. UU.) al filtro y se centrifugaron los tubos en las mismas condiciones descritas, deshaciéndonos de nuevo del filtrado. El lavado se repitió dos veces para eliminar totalmente las impurezas. A continuación, los cartuchos se transfirieron a un tubo nuevo y se añadieron 100 µl de tampón de elución (Invitrogen, ThermoFisher, EE. UU.). Se incubaron a temperatura ambiente durante 2 minutos para solubilizar el ARN extraído en el tampón de elución y, después, se centrifugaron a 12.000 xg y a temperatura ambiente durante 30 segundos. El filtrado obtenido contenía el ARN total de la muestra.
4.2.3. Cuantificación, pureza e integridad del ARN
Para determinar la concentración y pureza del ARN total obtenido, se utilizó un NanoDrop® One (ThermoFisher, EE. UU.). La pureza se obtiene a partir de las medidas de absorbancia a 260/280 nm (que nos da información sobre las posibles impurezas proteicas) y 260/230 nm (que nos indica si han quedado impurezas del disolvente orgánico). Cuanto más se acerquen estos dos cocientes a 2,0, mayor será la pureza del RNA obtenido. Tomamos nuestras muestras como válidas si su relación era de 1,8-2,0. Todas las mediciones se realizaron por duplicado.
Para comprobar la integridad del ARN, se realizó una electroforesis en un gel de agarosa. Para ello, se preparó un gel de agarosa al 2 % teñido con Red SafeTM (iNtRON Biotechnology, Corea del Sur), se cargó con 500 ng de ARN y se dejó a 90 V durante 20 minutos para la separación del ARN. Las bandas respectivas de ARN, 18s y 28s (principales ARN ribosomales de la célula), se observaron bien diferenciadas, lo que nos aseguró que el ARN estaba íntegro y no se había degradado (Figura 10).
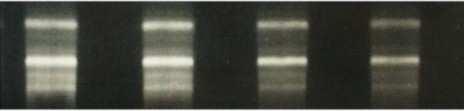
Figura 10. Gel de agarosa para verificar la integridad del ARN extraído. Tamaño esperado para el producto de amplificación: 236 pares de bases. Marcador molecular cuya primera banda es de 100 pb.
4.2.4. Tratamiento con DNasa
Después de la extracción con sales de guanidino y solventes orgánicos, las muestras fueron tratadas con un tratamiento riguroso con el kit Turbo DNA-freeTM (Invitrogen, ThermoFisher, EEUU), para eliminar las posibles trazas de ADN contaminante que podrían interferir con los ensayos realizados posteriormente. Brevemente, a 10 µg del ARN obtenido se le añadió 3 µL de 10X Turbo DNase Buffer, 3 µL de Turbo DNase (2 U/L) y agua libre de RNasas hasta un volumen final de 30 µL. Tras incubar la mezcla a 37 ºC durante 30 minutos, la DNasa fue inactivada añadiendo 6 µl de DNase Inactivation Reagent e incubando la mezcla a temperatura ambiente 2 minutos. Los tubos se centrifugaron a temperatura ambiente a 16.000 g durante 2 minutos para eliminar el reactivo de inactivación. El sobrenadante, que contiene el ARN libre de ADN, se recogió y se trasvasó a tubos nuevos, y finalmente se cuantificó la concentración de ARN siguiendo el mismo protocolo que en el apartado anterior.
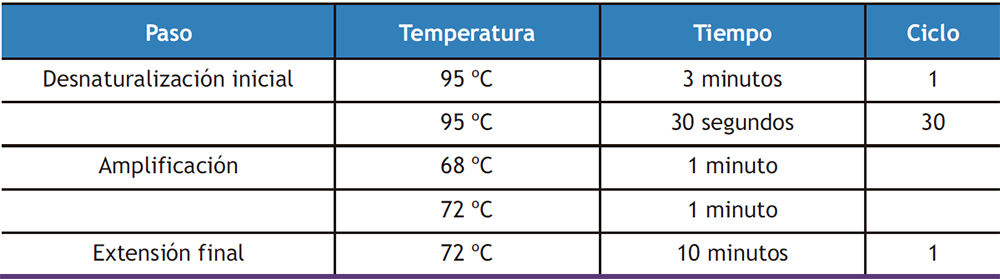
Para asegurarnos de que las muestras no estaban contaminadas con ADN, se llevó a cabo una reacción en cadena de la polimerasa (PCR). Si la muestra se encuentra completamente libre de ADN, la polimerasa no va a amplificar las regiones del gen de referencia RPS29 delimitadas por sus cebadores. Para asegurar que la PCR se había desarrollado correctamente, se procesó al mismo tiempo un control positivo (muestra de ADN complementario 5 ng/µL) junto con las muestras y un control negativo (agua destilada). Para llevar a cabo la PCR se utilizaron 125 ng de ARN a los que se les adicionó 12,5 µL de NZYTaq II 2x Green Master mix (NZYTech, Portugal), 2 µL de cebadores F y R de RPS29 (10 µM) y agua destilada hasta un volumen final de 25 µL. NZYTaq II 2X Green Master Mix (NZYTec, Portugal) es una mezcla lista para su uso que contiene Taq polimerasa, dNTPs, tampón para la PCR, MgCl2 y tampón de carga (con 2 colorantes, uno azul y otro amarillo). Las condiciones de la PCR se especifican en la Tabla 1 y fueron realizadas en un termociclador iCycler (Bio-Rad, California, EEUU).
Tabla 1. Protocolo de la PCR de verificación para el gen RPS29
Tras la realización de la PCR, las muestras se cargaron en un gel de agarosa al 2 % teñido con Red Safe TM (iNtRON Biotechnology, Corea del Sur) y fueron sometidas a electroforesis a 120 V. Los resultados se visualizaron en un transiluminador UVldoc System con cámara incorporada (Uvitec, Reino Unido), utilizando luz ultravioleta a 312 nm. La completa eliminación del ADN se aseguró con la ausencia de banda del producto de PCR en las muestras (Figura 11).

Figura 11. Gel de verificación de la eliminación completa de ADN contaminante.
4.2.5. Retrotranscripción del ARNm
Para poder realizar los estudios de expresión génica, es necesario que el ARN mensajero (ARNm) se retrotranscriba a ADN complementario (ADNc), y para ello se utilizó el kit SuperScript® II Reverse Transcriptase (Invitrogen, EE. UU.). Brevemente, se llevó a cabo el siguiente protocolo: a 2,5 ng de ARN tratado con DNasa se le adicionó 1 µL de oligo d(T) 70 µM (Sigma Chemical Co., EE. UU.) y agua ultrapura estéril hasta un volumen final de 12 µL. La mezcla se mantuvo a 72 ºC durante 10 minutos en un termociclador iCycler (Bio-Rad, EE. UU.). Posteriormente, se adicionaron 4 µl de First Buffer 5x (Invitrogen, EE. UU.), 2 µl de DTT (ditiotreitol) 0.1 M (Invitrogen, EE. UU.) y 1 µl de dNTPs 40 µM (Sigma Chemical Co., EE. UU.). La mezcla se incubó a 42 ºC durante 2 minutos en el termociclador. Finalmente, se adicionó 1 µL de transcriptasa inversa 100 U/µL (Invitrogen, Estados Unidos) y se mantuvo a 42 ºC durante 50 minutos, seguido de 15 minutos a 72 ºC para obtener el ADNc.
Una vez obtenido el ADNc, se llevó a cabo la comprobación de la funcionalidad de dicho ADNc mediante PCR, siguiendo el mismo protocolo que en el apartado anterior. En este caso, se debe obtener una banda correspondiente al gen de referencia RPS29 para asegurarnos de que las muestras de ADNc son funcionales.
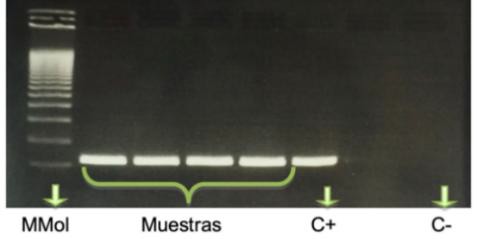
Figura 12. Gel de verificación de la funcionalidad del ADNc obtenido. Tamaño esperado del producto de amplificación RPS29: 236 pb. Marcador molecular (M Mol) cuya primera banda es de 100 pb.
4.2.6. Estudio de la expresión génica por PCR a tiempo real
La PCR a tiempo real es una técnica cualitativa y cuantitativa, que permite detectar en tiempo real la amplificación de una región específica del ADN según se va produciendo. Esto se consigue gracias a una molécula fluorescente, el SYBR-Green I, que se intercala en la doble hélice de ADN, aumentando unas 1.000 veces su fluorescencia cuando está unida a este (Figura 13). La cantidad de fluorescencia emitida será directamente proporcional al producto amplificado.
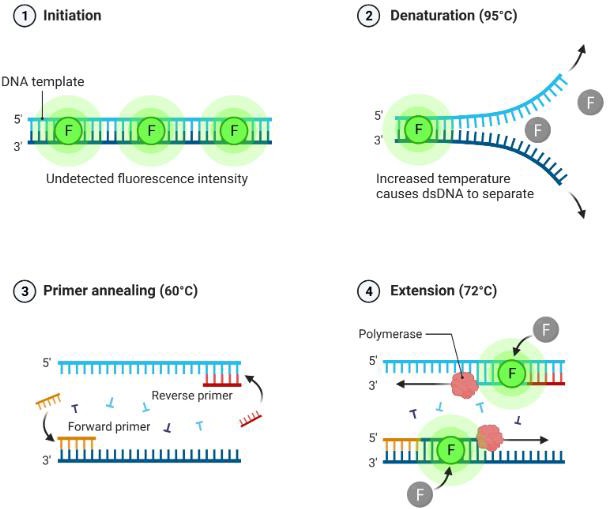
Figura 13. Proceso de amplificación y detección por fluorescencia del producto de amplificación mediante la unión del SYBR-Green I. Imagen realizada con Biorender (www.biorender.com).
El ciclo de amplificación consiste en 3 fases: background o fondo, exponencial y meseta. El proceso se basa en la medición del ciclo umbral (Cp), que es el ciclo en el que la señal de fluorescencia excede un umbral definido. Este valor Cp se correlaciona inversamente con la cantidad inicial del gen de interés; es decir, cuanto más bajo sea el valor de Cp, mayor será la expresión de ese gen en la muestra (34). Además, la eficiencia de la reacción y la linealidad del rango de detección se determinan mediante la construcción de una recta de calibrado (Figura 14), que se obtiene al realizar diluciones seriadas 1/10 de una muestra patrón del ADNc diana (35). La recta de calibrado obtenida relaciona el valor Cp de cada dilución con el logaritmo de la cantidad de ADN inicial. Utilizaremos la recta para obtener la eficiencia de los cebadores utilizados y para medir la expresión de los genes de interés.
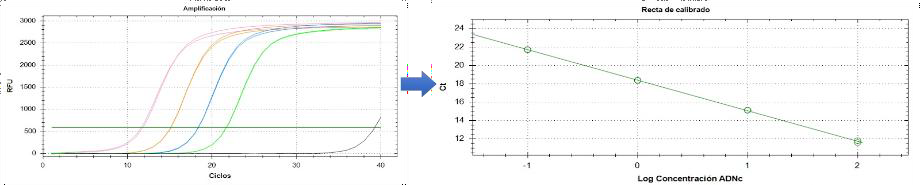
Figura 14. Curva de amplificación y recta de calibrado de una qPCR
4.2.6.1. Protocolo experimental de la PCR a tiempo real
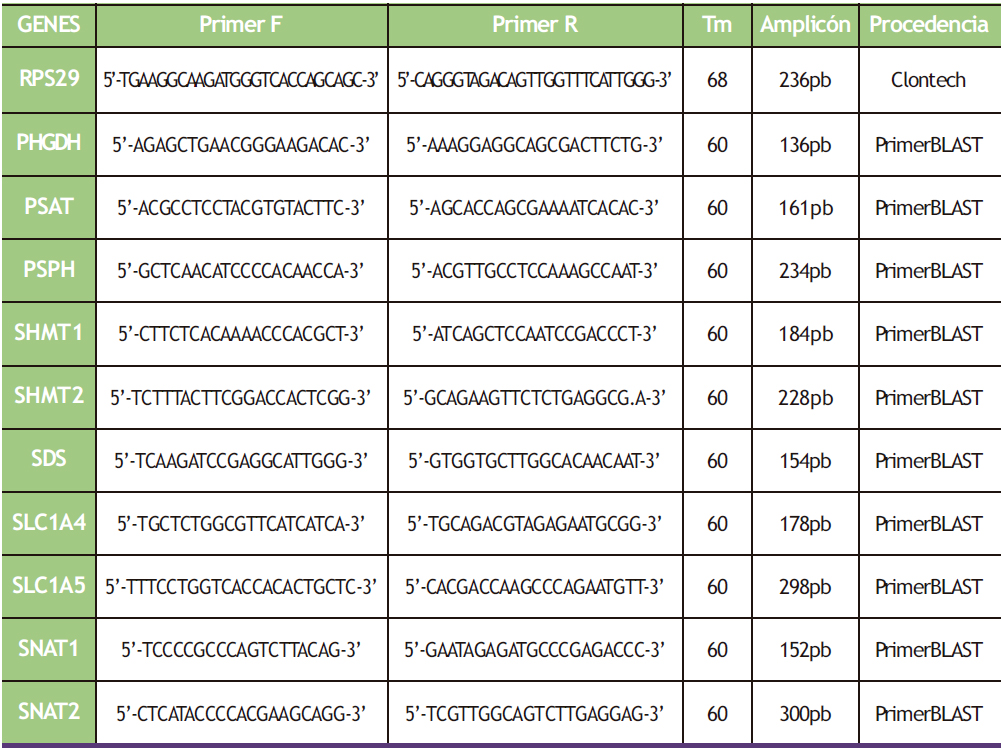
Para realizar la PCR a tiempo real se utilizó un termociclador de PCR a tiempo real CXF96® Touch (Bio-Rad, California, EE. UU.) y los cebadores que se detallan en la tabla 3 (IDT Technologies, EE. UU.). Los volúmenes de reactivos que se utilizaron en cada muestra fueron 10 µL de SYBR Premix Ex Taq (Takara Bio Inc., Japón), 1 µL de cebador F 10 µM (IDT Technologies, EE. UU.), 1 µL de cebador R 10 µM (IDT Technologies, EE. UU.), 2 µL de muestra de ADNc y 6 µL de agua mili-Q estéril. Las muestras de hígado y placenta fueron diluidas 1/30 para asegurarnos de que los valores Cp de las distintas muestras para todos los genes estuvieran dentro de la recta de calibrado (Cps < 30 ciclos), ya que, si no, hay mayor probabilidad de encontrar productos de amplificación inespecíficos, y por tanto la amplificación pierde eficiencia. Para hacer las diluciones se utilizó un ADNc con una concentración de 5 ng/µL.
Los parámetros programados para cada gen se obtuvieron de la base de datos de secuencias de cebadores RT-PCR Primer Sequence for Atlas Nylon Arrays (Clontech, EEUU) o de la aplicación para el diseño de cebadores Primer BLAST. El protocolo llevado a cabo para la amplificación de cada gen en el CXF96®, viene descrito en la tabla 2.
Tabla 2. Protocolo de PCR a tiempo real general utilizando para los estudios de expresión génica el CXF96
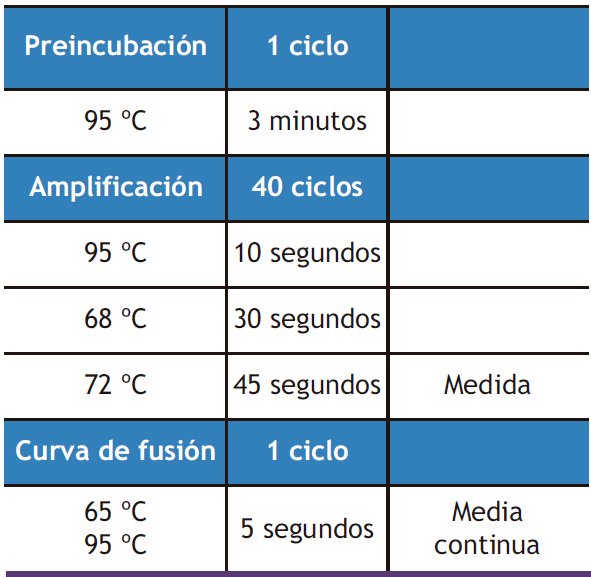
Tabla 3. Cebadores utilizados de cada gen específicos para rata.
4.2.7. Análisis estadístico
Los ensayos se realizaron con 5 animales por grupo. Los efectos del tratamiento se analizaron mediante análisis unidireccional de la varianza (ANOVA). Cuando estas condiciones resultaron estadísticamente significativas (p < 0,05), las diferencias se analizaron mediante el test de Tukey, utilizando el programa informático SPSS (versión 15). Cuando la varianza no era homogénea, se USÓ el test post hoc de Tamhane. Los resultados se han expresado como media ± error estándar. Cuando las diferencias entre los grupos fueron estadísticamente significativas (p < 0.05), se indicaron con diferentes letras.
5. Resultados y discusión
5.1. Efecto del consumo de fructosa en el metabolismo hepático de la serina en gestantes generación F0
Esta parte del estudio se realizó con el fin de averiguar si el consumo de fructosa o glucosa durante la gestación produce cambios en la expresión de los genes implicados en la síntesis de serina (PHGDH), su transformación a piruvato (SDS) o su interconversión en glicina (SHMT1).
El 3-fosfoglicerato es un intermediario de la glucólisis y sirve como precursor para la ruta de síntesis de la serina. La fructólisis y la glucólisis convergen en la formación de gliceraldehído-3-fosfato, que posteriormente es transformado a 3-fosfoglicerato. Por ello, el consumo de fructosa o glucosa podría alterar la expresión de la enzima PHGDH, que cataliza la etapa limitante de la ruta metabólica de síntesis de serina (28).
Como se puede ver en la figura 15, se observan diferencias significativas en la expresión de las tres enzimas en las ratas gestantes que consumieron ambos carbohidratos durante su gestación, respecto al grupo control.
En primer lugar, tanto el consumo de fructosa como el de glucosa en ratas gestantes condujo a un aumento significativo en la expresión de la enzima PHGDH (Figura 15A), por lo que se deduce que ambos azúcares están activando la ruta de la síntesis de serina durante la gestación. En cambio, la expresión de la enzima SDS que cataliza la conversión de serina en piruvato, el cual puede ser destinado a la gluconeogénesis (29), transformarse a alanina o lactato, o servir como precursor en la síntesis de ácidos grasos o colesterol (36), disminuye significativamente cuando se consume fructosa (Figura 15B). Estos resultados ponen de manifiesto que la ingesta de fructosa en ratas gestantes induce una mayor producción de serina y reduce su conversión a piruvato, por lo que cabría esperar un incremento en los niveles de serina. Sorprendentemente, en el grupo de ratas gestantes suplementadas con glucosa, se observa solamente una tendencia a una reducción en la expresión de SDS respecto al grupo control (figura 15B), por lo que es posible que el equilibrio serina/piruvato estuviera más controlado en este caso.
Por último, se evaluó la expresión del gen SHMT1, que es la enzima responsable de convertir serina en glicina en el citoplasma de la célula, y también de aportar una unidad de carbono al ciclo de ácido fólico (28). Como puede verse en la figura 15C, su expresión aumenta significativamente en las ratas gestantes que han consumido fructosa y glucosa. Estos datos implican una mayor interconversión de serina en glicina, lo que es de gran interés, ya que la glicina es un aminoácido indispensable para el correcto desarrollo del feto. De hecho, otros autores como Rivas-Ramírez et al. demostraron que la administración de glicina mejoró el desarrollo fetal y revirtió los efectos nocivos de la hiperglucemia en un modelo de embriopatía diabética (37).
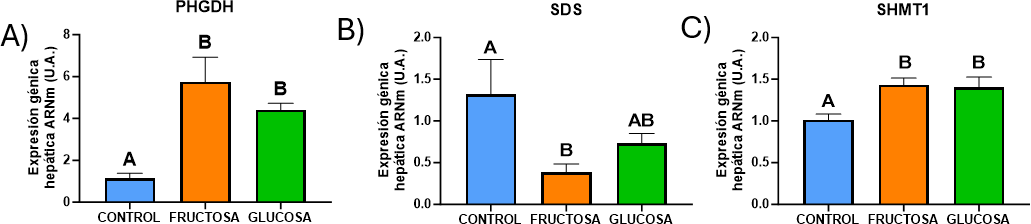
Figura 15. Expresión génica (ARNm) de las enzimas PHGDH (A), SDS (B), SHMT1 (C) en hígados de hembras gestantes descendientes de madres control (azul), madres-fructosa tras consumir durante 21 días agua con un 10% (p/v) de fructosa (naranja) y madres-glucosa con un 10% (p/v) de glucosa (verde). Los datos representan la media ± E.S. de 5 ratas. Las letras diferentes indican diferencias estadísticamente significativas entre hembras tratadas con diferente dieta.
Por otro lado, apoyando los efectos observados tras la ingesta de glucosa en el presente trabajo, Zhou et al. revelaron que el aumento en el consumo de glucosa activa la expresión de la enzima PHGDH, promoviendo así la síntesis de serina a partir de intermediarios glucolíticos (38). Sin embargo, no existen estudios, bajo nuestro conocimiento, que hayan evaluado previamente cómo el consumo de fructosa afecta a esta ruta metabólica.
Como hemos comentado en el apartado del metabolismo de la serina, la ruta de producción de serina funciona también como ruta anaplerótica, dado que permite la producción de α-cetoglutarato, un intermediario del ciclo del ácido cítrico, el cual se puede utilizar como precursor de múltiples compuestos, promoviendo así la proliferación celular y la biosíntesis de macromoléculas como triglicéridos y colesterol (36). Esto resulta de gran interés, ya que se ha observado que, en muchos procesos oncogénicos, el metabolismo alterado del α-KG conduce a hipermetilación del ADN y de histonas. Esto podría contribuir al silenciamiento de genes supresores de tumores, promoviendo así la progresión del tumor y la resistencia a tratamientos (39).
De acuerdo a los resultados anteriormente expuestos, observamos en la figura 16 cómo el consumo de fructosa aumenta significativamente los niveles de triglicéridos, siguiendo una tendencia muy similar a la observada en la expresión de la enzima PHGDH (figura 15A). Por tanto, estos datos sugieren que la ingesta de fructosa durante la gestación induce una mayor producción de serina que se convierte en glicina, la cual, debido a su interconexión con la ruta del metabolismo de un carbono (28), estaría proporcionando más sustratos para la síntesis de macromoléculas.
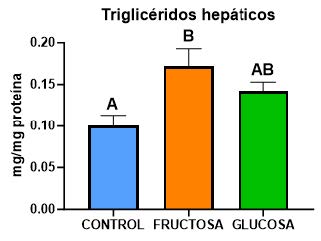
Figura 16. Niveles de triglicéridos hepáticos (mg/mg proteína) en hígados de hembras gestantes descendientes de madres control (azul), madres-fructosa tras consumir durante 21 días agua con un 10% (p/v) de fructosa (naranja) y madres-glucosa con un 10% (p/v) de glucosa (verde). Las letras diferentes indican diferencias estadísticamente significativas entre hembras tratadas con diferente dieta.
5.2. Efecto de la reexposición a fructosa durante la gestación en descendientes de madres-fructosa (generación F1): metabolismo hepático y placentario y transporte de serina de la madre al feto.
Una vez evaluado cómo el consumo de fructosa durante la gestación conduce a modificaciones en la expresión génica de diferentes genes implicados en la síntesis de serina, transformación en piruvato e interconversión en glicina (Figura 15), nos planteamos conocer si dichos cambios pudieran tener alguna repercusión a nivel de programación fetal en la descendencia hembra gestante.
5.2.1. Metabolismo de la serina en el hígado de gestantes de la generación F1
En primer lugar, se llevó a cabo un estudio de la expresión génica hepática de todos los genes involucrados en la síntesis de serina (Figura 17). Los datos obtenidos muestran un aumento en la expresión de la PHGDH en las descendientes F1 de madres expuestas a fructosa durante su gestación, que alcanza valores significativos en el grupo FF (Figura 17A). Estos datos sugieren un efecto de programación fetal, a través del cual la ingesta materna de fructosa induce en sus descendientes, una vez gestantes, la activación de la síntesis de serina. Sorprendentemente, este efecto se potencia cuando a las descendientes se les reexpone a fructosa durante su propia gestación (grupo FF).
En las otras dos enzimas de la ruta, la expresión de la PSAT1 (Figura 17B) no se ve alterada en los distintos grupos estudiados, y en la PSPH (Figura 17C) se observa una tendencia a una mayor expresión en los grupos FC y FF. Pese a que la expresión de la PSAT1 y la PSPH no cambie significativamente en los grupos FC y FF en comparación con el grupo control, al estar la PHGDH sobreexpresada, y siendo esta la enzima limitante de la ruta, sería lógico esperar que la concentración de serina se vea aumentada en el hígado de estos animales (29).
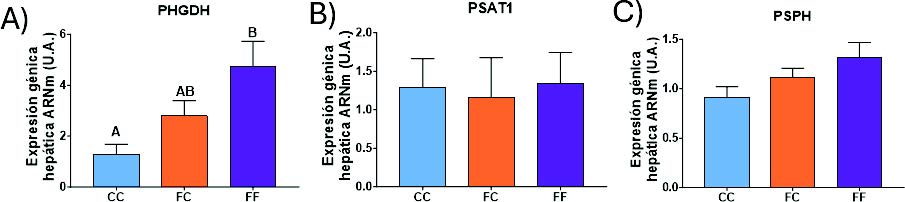
Figura 17. Expresión génica (ARNm) de las enzimas PHGDH (A), PSAT1 (B), PSPH (C) en hígados de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada). Las letras diferentes indican diferencias estadísticamente significativas entre hembras tratadas con diferente dieta.
Por otro lado, se evaluó la transformación de serina en piruvato a través de la enzima SDS. Como puede verse en la Figura 18, la SDS muestra una tendencia a una menor expresión en los grupos de descendientes de madres-fructosa (FC y FF), siendo esta reducción más acusada en el grupo FF.
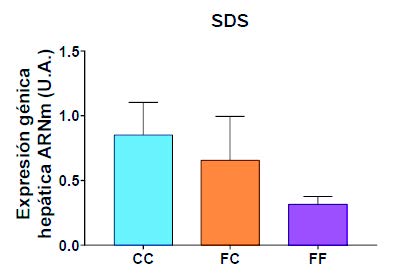
Figura 18. Expresión génica (ARNm) de la enzima SDS en hígados de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
Todos estos resultados podrían indicarnos un proceso de programación fetal a través del cual el consumo materno de fructosa promueve una mayor síntesis de serina en las descendientes hembra gestantes (Figura 17) y una posible mayor acumulación de esta en el hígado, ya que se observa una tendencia a una menor transformación de la serina en piruvato (Figura 18). Todos estos efectos se ven potenciados al reexponer a fructosa a las gestantes descendientes de madres-fructosa. Algunos autores han revelado cómo la hiperactivación de la biosíntesis de serina impulsa el desarrollo del cáncer, además de elevar la producción de glutatión, que ayuda a mantener el equilibrio redox, favoreciendo así la homeostasis tumoral (40).
A continuación, se determinó la expresión génica de la SHMT1 y SHMT2, enzimas implicadas en la interconversión de serina en glicina. A diferencia de la SHMT1, la enzima SHMT2 cataliza la reacción en las mitocondrias (Figura 6) (28). Sorprendentemente, no se encontraron diferencias significativas en la expresión de las enzimas SHMT1 (Figura 19A) ni SHMT2 (Figura 19B) en los distintos grupos experimentales estudiados. Aunque en el presente trabajo no hemos podido determinar los niveles hepáticos de serina ni de glicina, al existir una sobreexpresión de la PHGDH, enzima clave en la síntesis de serina, cabría esperar unos mayores niveles de serina, que por una mayor disponibilidad de sustrato desplazaría la reacción hacia una mayor interconversión a glicina. Meléndez-Hevia y de Paz-Lugo demostraron que cada mol de serina convertido genera exactamente un mol de glicina, de modo que cualquier incremento en la concentración de serina se traduce en un aumento equimolar de glicina (41).
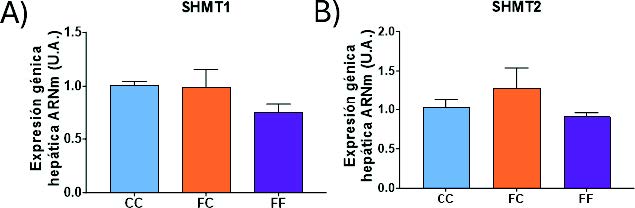
Figura 19. Expresión génica (ARNm) de las enzimas SHMT1 (A), SHMT2 (B) en hígados de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
Como hemos mencionado en el apartado anterior, la SHMT1 y SHMT2 son un punto de conexión entre el metabolismo de la serina y el metabolismo de un carbono (Figura 6). La MTHFR y la MTHFD2 son las dos enzimas limitantes de esta ruta (38). La primera actúa en el citoplasma y reduce el 5,10-metilentetrahidrofolato a 5-metil-tetrahidrofolato (5-Metil-THF). La segunda actúa en la mitocondria y cataliza la conversión de 5,10-metilen-THF a 10-formil-THF (28). Como se muestra en la figura 22, ambas enzimas presentan una tendencia muy similar. De esta manera, la MTHFD2 (Figura 20A) aumenta su expresión en las gestantes descendientes de madres-fructosa (FC y FF), llegando a ser este efecto significativo en el grupo FF. Además, esta misma tendencia, sin llegar a ser significativa, aparece en la expresión de la MTHFR (Figura 20B). Sorprendentemente, el metabolismo de la síntesis de serina (Figura 17) y el metabolismo de un carbono (Figura 20) muestran unos cambios de expresión similares. De esta manera, ambas rutas están activadas en aquellas descendientes cuyas madres han tomado fructosa durante la gestación. Estos datos refuerzan estudios previos en los cuales se indica la interconexión existente entre ambas rutas y la posible programación fetal asociada al consumo de fructosa (41,42).
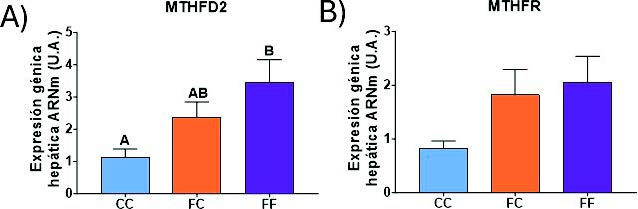
Figura 20. Expresión génica (ARNm) de las enzimas MTHF2 (A), MTHFR (B) en hígados de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada). Las letras diferentes indican diferencias estadísticamente significativas entre hembras tratadas con diferente dieta.
Como se ha explicado anteriormente, la activación de la síntesis de serina y, por consiguiente, del metabolismo de un carbono conduce a la producción de compuestos intermediarios que pueden ser utilizados en la síntesis de macromoléculas (28). De esta manera, la mayor síntesis de serina observada en el presente modelo experimental (Figura 17), unida a la activación del metabolismo de un carbono (Figura 20), puede ser una de las causas que explique que las descendientes de madres-fructosa contengan unos mayores niveles de triglicéridos y colesterol hepáticos (Figura 21).
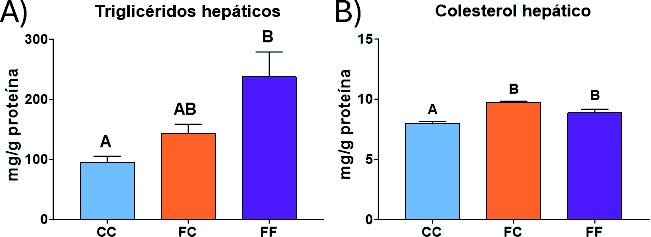
Figura 21. Niveles (mg/g proteína) de triglicéridos hepáticos (A) y colesterol hepático (B) en hígados de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada). Las letras diferentes indican diferencias estadísticamente significativas entre hembras tratadas con diferente dieta (44).
5.2.2. Efecto del consumo de fructosa en el metabolismo de la serina en la placenta de las gestantes de la generación F1 y su transporte de la madre al feto
En primer lugar, se llevó a cabo un estudio de los genes del metabolismo de síntesis de la serina en la placenta, ya que estudios previos han demostrado que dicho órgano es capaz de sintetizar serina (31). En las placentas, observamos que la expresión de las enzimas implicadas en la ruta de síntesis de serina no presentó diferencias significativas. Únicamente cabe destacar que en el grupo FC se observó una tendencia a una mayor expresión de la PHGDH (Figura 22A). Conviene mencionar que no pudo ser medida la expresión de PSAT1, ya que no era detectable por PCR a tiempo real en nuestro modelo experimental.
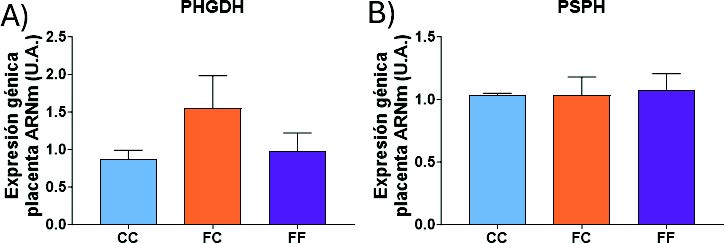
Figura 22. Expresión génica (ARNm) de las enzimas PHGDH (A) y PSPH (B) en placentas de hembras de 8 semanas de edad. Descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
Tampoco se detectaron cambios significativos en la expresión de la SHMT1 ni SHMT2 (Figura 23) en ninguno de los grupos experimentales estudiados.
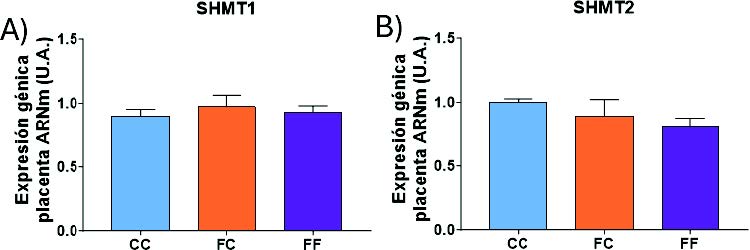
Figura 23. Expresión génica (ARNm) de las enzimas SHMT1 (A), SHMT2 (B), en placentas de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
A continuación, se evaluaron las enzimas implicadas en el metabolismo de un carbono, y observamos en el grupo FF que la expresión de MTHFD2 (Figura 24) tiende a bajar y la de MTHFR disminuye significativamente. Estos resultados podrían ser un efecto compensatorio a la mayor expresión encontrada en los hígados de las gestantes (Figura 20).
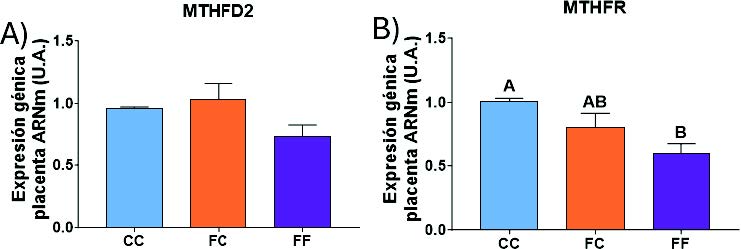
Figura 24. Expresión génica (ARNm) de las enzimas MTHFD2 (A) y MTHFR (B), en placentas de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada). Las letras diferentes indican diferencias estadísticamente significativas entre hembras tratadas con diferente dieta.
Finalmente, y dadas las diferencias observadas en la expresión de las enzimas implicadas en la síntesis de serina en los hígados de las gestantes de la generación F1 (figura 17), nos planteamos estudiar si se producía algún cambio en la expresión de los transportadores placentarios específicos de la serina, ya que este aminoácido es capaz de pasar de la madre al feto (31).
Resultó llamativo que el consumo de fructosa durante la gestación no alteró la expresión de ninguno de los transportadores estudiados (Figura 25). A pesar de estos resultados, dado que se ha comprobado (Figura 17) que la síntesis de serina está más activa en los hígados de gestantes descendientes de madres-fructosa, es posible que los niveles de dicho aminoácido sean mayores en el plasma, provocando un mayor flujo de serina de la madre al feto, sin necesidad de aumentar los niveles de los transportadores. Esta hipótesis estaría en concordancia con los trabajos descritos por diversos autores a través de un modelo computacional de la placenta, donde se ha visto que, aunque la expresión de ciertos transportadores disminuya, un mayor gradiente de concentración materna compensa esta reducción y aumenta el flujo neto de serina hacia el feto (43), lo que iría en consonancia también con los resultados del presente trabajo.
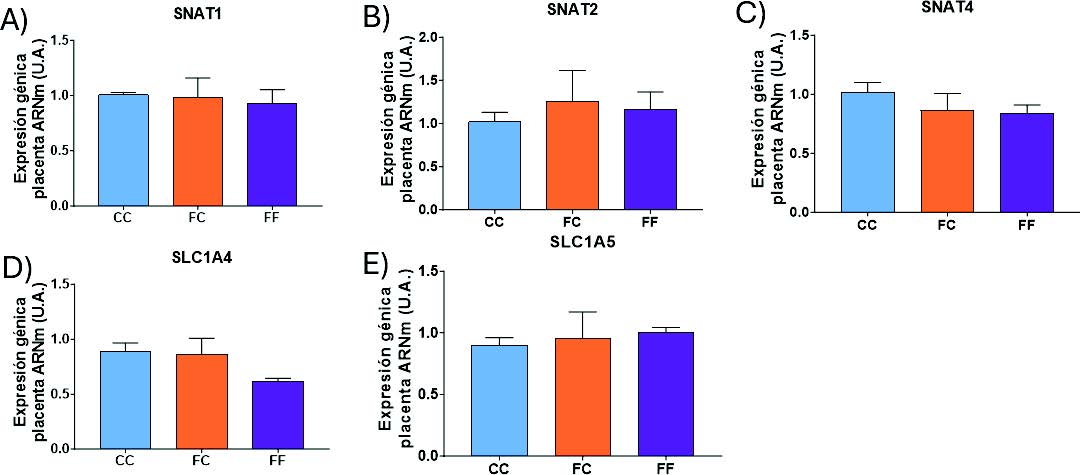
Figura 25. Expresión génica (ARNm) de los transportadores placentarios del sistema A, SNAT1 (A), SNAT2 (B), SNAT4 (C) y del sistema ASC, SLC1A4 (D), SLC1A5 (E) en placentas de hembras de 8 semanas de edad descendientes de madres-control (C) tras consumir agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
5.2.3. Metabolismo de la serina en el hígado de los fetos (futura generación F2) de gestantes F1.
En primer lugar, se estudió la expresión de las enzimas implicadas en el metabolismo de síntesis de serina en los hígados de los fetos. Como se muestra en la figura 26, no se encontraron diferencias significativas en ninguna de las enzimas estudiadas (PHGDH, PSAT1 y PSPH), aunque puede observarse una tendencia a una menor expresión en la PHGDH en el grupo FC (Figura 26A) y en la PSAT1 en el grupo FF (Figura 26B). En consecuencia, en los fetos se produce una tendencia contraria a la observada en sus madres, lo que podría sugerir un mecanismo compensatorio, como se ha descrito anteriormente.
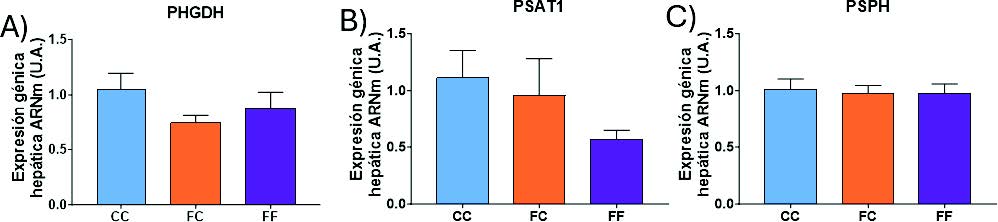
Figura 26. Expresión génica (ARNm) de las enzimas PHGDH(A), PSAT1(B) y PSPH(C) en hígados de fetos de hembras gestantes F1 descendientes de madres-control (C) tras consumir durante 21 días agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
En cuanto a la interconversión de la serina en glicina por la SHMT1 y la SHMT2, de nuevo no aparecen diferencias significativas en ninguno de los grupos estudiados, pero sí hay una tendencia a una menor expresión de la SHMT1 en el grupo FF (Figura 27A) y una tendencia a un aumento de la SHMT2 en el grupo FC (Figura 27B).
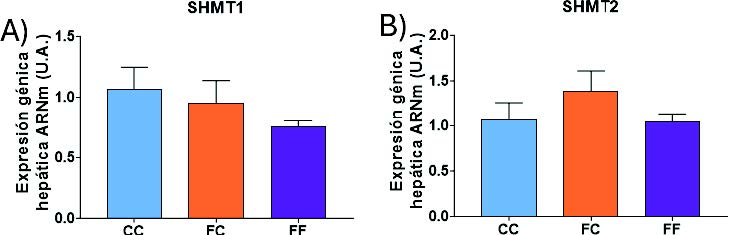
Figura 27. Expresión génica (ARNm) de las enzimas SHMT1(A) y SHMT2(B) en hígados de fetos de hembras gestantes F1 descendientes de madres-control (C) tras consumir durante 21 días agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
Finalmente, en el caso del metabolismo de un carbono, tampoco encontramos diferencias significativas en los grupos de fetos estudiados (Figura 28). No obstante, merece la pena destacar que la expresión de la MTHFR tiende a bajar su expresión en los fetos de gestantes (Figura 28B). Estos resultados observados en el grupo FF son similares a lo que ocurre en las placentas (Figura 24B) y opuestos a lo que se ha visto en los hígados de las gestantes (Figura 19B).
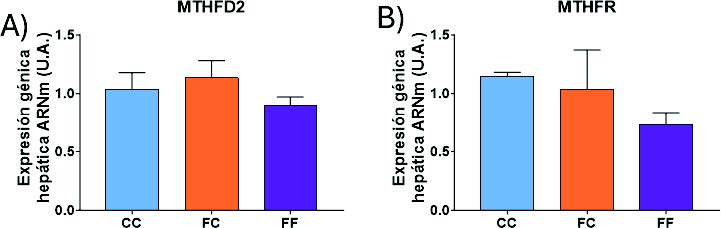
Figura 28. Expresión génica (ARNm) de las enzimas MTHF2(A) y MTHFR(B) en hígados de fetos de hembras gestantes F1 descendientes de madres-control (C) tras consumir durante 21 días agua sin aditivos durante su gestación (CC, barra azul), y hembras descendientes de madres-fructosa (F), tras consumir agua sin aditivo (FC, barra naranja) y agua con un 10% (p/v) de fructosa (FF, barra morada).
Como hemos podido comprobar en los resultados, la expresión génica de los genes del metabolismo de síntesis de serina (Figura 26) y del metabolismo de un carbono (Figura 28) en los hígados de los fetos procedentes de las gestantes F1 tiende a bajar, siguiendo una tendencia opuesta a la de sus madres, las gestantes F1 (Figura 17 y 20), lo que sugiere que los fetos podrían estar intentando compensar ese exceso de serina y compuestos intermediarios producidos por sus madres.
Estos hallazgos concuerdan con los resultados descritos por Rodríguez et al. Las madres que consumieron fructosa durante la gestación mostraron una activación de la lipogénesis hepática, reflejada en el incremento de la expresión de SREBP-1c, FAS y ACC, y una consiguiente acumulación de triglicéridos y colesterol en el hígado. Por el contrario, se observó una tendencia opuesta en los hígados de los fetos. La expresión de SREBP-1c, FAS y ACC disminuyó. Esto sugiere un efecto compensatorio de programación fetal, que frena la síntesis de lípidos y favorece su degradación para evitar la acumulación excesiva de grasa en el tejido fetal (45).
6. CONCLUSIONES
A partir de los resultados expuestos a lo largo del presente trabajo, podemos extraer las siguientes conclusiones:
El consumo de fructosa durante la gestación activa la ruta metabólica de la síntesis de serina y su conversión a glicina, la cual conecta con el metabolismo de un carbono. Todo ello favorecería la producción de precursores para la síntesis de macromoléculas, tales como los triglicéridos.
El consumo materno de fructosa conduce en sus descendientes, una vez gestantes, a la activación de la ruta de síntesis de la serina, así como a la del metabolismo de un carbono. Estos datos evidencian un claro mecanismo de programación fetal.
La reexposición a fructosa durante la gestación conduce en los descendientes de madres-fructosa a una mayor activación de ambas rutas metabólicas.
No se han obtenido resultados concluyentes que demuestren que el consumo de fructosa afecte al transporte de serina a través de la placenta hacia el feto, ya que no se han observado diferencias significativas en la expresión de los transportadores involucrados.
Se observó una tendencia a la reducción en la expresión de los genes de la ruta de síntesis de serina y del metabolismo de un carbono en las placentas y los hígados de los fetos procedentes de las ratas gestantes que a su vez son descendientes de madres-fructosa y fueron suplementadas con fructosa durante su propia gestación. Dicho efecto es opuesto al observado en la expresión de los mismos genes en sus madres, lo que sugiere un efecto compensatorio.
Abreviaturas
3PG: 3-fosfoglicerato
3-PHP: 3-fosfohidroxipiruvato5,10-CH₂-THF: 5,10-metilentetrahidrofolato 5-CH₃-THF: 5-metiltetrahidrofolato
ABCG5: ATP Binding Cassette Subfamily G Member 5
ADNc: ADN complementario
ADN: Ácido desoxirribonucleico
ADP: Adenosín difosfatoa
KG: Alfa-cetoglutarato
AMP: Adenosín monofosfato
AMPD: AMP desaminasa
ANOVA: Análisis unidireccional de la varianza
ARN: Ácido ribonucleico
ARNcirc: ARN circulares
ARNlnc: ARN no codificantes largos
ARNm: ARN mensajero
ARNnc: ARN no codificante
ASC: Transportadores alanina-serina-cisteína
ATP: Adenosín trifosfato
ChREBP: Proteína de unión al elemento respuesta de carbohidratos (Carbohydrate Responsive-Element Binding Protein)
CoA: Coenzima A
Cp: Ciclo umbral
CYP7A1: Colesterol 7-alfa hidroxilasa (citocromo p450 tipo 7 A1)
DNMT: ADN metiltransferasas
DTT: DitiotreitolENT: Enfermedades no transmisibles
HAT: Histonas acetiltransferasas
HDAC: Histonas deacetilasas
HDL: Lipoproteínas de alta densidad
HFCS: Jarabe de maíz rico en fructosa (High-Fructose Corn syrup)
IMP: Inosina monofosfato
KHK: Cetohexoquinasa
LDL: Lipoproteínas de baja densidad
LXRa: Receptor X hepático alfa
miARN: Micro-ácido ribonucleicos
mTOR: Diana de rapamicina en mamíferos (mammalian target of Rapamycin)
MTHFD2: Metilentetrahidrofolato deshidrogenasa 2
MTHFR: Metilentetrahidrofolato reductasa
MVM: Microvellosidades
NAD+: Dinucleótido de nicotinamida y adenina (oxidado)
NADH: Dinucleótido de nicotinamida y adenina (reducido)
NAFLD: Enfermedad del hígado graso no alcohólico
PCR: Reacción en cadena de la polimerasa PHGDH: 3-fosfoglicerato deshidrogenasa
PPARa: Receptor activado por proliferadores de peroxisomas alfa (Peroxisome proliferator-activated receptor alpha)
PSAT1: 3-fosfoserina aminotransferasa
PSPH: Fosfoserina fosfatasa
RCIU: Restricción del crecimiento intrauterino
SAM: S-adenosilmetionina
SDS: Serina deshidratasa
SHMT: Serina hidroximetiltransferasa
SLGT1: Cotransportador de sodio-glucosa 1 (sodium-glucose linked transporter)
SNAT: Transportadores de aminoácidos neutros acoplados a sodio (sodium-coupled neutral amino acid transporters)
SR-B1: Receptor Scavenger clase B tipo 1 (Scavenger receptor class B type 1)
SREBP-1c: Proteína de unión al elemento respuesta de esteroles tipo 1c (Sterol response element binding protein 1c)
THF: Tetrahidrofolato
VLDL: Lipoproteínas de muy baja densidad
4. Referencias
- Agarwal V, Das S, Kapoor N, Prusty B, Das B. Dietary Fructose: A Literature Review of Current Evidence and Implications on Metabolic Health. Cureus. 2022.
- Faruque S, Tong J, Lacmanovic V, Agbonghae C, Minaya DM, Czaja K. The Dose Makes the Poison: Sugar and Obesity in the United States – a Review. Pol J Food Nutr Sci. 2019;69(3):219-233.
- Herman MA, Birnbaum MJ. Molecular Aspects of Fructose Metabolism and Metabolic Disease. Cell Metab. 2020;32(3):383–396.
- Taskinen MR, Packard CJ, Borén J. Dietary Fructose and the Metabolic Syndrome. Nutrients. 2019;11(9):1987.
- Cena H, Calder PC. Defining a Healthy Diet: Evidence for The Role of Contemporary Dietary Patterns in Health and Disease. Nutrients. 2020;12(2):334.
- Merino B, Fernández-Díaz CM, Cózar-Castellano I, Perdomo G. Intestinal Fructose and Glucose Metabolism in Health and Disease. Nutrients. 2019;12(1):94.
- Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. 2018;128(2):545–555.
- Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond). 2005;2(1):5.
- Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–8.
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500.
- Wu YL, Lin ZJ, Li CC, Wu Y, Zhang B, Li C, et al. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduct Target Ther. 2023;8:98.
- Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019;29(5):1028–44.
- Marciniak A, Patro-Małysza J, Kimber-Trojnar Ż, Marciniak B, Oleszczuk J, Leszczyńska-Gorzelak B. Fetal programming of the metabolic syndrome. Taiwan J Obstet Gynecol. 2017;56(2):133–138.
- Rodríguez L, Otero P, Panadero MI, Alvarez-Millán JJ, Nimo-Rojas N, Roglans N, et al. Maternal fructose intake induces insulin resistance and oxidative stress in male, but not female, offspring. J Nutr Metab. 2015;2015:158091.
- Rodrigo S, Fauste E, de la Cuesta M, Panadero MI, Otero P, Rodríguez L, et al. Maternal fructose induces gender-dependent changes in both LXRα promoter methylation and cholesterol metabolism in progeny. J Nutr Biochem. 2018;61:163–172.
- Fisiopatología de la programación fetal y su repercusión en la salud futura (Internet). Ginec Obstet Mex; 2023 (citado 20 jun 2025). Disponible en:(https://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0300-90412023000800588).
- Zhu Z, Cao F, Li X. Epigenetic programming and fetal metabolic programming. Front Endocrinol (Lausanne). 2019;10:764.
- Lim DH, Maher ER. Genomic imprinting syndromes and cancer. Adv Genet. 2010;70:145–75.
- Unger JM, Vaidya R, Hershman DL, Minasian LM, Fleury ME. Systematic Review and Meta-Analysis of the Magnitude of Structural, Clinical, and Physician and Patient Barriers to Cancer Clinical Trial Participation. J Natl Cancer Inst. 2019;111(3):245–255.
- Gujral P, Mahajan V, Lissaman AC, Ponnampalam AP. Histone acetylation and the role of histone deacetylases in normal cyclic endometrium. Reprod Biol Endocrinol. 2020;18(1):84.
- Statello L, Guo CJ, Chen LL, Huarte M. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol. 2021;22(2):96–118.
- Yang K, Zeng L, Ge A, Wang S, Zeng J, Yuan X, et al. A systematic review of the research progress of non-coding RNA in neuroinflammation and immune regulation in cerebral infarction/ischemia-reperfusion injury. Front Immunol. 2022;13:930171.
- Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20(11):675–91.
- Asghar ZA, Thompson A, Chi M, Cusumano A, Scheaffer S, Al-Hammadi N, et al. Maternal fructose drives placental uric acid production leading to adverse fetal outcomes. Sci Rep. 2016;6:25091.
- Rodrigo S, Rodríguez L, Otero P, Panadero MI, García A, Barbas C, et al. Fructose during pregnancy provokes fetal oxidative stress: The key role of the placental heme oxygenase-1. Mol Nutr Food Res. 2016;60(12):2700–11.
- Liu S, Zhang H, Yan B, Zhao H, Wang Y, Gao T, et al. Maternal high-fructose consumption provokes placental oxidative stress resulting in asymmetrical fetal growth restriction in rats. J Clin Biochem Nutr. 2021;69(1):68–76.
- Rodríguez L, Panadero MI, Roglans N, Otero P, Alvarez-Millán JJ, Laguna JC, et al. Fructose during pregnancy affects maternal and fetal leptin signaling. J Nutr Biochem. 2013;24(10):1709–16.
- Lee CM, Hwang Y, Kim M, Park YC, Kim H, Fang S. PHGDH: a novel therapeutic target in cancer. Exp Mol Med. 2024;56:1513–22.
- Holeček M. Serine metabolism in health and disease and as a conditionally essential amino acid. Nutrients. 2022;14(9):1987.
- Li P, Wu X, Huang Y, Qin R, Xiong P, Qiu Y. L-serine metabolic regulation and host respiratory homeostasis. Front Cell Infect Microbiol. 2025;15:1518659.
- Cleal JK, Lewis RM. The mechanisms and regulation of placental amino acid transport to the human foetus. J Neuroendocrinol. 2008;20(4):419–426.
- Glazier JD, Cetin I, Perugino G, Ronzoni S, Grey AM, Mahendran D, et al. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the placenta and birth weight in human pregnancy. J Clin Invest. 1997;100(6):1424–33.
- Jansson T, Powell TL. Role of the placenta in fetal programming: underlying mechanisms and potential interventional approaches. Clin Sci (Lond). 2007;113(1):1–13.
- Huggett J, Dheda K, Bustin S, Zumla A. Real-time RT-PCR normalization; strategies and considerations. Genes Immun. 2005;6(4):279–84.
- Ruijter JM, Ramakers C, Hoogaars WM, Hube F, Smeele K, Vulto I, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37(6):e45.
- Nelson DL, Lehninger AL, Cox MM. Lehninger Principles of Biochemistry. 5th ed. New York: W.H. Freeman; 2008.
- Rivas-Ramírez L, Chirino G, Rivas-Farías A, Palomar-Morales M. El efecto embrioprotector de la glicina en un modelo de embriopatía diabética. Rev Bio Ciencias. 2025;12:e1730.
- Zhou J, Li X, Wang Y, Zhang C, Liu Y, Wang N, et al. High glucose induces PHGDH expression to drive serine synthesis in hepatocellular carcinoma. Cancer Metab. 2022;10:11.
- Xu W, Yang H, Liu Y, He F, Li Z, Wang J, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of $\alpha$-ketoglutarate–dependent dioxygenases. Cancer Cell. 2011;19(1):17–30.
- Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39(4):191-8.
- Meléndez-Hevia E, de Paz-Lugo P. Branch-point stoichiometry can generate weak links in metabolism: the case of glycine biosynthesis. J Biosci. 2008;33:771–780.
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13(8):572-83.
- Sengers BG, Please CP, Lewis RM. Computational modelling of amino acid transfer interactions in the placenta. Placenta. 2010;31(7):556–64.
- Fauste E, Panadero MI, Donis C, Otero P, Bocos C. Pregnancy is enough to provoke deleterious effects in descendants of fructose-fed mothers and their fetuses. Nutrients. 2021;13(10):3667.
- Rodríguez A, Panadero MI, Roglans N, Otero P, Álvarez-Millán JJ, Barroso E, et al. Maternal fructose consumption during gestation alters hepatic lipid metabolism and gene expression in rat fetuses. J Nutr Biochem. 2013;24(6):1057–1066.
